

Multidrug-resistant (MDR) A. baumannii is a difficult-to-treat health care-associated pathogen. Knowing the resistance genes present in isolates causing infection aids in empirical treatment selection. Furthermore, knowledge of the genetic background can assist in tracking patterns of transmission to limit the spread of infections in hospitals. The appearance of a new genetic background in A. baumannii strains with a different set of resistance genes and cell surface structures suggests that strong selective pressures exist, even in highly MDR pathogens. Because the new strains have levels of antimicrobial resistance similar to those of the strains that were displaced, we hypothesize that other features, including host colonization and infection, may confer additional selective advantages and contribute to their increased prevalence.
KEYWORDS: Acinetobacter, antibiotic resistance, genome analysis
ABSTRACT
The population structure of health care-associated pathogens reflects patterns of diversification, selection, and dispersal over time. Empirical data detailing the long-term population dynamics of nosocomial pathogens provide information about how pathogens adapt in the face of exposure to diverse antimicrobial agents and other host and environmental pressures and can inform infection control priorities. Extensive sequencing of clinical isolates from one hospital spanning a decade and a second hospital in the Cleveland, OH, metropolitan area over a 3-year time period provided high-resolution genomic analysis of the Acinetobacter baumannii metapopulation. Genomic analysis demonstrated an almost complete replacement of the predominant strain groups with a new, genetically distinct strain group during the study period. The new group, termed clade F, differs from other global clone 2 (GC2) strains of A. baumannii in several ways, including its antibiotic resistance and lipooligosaccharide biosynthesis genes. Clade F strains are part of a large phylogenetic group with broad geographic representation. Phylogenetic analysis of single-nucleotide variants in core genome regions showed that although the Cleveland strains are phylogenetically distinct from those isolated from other locations, extensive intermixing of strains from the two hospital systems was apparent, suggesting either substantial exchange of strains or a shared, but geographically restricted, external pool from which infectious isolates were drawn. These findings document the rapid evolution of A. baumannii strains in two hospitals, with replacement of the predominant clade by a new clade with altered lipooligosaccharide loci and resistance gene repertoires.
INTRODUCTION
Acinetobacter baumannii is generally considered to be a pathogen of the hospital environment, as community-acquired cases are rare in the United States (1–3). Before the early 2000s, A. baumannii infections were uncommon but readily treatable. Antimicrobial resistance has increased dramatically during the last 2 decades, with a majority of clinical isolates now being multidrug resistant (MDR), based on resistance to at least three classes of antibiotics (1), and extremely drug-resistant (XDR) isolates have been reported, which cannot be killed by any FDA-approved antimicrobial agents (4). As a result, considerable attention is focused upon reducing hospital-acquired A. baumannii infections and limiting patient-to-patient transmission (5).
Two large complex hospital systems (denoted hospital system A and hospital system B) serve the same metropolitan area in northeastern Ohio, and their main tertiary care hospitals are located within 1 mile of each other in Cleveland. Their physician networks are largely nonoverlapping, and there is limited patient flow between the two systems due to constraints of provider networks. We previously showed considerable genomic diversity of A. baumannii strains isolated in hospital system A from 2007 to 2008 (6). Most strains belonged to four major clades (clades A to D) that are subsets of the GC2 (global clone 2) lineage (7, 8), and a fifth clade belonged to multilocus sequence type (MLST) group ST79 (clade E). In the context of examining genome changes in sets of multiple isolates from individual patients obtained from 2007 to 2013, we found further support for five major clades, each of which exhibits significant strain-level variation in gene content (9).
In the present study, we expanded the genome survey from hospital system A to cover nearly a decade, from 2007 to 2016. This longitudinal sampling allowed us to examine how A. baumannii populations diverged, revealing a dramatic change with time in the predominant genetic background of strains causing infections. In addition, we sequenced 70 A. baumannii strains isolated at hospital system B between 2012 and 2015. We sought to address whether these two hospital systems had distinct A. baumannii populations and, if not, to what extent the populations in the two health systems overlapped. By comparing strains from the two hospital systems, we tested the hypothesis that the hospital is the primary site of pathogen transmission. If true, we would expect closer relationships among isolates within each hospital system than among those between systems. Remarkably, we found an extensive overlap of strains in the two systems, with limited clustering of very closely related strains, suggesting a reservoir of strains outside these hospital environments.
RESULTS
To place the metropolitan Cleveland isolates into a broader phylogenetic perspective, we combined 371 hospital system A and 70 hospital system B genome sequences with 354 additional genome sequences available in GenBank (see Materials and Methods). Phylogenetic analysis was performed with both the full set of 795 genomes and the 711 genomes that correspond to GC2 isolates, since these were the most common among the Cleveland strains (Fig. 1; see also Fig. S1 and Fig. S2 in the supplemental material). The GC2 isolates clustered into five major clades, four of which corresponded to those described previously (6). More than 90% of the Cleveland isolates belonged to GC2. Non-GC2 genomes were more diverse based on gene content and the extent of homologous recombination and tended to have fewer antibiotic resistance genes (Fig. S1).
FIG 1.

Phylogenetic tree of A. baumannii global clone 2 isolates. A phylogenetic tree was constructed using a core set of SNVs present in GC2 genomes. The rings of annotation extending outward from the inner ring depict clade, Oxford MLST type; year of isolation; K locus; OC locus; the presence of a pABUH1-like plasmid; OXA-23; the ISAba1-blaOXA insertion; Tn2006; AphA6; AphA1; the HUMC1-type integron; AadB; AadA2; the TCDC-AB7015-type integron; AacC1; AadA1; ArmA; two alternative PilA alleles, A-type PilA (GenBank accession number WP_000993717.1) and B-type PilA (GenBank accession number WP_000993718.1); two alternative PglC alleles, A-type PglC (GenBank accession number WP_000826956.1) and B-type PglC (GenBank accession number WP_000977704.1); and isolation location.
Phylogenetic tree of all A. baumannii strains from this study. The rings of annotation extending outward from the inner ring depict clade; OC locus; Pasteur MLST; Oxford MLST; genes encoding StrA, OXA-23, the ISAba1-blaOXA insertion, AphA6, AphA1, AadB, AadA2, AacC1, AadA1, A-type PilA (GenBank accession number WP_000993717.1), B-type PilA (GenBank accession number WP_000993718.1), A-type PglC (GenBank accession number WP_000826956.1), B-type PglC (GenBank accession number WP_000977704.1), and C-type PglC (GenBank accession number WP_000919783.1); and isolation location. Download FIG S1, PDF file, 0.7 MB (736.5KB, pdf) .
Copyright © 2019 Adams et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic tree of all clade F A. baumannii strains from this study. The rings of annotation extending outward from the inner ring depict Oxford MLST; year of isolation; K locus; OC locus; the presence of a pABUH1-like plasmid; genes encoding OXA-23, the ISAba1-blaOXA insertion, AphA6, AphA1, the HUMC1-type integron, AadB, AadA2, the TCDC-AB0715-type integron, AadC1, AadA1, ArmA, B-type PilA (GenBank accession number WP_000993718.1), and B-type PglC (GenBank accession number WP_000977704.1); and isolation location. Download FIG S2, PDF file, 1.4 MB (1.5MB, pdf) .
