

Abstract
Standard therapy for Acute Myeloid Leukemia (AML) is rarely curative, and several suggested improvements have had little success so far. We have reported that in an in vitro model of a potential therapeutic regimen for AML, the activity of cytarabine (AraC) is enhanced by a sequential treatment with a combination of the vitamin D2 analog Doxercalciferol (D2) and the plant-derived antioxidant carnosic acid (CA) Importantly, the enhancement occurred selectively in patient-derived AML blasts, but not in the normal bone marrow cells. We now demonstrate that TXNIP, previously known as Vitamin D up-regulated protein 1 (VDUP1) [PMID 808674] plays a part in signaling cell death (CD) in this regimen. This is shown by the reduced CD when TXNIP protein levels are decreased by the CRISPR/CAS9 or RNAi technology. Further, we show that direct activation of ASK1 kinase by TXNIP is required for the optimal transmission of the CD signal to apoptotic machinery, regulated by JNK and BIM. These studies provide a rationale for a projected clinical trial of this vitamin D-based new therapeutic regimen for AML.
Keywords: Vitamin D, Acute Myeloid leukemia, AraC, ASK1, TXNIP, Apoptosis, Carnosic Acid
Graphical Abstract
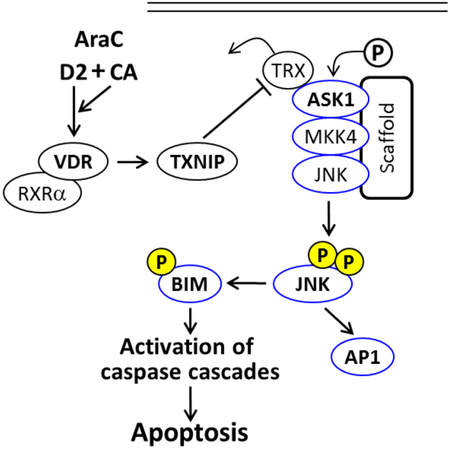
1. Introduction.
The clinical usefulness of vitamin D analogs in the therapy of malignant diseases is currently under question in spite of reported, largely anecdotal, successes. One not fully evaluated approach is the combination of vitamin D analogs approved for clinical use with other forms of therapy. We have reported that the addition of a combination of the vitamin D2 analog doxercalciferol (1-alpha-hydroxyvitamin D2;1-D2) and the plant-derived antioxidant carnosic acid (CA) to Acute Myeloid Leukemia (AML) cells pretreated with cytotoxic therapeutic drug cytarabine (aka, AraC) enhances the chemotherapy-induced cell death in AML cells. This was shown in AML cell lines and confirmed using ex vivo patient-derived leukemic blasts exposed to this new regimen, but not in mononuclear cells isolated from normal human bone marrow [1, 2]. We also demonstrated that this enhanced cell death (ECD) is vitamin D receptor (VDR)-dependent, and partially caspase-dependent, which was associated with an increased expression of the stress-related JNK proteins and the pro-apoptotic protein Bim [2, 3]. However, the intracellular signaling which results in the activation of JNK1/2 and their downstream cell death-inducing proteins was not clear.
In 1994 Chen and DeLuca reported the isolation of a novel cDNA from HL60 cells, the expression of which is upregulated by 1,25-alpha-dihydroxyvitamin D3 (1,25D), the activated form of vitamin D3, and encodes a 46 kDa protein. Since phorbol esters, which like 1,25D induce HL60 cell differentiation, did not regulate the expression of this protein, they dubbed it vitamin D upregulated protein-1 (VDUP1), implying vitamin D-specific regulation [4]. Further studies have revealed that VDUP1 plays multiple roles in a wide range of cellular processes, such as proliferation or apoptosis. Subsequently, Nishiyama et al [5] showed that a thioredoxin-binding protein identical to VUDP1 negatively regulates the expression and the activity of thioredoxin, and is thus involved in redox regulation. This protein is now generally known as TXNIP ("Entrez Gene:TXNIP Thioredoxin interacting protein".) In addition, TXNIP is known to interact with thioredoxin which inhibits the activity of Apoptosis Signal-regulating Kinase 1 (ASK1), also known as Mitogen-Activated Protein Kinase kinase kinase 5 (MAP3K5) [4, 6].
