

Abstract
A large body of evidence indicates that mitochondrial dysfunction has a major role in the pathogenesis of multiple cardiovascular disorders. Over the past 2 decades, extraordinary efforts have been focused on the development of agents that specifically target mitochondria for the treatment of cardiovascular disease. Despite such an intensive wave of investigation, no drugs specifically conceived to modulate mitochondrial functions are currently available for the clinical management of cardiovascular disease. In this Review, we discuss the therapeutic potential of targeting mitochondria in patients with cardiovascular disease, examine the obstacles that have restrained the development of mitochondria-targeting agents thus far, and identify strategies that might empower the full clinical potential of this approach.
Mitochondria occupy a central position in the biology of most eukaryotic cells, including all the cells of the cardiovascular system, because mitochondria have a major role in catabolic and anabolic metabolism, regulation of intracellular Ca2+ homeostasis, initiation of inflammatory reactions, and control of multiple pathways culminating in regulated cell death (RCD)1–4. In line with this notion, the mitochondrial network is constantly subjected to a tight qualitycontrol system that segregates dysfunctional mitochondria and delivers them to lysosomes for degradation5,6. Such a mechanism, commonly known as mitophagy, involves not only the core molecular machinery for autophagy7 but also a set of dedicated proteins that are required for the optimal recognition of damaged mitochondria8–10.
A tight control on mitochondrial fitness is paramount for the preservation of cardiovascular homeostasis for at least four reasons11. First, cardiomyocytes heavily rely on fatty aciddriven oxidative phosphorylation for ATP production, at least in physiological settings12. Thus, a decrease in the bioenergetic efficiency of the mitochondrial network can have a direct detrimental effect on the contractile capacity of cardiomyocytes. Second, Ca2+ fluxes are at the core of overall cardiac activity1. Therefore, defects in the capacity of the mitochondrial network (in conjunction with the endoplasmic reticulum) to regulate Ca2+ homeostasis can alter cardiac functions such as electrical conduction. Third, physiological inflammatory homeostasis is particularly important not only for normal cardiac functions13 but also for the preservation of vascular compartments14. Thus, damaged mitochondria accumulating in the cytosol of cardiomyocytes or endothelial cells can drive pathogenic inflammatory responses. Finally, the integrity of the cardiovascular system is crucial for optimal contractile and circulatory functions15. Severe mitochondrial dysfunction and/or the accumulation of permeabilized mitochondria (beyond a thresh old that depends on multiple parameters) can initiate several variants of RCD that culminate in pathological tissue loss (Fig. 1).
Fig. 1 |. Contribution of mitochondrial dysfunction to cardiovascular disease.

In physiological conditions, healthy mitochondria support the functions of virtually all cells from the cardiovascular system by ensuring optimal catabolic and anabolic metabolism and regulating the intracellular trafficking of Ca2+. Additionally, an intact mitochondrial network promotes the preservation of inflammatory homeostasis and tissue integrity by preventing the activation of signal transduction cascades that lead to the release of pro-inflammatory factors and regulated cell death. In addition to being accompanied by metabolic derangements and alterations in intracellular Ca2+ fluxes, mitochondrial dysfunction favours the establishment of an inflammatory milieu and facilitates regulated cell death, which culminates with tissue loss. By efficiently eliminating dysfunctional mitochondria that originate as a consequence of physiological cellular functions or accumulate in the context of pathological cues, mitophagy has a major role in the preservation of cardiovascular homeostasis.
In line with these observations, mitochondrial defects have been involved, at least to some extent, in the pathogenesis of a variety of cardiovascular disorders, including (but not limited to) myocardial infarction (MI), cardiomyopathies of different aetiology, some forms of arrhythmia, hypertension, atherosclerosis, and other vascular conditions16,17. Starting in the late 1990s, the identification of mitochondrial dysfunction as a central aetiological determinant of cardiovascular disease (CVD) drove an intensive wave of preclinical and clinical investigation aimed at the development of novel targeted therapies18. Thus far, the results of such an effort have been disappointing, as no molecules specifically conceived to target mitochondria are currently available for use against CVD in clinical settings19. In this Review, we discuss the rationale for using mitochondria targeting agents (MTAs) in the treatment of CVD, dissect the obstacles that have limited their development over the past 2 decades, and put forward strategies that might unleash the full potential of these promising — but hitherto unrealized — therapeutic tools.
Therapeutic potential of MTAs
Targeting mitochondria from multiple angles has been associated with beneficial effects in a variety of experimental CVD models (TABLES 1,2). However, limited benefits have been documented in clinical trials investigating the safety and efficacy of MTAs for the treatment of CVD, as discussed below.
Table 1 |.
Genetic studies implicating mitochondrial functions in cardiovascular physiology in mice
| Mouse model | Specificity | Phenotype | Refs |
|---|---|---|---|
| Atg5−/− | • Cardiomyocytes • In adults or nonregulated |
Cardiac hypertrophy and contractile dysfunction leading to premature death, accompanied by pronounced mitochondrial defects | 120,121 |
| Bnip3l−/− | •Whole body • Nonregulated |
Cardiac hypertrophy with reduced left ventricular contractile function at 60 weeks of age | 119 |
| Bnip3l−/−Bnip3−/− | • Cardiomyocytes (Bnip3l−/−) and whole body (Bnip3−/−) • Nonregulated |
Cardiac hypertrophy with reduced left ventricular contractile function at 30 weeks of age | 119 |
| Dnm1l−/− | • Cardiomyocytes • In adults |
Lethal dilated cardiomyopathy associated with PARK2 accumulation, which can be partially rescued by deletion of Park2 | 117 |
| DNM1L-C452F | • Whole body • Nonregulated |
Monogenic dilated cardiomyopathy associated with considerable mitophagic defects | 93 |
| Fbxo32−/− | • Whole body • Nonregulated |
Premature death due to cardiac degeneration associated with deficient autophagic responses | 122 |
| Lamp2−/− | • Whole body • Nonregulated |
Vacuolar myopathy affecting cardiac and skeletal muscle, similar to Danon disease | 123 |
| Mfn1−/−Mfn2−/− | • Cardiomyocytes • In adults |
Cardiomyocyte dysfunction associated with lethal dilated cardiomyopathy, attributed to defects in mitochondrial fusion | 90,91 |
| Mfn1−/−Mfn2−/−Dnm1l−/− | • Cardiomyocytes • In adults |
Cardiac hypertrophy associated with accumulation of mitochondria and severely distorted sarcomeric architecture | 91 |
| Mfn2−/− | • Cardiomyocytes • Nonregulated |
Progressive cardiomyopathy leading to premature death, associated with impaired cardiac contractility and insensitivity to β-adrenergic stimulation | 94,95 |
| MFN2-AA | • Cardiomyocytes • At birth |
Perinatal cardiomyopathy leading to premature death owing to a failure in the switch from fetal to adult mitochondria in cardiomyocytes | 97 |
| miR-212–132 cluster overexpression | • Cardiomyocytes • Nonregulated |
Cardiac hypertrophy leading to heart failure and premature death | 296 |
| Park2−/− | • Cardiomyocytes • At birth |
Perinatal cardiomyopathy leading to premature death owing to a failure in the switch from fetal to adult mitochondria in cardiomyocytes | 97 |
| • Cardiomyocytes • In adults |
No obvious phenotype | 117 | |
| Pink1−/− | • Whole body • Nonregulated |
Left ventricular dysfunction and cardiac hypertrophy at 2 months of age | 116 |
| Sirt1−/− | • Whole body • Nonregulated |
Developmental heart defect and perinatal lethality | 59 |
| Sirt1 overexpression | • Cardiomyocytes • Nonregulated |
Attenuated age-associated cardiac hypertrophy (with moderate Sirt1 overexpression) or spontaneous cardiomyopathy (with robust Sirt1 overexpression) | 62 |
| Sirt5−/− | • Whole body • Nonregulated |
Spontaneous hypertrophic cardiomyopathy linked to increased protein succinylation and altered β-oxidation | 66 |
| Sirt6−/− | • Cardiomyocytes • In adults |
Spontaneous cardiac hypertrophy and heart failure | 67 |
| Sirt7−/− | • Whole body • Nonregulated |
Hypertrophy and inflammatory cardiomyopathy characterized by extensive fibrosis and associated with premature death | 68 |
| Slc8b1−/− | • Cardiomyocytes • In adults |
Heart failure associated with left ventricular remodelling | 177 |
| Tfrc−/− | • Cardiomyocytes • Nonregulated |
Cardiac hypertrophy and premature death, accompanied by defects in mitochondrial respiration and ineffective mitophagy | 124 |
| Trp53−/− | • Whole body • Nonregulated |
Decelerated cardiac ageing associated with improved mitophagic responses | 125 |
| Txnrd2−/− | • Cardiomyocytes • Nonregulated |
Fatal dilated cardiomyopathy | 204 |
| • Cardiomyocytes • In adults |
Accelerated cardiac ageing linked with dysregulated autophagy | 205 | |
| Yme1l−/− | • Cardiomyocytes • Nonregulated |
Spontaneous cardiomyopathy associated with mitochondrial hyperfragmentation | 88 |
DNM1L, dynamin-1-like protein; MFN2, mitofusin 2; PARK2, parkin RBR E3 ubiquitin protein ligase.
Table 2 |.
Genetic studies implicating mitochondrial functions in cardiovascular pathology in mice
| Model | Specificity | Phenotype versus wild-type or control mice | Refs | ||||
|---|---|---|---|---|---|---|---|
| Atherosclerosis | |||||||
| Atg5−/− | • Monocytes • Nonregulated |
Accelerated atherosclerosis in mice fed a HFD and in Ldlr−/− mice | 127,128 | ||||
| Il1r1−/− | • Whole body • Nonregulated |
Reduced aortic atherosclerotic plaque areas in Apoe−/− mice fed a HFD | 245 | ||||
| Il1rn−/− | • Whole body • Nonregulated |
Synergized with the Apoe−/− genotype to cause aortic inflammation with destruction of the vascular architecture | 246 | ||||
| Il1rn overexpression | • Whole body • Nonregulated |
Marked protection against atherosclerosis | 246 | ||||
| Parp1−/− | • Whole body • Nonregulated |
Reduced aortic atherosclerotic plaque areas in Apoe−/− mice fed a HFD | 265 | ||||
| Sod2+/− | • Whole body • Nonregulated |
Accelerated progression of atherosclerosis in Apoe−/− mice fed a HFD | 202 | ||||
| Cardiomyopathy | |||||||
| Atg5+/− | • Whole body • Nonregulated |
Aggravated angiotensin-II-induced cardiac hypertrophy | 126 | ||||
| Atg7 overexpression | • Whole body • Nonregulated |
Decreased ventricular dysfunction and cardiac hypertrophy and improved survival in a model of desmin-related cardiomyopathy | 133 | ||||
| Becn1+/− | • Whole body • Nonregulated |
Accelerated heart failure in a model of desmin-related cardiomyopathy | 141 | ||||
| Lclat1−/− | • Whole body • Nonregulated |
Mitigated hypertrophic cardiomyopathy induced by thyroid hyperstimulation associated with improved mitophagic flux | 136 | ||||
| Ppif−/− | • Whole body • Nonregulated |
Protection against angiotensin-II-induced cardiac hypertrophy | 261 | ||||
| Sirt2−/− | • Whole body • Nonregulated |
Aggravated angiotensin-II-induced cardiac hypertrophy | 65 | ||||
| Sirt2 overexpression | •Cardiomyocytes • Nonregulated |
Mitigated angiotensin-II-induced cardiac hypertrophy | 65 | ||||
| Sirt3−/− | • Whole body • Nonregulated |
Aggravated angiotensin-II-induced cardiac hypertrophy | 63 | ||||
| Sirt3 overexpression | • Cardiomyocytes • Nonregulated |
Mitigated angiotensin-II-induced cardiac hypertrophy | 63 | ||||
| Sirt4−/− | • Whole body • Nonregulated |
Limited angiotensin-II-induced cardiac hypertrophy | 69 | ||||
| Sirt4 overexpression | • Cardiomyocytes • In adults |
Aggravated angiotensin-II-induced cardiac hypertrophy | 69 | ||||
| Cardiotoxicity | |||||||
| Becn1+/− | • Whole body • Nonregulated |
Reduced pathological cardiac remodelling after chronic doxorubicin administration | 144 | ||||
| • Cardiomyocytes • Nonregulated |
Accelerated decline in ventricular systolic function after chronic doxorubicin administration | 144 | |||||
| Ripk3−/− | • Whole body • Nonregulated |
Protected against doxorubicin-driven heart failure, coupled with impaired CaMKII activation and MPT desensitization | 266 | ||||
| Trp53−/− | • Whole body • Nonregulated |
Reduced sensitivity to doxorubicin, might be mediated by reduced mitophagic responses | 125 | ||||
| Myocardial infarction | |||||||
| Bcl2 overexpression | • Cardiomyocytes • Nonregulated |
Reduced infarct size after I/R injury | 258 | ||||
| Becn1+/− | • Whole body • Nonregulated |
Reduced cardiac damage at reperfusion | 143 | ||||
| Cgas−/− | • Whole body • Nonregulated |
Improved survival after I/R injury, coupled with diminished pathological remodelling, enhanced angiogenesis, and preserved ventricular contractile function | 240 | ||||
| • Whole body • Nonregulated |
Attenuated decline in cardiac function coupled with decreased production of inflammatory cytokines and chemokines and decreased inflammatory cell infiltration into the myocardium after left coronary artery ligation | 242 | |||||
| Myocardial infarction (cont.) | |||||||
| Dnm1l+/− | • Cardiomyocytes • Nonregulated |
Exacerbated heart failure associated with defective mitophagy and mitochondrial dysfunction after transverse aortic constriction | 99 | ||||
| • Cardiomyocytes • In adults |
Impaired autophagy and reduced left ventricular function after I/R injury | 100 | |||||
| Ifnar1−/− | • Whole body • Nonregulated |
Cardioprotective phenotype resembling that caused by the Cgas−/− genotype | 242 | ||||
| Irf3−/− | •Whole body • Nonregulated |
Cardioprotective phenotype resembling that caused by the Cgas−/− genotype | 242 | ||||
| Mcu−/− | • Cardiomyocytes • In adults |
Protected against Ca2+ overload-driven MPT, decreased infarct size, and preserved cardiac function | 174,175 | ||||
| • Whole body • Nonregulated |
Abolished sensitivity to cyclosporine A after I/R injury, with minimal effects on cardiac function | 173 | |||||
| MCU DN | • Cardiomyocytes • Nonregulated |
Preserved Δψm and limited ROS generation after I/R injury, but did not mediate overt cardioprotection | 173,176 | ||||
| Mfn1−/−Mfn2−/− | • Cardiomyocytes • In adults |
Reduced infarct size along with a decrease in mitochondrial Ca2+ overload and ROS generation | 92 | ||||
| miR-150−/− | • Whole body • Nonregulated |
Cardioprotection associated with reduced expression of genes associated with RCD and inflammation | 300 | ||||
| Slc8b1 overexpression | • Cardiomyocytes • In adults |
Reduced sensitivity to heart failure after I/R injury, at least partially dependent on reduced propensity to MPT | 177 | ||||
| Opa1+/− | • Whole body • Nonregulated |
Increased cardiac hypertrophy after transverse aortic constriction, associated with altered ejection fraction | 87 | ||||
| Parp1−/− | • Whole body • Nonregulated |
Decreased myocardial damage linked to reduced NF-κB signalling and general protection against RCD | 264 | ||||
| Pgam5−/− | • Whole body • Nonregulated |
Increased infarct size, correlating with inhibition of mitophagy and necrotic RCD | 139 | ||||
| Ppif−/− | • Whole body • Nonregulated |
Reduced sensitivity to I/R injury, mechanistically associated with reduced propensity to MPT-driven regulated necrosis | 259,260 | ||||
| Rheb overexpression | • Cardiomyocytes • Nonregulated |
Increased infarct size, which could be reversed by systemic administration of Rapamycin | 129 | ||||
| Ripk3−/− | • Whole body • Nonregulated |
Protected against heart failure after I/R injury, coupled with impaired CaMKII activation and MPT desensitization | 266 | ||||
| Sirt1+/− | • Whole body • Nonregulated |
Impaired IPC associated with hyperacetylation of cytoplasmic proteins and consequent autophagy inhibition | 60,62 | ||||
| Sirt1 overexpression | • Whole body • Nonregulated |
Cardioprotection associated with deacetylation of cytoplasmic proteins and consequent autophagy activation | 60,62 | ||||
| Sirt3−/− | • Whole body • Nonregulated |
Aggravated cardiac hypertrophy induced by transverse aortic constriction, potentially linked to MPT sensitization | 64 | ||||
| Sirt6 overexpression | • Cardiomyocytes • Nonregulated |
Inhibited cardiac hypertrophy induced by transverse aortic constriction, potentially linked to MPT desensitization | 67 | ||||
| Stk4−/− | • Whole body • Nonregulated |
Cardioprotection coupled to increased autophagic responses in the heart | 135 | ||||
| Pressure overload | |||||||
| Atg5−/− | • Cardiomyocytes • Nonregulated |
Increased sensitivity to pressure overload | 120 | ||||
| Becn1+/− | • Whole body • Nonregulated |
Reduced pathological cardiac remodelling | 142 | ||||
| Becn1 overexpression | • Whole body • Nonregulated |
Aggravated pathological cardiac remodelling | 142 | ||||
| Bnip3l−/−Bnip3−/− | • Cardiomyocytes (Bnip3l−/−) and whole body (Bnip3−/−) • Nonregulated |
Rapid functional cardiac decompensation | 119 | ||||
| Camk2a−/− | • Cardiomyocytes • Nonregulated |
Reduced ROS generation coupled with RCD inhibition and preserved systolic Function | 182 | ||||
| Dnase2a−/− | • Cardiomyocytes • Nonregulated |
Severe myocarditis and dilated cardiomyopathy associated with premature death | 131 | ||||
Δψm, mitochondrial transmembrane potential; CaMKII, calcium/calmodulin-dependent protein kinase II; DN, dominant-negative; HFD, high-fat diet; IPC, ischaemic preconditioning; I/R , ischaemia–reperfusion; MCU, calcium uniporter protein, mitochondrial; MPT, mitochondrial permeability transition; NF-κB, nuclear factor-κB; RCD, regulated cell death; ROS, reactive oxygen species; SIRT3, sirtuin 3.
