

ABSTRACT
Aspartyl proteases are a widely represented class of proteolytic enzymes found in eukaryotes and retroviruses. They have been associated with pathogenicity in a range of disease-causing microorganisms. The dimorphic human-pathogenic fungus Talaromyces marneffei has a large expansion of these proteases identified through genomic analyses. Here we characterize the expansion of these genes (pop – paralogue of pep) and their role in T. marneffei using computational and molecular approaches. Many of the genes in this monophyletic family show copy number variation and positive selection despite the preservation of functional regions and possible redundancy. We show that the expression profile of these genes differs and some are expressed during intracellular growth in the host. Several of these proteins have distinctive localization as well as both additive and epistatic effects on the formation of yeast cells during macrophage infections. The data suggest that this is a recently evolved aspartyl protease gene family which affects intracellular growth and contributes to the pathogenicity of T. marneffei.
KEYWORDS: Talaromyces marneffei, Penicillium marneffei, aspartyl protease, evolution, pathogenic fungi
Introduction
Proteases are a diverse and ubiquitously distributed group of enzymes that are important for a variety of biological processes including morphogenesis, cellular function, immunity, and nutrition. The aspartyl protease (aspartic peptidase) class of proteases similarly serve many roles but in particular have been implicated in virulence and pathogenicity of microorganisms such as HIV, Plasmodium, and Candida [1–3]. In several emerging and established fungal pathogens, genome sequencing has revealed unique expansions in the number of aspartyl protease-encoding genes and it is thought that this is commensurate with the shift towards a pathogenic lifestyle.
The acquisition or expansion of genes is a potential source of variation that can lead to the generation of new virulence factors. For example in the chytrid fungus Batrachochytrium dendrobatidis, responsible for amphibian infection and death worldwide, one aspartyl protease family is expanded to 77 genes (4.5 fold) relative to a closely related, less pathogenic, species [4]. Similarly, in the entomopathogenic genus Metarhizium, M. anisopliae, which has a broad host range, has significantly more aspartyl protease-encoding genes than its locust-specific relative, M. acridum [5]. In both instances the hypothesis is that this expansion plays an important role in virulence and pathogenicity; however, in neither case has this been tested functionally.
The most thoroughly studied families of aspartyl proteases belong to species in the Candida genus. These genes, known as secreted aspartyl proteases (SAPs), have been identified and/or characterized in C. glabrata, C. tropicalis, C. dubliniensis and C. albicans [3]. SAP genes were originally described in C. albicans, and several of the 10 members of this expansion have been associated with distinct cellular traits, including hyphal formation, adhesion, dimorphic switching and host cell apoptosis [6–8]. Despite some controversy due to complications with the genetic background of these mutants, and a revision of their importance to virulence in a murine model of hematogenously disseminated candidiasis [9], a number of these SAP genes have bona fide roles in immune responses, biofilm metabolism and mucosal infection [10–12]. In C. parapsilosis, production of three SAP proteins varies with respect to the tissue infected as well as between clinical isolates; thus multiple factors influence a single process such as infection [13,14].
Aspergillus fumigatus has two aspartyl protease-encoding genes and one of these, pep1, is expressed during infection in several animal models [15,16]. Mutant studies show that Pep1 is capable of degrading extracellular matrix proteins in the host, but loss does not affect tissue invasion or host mortality [17]. Therefore, while aspartyl proteases appear to have clear roles relevant to infection, both in species where these genes are expanded in number and those with only one to a few genes, loss does not always have a major impact on virulence. For both Candida species and A. fumigatus, this may be due to functional redundancy between members of the expanded families or other protease classes. An important distinction between Candida and many other morphologically complex fungi is that the latter also have other secreted protease classes besides the SAPs.
Despite these studies in Candida species and A. fumigatus, the function of aspartyl proteases in most other pathogens, including T. marneffei has not been examined. T. marneffei (formerly Penicillium marneffei) is a thermally dimorphic pathogen of humans that is endemic to Southeast Asia. The incidence of talaromycosis (formerly penicilliosis) peaked with the AIDS epidemic in this region [18]. T. marneffei is capable of growing as both multinucleate septate hyphae and uninucleate yeast cells dividing by fission [19]. The transition from hyphae to yeast and germination of conidia at 37°C in vitro occurs via an arthroconidial hyphal phase consisting of uninucleate, branched and septate cells. In vivo the transition from conidia to yeast cells is direct. The yeast form predominates during infection, where T. marneffei grows and proliferates within host phagocytic cells thereby avoiding exposure to, and clearance by, the immune system.