Here, we show that the regimen which results in the ECD of AML blasts is associated with a markedly increased expression of TXNIP, and that the regimen also upregulates and activates ASK1 in parallel with its downstream kinase effector JNK1. Further, we found that in ECD there is a significant increase in TXNIP binding to the ASK1-JNK1 signaling complex (the ASK1 signalosome). Importantly, VDR signaling appears essential for the optimal induction of cell death by the TXNIP-ASK1-JNK1 signalosome, as shown by CRISPR/Cas9- or siRNA-mediated VDR knockdown which results in parallel decreases in ECD and the expression of the components of the ASK1 signaling complex.
2. Materials and methods.
2.1. Chemicals and antibodies
Carnosic acid (CA) was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY). Arabinocytosine (Cytarbine, AraC) and Doxercalciferol (D2) were purchased from Sigma-Aldrich (St. Louis, MO). ASK1 inhibitor NQDI-1 was from Selleck Chemical Inc (Houston, TX). The antibodies that used for Western blotting as following: TXNIP (#14715), Phospho-ASK1 (Thr845, #3765), ASK1 (#8662), Phospho-MKK4 (Ser 257, #4514), MKK4 (#9152), Phospho-JNK (Thr183/Tyr185, #9255), JNK (#9252), Phospho-p38 MAPK (Thr180/Tyr182, #4511), Phospho-MEK1 (Ser217/221, #9121), MEK1 (#9122), cleaved caspase 9 (#9505), cleaved caspase 3 (#9661) and HRP-linked antirabbit (#7074) antibodies were purchased from Cell Signaling Technologies. BIM (sc-8625), VDR (sc-1008), p38 (sc-448) and CRK-L (sc-319) were obtained from Santa Cruz Biotechnology (Dallas, TX).
2.2. Cell culture
AML cell lines, HL60 and U937, were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated bovine calf serum (BCS) at 37°C in a 5% CO2 atmosphere. Cells were passaged two-three times a week to maintain log phase growth. For most experiments, the cells were seeded in 6-well plates at 1.5 × 105/mL followed by the addition of the various agents for the indicated times.
2.3. Enumeration of live and dead cells by the trypan blue exclusion assay
HL60 and U937 cells (1 χ 105 /ml) were incubated with either vehicle (sterile double-distilled water; Untreated) or 100 nM AraC, for 72 h. The number of live and dead cells was then determined on the basis of the trypan blue exclusion assay by counting in Vi-Cell XR cell viability analyzer (Beckman Coulter, Fullerton, CA, USA), as described previously [7]. Both the Untreated and AraC-treated cells were re-seeded at 0.4 χ 105 cells/ml and incubated with either vehicle (0.2% ethanol; Untreated-96 h) or the combination of 100 nM 1-D2 and 10 μM CA (D2/CA), for an additional 96 h. Cells were then enumerated as indicated above.
2.4. Isolation of mononuclear cells from a bone marrow sample of AML patient
A bone marrow specimen was obtained during a diagnostic procedure from a patient volunteer diagnosed with AML, according to the Institutional Review Board protocol. Mononuclear cells were isolated from the specimen by using Histopaque-1077 (Sigma-Aldrich) gradient centrifugation as previously described [8]. The mononuclear cells were divided into different groups that were exposed to AraC and other agents for the indicated times.