Copyright © 2019 Adams et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Emergence of a new GC2 group, clade F.
All five major GC2 clades belong to MLST group ST2 (Pasteur scheme [10]). The largest clade in the tree, termed clade F, includes 173 Cleveland strains, representing 32% of the isolates from the two hospital systems, and 250 genomes downloaded from GenBank, including 232 from a comparable health care system in Baltimore, MD (11) (Fig. S2). Clade F corresponds to two strain types in the Oxford (12) MLST scheme: ST281 and a single-locus variant, ST349. The ST349 and ST281 genomes are in separate phylogenetic groups.
The Cleveland and Baltimore clade F isolates are located in distinct branches of the tree, with the exception of UH7007, which was isolated 4 years before clade F strains began to appear more broadly. The few isolates from Pittsburgh (13) and Chicago (14) are located outside the Cleveland group on the tree. Considerable diversity of gene content was observed among clade F isolates, particularly those from Baltimore (Fig. 1). A few strains were isolated from distant geographic locations, including HUMC1, isolated in 2009 in Los Angeles, CA (15); ABIsac_ColiS and ABIsac_ColiR, isolated in France in 2011 (16, 17); and ORAB10, from Oregon in 2013 (18). Most clade F strains were isolated after 2011; however, this clade is likely much older than that based on the phylogenetic distances from other GC2 genomes and based on the variability in genome content, particularly among the Baltimore isolates. An average of 690 ± 110 single-nucleotide variant (SNV) differences separate core genome regions of clade F genomes from those in clades A to D, compared to generally fewer than 100 SNV differences within clades A to D.
Temporal patterns of A. baumannii.
Before 2013, the majority of strains from hospital system A originated from five distinct clades (Fig. S1A to E). The frequency of clade F began to rise in 2014 (Fig. 2). By the end of 2015, most of the strains belonged to clade F. This demonstrates a remarkable decrease in overall genetic diversity and a major restructuring of the population.
FIG 2.
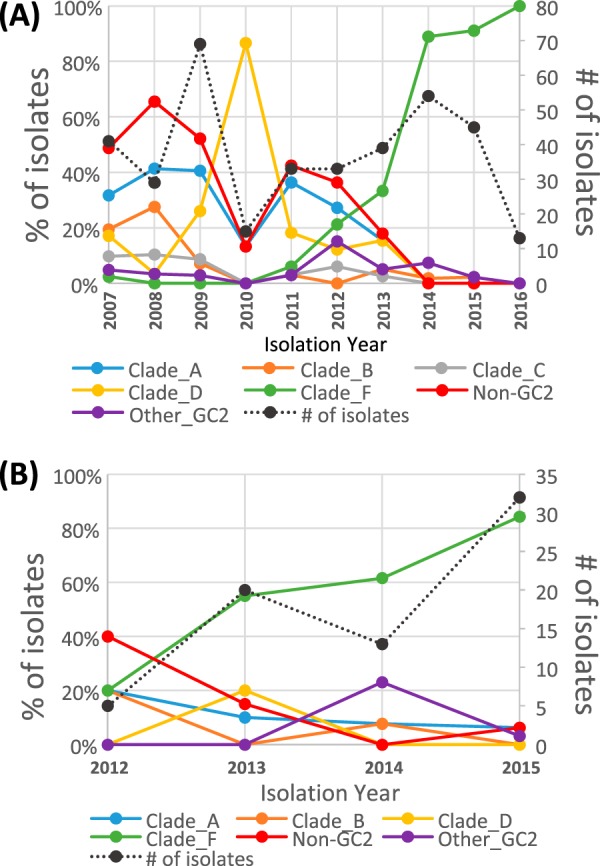
Change in clade abundance over time. The proportions of A. baumannii isolates from hospital system A (A) and hospital system B (B) belonging to each major clade are shown, grouped by the year in which they were isolated. The number of isolates per year is shown as a black dotted line, with units provided on the secondary axis (n = 443 for UH strains and n = 70 for CC strains).
Extensive overlap was evident in the phylogenetic placement for hospital system A and B strains isolated between 2012 and 2016. Each of the major clades contained genomes from isolates from both hospital systems. Approximately 80% of the strains isolated in 2014 to 2016 at both systems belonged to clade F. The average number of SNVs between clade F strains did not differ either within the hospital system genome sets or between the two sets (15 ± 7). The shared locations of insertion sequence (IS) elements were concordant with the SNV tree in supporting intermixing of strains between the two hospital systems (data not shown).
Clade F strains have very limited hospital-specific clustering.
The small number of SNV differences between strains makes precise inference of phylogenetic relationships difficult. However, two broad patterns were apparent in the distribution of Cleveland strains across the phylogenetic tree. First, extensive intermixing of genomes between the two hospital systems was revealed, indicating that strains diverged before entry into the hospital systems. This is most apparent in clade F, which was expected because it had the most isolates collected over similar time periods in both hospital systems. Second, there were small clusters of strains adjacent to one another on the tree that may represent local outbreaks within one hospital system. Sufficient information about patient locations was not available to support a more rigorous analysis of epidemiological patterns.
Antibiotic resistance gene features of clade F strains.
Overall, the antimicrobial resistance phenotypes of Cleveland strains from clade F are similar to those from other GC2 strains (Table 1). The antimicrobial resistance gene content of clade F strains differed in many significant ways from those of other GC2 isolates. The Tn1548 genomic island carries the armA gene, which confers resistance to most aminoglycoside antibiotics and is common in isolates from clades A and B. Tn1548 was absent from clade F strains, and the genome sequence where it is typically inserted adjacent to the homolog of ACICU_02399 was intact in these strains, with no evidence of an IS26 or transposon insertion at that location. Aminoglycoside resistance in clade F strains from Cleveland is likely to be mediated by genes carried in one of two alternative integron structures, as described below, and by a plasmid-borne aphA6 gene.
TABLE 1.
Antibiotic susceptibility phenotypes of A. baumannii isolatesa
| Clade | Amp/ sulbactam |
Cefepime |
Ceftazidime |
Imipenem |
Meropenem |
Amikacin |
Gentamicin |
Tobramycin |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of strains |
% non-S strains |
No. of strains |
% non-S strains |
No. of strains |
% non-S strains |
No. of strains |
% non-S strains |
No. of strains |
% non-S strains |
No. of strains |
% non-S strains |
No. of strains |
% non-S strains |
No. of strains |
% non-S strains |
|
| A | 82 | 96.3 | 82 | 97.6 | 82 | 100.0 | 56 | 100.0 | 82 | 97.6 | 54 | 79.6 | 82 | 96.3 | 82 | 87.8 |
| B | 23 | 73.9 | 25 | 100.0 | 25 | 100.0 | 18 | 83.3 | 25 | 100.0 | 18 | 77.8 | 25 | 96.0 | 25 | 88.0 |
| C | 15 | 93.3 | 16 | 100.0 | 16 | 100.0 | 13 | 53.8 | 16 | 43.8 | 11 | 18.2 | 16 | 93.8 | 16 | 25.0 |
| D | 56 | 75.0 | 56 | 98.2 | 55 | 98.2 | 36 | 94.4 | 55 | 94.5 | 42 | 78.6 | 56 | 96.4 | 55 | 96.4 |
| F | 169 | 81.7 | 155 | 95.5 | 157 | 100.0 | 155 | 99.4 | 169 | 99.4 | 161 | 95.0 | 169 | 93.5 | 167 | 80.8 |
| Other GC2 |
22 | 90.9 | 21 | 100.0 | 21 | 100.0 | 19 | 89.5 | 22 | 90.9 | 20 | 80.0 | 22 | 100.0 | 22 | 86.4 |
| Non- GC2 |
43 | 60.5 | 44 | 81.8 | 44 | 65.9 | 33 | 90.9 | 37 | 89.2 | 33 | 54.5 | 44 | 56.8 | 44 | 54.5 |
Shown are the numbers of strains in each group for which susceptibility data were available for the indicated antimicrobial agent. non-S, nonsusceptible; Amp, ampicillin.
