

Clostridioides difficile is an anaerobic, gastrointestinal pathogen of humans and other mammals. C. difficile produces two major toxins, TcdA and TcdB, which cause the symptoms of the disease, and forms dormant endospores to survive the aerobic environment outside the host. A recently discovered regulatory factor, RstA, inhibits toxin production and positively influences spore formation. Herein, we determine that RstA directly binds its own promoter DNA to repress its own gene transcription. In addition, our data demonstrate that RstA directly represses toxin gene expression and gene expression of two toxin gene activators, TcdR and SigD, creating a complex regulatory network to tightly control toxin production. This study provides a novel regulatory link between C. difficile sporulation and toxin production. Further, our data suggest that C. difficile toxin production is regulated through a direct, species-specific sensing mechanism.
KEYWORDS: Clostridium, Clostridium difficile, RNPP, RRNPP, TcdA, TcdB, helix-turn-helix, motility, spore, sporulation, toxin, transcriptional regulator
ABSTRACT
Clostridioides difficile infection (CDI) is a toxin-mediated diarrheal disease. Several factors have been identified that influence the production of the two major C. difficile toxins, TcdA and TcdB, but prior published evidence suggested that additional unknown factors were involved in toxin regulation. Previously, we identified a C. difficile regulator, RstA, that promotes sporulation and represses motility and toxin production. We observed that the predicted DNA-binding domain of RstA was required for RstA-dependent repression of toxin genes, motility genes, and rstA transcription. In this study, we further investigated the regulation of toxin and motility gene expression by RstA. DNA pulldown assays confirmed that RstA directly binds the rstA promoter via the predicted DNA-binding domain. Through mutational analysis of the rstA promoter, we identified several nucleotides that are important for RstA-dependent transcriptional regulation. Further, we observed that RstA directly binds and regulates the promoters of the toxin genes tcdA and tcdB, as well as the promoters for the sigD and tcdR genes, which encode regulators of toxin gene expression. Complementation analyses with the Clostridium perfringens RstA ortholog and a multispecies chimeric RstA protein revealed that the C. difficile C-terminal domain is required for RstA DNA-binding activity, suggesting that species-specific signaling controls RstA function. Our data demonstrate that RstA is a transcriptional repressor that autoregulates its own expression and directly inhibits transcription of the two toxin genes and two positive toxin regulators, thereby acting at multiple regulatory points to control toxin production.
INTRODUCTION
Clostridioides difficile infection (CDI) is a nosocomial and community-acquired gastrointestinal disease that affects individuals with dysbiotic gut microbiota, which commonly occurs after antibiotic treatment (1, 2). Clinical outcomes range from mild diarrhea to severe disease symptoms, including sepsis and death (1). The two glycosylating exotoxins, TcdA and TcdB, elicit CDI symptoms and are indispensable for C. difficile virulence (3). Environmental and intracellular signals, including nutrient availability and metabolic cues, strongly influence toxin production (4–7). There are numerous identified C. difficile factors that control toxin gene expression in response to these signals (8–12); however, the regulatory pathways and molecular mechanisms that directly control toxin gene expression are not fully understood (13).
Our previous work identified a novel regulator, RstA, which depresses C. difficile toxin production and motility (14). RstA inhibits transcription of the toxin genes tcdA and tcdB, the toxin-specific sigma factor, tcdR, and the flagellum-specific sigma factor, sigD, which is essential for motility and directs tcdR expression (11, 12, 14–16). In addition to repressing motility and toxin production, RstA positively influences C. difficile spore formation, which is critical for the survival of the bacterium outside of the host and for transmission from host to host, indicating that RstA regulates diverse phenotypes important for C. difficile pathogenesis. An rstA mutant exhibits increased toxin gene expression in vivo and is more virulent in the hamster model of CDI, demonstrating the impact RstA has on pathogenesis (14).
The predicted secondary structure of RstA reveals three apparent domains: an N-terminal conserved helix-turn-helix DNA-binding domain, followed by a series of multiple tetratricopeptide repeat (TPR) domains comprising a putative Spo0F-like protein-binding domain, and a C-terminal putative quorum-sensing-like domain (14). These characteristic features place RstA in the RRNPP (Rap/Rgg/NprR/PlcR/PrgX; formerly RNPP) family of proteins. RRNPP proteins are prevalent in Gram-positive organisms and regulate competence, sporulation, toxin production, and other important survival and virulence phenotypes (17–19). The DNA-binding or protein-binding activity of RRNPP proteins are controlled by the direct binding of small, quorum-sensing peptides (19). The precursor proteins encoding the quorum-sensing peptides are often adjacent to the regulatory RRNPP protein and are translated, exported, processed, and reinternalized at high cell densities (20–25). In addition, RRNPP proteins often autoregulate their own expression, as is observed for RstA (14). The presence of these conserved domains within RstA provides insight into how RstA may regulate C. difficile toxin production, motility, and sporulation.
To better understand the regulatory impact RstA exerts on C. difficile toxin production and sporulation, we examined the function of the conserved DNA-binding domain. Our previous study (14) had shown that the DNA-binding domain is required for RstA-dependent regulation of rstA expression and toxin gene expression but is expendable for sporulation regulation. Here, we demonstrate that RstA directly binds to its promoter via an imperfect inverted repeat and that it directly binds the sigD and toxin gene promoters. Further, our data demonstrate that RstA and SigD independently control toxin expression, creating a multitiered regulatory pathway by which RstA represses toxin production. Finally, we show that the Clostridium perfringens rstA ortholog does not complement toxin production or sporulation in a C. difficile rstA mutant. However, a chimeric RstA protein containing the C. perfringens DNA-binding domain and the C. difficile Spo0F-binding and quorum-sensing-binding domains restores sporulation and represses toxin production, providing evidence that the ability to respond to species-specific signaling is necessary for RstA DNA-binding activity.
RESULTS
RstA autoregulates its gene transcription via an inverted repeat overlapping the promoter.
Our previous work provided preliminary genetic evidence that the N-terminal putative helix-turn-helix DNA-binding domain was necessary for inhibition of toxin gene expression but was dispensable for sporulation initiation (14). However, further work with the recombinant His-tagged RstA proteins revealed that the constructs were expressed at low levels and were not detected by Western blotting of C. difficile lysates (data not shown). We created a new series of tagged proteins, possessing the 3×FLAG tag on the C-terminal end and found that these were stably expressed and easily detected in C. difficile rstA::erm lysates (see Fig. S2A in the supplemental material). Corroborating our previous data (14), expression of the wild-type RstA, the full-length FLAG-tagged RstA, and the truncated RstAΔHTH-FLAG-tagged allele complemented sporulation in the rstA mutant (Fig. S2B). As previously observed (14), only full-length RstA restored toxin production to wild-type levels in the rstA background (Fig. S2C and D), confirming that the helix-turn-helix motif within the DNA-binding domain is essential for RstA-dependent control of toxin production.
We hypothesized that RstA directly binds to DNA to control toxin gene expression and transcription of additional target genes. This interaction is predicted to occur via the putative DNA-binding domain, as observed for other RRNPP transcriptional regulators (26–28). Additionally, we previously observed that rstA expression remains relatively unchanged throughout growth in multiple conditions and that rstA transcription is increased in an rstA mutant (14), suggesting that expression of rstA may be autoregulated. To determine whether RstA is DNA-binding protein, we first defined the rstA promoter region and probed the DNA-binding capability of RstA within its own promoter. The transcriptional start of rstA was identified at −32 bp upstream from the translational start using 5′ RACE. Corresponding σA −10 and −35 consensus sequences were detected immediately upstream of this transcriptional start site (Fig. 1A and B). To verify the mapped promoter and to determine whether any additional promoters are present that drive rstA transcription, a series of promoter fragments fused to the phoZ reporter gene was created, and alkaline phosphatase (AP) activity was measured in the 630Δerm and rstA::erm mutants. As previously observed, the full-length 489-bp rstA promoter fragment exhibited a 1.8-fold increase in activity in the rstA mutant compared to the parent strain, indicating RstA-dependent repression (Fig. 1C) (14). The truncated promoter fragments, PrstA291 and PrstA231, produced similar fold changes in activity in the rstA mutant and parent strains, as observed for the full-length promoter. However, reporter activity was lower in the PrstA115 fragment compared to the longer fragments, suggesting that an enhancer sequence or an additional RstA-independent transcriptional activator is located between −231 bp to −115 bp upstream of the rstA open reading frame. A promoter fragment reporter fusion containing 380 bp of sequence upstream from the mapped rstA promoter [from −489 bp to −112 bp; intergenic region (IR) in Fig. 1A] was inactive, indicating that an additional promoter is not located within this region. We also tested whether RstA-dependent repression of the full-length PrstA reporter could be complemented. We expressed the rstA-FLAG construct from the nisin-inducible promoter, cprA (29), divergently from the PrstA::phoZ construct on the same plasmid in the rstA::erm background. PrstA reporter activity was reduced in a dose-dependent manner relative to the amount of nisin added to the medium (Fig. S3), further confirming the autoregulatory effect RstA exerts on its own expression. Altogether, the data demonstrate that the mapped σA-dependent promoter drives rstA expression and that RstA can repress transcription from this promoter.
FIG 1.

RstA controls its gene expression through an inverted repeat sequence overlapping the rstA promoter. (A) A schematic of the rstA promoter region denoting the general location of the putative RstA box, the transcriptional start (32 bp upstream from the start codon; represented by the bent arrow), and the rstA open reading frame (not to scale). The yellow boxes indicate the locations and sizes of promoter fragments constructed for the phoZ reporter fusions in panel C. (B) The rstA promoter, marked by +1, overlaps a 29-bp imperfect inverted repeat (shown in green). The asterisks above the sequence mark the mismatched nucleotides within the inverted repeat. The −10 and −35 consensus sequences and the ATG start codon are underlined. The nucleotides below the sequence represent the substitutions tested in panel D. (C and D) Alkaline phosphatase (AP) activity of the PrstA::phoZ reporter fusions of various lengths, including the upstream intergenic region (IR) (−489 bp to −112 relative to the translational start) of rstA (C) (PrstA115 [MC979/MC980]), PrstA231 [MC1010/MC1011], PrstA291 [MC1012/MC1013], PrstA489 [MC773/MC774], PrstAIR [MC1008/MC1009]) or of the full-length PrstA::phoZ promoter with various nucleotide substitutions (D) (PrstA489 [MC773/MC774], PrstAA-27T [MC830/MC831], PrstAA-27C [MC856/MC857], PrstAA-24G [MC858/MC859], PrstAT-23A [MC832/MC833], PrstAA-21C [MC860/MC861], PrstAT-19A [MC834/MC835], PrstAT-19G [MC862/MC863], PrstAT-19A/A-21C [MC1433/1434], PrstAA-18T [MC836/MC837], PrstAT-17A [MC838/MC839]) in strain 630Δerm and the rstA::erm mutant (MC391), respectively, grown on 70:30 sporulation agar at H8. The means ± standard errors of the means for four biological replicates are shown. Values that are significantly different (P < 0.05) by Student’s t test from the activity observed for the 630Δerm parent strain for each promoter construct are indicated by an asterisk.
The results obtained from the promoter-reporter fusions suggested that RstA binding was likely to occur within the 115 bp upstream of the translational start site. A 29-bp imperfect inverted repeat was identified within the predicted PrstA −10 consensus sequence, suggesting a possible regulatory binding site within this region (Fig. 1B). To determine whether this sequence serves as an RstA recognition site, we created a series of single nucleotide substitutions within the inverted repeat in the 489-bp PrstA reporter fusion, avoiding conserved residues required for RNAP-holoenzyme recognition (30). Most of the single nucleotide substitutions did not significantly alter reporter activity compared to the wild-type PrstA reporter (Fig. 1D). However, nucleotide substitutions in two positions, A-21 and T-19, abolished RstA repression in the parent strain, increasing reporter activity to match that of the rstA::erm mutant. These data suggest that the A-21 and T-19 nucleotides are important for RstA binding to the rstA promoter.
