

Abstract
The intrinsic rate of viral replication in HIV-infected patients treated with antiretroviral combination therapy is estimated by using a mathematical model of viral dynamics. This intrinsic replication is found to be episodic, varying considerably in quantity between patients (even among those achieving long-term undetectable levels of viremia) and is always reduced by increasing the potency of the antiviral drug regimen. The analysis reveals that even in conditions of perfect patient adherence and drug penetration a substantial level of residual viral replication is expected. The rate of evolution in the viral quasispecies, and thus also the probability of new drug-resistant viral strains being created, is proportional to the total amount of residual viral replication. Under most circumstances, the viral population continues to turn over rapidly during therapy, albeit at a much reduced level.
Keywords: antiviral therapy|residual replication|mathematical model
Treatment with combinations of antiretroviral compounds that inhibit viral replication is currently the main therapy used to slow progression to serious disease in HIV-infected patients. With the current available drugs, residual viral replication is often observed in otherwise successfully treated patients who show no overt signs of disease (1–4). In this study we quantify the capacity for HIV to replicate in treated patients, using a mathematical model to describe viral dynamics that generates predictions consistent with observed changes in viral load and CD4 abundance seen under therapy (5–10) and long-term trends in HIV pathogenesis (11). Residual replication is shown to be an intrinsic and expected feature of viral dynamics, where its episodic burst-like nature is found to reflect random patterns of host exposure to antigens that drive T cell activation. We quantify viral replication during therapy by using a dynamic measure: the average number of cells newly infected by HIV per day, which is proportional to the probability of emergence of viral resistance de novo. A wide range of residual replication rates are shown to be consistent with the apparently successful suppression of viral load, indicating that the viral population can be expected to continue to evolve even under therapy.
Methods
The Model.
The model of viral dynamics is defined by eight coupled ordinary differential equations:
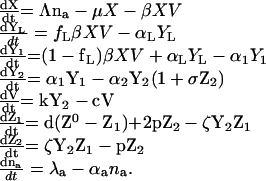 |
1 |
These equations describe changes over time in the number X of target cells, YL of long-lived infected cells, Y1 of early-stage infected cells, Y2 of late-stage infected cells [producing virus and susceptible to cytotoxic T lymphocyte (CTL) lysis], V of free virions, Z1 of resting HIV-specific CTLs, Z2 of activated anti-HIV CTLs, and finally na of distinct (HIV and non-HIV) antigens that activate CD4 cells. This model has 17 independent parameters, too many to simultaneously estimate by fitting to viral load decay curves, as in refs. 5–8. The parameters and their estimates or assumed values are presented in Table 1. This model builds on earlier related studies (11) and is consistent with long-term observed trends of HIV immunopathogenesis. It provides a realistic description of the viral set-point and its relation to disease progression rates and to the magnitude of the anti-HIV CTL response. Both deterministic and stochastic versions of the model were used in this analysis.
Table 1.
The parameters of the model
| Symbol/ equation | Description | Value | Unit | Comments/ref(s). |
|---|---|---|---|---|
| Λ | Cell activation per antigenic exposure | 2.5 × 107 | Cells per day | Approx 1:1000 cells activated per day is consistent with pathogenesis (11) |
| μ | Cell division rate | 1 | Per day | Activated CD4 cell takes approx. 1 day to divide |
| β | Viral infectivity | 4 × 10−10 | Per virion per cell per day | Derived from R0 below |
| fL | Fraction of infections resulting in a long-lived infected cell | 10−5 | Per infection | Chosen consistent with known first phase–second phase transitions in viral load decay curves after start of treatment (7) |
| αL | Rate of latent infected cell reactivation | 0.001 | Per day | Fastest observed long-term decay rate (2, 30) |