2.5. Knockdown of proteins by CRISPR/Cas9 and RNAi technologies
CRISPR/Cas9 knockdown cell lines were generated using a commercially available system (Santa Cruz Inc.) which comprised CRISPR/Cas9 knockout (KO) plasmids and homology-directed recombination (HDR) plasmids containing puromycin resistance gene. The following plasmids were used: VDR (sc-400171-KO-2, sc-400171-HDR-2), ASK1 (SC-400658-KO-2, SC-400658-HDR-2) and TXNIP (sc-400664, sc-400664-HDR). Each CRISPR/Cas9 KO plasmid system was represented by a pool of three plasmids each encoding the Cas9 nuclease and a site-specific 20 nt guide RNA (gRNA) designed for maximum knockout efficiency. Negative control cells were transfected with the control CRISPR/Cas9 plasmid encoding Cas9 nuclease and a non-specific 20 nt gRNA (sc-418922). Cell transfection was performed as previously described [9] using Amaxa Nucleofector (Lonza, Cologne, Germany) according to the manufacturer's protocol (program T-19). Briefly, 2 μg of CRISPR/Cas9 KO plasmid and 2 μg HDR plasmid were mixed in 300 μl plasmid transfection medium (sc-108062, Santa Cruz Inc) supplemented with 15 μl of UltraCruz transfection reagent (sc-395739). Prior to transfection, HL60 cells (2.5 × 106) were incubated in the transfection medium for 10 min on ice. The transfected cells were allowed to recover for 48 h, washed and resuspended in cell culture medium containing puromycin (1 μg/mL) for selection. Puromycin selection was performed in three independently transfected cultures, for seven days, replacing media every 2-3 days. The cultures showing the best knockdown of the specified target gene were chosen for experiments reported here.
VDR knockdown by RNAi was performed as described previously [10]. U937 cells were transfected with 10 μM of VDR siRNA (sc-106692, Santa Cruz Inc), which contains a pool of 3 target-specific 19-25 nt siRNAs designed to knockdown VDR expression, or scrambled Control siRNA (sc-37007). Transfection was performed by Endo-Porter delivery reagent from Gene Tools Inc (Philomath, OR) added 48-72 h before exposure to other agents. One part of the transfected cells was harvested to determine the extent of knockdown of VDR expression at both mRNA and protein levels. The remaining cells were allowed to recover for 24 h in RPMI 1640 medium with 10% FCS, followed by exposure to test agents.
2.6. Annexin V FITC assay
Experimental samples were collected, washed twice with 1xPBS, re-suspended in binding buffer, and stained using an Annexin V-FITC assay Kit (Fisher Scientific, Pittsburgh, PA). The cells were incubated with 50 μg/ml Annexin V and 10 μg/ml propidium iodide in 1x binding buffer at room temperature in the dark, for 15 min, and immediately analyzed by flow cytometry (EPICS XL). Annexin V-positive/PI-negative cells were considered to be early apoptotic, cells with both Annexin V and PI positive to be late apoptotic, and Annexin V negative but PI positive to be “necrotic” [11]. Percent of cell death was calculated as the sum of the percentages of both apoptotic and necrotic cells.
2.7. Western blotting
Western blotting was performed using 40 μg of whole cell extracts as described before [12]. Briefly, membranes were incubated with primary antibodies for 1-2 h according to the sensitivity of each antibody, and then blotted with HRP-linked secondary antibody for 1 h. The protein bands were visualized using a chemiluminescence detection system (Thermo Scientific, Waltham, MA). Each membrane was stripped and re-probed for CRK-L, which was used as an internal loading control. The relative OD of each band was quantitated and indicated in the Figures as a bar diagram above each band, or in a few cases, numerically below the bands in representative Western blot.
2.8. Real time quantitative PCR
Total RNA was extracted with RNeasy Mini kit (Qiagen, Germantown, MD) according to manufacturer’s protocol. Quantitative RT-PCR for CD14, VDR and CYP24 was carried out using an ABI 7500 Real-Time PCR System, as described previously [2]. Fold changes of mRNA levels in each target gene relative to the RNA polymerase II (RPII) control were calculated using the 2−ΔΔCt equation, where ΔΔCt = (Ct target gene – Ct RII)treated sample – (Ct target gene – Ct RPII)control sample. Primers used were as follows: CD14, forward (5′-AACTCCCTCA ATCTGTCGTTCGCT-3′) and reverse 5′-GGGCAAAGGGTTGAATTGGTCGAA-3′; VDR, forward (5′-CTTCAGGCGAAGCATGAAGC-3′) and reverse (5′-CCTTCATCATGC CGATGTCC-3′); CYP24A1, forward (5′ CAAACCGTGGAAGGCCTATC 3′) and reverse (5′ AGTCTTCCCCTTCCAGGATCA 3′); and for RP II, forward (5’-GCACCACGTCCAATG ACAT-3’) and reverse (5’-GTGCGGCTGCTTCCATAA-3’. The quality of all PCR products was monitored using post-PCR melting curve analysis.