Mitochondrial metabolism.
Healthy cardiomyocytes satisfy their elevated energy needs by catabolizing fatty acids (via β-oxidation), branchedchain amino acids, and, to a lesser extent, ketone bodies (via ketolysis) to fuel the tricarboxylic acid (TCA) cycle and drive ATP production via the mitochondrial respiratory chain (box 1). By comparison, pyruvate derived from glycolysis contributes minimally to ATP synthesis in the healthy heart11. Such a predominantly mitochondrial metabolic profile shifts in the course of numerous cardiac pathologies. Heart failure (HF) is accompanied by a gradual decline in the bioenergetic reserve capacity of the myocardium, which — beyond a specific threshold — can no longer be compensated for by endogenous mechanisms20. In multiple variants of cardiomyopathy culminating with HF, cardiomyocytes undergo metabolic reprogramming involving decreased βoxidation and branchedchain amino acid metabolism coupled with intracellular lipid deposition and increased glucose utilization21–24. The TCA cycle intermediate succinate accumulates in the ischaemic myocardium, and such an accumulation is mechanistically linked to oxidative damage at reperfusion25 (see below). Along similar lines, TCA cycle activity is impaired 6 weeks after MI26, potentially representing an early maladaptive phase of the surviving tissue.
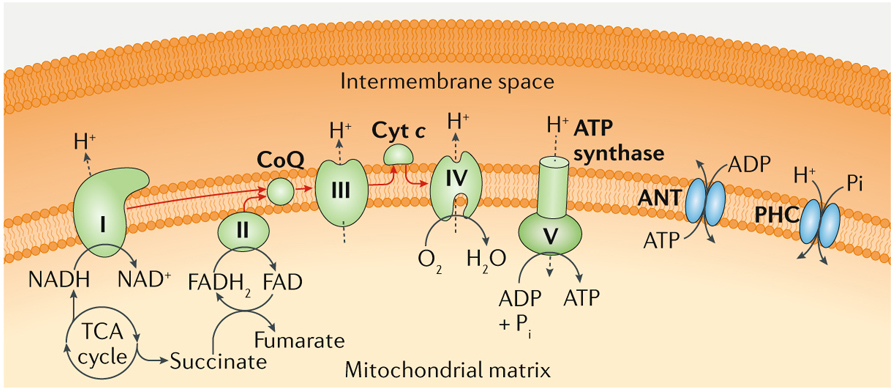
Box 1 |. Principles of oxidative phosphorylation.
Oxidative phosphorylation is a core bioenergetic process whereby reducing equivalents present in the mitochondrial matrix are sequentially used by four multiprotein complexes (generally referred to as respiratory complexes I–IV) and two electron shuttles (namely, coenzyme Q (CoQ) and cytochrome c (Cyt c)) to generate an electrochemical H+ gradient across the inner mitochondrial membrane that is harnessed in a controlled manner by the F1Fo ATP synthase (also known as respiratory complex V) to catalyse the phosphorylation of ADP into ATP. The main substrates for oxidative phosphorylation are NADH, which provides electrons to complex I (also known as NADH dehydrogenase), and succinate, which provides electrons to complex II (also known as succinate dehydrogenase) via FADH . Accordingly, FADH can also fuel oxidative phosphorylation at the level of complex II. Both complex I and II deliver electrons to complex III (also known as CoQ:Cyt c oxidoreductase) via CoQ. However, only complex I transfers electrons onto complex III while also extruding H+ ions from the mitochondrial matrix to the intermembrane space. Complex III transfers electrons to complex IV (also known as Cyt c oxidase) via Cyt c, culminating with the reduction of O2 into H2O. This last step is the reason why O2 is critical for oxidative phosphorylation. Both complex III and complex IV directly contribute to the generation of the mitochondrial transmembrane potential (Δψm). Finally, the F1Fo ATP synthase uses a well-described rotatory mechanism to dissipate the Δψm in a controlled manner, coupled with phosphorylation of ADP into ATP. This reaction requires ADP and inorganic phosphate (Pi), which are provided by the permeability transition pore components adenine nucleotide translocator (ANT) and phosphate carrier (PHC; also known as SLC25A3), respectively (see the figure; please note that stoichiometry is not respected for the sake of simplification). Importantly, the reaction catalysed by the F1Fo ATP synthase is reversible. This reversibility implies that in ischaemic conditions the capacity of oxidative phosphorylation to drive ATP synthesis is impaired, owing to limited oxygen availability, and that high amounts of ATP are consumed by the F1Fo ATP synthase to preserve the Δψm. All metabolic intermediates entering the tricarboxylic acid (TCA) cycle, including (but not limited to) glucose-derived pyruvate and branched-chain amino acid-derived and fatty acid-derived acetyl-CoA and succinyl-CoA, can drive the synthesis of NADH and succinate in the mitochondrial matrix, thereby supporting oxidative phosphorylation. Fatty acid oxidation also supports oxidative phosphorylation via FADH2 synthesis. Of note, the cellular efficiency of oxidative phosphorylation depends on a variety of parameters, including the number of mitochondria per cell and their fragmentation state, the amount of respiratory complexes per mitochondrion, the supramolecular organization of respiratory complexes, substrate and O2 availability, the expression of endogenous inhibitors, and local redox and pH conditions339,340.
The molecular mechanisms underlying metabolic reprogramming in the diseased myocardium remain to be fully elucidated, although a role for specific transcription factors has been postulated. For instance, nuclear receptor subfamily 2, group F, member 2 (NR2F2; also known as COUPTF2) is upregulated in patients with HF, and transgenedriven Nr2f2 over expression in mice favours dilated cardiomyopathy accompanied by pathological metabolic remodelling27. Similarly, hypoxiainducible factor 1α (HIF1α) initiates a transcriptional programme involving peroxisome proliferatoractivated receptorγ (PPARγ) that leads to increased glucose uptake and consequent lipid accumulation, apoptotic cell death, and contractile dysfunction21. Corroborating an aetiological role for this transcriptional module, ventricularspecific deletion of Hif1a prevents pressureoverloadinduced cardiomyopathy in mice21.
Additional metabolic functions ensured (at least in part) by mitochondria are relevant for CVD, including the folate cycle. An efficient folate cycle is indeed required for the optimal conversion of homocysteine into methionine, and defects in this pathway, including genetic variants in MTHFR (which encodes methyl enetetrahydrofolate reductase) are associated with an increased incidence of vascular disorders (such as thrombosis and atherosclerosis) secondary to, or at least paralleled by, homocysteine accumulation28. Of note, several mutations in mitochondrial or nuclear genes coding for components of the mitochondrial respiratory chain have been associated with familial cardiomyopathies in humans29. Moreover, experimental interventions inducing respiratory defects in myocardial cells, such as the tissuespecific deletion of Aifm1 (which encodes apoptosis inducing factor mitochondria associated 1)30 or Tfam (which encodes mitochondrial transcription factor A; TFAM)31, result in spontaneous, earlyonset cardiomyopathy. Taken together, these observations exemplify the involvement of mitochondrial metabolic dysfunction in CVD.
Early clinical trials testing lcarnitine supplementation, which (among other effects) favours the mitochondrial uptake of cytosolic fatty acids, in patients recovering from acute MI documented some degree of efficacy in reducing the incidence or severity of HF, left ventricular enlargement, arrhythmias, and cardiac death32,33. However, subsequent studies did not conclusively confirm these observations34,35. Moreover, oral lcarnitine can be metabolized by the gut microbiota into trimethylamine Noxide (TMAO), a proatherogenic molecule36. Accordingly, individuals with high lcarnitine levels and concurrently high TMAO levels in the blood are at increased risk of CVD and major adverse cardiac events36. Thus, the clinical development of lcarnitine for the treatment of CVD seems to be at an impasse.
The βoxidation inhibitor etomoxir has also been investigated in patients with congestive HF, with inconclusive results37,38. Conversely, perhexiline and trimetazidine — which resemble etomoxir in their capacity to inhibit βoxidation (although to different degrees) — are currently approved in multiple countries (including Australia and Canada) as antianginal agents39. The therapeutic efficacy of perhexiline and trimetazidine has been proposed not to reflect a switch from fatty aciddriven to glucosedriven catabolism40 but instead to entail an entire rebalancing of carbon and nucleotide phosphate fluxes41 linked to autophagy activation42 (see below). Perhexiline is also effective (at least to some extent) in a subset of patients with cardiomyopathy40,43, but not in patients with left ventricular hypertrophy undergoing cardiac surgery44,45. Trimetazidine has been tested in multiple cohorts of patients with distinct cardiovascular disorders beyond angina, with variable degrees of efficacy46–49. Nonetheless, in the USA (but not in other countries), the clinical development of perhexiline and trimetazidine has been discontinued, presumably owing to a fairly narrow therapeutic index39.
5Aminoimidazole4carboxamide ribonucleotide (AICAR; also known as acadesine) is an intermediate in the synthesis of inosine monophosphate that potently activates 5′AMPactivated protein kinase (AMPK), a metabolic sensor regulating mitochondrial biogenesis, dynamics, and metabolism50. Despite some promising preliminary results51,52, the clinical development of acadesine as a cardioprotective intervention in patients undergoing CABG surgery has been abandoned, at least in part owing to the lack of longterm efficacy53. In summary, despite a robust rationale to target mitochondrial metabolism for the prevention or treatment of CVD, this therapeutic strategy remains largely unrealized.
Sirtuins.
Sirtuins are a family of NAD+dependent deacetylases and deacylases that control multiple aspects of cellular metabolism, including mitochondrial function and redox balance54. The mammalian genome encodes seven different sirtuins, three of which (SIRT3, SIRT4, and SIRT5) are localized to mitochondria54. Pharmacological sirtuin activation mediates lifespanextending functions in multiple experimental models55–57, and defects in both mitochondrial and extramitochondrial sirtuins have been associated with a variety of cardiovascular disorders58. Sirt1−/− mice are viable but have considerable developmental heart defects59. In Sirt1+/− hearts, ischaemic preconditioning does not preserve cardiac function after ischaemia– reperfusion injury, potentially linked to hyperacetylation of cytosolic proteins and consequent inhibition of autophagy60,61, whereas myocardial Sirt1 overexpression has cardioprotective effects along with deacetylation of cytoplasmic proteins60,62. Sirt3−/− mice show signs of cardiac hypertrophy and interstitial fibrosis at 8 weeks of age, spontaneously develop agerelated cardiomyopathy, and are more sensitive than their wildtype littermates to hypertrophic stimuli, including aortic constriction63,64. Such a susceptibility to cardiac hypertrophy reflects, at least in part, an increased propensity of the Sirt3−/− myocardium to undergo regulated necrosis upon mitochondrial permeability transition (MPT) as a consequence of cyclophilin D (CypD; also known as PPIF) hyperacetylation63,64 (see below). Conversely, transgenic Sirt3 overexpression has robust cardioprotective effects in mice63. Similar results to those observed in Sirt3−/− mice have been obtained with Sirt2−/−, Sirt5−/−, Sirt6−/−, and Sirt7−/− mice, and as shown with Sirt3 overexpression, overexpression of Sirt2 specifically in the myocardium had cardioprotective effects65–68. By contrast, Sirt4−/− mice seem to be less susceptible to angiotensinIIinduced cardiac hypertrophy than their wildtype counterparts, whereas cardiomyocytespecific overexpression of Sirt4 reportedly mediates detrimental effects in this model69. However, these findings have not yet been confirmed. At least in part, the cardioprotective effects of sirtuin activation originate from an antioxidant transcriptional programme orchestrated by forkhead box protein O3A (FOXO3A; also known as FOXO3)63, proficient autophagic responses70, and potentially the inhibition of MPTdriven regulated necrosis64,71 (see below). Thus, sirtuins support cardiac fitness by affecting mitochondrial functions.
Sirtuins are activated by caloric restriction, which is also a potent inducer of autophagy, and a vast amount of literature is available on the multipronged beneficial effects of caloric restriction on cardiovascular health in humans, at least part of which are thought to depend mechanistically on sirtuins72. Additional sirtuin activators include the rather nonspecific natural polyphenols butein, honokiol, piceatannol, quercetin, and resveratrol73,74 as well as several synthetic sirtuinactivating compounds, including SRT1720, SRT2104, and SRT3025 (REF.57). All these molecules have been shown to mediate beneficial effects in rodent models of CVD, and both SRT1720 and SRT2104 extend mouse lifespan74–77. Similarly, dietary supplementation with nicotinamide mononucleotide (NMN; a precursor of NAD+) mediates potent cardioprotective effects in mouse models of cardiomyopathy and ischaemia–reperfusion injury via a SIRT1dependent or SIRT3dependent mechanism78–80. The capacity of dietary resveratrol to limit the incidence or severity of various cardiovascular disorders (mostly in the context of type 2 diabetes mellitus) has been investigated in multiple clinical trials81–83, with inconclusive findings (often due to problematic study design). Still, no fewer than 20 nonclosed (status: not terminated, suspended, or withdrawn) clinical trials are currently registered at clinicaltrials.gov to investigate dietary supplementation with resveratrol in individuals with ageassociated morbidities (mostly type 2 diabetes) and cardiovascular conditions including nonischaemic cardiomyopathy (NCT01914081), hypertension (NCT01842399), atherosclerosis (NCT02998918), and endothelial dysfunction (NCT02256540). Results from a small randomized clinical trial including 40 patients with psoriasis (NCT01154101) suggest that SRT2104 is well tolerated84. The safety of SRT3025 has been investigated in healthy volunteers (NCT01340911), but to the best of our knowledge the results of this study have not been disseminated. Finally, the effects of dietary NMN supplementation on cardiometabolic functions are currently being formally investigated (NCT03151239). Taken together, these observations suggest that, although multiple dietary interventions that activate sirtuins, including caloric restriction, resveratrol, and NMN (both ofwhich are available over the counter), might mediate robust cardioprotective effects, additional clinical testing is required for the establishment of official treatment protocols enabling the use of these agents for the treatment of CVD.
Mitochondrial dynamics.
The mitochondrial network constantly undergoes remodelling owing to the mutually antagonistic activity of multiple proteins that promote fission, such as mitochondrial fission factor (MFF), mitochondrial fission 1 protein (FIS1), and dynamin1 like protein (DNM1L), and fusion, such as mitofusin 1 (MFN1), MFN2, and optic atrophy protein 1 (OPA1)85 (Fig. 2). This process is paramount for the preservation of optimal mitochondrial functions in both physiological and pathological conditions, at least in part because fission enables the mitophagic disposal of dysfunctional mitochondria86. Accordingly, multiple genetic defects impairing mitochondrial dynamics have been linked to CVD in experimental models.
Fig. 2 |. overview of mitochondrial dynamics.

The mitochondrial network is constantly reshaped by the antagonistic activity of proteins that mediate fission, such as mitochondrial fission factor (MFF), mitochondrial fission 1 protein (FIS1), and dynamin 1-like protein (DNM1L), and proteins that promote fusion, such as mitofusin 1 (MFN1), MFN2, and optic atrophy protein 1 (OPA1). One of the essential roles of fission is to segregate dysfunctional mitochondria, thereby enabling their uptake by the autophagic machinery and consequent degradation in lysosomes. PARK2, parkin RBR E3 ubiquitin protein ligase; PINK1, PTEN-induced putative kinase protein 1.
The myocardium of Opa1+/− mice has clustered mitochondria with disorganized cristae and reduced mitochondrial DNA (mtDNA) content, and Opa1+/− mice are more susceptible to cardiac hypertrophy induced by transverse aortic constriction than their wildtype counterparts87. Cardiomyocytespecific deletion of Yme1l1 accelerates cardiac OPA1 proteolysis, thereby favouring mitochondrial hyperfragmentation and metabolic impairment, leading to HF88. Interestingly, angiotensinIIinduced cardiomyopathy leads to OPA1 acetylation and consequent mitochondrial fragmentation, a detrimental process that is inhibited by SIRT3 (REF.89). The codeletion of Mfn1 and Mfn2 from adult cardiomyocytes imposes a robust defect in mitochondrial fusion that drives cardiac dysfunction associated with rapidly progressive (and ultimately lethal) dilated cardiomyopathy90. Such a detrimental phenotype cannot be fully rescued by the concomitant deletion of Dnm1l, but the cardiomyopathy manifesting in Mfn1−/−Mfn2−/−Dnm1l−/− hearts progresses with different kinetics than in Mfn1−/−Mfn2−/− hearts and mostly reflects a mitophagic blockage91. However, Mfn1−/−Mfn2−/− hearts have reduced sensitivity to ischaemia–reperfusion injury compared with their wildtype counterparts, potentially as a consequence of mitigated Ca2+ overload92 (see below).
Transgenic expression of DNM1LC452F (a hyperactive DNM1L variant) also drives dilated cardiomyopathy accompanied by a considerable mitophagic defect93. Similarly, mouse Mfn2−/− hearts spontaneously develop dilated cardiomyopathy accompanied by mitochondrial hyperfragmentation, impaired contractile performance, and insensitivity to βadrenergic stimulation94,95. Further corroborating the importance of mitochondrial fusion for the preservation of cardiovascular homeostasis, adenovirusmediated delivery of Mfn2 to the mouse myocardium inhibits angiotensinII induced cardiomyopathy96. Interestingly, transgenedriven overexpression of a nonphosphorylatable MFN2 variant (MFN2AA) in the myocardium of newborn (but not adult) mice prevents normal mitochondrial maturation, accompanied by a switch from glucosedriven to fatty aciddriven metabolism, and leads to premature lethality, most probably as a consequence of impaired mitophagy97 (see below). Of note, physiological DNM1Ldependent mitochondrial fragmentation is critical for cardiac adaptation to increased energy demands98. Moreover, conditional deletion of one copy of Dnm1l from the myocardium exacerbates pressureoverloadinduced cardiomyopathy as well as ischaemia–reperfusion injury in mice as a consequence of mitophagy impairment99,100. Altogether, these observations suggest that a balanced interplay between fission and fusion is paramount for cardiovascular health as it preserves mitochondrial fitness in both physiological and pathological conditions. Further corroborating this notion, the levels of various factors involved in the regulation of mitochondrial dynamics, including FIS1, MFN2, and OPA1, are altered in the course of CVD101–103. Of note, MFN2 is also aetiologically involved in the proliferative arrest and death of vascular smooth muscle cells elicited by oxidative stress in rats104. In line with this notion, transgenedriven Mfn2 overexpression reportedly prevents vascular smooth muscle cell proliferation and restenosis in rat models of arterial injury induced by balloon denudation of the left common carotid artery105. However, these effects seem to be independent of the role of MFN2 in the regulation of mitochondrial dynamics104.
