

Summary
Interspecies cross‐feeding is a fundamental factor in anaerobic microbial communities. In the human colon, formate is produced by many bacterial species but is normally detected only at low concentrations. Ruminococcus bromii produces formate, ethanol and acetate in approximately equal molar proportions in pure culture on RUM‐RS medium with 0.2% Novelose resistant starch (RS3) as energy source. Batch co‐culturing on starch with the acetogen Blautia hydrogenotrophica however led to the disappearance of formate and increased levels of acetate, which is proposed to occur through the routing of formate via the Wood Ljungdahl pathway of B. hydrogenotrophica. We investigated these inter‐species interactions further using RNAseq to examine gene expression in continuous co‐cultures of R. bromii and B. hydrogenotrophica. Transcriptome analysis revealed upregulation of B. hydrogenotrophica genes involved in the Wood‐Ljungdahl pathway and of a 10 gene cluster responsible for increased branched chain amino acid fermentation in the co‐cultures. Cross‐feeding between formate‐producing species and acetogens may be a significant factor in short chain fatty acid formation in the colon contributing to high rates of acetate production. Transcriptome analysis also indicated competition for the vitamin thiamine and downregulation of dissimilatory sulfate reduction and key redox proteins in R. bromii in the co‐cultures, thus demonstrating the wide‐ranging consequences of inter‐species interactions in this model system.
Introduction
The anaerobic microbial communities found in the mammalian large intestine and rumen represent the most densely‐colonized microbial ecosystems in nature (Whitman et al., 1998). Metabolite cross‐feeding is recognized as a key feature of such systems that contributes to their complexity, stability and productivity (Sieber et al., 2012; Costliow and Degnan, 2017). Of particular importance are cross‐feeding interactions that can influence the metabolic pathways involved in fermentative metabolism and hence the energetics of bacterial metabolism. One of the first examples of this was provided by the rumen bacterium Ruminococcus albus; when grown on a carbohydrate energy source, R. albus derives ATP via acetate production from acetyl‐CoA, but has to route acetyl‐CoA partly to ethanol as a hydrogen sink. In the presence of a H2 ‐utilizing methanogen, however, R. albus is able to divert more carbon to acetate, and thus gains additional ATP (Wolin et al., 1997).
Recent work on the dominant Ruminococcus species found in the human colon, R. bromii, has shown that this organism has an important role in the degradation of dietary resistant starch (Walker et al., 2011; Ze et al., 2012). Rather little is known about R. bromii metabolism, although this bacterium is reported to have limited ability to synthesize required vitamins (Ze et al., 2012; Magnúsdóttir et al., 2015; Mukhopadhya et al., 2018). R. bromii is reported to produce ethanol, acetate and formate in pure culture with fructose as the carbon source (Herbeck and Bryant, 1974), suggesting that excess reducing equivalents are disposed largely through the production of formate and ethanol in this species. Methanogenic archaea are reported to utilize either formate or H2 and CO2 and have been shown to influence metabolism in hydrogen‐producing Firmicutes (Chassard and Bernalier‐Donadille, 2005) but are present in very low numbers in around 50% of the human population (Florin et al., 2000). On the other hand, another group of H2 and/or formate‐utilizing microorganisms, acetogenic bacteria, appear to be abundant and widespread in all healthy individuals (Doré et al., 1995; Rey et al., 2010). We decided therefore to explore the potential for such interactions using the acetogenic human colonic bacterium Blautia hydrogenotrophica and R. bromii as a model system. Co‐culture is shown to have wide‐ranging consequences for metabolic outputs and for gene expression in both species through interactions that involve vitamins, redox pathways, alternative energy sources and carbon flow.
Results and discussion
Co‐cultivation of R. bromii with B. hydrogenotrophica in batch culture
The major fermentation products of R. bromii L2‐63 when grown in batch culture on RUM‐RS medium with 0.2% Novelose resistant starch (RS3) as energy source were acetate, formate and ethanol (Fig. 1A). R. bromii utilized at least 70% of the RS3 substrate, based on the change in total hexose in the culture, although free reducing sugar accumulated during the incubation as reported previously (Fig. 1C) (Ze et al., 2012). The acetogenic bacterium B. hydogenotrophica DSM 10507 could not utilize starch, but grew on RUM‐G medium containing 0.2% glucose, producing acetate together with ethanol, formate and lactate. Growth of B. hydrogenotrophica on RUM‐G was found to be stimulated by the presence of formate, with the production of additional acetate (Supporting Information Fig. S1). Formate is assumed to be assimilated via the methyl branch of the Wood‐Ljungdahl pathway, as has been demonstrated for Bryantella formatexigenes (Wolin et al., 2003). B. hydrogenotrophica failed to grow in RUM medium in the absence of added glucose with or without added formate (Supporting Information Fig. S1).
Figure 1.
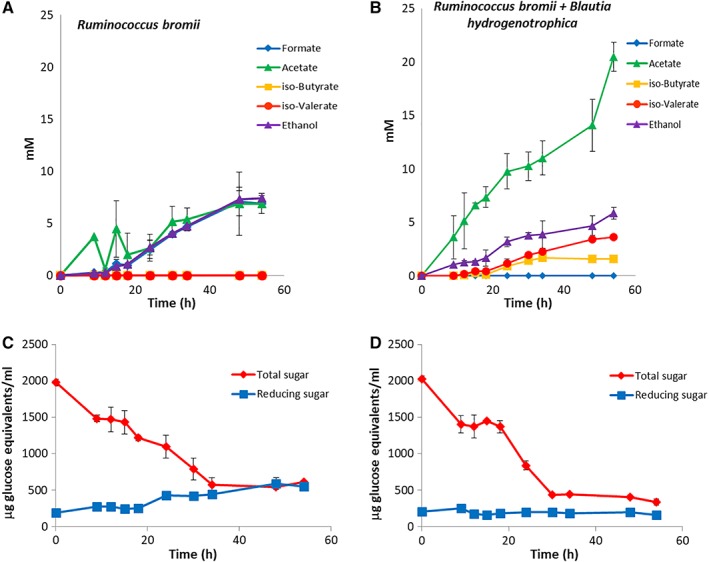
Fermentation products (A, B) and substrate utilization (C, D) for Ruminococcus bromii monocultures (A, C) and for co‐cultures with B. hydrogenotrophica grown in batch culture in RUM‐RS medium (B, D).
Results are shown as the means (with standard deviations) for triplicate cultures that were inoculated with 100 μl of overnight cultures simultaneously with the two bacteria at time 0. Cultures were sampled aseptically under anaerobic conditions at the time points shown (see Methods). In (C) and (D) reducing sugar refers to the culture supernatant only, whereas total sugar refers to the whole culture. Reducing sugar standard deviation values were small and hence are not visible on the figure.
Co‐cultivation of R. bromii and B. hydrogenotrophica in batch culture with resistant starch as the energy source (RUM‐RS) resulted in a major increase in the yield of acetate when compared with the R. bromii monoculture, while no formate was detected in co‐culture (Fig. 1B). The gradual increase in soluble reducing sugar that was seen in R. bromii monocultures was absent in co‐culture (Fig. 1C and D) suggesting that B. hydrogenotrophica was able to utilize sugars released from RS by R. bromii . In addition, the co‐culture gave rise to the branched chain fatty acids iso‐butyrate and iso‐valerate at concentrations not seen in the R. bromii monoculture on starch or in B. hydrogenotrophica monoculture on glucose (Fig. 1A and B). This indicates that there was an increase in fermentation of branched chain amino acids by one of the two species in the co‐culture. B. hydrogenotrophica can form the branched chain fatty acids, iso‐butyrate and iso‐valerate from branched chain amino acids (Rasmussen et al., 1988), in addition to the major fermentation products acetate, lactate and ethanol.