In this study, we describe a major expansion of aspartyl protease-encoding genes in T. marneffei, orthologous to the A. fumigatus pep1 gene. The expansion, named paralogue of pep (pop), was found to be monophyletic and is associated with repetitive elements and subtelomeric regions of the T. marneffei genome. This family shows evidence of significant positive selection as well as copy number variation amongst T. marneffei isolates. Despite these rapid evolutionary changes, the functional domains are conserved in the majority of the proteins. Based on their expression profiles, five pop genes were selected for functional analysis. Single and double deletions of these genes revealed cumulative and interdependent impacts on yeast cell formation during macrophage infection, while localization studies revealed unique cellular targeting during in vitro and ex vivo growth. The data suggest that the expansion of these genes may have been an important step in the evolution of pathogenicity in T. marneffei.
Results
The aspartyl protease expansion is monophyletic and variable
A comparative genomics study of 17 Eurotiomycetes revealed an increase in the number of genes encoding aspartyl proteases in T. marneffei (unpublished data). Reciprocal BLAST comparisons suggested that T. marneffei pepA (PMAA_090410) was least diverged from the A. fumigatus pep1 gene (Supplementary Table 1). To understand the nature of the expansion, genes found in this family and their orthologues in other species were used to generate a relatedness tree (Figure 1). The 23 genes in the expanded family in T. marneffei showed a monophyletic distribution and are therefore all paralogues in T. marneffei. The genes were named paralogues of pep (pop). Long branch lengths in the expansion relative to branch lengths of the Talaromycete orthologues point to a rapid generation of variation within the group.
Figure 1.
