

Abstract
The folding, stability, and oligomerization of helical membrane proteins depend in part on a precise set of packing interactions between transmembrane helices. To understand the energetic principles of these helix–helix interactions, we have used alanine-scanning mutagenesis and sedimentation equilibrium analytical ultracentrifugation to quantitatively examine the sequence dependence of the glycophorin A transmembrane helix dimerization. In all cases, we found that mutations to alanine at interface positions cost free energy of association. In contrast, mutations to alanine away from the dimer interface showed free energies of association that are insignificantly different from wild-type or are slightly stabilizing. Our study further revealed that the energy of association is not evenly distributed across the interface, but that there are several “hot spots” for interaction including both glycines participating in a GxxxG motif. Inspection of the NMR structure indicates that simple principles of protein–protein interactions can explain the changes in energy that are observed. A comparison of the dimer stability between different hydrophobic environments suggested that the hierarchy of stability for sequence variants is conserved. Together, these findings imply that the protein–protein interaction portion of the overall association energy may be separable from the contributions arising from protein–lipid and lipid–lipid energy terms. This idea is a conceptual simplification of the membrane protein folding problem and has implications for prediction and design.
Genome sequencing efforts reveal that approximately 20% of ORFs in complex organisms may encode proteins containing at least one helical transmembrane segment (1). Despite these numbers, as well as the fact that membrane proteins carry out many essential cell functions, our understanding of the sequence—structure–function relationships for this class of proteins lags far behind that of soluble proteins. These realities underscore the importance of biophysical and structural work aimed toward understanding chemical principles of helical membrane protein structural stability.
Because the phospholipid bilayer places structural constraints on a helical membrane protein, the folding of a polypeptide sequence into a helical membrane protein can be considered, experimentally and theoretically, in separable thermodynamic steps (2, 3). The usefulness of this framework arises from the fact that individual energetic processes can be independently studied. The principal features of a polypeptide sequence that will give rise to the formation of an independently stable transmembrane α-helix are generally known (3). This information has been used extensively in computational search algorithms with reasonable accuracy rates to identify potential helical transmembrane proteins (reviewed in ref. 3). Once this is accomplished, however, the helical membrane protein folding problem then becomes focused on understanding and predicting the side-to-side associations in which these preformed transmembrane α-helices will participate. It is this final thermodynamic step in helical membrane protein folding that we investigate in this study.
In a continuing effort to understand the structural and energetic principles of the side-to-side interactions of transmembrane α-helices, we have quantitatively examined the sequence dependence of the glycophorin A transmembrane helix dimerization. The propensity of the glycophorin A transmembrane domain to dimerize in a sequence-specific manner has been a paradigm for study of transmembrane helix–helix association in hydrophobic environments (4–9). An additional advantage for detailed thermodynamic analysis of the GpA transmembrane segment (TMS) dimerization is the fact that a solution NMR structure has been solved (10). Together with considerations of principles of stability of helices in membranes, the NMR structure provides a three-dimensional model for interpretation of potential structural consequences due to mutation.
Understanding the chemical principles driving the self-association of the glycophorin transmembrane α-helix is of particular interest because both the NMR structure and the exquisite sequence dependence determined by SDS/PAGE suggest that a detailed geometry of van der Waals interactions specify and stabilize the dimer (4, 8, 10). Only one residue with a polar side-chain, Thr87, is found at the dimer interface. The solution NMR structure of the glycophorin A transmembrane dimer in dodecylphosphocholine micelles reveals no interhelical hydrogen bond at this position (10), although the recent solid state NMR data from Smith and coworkers (11) hints that Thr87 might participate in an intermonomer hydrogen bond in lipid bilayers. Nevertheless, the energetic stabilization from such a hydrogen bond is uncertain. Recent studies on the introduction of polar side chains into model transmembrane peptides find that residues containing two polar side-chain atoms (such as asparagine) have a much greater tendency to drive transmembrane helix association than residues containing only one polar side-chain atom (threonine or serine; refs. 12 and 13). It has been proposed that side-chain rotamer entropy is not expected to play a large role in the self-association of the glycophorin A transmembrane α-helix (10). The interacting surface of the glycophorin A TMS contains only three residues with some rotamer freedom in an α-helix (Leu75, Ile76, and Thr87) according to the backbone dependent rotamer library of Dunbrack (14). Further, the side chain torsion angles in the glycophorin transmembrane domain are only slightly displaced from the ideal values of rotamers seen in helices (15–17). Thus, these side-chains do not undergo torsional strain on dimerization. Because the hydrophobic effect is thought to be minimal inside the phospholipid bilayer, the interaction of two helices composed primarily of apolar side-chain residues involves a balance of van der Waals interactions between the protein with itself and the protein with its hydrophobic environment. Here, we probe the energetics of the packing interactions involved in the glycophorin A transmembrane helix dimerization by examining the effect of single point mutations to alanine on the free energy of association.