Interactions between R. bromii and B. hydrogenotrophica in continuous culture
We decided to employ anaerobic continuous culture in order to explore these interactions further. It proved possible to grow R. bromii in continuous culture by supplying RUM‐S medium containing soluble starch as sole added energy source, although it was found necessary to renew the supply medium daily because of the lability of certain added vitamins (see Materials and Methods). Mean product concentrations for continuous monocultures of R. bromii grown with input of 0.5% starch were acetate 11.3 mM, ethanol 18.7 mM and formate 10.7 mM (means of 11 time points from two experiments – Fig. 2, Supporting Information Figs S2 and S3). This corresponds to an approximate ratio 1:2:1, as compared to a molar ratio closer to 1:1:1 in batch culture (Fig. 1).
Figure 2.
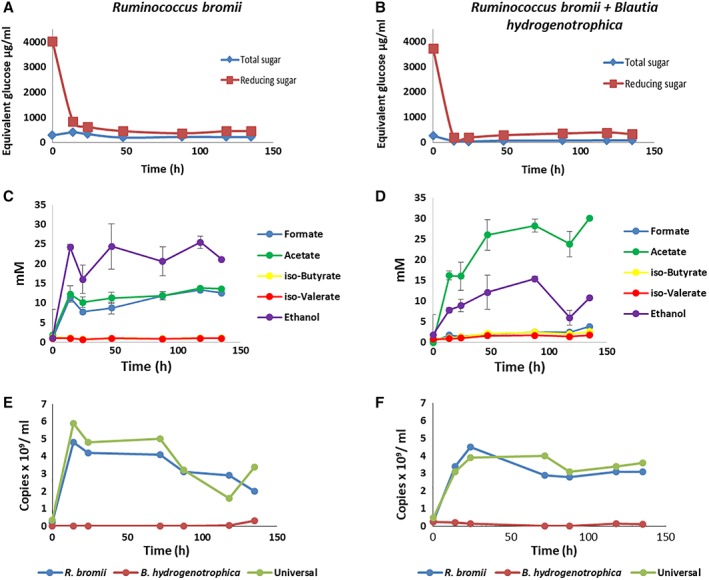
Interactions of R. bromii and B. hydrogenotrophica in continuous culture in RUM‐S medium containing 0.5% soluble starch.
Fermentors inoculated with R. bromii only and with R. bromii plus B. hydrogenotrophica were run in parallel. Substrate utilization (A, B), fermentation products (C, D) and bacterial populations (E, F) estimated by 16S rRNA gene‐targeted qPCR are shown for the mono‐culture (A, C, E) and co‐culture (B, D, F) respectively, based on single samples taken at each time point. Results shown here correspond to fermentors R2 and C2 (Supporting Information Figs S2 and S3); results from a second experiment (R1, C1) and for B. hydrogenotrophica monocultures grown in RUM‐G medium (with glucose as energy source) (B1, B2) are also shown in Supporting Information Figs S2 and S3 for comparison. Reducing and total sugar standard deviation values were small (ranging from 0.076 to 22.79 μg ml−1 for reducing sugar and 7.39 to 153.59 μg ml−1 for total sugar) and hence the error bars are not visible on the figure. For the qPCR data, each value plotted is based on mean of duplicate Cq values.
B. hydrogenotrophica was able to grow in continuous culture in RUM‐G medium with 0.2% glucose as energy source, producing acetate as the main fermentation acid. Supply of 0.5% glucose resulted in formation of lactate and ethanol in addition to acetate but did not increase cell numbers (Supporting Information Figs S2 and S3).
Continuous co‐cultures of R. bromii with B. hydrogenotrophica supplied with RUM‐S medium supported a dominant population of R. bromii together with a much smaller population (by a factor of log2 2–4 fold) of B. hydogenotrophica as assessed by qPCR analysis (Fig. 2E and F). The R. bromii 16S rRNA gene copy number did not change noticeably in the co‐culture relative to the monoculture. Co‐cultures gave rise to increased acetate (23.1 mM) and decreased ethanol (12.9 mM) and formate (2.2 mM) concentrations (means of 9 time points from two experiments) when compared with the R. bromii monoculture. In addition, concentrations of branched chain fatty acids (BCFA) were increased in co‐culture relative to monocultures of R. bromii on RUM‐S (iso‐butyrate 1.9 cf. 1.0; iso‐valerate 1.8 cf. 0.9) (Fig. 2C and D). The low concentrations of reducing sugar that were maintained in the R. bromii monoculture were further decreased in the co‐culture, indicating utilization by B. hydrogenotrophica (Fig. 2A and B).
A proposed explanation for the metabolic shifts seen in co‐cultures is provided by the metabolic scheme in Fig. 3A. When grown in monoculture, ethanol production by R. bromii provides a sink for reducing equivalents (NADH) from glycolysis and acetate production from acetyl‐CoA is limited to approximately one mol acetate per mol glucose fermented, while production of formate reduces the formation of gaseous hydrogen. In the co‐culture, B. hydrogenotrophica may be able to benefit from some glucose released from starch by R. bromii (Ze et al., 2012) (Fig. 1). In addition, however, based on evidence discussed further below, B. hydrogenotrophica appears able to utilize branched chain amino acids as energy sources in the co‐culture (Fig. 3B). We assume that formate produced by R. bromii in the co‐culture is routed into additional acetate via the Wood‐Ljungdahl pathway in B. hydrogenotrophica . The relatively low cell numbers achieved by B. hydrogenotrophica in the co‐culture were nevertheless sufficient to consume all of the formate produced by R. bromii and to greatly increase the yield of acetate. The metabolic scheme suggested in Fig. 3A predicts the formation of approximately 2 mol acetate and 1 mol ethanol per mol glucose consumed by the co‐cultures, with branched chain amino acid fermentation by B. hydrogenotrophica contributing additional ATP and NADH, with the formation of BCFA. Approximate carbon balances are shown in Supporting Information Table S1. An additional possibility, not shown in Fig. 3A, is that H2 is transferred from R. bromii to B. hydrogenotrophica in the co‐culture. Indeed, we do not rule out the possibility that R. bromii metabolism is modified in the co‐culture to produce more hydrogen and less formate.
Figure 3.
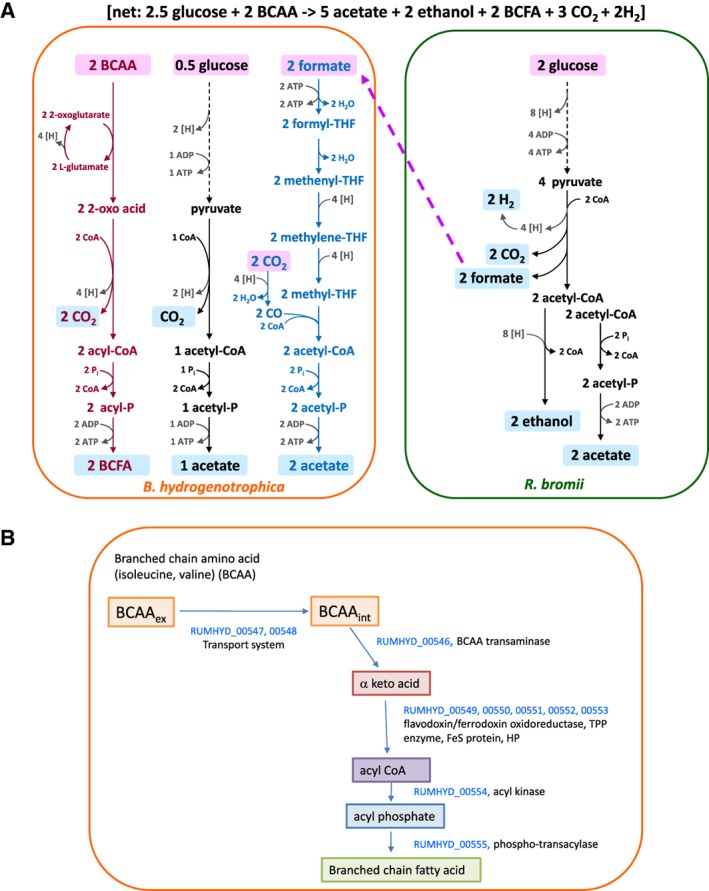
A. Scheme for proposed metabolic interactions between Ruminococcus bromii and Blautia hydrogenotrophica. This scheme approximates the fermentation stoichiometries observed in co‐cultures between R. bromii and B. hydrogenotrophica in batch culture on RUM‐RS medium. The relative amounts of glucose and branched chain amino acids fermented by B. hydrogenotrophica are not known precisely, but the assumption made here approximates to observed yield of BCFA in the co‐culture (see Fig. 1).