| α1 | Rate of progression from cell infection to viral expression and production | 1 | Per day | (16) |
| α2 | Death rate of productively infected cell | 1 | Per day | (16) |
| σ | Increase in death rate of productively infected cells caused by CTL activity | 10−5 | Per activated anti- HIV CTL per day | Chosen to match observed frequency of anti-HIV CTLs (15) |
| k/c | Ratio of free virus to infected cell numbers | 1,000 | Virions per cell | Conservative estimate based on ref. 32 |
| c | Clearance rate of free virus | 25 | Per day | Estimated from plasma apheresis experiment (33); precise value unimportant provided fast enough to decouple from model |
| p | Proliferation rate of activated anti-HIV CTLs | 1 | Per day | |
| d | Death rate of anti-HIV CTLs | 0.01 | Per day | (34) |
| λa | Rate of exposure to new antigens | 0.3334 | Per day | (11) |
| αa | Rate of antigen clearance | 0.03334 | Per day | Corresponding to an average of 10 concurrent distinct antigens, cleared in 30 days (11) |
| 〈R0〉 | Average basic reproduction number of HIV | 100 | Number | Higher than lower bound calculated in ref. 19, consistent with pathogenesis (11) |
| NPB | Factor used to convert figures from 1 ml of peripheral blood to whole body | 2.5 × 105 | Volume | (6) |
| ζ | Rate of CTL activation per productive infected cell | (i) 1.3334 × 10−8 (ii) 1.3334 × 10−9 (iii) 1.3334 × 10−10 | Per CTL per infected cell per day | Three example values determine set-point viral load |
| Steady-state values calculated from the model (for the pretreatment set-point) | ||||
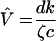 |
Viral load | (i) 7.5 × 108 (ii) 7.5 × 109 (iii) 7.5 × 1010 | Virions | Gives 3,000, 30,000, and 300,000 virions per ml of peripheral blood, chosen as low, median, and high baseline viral load (28) |
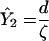 |
Number of productively infected cells | (i) 7.5 × 105 (ii) 7.5 × 106 (iii) 7.5 × 107 | Cells | |
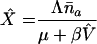 |
Number of susceptible cells | (i) 1.92 × 108 (ii) 6.25 × 107 (iii) 8.06 × 106 | Cells | |
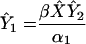 |
Number of early-stage infected cells | (i) 5.77 × 107 (ii) 1.88 × 108 (iii) 2.42 × 108 | Cells | |
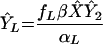 |
Number of long-lived infected cells | (i) 5.77 × 105 (ii) 1.88 × 106 (iii) 2.42 × 106 | Cells | Matches direct estimates (29) |
| Ẑ1 + Ẑ2 | Number of anti-HIV CTLs | (i) 7.87 × 108 (ii) 2.63 × 108 (iii) 4.27 × 107 | Cells | Matches direct estimates (15) |
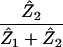 |
Percentage of CTLs activated | (i) 1% (ii) 1% (iii) 1% | Percent | |
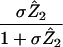 |
Percentage of infected cells killed by CTLs | (i) 98.7% (ii) 96.3% (iii) 80.9% | Percent | |
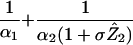 |
Average lifetime of an infected cell | (i) 1.01 (ii) 1.04 (iii) 1.19 | Days | |
| βX̂Ŷ2 | Number of new cells infected per day | (i) 5.77 × 107 (ii) 1.88 × 108 (iii) 2.42 × 108 | Infections per day | To be compared to same quantity calculated after therapy in Fig. 1 |
(i), (ii), and (iii) refer to parameters for patients with low, median, and high viral loads, respectively.
The first departure from the classic models of HIV dynamics (reviewed in ref. 8) is the explicit description of antigen driving the activation of susceptible target CD4+ T cells, and thus driving viral replication. Previous studies of viral treatment have usually described the CD4 pool as a uniform passive target for HIV [although the role of changing activation has been explored (10)]. This is unsatisfactory because HIV replicates primarily in activated cells (12, 13) that make up only a small but changing fraction of the CD4 pool. We choose the simplest possible description of antigen dynamics: antigens eliciting a new CD4 response are encountered by the patient at a Poisson rate λa and are then cleared at rate αa. Each new antigen activates CD4 cells at rate Λ. Because the average number of concurrent antigens is small (= λa/αa), it is important to model random variations in na over time: the long-term variance in na is given by its mean (λa/αa). These variations directly change the number of HIV target cells, affecting the efficacy of the whole viral replication cycle.