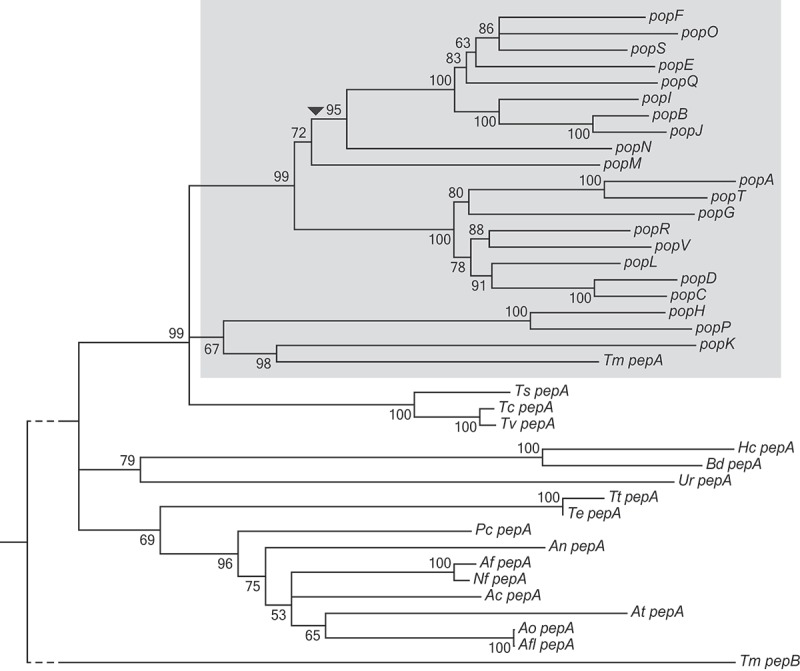
T. marneffei aspartyl protease encoding gene expansion.
A neighbor-joining relatedness tree of genes in the pop expansion as well as pepA orthologues in other Eurotiomycetidae species. Branch confidence values are bootstrap supports from 1000 pseudoreplicates. T. marneffei pepB is included as an outgroup. The grey shading denotes expanded pop clade in T. marneffei. The dashed line denotes shortened branch lengths. The arrowhead indicates the position of a predicted retrotransposition event. Species abbreviations are as follows: Ts, Talaromyces stipitatus; Tc, Talaromyces cellulolyticus; Tv, Talaromyces verruculosus; Te, Trichophyton equinum; Tt, Trichophyton tonsurans; An, Aspergillus nidulans; Pc, Penicillium chrysogenum; Af, Aspergillus fumigatus; Nf, Neosartorya fischeri; Ac, Aspergillus clavatus; At, Aspergillus terreus; Ao, Aspergillus oryzae; Aa, Aspergillus avus; Ur, Uncinocarpus reesei; Hc, Histoplasma capsulatum; Bd, Blastomyces dermatitidis.
In order to assess if changes in gene number were occurring over shorter evolutionary timescales, we examined the intra-species variation within the pop expansion. Using the genome sequencing data from a different study, which investigated 20 clinical and environmental isolates of T. marneffei (unpublished data), we mapped the depth of reads from these strains for each pop gene. This metric can be used as a proxy for the copy number of these genes in each strain (Figure 2). Eight of the 23 pop genes showed copy number variation. For example, popJ is lost in four strains while popD, popE, and popM are lost in one strain each. The popA, popB and popL genes are duplicated in three, seven and three strains, respectively, while popD, popE and popK show wide variations in copy number between strains.
Figure 2.
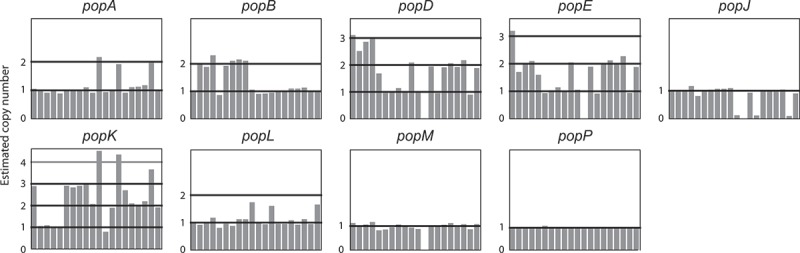
Aspartyl protease-encoding genes vary in copy number between strains.
The copy number of pepA and the pop genes in 20 strains of T. marneffei relative to the reference genome of FRR2161 is depicted. For each gene, the estimated copy number (vertical axis) in each strain (horizontal axis) is shown. Estimated copy number is based on read depth over each gene in each strain. The popP gene is an example of a gene with no copy number variation.
The pop genes are functionally conserved despite showing signatures of positive selection
To examine the evolutionary forces that have been operating on this gene expansion, we determined the non-synonymous/synonymous (dN/dS) substitution rates across this family. Codon-based alignments for each of these genes were generated, as were those of their orthologues in three closely related Talaromyces species (Figure 3). Selection between pepA orthologues in the three Talaromycetes is strongly negative indicating a conservation of function in these genes. While negative selection between these three Talaromyces spp. genes and the T. marneffei pop expansion is still maintained, it is less significant and in the case of popP and popH is absent. Selection within the pop expansion is either neutral or positive. This suggests that these genes are less likely to have a similar biochemical function as their ancestor but conceivably may have evolved new functions.
Figure 3.
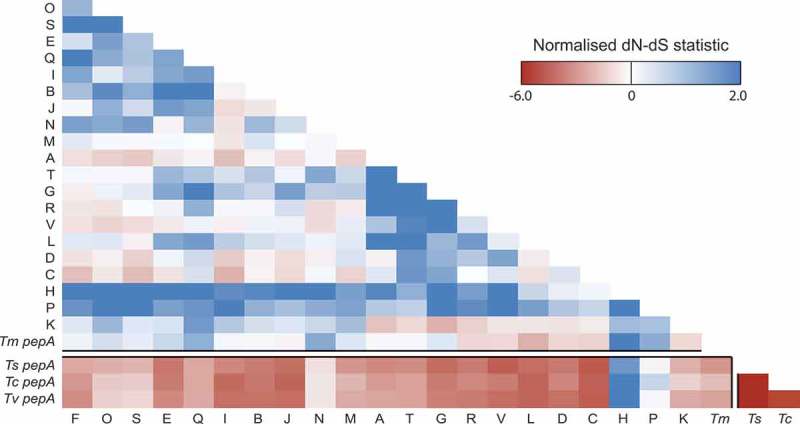
Both positive and negative selection is evident amongst the aspartyl protease-encoding genes.
