

Abstract
Background
HLA class I contributes to HIV immune control through antigen presentation to cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells. In contrast to investigations of CTL, studies of NK cells in HIV control through HLA-killer immunoglobulin-like receptor (KIR) interactions remain sparse in African cohorts.
Methods
Treatment-naive, chronically HIV-infected adults (N = 312) were recruited from South Africa, and the effects of HLA-KIR pairs on clinical outcome were analyzed.
Results
There was no significant difference in viral load among all subjects with HLA alleles from the HLA-C1 group (P = .1). However, differences in HLA-C type significantly influenced viremia among 247 KIR2DL3 positives (P = .04), suggesting that specific HLA-KIR interactions contribute to immune control. Higher viral load (P = .02) and lower CD4+ T-cell counts (P = .008) were observed in subjects with HLA-C*16:01+KIR2DL3+. Longitudinal analysis showed more rapid progression to AIDS among HLA-C*16:01+KIR2DL3+ subjects (adjusted hazard ratio 1.9, P = .03) than those without this genotype, independent of CD4+ T-cell count and viral load.
Conclusions
These results highlight the existence of unique anti-HIV innate immunity within distinct populations and the contribution of KIR on NK cells and some CTLs to the well-described HLA-mediated impact on HIV disease progression.
Keywords: HIV, HLA, KIR, South Africa, disease outcome
Association between HLA-KIR pair and HIV disease outcome among South African HIV patients was statistically analyzed, and HLA-C*16:01-KIR2DL3 was identified as a disease-susceptible pair, suggested existence of KIR-mediated innate immunity effecting on HIV disease progression.
class I human leukocyte antigen (HLA) molecules interact with both cytotoxic T lymphocytes (CTLs) through their T-cell receptors, and natural killer (NK) cells through their killer immunoglobulin-like receptors (KIRs). Associations between certain HLA-KIR pairs and clinical outcomes were previously reported in several diseases such as hematopoietic stem cell transplantation and its outcome in leukemia [1], fetal growth, preeclampsia, and miscarriage in reproductive diseases [2], and autoimmune diseases [3, 4].
In HIV infection, however, the contribution of NK cells to disease control in vivo is less clear compared with that of CTLs. NK cells are regulated by the combinatorial effect of many activating and inhibitory receptors, including KIRs [5]. Recently, HLA-KIR–associated viral sequence polymorphisms were demonstrated in chronically HIV-infected individuals, suggesting ongoing viral adaptation against NK cell-mediated immune pressure [6, 7]. KIRs contain 2 or 3 external immunoglobulin-like domains (2D, 3D) and have either long or short cytoplasmic tails, which determine whether they are inhibitory or activating receptors, respectively [8, 9]. Some KIRs recognize HLA class I molecules as their ligands: for example, the ligands for the KIR3DL1 subtypes are HLA-B molecules containing the Bw4 motif at residues 77–83 [10, 11]. The high level of expression of the KIR allotype KIR3DL1 and its ligands HLA-Bw4 alleles with isoleucine at residue 80 (Bw4-80I) has been associated with improved clinical outcome via modulation of antiviral NK activity [12, 13]. Interestingly, some of the most protective class I HLA alleles in HIV infection, such as HLA-B*57 in different ethnic groups [14–17] and HLA-B*58:01 in African populations [15], are included in the Bw4-80I group.
HLA-C alleles, which are ligands of KIR2D, are classified into either an HLA-C group 1 (HLA-C1) or group 2 (HLA-C2), depending on sequences at residue 77 and 80 [18, 19], such that HLA-C molecules expressing serine at residue 77 and asparagine at 80 belong to the HLA-C1 group, and HLA-C molecules expressing asparagine at residue 77 and lysine at 80 belong to the HLA-C2 group. The former has a binding affinity for KIR2DL2, KIR2DL3, and KIR2DS2, and the latter for KIR2DL1 and KIR2DS1 [5, 20]. However, whether there is an association between HLA-C–KIR2D interactions and HIV control, as has been demonstrated for HLA-Bw4–KIR3D, has not been fully investigated.
An additional aspect of HLA-mediated NK cell immunity involves the inhibitory HLA-E–NKG2A interaction [21]. The HLA-E–NKG2A interaction is mediated by HLA-E binding of an epitope within the signal peptide present at the N-terminus of classic class I HLA molecules, coded at residues −22 to −14 [22, 23]. Within this signal peptide epitope, methionine is a predominant amino acid at residue −21 (−21M), and all of HLA-A and C alleles share this −21M variant. However, some HLA-B alleles encode threonine at this position (−21T) [22, 23]. Compared to the signal peptide with −21T, the peptide with −21M binds with stronger affinity to HLA-E, consequently leading to higher expression of HLA-E, increased interaction with inhibitory NKG2A, and possibly reduced target cell lysis by NK cells [23]. HLA-E expression and its interaction with NKG2A are increased in diseases such as cytomegalovirus [24], HIV [25], and cancers [26–28], escaping from cell lysis by NK cells.