The second departure from published models is the decomposition of the interaction between CTLs and infected cells into multiple stages. We model CTL activation by viral antigens as preceding cell division, which occurs at a fixed rate p, and we assume that only activated CTLs can kill infected cells. Dynamically, such a model has several advantages. First, this response explains the large (1000-fold) variation in viral set-point between patients as a result of differences in the ability of CTLs to activate in response to viral antigens, rather than resulting from 1000-fold differences in CTL proliferation or death rates, or even 1000-fold differences in the availability of target cells. The model could be further increased in realism by allowing variation in multiple parameters to explain variations in viral load (as in ref. 14). Second, CTL proliferation is naturally rate limited, with a maximal doubling time of ln(2)/p, independent of other parameters. Third, the observed dynamics are stable, unlike previous models (8) that can exhibit prolonged oscillations in viral and cellular population sizes. Fourth, as seen in Table 1, the model predicts the correct inverse relation between the frequency of anti-HIV CTLs and set-point viral load observed in ref. 15. As highlighted in ref. 16, the division of the infected cell pool into subpopulations of early-stage (previral expression and production) and late-stage infected cells provides a natural explanation for the relative uniformity in infected cell lifetimes inferred from posttherapy decay curves (5, 6).
The equations are integrated in fixed time steps of Δt = 0.01 day. For the stochastic model, na is increased in each time step by a Poisson deviate of mean λaΔt, and reduced by binomial deviate with probability αaΔt and sample size na. For the deterministic model na is fixed = λa/αa. The other equations are integrated by using a fourth-order Runge–Kutta algorithm, assuming fixed na over the interval (17).
The Basic Reproduction Number.
The basic reproduction number is the number of new cells that an infected cell infects before it dies, in the absence of density-dependent constraints (8). For this model, it is approximately given by
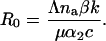 |
2 |
It is a function of the number of distinct antigens na and so fluctuates over time.
The average value of R0 in the stochastic model, denoted <R0>, coincides with the value of R0 in the deterministic model, and is given by
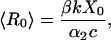 |
3 |
where X0 = Λλa/αa is the average number of susceptible target cells in the absence of HIV infection.
Steady States of the Deterministic Model.
The deterministic model has three equilibria. In the equations below, we approximate Z0 ≈ 0. The uninfected equilibrium is given by
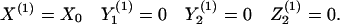 |
4 |
There are two infected equilibria. The first has no sustained CTL response, and is given by
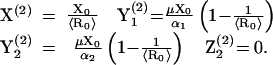 |
5 |
The second infected equilibrium is associated with a stable effective anti-HIV CTL response, and is given by
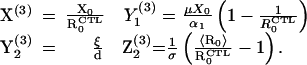 |
6 |
In the above, we define R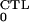 to be the
CTL-restricted reproduction number, as the number of secondary
infections caused by an infected cell before it dies, in the presence
of a mature CTL response. It is defined by analogy with
R0 and is given by
to be the
CTL-restricted reproduction number, as the number of secondary
infections caused by an infected cell before it dies, in the presence
of a mature CTL response. It is defined by analogy with
R0 and is given by
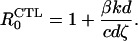 |
7 |
For all equilibria, the remaining densities are given by
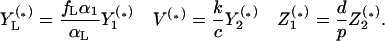 |
8 |
The uninfected steady state, defined in Eq. 4, is
stable if and only if 〈R0〉 < 1.
The CTL-free infected steady state, defined in Eq. 5, is
stable if and only if R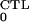 >
〈R0〉 > 1. The CTL-restricted
steady state, defined in Eq. 6, is stable if and only if
〈R0〉 >
R
>
〈R0〉 > 1. The CTL-restricted
steady state, defined in Eq. 6, is stable if and only if
〈R0〉 >
R > 1. The parameters that define the
boundaries between these stable states are threshold values.
> 1. The parameters that define the
boundaries between these stable states are threshold values.
Calculation of the Threshold Treatment Efficacies.
For the parameters we use (defined in Table 1), before antiviral treatment, the patients are in the CTL-restricted steady-state defined by Eq. 6. The effect of antiviral therapy is to reduce the ratio βk/c to (βk/c)(1 − ɛ), where ɛ is the treatment efficacy (7, 18); the thresholds separating equilibrium 6 from 5 and 5 from 4 are crossed sequentially with increasing ɛ. The threshold efficacies are given by
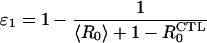 |
9 |
for the first crossing and
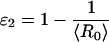 |
10 |
for the second.
The third threshold discussed arises from the reduction in the replenishment of the resting cell pool attributable to activated CD4 cells being destroyed as a result of HIV replication. The replenishment rate is proportional to the number of susceptible cells X(*), and thus it increases as treatment efficacy increases. If pS is the probability that an activated cell will successfully divide, then the threshold treatment efficacy is
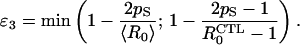 |
11 |
Stochastic effects do not remove, but rather blur, these threshold effects.