Materials and Methods
Sample Preparation and Analytical Ultracentrifugation.
Single point mutations to alanine were generated by using the Stratagene QuickChange mutagenesis kit. The DNA sequences for all mutants were confirmed. SNGpA99 fusion proteins were purified as previously described in detail (8). Immediately before sedimentation equilibrium analysis, samples were exchanged by column chromatography into buffer containing pentaoxylethylene-octylether (C8E5) as described previously (8).
Sedimentation equilibrium experiments were performed at 25°C by using a Beckman XL-I analytical ultracentrifuge as described previously in (8, 18, 19). The samples were centrifuged in three-compartment carbon-epoxy centerpieces with quartz windows for lengths of time sufficient to achieve equilibrium. Data obtained from absorbance at 230 nm were analyzed by nonlinear least-squares curve fitting of radial concentration profiles by using the windows version of nonlin (20) using the equations describing the reversible association in sedimentation equilibrium. For each global fit, nine data sets were collected. These consisted of three different initial protein concentrations analyzed at three significantly different speeds (20,000, 24,500, and 30,000; i.e., such that the speed factor ratios were minimally 1.0, 1.5, and 2.25). The monomeric molecular masses and partial specific volumes were calculated by using the program sedinterp (21). The calculated values of these parameters were held constant in fitting the absorbance versus radius profiles.
Comparison of Stability to SDS/PAGE and Genetic Assay.
A regression analysis was used to compare the relative stability of sequence variants between the sedimentation equilibrium results and the previously published SDS/PAGE stability and genetic assay results. We quantified the SDS/PAGE “dot” scale given in Lemmon et al. (22) by assigning numeric scores using the previously defined phenotypes as a guide. The phenotypes of “wild-type like,” “significant dimer,” “detectable dimer,” and “no dimer” were assigned fractional dimer values of 0.90, 0.5, 0.25, and 0.05, respectively. These fraction dimers were then used in a regression analysis against fraction dimer observed in C8E5 to show that a linear function could describe the relationship between the two environments. A similar analysis was carried for the genetic data out by using the fraction “wild-type signal” from Brosig and Langosch (23) and from Russ and Engelman (24).
Results
Analytical Ultracentrifugation.
To determine the free energy effect on glycophorin A transmembrane helix dimerization, we carried out sedimentation equilibrium analysis of GpATM sequence variants in the non-ionic detergent C8E5. As previously described in detail (8), conducting the experiments under density-matching conditions essentially eliminated the contribution of bound detergent. In this manner, the sedimentation curves can be analyzed in terms of the buoyant molecular weight of the protein alone. In all cases, the simplest model to globally describe the nine sedimentation profiles we collected for each mutant contained an equilibrating monomer and dimer species. In addition, we found that a small amount (<10%) of nonequilibrating tetrameric species was usually present. Fig. 1 shows typical sedimentation equilibrium data for the Gly83Ala mutant analyzed by using the global fitting procedure.
Figure 1.

Sedimentation equilibrium of the Gly83Ala SNGpA99 mutant in C8E5 micelles. Three typical data sets are shown for the Gly83Ala protein analyzed by sedimentation equilibrium at 25°C in 20 mM sodium phosphate (pH 7.0), 200 mM NaCl, and 33 mM C8E5. These data represent one-third of the total amount of information used in the determination of the equilibrium constant for this protein by using the global fitting procedure.
Fig. 2 shows the computed fraction dimer as a function of total molar concentration observed during sedimentation equilibrium. The difference in the free energy of dimerization, relative to the wild-type sequence, is shown in Fig. 3 (see also Table 1, which is published as supporting information on the PNAS web site, www.pnas.org). In all cases, we find that mutation to alanine at interface positions costs free energy of association. In contrast, mutations to alanine away from the dimer interface show free energies of association that are insignificantly different from wild-type or are slightly stabilizing. Visual inspection of the NMR structure indicates that mutation to alanine at the dimer interface disrupts the detailed geometry of favorable packing interactions that specify and stabilize the dimer.
Figure 2.

The variation in the fraction dimer as a function of total concentration. Curves were calculated by using dissociation constants determined by global fitting of sedimentation equilibrium data.
Figure 3.