Our objective here was to identify the effect of KIRs and their interaction with their HLA ligands on HIV viral control in a cohort of 312 chronically infected, treatment-naive adults from South Africa.
METHODS
Subjects and Data Collection
Chronically HIV-infected adults were recruited from outpatient clinics in Durban, South Africa, from March 2003 to March 2007 [29]. All the 312 study participants were antiretroviral treatment (ART) naive with absolute CD4+ T-cell counts of ≥200/mm3 at enrollment, measured by flow cytometry. Viral load was measured using the Roche Amplicor version 1.5 assay. Data regarding timing of ART introduction were also collected. For class I HLA genotyping, high-resolution genotyping for HLA class I loci was performed by polymerase chain reaction (PCR)–sequence-based typing, as recommended by the 13th International Histocompatibility Workshop (available at: http://www.ihwg.org/tmanual/TMcontents.htm). HLA sequences were analyzed using the ASSIGN software (Conexio Genomics, Fremantle, Western Australia, Australia). KIR genotyping for the presence or absence of each KIR gene was conducted by PCR with sequence-specific priming as described previously [30]. The presence and absence of specific PCR products was detected by agarose gel electrophoresis containing ethidium bromide and products were visualized under ultraviolet light. This research was approved by the ethical review boards at the University of KwaZulu-Natal, South Africa, and the University of Oxford, United Kingdom. All patients provided written informed consent for the collection of samples and subsequent analysis.
Statistical Analysis
Statistical analysis was performed using SPSS 21.0 (IBM, Armonk, New York). Kruskal-Wallis test was used to assess viral load differences among HLA-C1–comprising alleles, HLA-C2–comprising alleles, HLA-Bw4–comprising alleles, and HLA-Bw4 80I–comprising alleles. The effect of HLA-KIR pairs on viral load was tested with Mann–Whitney U test for univariate analysis, and linear regression model for multivariate analysis. To avoid overestimation of the CTL-related effect of an HLA allele on clinical outcome, in univariate analysis we analyzed viral load differences between subjects with a particular HLA allele-KIR pair versus subjects with the same HLA but lacking that KIR [31] (for example, difference in viral load between HLA-B*57:01+KIR3DL1+ subjects vs HLA-B*57:01+KIR3DL1− subjects). A Log-rank test was performed for the longitudinal analysis of ART introduction (time to ART initiation or CD4 count <200 cells/mm3 before ART initiation), and Cox hazard model was applied for multivariate analysis.
RESULTS
Characteristics of the Cohort
Of the 312 HIV-infected individuals, 253 (81%) were women and 59 (19%) were men (Table 1). HLA allele frequency and its linkage disequilibrium (Supplementary Table 1) and KIR frequency (Table 1) are shown in tables. Median absolute CD4+ T-cell count at enrollment was 394 cells/mm3 (interquartile range [IQR], 306–516), CD4/CD8 ratio was 0.4 (IQR, 0.3–0.6), and viral load was 4.6 log copies/mL (IQR, 3.8–5.1).
Table 1.
Characteristics of Subjects (N = 312)
| Characteristics | Value, No. (%) |
| CD4+ T cell, median (IQR), counts/μL | 394 (306–516) |
| CD4/CD8 ratio, median (IQR) | 0.4 (0.3–0.6) |
| Viral load, median (IQR) log copies/mL | 4.6 (3.8–5.1) |
| Sex, female | 253 (81) |
| HLA-Bw4 + | 175 (56) |
| HLA-Bw4 80I + | 134 (43) |
| HLA-Bw6 + | 273 (88) |
| HLA-C1 + | 201 (64) |
| HLA-C2 + | 254 (81) |
| HLA-C1C1 + | 58 (19) |
| HLA-C2C2 + | 111 (36) |
| KIR2DL1 + | 308 (99) |
| KIR2DL2 + | 220 (71) |
| KIR2DL3 + | 247 (79) |
| KIR2DL4 + | 310 (99) |
| KIR2DL5 + | 215 (69) |
| KIR2DS1 + | 49 (16) |
| KIR2DS2 + | 191 (61) |
| KIR2DS3 + | 92 (29) |
| KIR2DS4*219+ | 257 (82) |
| KIR2DS5 + | 160 (51) |
| KIR3DL1 + | 308 (99) |
| KIR3DL2 + | 312 (100) |
| KIR3DL3 + | 312 (100) |
| KIR3DS1 + | 31 (10) |
Abbreviations: HLA, human leukocyte antigen; IQR, interquartile range; KIR, killer immunoglobulin-like receptor.