RstA inhibits toxin and motility gene transcription.
Regulatory control of toxin gene expression in C. difficile involves multiple sigma factors and transcriptional regulators, which ensure that toxin production occurs in the appropriate environmental conditions (13). Our previous work (14) demonstrated that an rstA::erm mutant has increased transcription of the C. difficile toxin genes, tcdA and tcdB, the toxin-specific sigma factor, tcdR, and the flagellum-specific sigma factor, sigD, which is required for motility and directs tcdR transcription (11, 12). To determine whether RstA is involved directly in repressing transcription of these genes, we first constructed phoZ reporter fusions with the promoter regions for each gene and examined RstA-dependent transcriptional activity.
The tcdR promoter region contains four identified independent promoter elements: a σA-dependent promoter (−16 bp from the translational start), a σD-dependent promoter (−76 bp from the translational start), and two putative σTcdR promoters farther upstream (Fig. 2A) (11, 12, 31–33). Expression of the tcdR gene is relatively low in C. difficile (11, 32, 34), at least in part due to repression by CodY and CcpA binding throughout the tcdR promoter region under nutrient-rich conditions (8, 9, 33, 35, 36). We examined each of the promoter elements within PtcdR to determine whether RstA affects transcription from these promoters. A series of reporter fusions was created for each of the promoter elements, which were examined in the rstA::erm mutant and parent strain, and activity was measured after 24 h of growth in TY medium (Fig. 2A). A full-length 517-bp PtcdR::phoZ reporter and the two σTcdR-dependent promoter fusions exhibited similar low reporter activities in the parent and rstA strains (Fig. 2B). However, increased reporter activity was observed in the rstA mutant for the individual σA-dependent and σD-dependent promoter fusions. These results indicate that RstA impacts the function of these promoter elements and contributes to repression of tcdR transcription.
FIG 2.

RstA inhibits toxin gene expression. (A) A schematic of the promoter regions of tcdR, tcdA, and tcdB denoting the relative locations of the transcriptional start sites experimentally demonstrated (12, 32–34) and the open reading frames of all three genes (not drawn to scale). Pale red boxes approximate CodY- and CcpA-binding sites within the toxin gene promoters (8, 9, 36). The yellow boxes indicate the locations and sizes of the promoter fragments constructed for the phoZ reporter fusions in panels B to D. Alkaline phosphatase (AP) activity of the PtcdR::phoZ reporter fusions of various lengths (B) (promoterless phoZ [MC448], PtcdRσA [MC1285/MC1286], PtcdRσD [MC1145/MC1146], PtcdRσTcdR(P2) [MC1147/MC1148], and PtcdRσTcdR(P1) [MC1149/MC1150]) and the PtcdA::phoZ (C) (−511 bp to −1 bp upstream of transcriptional start; MC1249/MC1250) or PtcdB::phoZ (D) (−531 bp to −31 bp upstream of transcriptional start [MC1251/MC1252]) reporter fusions in strain 630Δerm and the rstA::erm mutant (MC391) grown in TY medium (pH 7.4) at H24. The means and standard errors of the means for four biological replicates are shown. *, P < 0.05, using Student’s t test compared to the activity observed in the 630Δerm parent strain for each promoter construct.
We also examined RstA-dependent regulation of tcdA and tcdB transcription, both of which are expressed solely from σTcdR-dependent promoters (Fig. 2A) (34, 37, 38). PtcdA reporter activity was increased 3.6-fold and PtcdB activity was 2.1-fold greater in the rstA strain compared to the parent (Fig. 2C and D). Altogether, these data indicate that RstA represses toxin gene transcription at the individual gene level and through repression of tcdR.
SigD, also known as FliA or σ28, is a sigma factor that coordinates flagellar gene expression and directly activates tcdR gene expression (32). The sigD gene is located in a large, early-stage flagellar operon that is transcribed from a σA-dependent promoter located 496 bp upstream from the first gene of the flgB operon (39). Interestingly, the flgB promoter sequences from two different C. difficile strains, the historical epidemic strain, strain 630, and a current epidemic strain, strain R20291, are identical to the σA promoter sequence through the translational start site but diverge considerably upstream of this region (Fig. S4). No additional promoter elements were identified in the strain 630 or R20291 sequences upstream of the σA-dependent promoter (Fig. 3A). To determine whether RstA influences sigD transcription through repression of PflgB, promoter reporter fusions representing each strain were constructed. As anticipated, activity of the strain 630Δerm and R20291 PflgB reporters were higher in the rstA mutant than in the parent strain (1.7-fold and 1.5-fold, respectively; Fig. 3B), indicating that RstA represses flgB and consequently, sigD transcription.
FIG 3.

RstA represses expression of flgB reporter fusions. (A) A schematic of the flgB promoter regions for C. difficile 630 and R20291 strains. The transcriptional start site for the σA-dependent promoter for strain 630 lies −496 bp upstream from the flgB translational start, while the R20291 strain initiates transcription −498 bp upstream (39, 56). (B) Alkaline phosphatase (AP) activity of the promoterless::phoZ vector in 630Δerm (MC1106) and PflgB630Δerm::phoZ (MC1294/MC1295) and PflgBR20291::phoZ (MC1296/MC1297) reporter fusions in 630Δerm and the rstA::erm mutant (MC391) grown in TY medium (pH 7.4) at T3 (three hours after the start of transition phase [OD600 of 1.0]). The means and standard errors of the means for three biological replicates are shown. *, P < 0.05, using Student’s t test compared to the activity observed in the 630Δerm parent strain for each promoter construct.
RstA directly binds the rstA, tcdR, flgB, tcdA, and tcdB promoters via the conserved helix-turn-helix DNA-binding domain.
To determine whether RstA directly binds target DNA, a variety of in vitro electrophoretic gel shift assays were attempted, but no binding was observed in any condition tested. We considered that the lack of RstA-DNA interaction by gel shift may occur because of the absence of a cofactor, such as a quorum-sensing peptide, or because of a transient complex or oligomerization state. To overcome this obstacle, we performed biotin-labeled DNA pulldown assays to assess the DNA-binding capacity of RstA under native conditions. Biotinylated DNA was coupled to streptavidin beads as bait and incubated with cell lysates expressing either full-length RstA-FLAG or RstAΔHTH-FLAG protein. Specifically bound proteins were eluted and analyzed by Western blotting using FLAG M2 antibody.
We first tested the ability of RstA to directly interact with its own promoter. RstA-FLAG protein was recovered using the wild-type rstA promoter region as bait, demonstrating specific interaction of the RstA protein (Fig. 4A). However, the PrstA fragment did not capture RstAΔHTH-FLAG protein, indicating that the conserved HTH domain of RstA is essential for DNA binding. In addition to the wild-type rstA promoter, the PrstA T-19A and PrstA A-21C variants that eliminated RstA-dependent regulation in vivo were used as bait (Fig. 1D). Both the PrstA T-19A and PrstA T-19A/A-21C variants captured significantly less RstA-FLAG than the wild-type promoter, suggesting that at least the T-19A nucleotide facilitates RstA interaction (Fig. 4A and Fig. S5A). The intergenic region upstream of the rstA promoter (Fig. 1A, IR) did not recover the full-length RstA-FLAG, indicating that RstA recognizes a specific DNA sequence within the promoter region. Finally, RstA-FLAG did not interact with unlabeled streptavidin beads nonspecifically (Fig. 4A and Fig. S5A). Altogether, these data demonstrate that RstA functions as a DNA-binding protein that directly and specifically binds its own promoter to repress transcription.
FIG 4.

RstA binds to the rstA, tcdR, flgB, tcdA, and tcdB promoters. Western blot analysis using FLAG M2 antibody to detect recombinant RstA-3XFLAG or RstAΔHTH-3XFLAG in cell lysates or following biotin-labeled DNA pulldown assays. As a control, cell lysate expressing the RstA-3XFLAG construct (MC1004) or the RstAΔHTH-3XFLAG construct (MC1028) is included in the first lane or two of each Western blot shown. Additional negative controls in each panel include unbiotinylated full-length rstA promoter (−) and beads-only controls to ensure that RstA does not interact with the beads nonspecifically. The biotin-labeled fragments used as bait are of the 115-bp wild-type, T-19A, A-21C, or T-19A/A-21C rstA promoters or of the 380-bp intergenic region upstream of the rstA promoter (IR; see Fig. 2; present in all panels) (A), the full-length tcdR (446-bp) or the 630Δerm or R20291 flgB (229-bp) promoters (B), the full-length tcdR (446-bp), σA-dependent (92-bp), σD-dependent (116-bp), σTcdRP2-dependent (188-bp), or σTcdRP1-dependent (112-bp) promoters (C), or the full-length tcdR (446-bp), tcdA (511-bp), or tcdB (501-bp) promoters (D). All promoter fragments were bound to streptavidin-coated magnetic beads and incubated with C. difficile cell lysates grown in TY medium (pH 7.4) supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin to mid-log phase (OD600 of 0.5 to 0.7), expressing either the RstA-3XFLAG construct (MC1004) or the RstAΔHTH-3XFLAG construct (MC1028).
To determine whether RstA directly binds DNA to repress the transcription of genes encoding toxin regulators, we examined RstA binding to the flgB and tcdR promoter regions. RstA-FLAG protein bound specifically to the full-length tcdR promoter region, as well as the 630 and R20291 flgB promoters (Fig. 4B and Fig. S5B). Again, the HTH domain was required for these RstA-promoter interactions. To identify which internal promoter elements directly interact with RstA, previously characterized tcdR promoter fragments were used as bait (Fig. 2B), with the exception of a longer σA-dependent promoter fragment (92 bp rather than 76 bp) to limit potential steric hindrance of RstA binding due to the 5ʹ biotin label. This longer 92-bp PtcdR(σA) fragment exhibited the same RstA-dependent regulation in reporter assays as the 76-bp reporter (Fig. S6). RstA-FLAG bound to the σA-dependent and σD-dependent tcdR promoter fragments but was not recovered from either of the σTcdR-dependent promoters (Fig. 4C and Fig. S5B), corroborating the reporter fusion results that demonstrated RstA repression of only the σA-dependent and σD-dependent tcdR promoter elements.
DNA pulldown assays were also performed to ascertain whether RstA directly binds to the tcdA and tcdB promoters. Both of the toxin promoters captured the full-length RstA-FLAG protein and failed to recover the RstAΔHTH-FLAG protein (Fig. 4D and Fig. S5B). These data provide direct biochemical evidence that RstA represses flgB, tcdR, tcdA, and tcdB transcription by binding to the promoter regions of these genes.
RstA represses toxin gene expression independently of SigD-mediated toxin regulation.
Our data indicate that RstA represses toxin gene expression directly by binding to the tcdA and tcdB promoter regions and indirectly by repressing transcription of the sigma factors tcdR and sigD, which activate toxin gene expression. The biotin pulldown data suggest that RstA represses toxin gene expression through a multitiered regulatory pathway. To test whether direct repression of tcdA and tcdB transcription by RstA is physiologically relevant and independent of SigD, we created an rstA sigD double mutant and examined the impact of each mutation on toxin production. To aid in construction of an rstA sigD double mutant, we utilized the recently developed CRISPR-Cas9 system modified for use in C. difficile to create an unmarked, nonpolar deletion of rstA in the 630Δerm and sigD::erm backgrounds (Fig. S7) (40). TcdA protein levels were ∼3-fold higher in the rstA sigD double mutant than in the sigD mutant (Fig. 5A; total protein loaded shown in Fig. S8A), indicating that RstA represses toxin production independently of SigD. Overexpression of rstA in the rstA sigD mutant returned TcdA protein to the levels found in the sigD mutant. Likewise, a previously characterized sigD overexpression construct (11, 41) restored TcdA to wild-type levels in the rstA sigD mutant, further supporting that SigD and RstA regulate toxin production independently (Fig. 5A). In addition, transcript levels of tcdA, tcdB, and tcdR were increased in the rstA sigD mutant compared to the levels in the sigD mutant (Fig. 5B), mirroring the TcdA protein results. Altogether, these data provide further evidence that RstA is major regulator of toxin production that directly and indirectly represses toxin gene expression independently of SigD.