The chemical DNM1L inhibitor mdivi1 mediates cardioprotective effects in rodent models of cardiac ischaemia–reperfusion injury106–108 and cardiomyopathy109,110, but the specificity of mdivi1 has been questioned111. Nonetheless, similar observations have been made with other DNM1L inhibitors such as P110 (REFs112,113) and dynasore114. A cellpermeant peptide enabling MFN2dependent mitochondrial fusion has also been developed115, but its biological activity in the cardiovascular system remains to be investigated. To the best of our knowledge, none of these agents has been tested in clinical settings thus far.
Mitophagy.
Mitophagy constitutes a pillar in the maintenance of mitochondrial homeostasis in both the healthy and diseased cardiovascular system5,6. Accordingly, multiple defects in the molecular apparatus underlying proficient mitophagic responses have been associated with spontaneous CVD in experimental models17. Pink1−/− mice (lacking a kinase involved in the recognition of depolarized mitochondria) develop left ventricular dysfunction and cardiac hypertrophy by 2 months of age116. Deletion of Park2 (also known as Prkn; encoding parkin RBR E3 ubiquitin protein ligase, a functional mitochondrial interactor of serine/threonine protein kinase PINK1, which is required for multiple variants of mitophagy) from the myocardium of adult mice causes a very mild cardiac phenotype in unstressed animals117. Conversely, Park2 ablation from the myocardium of neonate mice causes premature and rapidly lethal cardiomyopathy associated with failed mitochondria maturation (strikingly similar to the phenotype associated with MFN2AA expression)97. Similarly, knockout of park (the fly orthologue of Park2) in Drosophila melanogaster causes dilated cardiomyopathy that can be rescued by cardiomyocytespecific reexpression of park95,118. Bnip3l−/− mice lack a core component of the molecular apparatus for mitophagy and spontaneously develop cardiomegaly and contractile depression by 60 weeks of age, a pathological phenotype that is further accelerated by the concomitant deletion of Bnip3 (coding for yet another protein involved in mitophagy)119. Genetic defects affecting autophagy also compromise cardiovascular homeostasis owing to the accumulation of dysfunctional mitochondria. This observation holds true for: cardiomyocytespecific deletion of Atg5 in adult mice, which causes lethal cardiac hypertrophy accompanied by disorganized sarcomere structure as well as mitochondrial misalignment and aggregation120,121; wholebody deletion of Fbxo32 in mice, which is associated with premature death owing to cardiac degeneration associated with deficient autophagic responses122; and the Lamp2−/− genotype, which causes a major lysosomal dysfunction that, in mice, drives a vacuolar myopathy that affects cardiac and skeletal muscles, resembling Danon disease123. Of note, multiple genetic and pharmacological interventions that impair mitochondrial dynamics impose at least some degree of mitophagic incompetence86. These two processes are so intimately interconnected that mechanistically ascribing the phenotype to either of the alterations is difficult. Additional genetic alterations that trigger CVD in rodents, such as cardiac deletion of Tfrc (coding for the transferrin receptor)124, are associated with mitophagic defects. Moreover, genetic defects that improve mitophagic proficiency, such as wholebody absence of Trp53 (also known as Tp53; coding for a master regulator of cellular biology that inhibits autophagy in physiological settings), decelerate spontaneous cardiac ageing125. Taken together, these observations exemplify the critical role of mitophagy in the preservation of physiological cardiovascular homeostasis. That said, Park2 deletion seems to rescue, at least in part, the lethal phenotype of Dnm1l deletion in the adult myocardium117, suggesting a role for uncontrolled mitophagy in the detrimental phenotype imposed by defects in mitochondrial fission (see above).
Multiple genetic defects impairing mitophagic proficiency aggravate disease severity in experimental models of CVD17. Bnip3l−/−Bnip3−/− hearts are highly sensitive to decompensation induced by pressure overload119. Homozygous or heterozygous deletion of Atg5 from the mouse myocardium exacerbates cardiomyopathy driven by pressure overload120 and angiotensin II administration126. Similarly, mice bearing Atg5−/− monocytes are more susceptible to develop atherosclerotic lesions in response to a highfat diet or Ldlr deletion than mice with wildtype monocytes127,128. Mice engineered to overexpress Rheb, which encodes the endogenous autophagy inhibitor RAS homologue enriched in brain (RHEB), in the myocardium are more susceptible to cardiac ischaemia–reperfusion injury than wildtype mice, a detrimental phenotype that can be partially rescued by administration of the pharmacological autophagy activator rapamycin129,130. Dnase2a−/− mice, which lack a lysosomal nuclease (deoxyribonuclease 2α) that is involved in the autophagic degradation of mtDNA released upon mitochondrial damage, are extremely sensitive to pressureoverloadinduced cardiomyopathy, at least in part owing to exaggerated inflammatory responses in the myocardium131 (see below). Interestingly, cathelicidin antimicrobial peptide (CAMP) can bind mtDNA to limit its degradation by DNase 2α (DNASE2α), which has been associated with exacerbated atherosclerosis in Apoe−/− mice132.
Whole body overexpression of Atg7 (encoding a core component of the autophagic machinery) restrains cardiac hypertrophy and extends survival in a mouse model of desminrelated cardiomyopathy133. The Trp53−/− genotype limits both ischaemia–reperfusion injury and doxorubicin cardiotoxicity in mice, potentially owing to reduced myocardial susceptibility to RCD (see RCD section below), and improved mitophagy125,134. Multiple other genetic alterations that mediate beneficial effects in experimental models of CVD are associated with superior mitophagic responses (although precise mechanistic links are missing), including the Stk4−/− genotype, which limits cardiac ischaemia–reperfusion injury135, and the wholebody deletion of Lclat1, which mitigates hypertrophic cardiomyopathy induced by thyroid hyperstimulation136. Moreover, multiple cardioprotective interventions including hypothermia and the administration of glucagonlike peptide 1 receptor (GLP1R) agonists have been shown to promote autophagy (at least in some cell types), correlating with reduced amounts of RCD137,138. Conversely, Pgam5−/− mice are more susceptible to cardiac ischaemia–reperfusion injury than their wildtype littermates along with a wholebody defect in mitophagy, potentially linked to the capacity of phosphoglycerate mutase family member 5 (PGAM5) to regulate DNM1L dependent fission139. Similarly, mice with an endothelial cellspecific deletion of Pdcd10 spontaneously develop a syndrome resembling cerebral cavernous malformations, accompanied by robust autophagic defects140. Thus, the optimal elimination of damaged mitochondria by mitophagy is fundamental for the cardiovascular system to control potentially pathogenic challenges.
Interestingly, the role of beclin 1 (BECN1), a core component of the autophagic machinery that participates in multiple instances of mitophagy7, in the preservation of cardiovascular homeostasis in pathological settings is rather controversial. Indeed, whereas BECN1 has been attributed a cardioprotective role in some models of CVD99,141, Becn1+/− rodents consistently exhibited low sensitivity to potentially cardiotoxic challenges142–144. Although the reasons for this apparent discrepancy remain to be formally elucidated, linking them to emerging autophagyindependent functions of BECN1 in RCD regulation is tempting145. Further corroborating the critical role of mitophagy in cardiovascular homeostasis, ischaemic preconditioning has been associated with the translocation of PARK2 to depolarized mitochondria and consequent initiation of their autophagic disposal146. Moreover, the expression levels of components of the mitophagic apparatus such as PINK1 decrease in patients with CVD116, and HF is more frequent in individuals with mitophagy defects (as in patients with Parkinson disease)147.
Sirtuin activators such as caloric restriction and resveratrol are potent activators of autophagy, adding to multiple lines of evidence intimately linking the sirtuin system and autophagic responses. Additional pharmacological agents that promote mitophagy or autophagy have been shown to mediate beneficial effects in rodent models of CVD17. These include the natural polyamine spermidine, an inhibitor of the acetyltransferase E1Aassociated protein p300 (EP300)148–150, and the natural macrolide rapamycin (also known as sirolimus), which inhibits the master suppressor of autophagy mechanistic target of rapamycin (mTOR)151–154. Conversely, systemic administration of nonspecific inhibitors of autophagy such as 3methyladenine, which targets multiple variants of phosphatidylinositol 3kinase (PI3K), and bafilomycin A1, which suppresses lysosomal functions, generally increases disease severity in rodent models of CVD, including ischaemia– reperfusion injury153,155,156. Interestingly, sirolimus is largely employed in drugeluting stents to prevent restenosis after percutaneous coronary intervention157. Although this use originated from the potent antiproliferative and antiinflammatory activity of sirolimus158, it cannot be excluded that the therapeutic benefits of this strategy involve, at least in part, the induction of autophagy, which reportedly stimulates the degradation of oxidized LDL159 and might also favour the clearance of macrophages from the atherosclerotic plaque160,161. Moreover, multiple FDAapproved agents that mediate beneficial effects on the cardiovascular system, including aspirin (which is widely used as an antiinflammatory and anticoagulant)162, statins (which are currently used to lower circulating levels of cholesterol and triglycerides)163, and suberanilohydroxamic acid (SAHA; a histone deacetylase inhibitor used for the treatment of cutaneous T cell lymphoma)164,165, trigger proficient autophagic responses in the myocardium.
Despite the robust links between mitophagy and/or autophagy activation and improved cardiovascular homeostasis in health and disease, targeting the underlying molecular apparatus with specific pharmacological intervention has proved to be challenging130. Accordingly, no clinical trials are currently investigating the therapeutic potential of mitophagy and/or autophagy modulators beyond calorie restriction and sirolimus in patients with CVD.
Ca2+ homeostasis.
In cardiomyocytes, mitochondria participate (to some extent) in the buffering of cytosolic Ca2+ ions. Depolarization of the plasma membrane activates voltagedependent Ltype Ca2+ channels, and Ca2+ enters into the cytosol, which causes Ca2+induced Ca2+ release from the sarcoplasmic reticulum via ryanodine receptor 2 (RYR2); Ca2+ is removed from the cytosol predominantly by members of the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) family and by solute carrier family 8 member A1 (SLC8A1; also known as NCX1)166. In physiological conditions, mitochondrial Ca2+ uptake is mediated by calcium uniporter protein, mitochondrial (MCU)167,168. Conversely, Ca2+ efflux from the mitochondrial matrix relies primarily on the Na+/Ca2+ antiporter SLC8B1 (also known as NCLX)169. Although mild, transient elevations of mitochondrial Ca2+ levels support oxidative phosphorylation and ATP synthesis170, persistent Ca2+ overload favours MPT171. In line with this notion, the transgenedriven overexpression of a leaky variant of RYR2 in the mouse myocardium exacerbates the cardiotoxic effects of ischaemia–reperfusion injury and causes mitochondrial Ca2+ overload in cardiomyocytes172. Moreover, in multiple cell types, including cardiomyocytes, MCU deficiency confers resistance to MPT driven by mitochondrial Ca2+ overload173,174, and the conditional deletion of Mcu from adult cardiomyocytes mediates cardioprotective effects against ischaemia–reperfusion injury in vivo174,175. However, the hearts from Mcu−/− mice, as well as mouse hearts expressing a dominantnegative variant of MCU, are as susceptible to ischaemia–reperfusion injury ex vivo as their wildtype counterparts173,176. The reasons underlying this apparent discrepancy remain to be elucidated. As a possibility, the contribution of mitochondrial Ca2+ overload to MPT might be limited when ischaemia– reperfusion injury is imposed ex vivo. Irrespective of this conundrum, MCU seems to be required for optimal cardiac responses to acute physical demands174,175. Importantly, deletion of Slc8b1 from adult mouse cardiomyocytes provokes sudden death as a consequence of mitochondrial Ca2+ overload leading to widespread MPTdriven necrosis of the myocardium177. Conversely, Slc8b1 overexpression mediates robust cardioprotection in mouse models of cardiac ischaemia–reperfusion injury177. These observations exemplify the importance of mitochondrial Ca2+ fluxes for cardiovascular homeostasis in health and disease.
Further corroborating the crucial role for intracellular Ca2+ homeostasis in cardiac physiology, genetic defects in plasma membrane Ltype Ca2+ channels are known to impair cardiac signal conduction, potentially favouring the development of arrhythmia178. Moreover, hyperactivation of the cytosolic Ca2+responsive enzyme calcium/calmodulindependent protein kinase II (CaMKII) has been aetiologically linked to a variety of cardiovascular disorders, often reflecting the ability of CaMKII to regulate mitochondrial functions. Mice engineered to overexpress an endogenous inhibitor of CaMKII in cardiomyocytes areprotectedfromischaemia– reperfusion injury in vivo179, presumably reflecting the capacity of CaMKII to trigger MCUdependent mitochondrial Ca2+ overload, blunt antioxidant defences, and trigger DNM1Ldependent mitochondrial fragmentation179–182. Deletion of Camk2d (encoding one of the CaMKII subunits) attenuates pathological maladaptation in a genetic mouse model of decompensating cardiac hypertrophy182. Moreover, CaMKII seems to participate in the pathogenesis of atherosclerotic plaques183, although the underlying molecular mechanisms remain to be unveiled.
Although pharmacological regulators of cellular Ca2+ homeostasis are commonly available for the treatment of some cardiovascular disorders (for example, verapamil, a blocker of plasma membrane Ca2+ channels used virtually worldwide for the treatment of arrhythmia and some forms of hypertension)184, mitochondrial Ca2+ fluxes have been rather elusive drug targets. NCLX inhibitors such as CGP37157, KBR7943, and SEA0400 mediate promising cardioprotective effects in animal models of HF169,185. These results are at odds with the findings obtained with Nclx−/− mice177, most likely reflecting the capacity of chemical NCLX inhibitors such as CGP37157 to preserve redox homeostasis169. That said, NCLX inhibitors never entered clinical development, presumably owing to specificity issues, because these compounds also inhibit the plasma membrane Na+/Ca2+ antiporter SLC8A1186. Chemical inhibitors of MCU including DS16570511 have also been identified187, but whether MCU inhibition constitutes a valid therapeutic objective for the treatment of CVD remains controversial. Supporting caution over this approach, the anticancer agent mitoxantrone, which is associated with robust cardiotoxic effects in some patients, potently inhibits MCU (potentially contributing the adverse effects of this chemotherapeutic)188. The necroptosis inhibitor Necrox5 has also been suggested to mediate beneficial effects via MCU inhibition189, but the specificity of this molecule remains to be determined. Finally, a panel of CaMKII inhibitors is available for investigational purposes, including competitive and noncompetitive inhibitors of ATP or substrate binding, agents that disrupt calmodulin binding, and agents that mimic endogenous CaMKII blockers190. Although many of these agents mediate consistent beneficial effects in animal models of CVD (reviewed previously)190, none of them has entered clinical development.
Oxidative stress.
Mitochondria generate reactive oxygen species (ROS) as a normal byproduct of oxidative phosphorylation, and physiological ROS levels regulate multiple cardiovascular processes, including (but not limited to) metabolic functions in the myocardium and endothelial permeability in vessels191. However, mitochondrial dysfunction is generally associated with massive ROS overgeneration (box 2), which (especially when cellular antioxidant defences are lowered) causes oxidative damage to macromolecules, thereby favouring the establishment of local inflammation2 and initiating multiple variants of RCD including MPTdriven regulated necrosis and ferroptosis171,192. The human failing myocardium reportedly has more than twofold higher levels of superoxide anion than the healthy myocardium193. Similar observations have been made in the context of diabetic and hypertensive cardiomyopathy194,195. Moreover, markers of oxidative damage to lipids196, nucleic acids197, and proteins198 have been documented in the circulation or in the myocardial tissue of patients with MI or HF (and in animal models of these conditions)199. Finally, myocardial mitochondria exhibit increased oxidative damage in aged versus young rats200, and the mitochondrial network of rat endothelial cells produces increased levels of H O with ageing201. These observations suggest that oxidative stress is involved in multiple forms of CVD, including ageingassociated cardiovascular disorders. Corroborating an aetiological role for ROS overproduction in at least some variants of CVD, the absence of one copy of Sod2 (which encodes a mitochondrial superoxide dismutase) aggravates atherosclerosis progression in Apoe−/− mice202. Placing mice under progressive respiratory hypoxia after ischaemia–reperfusion limits ROS production because hypoxia induces a robust regenerative response with decreased myocardial fibrosis and improvement of left ventricular systolic function203. Moreover, cardiomyocytespecific deletion of Txnrd2 (which encodes thioredoxin reductase 2) from mouse embryos leads to fatal dilated cardiomyopathy204. Interestingly, imposing the same genetic defect on adult mice generates a much milder cardiac phenotype resembling accelerated cardiac ageing205. This finding suggests that the embryonic and neonatal myocardium and its adult counterpart have different sensitivity to oxidative stress.
Box 2 |. Mitochondrial generation of reactive oxygen species.