Contribution of NK Cells to HLA-Viral Load Association Through HLA-KIR Interaction
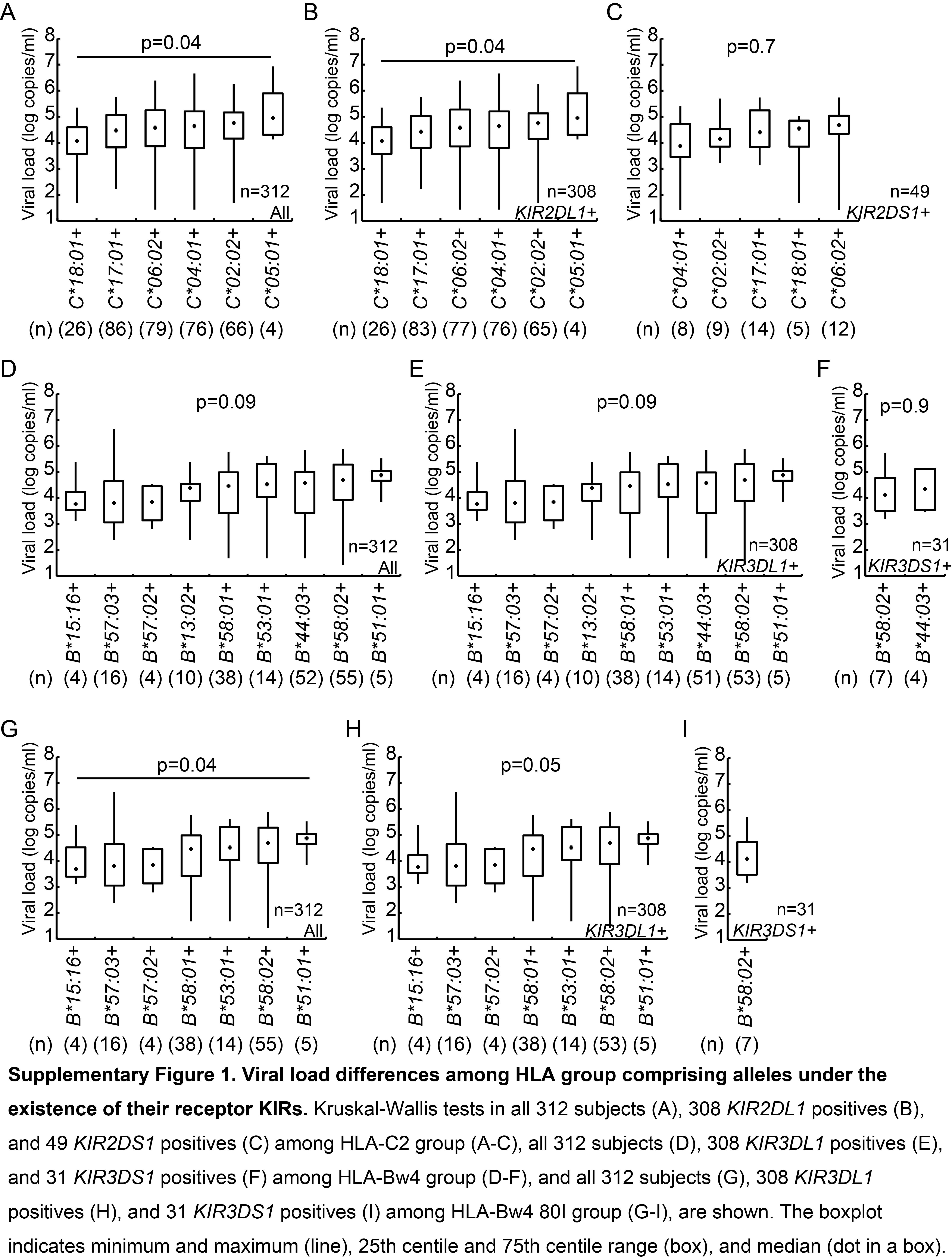
Previous population-specific studies conducted in various geographical regions have reported associations between HLA class I gene carriage and identified disease-protective and susceptible HLA alleles [14–17]. To investigate whether HLA-KIR interactions contribute to such associations, we first investigated whether viral load differed significantly among subjects with the combination of particular KIRs together their corresponding HLA ligands (Figure 1 and Supplementary Figure 1). Whilst viral load did not differ among subjects who were positive for 1 of the 8 HLA-C1 group alleles (P = .1) (Figure 1A) among KIR2DL3-positive subjects, but not KIR2DL2-positive or KIR2DS2-positive subjects, viral load differed significantly according to HLA-C1 allele (P = .04, P = .3, and P = .2, respectively) (Figure 1B–D). However, in post hoc analysis, there was no significant difference in viral load between HLA-C1 group alleles among KIR2DL3-positive subjects (Figure 1B), even including the subjects without HLA-C1 comprising alleles as control group (Supplementary Figure 2). This suggests that within HLA-viral load associations previously reported, NK cells might contribute to such an effect through HLA-KIR interactions, particularly through the HLA-C1–KIR2DL3 interaction in this population.