Most Cleveland clade F strains carried the blaOXA-23 gene on a pACICU2-like plasmid (19); however, it appears that the plasmid was lost from several isolates distributed throughout the clade F group. Upregulation of the endogenous OXA-51 family carbapenemase gene blaOXA-82 by insertion of an upstream ISAba1 element that carries a strong outward-facing promoter can also lead to high-level resistance to imipenem and meropenem (20, 21). All but two clade F strains had the ISAba1-blaOXA-82 structure, which may make the plasmid-borne blaOXA-23 gene redundant in certain growth environments. Most clade B isolates carried the ISAba1-blaOXA structure, along with several strains in clades C and D. No clear difference was seen in the patterns of susceptibility to carbapenem drugs for isolates with ISAba1-blaOXA-82 compared with isolates with blaOXA-23 or both genes. Fifteen clade F strains carry Tn2006, which harbors blaOXA-23 flanked by ISAba1 elements (22, 23). The genomes with Tn2006 are located at several distinct locations on the phylogenetic tree, suggesting multiple independent acquisitions.
The pACICU2-like plasmid that was the most common in clade F isolates from Cleveland is closely related to pABUH1 that carries both the blaOXA-23 gene in Tn2008 and aphA6 in TnaphA6 (7, 24). In genomes from Cleveland isolates, the absence of blaOXA-23 generally correlated with loss of the plasmid. Loss of the plasmid did not always result in loss of TnaphA6, however, suggesting that this element can mobilize to other genomic locations. Clade F isolates have about a dozen copies of ISAba125, making it impossible to precisely discern the location(s) of the TnaphA6 transposon in these draft genome sequences. There was more variability in plasmid sequences represented in the assemblies of genomes among the Baltimore isolates, and Tn2008 and TnaphA6 could not be linked to the plasmid in many cases.
Class 1 integrons carrying a range of antibiotic resistance genes are common in MDR A. baumannii (25). Clade F strains from Cleveland carry one of two distinct class 1 integrons. One integron carries a resistance gene cassette that has not been previously described in A. baumannii (Fig. 3A and B). This cassette includes the aadB gene encoding aminoglycoside (2′)-adenyltransferase ANT(2″)-Ia and aadA2 encoding streptomycin 3″-adenylyltransferase. A search by BLAST of the NCBI database revealed that a nearly identical integron sequence was present in Proteus mirabilis strain AR_0059 (GenBank accession number CP020052.1); several strains of Escherichia coli isolated from cow, pig, and chicken; and Salmonella enterica subsp. enterica (e.g., GenBank accession number CP014658.1). It appears that the integron was mobilized by a pair of IS26 elements in direct-repeat orientation. In strain HUMC1, an IS26-flanked Tn6020 element carrying aphA1 was located immediately adjacent to the integron. However, Tn6020 was not found in any other clade F strains. Previously, the aadB gene has been found most often associated with the small plasmid pRAY in A. baumannii (7). A few clade F strains have aadB in a different genomic context. Strains without aadB were more likely to be susceptible to tobramycin (data not shown).
FIG 3.

Integron structures in clade F strains. (A) A complex transposon structure was inserted into the HUMC1 genome at coordinates 1513187 to 1528810 (GenBank accession number LQRQ01000007.1). A partial deletion of this region was found at the corresponding location in the GC1 strain AB0057 and in other GC2 strains, including TYTH-1, ACICU, and the clade F strain UH7007. Most clade F genomes carried only the integron (labeled “b”) and not the Tn6020 segment (labeled “a”) from HUMC1. Yellow arrows indicate copies of IS26. (B) Gene diagram corresponding to the region in purple in panel A. (C) The complex transposon structure from ABUH585 and other clade F genomes. This region is similar to the integron present in the TCDC-AB0715 genome at bases 274173 to 281008 under GenBank accession number CP002522.2. Resistance genes are in purple. The * gene in panel B is qacEΔ1.
The second integron is closely related to a sequence from TCDC-AB0715 (26) (GenBank accession number CP002522.2), except that the qacEΔ gene is interrupted by an IS26 element (Fig. 3C). This structure includes the aacC1 and aadA1 genes, encoding aminoglycoside N(3′)-acetyltransferase and streptomycin 3″-adenylyltransferase, respectively. CCF48, CCF75, ABUH628, and HUMC1 appear to contain both integrons, whereas all other clade F genomes contain only one of them. The novel integron structure (Fig. 3B) is largely restricted to Cleveland isolates, whereas the TCDC-AB0715-like integron was present in more than half of the genomes of Baltimore isolates.
Most MDR GC2 A. baumannii genomes described to date have an AbaGRI1-type resistance island, also termed Tn6166, inserted into the comM gene (27–29). In contrast, most clade F strains lack an insertion at that location. CCF66 and CCF31 have an ∼20-kb island that is very similar to Tn6166 and includes strAB and a tetA gene (30). ABUH773 and eight other strains have a related structure at this location that lacks about 8 kb at the 3′ end of the element, including the strAB genes. In addition to resistance to antibiotic treatment, A. baumannii is subjected to selective pressure in the context of competition for resources with its host, immune evasion, and survival in the hospital environment. Genes involved in the host response and virulence include those related to motility, cell surface glycosylation, attachment to biotic and abiotic surfaces, micronutrient acquisition, and secretion systems (3). Genes related to these traits are also variable among clade F strains.
Two genetic loci carry genes whose products determine the structure of the lipooligosaccharide (LOS) structures on the cell surface (31, 32). Clade F strains were distinct from other GC2 strains by expressing the KL22 or KL13 variant at the K locus and an OCL3-like variant at the OC locus (Fig. 1). Fourteen clade F genomes have an intact OCL3 locus, but all other clade F strains carried one of several variants. An ISAba13 element interrupted the last gene in the cluster (gtrOC11), which encodes a glycosyltransferase (Fig. 4). Many clade F strains had additional modifications to the OCL3 locus, including deletions (adjacent to the ISAba13 insertion site) and insertions of ISAba125 and ISAba17. Kenyon et al. showed a similar pattern of IS element disruption of the OCL1 locus in GC2 isolates from Australian hospitals that resulted in truncated LOS structures (33). KL13 strains belong to ST349, while KL22 strains belong to ST281 in the Oxford MLST scheme.