Results
The key quantity that captures the capacity for viral replication within the patient is the basic reproduction number, R0, defined as the average number of secondary cell infections caused by the introduction of a single infected cell into a susceptible person (8, 19). The definition and derivation of R0, as well as the underlying biological assumptions incorporated into a mathematical model of HIV replication, are presented in Methods. An important feature of this model is that R0 is not fixed, but varies as a function of the rate of activation of CD4 cells; this in turn reflects the temporal pattern of exposure to HIV and non-HIV antigens. The extent to which antiretroviral therapy inhibits viral replication is simply the factor by which it reduces the magnitude of R0 (7, 18), thus providing both a precise definition for treatment efficacy and a measurement tool.
The long-term outcome of therapy depends crucially on the magnitude of the reduced value of R0 because this relates to the net rate of evolution of the virus. The dependence between the magnitude of R0 (determined by treatment efficacy) and the long-term outcome of treatment is predicted to be more complex than suggested by previous theoretical studies of HIV dynamics (20), in accord with observed patterns (9). In Methods, we describe how our model can be formulated as deterministic or stochastic, corresponding respectively to a fixed or a randomly fluctuating level of antigenic stimulation and subsequent T cell activation. We start by describing the deterministic model because it is simpler: antigenic stimulation, T cell activation, and thus R0 are all constant over time. We observe four qualitatively different outcomes to antiviral therapy, separated by three threshold values of treatment efficacy (derived in Methods). The short-term effect is always a reduction in viral load. The distinct dynamical regimes are as follows:
1. The short-term effect of low-efficacy treatment is to reduce the number of anti-HIV CTLs in circulation caused by a transient reduction in viral load. In the long term, viral load returns to its pretreatment value, whereas the number of CTLs remains reduced. The long-term effect of therapy is only a modest reduction in viral load because of the tradeoff between drug action and reduced CTL action.
2. As treatment efficacy is increased and R0 is reduced, a CTL stimulation threshold is crossed and the efficacy of the CTL response declines because of lower rates of activation caused by the reduced viral population (21).
3. At a second threshold, the probability that a CD4 cell can successfully divide without being either infected or suffering activation-related death reaches 0.5. Above this value therapy causes a change in viral load to a new and lower set-point but no continuing decay in viral load. However, the long-term depletion of CD4 cells is reversed (11), so overall this scenario represents a discordant response to therapy.
4. The third threshold is achieved when R0 falls below unity in value: below this value viral replication becomes unsustainable and a rapid biphasic decay in viral load is observed (7).
The order on which the thresholds are crossed depends on the parameter values, but is always either 1–2-3–4 or 1–3-2–4.
The effect of random variations in the number of antigens that are activating the CD4 cell pool is to cause R0, and concomitantly viral load, to fluctuate. When treatment efficacy is close to a threshold value, this introduces a degree of intermittency to the viral dynamics as the system bounces below and above the threshold, as seen in the viral load decay curves plotted in Fig. 1. Random antigenic exposures can therefore temporarily shift the patient back and forth between the states of viral decay and viral growth. It is thus a prediction of the model that short episodes of sustained viral replication are an intrinsic part of the dynamics of antiretroviral treatment. The duration of these intermittent episodes is determined by the average time taken to clear a typical antigen (given by 1/αa in the model: see Methods). The magnitude and frequency of these episodes is predicted to decrease with increasing treatment efficacy (Fig. 1), as clinically observed.
Figure 1.

Residual viral replication in treated HIV-positive patients. The figure illustrates results from the simulation for three sets of patients with high, median, and low set-point viral loads (300,000, 30,000, and 3,000 copies per ml in the Top, Middle, and Bottom graphs, respectively). The graphs to the Left show typical outcomes for a range of different treatment efficacies, as measured by the posttherapy average basic reproduction number R0. The dashed line indicates a limit of detection of 5 copies per ml. The graphs in the Center and Right columns illustrate the outcome of 1,000 separate simulations for 100 values of R0. For each simulation the average number of residual infections is recorded during the first 50 days (Center) and the subsequent 250 days (Right). The color of the graph illustrates the relative frequency of each outcome, as scaled by the color bar on the right. The dotted black line is the prediction calculated from the deterministic model in which antigenic exposure is assumed constant rather than random. The white line on these graphs is the fraction of the 1,000 simulated patients that reach levels of virus <5 copies per ml, after 50 days in the Center graphs, and after 24 weeks in the graphs to the Right.