B. Genes proposed to be involved in branched chain amino acid fermentation in Blautia hydrogenotrophica.
Transcriptome analysis
To determine the molecular pathways underpinning the observed shifts in fermentation products, transcriptomics analyses were carried out using continuous culture experiments under conditions that approximate to a steady state. Briefly, mRNA was extracted from a total of nine samples, three samples from co‐cultures and three samples from monocultures of each species (Supporting Information Fig. S3), and gene expression was measured by RNA‐seq as described. Gene expression data were analysed using the draft genomes of the two bacteria as a reference and the ratio of expression in the co‐culture relative to that in the monoculture was determined for transcripts of each species. A total of 235 million reads were produced from the nine samples, with an average of 26 million reads per sample (Supporting Information Table S2).
Principal‐component analysis (PCA) of the normalized read counts (Fig. 4A and B) showed that samples from the monocultures clustered closely, indicating good reproducibility of gene expression between independent replicates. Samples from the co‐cultures showed greater variance but clearly grouped separately from the monocultures. A volcano plot (Fig. 4C and D) shows fold changes and levels of significance for differential expression for all genes, with highly dysregulated genes appearing farther to the left and right sides and highly significant changes appearing higher on the plot. A total of 75 R. bromii genes and 420 B. hydrogenotrophica genes were significantly differentially expressed (FDR ≤ 0.05) in the co‐culture compared to the respective monocultures (Fig. 4C and F). For R. bromii, gene expression changes ranged from −3 to 2 log2 fold change, whereas in B. hydrogenotrophica larger changes in gene expression were found (ranged from −4 to 9 log2 fold change; Fig. 4C and D). Hierarchical cluster analysis of the differentially expressed genes (Fig. 4E and F) support the PCA analysis, with co‐culture or monoculture samples clustering together.
Figure 4.
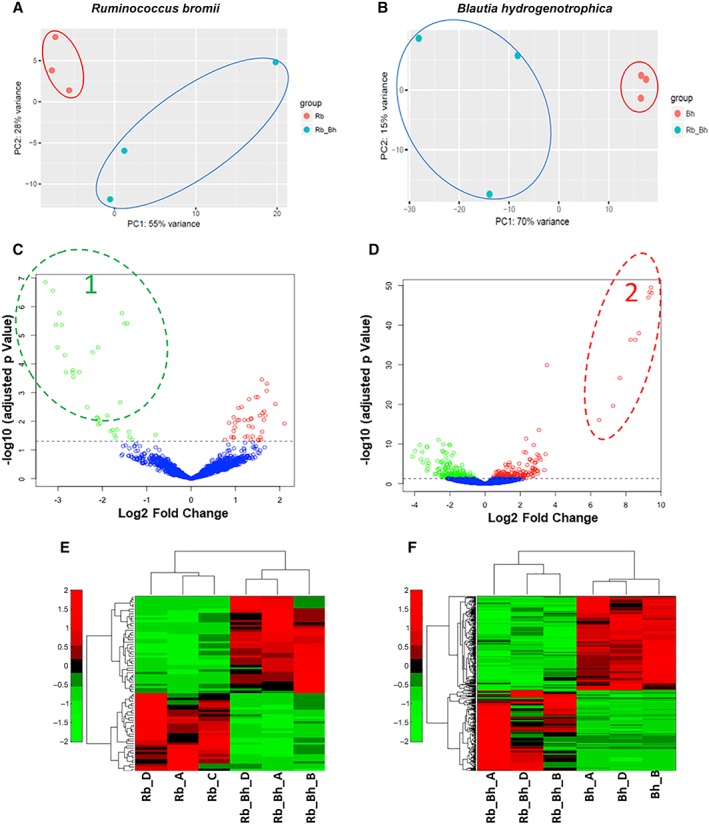
Principal‐component analysis, volcano plot and hierarchical cluster analysis of the RNA‐Seq samples examined in this study.
A and B. Principal‐component analysis of the RNA‐seq reads for the R. bromii and B. hydrogenotrophica monoculture and co‐culture experiments, comparing all RNA‐Seq samples analysed. Red circles show monoculture, whereas blue circle shows co‐culture samples. (A) R. bromii and (B) B. hydrogenotrophica. (c and d) Volcano plots showing fold change and levels of significance for differential expression for all genes. The up and down regulated genes (based on an adjusted p < 0.05) are shown in red and green respectively and the genes with no change are shown in blue. C. R. bromii and (D) B. hydrogenotrophica. E and F. Heat maps showing expression of differentially expressed genes in R. bromii and B. hydrogenotrophica. Red shows high expression and green shows low expression. (E) R. bromii and (F) B. hydrogenotrophica.
Changes in the R. bromii transcriptome in continuous co‐culture with B. hydrogenotrophica
Out of 75 R. bromii genes that showed differences in expression in the co‐culture relative to the monoculture (accounting for 3.5% of total R. bromii transcripts annotated in the reference genome used) 32 were upregulated and 43 were downregulated in the co‐culture. Seven differentially expressed genes were hypothetical proteins with no annotation matches (Supporting Information Table S3). Twenty R. bromii transcripts downregulated by co‐culture were present in a single cluster of linked genes (L2‐63_01134 – L2‐63_01158) consisting mainly of genes involved in sulfur and porphyrin metabolism (Table 1). These include a putative transcriptional regulator iscR, and genes coding for sulfate transport, sulfite reductase, thioredoxin and siroheme synthesis. This suggests that R. bromii shares with some other obligatory anaerobic bacteria the ability to reduce sulfate by the energy producing dissimilatory pathway in which sulfate (or sulfur) is the terminal electron acceptor (Wagner et al., 1998). This pathway starts with activation of sulfate by ATP to form adenylyl sulfate (APS), which is directly reduced to sulfite and then to sulfide by the dissimilatory sulfite reductase. The downregulation of the R. bromii sulfate reduction genes suggests that the activity of the acetogen, B. hydrogenotrophica in the co‐culture may provide an alternative electronic sink, thereby reducing the need for R. bromii to dispose of electrons by reducing sulfite, which may also benefit R. bromii energetically since sulfate reduction is ATP‐dependent (Rückert, 2016).
Table 1.