2.9. Co-immunoprecipitation and in vitro kinase assays
Co-immunoprecipitation (Co-IP) was done using the Thermo Scientific Pierce Co-IP kit following the manufacturer's protocol. Briefly, ASK1 or TXNIP antibody was first immobilized for 2 h using AminoLink Plus coupling resin. The resin was then washed, incubated with 200 ug cell lysate overnight, washed again, and the associated proteins were eluted using elution buffer. The eluted samples were analyzed by Western blotting or in-vitro kinase assay. Kinase reaction was performed in 30 μl 1× kinase buffer supplemented with 10 μM ATP and 1 μg of recombinant human MEK-1 (Santa Cruz, #sc-4025) or p38 (Abcam, ab45158) protein The samples were incubated at 30°C, for 30 min. Reaction was terminated by the addition of 20 μl 3× SDS sample buffer, followed by boiling for 5 min. The samples were separated on 10% SDS-PAGE gels and then transferred to nitrocellulose membranes. The membranes were probed with phospho-MEK1 (Ser217/Ser221) or phospho-p38 (Thr180/Tyr182) antibody, and after the stripping were re-probed with MEK1 or p38 antibody, as a loading control. Signal detection was accomplished using a chemiluminescence detection system (Thermo Scientific). The optical density (OD) of protein bands was quantified using ImageQuant 5.0 (Molecular Dynamics, Sunnyvale, CA) and indicated below each band in the Figures.
2.10. Statistical analysis
Each experiment was repeated 3-5 times. The results are presented as the Mean ± Standard Deviation. Significance of the differences between mean values was assessed by a 2-tailed Student's t-test using Microsoft EXCEL program.
3. Results
3.1. Effects of cytarabine (AraC) followed by the doxercalciferol-carnosic acid combination (D2/CA) on cell numbers and viability.
We have previously reported that sequential exposure of human AML blasts to AraC then D2/CA, results in the enhancement of AraC cytotoxicity, with cell death primarily by apoptosis [1-3]. In continuation of these studies we monitored cell death and now also cell growth in sequentially treated HL60 cells (Fig. 1) and U937 cells (Suppl. Fig. S1) using the Trypan Blue exclusion assay. In both cell lines, AraC-induced cell death was consistently increased by the addition of D2/CA percent of dead cells, demonstrating ECD (The percent of dead cells when D2/CA followed AraC, minus the percent of dead cells treated by AraC alone). In addition, the total cell number, which as expected was reduced by AraC, was further decreased by the subsequent exposure to D2/CA (Fig. 1A-D and Suppl. Figs S1A-D). The latter effect may reflect both disintegration of dead cells and inhibition of cell proliferation. In both HL60 cells and U937 cells the percent of ECD was small (18.4% and 13.8%, respectively) but was highly significant (p = 0.003; Fig. 1C and p = 0.007; Suppl. Fig. S1C). This is consistent with our previous studies performed in U937 and ex vivo AML blasts which showed that the ECD can range from approximately 20% to 50% [1-3], as can also be seen in Figs 2A and 3B below.
Figure 1. Effects of sequential treatment with AraC and D2/CA on the growth and viability of HL60 cells.

(A, B) Cell death and the total cell number at cell seeding (Untreated-0 h) and following incubation for 72 h with either vehicle (Untreated-72 h) or 100 nM AraC (AraC-72 h), as described in Material and Methods. (C, D) Cell death and the total cell number at seeding of the cells collected after 72 h, as in A and B, (Untreated-0 h; AraC-0 h) and following an additional incubation for 96 h of untreated control (Untreated-96 h) cells or AraC-treated (AraC-96 h) cells, with or without 100 nM 1-D2 and 10 μM CA (D2/CA), as indicated by the rectangles in the Figure. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001 vs. Untreated-72 h or −96 h. ♦, p < 0.05; ♦♦♦♦, p < 0.0001 vs. D2/CA alone; §§§, p < 0.001 vs. Untreated-0 h; #, p < 0.05; ##, p < 0.01; ###, p < 0.001, significant differences between the indicated groups.
Figure 2. Knock down of VDR reduced cell death in the ECD phase in parallel with the reduction in apoptosis-related protein levels in HL60 cells.