In physiological conditions, an estimated 0.2–2.0% of molecular O2 taken up by mitochondria is not used as a terminal electron acceptor in the respiratory chain (see box 1) but forms superoxide anion (O2 •−) at the level of complex I or complex III (a process known as electron leak). O2 •− can be rapidly metabolized by mitochondrial and mostly extramitochondrial variants of superoxide dismutase (SOD2 and SOD1, respectively), which catalyse the formation of hydrogen peroxide (H2O2) and O2. In turn, H2O2 can have different fates: it can be metabolized by catalase (CAT), resulting in H2O formation; it can be metabolized by multiple peroxidases (including glutathione peroxidase (GPx)), coupling the reduction of H2O2 to H2O with the oxidation of a nucleophilic species, such as reduced glutathione (GSH); and it can be converted into the hydroxyl radical (OH•) and hydroxyl anion (OH–) in the presence of Fe2+ or Cu1+ (Fenton reaction) (see figure; please note that stoichiometry is not respected for the sake of simplification). Physiological levels of reactive oxygen species (ROS) are involved in the regulation of several biological processes, including intracellular signalling, adaptation to hypoxia, autophagy, and both adaptive and innate immunity341. However, ROS levels that exceed endogenous antioxidant capacities cause extensive macromolecular damage to DNA, proteins, and lipids, generally leading to cellular senescence (the permanent proliferative inactivation of a cell damaged beyond repair) or regulated cell death.
In the hypoxic myocardium, electrons cannot flow normally through the respiratory chain because O2 availability is limited. This impairment favours the acquisition of a reduced state by respiratory complexes, which enables electron leak, O2 •− synthesis, and oxidative damage to the respiratory chain. At tissue reperfusion, restored oxygen availability drives an abrupt increase in electron flow through damaged respiratory complexes, which is associated with a burst in O2 •− production. Reperfusion is the phase at which mitochondria are most sensitive to ROS-mediated mitochondrial permeability transition because the low pH associated with ischaemia potently inhibits mitochondrial permeability transition. It has been proposed that uncoupling, the process whereby the transfer of electrons along the respiratory chain occurs in the absence of net extrusion of H+ ions from the mitochondrial matrix, leading to decreased mitochondrial transmembrane potential and therefore to reduced sensitivity of respiratory complexes to hypoxia-mediated reduction, might have evolved as a physiological barrier against oxidative damage rather than as a thermogenic process342.

The possibility to use antioxidants (including molecules available over the counter as dietary supplements) for the treatment of CVD drove an intense wave of preclinical and clinical investigation spanning the past 2 decades. Coenzyme Q10, αtocopherol (vitamin E), ascorbic acid (vitamin C), and βcarotene (the precursor of vitamin A) have all been clinically tested for the treatment or prophylaxis of HF206,207, highrisk heart surgery208, acute MI209–214, and atherosclerosis215,216. The majority of these studies confirmed that active levels of antioxidants can be achieved in the circulation of patients with CVD, although most often this is not associated with measurable clinical benefits, perhaps with the exception of coenzyme Q10 supplementation for the treatment of moderatetosevere HF206. Some clinical trials are ongoing to test the clinical activity of coenzyme Q10 or its reduced counterpart (ubiquinol) in patients with HF (NCT03133793, NCT01925937, NCT02779634, and NCT02847585), cardiac arrest (NCT02934555), and atherothrombosis (NCT02218476) as well as the capacity of ascorbic acid to prevent atrial fibrillation after CABG surgery (NCT03123107).
Promising preclinical results have been obtained with mitochondriatargeted antioxidants, including elamipretide (also known as Bendavia, MPT131, and SS31), mitoQ, and mitoTEMPO, in animal models of MI217, hypertensive cardiomyopathy195,218–220, HF221, ischaemia–reperfusion injury222–224, pathological tissue remodelling after MI225, and atherosclerosis226, fostering the initiation of multiple clinical trials. Both the EVOLVE (NCT01755858) and the EMBRACE STEMI (NCT01572909) studies, evaluating the capacity of elamipretide to limit restenosis after angioplasty of the renal or coronary artery, respectively, did not report clinical benefits227,228. Conversely, highdose elami pretide decreased left ventricular enddiastolic volume and endsystolic volume in HF218,221 with reduced ejection fraction, pointing to (at least some degree of) clinical efficacy229. Elamipretide is still being investigated in Europe for its therapeutic effects in patients with HF (NCT02914665 and NCT02788747), whereas in the USA, increased attention is being dedicated to the possibility of using elamipretide for the treatment of mitochondrial disorders (such as myopathies and retinopathies). Along similar lines, mitoQ is mostly being investigated in clinical settings other than CVD.
Inflammation.
The major role of mitochondria in the establishment of innate inflammatory responses that contribute to the pathogenesis of CVD is now clear230. This observation reflects the key contribution of mitochondrial metabolism and ROS production to multiple immune functions (which is beyond the scope of this Review)231, and the fact that mitochondria contain a large amount of endogenous molecules that can act as damage-associated molecular patterns (DAMPs) upon release232,233. These molecules include (but potentially are not limited to) ROS, mtDNA, ATP, and cardiolipin233. Both ROS and mtDNA (alone or complexed with TFAM) can stimulate inflammatory responses from the cytosol, owing to their capacity to stimulate the release of IL1β, IL18, and type I interferon upon activation of the inflammasome and the stimulator of interferon genes protein (STING; also known as TMEM173)234,235. Moreover, extracellular mtDNA can drive granulocyte degranulation upon binding to Toll like receptor 9 (TLR9)236. Extracellular ATP released in the course of RCD operates both as a chemoattractant and as an immunostimulant for myeloid cells237. Finally, cardiolipin can favour the activation of γδ T lymphocytes via a CD1Ddependent mechanism238. Although not all these processes have been implicated in the pathophysiology of CVD, these observations exemplify well how mitochondrial dysfunction in the cardiovascular system, especially in the presence of a mitophagic defect, can drive detrimental inflammatory responses.
In line with this notion, mice lacking the cytosolic DNA sensor cyclic GMPAMP synthase (CGAS)239 have improved early survival after MI along with reduced cardiac immune infiltration and consequent pathological tissue remodelling240. Cgas−/− mice, Irf3−/− mice (lacking an effector of CGAS signalling), and Ifnar1−/− mice (lacking one of the subunits of the type I interferon receptor)241 are protected against MI compared with wildtype mice, a cardioprotective phenotype that is accompanied by decreased cardiac expression of inflammatory cytokines and chemokines and decreased inflammatory cell infiltration in the myocardium242. Similar cardioprotective effects have been documented with hearts from Nlrp3−/− mice (which lack a core component of the inflammasome) subjected to ischaemia–reperfusion injury ex vivo243. Moreover, mtDNA escaping mitophagic degradation as a consequence of Dnase2 deletion aggravates disease symptoms and progression in a mouse model of pressureoverloadinduced cardiomyopathy, a detrimental phenotype that can be partially rescued by Tlr9 codeletion or administration of TLR9inhibiting oligodeoxynucleotides131. NLRP3, CGAS, and STING have also been aetiologically involved in the endothelial inflammatory response driven by dietinduced obesity and in some models of atherogenesis235,244. Moreover, atherogenesis caused by a highfat diet is inhibited in Apoe−/−Il1r1−/− mice (which lack both apolipoprotein E and the receptor for IL1β, IL1R1) compared with Apoe−/− mice245. By contrast, deletion of Il1rn (encoding an endogenous inhibitor of IL1R1) aggravates considerably the disease pathogenesis in Apoe−/− mice, whereas Apoe−/− mice engineered to overexpress Il1rn are largely protected from highfatdietinduced atherogenesis246. Finally, a common lossoffunction variant in P2RX7 (coding for one of the receptors for extracellular ATP) is associated with reduced risk of cardiovascular events in smokers247. These studies are only a few examples of how genetic defects in the proinflammatory signalling pathways elicited by mitochondrial DAMPs reduce disease incidence, severity, or progression in rodent models of CVD as a consequence of quenched inflammatory responses.
In line with this notion, pharmacological inhibitors of the signal transduction cascades activated by mitochondrial DAMPs provided beneficial effects in multiple experimental models of CVD. For instance, administration of a type I interferonneutralizing antibody protected mice against MI to a similar extent as the absence of Irf3 or Ifnar1 (REF.242). Similarly, wildtype mice subjected to ischaemia–reperfusion while receiving a pharmacological inhibitor of NLRP3 (16673340) had a significant reduction in infarct size afterwards compared with their control counterparts248,249. Some degree of cardioprotection has also been observed with the P2RX7 inhibitor Brilliant Blue in rat hearts subjected to ischaemia–reperfusion ex vivo250 as well as with the TLR9targeting oligodeoxynucleotide ODN 2088 in rats with spontaneous hypertension251. Interestingly, elamipretide binds to and prevents the peroxidation of cardiolipin252, and blocking γδ T lymphocytes with a monoclonal antibody specific for killer cell lectinlike receptor subfamily K member 1 (KLRK1) reportedly attenuates ischaemia– reperfusion injury in a cardiac transplantation model in rats253. However, whether elamipretide influences the capacity of cardiolipin to activate γδ T lymphocytes remains to be elucidated. Although multiple antiinflammatory agents are currently available for the treatment of CVD, they all operate by either inhibiting immune cell activation (as in the case of corticosteroids) or by blocking the production of proinflammatory eicosanoids (as in the case of NSAIDs)254,255. To the best of our knowledge, no therapeutic agent designed to intercept DAMP emission from mitochondria or specifically block the downstream signalling cascades has reached clinical development.
Regulated cell death.
A prominent aetiological component of multiple cardiovascular disorders, including HF, MI, and atherosclerosis, is the demise of cells damaged beyond recovery15, generally occurring via multiple, highly interconnected signalling cascades4 (box 3). Widespread and irreversible mitochondrial dysfunction culminating with the permeabilization of mitochondrial membranes has a central role in apoptosis, MPTdriven regulated necrosis, and parthanatos256, de facto contributing to pathological tissue loss in the context of CVD15. In line with this notion, mice bearing genetic alterations of the molecular apparatus for RCD are protected (at least to some degree) against multiple cardiovascular pathologies. For instance, Bbc3−/− mice lack one of the upstream activators of intrinsic apoptosis and have increased resistance to ischaemia–reperfusion injury compared with their wildtype littermates257. Similarly, mice overexpressing Bcl2, encoding the apoptosis regulator BCL2, have mitigated MI upon ischaemia–reperfusion injury258. Ppif−/− mice, which lack the crucial component for MPTdriven regulated necrosis CypD, are protected against cardiac ischaemia–reperfusion injury259,260, angiotensinIIinduced cardiomyopathy261, and arrhythmia (in this last case, perhaps also linked to preserved Ca2+ fluxes)262. The deletion of Parp1, which encodes poly(ADPribose) polymerase 1 (a nuclear DNA repair enzyme that is required for parthanatos), mediates beneficial effects in mouse models of ischaemia–reperfusion injury263,264 and atherogenesis265. Moreover, both Ripk3−/− mice (which lack a critical regulator of necroptosis) and mice engineered to overexpress dominantnegative CaMKII in the heart are protected against ischaemia–reperfusion injury and the cardiotoxic effects of doxorubicin266. These findings link the molecular machinery for necroptosis, which normally proceeds independently of mitochondria256, to mitochondrial dysfunction and consequent MPTdriven necrosis.
Box 3 |. Mechanistic notions on regulated cell death.
Mammalian cells exposed to very harsh microenvironmental conditions (such as extreme temperatures and elevated osmotic pressures) die in a virtually uncontrollable manner, reflecting the physical breakdown of the plasma membrane. However, this unregulated cell death is fairly uncommon in the context of human pathophysiology. Instead, human cells generally succumb to pathological cues in the context of failing adaptation to stress via regulated cell death (RCD), which ensues the activation of a genetically encoded machinery that determines the kinetics of the process and its immunological correlates. Indeed, according to current models, mammalian cell death is not caused by the activation of specific proteolytic or nucleolytic pathways, as was thought until the early 2010s, but rather by a lethal shortage of ATP coupled to the accumulation of unrepairable oxidative damage to macromolecules, leading to irreversible loss of plasma membrane integrity. Therefore, actual cytoprotection (that is, a reduction in the percentage of cells succumbing to a cytotoxic cue, as opposed to a simple delay in RCD) might not be achievable after cells are committed to death (that is, when cellular functions are compromised beyond recovery)4,330.
Irrespective of this (rather debated) point and its major therapeutic implications (see main text), multiple molecular cascades precipitating RCD in mammals have been identified. These signal transduction cascades rely on a dedicated molecular machinery, meaning that they can be retarded (or accelerated) by specific pharmacological or genetic interventions, and include the following:
Extrinsic and intrinsic variants of apoptosis: a caspase 3-dependent pathway optionally involving mitochondrial outer membrane permeabilization.
Mitochondrial permeability transition-driven necrosis: a cyclophilin D-dependent process elicited at the inner mitochondrial membrane.
Necroptosis: another form of regulated necrosis culminating with plasma membrane permeabilization dependent on mixed lineage kinase domain-like protein (MLKL).
Ferroptosis: an iron-dependent pathway mediated by uncontrolled lipid peroxidation.
Parthanatos: a poly(ADP-ribose) polymerase 1-dependent process resulting in a lethal bioenergetic crisis coupled to DNA degradation.
Pyroptosis: an inflammatory variant of RCD linked to plasma membrane permeabilization by gasdermin protein family members.
Lysosome-dependent cell death: RCD that is initiated by lysosomal breakdown and precipitated by lysosomal hydrolases.
Autophagy-dependent cell death: a form of RCD aetiologically linked to components of the molecular machinery for autophagy.
NETotic cell death: a reactive-oxygen-species-dependent form of RCD restricted to haematopoietic cells and linked to neutrophil extracellular trap (NET) production.
Entotic cell death: referring to the lysosomal degradation of living cells internalized by other, nonphagocytic cells via an actomyosin-dependent mechanism (entosis)4,330.
Extraordinary efforts have also been dedicated to the development of clinically useful inhibitors of RCD for cardioprotective purposes267, with rather dismal results. Indeed, although dozens of compounds targeting distinct modules of the molecular machinery for RCD have been successfully synthesized and demonstrated to mediate beneficial effects in experimental models of CVD267, none of these agents is currently approved for clinical use. Among other approaches, promising preclinical results in animal models of CVD have been obtained with caspase inhibitors, including: the pancaspase blockers ZVADFMK268 and MX1013 (REF.269); inhibitors of the core activator of intrinsic apoptosis, apoptosis regulator BAX (BAX), including a cellpermeant peptide derived from the endogenous BAX inhibitor BCL2like protein 1 (BCL2L1)270, as well as the two small molecules Bci1 and Bci2 (although these compounds were tested only in rodent models of brain ischaemia)271; inhibitors of serine protease HTRA2, mitochondrial (HTRA2), such as the small molecule UCF101 (REFs272,273); molecules that preserve the integrity of the respiratory chain in the course of RCD, including multiple 2sulfonylpyrimidinyl derivatives (although these compounds have been investigated only in rodent models of neurodegeneration)274,275; PARP1 inhibitors such as 3aminobenzamide263; and inhibitors of MPTdriven necrosis, including TRO40303 (a small molecule specific for translocator protein (TSPO)276), cinnamic anilides (the precise molecular target of which remains to be determined277), and the CypDtargeting compounds cyclosporine A, Debio025, NIM811, and sanglifehrin A (REF.278). Most of these molecules never reached clinical development, often owing to specificity or bioavailability issues267. Conversely, both TRO40303 and cyclosporine A have been investigated for their clinical benefits in patients undergoing percutaneous coronary intervention for acute MI279,280. However, despite considerable enthusiasm elicited by the release of efficacy data from the first randomized clinical trial to test cyclosporine A for this indication280, subsequent studies did not document clinical benefits281,282. Similarly, TRO40303 seems to be well tolerated but devoid of clinical efficacy283,284. To the best of our knowledge, the clinical development of TRO40303 has been discontinued. By contrast, a large number of clinical trials are ongoing to test the therapeutic effects of cyclosporine A. The vast majority of these studies, however, are aimed at investigating the activity of cyclosporine A as an immunosuppressant rather than as an MPT inhibitor. Indeed, cyclosporine A is approved by the FDA to prevent and treat graftversushost disease after bone marrow transplantation, the rejection of kidney, heart, and liver transplantation, and a panel of autoimmune disorders285,286. Of note, the vasodilator nicorandil, which is approved in several countries for the treatment of angina, reportedly potentiates ischaemic preconditioning, at least in some experimental models, by inhibiting MPT287. Clinical data from a few studies indicate that nicorandil (which was not conceived as an MTA) might confer cardioprotection after MI288–290, a possibility that remains under scrutiny.
Mitochondrial microRNAs.
Most (if not all) aspects of mitochondrial biology are now known to be subjected to epigenetic regulation by microRNAs (miRNAs)291. Importantly, this process occurs not only in the nucleus but also in the mitochondrial matrix, where all the components of the molecular apparatus for miRNAdependent gene silencing are present292. Both nuclear miRNAs and mitochondrial miRNAs (also known as mitomiRs) have been implicated in the pathogenesis of multiple cardiovascular disorders291. The codeletion of the sequences encoding miR181c and miR181d mediated cardioprotective effects in a mouse model of ischaemia–reperfusion injury, potentially linked to preserved levels of the mitochondrially encoded cytochrome c oxidase subunit 1 (MTCO1) and ameliorated respiratory functions293. Overexpression of miR30b in mouse cardiomyocytes decreases infarct size after ischaemia–reperfusion injury, reflecting the ability of miR30b to downregulate CypD levels and thereby impair MPT294. Similarly, miR2861 knockdown protects the mouse heart from ischaemia– reperfusion injury in vivo, a beneficial phenotype potentially linked to upregulation of solute carrier family 25 member 4 (SLC25A4)295. Codeletion of the genes encoding miR212 and miR132 provides cardioprotection against pressureoverloadinduced cardiomyopathy along with the activation of FOXO3Adependent autophagy296. Consistently, cardiomyocytespecific overexpression of miR132, miR199a, miR212, or miR421 in rodents triggers or aggravates CVD along with the induction of mitophagic defects296–298. Nanoparticlebased delivery of a miR181c coding vector also leads to cardiac dysfunction by provoking mitochondrial impairment299, as does the deletion of mir-150 and the codeletion of miR-181a and miR-181b293,300. Altogether, these observations exemplify the intimate links between the epigenetic regulation of gene expression at both mitochondrial and nuclear levels, mitochondrial biology, and CVD.