A heatmap showing the normalized non-synonymous/synonymous selection test statistic in pairwise comparison between all pop genes and pepA Talaromycete orthologues. The axes show the gene identifier (as a one letter abbreviation) for each T. marneffei pop gene or the species for each pepA gene (Tm, T. marneffei; Ts, T. stipitatus; Tc, T. cellulolyticus; Tv, T. verruculosus). Negative values suggest purifying selection is operating while positive values indicate relaxed selection.
In order to determine whether this positive selection was confined to particular amino acid residues or domains, the location of variable residues within the expansion was mapped on to the known 3D structure of the Aspergillus oryzae orthologue Pep1 [20] (Supplementary Figure 1(a)). This analysis revealed that surface residues were more likely to show variation between individual proteins of the pop expansion, while internal residues were significantly more conserved. To quantify the differences in variation between internal and external residues each residue was assigned a score for variation and surface exposure (solvent accessibility). A negative correlation was observed between surface exposure and variation indicating that positive selection occurring in these genes is predominantly associated with the protein surface (Supplementary Figure 1(b)).
Despite the changes in protein sequence due to positive selection the residues and domains that define aspartyl proteases are conserved in the pop expansion. These include the pre-pro peptide required for secretion, the activation peptide and the aspartic acid containing active site motifs. The secretion signal and activation peptide are widely conserved in this expansion with some important exceptions (Supplementary Figure 2(a)). The pepA product shows a moderate reduction in conservation in both sequences while popP and popH products show additional residues in the secretion signal as well as differences in the activation peptide compared to the other pop proteins. The popE activation peptide is disrupted which may impact function. Expression of popK has not been detected under any condition tested to date so the lack of conservation in this gene is likely to be unimportant. The core catalytic residues of aspartyl proteases are made up of two DTG or DSG motifs, which are highly conserved across taxa [21] (Supplementary Figure 2(b)). The first of these sites is conserved in 15 of 23 pop genes while the second is conserved in 19, with 15 showing conservation of both motifs. Five other proteins show conservative substitutions from G to A in the third residue of the first catalytic core, which is unlikely to affect function. As with the activation and secretion signals of popK the catalytic sites of this gene are disrupted. Cysteine residues, which form the structurally important C-terminal disulphide bridge, are absolutely conserved in pop proteins.
The pop genes are associated with flanking repetitive sequences
In order to understand the mechanism for the expansion of these genes in T. marneffei and the variation in copy number between strains, repetitive sequences were examined in the flanking regions surrounding each pop gene. In 17 of 23 pop genes, sequences within 10 kb of the genes were found elsewhere in the genome (Figure 4). In many cases, more than 10 other copies of the duplicated sequence were identified. Longer regions of duplicated DNA are often associated with transposable elements. Further analysis of these regions identified a number of different transposable element (TE) families including copia and gypsy retrotransposons as well as hAT and mariner DNA transposons, while other repetitive regions were not associated with any known TE (Table 1). Therefore, no single transposon family was uniquely associated with the expanded pop family. However, one clade of 10 pop genes lack introns entirely and are likely to have formed through an RNA-mediated retrotransposition event (Figure 1, event indicated by black arrowhead). As well as being associated with transposons pop genes are also over-represented in subtelomeric regions, with 39% of pops (9 of 23) located less than 100 kb from genome scaffold ends, while this was true for only 5.3% of all genes (594 of 10,036).
Figure 4.
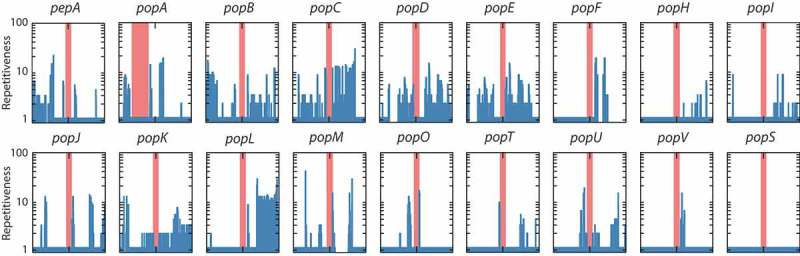
High prevalence of repetitive sequences flanking the aspartyl protease-encoding genes.