Figure 1.

Viral load differences among human leukocyte antigen-C1 (HLA-C1) group comprising alleles under the existence of their receptor killer immunoglobulin-like receptors (KIRs). Kruskal–Wallis tests are shown in all 312 subjects (A), 247 KIR2DL3 positives (B), 220 KIR2DL2 positives (C), and 191 KIR2DS2 positives (D). In a post hoc analysis, there were no significant differences between HLA alleles in KIR2DL3 positives, by the Steel-Dwass test (B). The boxplot indicates minimum and maximum (line), 25th centile and 75th centile range (box), and median (dot in a box).
To investigate the effect of NK cells on viral control, we next analyzed the differences in viral load and CD4+ T-cell count associated with individual HLA-KIR pairs (Supplementary Tables 2 and 3). We found that there was a significantly higher viral load in the individuals with KIR2DL3 compared to the individuals without KIR2DL3 (median 4.7 vs 4.4 log copies/mL; P = .01 by Mann–Whitney U test; Supplementary Table 2). The absolute CD4 count was a median of 45 cells/mm3 lower among subjects with KIR2DL3, although this difference did not reach statistical significance (median 432 versus 387 cells/mm3; P = .1; Supplementary Table 3). However, there was no significant viral load and CD4+ T-cell count difference between KIR-positive and KIR-negative subjects when the subjects were grouped by their ligands of HLA-C1, HLA-C2, or HLA-Bw4 groups (Supplementary Table 2 and 3). Two HLA-KIR combinations were significantly associated with changes in viral load: (1) there was lower viral load in subjects with HLA-C*02:02-KIR2DS1 (viral load median 4.2 log copies/mL in HLA-C*02:02+KIR2DS1+ vs 4.9 log copies/mL in HLA-C*02:02+KIR2DS1−; P = .04; Figure 2A); and (2) although there was a small number of subjects with HLA-C*16:01+KIR2DL3− (n = 2), there was higher viral load in HLA-C*16:01-KIR2DL3 (viral load median 5.0 log copies/mL in HLA-C*16:01+KIR2DL3+ vs 3.4 log copies/mL in HLA-C*16:01+KIR2DL3−; P = .04; Figure 2B and Supplementary Table 4). The HLA-C*16:01+KIR2DL3+ combination was also significantly associated with lower absolute CD4 count (median 357 cells/mm3 in HLA-C*16:01+KIR2DL3+ vs 674 cells/mm3 in HLA-C*16:01+KIR2DL3−; P = .03; Figure 2C and Supplementary Table 5). However, HLA-C*02:02-KIR2DS1 was not associated with a higher absolute CD4 count (median 363 cells/mm3 in HLA-C*02:02+KIR2DS1+ vs 376 cells/mm3 in HLA-C*02:02+KIR2DS1−; P = .6; Figure 2C and Supplementary Table 5). The HLA-C*16:01-KIR2DL3 associations with viral load and CD4 count remained significant after adjusting for the impact of KIR2DL2 and KIR2DS2, which are the other receptors for HLA-C*16:01 (P = .02 and P = .008, respectively; Table 2). These data suggest that significant clinical outcome differences are associated with certain HLA allele-KIR pairs identified in this South African cohort.
Figure 2.

A–C, Viral load and CD4+ T-cell count differences between killer immunoglobulin-like receptor (KIR) positives versus negatives under the existence of their ligand human leukocyte antigen (HLA) alleles. P values by Mann–Whitney U test are shown. The boxplot indicates minimum and maximum (line), 25th centile and 75th centile range (box), and median (dot in a box).
Table 2.