Two gene clusters that showed the greatest transcriptional responses to co‐culture.
| Gene | Log2 fold change | Annotation/likely function |
|---|---|---|
| 1. Clustered genes concerned with sulfur metabolism and siroheme synthesis in Ruminococcus bromii, downregulated when in co‐culture with Blautia hydrogenotrophica | ||
| L2‐63_01134 | −2.82 | sbp Sulfate starvation induced protein |
| L2‐63_01135 | −3.01 | cysW_1 Sulfate transport system permease protein |
| L2‐63_01136 | −3.29 | cysW_2 Sulfate transport system permease protein |
| L2‐63_01137 | −3.12 | cysA Sulfate/thiosulfate import ATP‐binding protein |
| L2‐63_01140 | −1.81 | hemA Glutamyl‐tRNA reductase |
| L2‐63_01141 | −1.7 | cysG_1 Siroheme synthase, precorrin‐2 dehydrogenase |
| L2‐63_01142 | −1.73 | hemC Porphobilinogen deaminase |
| L2‐63_01143 | −1.77 | cysG_2 Siroheme synthase, uroporphyrin‐III‐C‐methyltransferase |
| L2‐63_01144 | −1.89 | hemB Delta‐aminolevulinic acid dehydratase |
| L2‐63_01145 | −1.39 | hemL2 glutamate‐1‐semialdehyde aminotransferase |
| L2‐63_01146 | −2.65 | iscR_1 HTH‐type transcriptional regulator |
| L2‐63_01150 | −2.85 | trxA_3 Thioredoxin‐M |
| L2‐63_01151 | −3.05 | bifunctional sulfur carrier protein |
| L2‐63_01152 | −2.68 | moeZ probable adenyltraferase/sulfurtransferase |
| L2‐63_01153 | −2.67 | mec CysO‐cysteine peptidase |
| L2‐63_01154 | −2.35 | trxB_2 Thioredoxin reductase, alkyl hydroperoxide reductase subunit |
| L2‐63_01155 | −2.09 | ifcA fumarate reductase subunit precursor |
| L2‐63_01156 | −2.11 | Ferredoxin II, NADH‐plastoquinoine oxidoreductase subunit |
| L2‐63_01157 | −2.13 | cysD Sulfate adenylyl transferase subunit 2 |
| L2‐63_01158 | −2.52 | cysN Sulfate adenylyl transferase subunit 1 |
| 2. Putative branched chain amino acid fermentation gene cluster in Blautia hydrogenotrophica, upregulated when in co‐culture with Ruminococcus bromii | ||
| RUMHYD_00546 | 8.27 | ilvE branched chain amino acid transaminase |
| RUMHYD_00547 | 7.66 | branched chain amino acid transport protein |
| RUMHYD_00548 | 6.48 | brnQ branched chain amino acid transport system II carrier protein |
| RUMHYD_00549 | 8.54 | 2‐oxoacid:acceptor oxidoreductase gamma subunit |
| RUMHYD_00550 | 9.28 | thiamine pyrophosphate enzyme |
| RUMHYD_00551 | 9.34 | pyruvate flavodoxin/ferredoxin oxidoreductase, thiamine diP binding |
| RUMHYD_00552 | 7.27 | 4Fe‐4S binding domain protein |
| RUMHYD_00553 | 8.74 | hypothetical protein |
| RUMHYD_00554 | 9.42 | buk butyrate kinase (presumed acyl kinase) |
| RUMHYD_00555 | 9.46 | ptb phosphate butyryltransferase (presumed phosphate acyltransferase) |
| RUMHYD_00556 | 1.4 | transcriptional regulator, Gnt R family |
Transcripts correspond to those within the circles (1) and (2) in Fig. 4 C and D.
This cluster of linked genes also includes six upregulated genes (L2‐63_01140 to L2‐63_01145) encoding proteins involved in porphyrin/siroheme biosynthesis (hemA, cysG_1/MET8, hemC, cysG_2/cobA‐hemD, hemB, hemL) (Table 1, Supporting Information Table S3). Siroheme is a redox‐active cofactor and transcription of genes responsible for its biosynthesis is sensitive to cellular redox potential and/or the redox potential of the fermentation environment (Stroupe et al., 2003). Again, with the acetogen potentially providing an extra sink for cellular reductant through a process mediated by hydrogenases, there may be less overall demand for such R. bromii proteins mediating redox reactions. Broadly similar changes involving the IscR regulator have been reported in the transcriptome of Clostridium thermocellum in response to redox status (Sander et al., 2015).
Our data also reveal upregulation of four transcripts associated with thiamine salvage (the thiMDE cluster) and thiamine transport (thiT) in the co‐culture relative to the monoculture (L2‐63_00215 to L2‐63_00216, L2‐63_00218 and L2‐63_01258) (Supporting Information Table S3, Fig. 5). In most organisms, thiamine biosynthesis involves separate synthesis of thiazole and pyrimidine moieties that are coupled to form thiamin phosphate (Rodionov et al., 2002; Du et al., 2011). R. bromii however appears to possess the alternative salvage pathway (thiMDE) (Rodionov et al., 2002) (Fig. 5).
Figure 5.
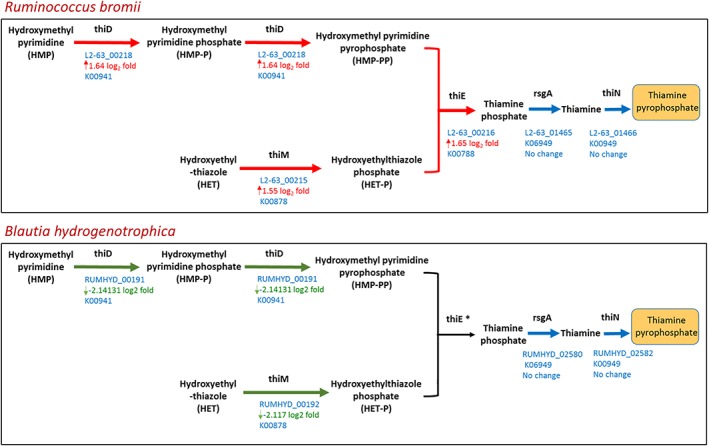
Thiamine salvage pathway in R. bromii and B. hydrogenotrophica.
Changes in expression of genes concerned with thiamine salvage in continuous co‐cultures between R. bromii and B. hydrogenotrophica. Upregulation is indicated by red arrows (R. bromii) and downregulation by green arrows (B. hydrogenotrophica). The asterisk indicates no transcript could be identified for thiE in B. hydrogenotrophica, but it cannot be ruled out that this activity is encoded by an unknown (hypothetical) protein.
Changes in the B. hydrogenotrophica transcriptome in continuous co‐culture with R. bromii
A total of 420 B. hydrogenotrophica genes showed differences in expression in the co‐culture relative to the monoculture (10.85% of the total genes annotated for B. hydrogenotrophica strain) of which 197 were upregulated and 223 were downregulated. One hundred and thirty six of the differentially expressed genes were hypothetical proteins (Supporting Information Table S3). KEGG‐based pathway analysis of the differentially expressed genes in B. hydrogenotrophica indicated that multiple pathways were significantly altered in response to co‐culture (enrichment p value ≤0.05).
Transcripts in B. hydrogenotrophica that showed the highest and most significant upregulation in the co‐culture relative to the monoculture are encoded by a cluster of 11 linked genes (RUMHYD_00546 to RUMHYD_00556) (Table 1, Supporting Information Table S3). These include homologues of butyrate kinase and phosphotransbutyrylase, together with a branched chain fatty acid transporter and a branched chain fatty acid transaminase and a keto acid oxidoreductase. This is in agreement with the observed formation of branched chain fatty acids in the co‐cultures (Fig. 3B). Thus, we propose that the upregulation of these genes enable B. hydrogenotrophica to form 2‐oxo acids by transamination that are then converted to branched chain fatty acids with the production of ATP in a manner directly analogous to the conversion of pyruvate to acetate. The two upregulated genes that were computationally annotated as butyrate kinase and phosphate butyryltransferase are predicted to encode branched chain fatty acid kinases and phosphorylases, respectively, since there is no evidence for butyrate production by B. hydrogenotrophica.