FIG 5.

RstA represses toxin gene expression independently of SigD-mediated regulation. (A) Western blot analysis of TcdA in 630Δerm pMC211 (MC282; vector control), rstA pMC211 (MC1224; vector control), sigD::erm pMC211 (MC506; vector control), rstA sigD::erm pMC211 (MC1281), rstA sigD::erm pPcprA-rstA (MC1282), rstA sigD::erm pPcprA-sigD (MC1283), and rstA pPcprA-rstA (MC1225) grown in TY medium (pH 7.4) supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin, at 24 h. The corresponding image showing total protein is shown in Fig. S8A in the supplemental material. (B) qRT-PCR analysis of tcdR, tcdA, and tcdB transcript levels in 630Δerm pMC211 (MC282; vector control), rstA pMC211 (MC1224; vector control), sigD::erm pMC211 (MC506; vector control), rstA sigD::erm pMC211 (MC1281), rstA sigD::erm pPcprA-rstA (MC1282), and rstA sigD::erm pPcprA-sigD (MC1283) grown in TY medium (pH 7.4) supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin, at T3 (three hours after the entry into stationary phase). The means and standard errors of the means for three biological replicates are shown. *, P < 0.05 by Student’s t test between sigD::erm pMC211 and rstA sigD::erm pMC211.
RstA DNA-binding activity requires the species-specific C-terminal domains.
The observation that RstA does not bind to target DNA in the tested in vitro conditions but does bind DNA in cell lysates suggests that a cofactor is required for RstA DNA-binding activity. We hypothesize that a small, quorum-sensing peptide serves as an activator for RstA DNA binding, as has been observed for other members of the RRNPP family (23–25, 42–44). To test this, we expressed RstA orthologs of other clostridial species (Fig. S9A) (45), including Clostridium acetobutylicum, Clostridium perfringens, and Clostridium (Paeniclostridium) sordellii in the C. difficile rstA mutant background. Only the C. perfringens RstA was stably produced in C. difficile (Fig. S9B). However, expression of the C. perfringens rstA ortholog failed to restore TcdA protein to wild-type levels (Fig. 6A; total protein loaded shown in Fig. S8B). C. perfringens RstA may be unable to repress C. difficile toxin production because the C. perfringens DNA-binding domain cannot recognize the C. difficile DNA target sequences and/or because the DNA-binding activity of C. perfringens RstA is not functional in C. difficile. To distinguish between these possibilities, we constructed a chimeric protein containing the C. perfringens DNA-binding domain (M1-Y51) fused to the C-terminal domains of the C. difficile RstA protein (herein known as CpHTH-CdCterminalFLAG) and examined the function of this chimeric RstA in the C. difficile rstA mutant. The RstA chimera restored C. difficile TcdA levels to those observed in the parent strain (Fig. 6A), indicating that the C. perfringens DNA-binding domain is functional in C. difficile. To confirm these results, we performed qRT-PCR analyses of tcdR, tcdA, and tcdB genes in these strains. The full-length C. perfringens RstA did not complement toxin gene expression in the C. difficile rstA mutant, while the CpHTH-CdCterminal-FLAG chimeric RstA restored toxin gene transcript levels back to those observed in the parent strain (Fig. 6B), corroborating our previous results. These data strongly suggest that the C-terminal portion of RstA responds to species-specific signals to control the N-terminal DNA-binding activity.
FIG 6.

A hybrid rstA construct containing the C. perfringens DNA-binding domain with the C. difficile Spo0F-like and quorum-sensing-like domains complements C. difficile rstA toxin production and sporulation. (A) Western blot analysis of TcdA in 630Δerm pMC211 (MC282; vector control), rstA::erm pMC211 (MC505; vector control), rstA::erm pPcprA-rstA3XFLAG (MC1004), rstA::erm pPcprA-Cp-rstA3XFLAG (MC1324), and rstA::erm pPcprA-CpHTHCdCterminal3XFLAG (MC1257) grown in TY medium, pH 7.4, supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin, at H24. The corresponding image showing total protein is shown in Fig. S8B. (B) qRT-PCR analysis of tcdR, tcdA, and tcdB transcript levels in 630Δerm pMC211 (MC282; vector control), rstA::erm pMC211 (MC505; vector control), rstA::erm pPcprA-rstA3XFLAG (MC1004), rstA::erm pPcprA-Cp-rstA3XFLAG (MC1324), and rstA::erm pPcprA-CpHTHCdCterminal3XFLAG (MC1257) grown in TY medium, pH 7.4, supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin, at T3 (three hours after the entry into stationary phase). (C) Ethanol-resistant spore formation of 630Δerm pMC211 (MC282; vector control), rstA::erm pMC211 (MC505; vector control), rstA::erm pPcprA-rstA3XFLAG (MC1004), rstA::erm pPcprA-Cp-rstA3XFLAG (MC1324), and rstA::erm pPcprA-CpHTHCdCterminal3XFLAG (MC1257) grown on 70:30 sporulation agar supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin. Sporulation frequency is calculated as the number of ethanol-resistant spores divided by the total number of cells enumerated at H24 as detailed in Materials and Methods. The means and standard errors of the means for at least three independent biological replicates are shown; asterisks represent P ≤ 0.05 by one-way ANOVA, followed by Dunnett’s multiple-comparison test compared to rstA pMC211 (MC505).
Finally, we assessed the ability of a C. perfringens RstA to complement the low sporulation frequency of the C. difficile rstA mutant. Overexpressing the full-length C. perfringens RstA did not complement sporulation in the C. difficile rstA mutant (Fig. 6C). Unexpectedly, a hypersporulation phenotype was observed when the CpHTH-CdC-terminalFLAG RstA chimera was expressed in the rstA mutant (Fig. 6C), indicating that the chimeric C. perfringens-C. difficile RstA promotes C. difficile sporulation to even higher levels than the native C. difficile RstA does. This hypersporulation phenotype suggests that the C. perfringens HTH portion of the chimeric RstA protein alters the structure or activity of RstA to increase the positive effect on early sporulation events. These data warrant further investigation into the molecular mechanisms by which the C-terminal domains of RstA cooperate with the DNA-binding domain to promote sporulation.
DISCUSSION
The production of exotoxins and the ability to form quiescent endospores are two essential features of C. difficile pathogenesis. The regulatory links between toxin production and spore formation are complex and poorly understood. Some conserved sporulation regulatory factors, including Spo0A, CodY, and CcpA, strongly influence toxin production, yet some of these regulatory effects appear to be dependent on the strain or are indirect (8, 36, 46–48). Further, additional environmental conditions and metabolic signals, such as temperature and proline, glycine, and cysteine availability (5, 6, 10, 49), impact toxin production independently of these regulators, revealing the possibility that additional unknown factors are directly involved in toxin regulation (13). The recently discovered RRNPP regulator, RstA, represses toxin production and promotes spore formation, potentially providing a direct and inverse link between C. difficile spore formation and toxin biogenesis (14).
In this study, we show that RstA is a major, direct transcriptional regulator of C. difficile toxin gene expression. RstA inhibits toxin production by directly binding to the tcdA and tcdB promoters and repressing their transcription. RstA reinforces this repression by directly downregulating gene expression of tcdR, which encodes the sole sigma factor that drives tcdA and tcdB transcription. Finally, RstA directly represses the flgB promoter, inhibiting gene expression of the flagellum-specific sigma factor, SigD. SigD activates motility gene transcription but is also required for full expression of tcdR (11, 12). RstA repression of each major component in the toxin regulatory pathway creates a multitiered network in which RstA directly and indirectly controls tcdA and tcdB gene expression (Fig. 7).
FIG 7.
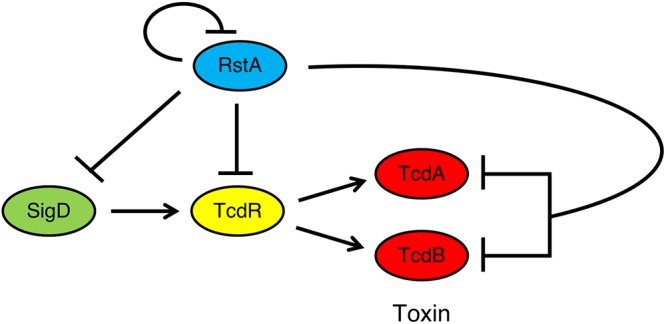
Model of RstA-mediated repression of C. difficile toxin production. SigD, the flagellum-specific sigma factor, directly induces gene transcription of tcdR, the toxin-specific sigma factor. Toxin gene expression is then directed by TcdR. RstA inhibits production of TcdA and TcdB and its own gene expression by directly binding to and repressing transcription of sigD, tcdR, tcdA, tcdB, and rstA, creating a complex, multitiered regulatory network to ensure that the toxin gene expression is appropriately timed in response to the signal(s) that activate RstA.
RstA is the third characterized transcriptional repressor that directly binds to promoter regions for tcdR, tcdA, and tcdB, following two other transcriptional repressors, CodY and CcpA (8, 9, 36), The in vivo contribution of this reinforced repression of tcdA and tcdB transcription by CodY, CcpA, and RstA remains unknown. Interestingly, recent evidence has demonstrated that tcdR gene expression serves as a bistable switch that determines whether individual C. difficile cells within a population produce TcdA and TcdB, creating a divided population of toxin-OFF and toxin-ON cells (50). TcdR governs this bistability state by maintaining low basal expression levels, allowing for small changes to result in stochastic gene expression, and by positively regulating its own expression, establishing a positive-feedback loop that bolsters the toxin-ON state (50). CodY was found to influence the population so that fewer cells produced toxin, but CcpA and RstA were not tested (50). We predict that both CcpA and RstA would bias the population of cells to a toxin-OFF state. Altogether, the tight control of tcdR transcription, reinforced by direct repression of tcdA and tcdB transcription by CcpA, CodY, and RstA, results in the convergence of multiple regulatory pathways at the bistable tcdR promoter to coordinate toxin production in response to nutritional and species-specific signals. This complex regulation ensures that the energy-intensive process of toxin production is initiated only to benefit the bacterium under the appropriate conditions.
Importantly, RstA is the first transcriptional regulator demonstrated to directly control flgB transcription initiation. To date, none of the previously identified regulators of flgB expression, including Spo0A, SigH, Agr, Hfq, SinR, and SinR′, have been shown to bind promoter DNA and regulate flagellar gene expression through transcription initiation (46, 51–54). flgB expression is further regulated posttranscriptionally via a c-di-GMP riboswitch and a flagellar switch, both of which are located within the large, 496-bp 5′ untranslated region (39, 55, 56); however, the impact of RstA-mediated repression of flgB gene expression through additional pathways has not yet been explored.
Although we have identified several direct RstA targets, the sequence required to recruit RstA to target promoters remains unclear. The rstA promoter contains a near-perfect inverted repeat; however, this sequence is AT rich, as is the case for many C. difficile promoters. Imperfect inverted repeats were also found overlapping the −35 consensus sequences of the tcdA, tcdB, flgB, and σA-dependent tcdR promoters, and immediately upstream of the σD-dependent tcdR promoter (Fig. S10) (57), suggesting that RstA inhibits transcription at these promoters by sterically obstructing RNA polymerase docking. No clear consensus sequence defining an RstA box is delineated from these sequences. Other RRNPP regulators have also been found to bind imperfect, palindromic repeats or specific, conserved sequences in target promoters, but to our knowledge, only PlcR has a defined binding motif (24, 58, 59). Exhaustive attempts at ChIP-seq analysis to identify the C. difficile RstA regulon proved unsuccessful; however, our data imply that RstA is a transcriptional repressor that directly controls multiple C. difficile phenotypes, and additional targets within the C. difficile genome seem likely.