Several miRNAtargeting strategies have been shown to mediate beneficial effects in preclinical models of CVD. The mitochondrial pool of miR378 increases in the course of diabetic cardiomyopathy in mice, and intraperitoneal administration of a miR378 antagonist mediates cardioprotection, linked with the preservation of mitochondrially encoded ATP synthase subunit a (MTATP6) synthesis301. The mitochondrial levels of mitochondrially encoded cytochrome b (MTCYB) are significantly lower in hearts from rats with spontaneous hypertension than in control hearts from Wistar rats, associated with an upregulation in the mitochondrial pool of miR21 (which promotes Cytb translation)302. In line with the hypothesis that miR21 upregulation constitutes a compensatory response to decreased MTCYB levels and consequent ROS overgeneration, intravenous delivery of an adenoviral vector for the overexpression of miR-21 in rats with spontaneous hypertension mediates shortterm beneficial effects on systolic blood pressure and longterm cardioprotection302. miR106a is robustly upregulated in the hypertrophic myocardium, along with a profound downregulation of MFN2, and data from cultured cardiomyocytes exposed to miR106a mimics or antagonists suggest that antagonizing miR106a might contribute to the restoration of MFN2 levels and conse quent rescue of mitochondrial functions303. miR3245p and miR761 are negative regulators of mitochondrial fission, and intravenous delivery of a miR3245p or miR761 mimic limits apoptotic RCD and tissue damage in the myocardium of mice exposed to ischaemia– reperfusion304,305. Similarly, administration of a miR499 antagonist (which also inhibits mitochondrial fission) exacerbates infarct size in mice exposed to ischaemia–reperfusion306. Expression of miR33a and miR33b is markedly increased in human carotid atherosclerotic plaques compared with normal arteries, and treatment of Apoe−/− mice with miR33 antagonists reduces arterial atherosclerotic lesions along with the normalization of mitochondrial functions307. Additional progress is required for miRNAtargeting agents to enter clinical development308.
Obstacles in the development of MTAs
Despite an extraordinary experimental effort spanning over the past 3 decades, virtually no MTAs are currently approved for use in patients with CVD. We surmise that such a dismal situation is linked (at least in part) to pharmacodynamic and pharmacokinetic issues, a hitherto fragmentary knowledge of the molecular mechanisms behind mitochondrial processes, and a rather simplistic appreciation of the pathophysiology of some cardiovascular disorders.
Pharmacological issues.
Multiple MTAs have limited pharmacological specificity for their mitochondrial targets. Cyclosporine A and other CypDtargeting agents are perhaps the most representative examples of this problem. Cyclosporine A and sanglifehrin A potently inhibit MPT by binding to CypD, de facto mediating robust cytoprotective effects in rodent models of CVD and other pathologies associated with MPTdependent tissue loss278. However, both cyclosporine A and sanglifehrin A also enable the binding of peptidylprolyl cistrans isomerase A (PPIA) to the heterodimeric phosphatase calcineurin, resulting in potent calcineurin inhibition and consequent complete blockage of T cell activation309. With systemic administration, the immunosuppressive effect of cyclosporine A and sanglifehrin A are prominent, as demonstrated by the fact that cyclosporine A is approved for use in various clinical settings as an immunosuppressant285,286. Novel CypD inhibitors that lack immunosuppressive activity such as Debio025 and NIM811 are currently being developed278. In addition, attention is being focused on strategies for the targeted delivery of cyclosporine A to the myocardium. In this setting, promising results have been obtained with poly(lacticcoglycolic acid) (PLGA) nanoparticles incorporating cyclosporine A, which were more potent than cyclosporine A at limiting ischaemia–reperfusion injury in mice in the absence of alterations in the myocardial recruitment of inflammatory monocytes310.
Untargeted antioxidants also have specificity issues because, on entering the cell, antioxidants can quench ROS from multiple (not necessarily mitochondrial) sources, which limits the purely mitochondrial activity of these compounds. Multiple strategies have been successfully used to target antioxidants specifically to mitochondria, most of which harness the capacity of cationic molecules to accumulate spontaneously within the mitochondrial matrix mediated by the mitochondrial transmembrane potential (Δψm)311,312. One of the major issues with this approach, potentially decreasing its therapeutic value, is that dysfunctional mitochondria often have decreased Δψm and, consequently, are unable to accumulate cationic molecules313. Alternative techniques for mitochondrial delivery, including the use of lipophilic cationic peptides314, also rely on the Δψm and, therefore, cannot circumvent this issue. Similarly, mitochondrial proteins encoded by the nuclear genome enter the mitochondrial matrix by a Δψmdependent mechanism315. Thus, devising a strategy for the targeted delivery of molecules to dysfunctional mitochondria will be important. The surface properties of permeabilized mitochondria (including PINK1 and PARK2 accumulation, as well as extensive ubiquitylation)6 could be useful but remain unexplored in this context.
Another pharmacological obstacle in the development of clinically useful MTAs relates to pharmacokinetics and biodistribution. In the absence of a tissuetargeting strategy, systemically administered MTAs are confronted by large numbers of mitochondria outside the cardiovascular system, which operate (at least to some degree) as a sink to limit bioavailability at diseased sites. Cardiomyocytes contain more mitochondria than many other cell types316, which could potentially favour MTA accumulation, but so do myocytes and neurons, and the skeletal muscle largely exceeds the myocardium in terms of mass. These considerations suggest that some MTAs delivered systemically at safe doses cannot reach bioactive levels at the mitochondrial compartment of diseased cells from the cardiovascular system. Strategies to target MTAs to specific cells of the cardiovascular system, such as PLGA nanoparticles310, might (at least partially) circumvent this obstacle.
Lack of precise mechanistic knowledge.
Despite considerable advances in the understanding of many mitochondrial processes involved in the pathogenesis of CVD, precise mechanistic knowledge is often lacking. Perhaps the best example of our lack of knowledge of mitochondrial processes comes from MPT317. The concept that MPT results from the activity of a supramolecular entity assembled at the interface between the inner and outer mitochondrial membranes, generally referred to as the permeability transition pore complex (PTPC), is widely accepted171. However, the precise molecular composition of the PTPC remains obscure, and multiple other aspects of the PTPC biology (including its potential links with the F1Fo ATP synthase) are still a matter of intense debate, despite >2 decades of experimental work on this topic317,318. This lack of precise mechanistic knowledge of mitochondrial processes reflects an intrinsic complexity of the system and the lack of good indicators of mitochondrial (dys)function for use in vivo.
Several mitochondrial proteins are strictly required for embryonic development or adult survival, generally owing to their essential bioenergetic functions. One notable example is cytochrome c, somatic (CYCS), which functions as an electron shuttle of the respiratory chain319. Because Cycs−/− mice die in utero, investigating the role of CYCS in RCD in vivo called for the development of refined genetic models320. Similar models have not yet been generated for the vast majority of mitochondrial proteins with a prominent vital function319. Another large group of mitochondrial proteins exists in multiple isoforms that have a large degree of genetic redundancy317. For instance, the mouse genome encodes at least three distinct variants of the PTPC component adenosine nucleotide translocase (Slc25a4, Slc25a5, and Slc25a31) and of the ATP synthase Fo complex subunit C (Atp5g1, Atp5g2, and Atp5g3)321,322. This genetic redundancy complicates considerably the generation of functional knockout models for in vivo studies, although it also presumably reflects the critical requirement for mitochondrial ATP synthesis for life (implying that complete knockout models might not be viable). In addition, some mitochondrial proteins have functional redundancy, meaning that they can substitute for each other in a specific activity. This functional redundancy seems to be the case for multiple components of the PTPC, at least in some experimental models317. These observations exemplify the intrinsic complexity of multiple mitochondrial processes.
Despite the existence of a variety of probes for in vitro use, monitoring mitochondrial function in vivo thus far has proved challenging. Carbonylation of circulating proteins or lipoproteins has been used to monitor oxidative stress in the context of CVD323. However, this technique per se does not enable the identification of the tissue experiencing oxidative damage, nor the precise source of ROS. Measuring the carbonylation of cardiac proteins, such as myosinbinding protein C, cardiactype (MYBPC), might constitute an improved alternative, although this approach also does not enable the identification of the ROS source and it can be performed only postmortem324. Massspectrometrybased profiling of energy metabolites in blood has been proposed as a surrogate biomarker of mitochondrial dysfunction in the context of HF325, but the wide applicability of these findings remains untested. One promising approach to monitor mitochondrial dysfunction in preclinical models of CVD is provided by the socalled MitoTimer mouse, a mouse strain engineered to express a mitochondriatargeted mutant of the DsRed fluorescent protein (which shifts to red fluorescence when oxidized) under the control of a cardiomyocytespecific promoter326,327. MitoTimer enables the study of mitochondrial structure, redox state, and mitophagic disposal by fluorescence microscopy on fixed tissue326,327. Finally, multiple radioactive tracers are being developed to monitor mitochondrial functions in real time in the setting of CVD26,328. These molecules, some of which are already approved for use in humans (for different applications), might constitute preferential tools to study the links between mitochondrial dysfunction and multiple forms of CVD in patients.
Limited appreciation of the multifactorial nature of CVD.
All cardiovascular disorders are complex pathological entities that develop in the context of multiple cellular, histological, and systemic processes including (but not limited to): an initial attempt of cells to cope with potentially detrimental perturbations of their microenvironment for the restoration of cellular homeostasis; the failure of such an adaptive mechanism, culminating with the initiation of RCD coupled to inflammatory responses; the establishment of acute local inflammation after the recruitment of immune cells, at least partly linked to the disposal of dead cells and cell remnants; and the initiation of repair processes, either culminating with resolved inflammation and fibrosis (if the initial perturbation of homeostasis is relieved) or proceeding chronically along with a continuous wave of RCD and lowdegree inflammation (if the initial perturbation of homeostasis persists).
This process is further complicated by at least four additional elements. First, the entire process involves not only cells from the cardiovascular system (the main target of clinically available drugs) but also stromal cells and, to a greater extent, immune cells230. Although the contribution of immune cells to some forms of CVD such as atherosclerosis was appreciated long ago14, the role of innate immune mechanisms such as dysregulated type I interferon signalling in HF has just begun to emerge242. Second, there is a critical, and we believe often underestimated, time component in the pathogenesis of most, if not all, cardiovascular disorders. As an example, ischaemia–reperfusion injury is often viewed (and experimentally modelled) as a rather uniform entity, and potential therapeutic interventions administered at reperfusion are tested for their capacity to decrease infarct size or improve survival. Although these models are widely viewed as clinically relevant (patients with acute MI indeed enter intensive care during the ischaemic phase), they are intrinsically unable to dissect the sequence of events initiated at reperfusion, many of which have a direct effect on patient survival. Third, CVD generally develops in elderly individuals, along with a variety of comorbidities, including (but not limited to) obesity, diabetes, and declining immune functions329. These disorders affect not only the natural progression of CVD but also its sensitivity to treatment329. However, only a few animal models of CVD that are currently available recapitulate such comorbidities. Fourth, many cellular processes involved in the pathogenesis of CVD have a considerable degree of redundancy. For instance, after mammalian cells commit to RCD, inhibiting one single variant of the process only delays (rather than prevents) cellular demise, and it has been argued that actual cytoprotection can be achieved only in the course of adaptive responses to perturbation of homeostasis330. This concept casts doubts on the hypothesis that pharmacologically blocking RCD in diseased cardiovascular cells provides clinical benefits (which has been intensively tested with dismal results) and suggests that improving the ability of healthy cells to cope with perturbations of homeostasis constitutes a robust prophylactic strategy. Interestingly, an abundant literature established a robust interconnection between various components of the molecular machineries for RCD and inflammation331. This finding opens the intriguing possibility that modulating RCD pathways in diseased cardiovascular cells might affect the consequent inflammatory responses, de facto mediating beneficial effects via cellextrinsic circuitries332. Such a possibility awaits urgent experimental validation. In support of this notion, cyclosporine A, one of the few MTAs currently approved for use in clinics (although not for the treatment of CVD), robustly inhibits MPT and mediates potent antiinflammatory effects.
Altogether, these observations indicate that improved pharmacodynamic and pharmacokinetic properties, a refined mechanistic knowledge of mitochondrial processes, and a reconsideration of the pathogenesis of (at least some) cardiovascular disorders, together with a redesigned pharmacological audit trail (Fig. 3), are instrumental for the development of novel MTAs with clinical use.
Fig. 3 |. Pharmacological audit trail for the development of novel mitochondria-targeting agents for clinical applications.

To develop novel, clinically useful mitochondria-targeting agents for the treatment or prevention of cardiovascular disease, it is paramount to delineate upfront: the therapeutic paradigms in which mitochondrial dysfunctions cause or aggravate cardiovascular disease; specific patient subsets in which such alterations might have a predominant role in disease pathogenesis; the cell populations that are affected by mitochondrial dysfunction (the diseased cells, which do not necessarily overlap with the cell populations that are commonly linked to disease pathogenesis); and the nature of mitochondrial dysfunction and how such a dysfunction affects the biology of diseased and/or other cells from the cardiovascular or immune system (bystander cells). This analysis will potentially enable the identification of a mitochondrial target for pharmacological interventions and a candidate drug. Delivery platforms tailored to the mitochondrial compartment of diseased cells will have to be developed and characterized in conventional pharmacokinetic and pharmacodynamic studies, followed by an assessment of mitochondrial, cellular, and microenvironmental parameters in both the diseased and bystander cell populations. In the absence of biological efficacy, the choice of molecular target, drug candidate, and/or delivery platforms will have to be re-evaluated, with particular attention for immunological disease correlates. Otherwise, a cardiovascular response followed by improved patient survival might emerge. In the absence of either or both, the entire therapeutic paradigm and/or patient selection should be fully reconsidered.
Conclusions
Robust genetic data demonstrated a crucial role for mitochondrial dysfunction in the pathogenesis of multiple cardiovascular disorders. Nonetheless, the development of MTAs for use in patients with CVD has been rather dismal. Thus far, great attention has been focused on modulating a single mitochondrial process in cells from cardiovascular compartments, and the immunological correlates of RCD and RCDdriven inflammation have been fairly overlooked. We firmly believe that systematically addressing CVD as a complex phenomenon that is intimately connected with inflammatory responses will be instrumental for the development of novel agents with clinical applications. Alongside, endowing MTAs with superior pharmacological specificity and acquiring additional knowledge on the precise molecular mechanisms linking mitochondrial dysfunction to CVD pathogenesis, potentially aiming at strategies that simultaneously modulate multiple aspects of the disease, will be paramount. In this context, it will be important to evaluate carefully the cardiovascular effects (or lack thereof) of precise genetic interventions targeting mitochondrial functions on the basis of the age and sex of the animals and the potential existence of compensatory pathways, especially based on functional (rather than genetic) redundancy, as well as evaluate the effects in the context of pathologically relevant comorbidities.
Deleting specific mitochondriarelevant genes from the embryonic myocardium has consequences that the same intervention does not provoke in the adult97, which is particularly relevant for the development of pharmacological interventions. Data accumulating over the past decade point to considerable differences in the sensitivity of male versus female rodents to experimental CVD, and epidemiological data in humans support similar conclusions333,334, but little work has been done with specific reference to mitochondrial dysfunction335. Moreover, whereas the effect of genetic redundancy on a specific mitochondrial pathway can be addressed with (relatively complex, but feasible) codeletion and/or depletion strategies336–338, identifying (and investigating) functional redundancy is far more complex. Finally, an unmet need exists for new rodent models that faithfully recapitulate the comorbidities that normally accompany CVD in humans329. In conclusion, although the route to the identification of clinically useful MTAs is long and tortuous, a large amount of evidence suggests that mitochondrial dysfunction remains a promising target for the treatment of multiple forms of CVD.
Regulated cell death.
(RCD). A form of cell death that relies on the activation of a genetically encoded machinery and which, therefore, can be retarded or accelerated with specific pharmacological or genetic interventions.
Autophagy.
Evolutionarily conserved cellular process that culminates with the lysosomal degradation of ectopic, supernumerary, dysfunctional, or potentially dangerous cytoplasmic entities (of endogenous or exogenous derivation).
β-Oxidation.
Biochemical pathway whereby fatty acids are converted into acetyl-CoA, which enters the TCA cycle, and NADH and FADH2, which fuel oxidative phosphorylation.
Ketolysis.
Biochemical pathway whereby ketone bodies are converted into acetyl-CoA, which enters the TCA cycle, and NADH, which fuels oxidative phosphorylation.
Folate cycle.
Biochemical pathway catalysing the cyclic conversion of tetrahydrofolate, 10-formyl-tetrahydrofolate (which feeds into purine synthesis), 5,10-methylenetetra-hydrofolate, and 5-methyl-tetrahydrofolate (which feeds into methionine metabolism).
Mitochondrial permeability transition.
(MPT). Rapid loss of the ionic barrier function of the inner mitochondrial membrane, culminating in mitochondrial breakdown and regulated necrosis.
Transferrin.
Iron-binding plasma glycoprotein that controls the level of free iron ions in biological fluids.
Cerebral cavernous malformations.
Cerebrovascular disease characterized by enlarged and leaky capillaries that predispose to seizures, focal neurological deficits, and fatal intracerebral haemorrhages.
Histone deacetylase inhibitor.
Member of a fairly new class of targeted anticancer agents that operate by derepressing histone acetylation, resulting in the epigenetic reconfiguration of multiple transcriptional modules.
Necroptosis.
Form of RCD that depends on mixed lineage kinase domain-like protein (MLKL), receptor-interacting serine/ threonine-protein kinase 3 (RiPK3), and, at least in some settings, the kinase activity of the RIPK3 homologue RIPK1
Ferroptosis.
Iron-dependent form of RCD that obligatorily relies on lipid peroxidation and is tonically inhibited by glutathione peroxidase 4 (gPx4).
Damage-associated molecular patterns.
(DAMPs). Endogenous molecules that exert potent immunomodulatory functions upon binding to cellular receptors that evolved to control microbial pathogens.
Inflammasome.
supramolecular complex containing caspase 1 (CAsP1), which, among other functions, catalyses the proteolytic processing of IL-1β and IL-18, thereby enabling their release in a bioactive form.
γδ T lymphocytes.
Small subsets of T cells expressing a rather invariant variant of the T cell receptor and mostly operating at the interface between innate and adaptive immunity.