Another cluster of 7 linked genes (RUMHYD _00315 to RUMHYD _00321) that showed significant upregulation in the co‐culture compared to B. hydrogenotrophica monoculture, was concerned with acetogenesis from formate and from CO2 (Supporting Information Table S3). KEGG analysis (Supporting Information Fig. S4) also indicated downregulation of several pathways involved in carbohydrate metabolism. This suggests a major shift from dependence on glucose utilization in the monoculture to the utilization of branched chain amino acids and acetogenesis in the co‐culture.
Interestingly, transcripts associated with thiamine biosynthesis (thiC, thiD, thiM, thiW, thiI) were downregulated in B. hydrogenotrophica in the co‐culture (Fig. 5) which contrasts with the parallel upregulation of the thiamine biosynthesis transcripts of R. bromii (Supporting Information Table S3, Fig. 5). Transcripts involved in biosynthesis of panthothenate and Co‐A were also detected in B. hydrogenotrophica (Supporting Information Fig. S5). B. hydrogenotrophica possesses most of the enzymes required for the generation of pantothenate (except for pantoate‐beta‐alanine ligase, E6.3.2.1, although there are unannotated hypothetical genes in the current draft genome and one of these may encode this function) and the three transcripts required to produce pantoate from valine were upregulated in the co‐culture (Supporting Information Fig. S5). R. bromii does not appear to have panthothenate biosynthesis capacity; as a result, it must rely on the available pantothenate provided in the growth medium, perhaps leading to upregulation of this pathway in B. hydrogenotrophica.
Conclusions
Acetate reaches the highest concentrations among the SCFA produced by anaerobic fermentation in the human large intestine and a ratio of approximately 3:1:1 acetate:propionate:butyrate is typically reported for faecal samples from healthy individuals. Acetate is utilized by many abundant species of butyrate‐producers in the human colon (Barcenilla et al., 2000; Louis and Flint, 2017) but its production is widespread among gut anaerobes. Many gut bacteria also produce formate as a major product in pure culture but little or no formate is normally detected in faecal samples. The observations reported here indicate that formate utilization by acetogenic bacteria may be a significant factor explaining the high acetate production and low formate levels that are seen in vivo. Formate is known to be utilized by some methanogenic archaea, but these organisms are present at very low levels in around 50% of the population (Florin et al., 2000). Acetogenic bacteria, however, appear to be widespread in the human colon (Rey et al., 2010). The Wood‐Ljungdahl pathway found in acetogens was estimated to contribute 30% of total acetate formation in human faecal incubations with 13CO2 incorporation into acetate exceeding its conversion to methane even in methanogenic individuals (Wolin et al., 1999). Interestingly isotope enrichment was found to be less in the methyl group than in the carboxyl group of acetate, which the authors suggested was likely to be due to the contribution of unlabelled formate derived from carbohydrate fermentation (Wolin et al., 1999). This is consistent with the type of cross‐feeding observed in the present study. As shown here, B. hydrogenotrophica can ferment carbohydrates and can grow ‘mixotrophically’ with net consumption of formate, in addition to being able to utilize H2 and CO2 to produce acetate (Leclerc et al., 1997). The phylogenetic distribution of acetogenesis has yet to be fully established, but it seems likely that a number of human colonic Lachnospiraceae in addition to Blautia hydrogenotrophica are able to convert formate into acetate via the Wood‐Ljungdahl pathway (Bernalier et al., 1996; Leclerc et al., 1997; Wolf et al., 2016). Many human colonic acetogens cannot grow with H2 and CO2 alone (Kamlage et al., 1997) and the metabolism of Bryantella formatexigens in the presence of fructose, for example, becomes increasingly acetogenic at high formate concentrations (Wolin et al., 2003). Recent theoretical models of human gut microbial metabolism are reported to underestimate acetate and overestimate formate concentrations relative to the measured outputs of complex communities studied in vitro (Kettle et al., 2015). Our findings suggest that this discrepancy might be resolved by incorporating formate utilization by acetogens into the models.
Ruminococcus bromii is a dominant member of the human intestinal microbiota whose population is readily increased by consumption of diets rich in resistant starch when this species can account for more than 10% of total faecal bacteria (Martínez et al., 2010; Walker et al., 2011). We can conclude that the metabolic consequences of an increased population of R. bromii are likely to be to boost acetate production, first through direct formation and second via conversion of formate to acetate through acetogens. In the case of the other major product seen in pure culture, ethanol, it remains to be established whether this is also subject to consumption by other species in the mixed community. It has been suggested that acetogens, such as B. hydrogenotrophica, may be favoured under conditions of mildly acidic pH and substrate availability that prevail in the proximal colon (Macfarlane and Gibson, 1997). The proximal colon would therefore be expected to favour this species and also to support high populations of R. bromii when fermentable resistant starch is being supplied from the diet. The co‐localisation of these two species seems likely, therefore, but this remains to be established experimentally. Ideally this would be done using techniques that allow in situ sampling of colonic lumen contents containing partially degraded starch particles.
In addition to formate cross‐feeding, our transcriptome analysis has revealed several other significant nutritional interactions that involve amino acid catabolism, electron disposal and vitamin synthesis. Intriguingly, genes concerned with thiamine synthesis were upregulated in R. bromii and downregulated in B. hydrogenotrophica in the co‐culture. Thiamine is required for the crucial pyruvate ferredoxin oxidoreductase reaction. It is not clear why its synthesis should be upregulated in R. bromii in the presence of very low numbers of B. hydrogenotrophica, but this may be a consequence of widespread changes in anaerobic metabolism triggered by the acetogen as evidenced by the gene expression changes affecting sulfate metabolism and siroheme in R. bromii. The greatly increased expression of genes that we infer to be responsible for branched chain amino acid fermentation by B. hydrogenotrophica in the co‐culture is likely to be a response to a limited supply of starch‐derived sugars released by R. bromii. In conclusion, this analysis has provided a unique insight into the complex nutritional interplay and interdependence of two of the dominant species that make up a healthy human gut microbiome.
Experimental procedures
Bacterial strains and growth conditions
Ruminococcus bromii L2‐63 is a Rowett isolate described previously (Ze et al., 2012). Blautia hydrogenotrophica DSM 10507 was purchased from Deutsche Sammlung von Mikrooganismen und Zellkulturen (DSMZ). Cultures were routinely maintained by growing for 16–18 h at 37 °C in 7.5 ml aliquots of anaerobic M2GSC medium (Miyazaki et al., 1997) under O2‐free CO2. Batch culture studies with R. bromii and B. hydrogenotrophica were carried out in RUM medium (Ze et al., 2015) that was simplified here by the omission of supplementary minerals to consist of (per 100 ml): casitone (1.0 g), yeast extract (0.25 g), NaHCO3 (0.4 g), cysteine (0.1 g), K2HPO4 (0.045 g), KH2PO4 (0.045 g) NaCl, (0.09 g) (NH4)2SO4 (0.09 g), MgSO4·7H2O (0.009 g), CaCl2, (0.009 g) resazurin (0.1 mg), biotin (1 μg), cobalamin (1 μg), p‐aminobenzoic acid (3 μg), folic acid (5 μg), pyridoxamine (15 μg). In addition, the following SCFA are included (final concentrations): acetate (33 mM), propionate (9 mM), iso‐butyrate, iso‐valerate and valerate (each 1 mM). Cysteine (0.1 g/100 ml) is added to the medium following boiling and dispensed into Hungate tubes while the tubes are flushed with CO2. After autoclaving, filter sterilized solutions of heat labile vitamins were added to give final concentrations of thiamine and riboflavin of 0.05 μg·ml−1 (each), pantothenate and nicotinamide of 1 μg·ml−1, pantethine of 50 μg·ml−1 and tetrahydrofolic acid of 0.1 μg·ml−1 as described previously (Ze et al., 2015). The final pH of the medium was adjusted to 6.8 ± 0.2. The starch (0.2% final concentration) used for the batch culture incubations was Novelose 330 (resistant starch type 3‐ RS3) which had been boiled for 10 min as described previously (Ze et al., 2012) referred to as RUM‐RS (Ze et al., 2015) or glucose (0.2%) referred to as RUM‐G.