FIG 4.

Variation in the OCL locus among clade F strains. The inferred genetic structure of the OCL locus is shown, with variants present in different genomes. The OCL3 locus from GenBank accession number KC118540.6 is shown. Asterisks denote the locations of IS element insertions in a subset of genomes. The OCL3 variants found among clade F genomes are depicted, along with the number of genomes in which the variant was observed.
Most A. baumannii strains express a type IV secretion system comprised of a complex cell surface pilus structure. The structure is encoded by multiple genes, and it has been noted that there is variation in the critical PilA structural pilin protein (34). Most GC2 strains carried a pilA gene homologous to ACICU_03380. Clade F strains harbored a different PilA family gene at this position, typified by the protein under GenBank accession number ETR04673.1 from UH7007. The two encoded proteins shared only 65% amino acid similarity. Interestingly, the gene replacement event in clade F strains also resulted in deletion of the adjacent gene downstream of the pilA-like gene, ACICU_03379, which encodes a Wzy_C (O-antigen ligase) domain-containing protein. The loss of this gene in clade F strains may result in additional differences in cell surface structure compared with other GC2 A. baumannii strains.
We also used a comprehensive pangenome analysis of the Cleveland isolates to identify broader patterns of genes that are shared and distinct among the clades. A neighbor-joining tree created using a distance matrix based on the number of shared genes largely supported the clades identified in the SNV tree (Fig. S3A). Fifty-three genes were specific to clade F, and 19 genes were present in all other clades but were missing from clade F. Most of these genes were annotated as encoding hypothetical proteins and may represent poorly annotated prophage. Within clade F, there was little correlation between the order of strains in the SNV tree and the shared-genes tree (Fig. S3B), indicating that there were recent changes in gene content, such as independent losses of the pACICU2-like plasmid and IS26-associated deletions. The clade A genomes comprised several subgroups in the pangenome analysis. One group of those clade A groups lacked the pACICU2-like plasmid and the blaOXA-23 gene. Clade D genomes had 109 clade-specific genes and lacked 51 genes that were present in all other clades. Clade D genomes encoded the KL42 capsular K locus cassette, and several of the differentially present genes were within this locus. Several metabolic enzymes were also encoded among the clade D-specific genes.
Pangenome shared gene content tree. (A) The number of genes shared by each strain was expressed as a distance matrix and used to create a neighbor-joining tree. The outer circles represent clade and hospital of origin. (B) Comparison of the order of strains in the SNV tree from Fig. 1 with the order in the gene content tree from panel A. Each point represents one genome, color-coded by clade. Download FIG S3, PDF file, 0.4 MB (405KB, pdf) .
Copyright © 2019 Adams et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Long-term population dynamics reflect the cumulative impact of dispersal, mutation, introgression of new genetic backgrounds, diversification through lateral gene transfer or homologous recombination, and selection in the local environment. Here we show that the predominant genetic background of A. baumannii strains shifted in a similar way during the course of several months, resulting in very closely related strains becoming predominant in two different hospital systems serving the same geographical area. Interleaving of the genomes from the two hospital systems in the phylogenetic tree and analysis of shared genomic features suggest multiple introductions of closely related but distinct A. baumannii strains with small local expansions.
The emergence and replacement of previous genetic backgrounds in both systems spanning the same time period could be due to several factors that may have contributed to shaping the set of strains causing infections. First, a common source of new strain backgrounds might exist in extended care facilities that exchange patients with both hospital systems (35, 36). The involvement of asymptomatic carriage and transmission of the Gram-negative pathogen Klebsiella pneumoniae was recently hypothesized as an explanation of genome variation observed in a large survey of isolates from multiple hospitals (37). A reservoir for A. baumannii outside health care settings has never been definitively demonstrated, and community-acquired infections are rare in the United States (38).
Second, the pattern of strains in Cleveland could reflect a broader shift in A. baumannii strains in the United States. Systematic genome-based surveys of A. baumannii strains do not presently exist in the United States, and so it is not possible to determine how representative the isolates from these Cleveland hospitals are. As shown in Fig. 1, isolates from Chicago, Baltimore, and Pittsburgh are all located in separate phylogenetic branches from the Cleveland isolates, suggesting geographical separation of the Cleveland isolates. There are, however, three non-Cleveland strains that cluster with the genomes from Cleveland in the core genome phylogeny: two strains isolated from the same patient in France (17) and one from Oregon. It seems likely that as additional isolates are sampled, there will be more examples of genomes that share a recent common ancestor with the Cleveland strains. This study along with those described above show that SNVs in the nonrecombinant core genome are only one view of shared genetic content. A full picture of relationships between strains needs to encompass SNVs, mobile elements, and homologous recombination events. To highlight this, the two genomes from France have only 3 ISAba1 copies, compared to 22 to 29 for the Cleveland isolates.
Finally, the clade F strains were not simply added to the population in the Cleveland hospital systems; over the course of about a year, they displaced the previously dominant lineages. The overall levels of antibiotic resistance are similar in strains from clade F and from other lineages (Table 1). Thus, while it is possible that antibiotic pressure contributed to the rise of clade F, we also hypothesize that these strains have some advantage in either survival or transmissibility. Harding et al. have summarized the current knowledge of virulence mechanisms in A. baumannii (3). Many of the mechanisms that they highlight are conserved across all A. baumannii genomes, including the type I and II secretion systems; the Ata protein, which is important for adherence to host surfaces; and the type IV pilus. However, several of the genes highlighted in the review by Harding et al. are absent or variable in the clade F genomes. The presence of a putative alternative major pilus protein, PilA, in the clade F genomes may indicate a functional difference of this structure that is important for twitching motility and adherence to epithelial cells (39, 40). The three-dimensional structures of three variant PilA proteins were recently described (34), providing insight into functional differences among these proteins. The pilA allele in clade F genomes is distinct from all of those, with <80% amino acid identity of the encoded protein.
Lipopolysaccharide, lipooligosaccharide, and capsule structures contribute to evasion of innate immune responses in several pathogens, including Escherichia coli (41). Capsule replacement has also been suggested to have contributed to the success of the ST258 lineage of Klebsiella pneumoniae (42). We also found an enrichment in variants affecting cell surface structures among longitudinally sampled A. baumannii strains from individual patients (9). Most GC2 A. baumannii strains carry the OCL1 locus, whereas clade F strains carry several variants of the OCL3 locus. These variants are characterized by multiple intragenic IS element insertions and deletions, suggesting the loss of some glycosyltransferase activities. Multiple novel variants of the phosphoglycosyltransferase gene pglC that catalyzes the initial step of capsule synthesis (43) were also found among the GC2 genomes. The functional impact of variation at these loci bears further investigation.
Among genome features of the clade F strains, relatively few are unique to the Cleveland isolates. The HUMC1-type integron (Fig. 3B) is restricted to HUMC1 and the Cleveland isolates, among the genomes that have so far been described. This integron carries aadB and aadA2, which confer aminoglycoside resistance, and while this structure is novel, those genes are present in different genetic contexts in other A. baumannii genomes. The alternative pilA allele is shared by Cleveland and non-Cleveland clade F genomes, as are the OC and K locus variants.