Eicosanoids.
Large family of arachidonic acid derivatives involved in the regulation of multiple biological processes, including the recruitment and activation of immune cells.
Apoptosis.
Form of RCD initiated by extracellular or intracellular cues that is precipitated by the sequential activation of various members of the caspase protein family.
Parthanatos.
Necrotic variant of RCD driven by PARP1 hyperactivation and precipitated by the consequent bioenergetic catastrophe coupled to enzymatic DNA degradation.
microRNAs.
(miRNAs). small non-coding RNA molecules that regulate the expression of target genes at the transcriptional or post-transcriptional level.
Nanoparticle.
Particle of 1–100 nm in size surrounded by an interfacial layer consisting of ions, inorganic molecules, or organic molecules that determines the biological and biophysical properties of the particle.
Mitochondrial transmembrane potential.
(Δψm). Electrochemical gradient built across the inner mitochondrial membrane by the respiratory chain. The Δψm drives multiple mitochondrial functions, including ATP synthesis and protein transport.
Carbonylation.
Term generally referring to the metal-catalysed oxidation (primary carbonylation) or addition of reactive aldehydes (secondary carbonylation) to amino acid side chains.
Pharmacological audit trail.
Rational framework to guide the development of novel therapeutic agents that involves assessing the risk of failure at any specific stage.
Key points.
Mitochondrial dysfunction is involved in the pathogenesis of multiple cardiovascular disorders, including myocardial infarction, cardiomyopathies of various aetiologies, arrhythmias, hypertension, and atherosclerosis.
Mitochondria are essential for the physiological activity of the cardiovascular system owing to their crucial role in bioenergetic and anabolic metabolism and their central position in intracellular Ca2+ fluxes.
In addition to losing their physiological functions, damaged mitochondria actively drive inflammatory responses and waves of regulated cell death that contribute to the pathogenesis of cardiovascular disease.
An intensive wave of investigation attempted to develop mitochondria-targeting agents for preventing or treating cardiovascular disorders in patients, with rather dismal results.
Molecules with improved pharmacological features, precise mechanistic insights into mitochondrial processes, and reconsidering the pathogenesis of some cardiovascular disorders are instrumental for the development of mitochondria-targeting agents with clinical use.
Acknowledgements
The authors apologize to the authors of several high-quality articles on mitochondria as a therapeutic target for cardiovascular disorders that could not be discussed and cited owing to space limitations. M.R.W. is supported by a Polish National Science Centre grant (UMO-2014/15/B/NZ1/00490). D.A.S. receives support from the Glenn Foundation for Medical Research, the Sinclair Gift Fund, and the US NIH/National Institute on Aging (R01 AG028730 and R01 DK100263). G.K. receives support from the Ligue Nationale contre le Cancer Comité de Charente-Maritime (équipe labellisée); the Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; the Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), the Fondation pour la Recherche Médicale (FRM); a donation by Elior; the European Commission (ArtForce); the European Research Council (ERC); Fondation Carrefour; the Institut National du Cancer (INCa); INSERM (HTE); the Institut Universitaire de France; the LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI). P.P. is grateful to Camilla degli Scrovegni for continuous support; P.P. receives support from the Italian Ministry of Education, the University and Research; Telethon (GGP15219/B); the Italian Association for Cancer Research (AIRC; IG-18624); and by local funds from the University of Ferrara (Ferrara, Italy). L.G. is supported by a start-up grant from the Department of Radiation Oncology at Weill Cornell Medicine (New York, NY, USA) and by donations from Sotio a.s. (Prague, Czech Republic), Phosplatin (New York, NY, USA), and the Luke Heller TECPR2 Foundation (Boston, MA, USA).
Related links
ClinicalTrials.gov: https://clinicaltrials.gov
Footnotes
Competing interests
D.A.S. is a consultant to and inventor on patents licensed to CohBar, GlaxoSmithKline, Jumpstart Fertility, Liberty Biosecurity, Life Biosciences, and MetroBiotech. The other authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Cardiology thanks R. Gottlieb, M. Hirschey, M. Sack, and the other anonymous reviewer(s) for their contribution to the peer review of this work.
References
- 1.Lopez-Crisost C. et al. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol 14, 342–360 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Mehta MM, Weinberg SE & Chandel NS Mitochondrial control of immunity: beyond ATP. Nat. Rev. Immunol 17, 608–620 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Kepp O, Trojel-Hansen C & Kroemer G Mitochondrial control of cellular life, stress, and death. Circ. Res 111, 1198–1207 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors of this paper provide a comprehensive review of the pathways through which mammalian cells die in response to microenvironmental perturbations and propose a unified nomenclature for cell death on the basis of mechanistic and essential (as opposed to morphological and dispensable) aspects of the process.
- 5.Delbridge LMD, Mellor KM, Taylor DJ & Gottlieb RA Myocardial stress and autophagy: mechanisms and potential therapies. Nat. Rev. Cardiol 14, 412–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green DR, Galluzzi L & Kroemer G Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333, 1109–1112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galluzzi L et al. Molecular definitions of autophagy and related processes. EMBO J 36, 1811–1836 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stolz A, Ernst A & Dikic I Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol 16, 495–501 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Sica V et al. Organelle-specific initiation of autophagy. Mol. Cell 59, 522–539 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Harper JW, Ordureau A & Heo JM Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol 19, 93–108 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Murphy E et al. Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circ. Res 118, 1960–1991 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulze PC, Drosatos K & Goldberg IJ Lipid use and misuse by the heart. Circ. Res 118, 1736–1751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu YF, Chen YJ, Lin YJ & Chen SA Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol 12, 230–243 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Gistera A & Hansson GK The immunology of atherosclerosis. Nat. Rev. Nephrol 13, 368–380 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Amgalan D, Chen Y & Kitsis RN Death receptor signaling in the heart: cell survival, apoptosis, and necroptosis. Circulation 136, 743–746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suomalainen A & Battersby BJ Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol 19, 77–92 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Bravo-San Pedro JM, Kroemer G & Galluzzi L Autophagy and mitophagy in cardiovascular disease. Circ. Res 120, 1812–1824 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Brown DA et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol 14, 238–250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreux PA, Houtkooper RH & Auwerx J Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov 12, 465–483 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill BG & Schulze PC Insights into metabolic remodeling of the hypertrophic and failing myocardium. Circ. Heart Fail 7, 874–876 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnan J et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab 9, 512–524 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Sun H et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 133, 2038–2049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sansbury BE et al. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ. Heart Fail 7, 634–642 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi YS et al. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J. Mol. Cell. Cardiol 100, 64–71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chouchani ET et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides an elegant demonstration that succinate accumulating in the ischaemic myocardium drives an intense oxidative burst at respiratory complex I during reperfusion, thereby contributing to the pathogenesis of ischaemia–reperfusion injury.
- 26.Dodd MS et al. Impaired in vivo mitochondrial Krebs cycle activity after myocardial infarction assessed using hyperpolarized magnetic resonance spectroscopy. Circ. Cardiovasc. Imaging 7, 895–904 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu SP et al. Increased COUP-TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat. Commun 6, 8245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moll S & Varga EA Homocysteine and MTHFR mutations. Circulation 132, e6–e9 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Rodenburg RJ Mitochondrial complex I-linked disease. Biochim. Biophys. Acta 1857, 938–945 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Joza N et al. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol. Cell. Biol 25, 10261–10272 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H et al. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc. Natl Acad. Sci. USA 97, 3467–3472 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iliceto S et al. Effects of L-carnitine administration on left ventricular remodeling after acute anterior myocardial infarction: the L-Carnitine Ecocardiografia Digitalizzata Infarto Miocardico (CEDIM) Trial. J. Am. Coll. Cardiol 26, 380–387 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Davini P, Bigalli A, Lamanna F & Boem A Controlled study on L-carnitine therapeutic efficacy in post-infarction. Drugs Exp. Clin. Res 18, 355–365 (1992). [PubMed] [Google Scholar]
- 34.Iyer R, Gupta A, Khan A, Hiremath S & Lokhandwala Y Does left ventricular function improve with L-carnitine after acute myocardial infarction? J. Postgrad. Med 45, 38–41 (1999). [PubMed] [Google Scholar]
- 35.Tarantini G et al. Metabolic treatment with L-carnitine in acute anterior ST segment elevation myocardial infarction. A randomized controlled trial. Cardiology 106, 215–223 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Koeth RA et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med 19, 576–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt-Schweda S & Holubarsch C First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin. Sci. (Lond.) 99, 27–35 (2000). [PubMed] [Google Scholar]
- 38.Holubarsch CJ et al. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. (Lond.) 113, 205–212 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Chong CR, Ong GJ & Horowitz JD Emerging drugs for the treatment of angina pectoris. Expert Opin. Emerg. Drugs 21, 365–376 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Beadle RM et al. Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail 3, 202–211 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Yin X et al. Effects of perhexiline-induced fuel switch on the cardiac proteome and metabolome. J. Mol. Cell. Cardiol 55, 27–30 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marino G et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell 53, 710–725 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Abozguia K et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 122, 1562–1569 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Senanayake EL et al. Multicentre double-blind randomized controlled trial of perhexiline as a metabolic modulator to augment myocardial protection in patients with left ventricular hypertrophy undergoing cardiac surgery. Eur. J. Cardiothorac. Surg 48, 354–362 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Drury NE et al. The effect of perhexiline on myocardial protection during coronary artery surgery: a two-centre, randomized, double-blind, placebo-controlled trial. Eur. J. Cardiothorac. Surg 47, 464–472 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang N et al. The effectiveness of preoperative trimetazidine on myocardial preservation in coronary artery bypass graft patients: a systematic review and meta-analysis. Cardiology 131, 86–96 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Tuunanen H et al. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation 118, 1250–1258 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Vitale C et al. Trimetazidine improves exercise performance in patients with peripheral arterial disease. Pharmacol. Res 63, 278–283 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Rosano GM, Vitale C, Sposato B, Mercuro G & Fini M Trimetazidine improves left ventricular function in diabetic patients with coronary artery disease: a double-blind placebo-controlled study. Cardiovasc. Diabetol 2, 16 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marin TL et al. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal 10, eaaf7478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leung JM et al. An initial multicenter, randomized controlled trial on the safety and efficacy of acadesine in patients undergoing coronary artery bypass graft surgery. SPI Research Group. Anesth. Analg 78, 420–434 (1994). [DOI] [PubMed] [Google Scholar]
- 52.Menasche P, Jamieson WR, Flameng W & Davies MK Acadesine: a new drug that may improve myocardial protection in coronary artery bypass grafting. Results of the first international multicenter study. Multinational Acadesine Study Group. J. Thorac. Cardiovasc. Surg 110, 1096–1106 (1995). [DOI] [PubMed] [Google Scholar]
- 53.Newman MF et al. Effect of adenosine-regulating agent acadesine on morbidity and mortality associated with coronary artery bypass grafting: the RED-CABG randomized controlled trial. JAMA 308, 157–164 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Houtkooper RH, Pirinen E & Auwerx J Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol 13, 225–238 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howitz KT et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Anderson RM, Bitterman KJ, Wood JG, Medvedik O & Sinclair DA Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 55 and 56 are among the first studies to demonstrate that sirtuin activation by calorie restriction or specific pharmacological agents extends the lifespan of Saccharomyces cerevisiae.
- 57.Bonkowski MS & Sinclair DA Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol 17, 679–690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hershberger KA, Martin AS & Hirschey MD Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol 13, 213–225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng HL et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl Acad. Sci. USA 100, 10794–10799 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nadtochiy SM et al. SIRT1-mediated acute cardioprotection. Am. J. Physiol. Heart Circ. Physiol 301, H1506–H1512 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F & Kroemer G Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab 21, 805–821 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Alcendor RR et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res 100, 1512–1521 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Sundaresan NR et al. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest 119, 2758–2771 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hafner AV et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2, 914–923 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang X et al. SIRT2 acts as a cardioprotective deacetylase in pathological cardiac hypertrophy. Circulation 136, 2051–2067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sadhukhan S et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl Acad. Sci. USA 113, 4320–4325 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sundaresan NR et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med 18, 1643–1650 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vakhrusheva O et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res 102, 703–710 (2008). [DOI] [PubMed] [Google Scholar]
- 69.Luo YX et al. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur. Heart J 38, 1389–1398 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Hariharan N et al. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res 107, 1470–1482 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang W et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell 167, 985–1000. e21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article elucidates the interactome of mitochondrial sirtuins and identifies a physical interaction between SIRT3 and the F1Fo ATP synthase that supports physiological metabolism in healthy cells.
- 72.Cohen HY et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Baur JA & Sinclair DA Therapeutic potential of resveratrol: the in vivo evidence. Nat. Rev. Drug Discov 5, 493–506 (2006). [DOI] [PubMed] [Google Scholar]
- 74.Pillai VB et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun 6, 6656 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen YX, Zhang M, Cai Y, Zhao Q & Dai W The Sirt1 activator SRT1720 attenuates angiotensin II-induced atherosclerosis in apoE(−)/(−) mice through inhibiting vascular inflammatory response. Biochem. Biophys. Res. Commun 465, 732–738 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Chan AY et al. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J. Biol. Chem 283, 24194–24201 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sulaiman M et al. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol 298, H833–H843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martin AS et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight 2, 93885 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang R et al. Short-term administration of nicotinamide mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J. Mol. Cell. Cardiol 112, 64–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yamamoto T et al. Nicotinamide mononucleotide, an intermediate of NAD + synthesis, protects the heart from ischemia and reperfusion. PLoS ONE 9, e98972 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bo S et al. Six months of resveratrol supplementation has no measurable effect in type 2 diabetic patients. A randomized, double blind, placebo-controlled trial. Pharmacol. Res 111, 896–905 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Marques B et al. Beneficial effects of acute trans-resveratrol supplementation in treated hypertensive patients with endothelial dysfunction. Clin. Exp. Hypertens 40, 218–223 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Magyar K et al. Cardioprotection by resveratrol: a human clinical trial in patients with stable coronary artery disease. Clin. Hemorheol. Microcirc 50, 179–187 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Krueger JG et al. A randomized, placebo-controlled study of SRT2104, a SIRT1 activator, in patients with moderate to severe psoriasis. PLoS ONE 10, e0142081 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mishra P & Chan DC Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol 15, 634–646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Burte F, Carelli V, Chinnery PF & Yu-Wai-Man P Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol 11, 11–24 (2015). [DOI] [PubMed] [Google Scholar]
- 87.Piquereau J et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc. Res 94, 408–417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wai T et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350, aad0116 (2015). [DOI] [PubMed] [Google Scholar]; The authors of this paper elegantly demonstrate that genetic interventions enabling accelerated OPA1 proteolysis profoundly alter mitochondrial metabolism to cause dilated cardiomyopathy and HF.
- 89.Samant SA et al. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell. Biol 34, 807–819 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen Y, Liu Y & Dorn GW II. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res 109, 1327–1331 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Song M, Franco A, Fleischer JA, Zhang L & Dorn GW II. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab 26, 872–883.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hall AR et al. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis 7, e2238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cahill TJ et al. Resistance of dynamin-related protein 1 oligomers to disassembly impairs mitophagy, resulting in myocardial inflammation and heart failure. J. Biol. Chem 290, 25907–25919 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Papanicolaou KN et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol 31, 1309–1328 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen Y & Dorn GW II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu H et al. Mitofusin 2 inhibits angiotensin II-induced myocardial hypertrophy. J. Cardiovasc. Pharmacol. Ther 16, 205–211 (2011). [DOI] [PubMed] [Google Scholar]
- 97.Gong G et al. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350, aad2459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reports that cardiac deletion of Park2 or overexpression of MFN2-AA from birth (but not after weaning) blocks postnatal mitophagy-dependent mitochondrial maturation, which is essential for survival.
- 98.Coronado M et al. Physiological mitochondrial fragmentation is a normal cardiac adaptation to increased energy demand. Circ. Res 122, 282–295 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shirakabe A et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation 133, 1249–1263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ikeda Y et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res 116, 264–278 (2015). [DOI] [PubMed] [Google Scholar]
- 101.Fang L et al. Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo. Life Sci 80, 2154–2160 (2007). [DOI] [PubMed] [Google Scholar]
- 102.Javadov S et al. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: the effect of NHE-1 inhibition. Basic Res. Cardiol 106, 99–109 (2011). [DOI] [PubMed] [Google Scholar]
- 103.Chen L, Gong Q, Stice JP & Knowlton AA Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res 84, 91–99 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guo X et al. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ. Res 101, 1113–1122 (2007). [DOI] [PubMed] [Google Scholar]
- 105.Chen KH et al. Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Biol 6, 872–883 (2004). [DOI] [PubMed] [Google Scholar]
- 106.Sharp WW et al. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J 28, 316–326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ong SB et al. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121, 2012–2022 (2010). [DOI] [PubMed] [Google Scholar]
- 108.Ishikita A et al. Nanoparticle-mediated delivery of mitochondrial division inhibitor 1 to the myocardium protects the heart from ischemia-reperfusion injury through inhibition of mitochondria outer membrane permeabilization: a new therapeutic modality for acute myocardial infarction. J. Am. Heart Assoc 5, e003872 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Givvimani S et al. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS ONE 7, e32388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gharanei M, Hussain A, Janneh O & Maddock H Attenuation of doxorubicin-induced cardiotoxicity by mdivi-1: a mitochondrial division/mitophagy inhibitor. PLoS ONE 8, e77713 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bordt EA et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev. Cell 40, 583–594.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tian L et al. Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J. Mol. Med. (Berl.) 95, 381–393 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Disatnik MH et al. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc 2, e000461 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gao D et al. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS ONE 8, e60967 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Franco A et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540, 74–79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors generate a cell-permeant small peptide that can restore the ability of mutant MFN2 to mediate mitochondrial fusion.