The inability to recapitulate RstA-DNA binding with purified RstA in vitro together with the functional analysis of full-length and chimeric C. difficile and C. perfringens proteins suggest that (i) RstA DNA-binding activity requires a cofactor and (ii) this cofactor is species specific. Most RRNPP members are cotranscribed with their cognate quorum-sensing peptide precursor (19), but there are notable exceptions, including those encoded by unlinked genes (42, 60) and orphan receptors whose cognate ligands have not yet been discovered (61–63). RstA falls into this latter category, as there are no open reading frames adjacent to rstA that encode an apparent quorum-sensing peptide precursor. Importantly, no type of ligand other than small, quorum-sensing peptides has been identified for RRNPP proteins. In addition to RstA, other quorum-sensing factors have been implicated in C. difficile toxin production. The incomplete Agr1 and conserved Agr2 quorum-sensing systems induce toxin production through the production of a cyclic autoinducer peptide (AIP) that is sensed extracellularly (52, 64, 65); however, it is highly unlikely that the extracellular AIP molecule directly interacts with the cytosolic RstA protein. In addition, the interspecies LuxS-derived autoinducer-2 (AI-2) quorum-sensing molecule was found to increase C. difficile tcdA and tcdB gene expression, but not tcdR gene expression (66), indicating that AI-2 does not signal through RstA either. Identification of the cofactor that controls RstA activity is a high priority, as this will likely provide insight into the physiological conditions and/or metabolites that influence C. difficile TcdA and TcdB production.
Finally, as RstA is necessary for efficient C. difficile spore formation, the possibility remains that species-specific signaling is required for RstA-dependent control of early sporulation and that RstA coordinates C. difficile toxin production and spore formation in response to the same signal(s). Elucidating the molecular mechanisms that govern RstA activity will provide important insights into the regulatory control between sporulation and toxin production, reveal host cues and conditions that lead to increased toxin production, and help delineate the early sporulation events that control C. difficile Spo0A phosphorylation and activation.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in the study are listed in Table 1. Clostridioides difficile strains were routinely cultured in BHIS or TY medium (pH 7.4) supplemented with 2 to 5 µg/ml thiamphenicol and/or 1 µg/ml nisin throughout growth as needed (67). Overnight cultures of C. difficile were supplemented with 0.1% taurocholate and 0.2% fructose to promote spore germination and prevent sporulation, respectively, as indicated (67, 68). C. difficile strains were cultured in a 37°C anaerobic chamber with an atmosphere of 10% H2, 5% CO2, and 85% N2, as previously described (69). Escherichia coli strains were grown at 37°C in LB (70) with 100 µg/ml ampicillin and/or 20 µg/ml chloramphenicol as needed. Kanamycin (50 µg/ml) was used for counterselection against E. coli HB101 pRK24 after conjugation with C. difficile, as previously described (71).
TABLE 1.
Bacterial strains and plasmids used in this study
| Plasmid or strain | Relevant genotype or feature(s) | Source, construction, or reference |
|---|---|---|
| Plasmids | ||
| pRK24 | Tra+ Mob+ bla, tet | 78 |
| pJK02 | E. coli-C. difficile shuttle vector; catP, cas9, pyrE sgRNA, pyrE homology region | 40 |
| pMC123 | E. coli-C. difficile shuttle vector; bla catP | 29 |
| pMC211 | pMC123 PcprA | 77 |
| pMC358 | pMC123 ::phoZ | 75 |
| pMC367 | pMC123 PcprA-rstA (CD3668) | 14 |
| pMC533 | pMC123 PcprA-rstA (C. sordellii ATCC 9714) | This study |
| pMC543 | pMC123 PrstA489::phoZ | 14 |
| pMC559 | pMC123 PrstAA-27T::phoZ | This study |
| pMC560 | pMC123 PrstAT-23A::phoZ | This study |
| pMC561 | pMC123 PrstAT-19A::phoZ | This study |
| pMC562 | pMC123 PrstAA-18T::phoZ | This study |
| pMC563 | pMC123 PrstAT-17A::phoZ | This study |
| pMC573 | pMC123 PrstAA-27C::phoZ | This study |
| pMC574 | pMC123 PrstAA-24G::phoZ | This study |
| pMC575 | pMC123 PrstAA-21C::phoZ | This study |
| pMC576 | pMC123 PrstAT-19G::phoZ | This study |
| pMC660 | pMC123 PrstA115::phoZ | This study |
| pMC675 | pMC123 PcprA-rstA-3XFLAG | This study |
| pMC676 | pMC123 PrstAIR(380 bp)::phoZ | This study |
| pMC677 | pMC123 PrstA231::phoZ | This study |
| pMC678 | pMC123 PrstA291::phoZ | This study |
| pMC682 | pMC123 PcprA-rstAΔHTH-3XFLAG | This study |
| pMC713 | pMC123 PtcdR::phoZ | This study |
| pMC726 | pJK02 with rstA homology region | This study |
| pMC729 | pMC726 with rstA sgRNA (oMC1724) | This study |
| pMC752 | pMC123 PtcdR(σA-92 bp)::phoZ | This study |
| pMC753 | pMC123 PtcdR(σD)::phoZ | This study |
| pMC754 | pMC123 PtcdR(P2 σTcdR)::phoZ | This study |
| pMC755 | pMC123 PtcdR(P1 σTcdR)::phoZ | This study |
| pMC780 | pMC123 PcprA-rstA (C. perfringens S13) | This study |
| pMC787 | pMC123 PcprA-rstA (C. acetobutylicum ATCC 824) | This study |
| pMC795 | pMC123 PtcdA::phoZ | This study |
| pMC796 | pMC123 PtcdB::phoZ | This study |
| pMC798 | pMC123 PcprA-rstACpHTHCdCterminal-3XFLAG | This study |
| pMC812 | pMC123 PtcdR(σA-76 bp)::phoZ | This study |
| pMC817 | pRT1824 PflgB (630)::phoZ | This study |
| pMC818 | pRT1824 PflgB (R20291)::phoZ | This study |
| pMC828 | pMC123 PcprA-rstA-3XFLAG (C. acetobutylicum ATCC 824) | This study |
| pMC829 | pMC123 PcprA-rstA-3XFLAG (C. perfringens S13) | This study |
| pMC830 | pMC123 PcprA-rstA-3XFLAG (C. sordellii ATCC 9714) | This study |
| pMC888 | pMC123 PrstA::phoZ PcprA-rstA-3XFLAG | This study |
| pMC889 | pMC123 PrstAT-19A/A-21C::phoZ | This study |
| pRPF144 | pMLT960 Pcwp2-gusA | 79 |
| pRT1824 | pMLT960 ::phoZ | This study |
| pSigD | pMC123 PcprA-sigD | 11 |
| E. coli strains | ||
| HB101 pRK24 | F− mcrB mrr hsdS20(rB− mB−) recA13 leuB6 ara-14 proA2 lacY1 galK2 xyl-5 mtl-1 rpsL20 pRK24 | B. Dupuy |
| C. difficile strains | ||
| 630Δerm | Erms derivative of strain 630 | Nigel Minton; 80 |
| MC282 | 630Δerm pMC211 | 77 |
| MC310 | 630Δerm spo0A::erm | 77 |
| MC391 | 630Δerm rstA::erm | 14 |
| MC448 | 630Δerm pMC358 | 75 |
| MC480 | 630Δerm rstA::erm pMC367 | 14 |
| MC505 | 630Δerm rstA::erm pMC211 | 14 |
| MC506 | 630Δerm sigD::erm pMC211 | This study |
| MC762 | 630Δerm rstA::erm pMC533 | This study |
| MC773 | 630Δerm pMC543 | 14 |
| MC774 | 630Δerm rstA::erm pMC543 | 14 |
| MC830 | 630Δerm pMC559 | This study |
| MC831 | 630Δerm rstA::erm pMC559 | This study |
| MC832 | 630Δerm pMC560 | This study |
| MC833 | 630Δerm rstA::erm pMC560 | This study |
| MC834 | 630Δerm pMC561 | This study |
| MC835 | 630Δerm rstA::erm pMC561 | This study |
| MC836 | 630Δerm pMC562 | This study |
| MC837 | 630Δerm rstA::erm pMC562 | This study |
| MC838 | 630Δerm pMC563 | This study |
| MC839 | 630Δerm rstA::erm pMC563 | This study |
| MC856 | 630Δerm pMC573 | This study |
| MC857 | 630Δerm rstA::erm pMC573 | This study |
| MC858 | 630Δerm pMC574 | This study |
| MC859 | 630Δerm rstA::erm pMC574 | This study |
| MC860 | 630Δerm pMC575 | This study |
| MC861 | 630Δerm rstA::erm pMC575 | This study |
| MC862 | 630Δerm pMC576 | This study |
| MC863 | 630Δerm rstA::erm pMC576 | This study |
| MC979 | 630Δerm pMC660 | This study |
| MC980 | 630Δerm rstA::erm pMC660 | This study |
| MC1004 | 630Δerm rstA::erm pMC675 | This study |
| MC1008 | 630Δerm pMC676 | This study |
| MC1009 | 630Δerm rstA::erm pMC676 | This study |
| MC1010 | 630Δerm pMC677 | This study |
| MC1011 | 630Δerm rstA::erm pMC677 | This study |
| MC1012 | 630Δerm pMC678 | This study |
| MC1013 | 630Δerm rstA::erm pMC678 | This study |
| MC1028 | 630Δerm rstA::erm pMC682 | This study |
| MC1088 | 630Δerm pMC713 | This study |
| MC1089 | 630Δerm rstA::erm pMC713 | This study |
| MC1118 | 630Δerm ΔrstA | This study |
| MC1133 | 630Δerm pMC729 | This study |
| MC1143 | 630Δerm pMC752 | This study |
| MC1144 | 630Δerm rstA::erm pMC752 | This study |
| MC1145 | 630Δerm pMC753 | This study |
| MC1146 | 630Δerm rstA::erm pMC753 | This study |
| MC1147 | 630Δerm pMC754 | This study |
| MC1148 | 630Δerm rstA::erm pMC754 | This study |
| MC1149 | 630Δerm pMC755 | This study |
| MC1150 | 630Δerm rstA::erm pMC755 | This study |
| MC1193 | 630Δerm sigD::erm pMC729 | This study |
| MC1224 | 630Δerm ΔrstA pMC211 | This study |
| MC1225 | 630Δerm ΔrstA pMC367 | This study |
| MC1249 | 630Δerm pMC795 | This study |
| MC1250 | 630Δerm rstA::erm pMC795 | This study |
| MC1251 | 630Δerm pMC796 | This study |
| MC1252 | 630Δerm rstA::erm pMC796 | This study |
| MC1257 | 630Δerm rstA::erm pMC798 | This study |
| MC1278 | 630Δerm ΔrstA sigD::erm | This study |
| MC1281 | 630Δerm ΔrstA sigD::erm pMC211 | This study |
| MC1282 | 630Δerm ΔrstA sigD::erm pMC367 | This study |
| MC1283 | 630Δerm ΔrstA sigD::erm pSigD | This study |
| MC1285 | 630Δerm pMC812 | This study |
| MC1286 | 630Δerm rstA::erm pMC812 | This study |
| MC1294 | 630Δerm pMC817 | This study |
| MC1295 | 630Δerm rstA::erm pMC817 | This study |
| MC1296 | 630Δerm pMC818 | This study |
| MC1297 | 630Δerm rstA::erm pMC818 | This study |
| MC1323 | 630Δerm rstA::erm pMC828 | This study |
| MC1324 | 630Δerm rstA::erm pMC829 | This study |
| MC1325 | 630Δerm rstA::erm pMC830 | This study |
| MC1433 | 630Δerm pMC889 | This study |
| MC1434 | 630Δerm rstA::erm pMC889 | This study |
| MC1435 | 630Δerm rstA::erm pMC888 | This study |
| RT1075 | 630Δerm sigD::erm | 81 |
| Other strains | ||
| ATCC 824 | Clostridium acetobutylicum | ATCC |
| ATCC 9714 | Clostridium sordellii | ATCC |
Strain and plasmid construction and accession numbers.