A number of studies have used genomic approaches to enhance epidemiological studies of A. baumannii in hospital environments (44–46). We previously found that almost one-third of patients with multiple A. baumannii isolates were infected by more than one genetically distinct strain (9). Others have seen a similar diversity of cocirculating strains. Schultz et al. found multiple distinct lineages of carbapenem-resistant A. baumannii in a Vietnamese hospital and documented the transposon-mediated spread of a blaOXA-23 gene to new genetic backgrounds (47). These investigators also observed switching of the capsule locus genes, likely by homologous recombination in flanking regions. A yearlong survey of a German intensive care unit found that half of the A. baumannii isolates belonged to one of two clusters but that all others were genetically distinct from each other (48).
A somewhat different picture emerged in an analysis of 85 strains from Mexico (49). The Mexico strains had very few SNV differences compared to a broad set of reference genomes and thus appeared to be clonally related. However, they exhibited extensive variation in gene content, with scores of strain-specific genes and with few genomes highly similar in gene content. Chromosomal and plasmid-mediated mobile elements were a chief source of the variation in gene number, but the origins of the variable gene content could not be determined. Feng et al. characterized A. baumannii isolates from East Asia and also found strains that were very closely related in their core gene phylogeny despite being isolated in hospitals that are more than 2,500 km apart (50). Even closely related genomes had differences in mobile elements related to antibiotic resistance. A common theme of these studies and the current survey of A. baumannii genomes from Cleveland is that A. baumannii genomes exhibit a high degree of genomic variation and that apparently closely related strains can nonetheless differ in genome composition, leading to phenotypic differences.
Knowledge regarding mechanisms by which MDR pathogens are transmitted among and within hospitals is important for understanding how new resistance genes or virulent lineages disperse from local to global scales. The rapid shift in genetic background described here highlights the importance of genetic monitoring of strains causing infection as part of an ongoing effort to optimize treatment and prevention strategies. Extensive genic diversity is a hallmark of A. baumannii isolates. While the antimicrobial resistance genotypes are certainly an important component of this variation, the breadth of functions represented among genes that are acquired and lost suggests that other selective forces, such as host interaction and survival, also play a significant role in shaping the genome evolution of A. baumannii and that these areas are worthy of further exploration in developing an improved understanding of the basis for the clinical impact of this pathogen.
MATERIALS AND METHODS
Strains.
Clinical isolates of A. baumannii were archived by the clinical microbiology departments under protocols approved by institutional review boards at each institution. Isolates from hospital system A were collected between 2007 and 2016 and stored at −80°C. Samples were randomly selected for sequencing to span the time frame of collection. In cases where more than one strain was isolated from a patient, only one isolate was included in the analysis presented here.
Isolates from hospital system B were obtained from bloodstream infections during 2012 and 2016. Only meropenem-resistant strains were selected; these represented 51% of all A. baumannii bloodstream infections during this period. A total of 371 strains from hospital system A and 70 from hospital system B were included. Genome sequences for 344 of those strains were determined in this study, and the remainder have been reported previously (6, 9). Accession numbers for all genomes are available in Data Set S1 in the supplemental material.
Strain list with accession numbers and antimicrobial susceptibility information. Download Data Set S1, XLSX file, 0.1 MB (82.7KB, xlsx) .
Copyright © 2019 Adams et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genome sequencing, annotation, and comparative analysis.
DNA was isolated using mechanical lysis with glass beads (51). Libraries were prepared for sequencing using NexteraXT kits and sequenced on an Illumina NextSeq 500 DNA sequencer as paired-end, 150-base reads. Read sets were assembled using velvet (52) and submitted for annotation to the Prokaryotic Genome Annotation Pipeline (PGAP) at the National Center for Biotechnology Information (53). Antibiotic resistance genes were identified using the Comprehensive Antibiotic Resistance Database (CARD) (http://arpcard.mcmaster.ca) (54). In silico MLST typing was performed using LOCUST (55). The locations of insertion sequence elements in draft genomes were mapped using ISseeker (56). ISseeker finds partial IS matches at contig edges and maps the flanking sequence to a reference genome, in this case ACICU (GenBank accession number NC_010611.1). To define the structures of integrons, we began by identifying antibiotic resistance genes of interest (using a tBLASTn search with protein sequence queries of the draft genomes). Contigs containing these genes were then compared to a collection of previously described genomic islands and other mobile elements from A. baumannii, with particular attention being paid to well-characterized elements in reported genomes. Matching elements were then aligned to the draft genome, and an accounting was made of regions of identity and of difference. A set of nucleotide query sequences was then used to search the full set of genome sequences to identify all genomes with highly similar content. This back-and-forth strategy was used to refine the interpretation of the draft genome content. OC and K loci were mapped by BLAST of contigs from each genome to a database comprised of previously reported sequences (31, 32). An OC or K locus was considered a match if >98% of the locus was represented by sequences in the query genome. Pangenome analysis was performed using PanOCT (57) with the Cleveland genomes plus the 70 complete genomes from GenBank. Pairwise comparisons between the nucleotide sequences of all protein-coding genes were performed using an iterative approach to building the distance matrix. Fourteen groups were used in the first round of distance matrix construction, selected based on proximity on the phylogenetic tree. A gene presence/absence matrix was constructed, and the matrix was used to construct a neighbor-joining tree based on shared gene content.
Phylogeny.
Genome sequences for 70 strains, representing complete genome sequences of all strains available in GenBank as of 28 June 2017, were included in the analysis to provide phylogenetic context for the Cleveland strains. In addition, all genomes that fall into the same clade as HUMC1 in the GenBank genomes division were downloaded and included in certain analyses of clade F to provide as broad a context as possible for the Cleveland genomes (250 additional genomes after removing 10 assemblies with more than 300 contigs). Single-nucleotide variants in the genome sequences were called using Parsnp (58), using the genome sequence of strain HUMC1 (15) as the reference. Three different genome groups were analyzed: (i) all the genomes described above, (ii) all GC2 genomes, and (iii) all clade F genomes. Parsnp calls SNVs in core genome regions, so the number of informative variants increases as the number of genes shared across the data set increases and as the genome diversity increases. To identify regions of recombination, SNVs were imputed onto the HUMC1 genome sequences using the FastaAlternateReferenceMaker feature of the Genome Analysis Toolkit (GATK) (59) to create the input sequences for Gubbins (60). Gubbins iteratively identifies regions of recombination based on the density of SNVs and simultaneously constructs a maximum likelihood tree based on the filtered polymorphisms using RAxML (61). For the all-GC2 tree in Fig. 1, 19,551 SNVs identified by Parsnp were reduced to 8,868 in nonrecombining regions by Gubbins. For the clade F tree in Fig. S2, 15,725 SNVs identified by Parsnp were reduced to 9,090 in nonrecombining regions.
Data availability.
Data Set S1 in the supplemental material contains information about each strain, including GenBank accession numbers and antimicrobial susceptibility patterns, when available. Newly sequenced isolates have been registered at the NCBI under BioProject accession numbers PRJNA352251, PRJNA316619, and PRJNA271775.
ACKNOWLEDGMENTS
We thank the JCVI Sequencing Core Facility and Informatics teams for their contributions to this project.