- 116.Billia F et al. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl Acad. Sci. USA 108, 9572–9577 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Song M et al. Interdependence of parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ. Res 117, 346–351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bhandari P, Song M, Chen Y, Burelle Y & Dorn GW II. Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ. Res 114, 257–265 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dorn GW II. Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J. Cardiovasc. Transl Res 3, 374–383 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nakai A et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med 13, 619–624 (2007). [DOI] [PubMed] [Google Scholar]
- 121.Taneike M et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 6, 600–606 (2010). [DOI] [PubMed] [Google Scholar]
- 122.Zaglia T et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J. Clin. Invest 124, 2410–2424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nishino I et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906–910 (2000). [DOI] [PubMed] [Google Scholar]
- 124.Xu W et al. Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. Cell Rep 13, 533–545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hoshino A et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun 4, 2308 (2013). [DOI] [PubMed] [Google Scholar]
- 126.Zhao W et al. Atg5 deficiency-mediated mitophagy aggravates cardiac inflammation and injury in response to angiotensin II. Free Radic. Biol. Med 69, 108–115 (2014). [DOI] [PubMed] [Google Scholar]
- 127.Liao X et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 15, 545–553 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Razani B et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 15, 534–544 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sciarretta S et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation 125, 1134–1146 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR & Kroemer G Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov 16, 487–511 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Oka T et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is the first demonstration that defects in the autophagic degradation of mtDNA result in the activation of a TLR9-dependent inflammatory response that contributes to the pathogenesis of dilated cardiomyopathy.
- 132.Zhang Z et al. Mitochondrial DNA-LL-37 complex promotes atherosclerosis by escaping from autophagic recognition. Immunity 43, 1137–1147 (2015). [DOI] [PubMed] [Google Scholar]
- 133.Bhuiyan MS et al. Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest 123, 5284–5297 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hoshino A et al. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J. Mol. Cell. Cardiol 52, 175–184 (2012). [DOI] [PubMed] [Google Scholar]
- 135.Maejima Y et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med 19, 1478–1488 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liu X et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol. Cell. Biol 32, 4493–4504 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zummo FP et al. Glucagon-like peptide 1 protects pancreatic beta-cells from death by increasing autophagic flux and restoring lysosomal function. Diabetes 66, 1272–1285 (2017). [DOI] [PubMed] [Google Scholar]
- 138.Krech J et al. Moderate therapeutic hypothermia induces multimodal protective effects in oxygen-glucose deprivation/reperfusion injured cardiomyocytes. Mitochondrion 35, 1–10 (2017). [DOI] [PubMed] [Google Scholar]
- 139.Lu W et al. Mitochondrial protein PGAM5 regulates mitophagic protection against cell necroptosis. PLoS ONE 11, e0147792 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Marchi S et al. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol. Med 7, 1403–1417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tannous P et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc. Natl Acad. Sci. USA 105, 9745–9750 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhu H et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest 117, 1782–1793 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Matsui Y et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res 100, 914–922 (2007). [DOI] [PubMed] [Google Scholar]
- 144.Li DL et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 133, 1668–1687 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Elgendy M et al. Beclin 1 restrains tumorigenesis through Mcl-1 destabilization in an autophagy-independent reciprocal manner. Nat. Commun 5, 5637 (2014). [DOI] [PubMed] [Google Scholar]
- 146.Huang C et al. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE 6, e20975 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zesiewicz TA et al. Heart failure in Parkinson’s disease: analysis of the United States medicare current beneficiary survey. Parkinsonism Relat. Disord 10, 417–420 (2004). [DOI] [PubMed] [Google Scholar]
- 148.Eisenberg T et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med 22, 1428–1438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that oral supplementation of the natural polyamine spermidine extends the lifespan of mice as it exerts cardioprotective effects in the ageing myocardium that depend on the core autophagy protein ATG5.
- 149.Pietrocola F et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ 22, 509–516 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Eisenberg T et al. Dietary spermidine for lowering high blood pressure. Autophagy 13, 767–769 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dai DF et al. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 13, 529–539 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yan L et al. Calorie restriction can reverse, as well as prevent, aging cardiomyopathy. Age (Dordr.) 35, 2177–2182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wu X et al. Impaired autophagy contributes to adverse cardiac remodeling in acute myocardial infarction. PLoS ONE 9, e112891 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Goh SS et al. The red wine antioxidant resveratrol prevents cardiomyocyte injury following ischemia-reperfusion via multiple sites and mechanisms. Antioxid. Redox Signal 9, 101–113 (2007). [DOI] [PubMed] [Google Scholar]
- 155.Kanamori H et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am. J. Physiol. Heart Circ. Physiol 300, H2261–H2271 (2011). [DOI] [PubMed] [Google Scholar]
- 156.Liu B et al. Puerarin prevents cardiac hypertrophy induced by pressure overload through activation of autophagy. Biochem. Biophys. Res. Commun 464, 908–915 (2015). [DOI] [PubMed] [Google Scholar]
- 157.Garg S, Bourantas C & Serruys PW New concepts in the design of drug-eluting coronary stents. Nat. Rev. Cardiol 10, 248–260 (2013). [DOI] [PubMed] [Google Scholar]
- 158.Marx SO & Marks AR Bench to bedside: the development of rapamycin and its application to stent restenosis. Circulation 104, 852–855 (2001). [DOI] [PubMed] [Google Scholar]
- 159.Zhang Y et al. Rapamycin promotes the autophagic degradation of oxidized low-density lipoprotein in human umbilical vein endothelial cells. J. Vasc. Res 52, 210–219 (2015). [DOI] [PubMed] [Google Scholar]
- 160.Martinet W, Verheye S & De Meyer GR Everolimus-induced mTOR inhibition selectively depletes macrophages in atherosclerotic plaques by autophagy. Autophagy 3, 241–244 (2007). [DOI] [PubMed] [Google Scholar]
- 161.Verheye S et al. Selective clearance of macrophages in atherosclerotic plaques by autophagy. J. Am. Coll. Cardiol 49, 706–715 (2007). [DOI] [PubMed] [Google Scholar]
- 162.Pietrocola F et al. Aspirin recapitulates features of caloric restriction. Cell Rep 22, 2395–2407 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Andres AM et al. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid. Redox Signal 21, 1960–1973 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.McLendon PM et al. Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy. Proc. Natl Acad. Sci. USA 111, E5178–E5186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Xie M et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129, 1139–1151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kho C, Lee A & Hajjar RJ Altered sarcoplasmic reticulum calcium cycling—targets for heart failure therapy. Nat. Rev. Cardiol 9, 717–733 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Baughman JM et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.De Stefani D, Raffaello A, Teardo E, Szabo I & Rizzuto R A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 167 and 168 are the first studies to identify and characterize the long-sought protein responsible for ruthenium red-sensitive mitochondrial Ca2+ import, which was named MCU.
- 169.Boyman L, Williams GS, Khananshvili D, Sekler I & Lederer WJ NCLX: the mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol 59, 205–213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Williams GS, Boyman L & Lederer WJ Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell. Cardiol 78, 35–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bonora M et al. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34, 1475–1486 (2015). [DOI] [PubMed] [Google Scholar]
- 172.Santulli G, Xie W, Reiken SR & Marks AR Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl Acad. Sci. USA 112, 11389–11394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors provide robust evidence in support of the notion that diastolic Ca2+ leaks from the sarcoplasmic reticulum lead to mitochondrial Ca2+ overload, which contributes to the pathogenesis of HF.
- 173.Pan X et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol 15, 1464–1472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Luongo TS et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 12, 23–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kwong JQ et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep 12, 15–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rasmussen TP et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl Acad. Sci. USA 112, 9129–9134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Luongo TS et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545, 93–97 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that deletion of Slc8b1 from the adult heart causes sudden death as a consequence of severe myocardial dysfunction and fulminant HF.
- 178.Venetucci L, Denegri M, Napolitano C & Priori SG Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat. Rev. Cardiol 9, 561–575 (2012). [DOI] [PubMed] [Google Scholar]
- 179.Joiner ML et al. CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors identify a functional link between CaMKII and MPT-driven regulated necrosis, namely, MCU-dependent Ca2+ overload, which contributes to ischaemia–reperfusion injury in the mouse myocardium.
- 180.Xu S et al. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat. Commun 7, 13189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fazal L et al. Multifunctional mitochondrial Epac1 controls myocardial cell death. Circ. Res 120, 645–657 (2017). [DOI] [PubMed] [Google Scholar]
- 182.Westenbrink BD et al. Mitochondrial reprogramming induced by CaMKIIdelta mediates hypertrophy decompensation. Circ. Res 116, e28–e39 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Maione AS et al. Cellular subtype expression and activation of CaMKII regulate the fate of atherosclerotic plaque. Atherosclerosis 256, 53–61 (2017). [DOI] [PubMed] [Google Scholar]
- 184.Macle L & Nattel S Arrhythmias in 2015: advances in drug, ablation, and device therapy for cardiac arrhythmias. Nat. Rev. Cardiol 13, 67–68 (2016). [DOI] [PubMed] [Google Scholar]
- 185.Liu T et al. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res 115, 44–54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tanaka H et al. Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br. J. Pharmacol 135, 1096–1100 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kon N et al. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov 3, 17045 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Arduino DM et al. Systematic identification of MCU modulators by orthogonal interspecies chemical screening. Mol. Cell 67, 711–723.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Thu VT et al. NecroX-5 prevents hypoxia/ reoxygenation injury by inhibiting the mitochondrial calcium uniporter. Cardiovasc. Res 94, 342–350 (2012). [DOI] [PubMed] [Google Scholar]
- 190.Pellicena P & Schulman H CaMKII inhibitors: from research tools to therapeutic agents. Front. Pharmacol 5, 21 (2014).24600394 [Google Scholar]
- 191.Shadel GS & Horvath TL Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Galluzzi L, Kepp O, Krautwald S, Kroemer G & Linkermann A Molecular mechanisms of regulated necrosis. Semin. Cell Dev. Biol 35, 24–32 (2014). [DOI] [PubMed] [Google Scholar]
- 193.Sam F et al. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J. Card. Fail 11, 473–480 (2005). [DOI] [PubMed] [Google Scholar]
- 194.Montaigne D et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130, 554–564 (2014). [DOI] [PubMed] [Google Scholar]
- 195.Dai DF et al. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol 58, 73–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mallat Z et al. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 97, 1536–1539 (1998). [DOI] [PubMed] [Google Scholar]
- 197.Kono Y et al. Elevated levels of oxidative DNA damage in serum and myocardium of patients with heart failure. Circ. J 70, 1001–1005 (2006). [DOI] [PubMed] [Google Scholar]
- 198.Canton M et al. Oxidation of myofibrillar proteins in human heart failure. J. Am. Coll. Cardiol 57, 300–309 (2011). [DOI] [PubMed] [Google Scholar]
- 199.Ide T et al. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res 85, 357–363 (1999). [DOI] [PubMed] [Google Scholar]
- 200.Judge S, Jang YM, Smith A, Hagen T & Leeuwenburgh C Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J 19, 419–421 (2005). [DOI] [PubMed] [Google Scholar]
- 201.Ungvari Z et al. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am. J. Physiol. Heart Circ. Physiol 293, H37–H47 (2007). [DOI] [PubMed] [Google Scholar]
- 202.Vendrov AE et al. Attenuated superoxide dismutase 2 activity induces atherosclerotic plaque instability during aging in hyperlipidemic mice. J. Am. Heart Assoc 6, e006775 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nakada Y et al. Hypoxia induces heart regeneration in adult mice. Nature 541, 222–227 (2017). [DOI] [PubMed] [Google Scholar]; This study is the first report demonstrating that gradual respiratory hypoxia, resulting in severe systemic hypoxaemia, after MI induces a robust regenerative response that ameliorates disease outcome, at least in mice.
- 204.Conrad M et al. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol. Cell. Biol 24, 9414–9423 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kiermayer C et al. Heart-specific knockout of the mitochondrial thioredoxin reductase (Txnrd2) induces metabolic and contractile dysfunction in the aging myocardium. J. Am. Heart Assoc 4, e002153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Mortensen SA et al. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC Heart Fail 2, 641–649 (2014). [DOI] [PubMed] [Google Scholar]
- 207.Oleck S & Ventura HO Coenzyme Q10 and utility in heart failure: just another supplement? Curr. Heart Fail. Rep 13, 190–195 (2016). [DOI] [PubMed] [Google Scholar]
- 208.Judy WV, Stogsdill WW & Folkers K Myocardial preservation by therapy with coenzyme Q10 during heart surgery. Clin. Investig 71, S155–S161 (1993). [DOI] [PubMed] [Google Scholar]
- 209.Singh RB et al. Randomized, double-blind placebo-controlled trial of coenzyme Q10 in patients with acute myocardial infarction. Cardiovasc. Drugs Ther 12, 347–353 (1998). [DOI] [PubMed] [Google Scholar]
- 210.Brown BG et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N. Engl. J. Med 345, 1583–1592 (2001). [DOI] [PubMed] [Google Scholar]
- 211.Heart Protection Study Collaborative Group. MRC/ BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 360, 23–33 (2002).12114037 [Google Scholar]; This large, randomized, placebo-controlled clinical trial demonstrates that antioxidant vitamin supplementation does not produce any significant reduction in 5-year mortality from, or incidence of, any type of CVD.
- 212.Stephens NG et al. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 347, 781–786 (1996). [DOI] [PubMed] [Google Scholar]
- 213.Sesso HD et al. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians’ Health Study II randomized controlled trial. JAMA 300, 2123–2133 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lee IM et al. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women’s Health Study: a randomized controlled trial. JAMA 294, 56–65 (2005). [DOI] [PubMed] [Google Scholar]
- 215.Magliano D et al. The Melbourne Atherosclerosis Vitamin E Trial (MAVET): a study of high dose vitamin E in smokers. Eur. J. Cardiovasc. Prev. Rehabil 13, 341–347 (2006). [DOI] [PubMed] [Google Scholar]
- 216.Singh RB et al. Effect of coenzyme Q10 on risk of atherosclerosis in patients with recent myocardial infarction. Mol. Cell. Biochem 246, 75–82 (2003). [PubMed] [Google Scholar]
- 217.Cho J et al. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron. Artery Dis 18, 215–220 (2007). [DOI] [PubMed] [Google Scholar]
- 218.Eirin A et al. Restoration of mitochondrial cardiolipin attenuates cardiac damage in swine renovascular hypertension. J. Am. Heart Assoc 5, e003118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lu HI et al. Administration of antioxidant peptide SS-31 attenuates transverse aortic constriction-induced pulmonary arterial hypertension in mice. Acta Pharmacol. Sin 37, 589–603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kroller-Schon S et al. Peroxisome proliferator-activated receptor gamma, coactivator 1alpha deletion induces angiotensin II-associated vascular dysfunction by increasing mitochondrial oxidative stress and vascular inflammation. Arterioscler. Thromb. Vasc. Biol 33, 1928–1935 (2013). [DOI] [PubMed] [Google Scholar]
- 221.Ribeiro Junior RF et al. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med 117, 18–29 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Adlam VJ et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J 19, 1088–1095 (2005). [DOI] [PubMed] [Google Scholar]
- 223.Sloan RC et al. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J. Mol. Cell. Cardiol 52, 1009–1018 (2012). [DOI] [PubMed] [Google Scholar]
- 224.Brown DA et al. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J. Cardiovasc. Pharmacol. Ther 19, 121–132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Shi J et al. Bendavia restores mitochondrial energy metabolism gene expression and suppresses cardiac fibrosis in the border zone of the infarcted heart. Life Sci 141, 170–178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zhang M et al. Chronic administration of mitochondrion-targeted peptide SS-31 prevents atherosclerotic development in ApoE knockout mice fed Western diet. PLoS ONE 12, e0185688 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Saad A et al. Phase 2a clinical trial of mitochondrial protection (elamipretide) during stent revascularization in patients with atherosclerotic renal artery stenosis. Circ. Cardiovasc. Interv 10, e005487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Gibson CM et al. EMBRACE STEMI study: a Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur. Heart J 37, 1296–1303 (2016). [DOI] [PubMed] [Google Scholar]
- 229.Daubert MA et al. Novel mitochondria-targeting peptide in heart failure treatment: a randomized, placebo-controlled trial of elamipretide. Circ. Heart Fail 10, e004389 (2017). [DOI] [PubMed] [Google Scholar]
- 230.Ruparelia N, Chai JT, Fisher EA & Choudhury RP Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat. Rev. Cardiol 14, 133–144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Buck MD, Sowell RT, Kaech SM & Pearce EL Metabolic instruction of immunity. Cell 169, 570–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Mills EL, Kelly B & O’Neill LAJ Mitochondria are the powerhouses of immunity. Nat. Immunol 18, 488–498 (2017). [DOI] [PubMed] [Google Scholar]
- 233.Galluzzi L, Kepp O & Kroemer G Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol 13, 780–788 (2012). [DOI] [PubMed] [Google Scholar]
- 234.Durga Devi T et al. Aggravated postinfarct heart failure in type 2 diabetes is associated with impaired mitophagy and exaggerated inflammasome activation. Am. J. Pathol 187, 2659–2673 (2017). [DOI] [PubMed] [Google Scholar]
- 235.Mao Y et al. STING-IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler. Thromb. Vasc. Biol 37, 920–929 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhang Q et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kepp O, Loos F, Liu P & Kroemer G Extracellular nucleosides and nucleotides as immunomodulators. Immunol. Rev 280, 83–92 (2017). [DOI] [PubMed] [Google Scholar]
- 238.Dieude M et al. Cardiolipin binds to CD1d and stimulates CD1d-restricted gammadelta T cells in the normal murine repertoire. J. Immunol 186, 4771–4781 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Galluzzi L, Vanpouille-Box C, Bakhoum SF & Demaria S Snapshot: CGAS-STING signaling. Cell 173, 276–276.e1 (2018). [DOI] [PubMed] [Google Scholar]
- 240.Cao D et al. Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation 137, 2613–2634 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ & Kroemer G Type I interferons in anticancer immunity. Nat. Rev. Immunol 15, 405–414 (2015). [DOI] [PubMed] [Google Scholar]
- 242.King KR et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med 23, 1481–1487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is an elegant demonstration that interferon regulatory factor 3 (IRF3)-dependent type I interferon synthesis leads to the establishment of an inflammatory response that aggravates disease outcome after MI.