Continuous culture
Single‐stage fermentor systems, with working volumes of 250 ml were set‐up as described previously (Chung et al., 2016) at a constant pH (6.5 ± 0.2) with a continuous flow of the semi defined RUM medium described above with either boiled (100 °C for 10 min) soluble potato starch (Sigma, S2004) or glucose as the carbon source. The fermentor culture vessels were maintained under a stream of CO2 at a constant temperature of 37°C using thermal jackets. The medium reservoir and fermentor culture vessel were mixed by internal stirrer bars powered by external stirring units. The volume of the culture was kept constant at 250 ml with a constant flow of fresh medium at a turnover of 250 ml day−1 with fresh heat labile vitamins added daily to give final concentrations of thiamine and riboflavin of 0.05 μg·ml−1 (each), pantothenate and nicotinamide of 1 μg·ml−1, pantethine of 50 μg·ml−1 and tetrahydrofolic acid of 0.1 μg·ml−1. The pH values of the fermentor vessels were monitored and controlled to 6.5 ± 0.2 using a pH controller which delivers either 0.1 M HCl or 0.1 M NaOH solutions to maintain a constant pH for the full period of the study. Fresh 10 ml cultures of R. bromii L2‐63 and B. hydrogenotrophica DSM 10507, grown to stationary phase in RUM medium with either soluble potato starch or glucose respectively as energy source, were used to inoculate the fermentors.
Total sugar and reducing sugar analysis
Total sugars were determined by the phenol sulfuric assay as described previously (Dubois et al., 1956). Briefly, 0.5 ml of samples or glucose standards were mixed with 0.5 ml of phenol solution (5%, w/v in d.H2O), then 2.5 ml of concentrated sulfuric acid (95%–98%) was added to the tubes were allowed to cool, then 200 μl were transferred into 96‐well flat bottom plate and OD490 was read using a Safire2 microplate reader (Tecan Group, Männedorf, Switzerland). Reducing sugars were measured separately (Lever, 1977) by transferring 100 μl of culture supernatants or glucose standards into 2.5 ml of 4‐hydroxybenzhydrazide (PAHBAH) reagent. After incubation at 70 °C for 10 min, 200 μl were transferred into 96‐well flat bottom plate and OD415 read on a Safire2 microplate reader (Tecan Group, Männedorf, Switzerland).
SCFA and ethanol analysis
SCFA concentrations were measured by gas chromatography as described previously (Richardson et al., 1989). Following derivatization of the samples using N‐tert‐butyldimethylsilyl‐N‐methyltrifluoroacetamide, the samples were analysed using a Hewlett Packard gas chromatograph (GC) fitted with a silica capillary column using helium as the carrier gas. Ethanol concentrations were also measured by gas chromatography using the same GC but fitted with a ZB WAX column.
Quantitative PCR
Real time qPCR was performed in duplicate with iTaq™ Universal SYBR® Green Supermix (Bio‐Rad, Laboratories Ltd, Hertfordshire, UK) in a total volume of 10 μl and 2 ng of DNA in a CFX384TM Real‐time System (Bio‐Rad) as described previously (Chung et al., 2016). The primer pairs used are described in Supporting Information Table S4. Annealing temperature was 60 °C for universal and R. bromii primers and 63 °C for B. hydrogenotrophica primers. Standard curves consisted of 10‐fold dilution series of amplified bacterial 16S rRNA genes from the two strains. Samples were amplified with universal primers against total bacteria and specific primers against R. bromii and B. hydrogenotrophica. Relative bacterial concentrations in each sample were estimated by comparing the gene copy numbers calculated using standard curves. Data were analysed using BioRad CFX manager software and the detection limit was determined with negative controls containing only herring sperm DNA.
RNA extraction
Total RNA was extracted from 10 ml of culture using phenol‐chloroform extraction followed by ethanol precipitation. Any residual DNA was removed using DNase‐treatment with TURBO DNA‐free kit (Invitrogen). RNA concentration and purity were determined spectrophotometrically using Nano drop Spectrophotometer ND‐1000, BioAnalyzer 2100 (Agilent Technologies UK Ltd, Edinburgh, Scotland, UK) and Qubit (Thermo Fisher Scientific, Perth, Scotland, UK). Ribosomal RNA was depleted in the samples using the Ribo‐Zero rRNA removal kit for bacteria (Illumina, San Diego, CA, USA) according to manufacturer's protocol. mRNA was then purified using RNeasy MinElute kit (Qiagen, Crawley, UK).
RNA‐seq
A total of nine RNA libraries (three replicates each of R. bromii monoculture, B. hydrogenotrophica monoculture and R. bromii – B. hydrogenotrophica co‐culture) from the fermenter systems were prepared using the adapted TruSeq RNA protocol (Illumina 15026495 Rev.B). The library preparation involved QC of the RNA (Pico kit, Bioanalyzer 2100, Agilent Technologies) to detect any potential rRNA contamination. The ribo‐depleted RNA was enzymatically fragmented and double strand (ds) cDNA was synthesized using random hexamers in the presence of reverse transcriptase. The samples were end repaired, A‐tailed, adaptor‐ligated, fractionated, purified and enriched. The insert size of the libraries was verified by running an aliquot of the DNA library on a PerkinElmer GX using the High Sensitivity DNA chip (PerkinElmer CLS760672, Perkin Elmer Ltd, Beaconsfield, UK) and the concentration was determined using a High Sensitivity Qubit assay. Libraries were sequenced on an Illumina HiSeq 2500 instrument to generate 100 bp paired end reads. Sequence reads were subjected to post processing to trim Illumina adapters and primer sequences. Sequencing was done at the Earlham Institute (BBSRC Genome Analysis Centre), Norwich, UK.
Transcriptome analysis
High quality reads for each sample were aligned against the combined reference genomes of R. bromii L2‐63 and B. hydrogenotrophica DSM 10507, downloaded from GenBank repository (GCA_000209875.1 and GCA_000157975.1). Associated annotation file in gff3 format was used to obtain information for downstream analysis. Alignments were generated using TopHat (version 2.1.0 release 6/29/2015) with the ‐‐max‐multihits 1 option (Trapnell et al., 2009). Read counts were generated using HTSeq‐count (version 0.6.1) (Anders et al., 2015). Gene expression for each sample was computed as a measure of the total number of reads uniquely aligning to the reference, binned by genic coordinates (information acquired from the annotation file). Differential gene expression analysis was performed using the Bioconductor package DESeq2 (version 1.14.0) (Anders and Huber, 2010). Raw read counts thus obtained were normalized to account for differences in sequencing depth and composition using methods implemented within DESeq. Differential expression of pairwise comparisons (of the different groups) was assessed using the negative binomial test with Benjamani–Hochberg false discovery rate (FDR) adjustment (Hochberg and Benjamini, 1990) applied for multiple testing corrections. For this study, an FDR of 0.05 was applied and any candidate that had a p‐adjusted value of ≤ 0.05 was considered to be significantly up‐ or downregulated. To gain further insights into the effects of co‐cultivation, we performed a KEGG pathway analysis. KEGG orthology was assigned to all R. bromii and B. hydrogenotrophica genes using KAAS‐KEGG (Moriya et al., 2007). KAAS‐KEGG outputs were then converted to GMT format and combined with KEGG pathway information gathered using the KEGG Rest API (http://www.kegg.jp/kegg/rest/). Analysis of significant enrichment of KEGG pathways in the differentially expressed gene lists was then carried out using the GO Enrichment function of the software package Partek® Genomics Suite® software version 6.6 Copyright ©; 2016. The Fisher Exact test function was used only analysing pathways with more than two genes present and pathways with a p < 0.05 were deemed to be significantly enriched.