Oligonucleotides used in this study are listed in Table 2. Details of vector construction are described in the supplemental material (see Fig. S1 in the supplemental material). C. difficile strains 630 (GenBank accession no. NC_009089.1) and R20291 (GenBank accession no. FN545816.1), Clostridium acetobutylicum ATCC 824 (GenBank accession no. NC_003030.1), Clostridium sordellii ATCC 9714 (GenBank accession no. APWR00000000), and Clostridium perfringens S13 (GenBank accession no. BA000016.3) were used as the templates for primer design and PCR amplification. The rstA ortholog from C. acetobutylicum was synthesized by Genscript (Piscataway, NJ). The Streptococcus pyogenes CRISPR-Cas9 system, which has been modified for use in C. difficile (40), was used to create a nonpolar deletion of the rstA gene. The 630Δerm and RT1075 (sigD::erm) strains containing the rstA-targeted CRISPR-Cas9 plasmid (MC1133 and MC1193, respectively) were grown overnight in TY medium with 5 μg/ml thiamphenicol. The next morning, the cultures were backdiluted into fresh TY medium supplemented with 5 μg/ml thiamphenicol and 100 ng/ml anhydrous tetracycline for 24 h to induce expression of the CRISPR-Cas9 system. A small aliquot of this culture was streaked onto BHIS plates, and colonies were screened by PCR for the presence or absence of the rstA allele.
TABLE 2.
Oligonucleotides used in this studya



Underlined nucleotides denote the restriction sites used for vector construction. Boldface red nucleotides indicate the bases mutated within the inverted repeat overlapping the rstA promoter.
DNA cloning and vector details. Download FIG S1, PDF file, 0.06 MB (60.4KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The conserved helix-turn-helix (HTH) DNA-binding domain of RstA is not necessary for positively controlling sporulation but is required for the regulation of toxin production and PrstA expression. (A) Western blot analysis using FLAG M2 antibody in rstA pPcprA-rstA-3XFLAG (MC1004) and rstA pPcprA-rstAΔHTH-3XFLAG (MC1028) grown in TY medium, pH 7.4, at log phase (OD600 of 0.5). (B) Ethanol-resistant spore formation of 630Δerm pMC211 (MC282; vector control), rstA::erm pMC211 (MC505; vector control), rstA::erm pPcprA-rstA (MC480), rstA::erm pPcprA-rstA-3XFLAG (MC1004), and rstA::erm pPcprA-rstAΔHTH-3XFLAG (MC1028) grown on 70:30 sporulation agar supplemented with 2 µg/ml thiamphenicol in the presence or absence of 1 µg/ml nisin. Sporulation frequency is calculated as the number of ethanol-resistant spores divided by the total number of cells enumerated at H24. The limit of detection for ethanol-resistant spores is 20 CFU/ml. Western blot analysis of TcdA (C) and the corresponding stain-free image of total protein (D) in 630Δerm pMC211 (MC282; vector control), rstA::erm pMC211 (MC505; vector control), rstA::erm pPcprA-rstA (MC480), rstA::erm pPcprA-rstA-3XFLAG (MC1004), and rstA::erm pPcprA-rstAΔHTH-3XFLAG (MC1028) grown in TY medium, pH 7.4, at 24 h. The means and standard errors of the means for at least three independent biological replicates are shown; asterisks represent P ≤ 0.05 by one-way ANOVA followed by Dunnett’s multiple-comparison test compared to the rstA::erm pMC211 strain (MC505). Download FIG S2, PDF file, 1.3 MB (1.3MB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Overexpression of RstA represses PrstA::phoZ alkaline phosphatase activity in a dose-dependent manner. Alkaline phosphatase (AP) activity of a promoterless::phoZ construct in the 630Δerm background (MC448), the PrstA::phoZ construct expressed in 630Δerm (MC773) and the rstA::erm mutant (MC774), and the PrstA::phoZ construct divergently expressed from the nisin-inducible PcprA-rstA-3XFLAG construct in the rstA::erm mutant (MC1435) grown on 70:30 agar supplemented with the indicated concentration of nisin at H8. The means and standard errors of the means for three biological replicates are shown. *, P < 0.05 by one-way ANOVA followed by Tukey’s multiple-comparison test between the comparisons indicated in the figure. Download FIG S3, PDF file, 0.6 MB (589.9KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Sequence alignment of the flgB promoters from C. difficile 630 and R20291 (229 bp total). The PflgB sequence is identical between strain 630 and its derivative 630Δerm. The asterisks below the sequence indicate identical nucleotides. The −35 and −10 consensus sequence of the σA-dependent promoter are marked, which are identical between both the 630 and R20291 strains (39). Download FIG S4, PDF file, 0.9 MB (880.4KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Densitometry of RstA binding to promoter DNA in biotin pulldown assays. Intensity of RstA-FLAG detected by anti-FLAG Western blotting was measured using the densitometry tools provided in Image Lab (Bio-Rad). The adjusted total band volumes were normalized to RstA-FLAG recovered using either the PrstA promoter DNA (A) or the PtcdR promoter DNA (B) as bait. Representative Western blots of each biotin pulldown are shown in Fig. 4; IR is a 380-bp intergenic region upstream of the mapped rstA promoter that contains no promoter elements or identified RstA binding sequences (Fig. 1). The means and standard errors of the means are shown for at least three individual pulldowns for each condition. *, P < 0.05 by one-way ANOVA followed by Dunnett’s multiple-comparison test compared to either PrstA or PtcdR. Download FIG S5, PDF file, 0.7 MB (751.8KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RstA-mediated repression of two PtcdR σA-dependent promoter fragments of different lengths is similar. Alkaline phosphatase (AP) activity of a promoterless::phoZ construct in the 630Δerm background (MC448) and a longer 92-bp σA-dependent PtcdR::phoZ reporter fusion in 630Δerm (MC1143) and the rstA::erm mutant (MC1144) grown in TY medium, pH 7.4 at H24. The means and standards error of the means for four biological replicates are shown. *, P < 0.05, using Student’s t test compared to the activity observed in the 630Δerm parent strain for each promoter construct. The measured AP activity and calculated fold change between the rstA background versus the parent is the same as the 76-bp promoter region (Fig. 2B; no statistically significant difference between the 92-bp and 76-bp reporters). Download FIG S6, PDF file, 0.5 MB (554.1KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PCR verification of the rstA and sigD mutations in C. difficile 630Δerm. PCR amplification from overnight cultures of 630Δerm, rstA::erm (MC391), sigD::erm (RT1075), rstA (MC1118), and rstA sigD::erm (MC1278) strains using primers sigDqF and oMC1006 to verify the sigD alleles (A) (the expected sizes for the PCR products are 449 bp for the sigD wild-type allele and ∼2,449 bp for the sigD::erm insertion) and primers oMC352 and oMC1204 to verify the rstA alleles (B) (the expected sizes for the PCR products are 2,654 bp for the wild-type allele, ∼4,654 bp for the rstA::erm allele and 1,361 bp for the ΔrstA allele). The NEB 1-kb DNA ladder serves as the molecular marker. (C and D) A schematic representing the chromosomal organization of sigD (C) and rstA (D). The lines above the genes represent the locations of the wild-type product amplified with the indicated primers. The rstA gene is encoded on the complement strand. Download FIG S7, PDF file, 1.0 MB (994.7KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Total protein (8 µg) transferred to nitrocellulose for TcdA Western blotting. The corresponding TGX stain-free gels used for the indicated Western blots shown in Fig. 5A (panel A above) and Fig. 6A (panel B above). For each strain tested, 8 µg of total protein was loaded onto a 4 to 15% TGX stain-free gel and imaged by a ChemiDoc (Bio-Rad) after electrophoresis. The protein was then transferred to nitrocellulose, and the Western blots were performed as described in Materials and Methods. Download FIG S8, PDF file, 1.1 MB (1.1MB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Alignment and stability of RstA orthologs encoded in C. difficile 630, C. sordellii ATCC 9714, C. perfringens S13, C. acetobutylicum ATCC 824. (A) Multiple-sequence alignment performed by EMBL-EMI Clustal Omega tool (45). The regions denoting the DNA-binding domain (from M1 to Y51), which were used in the RstA C. perfringens DNA-binding domain-C. difficile C-terminal domains hybrid construct, and the residues deleted in conserved helix-turn-helix motif for the rstAΔHTH-3XFLAG construct are marked. Identical residues (*), strongly conserved residues (:), and weakly conserved residues (.) are indicated. The colors group the amino acid residues based on their physiochemical properties as follows: red, small, hydrophobic residues; blue, acidic residues; magenta, basic residues; green, hydroxyl-, sulfhydryl-, or amine-containing residue. (B) Western blot analysis using FLAG M2 antibody to detect recombinant RstA-3XFLAG proteins (∼51 kDa) expressed in 630Δerm pMC211 (MC282; vector control), rstA::erm pMC211 (MC505; vector control), rstA::erm pMC828 (MC1323; Clostridium acetobutylicum rstA [Ca_C0957]), rstA::erm pMC675 (MC1004; Clostridium difficile rstA), rstA::erm pMC829 (MC1324; Clostridium perfringens rstA [CPE1448]), rstA::erm pMC830 (MC1325; Clostridium sordelii rstA [ATCC9714_3891]), and rstA::erm pMC798 (MC1257; pPcprA-rstACpHTHCdCterminal-3XFLAG) grown in TY medium supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin at H24. Download FIG S9, PDF file, 2.5 MB (2.5MB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Alignment of inverted repeats within the promoters of direct RstA targets. The inverted repeats identified by EMBOSS Palindrome Finder (57) within each promoter that RstA directly binds are shown. The conserved −10 or −35 elements within each promoter are underlined. The predicted −35 element for PtcdR(σD) begins immediately following the sequence shown here. The two nucleotides that are important for RstA binding to PrstA DNA are marked in red. Download FIG S10, PDF file, 0.5 MB (494.7KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mapping the rstA transcriptional start with 5ʹ rapid amplification of cDNA ends (5ʹ RACE).
DNase I-treated RNA from the rstA::erm mutant (MC391) was obtained as described above. 5ʹ RACE was performed using the 5ʹ/3ʹ RACE kit, Second Generation (Roche), following the manufacturer’s instructions as previously reported (72). Briefly, first strand cDNA synthesis was performed using the rstA-specific primer oMC982, followed by purification with the High Pure PCR Product purification kit (Roche). After subsequent poly(A) tailing of first strand cDNA, PCR amplification was performed using an oligo(T) primer and the rstA-specific primer oMC983 with Phusion DNA Polymerase (NEB). The resulting PCR products were purified from a 0.7% agarose gel (Qiagen) and TA cloned into pCR2.1 (Invitrogen) using the manufacturer’s supplied protocols. Plasmids were isolated and sequenced (Eurofins MWG Operon) to determine the transcriptional start site (−32 bp from translational start site; n = 7).
Sporulation assays.