The model was further used to quantify the amount of residual replication expected in patients where treatment efficacy is on average sufficient to render viral replication unsustainable (R0 < 1 on average). Two measures were estimated: one for the first 50 days of treatment, and one for the subsequent 250 days. This approach was adopted because a high rate of target cell infection is expected in the early parts of treatment when viral loads are still high, and a low rate once treatment effectively suppresses viral load. The measure of viral replication used was the average number of cells infected by HIV per day, as plotted in Fig. 1, as a function of the value of the average posttherapy R0. Because the average pretherapy R0 is fixed in the simulation, posttherapy R0 and treatment efficacy are interchangeable: the lower R0, the higher the efficacy. The simulation was repeated 1,000 times for 100 different values of R0, separately for patients with high, median, or low set-point viral loads (as defined in Table 1). Comparative values are given for the deterministic model to illustrate the effect of intermittency on residual replication. The predicted fraction of patients achieving undetectable levels of virus after 50 days and after 24 weeks [gauged at <5 copies per ml of peripheral blood, in line with current ultrasensitive assays (7)], are also plotted in Fig. 1.
The analysis shows that for high-efficacy drug regimens, there is considerably more residual replication in the early part of treatment. For marginally effective treatments (R0 close to 1), antigen-driven intermittency was found to increase long-term residual replication substantially above that predicted by the deterministic model, up to levels equivalent to the first 50 days of treatment, and the number of patients failing to reach undetectable levels of virus dropped accordingly (independent of other factors such as poor compliance or the emergence of viral resistance to treatment). Outside this zone, the outcomes of the simulation are centered on the prediction of the deterministic model, thus randomly varying antigenic exposure does not in general predict more residual replication to occur than the model where this is constant. In identical simulations, a minimum 2-fold variation in the average number of cells newly infected per day was found.
The measure used was approximately equal to the number of new mutations generated in the viral population, and thus also to the probability of emergence of de novo drug resistance (8, 22, 23). This probability is expected to relate to the likelihood of treatment failure in the long term (20). The variation reflects the randomness of antigenic encounters in simulated patients with identical parameters. A millionfold variation in viral replication was observed in the range 0.1 < R0 < 1 (corresponding to 0.9% variation in treatment efficacy). Even when 100% of the patients are predicted to reach undetectable viral loads (R0 < ≈0.5), more than a hundredfold variation in residual viral replication was observed. The degree of residual replication was found to be intrinsically variable, but is always reduced by increasing treatment efficacy.
Discussion
Persistent viral replication, even at low levels, compromises the long-term success of therapy by providing opportunities for the evolution of drug-resistant variants of the virus (the net rate of evolution is directly proportional to the net rate of viral replication) and concomitant treatment failure. There are many factors that can cause residual replication in treated HIV-infected patients (24). Poor adherence of patients to treatment protocols has been shown to be predictive of drug failure (25). Cellular reservoirs or physiological compartments (such as the central nervous system) where drug penetration is poor have also been implicated (24). Whereas the reality of in vivo treatment is multifactorial, the use of a mathematical model allows us to separately analyze and dissect these factors, performing a “virtual experiment” to estimate how much residual replication is intrinsic to the stochastic dynamics of viral replication and antigenic exposure. The estimates are subject to the caveat that they extrapolate information from peripheral blood measurements to the whole body, whereas there is an additional contribution to viral decay and diversity from virus attached to follicular dendritic cells in lymphatic tissues (26).
The mathematical model is founded on a substantial experimental, observational, and theoretical literature (recently reviewed in ref. 8). The model captures both the role of antigenic exposure in regulating viral replication by limiting the number of available target cells for HIV to those that are in the dividing stage (12), and the impact of an active CTL response (27). It predicts a pattern of intermittency in viral replication during therapy, and thus offers an explanation for the episodic nature of viral replication in patients known to adhere well to treatment regimens (4). The model also helps explain the wide variations in pretreatment viral load seen between patients (28). The description of a multistage model (i.e., resting and activated HIV antigen-specific cells) to describe the anti-HIV CTL response generates predictions consistent both with long-term trends in HIV pathogenesis (11) and with the relative constancy in infected cell lifetimes (16).