The differences in free energies of dimerization for each of the alanine mutants relative to the wild-type. Error bars represent proper propagation (33) of the standard deviation of the global sedimentation equilibrium fits for each protein. The error bar on the wild-type sample indicates the precision of this measurement determined by averaging the results from several independent experiments.
Comparison to SDS/PAGE.
The sequence-dependence of the glycophorin A transmembrane segment dimerization was initially characterized by SDS/PAGE (4). Because we now have a number of mutants whose association has been measured in a different hydrophobic environment, we sought to compare the relative stability for the sequence variants between the two environments. Fig. 4A shows a comparison of fraction dimer observed in C8E5 vs. SDS. We found a linear correlation between the hierarchy of stability for sequence variants between these two hydrophobic environments.
Figure 4.

Comparison of fraction dimer in C8E5 with other hydrophobic environments. (A) Comparison to fraction dimer observed in Lemmon et al. (4). (B) Comparison with fraction wild-type activity observed by Langosch et al. (25). The dispersion of the data in A may be due to the available precision of the scoring system derived from SDS/PAGE.
Comparison with Genetic Assay.
Two groups have used a genetic assay to quantify in vivo the relative stability of glycophorin A TMS sequence variants (24–26). Overall, the results of Langosch suggested that the glycophorin A TMS is more associated in vivo, because mildly disruptive mutants (according to our assay) do not show association values significantly different from wild-type under their conditions. However, the most disruptive mutants do differ significantly from wild-type in the biological assay. As shown in Fig. 4B, the dimer stability of these mutants (relative to the wild-type stability in this assay) are correlated with the relative stability that is measured by using sedimentation equilibrium in C8E5. The linear trend between the biological environment and the C8E5 environment in vitro is also found on comparison with the genetic data observed by Russ and Engelman (26) (data not shown).
Discussion
Previously, we described in detail a protocol for measurement of reversible association by using sedimentation equilibrium in detergent solutions. Here, we extend these studies to include single point mutations to alanine along the length of the glycophorin A TMS encompassing the dimerization interface. In all cases, we find that mutation to alanine at interface positions costs free energy of association. In contrast, mutations to alanine away from the dimer interface show free energies of association that are insignificantly different from wild type or are slightly stabilizing.
Interaction “Hot Spots.”
The interaction energy is not evenly distributed across the interface. Among the most dramatic changes in free energy of association are those seen by mutations to alanine at Gly79 and Gly83, which cost 1.7 and 3.2 kcal⋅mol−1, respectively, and which involve the introduction of a steric clash. It is of interest to find that these two glycines are energetic “hot spots”. This result is consistent with the recent genetic and statistical data suggesting that the GxxxG motif is a framework for transmembrane helix–helix association (24, 27). Occluded surface analysis of internal packing of helical membrane proteins also showed that glycine is among the residues with the highest packing values (28, 29), suggesting a special role for glycines in mediating helix–helix interactions.
According to the statistical analysis of sequence pairs in transmembrane helices (Senes et al., ref. 27), the glycophorin transmembrane domain contains 40 combinations with an odds ratio >1. Of these statistically over-represented sets of amino acid combinations, 34 involve the helix–helix interface residues that we show are destabilized on mutation to alanine. Two-thirds of these statistically over-represented sites (i.e., 23 of 34) involve residues for which we find that mutation to alanine costs greater than 1 kcal per mole (Leu75Ala, Ile76Ala, Gly79Ala, and Gly83Ala). Emphasizing further the potentially important role played by glycines in helix–helix interactions, it is noteworthy that 15 of these 34 over-represented sequence pairs that involve either Gly79Ala or Gly83Ala.
Structural Consequences.
Inspection of the NMR structure reveals that simple principles of protein–protein interactions can be hypothesized to explain the changes in energy that are observed (8, 30). The expectation was that mutation to alanine at interface positions should destabilize the dimer by disrupting the favorable packing interactions that specify the GpATM dimer interface. Energetically, this hypothesis is validated, because we find that all mutations at helix–helix interface positions are destabilizing. Examination of the individual sites suggests that the probable structural consequences for alanine substitutions can be explained by the introduction of steric clashes or by the removal of favorable van der Waals interactions. The introduction of a steric clash is expected to be the physical consequence at the glycine hot spots, Gly79 and Gly83. If steric clashes at these sites force the helices to move apart, then mutation at these sites may be compounded by a loss of favorable van der Waals interactions at other interface residues. In addition to the favorable energy provided by close packing, it may be that weak H-bonds between alpha carbon hydrogens and carbonyl groups are facilitated across the interface by the close approach of the backbones at the glycine positions (31). Such interactions would also be reduced by the addition of groups that force the helices apart, particularly the Gly to Ala substitutions. Mutation to alanine at all other sites can be accommodated by the helices without large backbone movements and can be rationalized as a loss of favorable packing interactions.