A. VDR was knocked down by CRISPR/Cas9 in HL60 cells. Then the cells were pretreated with AraC (100 nM) for 72 h, followed by 100 nM 1-D2 and 10 μM CA (D2/CA) for 96 h. Percent of cell death determined by either Trypan blue exclusion or Annexin V/PI staining is shown in the bar chart. B. mRNA levels of VDR and its target genes CD14 and CYP24A1 in VDR KD cells (VDR CRISPR) as compared to the negative control cells (CTL CRISPR). C. A Western blot performed for the indicated apoptosis-related proteins. CRK-L was stripped and used as a loading control. The average relative signal intensities from three separate experiments are shown in bar charts above each blot. *, p<0.05 vs the AraC group; #, p<0.01 when comparing CTL CRISPR-ECD with VDR CRISPR-ECD.
Figure 3. Knock down of TXNIP by CRISPR/Cas9 in HL60 cells reduces ECD.

A. Depletion of TXNIP by CRISPR/Cas9 in HL60. The pretreatment with AraC for 72 h, then addition of D2/CA for 96 h. The percentages of cell death, as determined by trypan blue exclusion or annexin V/PI staining, are shown in the table. B. Western blotting to detect the TXNIP downstream targets, ASK1, MKK4 and JNK, as well as the apoptosis-related proteins, BIM and caspase 3. CRK-L was used as a loading control. The average of relative signal intensities from three separate experiments are shown in bar charts above each blot. *, p<0.05; **, p < 0.01 vs the AraC group; #, p<0.01 when comparing CTL CRISPR-ECD with VDR CRISPR-ECD.
3.2. Knock down of VDR by CRISPR/Cas9 or siRNA in AML cell lines reduces enhanced cell death (ECD) in parallel with the reduction in the associated increases in TXNIP protein levels, as well as the increased phosphorylation of ASK1 and its downstream targets.
TXNIP, originally named VDUP1 by DeLuca [4], is known to be regulated by vitamin D in HL60 cells [13], and was studied under that name by many groups [14, 15]. We therefore hypothesized that the ECD is at least in part mediated by TXNIP because of the presence of 1-D2 in the D2/CA post-AraC treatment. Accordingly, we confirmed that the depletion of VDR by partial knock down (KD) by either CRISPR/Cas9 or siRNA reduces ECD in HL60 cells (Fig. 2A, and previous publications [2, 16]) and in U937 cells (Suppl. Figs S2A, B).
Examination of AML cells by qRT-PCR showed that the KD of VDR reduced VDR mRNA expression in all treatment groups by at least 50%, but in the ECD group down to ~35% in HL60 cells (Fig. 2B) and to ~60% in U937 cells (Suppl. Fig. S2B). In parallel, the downstream genes regulated by VDR, CYP24A1 and CD14, also showed marked down-regulation by the VDR KD in most groups in both HL60 (Fig. 2B) and U937 cells (Suppl. Fig. S2).
The above mRNA data were corroborated by Western blotting which showed that the VDR KD reduced protein levels of VDR and several ECD-related signaling molecules to an approximately same extent as the VDR KD reduction in mRNA expression of VDR and its target genes (compare Fig. 2B with Fig. 2C). Particularly, CRISPR/Cas9-mediated KD of VDR reduced its protein level to ~30% of the negative CRISPR/Cas9 control levels in all treatment groups. Also, as expected, the ECD-associated increases in TXNIP levels were markedly reduced in parallel with VDR reduction.
Previous literature indicates that TXNIP can influence cellular REDOX state and regulates cell death by activating ASK1 [8, 17]. Consistent with our hypothesis, VDR also appears to regulate the activation of ASK1-regulated apoptotic machinery, as shown by activating phosphorylation of ASK1 and JNK, and the expression of BIM (Fig. 2C) Thus, the control of TXNIP by VDR can lead to increased cell death in this potentially therapeutic sequential protocol.
3.3. Knock down of TXNIP by CRISPR/Cas9 in HL60 cells reduces ECD in parallel with attenuation of the ASK1-regulated apoptotic cascade.