Viral Load and CD4+ T-Cell Count Differences Between KIR Positives versus Negatives Under the Existence of Their Ligand HLA Alleles
| R square | B (95% CI) | β | P | |
| Viral load, log copies/mL | ||||
| HLA-C*02:02 + | 0.07 | |||
| KIR2DL1 + | −1.7 (−3.7 to 0.3) | −0.2 | .1 | |
| KIR2DS1 + | −0.6 (−1.3 to 0.08) | −0.2 | .08 | |
| HLA-C*16:01 + | 0.2 | |||
| KIR2DL2 + | 0.7 (−1.2 to 2.6) | 0.4 | .5 | |
| KIR2DL3 + | 1.6 (0.2 to 3.1) | 0.5 | .02 | |
| KIR2DS2 + | −0.3 (−2.2 to 1.6) | −0.1 | .8 | |
| CD4+ T cell, counts/mm3 | ||||
| HLA-C*16:01 + | 0.3 | |||
| KIR2DL2 + | −149 (−417 to 119) | −0.5 | .3 | |
| KIR2DL3 + | −276 (−473 to −78) | −0.5 | .008 | |
| KIR2DS2 + | 119 (−150 to 388) | 0.4 | .4 |
P values by multivariate linear regression model are shown.
Abbreviations: CI, confidence interval; HLA, human leukocyte antigen; KIR, killer immunoglobulin-like receptor.
Linkage Disequilibrium Between HLA-C*16:01 and HLA-B*45:01, but Less Impact of HLA-E–NKG2A Interaction on Disease Outcome Among HLA-C*16:01 Positives
As another aspect of HLA-mediated NK cell immunity, HLA-E–NKG2A interaction was considered [21]. In this study population, HLA-C*16:01 is in linkage disequilibrium with HLA-B*45:01 (Supplementary Table 1). However, HLA-B*45:01 expresses the −21T HLA-E variant within the signal peptide, suggesting less impact of HLA-E–NKG2A interaction on disease outcome among HLA-C*16:01 positives.
More Rapid Progression to ART Initiation Among Subjects With Certain HLA Allele-KIR Pairs
Next, we assessed the impact of HLA allele-KIR pairs on time to meeting CD4 criteria for ART initiation prevailing at this time (absolute CD4 count of <200 cells/mm3) among 289 out of 312 patients with follow-up data. In detail, 134 out of 289 patients (46%) initiated ART during the period of follow up, of whom 118 had absolute CD4 counts of <200 cells/mm3, and 16 initiated ART before drop of CD4 <200 cells/mm3 (at a median 232 cells/mm3). First we found that the subjects with HLA-C*06:02+KIR2DL1+ (P = .02 by log rank test; Figure 3A), HLA-C*16:01+KIR2DL3+ (P = .004; Figure 3B), or HLA-B*58:02+KIR3DL1+ (P = .001; Figure 3C) met CD4 criteria to start ART earlier than subjects without those HLA-KIR pairs. On the other hand, the subjects with HLA-B*44:03+KIR3DL1+ met CD4 criteria to start ART later than subjects without this HLA-KIR pair (P = .04; Figure 3D). These results remained significant when the analysis was undertaken using an endpoint of ART initiation at an absolute CD4 count of <200 cells/mm3, therefore excluding these 16 individuals (Supplementary Figure 3). In order to control for the effect of other factors on rate of progression to meet CD4 criteria for ART initiation, a multivariate analysis was performed. Included in the model were sex, CD4 counts, CD4/8 ratio, viral load, HLA-C*06:02+KIR2DL1+, HLA-C*16:01+KIR2DL3+, HLA-B*44:03+KIR3DL1+, and HLA-B*58:02+KIR3DL1+ (Table 3). In the multivariate analysis, carriage of the HLA-C*16:01+KIR2DL3+ (adjusted hazard ratio [aHR], 1.9; 95% confidence interval [CI], 1.1–3.5; P = .02) pair remained a significant factor associated with higher rates of progression to an absolute CD4 count of <200 cells/mm3, which was independent of CD4+ T-cell count (aHR, 0.9; 95% CI, 0.9–0.9; P < .001) and viral load (aHR, 2.0; 95% CI, 1.5–2.6; P < .001) (Table 3). This result strongly suggests the deleterious effect of HLA-C*16:01+KIR2DL3+ on HIV clinical outcome in the studied South African population.
Figure 3.

A–D, CD4+ T-cell counts ≥200/mm3 and antiretroviral treatment (ART)-free rate differences between human leukocyte antigen-killer immunoglobulin-like receptor (HLA-KIR) pair positives and negatives. Log-rank tests are shown.
Table 3.
CD4+ T-Cell Counts <200/mm3 or ART Initiation Rate Difference Among HLA-KIR Pairs
| Variables | aHR (95% CI) | P |
| Sex, female | 1.2 (0.7–1.9) | .5 |
| CD4+T cell, counts/mm3 | 0.9 (0.9–0.9) | <.001 |
| CD4/CD8 ratio | 0.4 (0.1–1.0) | .05 |
| Viral load, log copies/mL | 2.0 (1.5–2.6) | <.001 |
| HLA-C*06:02 + KIR2DL1 + | 0.9 (0.5–1.7) | .7 |
| HLA-C*16:01 + KIR2DL3 + | 1.9 (1.1–3.5) | .02 |
| HLA-B*44:03 + KIR3DL1 + | 1.0 (0.4–2.6) | .9 |
| HLA-B*58:02 + KIR3DL1 + | 1.5 (0.7–3.0) | .3 |
Multivariate Cox hazard model analysis is shown.