These results suggest that the optimized detailed geometry of packing found in the wild-type glycophorin A TMS dimer can involve at least as much energy as is seen in model transmembrane peptides driven to associate by the introduction of polar side-chains (12). Our observation that mutation to alanine at sites away from the interface revealed essentially no effect on the free energy of association is consistent with previous studies, which showed that mutation of all non-motif residues to leucine has no effect in SDS/PAGE (32) or in genetic assays (26).
Role of the Hydrophobic Environment.
With quantitative energetic information on a set of point mutants in hand, we sought to address the role played by the hydrophobic environment in the dimerization of glycophorin A TMS. The association equilibrium constant for the wild-type sequence in three different detergent environments has been measured by fluorescence resonance energy transfer techniques (9). This study showed that the hydrophobic environment can influence the overall association level by ≈2 kcal⋅mol−1. The SDS micelle environment was found to be the most destabilizing to the wild-type dimerization. This effect can be understood by consideration of the energetic terms contributing to the observed overall free energy of association of transmembrane α-helices in a given hydrophobic environment. This free energy can be expressed (very simply) as:
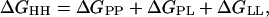 |
1 |
where the term lipid here is used to indicate the hydrophobic environment in a general sense, ΔGHH indicates the observed free energy of transmembrane helix association for a given set of conditions, ΔGPP indicates the contribution from protein–protein interactions, ΔGPL indicates the contribution from protein–lipid interactions, and ΔGLL indicates the contribution from lipid–lipid (or detergent-detergent) interactions.
Even though we anticipated that the overall level of dimerization would be reduced in SDS (4) vs. C8E5, we asked the question of whether or not sequence variants affect the stability differently in SDS compared with C8E5. We carried this out by comparing the fraction dimer for sequence variants observed on SDS/PAGE with the fraction dimer observed in C8E5 by sedimentation equilibrium. Shown in Fig. 4A, this analysis reveals a linear correlation of fraction dimer for the sequence variants between the two environments. This finding suggests that the hierarchy of stability of sequence variants is conserved between the two environments: i.e., the wild-type sequence defines the most stable dimer in both environments, the Gly83Ala mutant defines the least stable dimer in both environments, and other sequence variants fall into line with intermediate values.
We further compared the relative stability for sequence variants measured in C8E5 with those measured in biological membranes by using a genetic assay. Two groups have carried out genetic assays of association by using glycophorin A TMS variants. The largest amount of data is available for the study by Langosch and coworkers (25), and a comparison of fraction dimer between their study and the current one is shown in Fig. 4B. The linear trend of conservation of hierarchy of stability persists in this analysis. This result is quite important because it shows that our measurements carried out in vitro reflect the potential for association in vivo.
The fact that we observed a conserved hierarchy of stability between three chemically different hydrophobic environments suggests the idea that the protein–protein portion of the overall energy term may be experimentally separable from the protein–lipid and lipid–lipid energy terms. This result validates the separation of the ΔGPP term from the other terms in Eq. 1. The notion that we can explain the changes in free energy as a function of sequence principally in terms of ΔGPP is further supported by visual inspection of the NMR structure. No explicit hydrophobic solvent is observed in the NMR structure, where only simple principles of protein–protein interaction need to be invoked to explain the change in energy as a function of sequence.
This separation of energy terms would reflect a conceptual simplification of the membrane protein-folding problem and would have implications for prediction and design. The potential payoff may be great because this class of proteins have historically been excellent targets for therapeutics. Advances in our ability to understand and manipulate membrane proteins may lead to the discovery or design of pharmaceutical agents that can modulate their functions.
Supplementary Material
Acknowledgments
We thank Yang Chen and Ellen Hibbard for technical assistance. This work was supported by National Institutes of Health Grants GM57534 (to K.G.F.) and GM54160 (to D.M.E.).