The results shown above strongly suggest that TXNIP targets ASK1 to increase the AraC-induced cell kill. To confirm, we depleted TXNIP by CRISPR/Cas9 in HL60 cells and showed that this reduced ECD by ~ 40% (Fig. 3A). This was further validated by the higher level of phosphorylated ASK1 in ECD, which was reduced by TXNIP KD (Fig. 3B). In addition, we show the activation and TXNIP-dependence of the ASK1 apoptotic pathway, which includes MKK4, an immediate target of P-ASK1), the MKK4 target JNK, and the upregulation of BIM expression (Fig. 3B). We also demonstrate the activating cleavage of caspase 3 in ECD, which is also TXNIP-dependent in this system (Fig. 3B).
3.4. ASK1 directly contributes to ECD in AML blasts.
Phosphorylation of ASK1 on threonine 845 has been reported to activate its kinase activity [18], but to establish that this is the case in human AML blasts we also demonstrated by two separate approaches that ASK1 enzyme activity is increased in ECD (Fig. 4), and this correlated well with the upregulation of P-ASK1 (Thr-845) in ECD (Fig. 3B). We then showed that the inhibition of ASK1 enzyme activity by a specific inhibitor NQDI [19, 20] reduces ECD in HL60 cells (Fig. 5A) as well as in a sample of human ex vivo AML blasts (Fig. 6),PI though the amount of the blasts available was insufficient to repeat the Annexin V/PI cell death assay. This was confirmed by the KD of ASK1 in HL60 cells, resulting in the reduction in ECD (Fig. 5B). The KD of ASK1 was also accompanied by the reduced downstream activation of several regulators of apoptosis in both HL60 and in ex vivo AML cells (Figs 5C and 6B).
Figure 4.

In vitro assays of ASK1 kinase activity showing increased enzyme activity in ECD using two different substrates (note the fourth lane in A and B) to demonstrate consistency. HL60 cell lysates (100 μg) were immunoprecipitated using ASK1 cross-linked column, then incubated with 1 μg of recombinant human p38 or MEK1 proteins as substrates for ASK1-immunoprecipitated kinase assay in the presence of ATP. Western blots were run and probed for Phospho-p38 (Thr180/Tyr182) or for Total-p38, and Phospho-MEK1 (Ser217/221) or Total-MEK1.
Figure 5.

A. Inhibition of ASK1 by specific pharmacological inhibitor NQDI-1 or B. Knock down of ASK1 by CRISPR/Cas9 in HL60 cells reduces AraC/D2/CA induced cell death in ECD. *, p<0.05, **, p<0.01 vs the Control group; ♦; p<0.01 vs the NQDI or ASK1 CRISPR Control group; #, p<0.01 when comparing CTL CRISPR-ECD with VDR CRISPR-ECD, as indicated in the bar charts. C. Western blots were performed for ASK1, and its direct downstream targets, phosphorylated MKK4 and JNK, and the apoptosis regulator BIM. Note the enhanced ECD-associated increase in these markers, and their decrease by ASK1 knock down. CRK-L was used as a loading control. The average of relative signal intensities from three separate experiments are shown in bar charts above each blot. *, p<0.05; **, p < 0.01 vs the AraC group; #, p<0.01 when comparing CTL CRISPR-ECD with VDR CRISPR-ECD.
Figure 6. ASK1 contributes to ECD in AML ex vivo blasts.

A. Mononuclear cells were isolated from the bone marrow of a female AML-M4 patient. Cells were pretreated with either vehicle (Control) or 100 nM AraC, for 72 h. Both Control and AraC-treated cells were then post-treated for 96 h with ASK1 inhibitor NQDI-1 while D2/CA was added to a portion of AraC-treated cells during the post-treatment period, as indicted in the Figure. The percentages of dead cells determined by Trypan blue exclusion or annexin V/PI staining are shown in the bar chart. Since the ex vivo blast sample had limited size, we were not able to perform statistical analysis for Annexin V/PI assay. For Trypan blue exclusion assay, *, p<0.05 when compared to the AraC group; #, p<0.05 for the comparisons between the groups indicated in the chart. B. Western blots were performed for the indicated ASK1-related apoptosis signaling proteins. CRK-L was used as a loading control.