Data availability
RNA‐seq data have been deposited in the ArrayExpress database at EMBL‐EBI (www.ebi.ac.uk/arrayexpress) under accession number E‐MTAB‐ 210350.
Author contributions
HJF devised the study. JLG, IM, SD performed the experiments. JLG, IM, HJF, PL, SD, SS and EDG analysed the data and prepared figures and tables. EC and NJ provided critical resources and support. HJF, IM, SD wrote the paper. All authors read and approved the final manuscript.
Supporting information
Fig. S1. Effect of formate on growth and metabolism of Blautia hydrogenotrophica in RUM medium. a) shows growth (OD650) with or without the addition of 10 mM formate and/or 0.2% (11 mM) glucose to the basal medium; b) and c) Metabolites formed and utilized during growth on glucose with or without formate (for the experiment shown in a). Values represent means of triplicate cultures.
Fig. S2. Growth (OD650) of Ruminococcus bromii, Blautia hydrogenotrophica and co‐cultures in continuous culture. Results are shown for six fermentors. Vessels R1 and C1 were inoculated simultaneously with R. bromii only (R1) or with both bacteria (C1) and supplied with RUM‐S medium containing 0.5% soluble starch. R2 and C2 refer to a repeat of this experiment. B1 and B2 vessels were run separately and supplied with RUM‐G medium containing 0.5% glucose (B1) or 0.2% glucose (B2); these vessels were inoculated with B. hydrogenotrophica only.
Fig. S3. Metabolite concentrations for the six fermentor experiments shown in Fig. S2. Blue arrows indicate the sampling points used for RNAseq analysis.
Fig. S4. KEGG Carbon metabolism pathway map for Blautia hydrogenotrophica. Genes present in the differentially expressed gene (DEG) list are highlighted with those with up regulated expression in the co‐culture compared to the mono‐culture in red and those with down regulated expression in green. Genes with KEGG orthologs found by KAAS‐KEGG, but no significant change in expression, are highlighted in blue. Numbers inside boxes are enzyme commission numbers.
Fig. S5. KEGG pathway map for metabolism of pantothenate and CoA in Blautia hydrogenotrophica. Genes present in the differentially expressed gene (DEG) list are highlighted with those with up regulated expression in the co‐culture compared to the mono‐culture in red and those with down regulated expression in green. Genes with KEGG orthologs found by KAAS‐KEGG, but no significant change in expression, are highlighted in blue.
Table S1. Calculated carbon balances for monocultures and co‐cultures.
Table S2. RNAseq read counts.
Table S3. Differentially expressed genes (DEGs) list from Ruminococcus bromii and Blautia hydrogenotrophica.
Table S4. Primers for qPCR.
Acknowledgements
We acknowledge support from BBSRC grant no. BB/L009951/1, from the Scottish government Food, Land and People program, and from the Society for Applied Microbiology. We thank Andrea Telatin (Quadram Institute Bioscience, Norwich, UK) for helping with RNAseq data submission to EMBL‐EBI. The authors declare no conflict of interest.
References
- Anders, S. , and Huber, W. (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, S. , Pyl, P. T. , and Huber, W. (2015) HTSeq—a python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcenilla, A. , Pryde, S. E. , Martin, J. C. , Duncan, S. H. , Stewart, C. S. , Henderson, C. , and Flint, H. J. (2000) Phylogenetic relationships of butyrate‐producing bacteria from the human gut. Appl Environ Microbiol 66: 1654–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernalier, A. , Willems, A. , Leclerc, M. , Rochet, V. , and Collins, M. D. (1996) Ruminococcus hydrogenotrophicus sp. nov., a new H2/CO2‐utilizing acetogenic bacterium isolated from human feces. Arch Microbiol 166: 176–183. [DOI] [PubMed] [Google Scholar]
- Chassard, C. , and Bernalier‐Donadille, A. (2005) H2 and acetate transfers during xylan fermentation between a butyrate‐producing xylanolytic species and hydrogenotrophic microorganisms from the human gut. FEMS Microbiol Lett 254: 116–122. [DOI] [PubMed] [Google Scholar]
- Chung, W. S. , Walker, A. W. , Louis, P. , Parkhill, J. , Vermeiren, J. , Bosscher, D. , et al (2016) Modulation of the human gut microbiota by dietary fibres occurs at the species level. BMC Biol 14: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costliow, Z. A. , and Degnan, P. H. (2017) Thiamine acquisition strategies impact metabolism and competition in the gut microbe Bacteroides thetaiotaomicron. MSystems 2: e00116–e00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré, J. , Pochart, P. , Bernalier, A. , Goderel, I. , Morvan, B. , and Rambaud, J. (1995) Enumeration of H2‐utilizing methanogenic archaea, acetogenic and sulfate‐reducing bacteria from human feces. FEMS Microbiol Ecol 17: 279–284. [Google Scholar]
- Du, Q. , Wang, H. , and Xie, J. (2011) Thiamin (vitamin B1) biosynthesis and regulation: a rich source of antimicrobial drug targets? Int J Biol Sci 7: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois, M. , Gilles, K. , Hamilton, J. , Rebers, P. , and Smith, F. (1956) Colorimetric method for determination of sugars and related substances. Anal Chem 28: 350–356. [Google Scholar]
- Florin, T. H. , Zhu, G. , Kirk, K. M. , and Martin, N. G. (2000) Shared and unique environmental factors determine the ecology of methanogens in humans and rats. Am J Gastroenterol 95: 2872. [DOI] [PubMed] [Google Scholar]
- Herbeck, J. L. , and Bryant, M. (1974) Nutritional features of the intestinal anaerobe Ruminococcus bromii . Appl Microbiol 28: 1018–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg, Y. , and Benjamini, Y. (1990) More powerful procedures for multiple significance testing. Stat Med 9: 811–818. [DOI] [PubMed] [Google Scholar]
- Kamlage, B. , Gruhl, B. , and Blaut, M. (1997) Isolation and characterization of two new homoacetogenic hydrogen‐utilizing bacteria from the human intestinal tract that are closely related to Clostridium coccoides . Appl Environ Microbiol 63: 1732–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettle, H. , Louis, P. , Holtrop, G. , Duncan, S. H. , and Flint, H. J. (2015) Modelling the emergent dynamics and major metabolites of the human colonic microbiota. Environ Microbiol 17: 1615–1630. [DOI] [PubMed] [Google Scholar]
- Leclerc, M. , Bernalier, A. , Donadille, G. , and Lelait, M. (1997) H 2/CO 2 metabolism in acetogenic bacteria isolated from the human colon. Anaerobe 3: 307–315. [DOI] [PubMed] [Google Scholar]
- Lever, M. (1977) Carbohydrate determination with 4‐hydroxybenzoic acid hydrazide (PAHBAH): effect of bismuth on the reaction. Anal Biochem 81: 21–27. [DOI] [PubMed] [Google Scholar]
- Louis, P. , and Flint, H. J. (2017) Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol 19: 29–41. [DOI] [PubMed] [Google Scholar]
- Macfarlane, G. T. , and Gibson, G. R. (1997) Carbohydrate fermentation, energy transduction and gas metabolism in the human large intestine . In Gastrointestinal Microbiology, Mackie R. L., and White B. A. (eds). Boston, MA: Springer, pp. 269–318. [Google Scholar]