C. difficile cultures were grown in BHIS medium supplemented with 0.1% taurocholate and 0.2% fructose until mid-exponential phase (i.e., an optical density at 600 nm [OD600] of 0.5), and 0.25-ml portions were spotted onto 70:30 sporulation agar supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin as a lawn (68). After 24 h growth, ethanol resistance assays were performed as previously described (73, 74). Briefly, the cells were scraped from plates after 24 h (H24) and suspended in BHIS medium to an OD600 of 1.0. The total number of vegetative cells per milliliter was determined by immediately serially diluting and applying the resuspended cells to BHIS plates. Simultaneously, a 0.5-ml aliquot was mixed with 0.3 ml of 95% ethanol and 0.2 ml of dH2O to achieve a final concentration of 28.5% ethanol, vortexed, and incubated for 15 min to eliminate all vegetative cells; ethanol-treated cells were subsequently serially diluted in 1× PBS plus 0.1% taurocholate and applied to BHIS plus 0.1% taurocholate plates to determine the total number of spores. After at least 36 h of growth, CFU were enumerated, and the sporulation frequency was calculated as the total number of spores divided by the total number of viable cells (spores plus vegetative cells). A spo0A mutant (MC310) was used as a negative sporulation control. Statistical significance was determined using a one-way ANOVA, followed by Dunnett’s multiple-comparison test (GraphPad Prism v6.0), to compare sporulation efficiency to that of the rstA mutant.
Alkaline phosphatase activity assays.
C. difficile strains containing the reporter fusions listed in Table 1 were grown and harvested on either 70:30 sporulation agar at H8, defined as eight hours after the cultures are applied to the plates (early stationary phase), or from TY liquid medium in stationary phase (T3, defined as three hours after the start of transition phase [approximately equivalent to H8 on plates; early stationary phase], or H24, defined as 24 h after the cultures are inoculated [late stationary phase]). Alkaline phosphatase assays were performed as described previously (75) with the exception that no chloroform was used for cell lysis. Technical duplicates were averaged, and the results are presented as the means and standard errors of the means for three biological replicates. The two-tailed Student’s t test was used to compare the activity in the rstA mutant to the activity in the parent strain.
Biotin pulldown assays.
Biotin pulldown assays were performed as described by Jutras et al. (76). Briefly, a threefold excess of biotin-labeled DNA bait (30 µg) was coupled to streptavidin-coated magnetic beads (Invitrogen; binding capacity of 10 µg) in B/W buffer, and the bead-DNA complexes were washed with TE buffer to remove unbound DNA. In addition, an unbiotinylated PrstA (30 µg) negative control and a beads-only (dH2O) negative control were treated alongside the test DNA fragments to ensure that RstA did not interact nonspecifically with the streptavidin-coated magnetic beads. To determine the total amount of biotinylated-DNA bound to each bead preparation, each incubation and subsequent washes were quantitated via a Nanodrop 1000 and subtracted from the initial amount of DNA. To prepare cell lysates, C. difficile expressing either RstA-FLAG (MC1004) or RstAΔHTH-FLAG (MC1028) in the rstA background were grown to mid-log phase (OD600 of 0.5 to 0.7) in 500 ml TY medium (pH 7.4) supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin, pelleted, rinsed with sterile water, and stored at −80°C overnight. The pellets were suspended in 4.5 ml BS/THES buffer and lysed by cycling between a dry ice/ethanol bath and a 37°C water bath. The cell lysates were vortexed for 1 min to shear genomic DNA, and cell debris was pelleted at 15K rpm for 15 min at 4°C. The supernatant, along with 10 µg salmon sperm DNA as a nonspecific competitor, was then applied to the bead-DNA complexes and rotated for 30 min at room temperature. This incubation was repeated once with additional supernatant and 10 µg salmon sperm DNA for two total incubations. The bead-DNA-protein complexes were washed seven times with BS/THES buffer supplemented with 10 µg/ml salmon sperm DNA and then without salmon sperm DNA to remove nonspecific proteins. The beads were transferred to clean microcentrifuge tubes twice during the washes to eliminate carry-over contamination. The remaining bound protein was eluted with 250 mM NaCl in Tris-HCl, pH 7.4, and the eluates were immediately analyzed by SDS-PAGE and Western blotting using FLAG M2 antibody (Sigma; see below). Each DNA bait fragment was tested in at least three independent experiments. As a control following each experiment, bait DNA was recovered by incubating the labeled beads in dH2O at 70°C for 10 min and analyzed on a 1.5% agarose gel to ensure that no cross-contamination occurred (data not shown). Densitometry was performed using Image Lab Software (Bio-Rad), and subsequent statistical analyses included a one-way ANOVA, followed by Dunnett’s multiple-comparison test (GraphPad Prism v6.0).
Western blot analysis.
The indicated C. difficile strains were grown in TY medium (pH 7.4) supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin at 37°C and harvested at H24 (24 h) (74). Total protein was quantitated using the Pierce Micro BCA protein assay kit (Thermo Scientific), and 8 µg of total protein was separated by electrophoresis on a precast 4 to 15% TGX stain-free gradient gel (Bio-Rad), and total protein was imaged using a ChemiDoc (Bio-Rad). Corresponding gel images for each Western blot are included in the supplemental material as indicated in the text. Protein was then transferred to a 0.45-µm nitrocellulose membrane, and Western blot analysis was conducted with either mouse anti-TcdA (Novus Biologicals) or mouse anti-FLAG (Sigma) primary antibody, followed by goat anti-mouse Alexa Fluor 488 (Life Technologies) secondary antibody. Imaging and densitometry were performed with a ChemiDoc and Image Lab software (Bio-Rad), and a one-way ANOVA, followed by Dunnett’s multiple-comparison test, was performed to assess statistical differences in TcdA protein levels between the rstA mutant and each rstA overexpression strain (GraphPad Prism v6.0). At least three biological replicates were analyzed for each strain, and a representative Western blot image is shown.
Quantitative reverse transcription-PCR analysis.
C. difficile was cultivated in TY medium (pH 7.4) supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin and harvested at T3 (defined as three hours after the start of transition phase; OD600 of 1.0 [approximately equivalent to H8 on plates]). Aliquots (3 ml) of culture were immediately mixed with 3 ml of ice-cold ethanol-acetone (1:1) and stored at −80°C. RNA was purified and DNase I treated (Ambion) as previously described (29, 35, 77), and cDNA was synthesized using random hexamers (77). Quantitative reverse transcription-PCR (qRT-PCR) analysis, using 50 ng cDNA per reaction and the SensiFAST SYBR & Fluorescein kit (Bioline), was performed in technical triplicates on a Roche Lightcycler 96. cDNA synthesis reaction mixtures containing no reverse transcriptase were included as a negative control to ensure that no genomic DNA contamination was present. Results are presented as the means and standard errors of the means for three biological replicates. Statistical significance was determined using a one-way ANOVA, followed by Dunnett’s multiple-comparison test (GraphPad Prism v6.0), to compare transcript levels between the rstA mutant and each rstA overexpression strain.
ACKNOWLEDGMENTS
We are grateful for the gift of C. perfringens S13 genomic DNA from Jorge Vidal. We give special thanks to Charles Moran and the members of McBride lab for helpful suggestions and discussions during the course of this work.
This research was supported by the U.S. National Institutes of Health through research grants AI116933 and AI121684 to S.M.M., AI107029 to R.T., and AI120613 to B.R.A.-F.
The contents of this article are solely the responsibility of the authors and do not necessarily reflect the official views of the National Institutes of Health.
Footnotes
Citation Edwards AN, Anjuwon-Foster BR, McBride SM. 2019. RstA is a major regulator of Clostridioides difficile toxin production and motility. mBio 10:e01991-18. https://doi.org/10.1128/mBio.01991-18.
REFERENCES
- 1.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 2.Martin JS, Monaghan TM, Wilcox MH. 2016. Clostridium difficile infection: epidemiology, diagnosis and understanding transmission. Nat Rev Gastroenterol Hepatol 13:206–216. doi: 10.1038/nrgastro.2016.25. [DOI] [PubMed] [Google Scholar]
- 3.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 4.Onderdonk AB, Lowe BR, Bartlett JG. 1979. Effect of environmental stress on Clostridium difficile toxin levels during continuous cultivation. Appl Environ Microbiol 38:637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karlsson S, Burman LG, Akerlund T. 1999. Suppression of toxin production in Clostridium difficile VPI 10463 by amino acids. Microbiology 145:1683–1693. doi: 10.1099/13500872-145-7-1683. [DOI] [PubMed] [Google Scholar]
- 6.Karlsson S, Lindberg A, Norin E, Burman LG, Akerlund T. 2000. Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect Immun 68:5881–5888. doi: 10.1128/IAI.68.10.5881-5888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karlsson S, Burman LG, Akerlund T. 2008. Induction of toxins in Clostridium difficile is associated with dramatic changes of its metabolism. Microbiology 154:3430–3436. doi: 10.1099/mic.0.2008/019778-0. [DOI] [PubMed] [Google Scholar]
- 8.Dineen SS, Villapakkam AC, Nordman JT, Sonenshein AL. 2007. Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol 66:206–219. doi: 10.1111/j.1365-2958.2007.05906.x. [DOI] [PubMed] [Google Scholar]
- 9.Antunes A, Martin-Verstraete I, Dupuy B. 2011. CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol 79:882–899. doi: 10.1111/j.1365-2958.2010.07495.x. [DOI] [PubMed] [Google Scholar]
- 10.Dubois T, Dancer-Thibonnier M, Monot M, Hamiot A, Bouillaut L, Soutourina O, Martin-Verstraete I, Dupuy B. 2016. Control of Clostridium difficile physiopathology in response to cysteine availability. Infect Immun 84:2389–2405. doi: 10.1128/IAI.00121-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKee RW, Mangalea MR, Purcell EB, Borchardt EK, Tamayo R. 2013. The second messenger cyclic di-GMP regulates Clostridium difficile toxin production by controlling expression of sigD. J Bacteriol 195:5174–5185. doi: 10.1128/JB.00501-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Meouche I, Peltier J, Monot M, Soutourina O, Pestel-Caron M, Dupuy B, Pons JL. 2013. Characterization of the SigD regulon of C. difficile and its positive control of toxin production through the regulation of tcdR. PLoS One 8:e83748. doi: 10.1371/journal.pone.0083748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin-Verstraete I, Peltier J, Dupuy B. 2016. The regulatory networks that control Clostridium difficile toxin synthesis. Toxins (Basel) 8:153. doi: 10.3390/toxins8050153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards AN, Tamayo R, McBride SM. 2016. A novel regulator controls Clostridium difficile sporulation, motility and toxin production. Mol Microbiol 100:954–971. doi: 10.1111/mmi.13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mani N, Dupuy B. 2001. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc Natl Acad Sci U S A 98:5844–5849. doi: 10.1073/pnas.101126598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aubry A, Hussack G, Chen W, KuoLee R, Twine SM, Fulton KM, Foote S, Carrillo CD, Tanha J, Logan SM. 2012. Modulation of toxin production by the flagellar regulon in Clostridium difficile. Infect Immun 80:3521–3532. doi: 10.1128/IAI.00224-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rocha-Estrada J, Aceves-Diez AE, Guarneros G, de la Torre M. 2010. The RNPP family of quorum-sensing proteins in Gram-positive bacteria. Appl Microbiol Biotechnol 87:913–923. doi: 10.1007/s00253-010-2651-y. [DOI] [PubMed] [Google Scholar]