This work has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH), Department of Health and Human Services, under award number U19AI110819 (to JCVI). Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the NIH under award numbers R01AI100560, R01AI063517, and R01AI072219 to R.A.B. This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, award number 1I01BX001974 to R.A.B. from the Biomedical Laboratory Research and Development Service of the VA Office of Research and Development, and Geriatric Research Education and Clinical Center VISN 10. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Adams MD, Wright MS, Karichu JK, Venepally P, Fouts DE, Chan AP, Richter SS, Jacobs MR, Bonomo RA. 2019. Rapid replacement of Acinetobacter baumannii strains accompanied by changes in lipooligosaccharide loci and resistance gene repertoire. mBio 10:e00356-19. https://doi.org/10.1128/mBio.00356-19.
Contributor Information
Jason W. Sahl, Northern Arizona University.
Paul Keim, Northern Arizona University.
REFERENCES
- 1.Peleg AY, de Breij A, Adams MD, Cerqueira GM, Mocali S, Galardini M, Nibbering PH, Earl AM, Ward DV, Paterson DL, Seifert H, Dijkshoorn L. 2012. The success of Acinetobacter species; genetic, metabolic and virulence attributes. PLoS One 7:e46984. doi: 10.1371/journal.pone.0046984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Potron A, Poirel L, Nordmann P. 2015. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: mechanisms and epidemiology. Int J Antimicrob Agents 45:568–585. doi: 10.1016/j.ijantimicag.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Harding CM, Hennon SW, Feldman MF. 2018. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat Rev Microbiol 16:91–102. doi: 10.1038/nrmicro.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Viehman JA, Nguyen MH, Doi Y. 2014. Treatment options for carbapenem-resistant and extensively drug-resistant Acinetobacter baumannii infections. Drugs 74:1315–1333. doi: 10.1007/s40265-014-0267-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thom KA, Rock C, Jackson SS, Johnson JK, Srinivasan A, Magder LS, Roghmann MC, Bonomo RA, Harris AD. 2017. Factors leading to transmission risk of Acinetobacter baumannii. Crit Care Med 45:e633–e639. doi: 10.1097/CCM.0000000000002318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright MS, Haft DH, Harkins DM, Perez F, Hujer KM, Bajaksouzian S, Benard MF, Jacobs MR, Bonomo RA, Adams MD. 2014. New insights into dissemination and variation of the health care-associated pathogen Acinetobacter baumannii from genomic analysis. mBio 5:e00963-13. doi: 10.1128/mBio.00963-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nigro SJ, Post V, Hall RM. 2011. Aminoglycoside resistance in multiply antibiotic-resistant Acinetobacter baumannii belonging to global clone 2 from Australian hospitals. J Antimicrob Chemother 66:1504–1509. doi: 10.1093/jac/dkr163. [DOI] [PubMed] [Google Scholar]
- 8.Nemec A, Krizova L, Maixnerova M, Diancourt L, van der Reijden TJ, Brisse S, van den Broek P, Dijkshoorn L. 2008. Emergence of carbapenem resistance in Acinetobacter baumannii in the Czech Republic is associated with the spread of multidrug-resistant strains of European clone II. J Antimicrob Chemother 62:484–489. doi: 10.1093/jac/dkn205. [DOI] [PubMed] [Google Scholar]
- 9.Wright MS, Iovleva A, Jacobs MR, Bonomo RA, Adams MD. 2016. Genome dynamics of multidrug-resistant Acinetobacter baumannii during infection and treatment. Genome Med 8:26. doi: 10.1186/s13073-016-0279-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diancourt L, Passet V, Nemec A, Dijkshoorn L, Brisse S. 2010. The population structure of Acinetobacter baumannii: expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS One 5:e10034. doi: 10.1371/journal.pone.0010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace L, Daugherty SC, Nagaraj S, Johnson JK, Harris AD, Rasko DA. 2016. Use of comparative genomics to characterize the diversity of Acinetobacter baumannii surveillance isolates in a health care institution. Antimicrob Agents Chemother 60:5933–5941. doi: 10.1128/AAC.00477-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartual SG, Seifert H, Hippler C, Luzon MA, Wisplinghoff H, Rodriguez-Valera F. 2005. Development of a multilocus sequence typing scheme for characterization of clinical isolates of Acinetobacter baumannii. J Clin Microbiol 43:4382–4390. doi: 10.1128/JCM.43.9.4382-4390.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mustapha MM, Li B, Pacey MP, Mettus RT, McElheny CL, Marshall CW, Ernst RK, Cooper VS, Doi Y. 2018. Phylogenomics of colistin-susceptible and resistant XDR Acinetobacter baumannii. J Antimicrob Chemother 73:2952–2959. doi: 10.1093/jac/dky290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fitzpatrick MA, Ozer EA, Hauser AR. 2016. Utility of whole-genome sequencing in characterizing Acinetobacter epidemiology and analyzing hospital outbreaks. J Clin Microbiol 54:593–612. doi: 10.1128/JCM.01818-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo G, Lin L, Ibrahim AS, Baquir B, Pantapalangkoor P, Bonomo RA, Doi Y, Adams MD, Russo TA, Spellberg B. 2012. Active and passive immunization protects against lethal, extreme drug resistant-Acinetobacter baumannii infection. PLoS One 7:e29446. doi: 10.1371/journal.pone.0029446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rolain JM, Roch A, Castanier M, Papazian L, Raoult D. 2011. Acinetobacter baumannii resistant to colistin with impaired virulence: a case report from France. J Infect Dis 204:1146–1147. doi: 10.1093/infdis/jir475. [DOI] [PubMed] [Google Scholar]
- 17.Rolain JM, Diene SM, Kempf M, Gimenez G, Robert C, Raoult D. 2013. Real-time sequencing to decipher the molecular mechanism of resistance of a clinical pan-drug-resistant Acinetobacter baumannii isolate from Marseille, France. Antimicrob Agents Chemother 57:592–596. doi: 10.1128/AAC.01314-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buser GL, Cassidy PM, Cunningham MC, Rudin S, Hujer AM, Vega R, Furuno JP, Marshall SH, Higgins PG, Jacobs MR, Wright MS, Adams MD, Bonomo RA, Pfeiffer CD, Beldavs ZG. 2017. Failure to communicate: transmission of extensively drug-resistant bla OXA-237-containing Acinetobacter baumannii—multiple facilities in Oregon, 2012-2014. Infect Control Hosp Epidemiol 38:1335–1341. doi: 10.1017/ice.2017.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamidian M, Hall RM. 2014. pACICU2 is a conjugative plasmid of Acinetobacter carrying the aminoglycoside resistance transposon TnaphA6. J Antimicrob Chemother 69:1146–1148. doi: 10.1093/jac/dkt488. [DOI] [PubMed] [Google Scholar]