Abbreviations: aHR, adjusted hazard ratio; ART, antiretroviral treatment; CI, confidence interval; HLA, human leukocyte antigen; KIR, killer immunoglobulin-like receptor.
DISCUSSION
This study systematically investigated the effect of particular KIRs and their HLA ligands on clinical outcome in a South African cohort of chronically HIV-infected ART-naive adults. Here, in both cross-sectional and longitudinal analyses, we identified the deleterious effect of HLA-C allele-KIR2D pairs, in particular of HLA-C*16:01+KIR2DL3+, on HIV clinical outcome.
The protective effect of HLA-Bw4 in HIV infection was first described in 2001 [32], and the subsequently advantageous effect of high cellular surface expression allotype of KIR3DL1*h/*y in combination with HLA-Bw4-80I was reported [13, 33]. In contrast, whether the HLA-C–KIR2D pair confers protection against HIV infection has remained unclear. The deleterious effect of the HLA-C1–KIR2DL3 pair on HIV disease outcome observed here is consistent with 2 previous studies, in Cote d’Ivoire [34] and in Thailand [35]. However, the HLA-C1–KIR2DL3 combination has also been reported as protective against mother-to-child transmission in South Africa [36], and against HIV infection among intravenous drug users (IDUs) in Vietnam [37]. A possible explanation for the discrepancy between the reported effects of HLA-C1–KIR2DL3 gene carriage in HIV infection may be found in the study examining the impact of KIRs in hepatitis C virus (HCV) infection [38]. This study showed that the HLA-C1–KIR2DL3 pair was advantageous for HCV resolution in cases of low-dose viral transmission such as needle prick injuries and IDU injections, but not in those with high-dose exposure, such as through blood transfusions. Indeed, in the mother-to-child transmission study [36], the protective impact of the HLA-C1–KIR2DL3 pair was significantly elevated in the group of infants with lower maternal viral load, whereas high viral load masked the protective effect of this allele combination. Also, no HLA-KIR pair was reported as a significant factor contributing to HIV infection among highly exposed uninfected hemophilia A patients in the United Kingdom [39]. Such differences in HIV transmission route and inoculum dose may thus explain the discrepancy in the relative contribution of HLA-KIR pairs to viral control in HIV infection.
In terms of the potential mechanisms underlying the association between HLA-KIR pair and susceptibility to HIV infection, Alter et al [7] and Hölzemer et al [6] reported that inhibitory KIR-associated viral mutation sites can enhance binding of NK cells through inhibitory KIRs, causing an inhibition of NK-cell activation and subsequent escape from NK cell-mediated lysis of infected cells. These findings suggest that detection of HLA-KIR pairs that affect clinical outcomes, and definition of the critical viral residues that influence the HLA-KIR interactions would be warranted for further understanding of mechanism of class I HLA-induced antiviral immunity.
HLA-C*16:01 is frequent among African populations (8%–17% population frequency and 9% in this study; Supplementary Table 1) and Caucasians (5%–10%), but rare among Asian populations [40]. HLA-C*16 alleles, including HLA-C*16:01, were also previously reported to be among disease-susceptible HLA-C alleles associated with high viral loads in South Africa [15], and also in a European American population in the United States [41]. Apps et al also reported [41] that HLA-C alleles contribute to HIV disease outcome, with higher cellular surface expression level associated with better CTL activity and improved disease outcome. However, the cellular surface expression level of HLA-C*16:01 was at an intermediate level among HLA-C alleles [41], suggesting that the interaction observed here between HLA-C*16:01 and KIR2DL3 may be one of the mechanisms contributing to a deleterious effect on HIV disease outcome mediated by this allele.
A limitation of this study is the number of participants enrolled, especially the small number with HLA-C*16:01+KIR2DL3− (n = 2), which limited the analyses available. Although we identified statistically significant associations of several HLA allele-KIR pairs and clinical outcome, further analysis with a larger number of participants, greater genetic diversity, and more clinical data, such as ART regimen, its adherence, and viral control rate by multivariate analyses, would be necessary to confirm these findings.