- Magnúsdóttir, S. , Ravcheev, D. , de Crécy‐Lagard, V. , and Thiele, I. (2015) Systematic genome assessment of B‐vitamin biosynthesis suggests co‐operation among gut microbes. Front Genet 6: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez, I. , Kim, J. , Duffy, P. R. , Schlegel, V. L. , and Walter, J. (2010) Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS One 5: e15046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki, K. , Martin, J. C. , Marinsek‐Logar, R. , and Flint, H. J. (1997) Degradation and utilization of xylans by the rumen anaerobe Prevotella bryantii (formerly P. ruminicola subsp. brevis) B(1)4. Anaerobe 3: 373–381. [DOI] [PubMed] [Google Scholar]
- Moriya, Y. , Itoh, M. , Okuda, S. , Yoshizawa, A. C. , and Kanehisa, M. (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35: W182–W185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhya, I. , Moraïs, S. , Laverde‐Gomez, J. , Sheridan, P. O. , Walker, A. W. , Kelly, W. , et al (2018) Sporulation capability and amylosome conservation among diverse human colonic and rumen isolates of the keystone starch‐degrader Ruminococcus bromii . Environ Microbiol 20: 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen, H. S. , Holtug, K. , and Mortensen, P. (1988) Degradation of amino acids to short‐chain fatty acids in humans: an in vitro study. Scand J Gastroenterol 23: 178–182. [DOI] [PubMed] [Google Scholar]
- Rey, F. E. , Faith, J. J. , Bain, J. , Muehlbauer, M. J. , Stevens, R. D. , Newgard, C. B. , and Gordon, J. I. (2010) Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem 285: 22082–22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, A. , Calder, A. , Stewart, C. , and Smith, A. (1989) Simultaneous determination of volatile and non‐volatile acidic fermentation products of anaerobes by capillary gas chromatography. Lett Appl Microbiol 9: 5–8. [Google Scholar]
- Rodionov, D. A. , Vitreschak, A. G. , Mironov, A. A. , and Gelfand, M. S. (2002) Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J Biol Chem 277: 48949–48959. [DOI] [PubMed] [Google Scholar]
- Rückert, C. (2016) Sulfate reduction in microorganisms—recent advances and biotechnological applications. Curr Opin Microbiol 33: 140–146. [DOI] [PubMed] [Google Scholar]
- Sander, K. , Wilson, C. M. , Rodriguez, M. , Klingeman, D. M. , Rydzak, T. , Davison, B. H. , and Brown, S. D. (2015) Clostridium thermocellum DSM 1313 transcriptional responses to redox perturbation. Biotechnol Biofuels 8: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber, J. R. , McInerney, M. J. , and Gunsalus, R. P. (2012) Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. Annu Rev Microbiol 66: 429–452. [DOI] [PubMed] [Google Scholar]
- Stroupe, M. E. , Leech, H. K. , Daniels, D. S. , Warren, M. J. , and Getzoff, E. D. (2003) CysG structure reveals tetrapyrrole‐binding features and novel regulation of siroheme biosynthesis. Nat Struct Mol Biol 10: 1064. [DOI] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. , and Salzberg, S. L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, M. , Roger, A. J. , Flax, J. L. , Brusseau, G. A. , and Stahl, D. A. (1998) Phylogeny of dissimilatory sulfite reductases supports an early origin of sulfate respiration. J Bacteriol 180: 2975–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, A. W. , Ince, J. , Duncan, S. H. , Webster, L. M. , Holtrop, G. , Ze, X. , et al (2011) Dominant and diet‐responsive groups of bacteria within the human colonic microbiota. ISME J 5: 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman, W. B. , Coleman, D. C. , and Wiebe, W. J. (1998) Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 95: 6578–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, P. G. , Biswas, A. , Morales, S. E. , Greening, C. , and Gaskins, H. R. (2016) H2 metabolism is widespread and diverse among human colonic microbes. Gut microbes 7: 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin, M. , Miller, T. , and Stewart, C. (1997)Microbe‐microbe interactions . In The Rumen Microbial Ecosystem. London, UK: Springer, pp. 467–491. [Google Scholar]
- Wolin, M. J. , Miller, T. L. , Collins, M. D. , and Lawson, P. A. (2003) Formate‐dependent growth and homoacetogenic fermentation by a bacterium from human feces: description of Bryantella formatexigens gen. Nov., sp. nov. Appl Environ Microbiol 69: 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin, M. J. , Miller, T. L. , Yerry, S. , Zhang, Y. , Bank, S , and Weaver, G. A. (1999) Changes of fermentation pathways of fecal microbial communities associated with a drug treatment that increases dietary starch in the human colon. Appl Environ Microbiol 65: 2807–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ze, X. , Duncan, S. H. , Louis, P. , and Flint, H. J. (2012) Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J 6: 1535–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ze, X. , Ben David, Y. , Laverde‐Gomez, J. A. , Dassa, B. , Sheridan, P. O. , Duncan, S. H. , et al (2015) Unique organization of extracellular amylases into amylosomes in the resistant starch‐utilizing human colonic Firmicutes bacterium Ruminococcus bromii . MBio 6: e01058–e01015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effect of formate on growth and metabolism of Blautia hydrogenotrophica in RUM medium. a) shows growth (OD650) with or without the addition of 10 mM formate and/or 0.2% (11 mM) glucose to the basal medium; b) and c) Metabolites formed and utilized during growth on glucose with or without formate (for the experiment shown in a). Values represent means of triplicate cultures.
Fig. S2. Growth (OD650) of Ruminococcus bromii, Blautia hydrogenotrophica and co‐cultures in continuous culture. Results are shown for six fermentors. Vessels R1 and C1 were inoculated simultaneously with R. bromii only (R1) or with both bacteria (C1) and supplied with RUM‐S medium containing 0.5% soluble starch. R2 and C2 refer to a repeat of this experiment. B1 and B2 vessels were run separately and supplied with RUM‐G medium containing 0.5% glucose (B1) or 0.2% glucose (B2); these vessels were inoculated with B. hydrogenotrophica only.
Fig. S3. Metabolite concentrations for the six fermentor experiments shown in Fig. S2. Blue arrows indicate the sampling points used for RNAseq analysis.
Fig. S4. KEGG Carbon metabolism pathway map for Blautia hydrogenotrophica. Genes present in the differentially expressed gene (DEG) list are highlighted with those with up regulated expression in the co‐culture compared to the mono‐culture in red and those with down regulated expression in green. Genes with KEGG orthologs found by KAAS‐KEGG, but no significant change in expression, are highlighted in blue. Numbers inside boxes are enzyme commission numbers.
Fig. S5. KEGG pathway map for metabolism of pantothenate and CoA in Blautia hydrogenotrophica. Genes present in the differentially expressed gene (DEG) list are highlighted with those with up regulated expression in the co‐culture compared to the mono‐culture in red and those with down regulated expression in green. Genes with KEGG orthologs found by KAAS‐KEGG, but no significant change in expression, are highlighted in blue.
Table S1. Calculated carbon balances for monocultures and co‐cultures.
Table S2. RNAseq read counts.
Table S3. Differentially expressed genes (DEGs) list from Ruminococcus bromii and Blautia hydrogenotrophica.
Table S4. Primers for qPCR.
Data Availability Statement
RNA‐seq data have been deposited in the ArrayExpress database at EMBL‐EBI (www.ebi.ac.uk/arrayexpress) under accession number E‐MTAB‐ 210350.