- 20.Figueiredo S, Poirel L, Croize J, Recule C, Nordmann P. 2009. In vivo selection of reduced susceptibility to carbapenems in Acinetobacter baumannii related to ISAba1-mediated overexpression of the natural blaOXA-66 oxacillinase gene. Antimicrob Agents Chemother 53:2657–2659. doi: 10.1128/AAC.01663-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zander E, Chmielarczyk A, Heczko P, Seifert H, Higgins PG. 2013. Conversion of OXA-66 into OXA-82 in clinical Acinetobacter baumannii isolates and association with altered carbapenem susceptibility. J Antimicrob Chemother 68:308–311. doi: 10.1093/jac/dks382. [DOI] [PubMed] [Google Scholar]
- 22.Nigro SJ, Hall RM. 2016. Structure and context of Acinetobacter transposons carrying the oxa23 carbapenemase gene. J Antimicrob Chemother 71:1135–1147. doi: 10.1093/jac/dkv440. [DOI] [PubMed] [Google Scholar]
- 23.Mugnier P, Poirel L, Pitout M, Nordmann P. 2008. Carbapenem-resistant and OXA-23-producing Acinetobacter baumannii isolates in the United Arab Emirates. Clin Microbiol Infect 14:879–882. doi: 10.1111/j.1469-0691.2008.02056.x. [DOI] [PubMed] [Google Scholar]
- 24.Nigro SJ, Holt KE, Pickard D, Hall RM. 2015. Carbapenem and amikacin resistance on a large conjugative Acinetobacter baumannii plasmid. J Antimicrob Chemother 70:1259–1261. doi: 10.1093/jac/dku486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poirel L, Bonnin RA, Nordmann P. 2011. Genetic basis of antibiotic resistance in pathogenic Acinetobacter species. IUBMB Life 63:1061–1067. doi: 10.1002/iub.532. [DOI] [PubMed] [Google Scholar]
- 26.Chen CC, Lin YC, Sheng WH, Chen YC, Chang SC, Hsia KC, Liao MH, Li SY. 2011. Genome sequence of a dominant, multidrug-resistant Acinetobacter baumannii strain, TCDC-AB0715. J Bacteriol 193:2361–2362. doi: 10.1128/JB.00244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nigro SJ, Farrugia DN, Paulsen IT, Hall RM. 2013. A novel family of genomic resistance islands, AbGRI2, contributing to aminoglycoside resistance in Acinetobacter baumannii isolates belonging to global clone 2. J Antimicrob Chemother 68:554–557. doi: 10.1093/jac/dks459. [DOI] [PubMed] [Google Scholar]
- 28.Hamidian M, Hall RM. 2011. AbaR4 replaces AbaR3 in a carbapenem-resistant Acinetobacter baumannii isolate belonging to global clone 1 from an Australian hospital. J Antimicrob Chemother 66:2484–2491. doi: 10.1093/jac/dkr356. [DOI] [PubMed] [Google Scholar]
- 29.Fournier PE, Richet H. 2006. The epidemiology and control of Acinetobacter baumannii in health care facilities. Clin Infect Dis 42:692–699. doi: 10.1086/500202. [DOI] [PubMed] [Google Scholar]
- 30.Nigro SJ, Hall RM. 2012. Tn6167, an antibiotic resistance island in an Australian carbapenem-resistant Acinetobacter baumannii GC2, ST92 isolate. J Antimicrob Chemother 67:1342–1346. doi: 10.1093/jac/dks037. [DOI] [PubMed] [Google Scholar]
- 31.Kenyon JJ, Hall RM. 2013. Variation in the complex carbohydrate biosynthesis loci of Acinetobacter baumannii genomes. PLoS One 8:e62160. doi: 10.1371/journal.pone.0062160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenyon JJ, Nigro SJ, Hall RM. 2014. Variation in the OC locus of Acinetobacter baumannii genomes predicts extensive structural diversity in the lipooligosaccharide. PLoS One 9:e107833. doi: 10.1371/journal.pone.0107833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kenyon JJ, Holt KE, Pickard D, Dougan G, Hall RM. 2014. Insertions in the OCL1 locus of Acinetobacter baumannii lead to shortened lipooligosaccharides. Res Microbiol 165:472–475. doi: 10.1016/j.resmic.2014.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ronish LA, Lillehoj E, Fields JK, Sundberg EJ, Piepenbrink KH. 2019. The structure of PilA from Acinetobacter baumannii AB5075 suggests a mechanism for functional specialization in Acinetobacter type IV pili. J Biol Chem 294:218–230. doi: 10.1074/jbc.RA118.005814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchaim D, Chopra T, Bogan C, Bheemreddy S, Sengstock D, Jagarlamudi R, Malani A, Lemanek L, Moshos J, Lephart PR, Ku K, Hasan A, Lee J, Khandker N, Blunden C, Geffert SF, Moody M, Hiro R, Wang Y, Ahmad F, Mohammadi T, Faruque O, Patel D, Pogue JM, Hayakawa K, Dhar S, Kaye KS. 2012. The burden of multidrug-resistant organisms on tertiary hospitals posed by patients with recent stays in long-term acute care facilities. Am J Infect Control 40:760–765. doi: 10.1016/j.ajic.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 36.Mody L, Gibson KE, Horcher A, Prenovost K, McNamara SE, Foxman B, Kaye KS, Bradley S. 2015. Prevalence of and risk factors for multidrug-resistant Acinetobacter baumannii colonization among high-risk nursing home residents. Infect Control Hosp Epidemiol 36:1155–1162. doi: 10.1017/ice.2015.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerqueira GC, Earl AM, Ernst CM, Grad YH, Dekker JP, Feldgarden M, Chapman SB, Reis-Cunha JL, Shea TP, Young S, Zeng Q, Delaney ML, Kim D, Peterson EM, O’Brien TF, Ferraro MJ, Hooper DC, Huang SS, Kirby JE, Onderdonk AB, Birren BW, Hung DT, Cosimi LA, Wortman JR, Murphy CI, Hanage WP. 2017. Multi-institute analysis of carbapenem resistance reveals remarkable diversity, unexplained mechanisms, and limited clonal outbreaks. Proc Natl Acad Sci U S A 114:1135–1140. doi: 10.1073/pnas.1616248114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong D, Nielsen TB, Bonomo RA, Pantapalangkoor P, Luna B, Spellberg B. 2017. Clinical and pathophysiological overview of Acinetobacter infections: a century of challenges. Clin Microbiol Rev 30:409–447. doi: 10.1128/CMR.00058-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harding CM, Tracy EN, Carruthers MD, Rather PN, Actis LA, Munson RS Jr.. 2013. Acinetobacter baumannii strain M2 produces type IV pili which play a role in natural transformation and twitching motility but not surface-associated motility. mBio 4:e00360-13. doi: 10.1128/mBio.00360-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piepenbrink KH, Lillehoj E, Harding CM, Labonte JW, Zuo X, Rapp CA, Munson RS Jr, Goldblum SE, Feldman MF, Gray JJ, Sundberg EJ. 2016. Structural diversity in the type IV pili of multidrug-resistant Acinetobacter. J Biol Chem 291:22924–22935. doi: 10.1074/jbc.M116.751099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miajlovic H, Smith SG. 2014. Bacterial self-defence: how Escherichia coli evades serum killing. FEMS Microbiol Lett 354:1–9. doi: 10.1111/1574-6968.12419. [DOI] [PubMed] [Google Scholar]
- 42.Chen L, Mathema B, Pitout JD, DeLeo FR, Kreiswirth BN. 2014. Epidemic Klebsiella pneumoniae ST258 is a hybrid strain. mBio 5:e01355-14. doi: 10.1128/mBio.01355-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harding CM, Haurat MF, Vinogradov E, Feldman MF. 2018. Distinct amino acid residues confer one of three UDP-sugar substrate specificities in Acinetobacter baumannii PglC phosphoglycosyltransferases. Glycobiology 28:522–533. doi: 10.1093/glycob/cwy037. [DOI] [PubMed] [Google Scholar]