Several recent studies have found evidence for active viral transcription in patients where viral load is successfully suppressed (1–3). Such residual replication is thought to replenish stocks of long-lived infected CD4 cells (4), which are held responsible for long-term viral persistence in infected patients (29, 30). The current study shows unambiguously that a substantial amount of residual viral replication is an intrinsic feature of HIV dynamics during antiretroviral therapy, and that the viral population continues to turn over rapidly, albeit at a much-reduced net rate. Even when currently undetectable levels of virus are reached, there remains scope to reduce residual replication by intensifying the treatment [e.g., five-drug regimens (7)]. Intensifying treatment by using drugs with better pharmacodynamic properties (i.e., a greater ability to suppress viral replication and longer half-lives) and attempting to minimize poor patient adherence by education and counseling is essential to reduce current rates of treatment failure (31). However, a recent re-appraisal of pharmacodynamic measures of drug efficacy indicates that complete blocking of viral replication by the combinations of the currently available drugs is more difficult than previously thought (18).
Acknowledgments
C.F. and R.M.A. thank the Wellcome Trust for funding and N.M.F. thanks the Royal Society.
Abbreviation
- CTL
cytotoxic T lymphocyte
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Furtado M R, Callaway D S, Phair J P, Kunstman K J, Stanton J L, Macken C A, Perelson A S, Wolinsky S M. N Engl J Med. 1999;340:1614–1622. doi: 10.1056/NEJM199905273402102. [DOI] [PubMed] [Google Scholar]
- 2.Zhang L, Ramratnam B, Tenner-Racz K, He Y, Vesanen M, Lewin S, Talal A, Racz P, Perelson A S, Korber B T, et al. N Engl J Med. 1999;340:1605–1613. doi: 10.1056/NEJM199905273402101. [DOI] [PubMed] [Google Scholar]
- 3.Sharkey M E, Teo I, Greenough T, Sharova N, Luzuriaga K, Sullivan J L, Bucy R P, Kostrikis L G, Haase A, Veryard C, et al. Nat Med. 2000;6:76–81. doi: 10.1038/71569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramratnam B, Mittler J E, Zhang L Q, Boden D, Hurley A, Fang F, Macken C A, Perelson A S, Markowitz M, Ho D D. Nat Med. 2000;6:82–85. doi: 10.1038/71577. [DOI] [PubMed] [Google Scholar]
- 5.Wei X P, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, et al. Nature (London) 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 6.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Nature (London) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 7.Ferguson N M, de Wolf F, Ghani A C, Fraser C, Donnelly C A, Reiss P, Lange J M A, Danner S A, Garnett G P, Goudsmit J, Anderson R M. Proc Natl Acad Sci USA. 1999;96:15167–15172. doi: 10.1073/pnas.96.26.15167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nowak M A, May R M. Virus Dynamics: Mathematical Principles of Immunology and Virology. Oxford: Oxford Univ. Press; 2000. [Google Scholar]
- 9.Kaufmann D, Pantaleo G, Sudre P, Telenti A. Lancet. 1998;351:723–724. doi: 10.1016/s0140-6736(98)24010-4. [DOI] [PubMed] [Google Scholar]
- 10.Grossman Z, Polis M, Feinberg M B, Levi I, Jankelevich S, Yarchoan R, Boon J, de Wolf F, Lange J M A, Goudsmit J, et al. Nat Med. 1999;5:1099–1104. doi: 10.1038/13410. [DOI] [PubMed] [Google Scholar]
- 11.Fraser C, Ferguson N M, de Wolf F, Anderson R M. Proc R Soc London Ser B. 2001;268:2085–2095. doi: 10.1098/rspb.2001.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevenson M, Stanwick T L, Dempsey M P, Lamonica C A. EMBO J. 1990;9:1551–1560. doi: 10.1002/j.1460-2075.1990.tb08274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orendi J M, Bloem A C, Borleffs J C C, Wijnholds F J, deVos N M, Nottet H S L M, Visser M R, Snippe H, Verhoef J, Boucher C A B. J Infect Dis. 1998;178:1279–1287. doi: 10.1086/314451. [DOI] [PubMed] [Google Scholar]