- 18.Do H, Kumaraswami M. 2016. Structural mechanisms of peptide recognition and allosteric modulation of gene regulation by the RRNPP family of quorum-sensing regulators. J Mol Biol 428:2793–2804. doi: 10.1016/j.jmb.2016.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neiditch MB, Capodagli GC, Prehna G, Federle MJ. 2017. Genetic and structural analyses of RRNPP intercellular peptide signaling of Gram-positive bacteria. Annu Rev Genet 51:311–333. doi: 10.1146/annurev-genet-120116-023507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perego M, Hoch JA. 1996. Cell-cell communication regulates the effects of protein aspartate phosphatases on the phosphorelay controlling development in Bacillus subtilis. Proc Natl Acad Sci U S A 93:1549–1553. doi: 10.1073/pnas.93.4.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perego M. 1997. A peptide export-import control circuit modulating bacterial development regulates protein phosphatases of the phosphorelay. Proc Natl Acad Sci U S A 94:8612–8617. doi: 10.1073/pnas.94.16.8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang M, Grau R, Perego M. 2000. Differential processing of propeptide inhibitors of Rap phosphatases in Bacillus subtilis. J Bacteriol 182:303–310. doi: 10.1128/JB.182.2.303-310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slamti L, Lereclus D. 2002. A cell-cell signaling peptide activates the PlcR virulence regulon in bacteria of the Bacillus cereus group. EMBO J 21:4550–4559. doi: 10.1093/emboj/cdf450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perchat S, Dubois T, Zouhir S, Gominet M, Poncet S, Lemy C, Aumont-Nicaise M, Deutscher J, Gohar M, Nessler S, Lereclus D. 2011. A cell-cell communication system regulates protease production during sporulation in bacteria of the Bacillus cereus group. Mol Microbiol 82:619–633. doi: 10.1111/j.1365-2958.2011.07839.x. [DOI] [PubMed] [Google Scholar]
- 25.Chang JC, LaSarre B, Jimenez JC, Aggarwal C, Federle MJ. 2011. Two group A streptococcal peptide pheromones act through opposing Rgg regulators to control biofilm development. PLoS Pathog 7:e1002190. doi: 10.1371/journal.ppat.1002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lereclus D, Agaisse H, Gominet M, Salamitou S, Sanchis V. 1996. Identification of a Bacillus thuringiensis gene that positively regulates transcription of the phosphatidylinositol-specific phospholipase C gene at the onset of the stationary phase. J Bacteriol 178:2749–2756. doi: 10.1128/jb.178.10.2749-2756.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lazazzera BA, Kurtser IG, McQuade RS, Grossman AD. 1999. An autoregulatory circuit affecting peptide signaling in Bacillus subtilis. J Bacteriol 181:5193–5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mashburn-Warren L, Morrison DA, Federle MJ. 2010. A novel double-tryptophan peptide pheromone controls competence in Streptococcus spp. via an Rgg regulator. Mol Microbiol 78:589–606. doi: 10.1111/j.1365-2958.2010.07361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McBride SM, Sonenshein AL. 2011. Identification of a genetic locus responsible for antimicrobial peptide resistance in Clostridium difficile. Infect Immun 79:167–176. doi: 10.1128/IAI.00731-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helmann JD. 1995. Compilation and analysis of Bacillus subtilis sigma A-dependent promoter sequences: evidence for extended contact between RNA polymerase and upstream promoter DNA. Nucleic Acids Res 23:2351–2360. doi: 10.1093/nar/23.13.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moncrief JS, Barroso LA, Wilkins TD. 1997. Positive regulation of Clostridium difficile toxins. Infect Immun 65:1105–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mani N, Lyras D, Barroso L, Howarth P, Wilkins T, Rood JI, Sonenshein AL, Dupuy B. 2002. Environmental response and autoregulation of Clostridium difficile TxeR, a sigma factor for toxin gene expression. J Bacteriol 184:5971–5978. doi: 10.1128/JB.184.21.5971-5978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouillaut L, Dubois T, Sonenshein AL, Dupuy B. 2015. Integration of metabolism and virulence in Clostridium difficile. Res Microbiol 166:375–383. doi: 10.1016/j.resmic.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dupuy B, Sonenshein AL. 1998. Regulated transcription of Clostridium difficile toxin genes. Mol Microbiol 27:107–120. doi: 10.1046/j.1365-2958.1998.00663.x. [DOI] [PubMed] [Google Scholar]
- 35.Dineen SS, McBride SM, Sonenshein AL. 2010. Integration of metabolism and virulence by Clostridium difficile CodY. J Bacteriol 192:5350–5362. doi: 10.1128/JB.00341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antunes A, Camiade E, Monot M, Courtois E, Barbut F, Sernova NV, Rodionov DA, Martin-Verstraete I, Dupuy B. 2012. Global transcriptional control by glucose and carbon regulator CcpA in Clostridium difficile. Nucleic Acids Res 40:10701–10718. doi: 10.1093/nar/gks864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammond GA, Lyerly DM, Johnson JL. 1997. Transcriptional analysis of the toxigenic element of Clostridium difficile. Microb Pathog 22:143–154. doi: 10.1006/mpat.1996.0100. [DOI] [PubMed] [Google Scholar]
- 38.Hundsberger T, Braun V, Weidmann M, Leukel P, Sauerborn M, von Eichel-Streiber C. 1997. Transcription analysis of the genes tcdA-E of the pathogenicity locus of Clostridium difficile. Eur J Biochem 244:735–742. doi: 10.1111/j.1432-1033.1997.t01-1-00735.x. [DOI] [PubMed] [Google Scholar]
- 39.Soutourina OA, Monot M, Boudry P, Saujet L, Pichon C, Sismeiro O, Semenova E, Severinov K, Le Bouguenec C, Coppee JY, Dupuy B, Martin-Verstraete I. 2013. Genome-wide identification of regulatory RNAs in the human pathogen Clostridium difficile. PLoS Genet 9:e1003493. doi: 10.1371/journal.pgen.1003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McAllister KN, Bouillaut L, Kahn JN, Self WT, Sorg JA. 2017. Using CRISPR-Cas9-mediated genome editing to generate C. difficile mutants defective in selenoproteins synthesis. Sci Rep 7:14672. doi: 10.1038/s41598-017-15236-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKee RW, Aleksanyan N, Garrett EM, Tamayo R. 2018. Type IV pili promote Clostridium difficile adherence and persistence in a mouse model of infection. Infect Immun 86:e00943-17. doi: 10.1128/IAI.00943-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozlowicz BK, Shi K, Gu ZY, Ohlendorf DH, Earhart CA, Dunny GM. 2006. Molecular basis for control of conjugation by bacterial pheromone and inhibitor peptides. Mol Microbiol 62:958–969. doi: 10.1111/j.1365-2958.2006.05434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zouhir S, Perchat S, Nicaise M, Perez J, Guimaraes B, Lereclus D, Nessler S. 2013. Peptide-binding dependent conformational changes regulate the transcriptional activity of the quorum-sensor NprR. Nucleic Acids Res 41:7920–7933. doi: 10.1093/nar/gkt546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cabrera R, Rodriguez-Romero A, Guarneros G, de la Torre M. 2016. New insights into the interaction between the quorum-sensing receptor NprR and its DNA target, or the response regulator Spo0F. FEBS Lett 590:3243–3253. doi: 10.1002/1873-3468.12371. [DOI] [PubMed] [Google Scholar]
- 45.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deakin LJ, Clare S, Fagan RP, Dawson LF, Pickard DJ, West MR, Wren BW, Fairweather NF, Dougan G, Lawley TD. 2012. The Clostridium difficile spo0A gene is a persistence and transmission factor. Infect Immun 80:2704–2711. doi: 10.1128/IAI.00147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nawrocki KL, Edwards AN, Daou N, Bouillaut L, McBride SM. 2016. CodY-dependent regulation of sporulation in Clostridium difficile. J Bacteriol 198:2113–2130. doi: 10.1128/JB.00220-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mackin KE, Carter GP, Howarth P, Rood JI, Lyras D. 2013. Spo0A differentially regulates toxin production in evolutionarily diverse strains of Clostridium difficile. PLoS One 8:e79666. doi: 10.1371/journal.pone.0079666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karlsson S, Dupuy B, Mukherjee K, Norin E, Burman LG, Akerlund T. 2003. Expression of Clostridium difficile toxins A and B and their sigma factor TcdD is controlled by temperature. Infect Immun 71:1784–1793. doi: 10.1128/IAI.71.4.1784-1793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ransom EM, Kaus GM, Tran PM, Ellermeier CD, Weiss DS. 2018. Multiple factors contribute to bimodal toxin gene expression in Clostridioides (Clostridium) difficile. Mol Microbiol 110:533–549. doi: 10.1111/mmi.14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saujet L, Monot M, Dupuy B, Soutourina O, Martin-Verstraete I. 2011. The key sigma factor of transition phase, SigH, controls sporulation, metabolism, and virulence factor expression in Clostridium difficile. J Bacteriol 193:3186–3196. doi: 10.1128/JB.00272-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin MJ, Clare S, Goulding D, Faulds-Pain A, Barquist L, Browne HP, Pettit L, Dougan G, Lawley TD, Wren BW. 2013. The agr locus regulates virulence and colonization genes in Clostridium difficile 027. J Bacteriol 195:3672–3681. doi: 10.1128/JB.00473-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boudry P, Gracia C, Monot M, Caillet J, Saujet L, Hajnsdorf E, Dupuy B, Martin-Verstraete I, Soutourina O. 2014. Pleiotropic role of the RNA chaperone protein Hfq in the human pathogen Clostridium difficile. J Bacteriol 196:3234–3248. doi: 10.1128/JB.01923-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Girinathan BP, Ou J, Dupuy B, Govind R. 2018. Pleiotropic roles of Clostridium difficile sin locus. PLoS Pathog 14:e1006940. doi: 10.1371/journal.ppat.1006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. 2008. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anjuwon-Foster BR, Tamayo R. 2017. A genetic switch controls the production of flagella and toxins in Clostridium difficile. PLoS Genet 13:e1006701. doi: 10.1371/journal.pgen.1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 58.Gohar M, Faegri K, Perchat S, Ravnum S, Okstad OA, Gominet M, Kolsto AB, Lereclus D. 2008. The PlcR virulence regulon of Bacillus cereus. PLoS One 3:e2793. doi: 10.1371/journal.pone.0002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lasarre B, Aggarwal C, Federle MJ. 2013. Antagonistic Rgg regulators mediate quorum sensing via competitive DNA binding in Streptococcus pyogenes. mBio 3:e00333-12. doi: 10.1128/mBio.00333-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Folli C, Mangiarotti L, Folloni S, Alfieri B, Gobbo M, Berni R, Rivetti C. 2008. Specificity of the TraA-DNA interaction in the regulation of the pPD1-encoded sex pheromone response in Enterococcus faecalis. J Mol Biol 380:932–945. doi: 10.1016/j.jmb.2008.05.058. [DOI] [PubMed] [Google Scholar]
- 61.Even-Tov E, Omer Bendori S, Pollak S, Eldar A. 2016. Transient duplication-dependent divergence and horizontal transfer underlie the evolutionary dynamics of bacterial cell-cell signaling. PLoS Biol 14:e2000330. doi: 10.1371/journal.pbio.2000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Makthal N, Gavagan M, Do H, Olsen RJ, Musser JM, Kumaraswami M. 2016. Structural and functional analysis of RopB: a major virulence regulator in Streptococcus pyogenes. Mol Microbiol 99:1119–1133. doi: 10.1111/mmi.13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parashar V, Konkol MA, Kearns DB, Neiditch MB. 2013. A plasmid-encoded phosphatase regulates Bacillus subtilis biofilm architecture, sporulation, and genetic competence. J Bacteriol 195:2437–2448. doi: 10.1128/JB.02030-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Darkoh C, DuPont HL, Norris SJ, Kaplan HB. 2015. Toxin synthesis by Clostridium difficile is regulated through quorum signaling. mBio 6:e02569-14. doi: 10.1128/mBio.02569-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Darkoh C, Odo C, DuPont HL. 2016. Accessory gene regulator-1 locus is essential for virulence and pathogenesis of Clostridium difficile. mBio 7:e01237-16. doi: 10.1128/mBio.01237-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee AS, Song KP. 2005. LuxS/autoinducer-2 quorum sensing molecule regulates transcriptional virulence gene expression in Clostridium difficile. Biochem Biophys Res Commun 335:659–666. doi: 10.1016/j.bbrc.2005.07.131. [DOI] [PubMed] [Google Scholar]