In conclusion, this study investigated the effect of HLA and KIR pairs on viral control in a chronically HIV-infected ART-naive South African population. We have found that certain HLA allele-KIR pairs affects clinical outcome in this population. These data are consistent with the studies demonstrating that the activity of NK cells can make a significant contribution of the impact of HLA on viral load. Our data highlight geographical differences in the frequency and distribution of HLA-KIR pairs, which in turn may affect the NK-cell response and, consequently, the selection pressure exerted on HIV in each endemic area. The contribution of KIR-mediated viral control will most likely be determined by host differences such as HLA class I allele and KIR frequencies in different geographical regions, viral transmission route and inoculum volume, as well as the viral pathogen in question. The identification of uniquely protective and susceptible HLA-KIR pairs in each endemic area will provide the opportunity to better define the nature of HLA-associated immune control of HIV.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Supplementary Material
{kind=link}
Notes
Disclaimer. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust or the UK government. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Financial support. This work was supported by the Japan Society for the Promotion of Science (grant number 16K08843 to M. M.); the Department of Science and Technology Republic of South Africa (South African Research Chair in Systems Biology of HIV/AIDS awarded to T. N.); the DELTAS Africa Initiative (grant number DEL-15-006 to the Sub-Saharan African Network for TB/HIV Research Excellence) supported by the Wellcome Trust (grant number 107752/Z/15/Z to T. N.) and the UK Government; the Frederick National Laboratory for Cancer Research (grant number HHSN261200800001E to M. C.); the Wellcome Trust (grant number WT104748MA to P. G.); and National Institutes of Health (grant number RO1AI133673 to P. G.).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Venstrom JM, Pittari G, Gooley TA, et al. . HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med 2012; 367:805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moffett A, Colucci F. Co-evolution of NK receptors and HLA ligands in humans is driven by reproduction. Immunol Rev 2015; 267:283–97. [DOI] [PubMed] [Google Scholar]
- 3. Khakoo SI, Carrington M. KIR and disease: a model system or system of models? Immunol Rev 2006; 214:186–201. [DOI] [PubMed] [Google Scholar]
- 4. Martin MP, Nelson G, Lee JH, et al. . Cutting edge: susceptibility to psoriatic arthritis: influence of activating killer Ig-like receptor genes in the absence of specific HLA-C alleles. J Immunol 2002; 169:2818–22. [DOI] [PubMed] [Google Scholar]
- 5. Thielens A, Vivier E, Romagné F. NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. Curr Opin Immunol 2012; 24:239–45. [DOI] [PubMed] [Google Scholar]
- 6. Hölzemer A, Thobakgale CF, Jimenez Cruz CA, et al. . Selection of an HLA-C*03:04-restricted HIV-1 p24 gag sequence variant is associated with viral escape from KIR2DL3+ natural killer cells: data from an observational cohort in South Africa. PLoS Med 2015; 12:e1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alter G, Heckerman D, Schneidewind A, et al. . HIV-1 adaptation to NK-cell-mediated immune pressure. Nature 2011; 476:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parham P, Moffett A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat Rev Immunol 2013; 13:133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol 2002; 20:217–51. [DOI] [PubMed] [Google Scholar]
- 10. Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 public epitope of HLA-B molecules confers reactivity with natural killer cell clones that express NKB1, a putative HLA receptor. J Exp Med 1995; 181:1133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cella M, Longo A, Ferrara GB, Strominger JL, Colonna M. NK3-specific natural killer cells are selectively inhibited by Bw4-positive HLA alleles with isoleucine 80. J Exp Med 1994; 180:1235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boulet S, Song R, Kamya P, et al. . HIV protective KIR3DL1 and HLA-B genotypes influence NK cell function following stimulation with HLA-devoid cells. J Immunol 2010; 184:2057–64. [DOI] [PubMed] [Google Scholar]
- 13. Martin MP, Qi Y, Gao X, et al. . Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet 2007; 39:733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goulder PJ, Walker BD. HIV and HLA class I: an evolving relationship. Immunity 2012; 37:426–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leslie A, Matthews PC, Listgarten J, et al. . Additive contribution of HLA class I alleles in the immune control of HIV-1 infection. J Virol 2010; 84:9879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O’Brien SJ, Gao X, Carrington M. HLA and AIDS: a cautionary tale. Trends Mol Med 2001; 7:379–81. [DOI] [PubMed] [Google Scholar]