- 14.Muller V, Maree A F M, De Boer R J. Proc R Soc London Ser B. 2001;268:235–242. doi: 10.1098/rspb.2000.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogg G S, Jin X, Bonhoeffer S, Dunbar P R, Nowak M A, Monard S, Segal J P, Cao Y Z, Rowland-Jones S L, Cerundolo V, et al. Science. 1998;279:2103–2106. doi: 10.1126/science.279.5359.2103. [DOI] [PubMed] [Google Scholar]
- 16.Klenerman P, Phillips R E, Rinaldo C R, Wahl L M, Ogg G, May R M, McMichael A J, Nowak M A. Proc Natl Acad Sci USA. 1996;93:15323–15328. doi: 10.1073/pnas.93.26.15323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Press W H, Teukolsky S A, Vetterling W T, Flannery B P. Numerical Recipes in C: The Art of Scientific Computing. Cambridge, U.K.: Cambridge Univ. Press; 1992. [Google Scholar]
- 18.Ferguson N M, Fraser C, Anderson R M. Trends Pharmacol Sci. 2001;22:97–100. doi: 10.1016/s0165-6147(00)01615-1. [DOI] [PubMed] [Google Scholar]
- 19.Little S J, McLean A R, Spina C A, Richman D D, Havlir D V. J Exp Med. 1999;190:841–850. doi: 10.1084/jem.190.6.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonhoeffer S, May R M, Shaw G M, Nowak M A. Proc Natl Acad Sci USA. 1997;94:6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wodarz D, May R M, Nowak M A. Int Immunol. 2000;12:467–477. doi: 10.1093/intimm/12.4.467. [DOI] [PubMed] [Google Scholar]
- 22.Nowak M A, Anderson R M, McLean A R, Wolfs T F W, Goudsmit J, May R M. Science. 1991;254:963–969. doi: 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]
- 23.Ribeiro R M, Bonhoeffer S. Proc Natl Acad Sci USA. 2000;97:7681–7686. doi: 10.1073/pnas.97.14.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deeks S G. Clin Infect Dis. 2000;30:S177–S184. doi: 10.1086/313855. [DOI] [PubMed] [Google Scholar]
- 25.Paterson D L, Swindells S, Mohr J, Brester M, Vergis E N, Squier C, Wagener M M, Singh N. Ann Intern Med. 2000;133:21–30. doi: 10.7326/0003-4819-133-1-200007040-00004. [DOI] [PubMed] [Google Scholar]
- 26.Hlavacek W S, Stilianakis N I, Notermans D W, Danner S A, Perelson A S. Proc Natl Acad Sci USA. 2000;97:10966–10971. doi: 10.1073/pnas.190065897. . (First Published September 19, 2000; 10.1073/pnas.190065897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin X, Bauer D E, Tuttleton S E, Lewin S, Gettie A, Blanchard J, Irwin C E, Safrit J T, Mittler J, Weinberger L, et al. J Exp Med. 1999;189:991–998. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellors J W, Rinaldo C R, Gupta P, White R M, Todd J A, Kingsley L A. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 29.Chun T W, Carruth L, Finzi D, Shen X F, DiGiuseppe J A, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn T C, et al. Nature (London) 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 30.Finzi D, Blankson J, Siliciano J D, Margolick J, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, et al. Nat Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 31.Mocroft A, Miller V, Chiesi A, Blaxhult A, Katlama C, Clotet B, Barton S, Lundgren J D. Antiviral Ther. 2000;5:107–112. [PubMed] [Google Scholar]
- 32.Hockett R D, Kilby J M, Derdeyn C A, Saag M S, Sillers M, Squires K, Chiz S, Nowak M A, Shaw G M, Bucy R P. J Exp Med. 1999;189:1545–1554. doi: 10.1084/jem.189.10.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramratnam B, Bonhoeffer S, Binley J, Hurley A, Zhang L Q, Mittler J E, Markowitz M, Moore J P, Perelson A S, Ho D D. Lancet. 1999;354:1782–1785. doi: 10.1016/S0140-6736(99)02035-8. [DOI] [PubMed] [Google Scholar]
- 34.Ogg G S, Jin X, Bonhoeffer S, Moss P, Nowak M A, Monard S, Segal J P, Cao Y, Rowland-Jones S L, Hurley A, et al. J Virol. 1999;73:797–800. doi: 10.1128/jvi.73.1.797-800.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]