- 243.Sandanger O et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res 99, 164–174 (2013). [DOI] [PubMed] [Google Scholar]
- 244.Duewell P et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Chamberlain J et al. Interleukin-1 regulates multiple atherogenic mechanisms in response to fat feeding. PLoS ONE 4, e5073 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Merhi-Soussi F et al. Interleukin-1 plays a major role in vascular inflammation and atherosclerosis in male apolipoprotein E-knockout mice. Cardiovasc. Res 66, 583–593 (2005). [DOI] [PubMed] [Google Scholar]
- 247.Gidlof O et al. A common missense variant in the ATP receptor P2X7 is associated with reduced risk of cardiovascular events. PLoS ONE 7, e37491 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Marchetti C et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol 63, 316–322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Marchetti C et al. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. J. Cardiovasc. Pharmacol 66, 1–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Granado M et al. Altered expression of P2Y2 and P2X7 purinergic receptors in the isolated rat heart mediates ischemia-reperfusion injury. Vascul. Pharmacol 73, 96–103 (2015). [DOI] [PubMed] [Google Scholar]
- 251.McCarthy CG et al. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc. Res 107, 119–130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Szeto HH First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol 171, 2029–2050 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Shen B, Li J & Yang B NKG2D blockade significantly attenuates ischemia-reperfusion injury in a cardiac transplantation model. Transplant. Proc 45, 2513–2516 (2013). [DOI] [PubMed] [Google Scholar]
- 254.von Lueder TG & Krum H New medical therapies for heart failure. Nat. Rev. Cardiol 12, 730–740 (2015). [DOI] [PubMed] [Google Scholar]
- 255.Back M & Hansson GK Anti-inflammatory therapies for atherosclerosis. Nat. Rev. Cardiol 12, 199–211 (2015). [DOI] [PubMed] [Google Scholar]
- 256.Galluzzi L, Kepp O & Kroemer G Mitochondrial regulation of cell death: a phylogenetically conserved control. Microb. Cell 3, 101–108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Toth A et al. Targeted deletion of Puma attenuates cardiomyocyte death and improves cardiac function during ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol 291, H52–H60 (2006). [DOI] [PubMed] [Google Scholar]
- 258.Kristen AV et al. Inhibition of apoptosis by the intrinsic but not the extrinsic apoptotic pathway in myocardial ischemia-reperfusion. Cardiovasc. Pathol 22, 280–286 (2013). [DOI] [PubMed] [Google Scholar]
- 259.Nakagawa T et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658 (2005). [DOI] [PubMed] [Google Scholar]
- 260.Baines CP et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 (2005). [DOI] [PubMed] [Google Scholar]; References 259 and 260 are among the first articles to provide robust genetic evidence in support of a non-redundant role for CypD in the regulation of MPT-driven regulated necrosis.
- 261.Itani HA et al. Mitochondrial cyclophilin D in vascular oxidative stress and hypertension. Hypertension 67, 1218–1227 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Gordan R, Fefelova N, Gwathmey JK & Xie LH Involvement of mitochondrial permeability transition pore (mPTP) in cardiac arrhythmias: evidence from cyclophilin D knockout mice. Cell Calcium 60, 363–372 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Zhou HZ et al. Poly(ADP-ribose) polymerase-1 hyperactivation and impairment of mitochondrial respiratory chain complex I function in reperfused mouse hearts. Am. J. Physiol. Heart Circ. Physiol 291, H714–H723 (2006). [DOI] [PubMed] [Google Scholar]
- 264.Zingarelli B et al. Absence of poly(ADP-ribose) polymerase-1 alters nuclear factor-kappa B activation and gene expression of apoptosis regulators after reperfusion injury. Mol. Med 9, 143–153 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wei SJ et al. Poly(ADP-ribose) polymerase 1 deficiency increases nitric oxide production and attenuates aortic atherogenesis through downregulation of arginase II. Clin. Exp. Pharmacol. Physiol 44, 114–122 (2017). [DOI] [PubMed] [Google Scholar]
- 266.Zhang T et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med 22, 175–182 (2016). [DOI] [PubMed] [Google Scholar]; The authors characterize a CaMKII-dependent mechanism whereby necroptotic receptor-interacting serine/threonine-protein kinase 3 (RIPK3) activation feeds into MPT-driven regulated necrosis, thereby contributing to HF driven by ischaemia–reperfusion or doxorubicin administration.
- 267.Oerlemans MI et al. Targeting cell death in the reperfused heart: pharmacological approaches for cardioprotection. Int. J. Cardiol 165, 410–422 (2013). [DOI] [PubMed] [Google Scholar]
- 268.Yaoita H, Ogawa K, Maehara K & Maruyama Y Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 97, 276–281 (1998). [DOI] [PubMed] [Google Scholar]
- 269.Yang W et al. MX1013, a dipeptide caspase inhibitor with potent in vivo antiapoptotic activity. Br. J. Pharmacol 140, 402–412 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Ono M et al. BH4 peptide derivative from Bcl-xL attenuates ischemia/reperfusion injury thorough anti-apoptotic mechanism in rat hearts. Eur. J. Cardiothorac. Surg 27, 117–121 (2005). [DOI] [PubMed] [Google Scholar]
- 271.Hetz C et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J. Biol. Chem 280, 42960–42970 (2005). [DOI] [PubMed] [Google Scholar]
- 272.Liu HR et al. Role of Omi/HtrA2 in apoptotic cell death after myocardial ischemia and reperfusion. Circulation 111, 90–96 (2005). [DOI] [PubMed] [Google Scholar]
- 273.Bhuiyan MS & Fukunaga K Inhibition of HtrA2/Omi ameliorates heart dysfunction following ischemia/ reperfusion injury in rat heart in vivo. Eur. J. Pharmacol 557, 168–177 (2007). [DOI] [PubMed] [Google Scholar]
- 274.Jiang X et al. A small molecule that protects the integrity of the electron transfer chain blocks the mitochondrial apoptotic pathway. Mol. Cell 63, 229–239 (2016). [DOI] [PubMed] [Google Scholar]
- 275.Li L et al. Discovery of highly potent 2-sulfonyl-pyrimidinyl derivatives for apoptosis inhibition and ischemia treatment. ACS Med. Chem. Lett 8, 407–412 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Schaller S et al. TRO40303, a new cardioprotective compound, inhibits mitochondrial permeability transition. J. Pharmacol. Exp. Ther 333, 696–706 (2010). [DOI] [PubMed] [Google Scholar]
- 277.Fancelli D et al. Cinnamic anilides as new mitochondrial permeability transition pore inhibitors endowed with ischemia-reperfusion injury protective effect in vivo. J. Med. Chem 57, 5333–5347 (2014). [DOI] [PubMed] [Google Scholar]
- 278.Conrad M, Angeli JP, Vandenabeele P & Stockwell BR Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov 15, 348–366 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.MITOCARE Study Group. Rationale and design of the ‘MITOCARE’ Study: a phase II, multicenter, randomized, double-blind, placebo-controlled study to assess the safety and efficacy of TRO40303 for the reduction of reperfusion injury in patients undergoing percutaneous coronary intervention for acute myocardial infarction. Cardiology 123, 201–207 (2012). [DOI] [PubMed] [Google Scholar]
- 280.Piot C et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med 359, 473–481 (2008). [DOI] [PubMed] [Google Scholar]
- 281.Cung TT et al. Cyclosporine before PCI in patients with acute myocardial infarction. N. Engl. J. Med 373, 1021–1031 (2015). [DOI] [PubMed] [Google Scholar]; This multicentre, double-blind, randomized clinical trial documents no cardioprotective role for cyclosporine A administered before PCI in patients with acute anterior ST-segment elevation MI.
- 282.Ottani F et al. Cyclosporine A in reperfused myocardial infarction: the multicenter, controlled, open-label CYCLE Trial. J. Am. Coll. Cardiol 67, 365–374 (2016). [DOI] [PubMed] [Google Scholar]
- 283.Le Lamer S et al. Translation of TRO40303 from myocardial infarction models to demonstration of safety and tolerance in a randomized Phase I trial. J. Transl Med 12, 38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Atar D et al. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur. Heart J 36, 112–119 (2015). [DOI] [PubMed] [Google Scholar]
- 285.van Gelder T, van Schaik RH & Hesselink DA Pharmacogenetics and immunosuppressive drugs in solid organ transplantation. Nat. Rev. Nephrol 10, 725–731 (2014). [DOI] [PubMed] [Google Scholar]
- 286.Choi SW & Reddy P Current and emerging strategies for the prevention of graft-versus-host disease. Nat. Rev. Clin. Oncol 11, 536–547 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Hausenloy DJ, Yellon DM, Mani-Babu S & Duchen MR Preconditioning protects by inhibiting the mitochondrial permeability transition. Am. J. Physiol. Heart Circ. Physiol 287, H841–H849 (2004). [DOI] [PubMed] [Google Scholar]
- 288.Ishii H et al. Impact of a single intravenous administration of nicorandil before reperfusion in patients with ST-segment-elevation myocardial infarction. Circulation 112, 1284–1288 (2005). [DOI] [PubMed] [Google Scholar]
- 289.Kitakaze M et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet 370, 1483–1493 (2007). [DOI] [PubMed] [Google Scholar]
- 290.Lee HC et al. Effect of intra-coronary nicorandil administration prior to reperfusion in acute ST segment elevation myocardial infarction. Circ. J 72, 1425–1429 (2008). [DOI] [PubMed] [Google Scholar]
- 291.Wojciechowska A, Braniewska A & Kozar-Kaminska K MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med 26, 865–874 (2017). [DOI] [PubMed] [Google Scholar]
- 292.Geiger J & Dalgaard LT Interplay of mitochondrial metabolism and microRNAs. Cell. Mol. Life Sci 74, 631–646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Das S et al. Divergent effects of miR-181 family members on myocardial function through protective cytosolic and detrimental mitochondrial microRNA targets. J. Am. Heart Assoc 6, e004694 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Wang K et al. E2F1-regulated miR-30b suppresses Cyclophilin D and protects heart from ischemia/ reperfusion injury and necrotic cell death. Cell Death Differ 22, 743–754 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Wang K et al. MicroRNA-2861 regulates programmed necrosis in cardiomyocyte by impairing adenine nucleotide translocase 1 expression. Free Radic. Biol. Med 91, 58–67 (2016). [DOI] [PubMed] [Google Scholar]
- 296.Ucar A et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun 3, 1078 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Li Z et al. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ 24, 1205–1213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Wang K et al. E2F1-dependent miR-421 regulates mitochondrial fragmentation and myocardial infarction by targeting Pink1. Nat. Commun 6, 7619 (2015). [DOI] [PubMed] [Google Scholar]
- 299.Das S et al. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS ONE 9, e96820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Tang Y et al. MicroRNA-150 protects the mouse heart from ischaemic injury by regulating cell death. Cardiovasc. Res 106, 387–397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Jagannathan R et al. Translational regulation of the mitochondrial genome following redistribution of mitochondrial microRNA in the diabetic heart. Circ. Cardiovasc. Genet 8, 785–802 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Li H et al. MicroRNA-21 lowers blood pressure in spontaneous hypertensive rats by upregulating mitochondrial translation. Circulation 134, 734–751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Guan X et al. miR-106a promotes cardiac hypertrophy by targeting mitofusin 2. J. Mol. Cell. Cardiol 99, 207–217 (2016). [DOI] [PubMed] [Google Scholar]
- 304.Wang K et al. NFAT4-dependent miR-324–5p regulates mitochondrial morphology and cardiomyocyte cell death by targeting Mtfr1. Cell Death Dis 6, e2007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Long B et al. miR-761 regulates the mitochondrial network by targeting mitochondrial fission factor. Free Radic. Biol. Med 65, 371–379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Wang JX et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med 17, 71–78 (2011). [DOI] [PubMed] [Google Scholar]
- 307.Karunakaran D et al. Macrophage mitochondrial energy status regulates cholesterol efflux and is enhanced by anti-miR33 in atherosclerosis. Circ. Res 117, 266–278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Rincon MY, VandenDriessche T & Chuah MK Gene therapy for cardiovascular disease: advances in vector development, targeting, and delivery for clinical translation. Cardiovasc. Res 108, 4–20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Rusnak F & Mertz P Calcineurin: form and function. Physiol. Rev 80, 1483–1521 (2000). [DOI] [PubMed] [Google Scholar]
- 310.Ikeda G et al. Nanoparticle-mediated targeting of cyclosporine a enhances cardioprotection against ischemia-reperfusion injury through inhibition of mitochondrial permeability transition pore opening. Sci. Rep 6, 20467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Kelso GF et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem 276, 4588–4596 (2001). [DOI] [PubMed] [Google Scholar]
- 312.Jiang J et al. A mitochondria-targeted triphenylphosphonium-conjugated nitroxide functions as a radioprotector/mitigator. Radiat. Res 172, 706–717 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Galluzzi L et al. Methods to dissect mitochondrial membrane permeabilization in the course of apoptosis. Methods Enzymol 442, 355–374 (2008). [DOI] [PubMed] [Google Scholar]
- 314.Horton KL, Stewart KM, Fonseca SB, Guo Q & Kelley SO Mitochondria-penetrating peptides. Chem. Biol 15, 375–382 (2008). [DOI] [PubMed] [Google Scholar]
- 315.Schmidt O, Pfanner N & Meisinger C Mitochondrial protein import: from proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol 11, 655–667 (2010). [DOI] [PubMed] [Google Scholar]
- 316.Barth E, Stammler G, Speiser B & Schaper J Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol 24, 669–681 (1992). [DOI] [PubMed] [Google Scholar]
- 317.Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G & Galluzzi L Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol 26, 655–667 (2016). [DOI] [PubMed] [Google Scholar]
- 318.Bonora M et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12, 674–683 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is the first demonstration that the c subunit of the F1Fo ATP synthase is mechanistically involved in MPT-driven regulated necrosis, at least in some experimental models.
- 319.Galluzzi L et al. No death without life: vital functions of apoptotic effectors. Cell Death Differ 15, 1113–1123 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Hao Z et al. Specific ablation of the apoptotic functions of cytochrome C reveals a differential requirement for cytochrome C and Apaf-1 in apoptosis. Cell 121, 579–591 (2005). [DOI] [PubMed] [Google Scholar]; The authors elegantly uncouple the vital and lethal roles of cytochrome c by generating a mutant cytochrome c that functions normally within the respiratory chain but fails to activate apoptotic protease-activating factor 1 (APAF1).
- 321.Zhivotovsky B, Galluzzi L, Kepp O & Kroemer G Adenine nucleotide translocase: a component of the phylogenetically conserved cell death machinery. Cell Death Differ 16, 1419–1425 (2009). [DOI] [PubMed] [Google Scholar]
- 322.De Grassi A, Lanave C & Saccone C Evolution of ATP synthase subunit c and cytochrome c gene families in selected Metazoan classes. Gene 371, 224–233 (2006). [DOI] [PubMed] [Google Scholar]
- 323.Mutlu-Turkoglu U et al. Increased plasma malondialdehyde and protein carbonyl levels and lymphocyte DNA damage in patients with angiographically defined coronary artery disease. Clin. Biochem 38, 1059–1065 (2005). [DOI] [PubMed] [Google Scholar]
- 324.Aryal B, Jeong J & Rao VA Doxorubicin-induced carbonylation and degradation of cardiac myosin binding protein C promote cardiotoxicity. Proc. Natl Acad. Sci. USA 111, 2011–2016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Ruiz M et al. Circulating acylcarnitine profile in human heart failure: a surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am. J. Physiol. Heart Circ. Physiol 313, H768–H781 (2017). [DOI] [PubMed] [Google Scholar]
- 326.Stotland A & Gottlieb RA α-MHC MitoTimer mouse: in vivo mitochondrial turnover model reveals remarkable mitochondrial heterogeneity in the heart. J. Mol. Cell. Cardiol 90, 53–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wilson RJ et al. Conditional MitoTimer reporter mice for assessment of mitochondrial structure, oxidative stress, and mitophagy. Mitochondrion 10.1016/j.mito.2017.12.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Li J, Lu J & Zhou Y Mitochondrial-targeted molecular imaging in cardiac disease. BioMed. Res. Int 2017, 5246853 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Sack MN & Murphy E The role of comorbidities in cardioprotection. J. Cardiovasc. Pharmacol. Ther 16, 267–272 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Galluzzi L et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 22, 58–73 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Weinlich R, Oberst A, Beere HM & Green DR Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol 18, 127–136 (2017). [DOI] [PubMed] [Google Scholar]
- 332.Galluzzi L, Lopez-Soto A, Kumar S & Kroemer G Caspases connect cell-death signaling to organismal homeostasis. Immunity 44, 221–231 (2016). [DOI] [PubMed] [Google Scholar]
- 333.Dean J, Cruz SD, Mehta PK & Merz CN Coronary microvascular dysfunction: sex-specific risk, diagnosis, and therapy. Nat. Rev. Cardiol 12, 406–414 (2015). [DOI] [PubMed] [Google Scholar]
- 334.Pagidipati NJ & Peterson ED Acute coronary syndromes in women and men. Nat. Rev. Cardiol 13, 471–480 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Hu XX et al. The cardioprotective effect of vitamin E (alpha-tocopherol) is strongly related to age and gender in mice. PLoS ONE 10, e0137405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Baines CP, Kaiser RA, Sheiko T, Craigen WJ & Molkentin JD Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol 9, 550–555 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Galluzzi L & Kroemer G Mitochondrial apoptosis without VDAC. Nat. Cell Biol 9, 487–489 (2007). [DOI] [PubMed] [Google Scholar]
- 338.Kokoszka JE et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Gustafsson AB & Gottlieb RA Heart mitochondria: gates of life and death. Cardiovasc. Res 77, 334–343 (2008). [DOI] [PubMed] [Google Scholar]
- 340.Green DR, Galluzzi L & Kroemer G Cell biology. Metabolic control of cell death. Science 345, 1250256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Sena LA & Chandel NS Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Schwarz K et al. The breathing heart — mitochondrial respiratory chain dysfunction in cardiac disease. Int. J. Cardiol 171, 134–143 (2014). [DOI] [PubMed] [Google Scholar]