3.5. Binding to ASK1 of TXNIP, P-MKK4, P-JNK1 but not JNK2 is increased in the ECD phase.
To establish that TXNIP can directly influence the activation of ASK1 it is necessary to demonstrate a direct contact between these two proteins. Using Co-IP, we show that TXNIP forms a complex with ASK1 and its immediate target for activation of the signaling cascade, MKK4 (Fig. 7A, B). Interestingly, only JNK1 is also a part of this complex but JNK2 is not. This selectivity provides evidence of binding specificity, as is also provided by the absence in the ASK1 complex of CRK-L, an adaptor protein [21], which we use to demonstrate equal loading of the lanes in Western immunoblots.
Figure 7. TXNIP and P-JNK protein association with ASK1 increases in the ECD phase.

Co-IPs of ASK1 in both (A) HL60 and (B) U937 cells followed by determination of proteins in the complex by Western blotting with different antibodies. This showed that in addition to TXNIP and MKK4, P-JNK1 and P-p38 also bind to ASK1 in all treatment groups, and their levels are markedly increased in the ECD phase (lanes 4 and 8). Note the absence of JNK2 binding to ASK1, and the specificity of the Co-IP is also shown by the lack CRK-L binding to ASK1. ftable
The results obtained with HL60 cells (Fig. 7A) were very similar with the results of experiments using U937 cells (Fig. 7B) and ex vivo AML blasts (Fig. 6), as also were the results of ECD determinations (not shown here for U937 cells), strongly suggesting that the conclusions derived here regarding the VDR-TXNIP-ASK1 axis can apply to other subtypes of human AML.
Supplementary Material
Highlights:
A vitamin D2 analog (D2) combined with a plant-derived antioxidant carnosic acid (CA) can potentiate the therapeutic effect of Cytarabine (AraC) on myeloid leukemia blasts.
D2/CA activates apoptotic signals in cells with DNA damage resulting from AraC treatment.
Vitamin D-upregulated protein TXNIP is an upstream molecular regulator of the apoptotic cascade triggered by the differentiation agents (D2/CA) in this system.
TXNIP targets the apoptosis signal regulating kinase ASK1, which then sequentially activates MKK4 and JNK and the executioners of apoptosis.
These data may provide a paradigm for future improvements of Cytarabine-based therapy of patients with AML.
Acknowledgements.
This research was supported in part by the National Cancer Institute, NIH, grant R01CA044722-27 to GPS, by the Israel Science Foundation grant 226/16 to MD, and by the US-Israel Binational Science Foundation grant 2017112 to MD and GPS.
Abbreviations.
- AML
Acute myeloid leukemia
- AraC
Cytarabine
- CA
carnosic acid
- CD
cell death
- ECD
Enhancement of cell death by adding D2/CA to AraC-treated cells
- D2
Doxercalciferol
- OD
optical density
Footnotes
All authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Wang X, Harrison JS, Studzinski GP, Enhancement of arabinocytosine (AraC) toxicity to AML cells by a differentiation agent combination, J Steroid Biochem Mol Biol, 164 (2016) 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang X, Harrison JS, Studzinski GP, BRAF signals to pro-apoptotic BIM to enhance AraC cytotoxicity induced in AML cells by Vitamin D-based differentiation agents, J Steroid Biochem Mol Biol, 173 (2017) 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wang X, Beute WK, Harrison JS, Studzinski GP, JNK1 as a signaling node in VDR-BRAF induction of cell death in AML, J Steroid Biochem Mol Biol, 177 (2018) 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen KS, DeLuca HF, Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3, Biochim Biophys Acta, 1219 (1994) 26–32. [DOI] [PubMed] [Google Scholar]
- [5].Nishiyama A, Masutani H, Nakamura H, Nishinaka Y, Yodoi J, Redox regulation by thioredoxin and thioredoxin-binding proteins, IUBMB life, 52 (2001) 29–33. [DOI] [PubMed] [Google Scholar]