The prevalence of repetitive sequences in the upstream and downstream 10kb regions flanking pepA and pop genes is depicted. The red bar denotes each gene locus while the repetitiveness of flanking regions is shown by blue bars. A log scale of 1 to 100 indicates repetitiveness scores for the flanking regions. For example, a score of 5 at a given position indicated that the 30mer at that position is found in five other locations in the genome. The Y-axis is logarithmic. The popA gene is offset from the center due to its proximity to the end of a genome assembly scaffold. Repetitiveness of zero is due to unknown sequences in the incomplete scaffold assembly. The popS gene is an example of a gene with no repetitive flanking sequences.
Table 1.
Transposons and repetitive elements in 10kb region surrounding the pop genes.
| Homologue | 5ʹ flanking region | Transposon type | 3ʹ flanking region | Transposon type |
|---|---|---|---|---|
| pepA | 1.2kb repetitive DNA | - | Gypsy 3–1 AF (+1000bp) | Retrotransposon |
| popH | Mariner 1-AF | DNA transposon | - | - |
| popO | 1kb repetitive DNA | - | 200bp repetitive DNA | - |
| popP | - | - | - | - |
| popS | Mariner 1-AF | DNA transposon | - | - |
| popA | flanking simple repeats | - | flanking simple repeats | - |
| popB | Copia-1 AT e-1 | Retrotransposon | I-5 AO | Retrotransposon |
| popC | Copia-1 Va-1 | Retrotransposon | Mariner 1-AF | DNA transposon |
| popD | popE gene directly upstream | - | Ribosomal RNA encoding gene | |
| popE | popD gene directly upstream | - | Copia 71 BG1 | Retrotransposon |
| popF | Copia 71 BG1 | Retrotransposon | - | - |
| popG | Mariner fragment | DNA transposon | - | - |
| popI | - | - | 2.5kb repetitive DNA | - |
| popJ | flanking simple repeats | - | I-5 AO | Retrotransposon |
| popK | - | - | hAT-1 AN | DNA transposon |
| popL | - | - | 1 kb repetitive DNA Copia-1 VA1 | Retrotransposon |
| popM | I-5 AO | Retrotransposon | 100bp repetitive DNA | |
| popN | hAT 1-AN | DNA transposon | - | - |
| popQ | - | - | - | - |
| popR | 2× Gypsy (3–1 AF and 5PGr-1) |
Retrotransposon | - | - |
| popT | - | - | 200bp repetitive DNA | - |
| popU | I-5 AO | Retrotransposon | Mariner 1-AF | DNA transposon |
| popV | flanking simple repeats | - | - | - |
“Simple repeats” are local and sequential and identified by repeatmasker.[22]
“Repetitive DNA” are regions identified in more than one location in the genome that is un-annotated.
Pop genes are important for yeast cell morphogenesis during macrophage infection
In order to determine which, if any, of the pop genes are important for pathogenicity of T. marneffei, we interrogated a previously generated RNAseq dataset from saprophytic (25°C hyphal in vitro) and pathogenic (37°C in vitro and ex vivo human and murine macrophage infections) growth forms (unpublished data). Expression patterns for the 16 pop genes showed a strong bias towards the pathogenic growth form with the remainder showing minimal expression in the conditions tested (Figure 5). In particular, popP and popH showed strong expression in all conditions with yeast cell morphology. The remaining 14 pop genes showed varying degrees of expression specifically during intracellular growth in both human and mouse macrophage lines. In the majority of cases, expression was higher during infection of human cells (Figure 5).
Figure 5.