- 44.Halachev MR, Chan JZ, Constantinidou CI, Cumley N, Bradley C, Smith-Banks M, Oppenheim B, Pallen MJ. 2014. Genomic epidemiology of a protracted hospital outbreak caused by multidrug-resistant Acinetobacter baumannii in Birmingham, England. Genome Med 6:70. doi: 10.1186/s13073-014-0070-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willems S, Kampmeier S, Bletz S, Kossow A, Kock R, Kipp F, Mellmann A. 2016. Whole-genome sequencing elucidates epidemiology of nosocomial clusters of Acinetobacter baumannii. J Clin Microbiol 54:2391–2394. doi: 10.1128/JCM.00721-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lewis T, Loman NJ, Bingle L, Jumaa P, Weinstock GM, Mortiboy D, Pallen MJ. 2010. High-throughput whole-genome sequencing to dissect the epidemiology of Acinetobacter baumannii isolates from a hospital outbreak. J Hosp Infect 75:37–41. doi: 10.1016/j.jhin.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 47.Schultz MB, Pham Thanh D, Tran Do Hoan N, Wick RR, Ingle DJ, Hawkey J, Edwards DJ, Kenyon JJ, Phu Huong Lan N, Campbell JI, Thwaites G, Thi Khanh Nhu N, Hall RM, Fournier-Level A, Baker S, Holt KE. 2016. Repeated local emergence of carbapenem-resistant Acinetobacter baumannii in a single hospital ward. Microb Genom 2:e000050. doi: 10.1099/mgen.0.000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wendel AF, Malecki M, Otchwemah R, Tellez-Castillo CJ, Sakka SG, Mattner F. 2018. One-year molecular surveillance of carbapenem-susceptible A. baumannii on a German intensive care unit: diversity or clonality. Antimicrob Resist Infect Control 7:145. doi: 10.1186/s13756-018-0436-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grana-Miraglia L, Lozano LF, Velazquez C, Volkow-Fernandez P, Perez-Oseguera A, Cevallos MA, Castillo-Ramirez S. 2017. Rapid gene turnover as a significant source of genetic variation in a recently seeded population of a healthcare-associated pathogen. Front Microbiol 8:1817. doi: 10.3389/fmicb.2017.01817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng Y, Ruan Z, Shu J, Chen CL, Chiu CH. 2016. A glimpse into evolution and dissemination of multidrug-resistant Acinetobacter baumannii isolates in East Asia: a comparative genomics study. Sci Rep 6:24342. doi: 10.1038/srep24342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koser CU, Fraser LJ, Ioannou A, Becq J, Ellington MJ, Holden MT, Reuter S, Torok ME, Bentley SD, Parkhill J, Gormley NA, Smith GP, Peacock SJ. 2014. Rapid single-colony whole-genome sequencing of bacterial pathogens. J Antimicrob Chemother 69:1275–1281. doi: 10.1093/jac/dkt494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Ciufo S, Li W. 2013. Prokaryotic genome annotation pipeline, p 173–186. In Beck J, Benson D, Coleman J, Hoeppner M, Johnson M, Maglott D, Mizrachi I, Morris R, Ostell J, Pruitt K, Rubinstein W, Sayers E, Sirotkin K, Tatusova T (ed), NCBI handbook, 2nd ed National Center for Biotechnology Information, Bethesda, MD: https://www.ncbi.nlm.nih.gov/books/NBK174280. [Google Scholar]
- 54.Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN, Doshi S, Courtot M, Lo R, Williams LE, Frye JG, Elsayegh T, Sardar D, Westman EL, Pawlowski AC, Johnson TA, Brinkman FS, Wright GD, McArthur AG. 2017. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 45:D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brinkac LM, Beck E, Inman J, Venepally P, Fouts DE, Sutton G. 2017. LOCUST: a custom sequence locus typer for classifying microbial isolates. Bioinformatics 33:1725–1726. doi: 10.1093/bioinformatics/btx045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adams MD, Bishop B, Wright MS. 2016. Quantitative assessment of insertion sequence impact on bacterial genome architecture. Microb Genom 2:e000062. doi: 10.1099/mgen.0.000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fouts DE, Brinkac L, Beck E, Inman J, Sutton G. 2012. PanOCT: automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species. Nucleic Acids Res 40:e172. doi: 10.1093/nar/gks757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Treangen TJ, Ondov BD, Koren S, Phillippy AM. 2014. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, Parkhill J, Harris SR. 2015. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 43:e15. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic tree of all A. baumannii strains from this study. The rings of annotation extending outward from the inner ring depict clade; OC locus; Pasteur MLST; Oxford MLST; genes encoding StrA, OXA-23, the ISAba1-blaOXA insertion, AphA6, AphA1, AadB, AadA2, AacC1, AadA1, A-type PilA (GenBank accession number WP_000993717.1), B-type PilA (GenBank accession number WP_000993718.1), A-type PglC (GenBank accession number WP_000826956.1), B-type PglC (GenBank accession number WP_000977704.1), and C-type PglC (GenBank accession number WP_000919783.1); and isolation location. Download FIG S1, PDF file, 0.7 MB (736.5KB, pdf) .
Copyright © 2019 Adams et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic tree of all clade F A. baumannii strains from this study. The rings of annotation extending outward from the inner ring depict Oxford MLST; year of isolation; K locus; OC locus; the presence of a pABUH1-like plasmid; genes encoding OXA-23, the ISAba1-blaOXA insertion, AphA6, AphA1, the HUMC1-type integron, AadB, AadA2, the TCDC-AB0715-type integron, AadC1, AadA1, ArmA, B-type PilA (GenBank accession number WP_000993718.1), and B-type PglC (GenBank accession number WP_000977704.1); and isolation location. Download FIG S2, PDF file, 1.4 MB (1.5MB, pdf) .
Copyright © 2019 Adams et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Pangenome shared gene content tree. (A) The number of genes shared by each strain was expressed as a distance matrix and used to create a neighbor-joining tree. The outer circles represent clade and hospital of origin. (B) Comparison of the order of strains in the SNV tree from Fig. 1 with the order in the gene content tree from panel A. Each point represents one genome, color-coded by clade. Download FIG S3, PDF file, 0.4 MB (405KB, pdf) .
Copyright © 2019 Adams et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strain list with accession numbers and antimicrobial susceptibility information. Download Data Set S1, XLSX file, 0.1 MB (82.7KB, xlsx) .
Copyright © 2019 Adams et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Data Set S1 in the supplemental material contains information about each strain, including GenBank accession numbers and antimicrobial susceptibility patterns, when available. Newly sequenced isolates have been registered at the NCBI under BioProject accession numbers PRJNA352251, PRJNA316619, and PRJNA271775.