- [6].Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H, Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1, The EMBO journal, 17 (1998) 2596–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pesakhov S, Nachliely M, Barvish Z, Aqaqe N, Schwartzman B, Voronov E, Sharoni Y, Studzinski GP, Fishman D, Danilenko M, Cancer-selective cytotoxic Ca2+ overload in acute myeloid leukemia cells and attenuation of disease progression in mice by synergistically acting polyphenols curcumin and carnosic acid, Oncotarget, 7 (2016) 31847–31861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jia Y, Xu H, Li Y, Wei C, Guo R, Wang F, Wu Y, Liu J, Jia J, Yan J, Qi X, Li Y, Gao X, A Modified Ficoll-Paque Gradient Method for Isolating Mononuclear Cells from the Peripheral and Umbilical Cord Blood of Humans for Biobanks and Clinical Laboratories, Biopreservation and biobanking, 16 (2018) 82–91. [DOI] [PubMed] [Google Scholar]
- [9].Wang X, Gocek E, Novik V, Harrison JS, Danilenko M, Studzinski GP, Inhibition of Cot1/Tlp2 oncogene in AML cells reduces ERK5 activation and up-regulates p27Kip1 concomitant with enhancement of differentiation and cell cycle arrest induced by silibinin and 1,25-dihydroxyvitamin D(3), Cell Cycle, 9 (2010) 4542–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gocek E, Wang X, Liu X, Liu CG, Studzinski GP, MicroRNA-32 upregulation by 1,25-dihydroxyvitamin D3 in human myeloid leukemia cells leads to Bim targeting and inhibition of AraC-induced apoptosis, Cancer Res, 71 (2011) 6230–6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nuñez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G, Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012, Cell Death Differ, 19 (2012) 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Danilenko M, Wang X, Studzinski GP, Carnosic acid and promotion of monocytic differentiation of HL60-G cells initiated by other agents, J Natl Cancer Inst, 93 (2001) 1224–1233. [DOI] [PubMed] [Google Scholar]
- [13].Chung JW, Jeon JH, Yoon SR, Choi I, Vitamin D3 upregulated protein 1 (VDUP1) is a regulator for redox signaling and stress-mediated diseases, The Journal of dermatology, 33 (2006) 662–669. [DOI] [PubMed] [Google Scholar]
- [14].Abu El Maaty MA, Almouhanna F, Wolfl S, Expression of TXNIP in Cancer Cells and Regulation by 1,25(OH)(2)D(3): Is It Really the Vitamin D(3) Upregulated Protein?, International journal of molecular sciences, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hamilton JP, Potter JJ, Koganti L, Meltzer SJ, Mezey E, Effects of vitamin D3 stimulation of thioredoxin-interacting protein in hepatocellular carcinoma, Hepatology research : the official journal of the Japan Society of Hepatology, 44 (2014) 1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Harrison JS, Wang X, Studzinski GP, The role of VDR and BIM in potentiation of cytarabine-induced cell death in human AML blasts, Oncotarget, 7 (2016) 36447–36460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shi Y, Ren Y, Zhao L, Du C, Wang Y, Zhang Y, Li Y, Zhao S, Duan H, Knockdown of thioredoxin interacting protein attenuates high glucose-induced apoptosis and activation of ASK1 in mouse mesangial cells, FEBS letters, 585 (2011) 1789–1795. [DOI] [PubMed] [Google Scholar]
- [18].Tobiume K, Saitoh M, Ichijo H, Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer, Journal of cellular physiology, 191 (2002) 95–104. [DOI] [PubMed] [Google Scholar]
- [19].Volynets GP, Chekanov MO, Synyugin AR, Golub AG, Kukharenko OP, Bdzhola VG, Yarmoluk SM, Identification of 3H-naphtho[1,2,3-de]quinoline-2,7-diones as inhibitors of apoptosis signal-regulating kinase 1 (ASK1), Journal of medicinal chemistry, 54 (2011) 2680–2686. [DOI] [PubMed] [Google Scholar]
- [20].El Eter E, NQDI 1, an inhibitor of ASK1 attenuates acute ischemic renal injury by modulating oxidative stress and cell death, Cardiovascular & hematological agents in medicinal chemistry, 11 (2013) 179–186. [DOI] [PubMed] [Google Scholar]
- [21].Birge RB, Kalodimos C, Inagaki F, Tanaka S, Crk and CrkL adaptor proteins: networks for physiological and pathological signaling, Cell communication and signaling : CCS, 7 (2009) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.