Abbreviations
- TMS
transmembrane segment
- C8E5
pentaoxylethylene-octylether
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Liu J, Rost B. Protein Sci. 2001;10:1970–1979. doi: 10.1110/ps.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Popot J-L, Engelman D M. Biochemistry. 1990;29:4032–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 3.White S H, Wimley W C. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 4.Lemmon M A, Flanagan J M, Treutlein H R, Zhang J, Engelman D M. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 5.Mingarro I, Elofsson A, von Heijne G. J Mol Biol. 1997;272:633–641. doi: 10.1006/jmbi.1997.1276. [DOI] [PubMed] [Google Scholar]
- 6.Bu Z, Engelman D M. Biophys J. 1999;77:1064–1073. doi: 10.1016/S0006-3495(99)76956-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adair B D, Engelman D M. Biochemistry. 1994;33:5539–5544. doi: 10.1021/bi00184a024. [DOI] [PubMed] [Google Scholar]
- 8.Fleming K G, Ackerman A L, Engelman D M. J Mol Biol. 1997;272:266–275. doi: 10.1006/jmbi.1997.1236. [DOI] [PubMed] [Google Scholar]
- 9.Fisher L E, Engelman D, Sturgis J. J Mol Biol. 1999;293:639–651. doi: 10.1006/jmbi.1999.3126. [DOI] [PubMed] [Google Scholar]
- 10.MacKenzie K R, Prestegard J H, Engelman D M. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 11.Smith S O, Song D, Shekar S, Groesbeek M, Ziliox M, Aimoto S. Biochemistry. 2001;40:6553–6558. doi: 10.1021/bi010357v. [DOI] [PubMed] [Google Scholar]
- 12.Gratkowski H, Lear J D, DeGrado W F. Proc Natl Acad Sci USA. 2001;98:880–885. doi: 10.1073/pnas.98.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou F X, Merianos H J, Brunger A T, Engelman D M. Proc Natl Acad Sci USA. 2001;98:2250–2255. doi: 10.1073/pnas.041593698. . (First Published February 13, 2001; 10.1073/pnas.041593698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunbrack R L, Karplus M. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 15.Dunbrack R L, Cohen F E. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacKenzie K R. Molecular Biophysics and Biochemistry. New Haven, CT: Yale Univ. Press; 1996. pp. 1–349. [Google Scholar]
- 17.Ponder J W, Richards F M. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 18.Fleming K G. In: Chemtracts: Biological Applications of the Analytical Ultracentrifuge. Hanson J C, editor. Vol. 11. New York: Springer; 1998. pp. 985–990. [Google Scholar]
- 19.Fleming K G. Methods Enzymol. 2000;323:63–77. doi: 10.1016/s0076-6879(00)23361-2. [DOI] [PubMed] [Google Scholar]
- 20.Johnson M L, Correia J J, Yphantis D A, Halvorson H R. Biophys J. 1981;36:575–588. doi: 10.1016/S0006-3495(81)84753-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laue T M, Shah B, Ridgeway T M, Pelletier S L. In: Analytical Ultracentrifugation in Biochemistry and Polymer Science. Harding S E, Rowe A J, Horton J C, editors. Cambridge, U.K.: R. Soc. Chem.; 1992. pp. 90–125. [Google Scholar]
- 22.Lemmon M A, Flanagan J M, Hunt J F, Adair B D, Bormann B J, Dempsey C E, Engelman D M. J Biol Chem. 1992;267:7683–7689. [PubMed] [Google Scholar]
- 23.Brosig B, Langosch D. Protein Sci. 1998;7:1052–1056. doi: 10.1002/pro.5560070423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russ W P, Engelman D M. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 25.Langosch D, Brosig B, Kolmar H, Fritz H-J. J Mol Biol. 1996;263:525–530. doi: 10.1006/jmbi.1996.0595. [DOI] [PubMed] [Google Scholar]
- 26.Russ W P, Engelman D M. Proc Natl Acad Sci USA. 1999;96:863–868. doi: 10.1073/pnas.96.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Senes A, Gerstein M, Engelman D M. J Mol Biol. 2000;296:921–936. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- 28.Eilers M, Shekar S C, Shieh T, Smith S O, Fleming P J. Proc Natl Acad Sci USA. 2000;97:5796–5801. doi: 10.1073/pnas.97.11.5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Javadpour M M, Eilers M, Groesbeek M, Smith S O. Biophys J. 1999;77:1609–1618. doi: 10.1016/S0006-3495(99)77009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacKenzie K R, Engelman D M. Proc Natl Acad Sci USA. 1998;95:3583–3590. doi: 10.1073/pnas.95.7.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Senes A, Ubarretxena-Belandia I, Engelman D M. Proc Natl Acad Sci USA. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemmon M A, Treutlein H R, Adams P D, Brunger A T, Engelman D M. Nat Struct Biol. 1994;1:157–163. doi: 10.1038/nsb0394-157. [DOI] [PubMed] [Google Scholar]
- 33.Bevington P R. Data Reduction and Error Analysis for the Physical Sciences. New York: McGraw–Hill; 1969. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.