- 17. Mori M, Wichukchinda N, Miyahara R, et al. . HLA-B*35: 05 is a protective allele with a unique structure among HIV-1 CRF01_AE-infected Thais, in whom the B*57 frequency is low. AIDS 2014; 28:959–67. [DOI] [PubMed] [Google Scholar]
- 18. Mandelboim O, Reyburn HT, Sheu EG, et al. . The binding site of NK receptors on HLA-C molecules. Immunity 1997; 6:341–50. [DOI] [PubMed] [Google Scholar]
- 19. Mandelboim O, Reyburn HT, Valés-Gómez M, et al. . Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. J Exp Med 1996; 184:913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Winter CC, Gumperz JE, Parham P, Long EO, Wagtmann N. Direct binding and functional transfer of NK cell inhibitory receptors reveal novel patterns of HLA-C allotype recognition. J Immunol 1998; 161:571–7. [PubMed] [Google Scholar]
- 21. Braud VM, Allan DS, O’Callaghan CA, et al. . HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998; 391:795–9. [DOI] [PubMed] [Google Scholar]
- 22. Braud V, Jones EY, McMichael A. The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur J Immunol 1997; 27:1164–9. [DOI] [PubMed] [Google Scholar]
- 23. Merino AM, Sabbaj S, Easlick J, Goepfert P, Kaslow RA, Tang J. Dimorphic HLA-B signal peptides differentially influence HLA-E- and natural killer cell-mediated cytolysis of HIV-1-infected target cells. Clin Exp Immunol 2013; 174:414–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ulbrecht M, Martinozzi S, Grzeschik M, et al. . Cutting edge: the human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J Immunol 2000; 164:5019–22. [DOI] [PubMed] [Google Scholar]
- 25. Nattermann J, Nischalke HD, Hofmeister V, et al. . HIV-1 infection leads to increased HLA-E expression resulting in impaired function of natural killer cells. Antivir Ther 2005; 10:95–107. [DOI] [PubMed] [Google Scholar]
- 26. Sun C, Xu J, Huang Q, et al. . High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology 2017; 6:e1264562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wieten L, Mahaweni NM, Voorter CE, Bos GM, Tilanus MG. Clinical and immunological significance of HLA-E in stem cell transplantation and cancer. Tissue Antigens 2014; 84:523–35. [DOI] [PubMed] [Google Scholar]
- 28. Levy EM, Bianchini M, Von Euw EM, et al. . Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. Int J Oncol 2008; 32:633–41. [PubMed] [Google Scholar]
- 29. Kiepiela P, Ngumbela K, Thobakgale C, et al. . CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 2007; 13:46–53. [DOI] [PubMed] [Google Scholar]
- 30. Martin MP, Carrington M. KIR locus polymorphisms: genotyping and disease association analysis. Methods Mol Biol 2008; 415:49–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mori M, Wichukchinda N, Miyahara R, et al. . Impact of HLA allele-KIR pairs on disease outcome in HIV-infected Thai population. J Acquir Immune Defic Syndr 2018; 78:356–61. [DOI] [PubMed] [Google Scholar]
- 32. Flores-Villanueva PO, Yunis EJ, Delgado JC, et al. . Control of HIV-1 viremia and protection from AIDS are associated with HLA-Bw4 homozygosity. Proc Natl Acad Sci USA 2001; 98:5140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Altfeld M, Goulder P. ‘Unleashed’ natural killers hinder HIV. Nat Genet 2007; 39:708–10. [DOI] [PubMed] [Google Scholar]
- 34. Jennes W, Verheyden S, Demanet C, et al. . Cutting edge: resistance to HIV-1 infection among African female sex workers is associated with inhibitory KIR in the absence of their HLA ligands. J Immunol 2006; 177:6588–92. [DOI] [PubMed] [Google Scholar]
- 35. Mori M, Wichukchinda N, Miyahara R, et al. . The effect of KIR2D-HLA-C receptor-ligand interactions on clinical outcome in a HIV-1 CRF01_AE-infected Thai population. AIDS 2015; 29:1607–15. [DOI] [PubMed] [Google Scholar]
- 36. Paximadis M, Minevich G, Winchester R, et al. . KIR-HLA and maternal-infant HIV-1 transmission in sub-Saharan Africa. PLoS One 2011; 6:e16541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ravet S, Scott-Algara D, Bonnet E, et al. . Distinctive NK-cell receptor repertoires sustain high-level constitutive NK-cell activation in HIV-exposed uninfected individuals. Blood 2007; 109:4296–305. [DOI] [PubMed] [Google Scholar]
- 38. Khakoo SI, Thio CL, Martin MP, et al. . HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 2004; 305:872–4. [DOI] [PubMed] [Google Scholar]
- 39. Vince N, Bashirova AA, Lied A, et al. . HLA class I and KIR genes do not protect against HIV type 1 infection in highly exposed uninfected individuals with hemophilia A. J Infect Dis 2014; 210:1047–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. González-Galarza FF, Takeshita LY, Santos EJ, et al. . Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res 2015; 43:D784–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Apps R, Qi Y, Carlson JM, et al. . Influence of HLA-C expression level on HIV control. Science 2013; 340:87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.