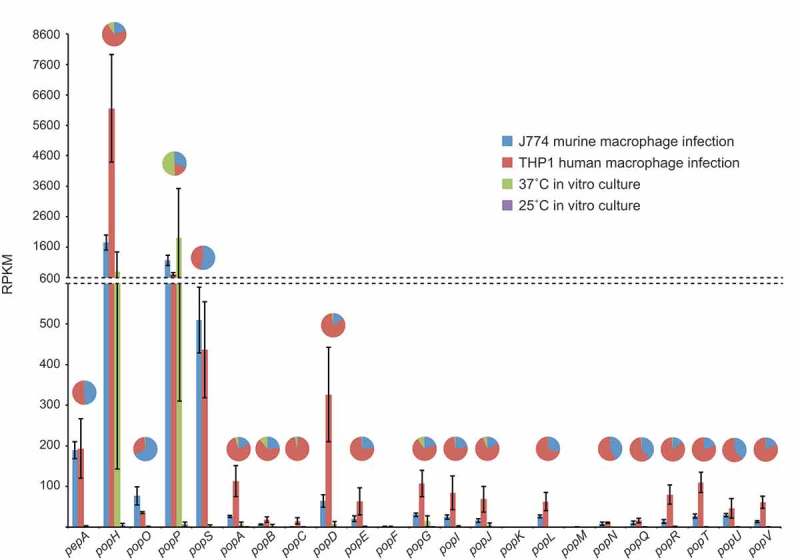
Aspartyl protease genes show specific, differential expression patterns.
Gene expression profiles for the pepA and pop genes during in vitro and ex vivo growth are shown based on RPKM. The in vitro conditions were vegetative growth in the hyphal form at 25 °C and yeast form at 37 °C in BHI medium after four and six days, respectively. The ex vivo conditions were growth in the yeast form inside murine J774 and human THP-1 macrophages for 24-h. The pie charts depict a proportional representation of the expression under each condition for each gene. Dashed line denotes a change in the y-axis scale.
Two groups of genes were selected for molecular genetic analysis based on their expression profiles (Figure 1, genes highlighted in bold). The first group was popP and popH which show yeast morphology specific expression. The second includes popO, popS, and pepA which all show ex vivo macrophage-specific expression. Deletion strains for each of these five genes were generated using a gene replacement strategy, in order to interrogate the contribution of each to growth and pathogenicity. These strains were also complemented through targeted integration of the wild-type alleles at a specific ectopic genomic locus to verify the mutant phenotype. Double deletion strains were also generated through subsequent rounds of gene replacement. These strains included ∆popH/∆popP, ∆popP/∆pepA, ∆popS/∆pepA, ∆popS/∆popP, ∆popS/∆popO, ∆popO/∆pepA, and ∆popO/∆popP.
All strains were examined for phenotypic variation in vitro and ex vivo to assess the effects during germination and hyphal and yeast growth. No difference in growth, morphology or development was observed for any mutant strain relative to the parental strain in vitro (Supplementary Figure 3). At 24-h post infection, wild-type T. marneffei within macrophages produced numerous yeast cells that divide by fission. Similar to the wild-type, the ∆popH strain showed no phenotypic effect while the ∆pepA strain had a minor but insignificant effect during growth in J774 murine macrophages (Figure 6). In contrast, the ∆popP, ∆popS and ∆popO mutants showed a reduction in yeast cell formation to 85.0 ± 2.4%, 60.7 ± 3.5%, and 66.8 ± 8.8% of wild-type levels, respectively. The complemented strains for each of these single mutants restored wild-type levels of yeast cell formation (Table 2). As popH and popP have similar expression profiles, we examined the possibility of synthetic phenotypes in the double ∆popH/∆popP mutant. Yeast cell formation in the double mutant was 85.6 ± 3.0% of the wild-type and not significantly different from the ∆popP mutant. Similarly, pepA mutants lacked significant effects on germination in infected macrophages, despite showing ex vivo specific expression, and when examined in combination with ∆popO (∆popO/∆pepA) showed no significant difference from the ∆popO single mutant (60.7 ± 2.4% compared to 66.8 ± 8.8%). In contrast, the ∆popS/∆popP and ∆popS/∆popO mutants showed additive to near-additive effects (40.7 ± 2.8% and 43.4 ± 3.7%, respectively) while the ∆popS/∆pepA mutant exhibited synergistic effects (39.6 ± 6.2%) with respect to the single ∆popS mutant (60.7 ± 3.5%). The ∆popO/∆popP double mutant shows no significant difference from ∆popO (65.2 ± 2.0% compared to 66.8 ± 8.8%) in yeast cell formation despite the fact that both single mutants show significant independent effects. These data clearly demonstrate a complex relationship with both overlapping and unique functions for these pop genes.