- 67.Sorg JA, Dineen SS. 2009. Laboratory maintenance of Clostridium difficile. Curr Protoc Microbiol Chapter9:Unit9A.1. doi: 10.1002/9780471729259.mc09a01s12. [DOI] [PubMed] [Google Scholar]
- 68.Putnam EE, Nock AM, Lawley TD, Shen A. 2013. SpoIVA and SipL are Clostridium difficile spore morphogenetic proteins. J Bacteriol 195:1214–1225. doi: 10.1128/JB.02181-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edwards AN, Suarez JM, McBride SM. 2013. Culturing and maintaining Clostridium difficile in an anaerobic environment. J Vis Exp 2013(79):50787. doi: 10.3791/50787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luria SE, Burrous JW. 1957. Hybridization between Escherichia coli and Shigella. J Bacteriol 74:461–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Purcell EB, McKee RW, McBride SM, Waters CM, Tamayo R. 2012. Cyclic diguanylate inversely regulates motility and aggregation in Clostridium difficile. J Bacteriol 194:3307–3316. doi: 10.1128/JB.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Woods EC, Nawrocki KL, Suarez JM, McBride SM. 2016. The Clostridium difficile Dlt pathway is controlled by the ECF sigma factor, sigmaV, in response to lysozyme. Infect Immun 84:1902–1916. doi: 10.1128/IAI.00207-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edwards AN, McBride SM. 2017. Determination of the in vitro sporulation frequency of Clostridium difficile. Bio Protocol 7:e2125. doi: 10.21769/BioProtoc.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Childress KO, Edwards AN, Nawrocki KL, Woods EC, Anderson SE, McBride SM. 2016. The phosphotransfer protein CD1492 represses sporulation initiation in Clostridium difficile. Infect Immun 84:3434–3444. doi: 10.1128/IAI.00735-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Edwards AN, Pascual RA, Childress KO, Nawrocki KL, Woods EC, McBride SM. 2015. An alkaline phosphatase reporter for use in Clostridium difficile. Anaerobe 32:98–104. doi: 10.1016/j.anaerobe.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jutras BL, Verma A, Stevenson B. 2012. Identification of novel DNA-binding proteins using DNA-affinity chromatography/pull down. Curr Protoc Microbiol Chapter1:Unit1F.1. doi: 10.1002/9780471729259.mc01f01s24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Edwards AN, Nawrocki KL, McBride SM. 2014. Conserved oligopeptide permeases modulate sporulation initiation in Clostridium difficile. Infect Immun 82:4276–4291. doi: 10.1128/IAI.02323-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thomas CM, Smith CA. 1987. Incompatibility group P plasmids: genetics, evolution, and use in genetic manipulation. Annu Rev Microbiol 41:77–101. doi: 10.1146/annurev.mi.41.100187.000453. [DOI] [PubMed] [Google Scholar]
- 79.Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J Biol Chem 286:27483–27493. doi: 10.1074/jbc.M111.263889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hussain HA, Roberts AP, Mullany P. 2005. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Δerm) and demonstration that the conjugative transposon Tn916ΔE enters the genome of this strain at multiple sites. J Med Microbiol 54:137–141. doi: 10.1099/jmm.0.45790-0. [DOI] [PubMed] [Google Scholar]
- 81.Bordeleau E, Purcell EB, Lafontaine DA, Fortier LC, Tamayo R, Burrus V. 2015. Cyclic di-GMP riboswitch-regulated type IV pili contribute to aggregation of Clostridium difficile. J Bacteriol 197:819–832. doi: 10.1128/JB.02340-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DNA cloning and vector details. Download FIG S1, PDF file, 0.06 MB (60.4KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The conserved helix-turn-helix (HTH) DNA-binding domain of RstA is not necessary for positively controlling sporulation but is required for the regulation of toxin production and PrstA expression. (A) Western blot analysis using FLAG M2 antibody in rstA pPcprA-rstA-3XFLAG (MC1004) and rstA pPcprA-rstAΔHTH-3XFLAG (MC1028) grown in TY medium, pH 7.4, at log phase (OD600 of 0.5). (B) Ethanol-resistant spore formation of 630Δerm pMC211 (MC282; vector control), rstA::erm pMC211 (MC505; vector control), rstA::erm pPcprA-rstA (MC480), rstA::erm pPcprA-rstA-3XFLAG (MC1004), and rstA::erm pPcprA-rstAΔHTH-3XFLAG (MC1028) grown on 70:30 sporulation agar supplemented with 2 µg/ml thiamphenicol in the presence or absence of 1 µg/ml nisin. Sporulation frequency is calculated as the number of ethanol-resistant spores divided by the total number of cells enumerated at H24. The limit of detection for ethanol-resistant spores is 20 CFU/ml. Western blot analysis of TcdA (C) and the corresponding stain-free image of total protein (D) in 630Δerm pMC211 (MC282; vector control), rstA::erm pMC211 (MC505; vector control), rstA::erm pPcprA-rstA (MC480), rstA::erm pPcprA-rstA-3XFLAG (MC1004), and rstA::erm pPcprA-rstAΔHTH-3XFLAG (MC1028) grown in TY medium, pH 7.4, at 24 h. The means and standard errors of the means for at least three independent biological replicates are shown; asterisks represent P ≤ 0.05 by one-way ANOVA followed by Dunnett’s multiple-comparison test compared to the rstA::erm pMC211 strain (MC505). Download FIG S2, PDF file, 1.3 MB (1.3MB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Overexpression of RstA represses PrstA::phoZ alkaline phosphatase activity in a dose-dependent manner. Alkaline phosphatase (AP) activity of a promoterless::phoZ construct in the 630Δerm background (MC448), the PrstA::phoZ construct expressed in 630Δerm (MC773) and the rstA::erm mutant (MC774), and the PrstA::phoZ construct divergently expressed from the nisin-inducible PcprA-rstA-3XFLAG construct in the rstA::erm mutant (MC1435) grown on 70:30 agar supplemented with the indicated concentration of nisin at H8. The means and standard errors of the means for three biological replicates are shown. *, P < 0.05 by one-way ANOVA followed by Tukey’s multiple-comparison test between the comparisons indicated in the figure. Download FIG S3, PDF file, 0.6 MB (589.9KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Sequence alignment of the flgB promoters from C. difficile 630 and R20291 (229 bp total). The PflgB sequence is identical between strain 630 and its derivative 630Δerm. The asterisks below the sequence indicate identical nucleotides. The −35 and −10 consensus sequence of the σA-dependent promoter are marked, which are identical between both the 630 and R20291 strains (39). Download FIG S4, PDF file, 0.9 MB (880.4KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Densitometry of RstA binding to promoter DNA in biotin pulldown assays. Intensity of RstA-FLAG detected by anti-FLAG Western blotting was measured using the densitometry tools provided in Image Lab (Bio-Rad). The adjusted total band volumes were normalized to RstA-FLAG recovered using either the PrstA promoter DNA (A) or the PtcdR promoter DNA (B) as bait. Representative Western blots of each biotin pulldown are shown in Fig. 4; IR is a 380-bp intergenic region upstream of the mapped rstA promoter that contains no promoter elements or identified RstA binding sequences (Fig. 1). The means and standard errors of the means are shown for at least three individual pulldowns for each condition. *, P < 0.05 by one-way ANOVA followed by Dunnett’s multiple-comparison test compared to either PrstA or PtcdR. Download FIG S5, PDF file, 0.7 MB (751.8KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RstA-mediated repression of two PtcdR σA-dependent promoter fragments of different lengths is similar. Alkaline phosphatase (AP) activity of a promoterless::phoZ construct in the 630Δerm background (MC448) and a longer 92-bp σA-dependent PtcdR::phoZ reporter fusion in 630Δerm (MC1143) and the rstA::erm mutant (MC1144) grown in TY medium, pH 7.4 at H24. The means and standards error of the means for four biological replicates are shown. *, P < 0.05, using Student’s t test compared to the activity observed in the 630Δerm parent strain for each promoter construct. The measured AP activity and calculated fold change between the rstA background versus the parent is the same as the 76-bp promoter region (Fig. 2B; no statistically significant difference between the 92-bp and 76-bp reporters). Download FIG S6, PDF file, 0.5 MB (554.1KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PCR verification of the rstA and sigD mutations in C. difficile 630Δerm. PCR amplification from overnight cultures of 630Δerm, rstA::erm (MC391), sigD::erm (RT1075), rstA (MC1118), and rstA sigD::erm (MC1278) strains using primers sigDqF and oMC1006 to verify the sigD alleles (A) (the expected sizes for the PCR products are 449 bp for the sigD wild-type allele and ∼2,449 bp for the sigD::erm insertion) and primers oMC352 and oMC1204 to verify the rstA alleles (B) (the expected sizes for the PCR products are 2,654 bp for the wild-type allele, ∼4,654 bp for the rstA::erm allele and 1,361 bp for the ΔrstA allele). The NEB 1-kb DNA ladder serves as the molecular marker. (C and D) A schematic representing the chromosomal organization of sigD (C) and rstA (D). The lines above the genes represent the locations of the wild-type product amplified with the indicated primers. The rstA gene is encoded on the complement strand. Download FIG S7, PDF file, 1.0 MB (994.7KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Total protein (8 µg) transferred to nitrocellulose for TcdA Western blotting. The corresponding TGX stain-free gels used for the indicated Western blots shown in Fig. 5A (panel A above) and Fig. 6A (panel B above). For each strain tested, 8 µg of total protein was loaded onto a 4 to 15% TGX stain-free gel and imaged by a ChemiDoc (Bio-Rad) after electrophoresis. The protein was then transferred to nitrocellulose, and the Western blots were performed as described in Materials and Methods. Download FIG S8, PDF file, 1.1 MB (1.1MB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Alignment and stability of RstA orthologs encoded in C. difficile 630, C. sordellii ATCC 9714, C. perfringens S13, C. acetobutylicum ATCC 824. (A) Multiple-sequence alignment performed by EMBL-EMI Clustal Omega tool (45). The regions denoting the DNA-binding domain (from M1 to Y51), which were used in the RstA C. perfringens DNA-binding domain-C. difficile C-terminal domains hybrid construct, and the residues deleted in conserved helix-turn-helix motif for the rstAΔHTH-3XFLAG construct are marked. Identical residues (*), strongly conserved residues (:), and weakly conserved residues (.) are indicated. The colors group the amino acid residues based on their physiochemical properties as follows: red, small, hydrophobic residues; blue, acidic residues; magenta, basic residues; green, hydroxyl-, sulfhydryl-, or amine-containing residue. (B) Western blot analysis using FLAG M2 antibody to detect recombinant RstA-3XFLAG proteins (∼51 kDa) expressed in 630Δerm pMC211 (MC282; vector control), rstA::erm pMC211 (MC505; vector control), rstA::erm pMC828 (MC1323; Clostridium acetobutylicum rstA [Ca_C0957]), rstA::erm pMC675 (MC1004; Clostridium difficile rstA), rstA::erm pMC829 (MC1324; Clostridium perfringens rstA [CPE1448]), rstA::erm pMC830 (MC1325; Clostridium sordelii rstA [ATCC9714_3891]), and rstA::erm pMC798 (MC1257; pPcprA-rstACpHTHCdCterminal-3XFLAG) grown in TY medium supplemented with 2 µg/ml thiamphenicol and 1 µg/ml nisin at H24. Download FIG S9, PDF file, 2.5 MB (2.5MB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Alignment of inverted repeats within the promoters of direct RstA targets. The inverted repeats identified by EMBOSS Palindrome Finder (57) within each promoter that RstA directly binds are shown. The conserved −10 or −35 elements within each promoter are underlined. The predicted −35 element for PtcdR(σD) begins immediately following the sequence shown here. The two nucleotides that are important for RstA binding to PrstA DNA are marked in red. Download FIG S10, PDF file, 0.5 MB (494.7KB, pdf) .
Copyright © 2019 Edwards et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.