Figure 6.
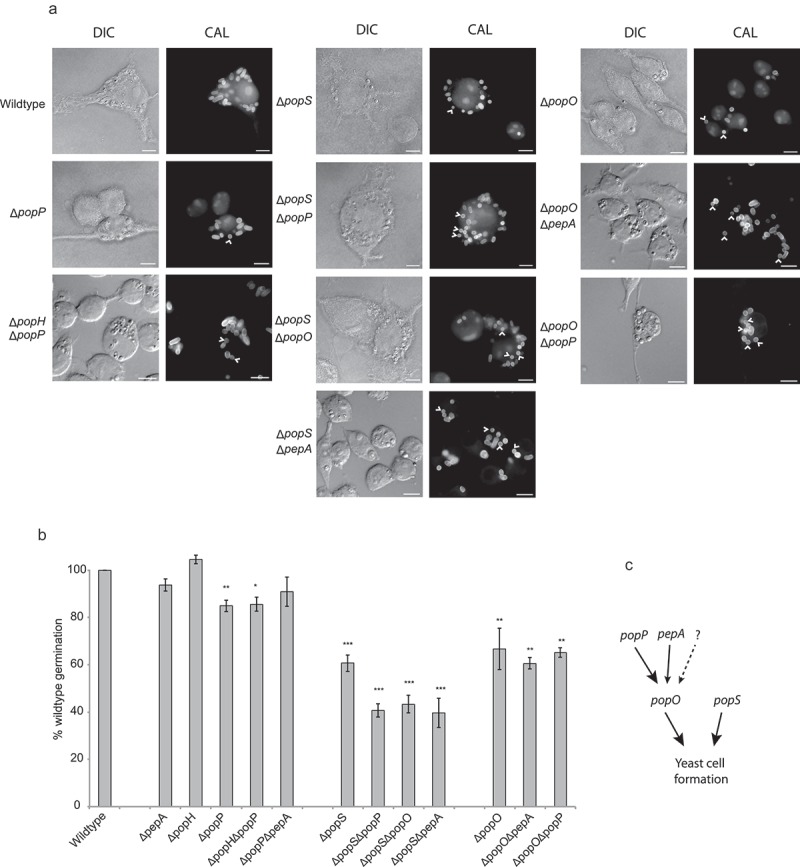
Aspartyl protease-encoding genes are required for appropriate morphogenesis inside host macrophages.
(a) Strains bearing mutations in the pop genes showing yeast cell-specific expression were used to infect murine J774 macrophages and assayed for yeast cell morphogenesis after 24-h post infection. The popP, popO and popS single deletion mutant strains show decreased yeast cell formation in macrophages compared to the wild-type. Double deletion mutant strain combinations for these genes show varying levels of reduction in yeast cell formation with popS combinations showing the greatest change. Representative images using DIC and epifluorescence (CAL, calcofluor staining) are shown. Arrows highlight spores that failed to produce yeast cells. Scale bars = 10 µm. (b) Conversion of dormant conidia into yeast cells (germination) inside J774 macrophages was quantified for each of the mutant strains. Approximately 100 infected J774 macrophage cells infected with each strain in three biological repeat experiments were used to determine germination, and then plotted as a percentage of the wild-type levels. The error bars represent SEM with t-test values falling in the following range * = <0.05, ** = <0.01, *** = <0.001. (c) A model of the postulated genetic interaction pathway based on the severity of their phenotypes and non-additive, epistatic interactions noted for the various combinations of gene deletions and the single mutants. The dotted arrow and question mark denote possible unidentified pathways or proteases of the pop family that may contribute to the formation of yeast cells in T. marneffei during intracellular growth.
Table 2.
Germination of pop mutants and complemented strains.
| Strain | Germination (%)a | t-Testb |
|---|---|---|
| wild-type | 100 ± 0 | c |
| ΔpepA | 93.79 ± 2.5 | |
| ΔpepA pepA+ | 103.17 ± 4.4 | |
| ΔpopH | 104.53 ± 1.9 | |
| ΔpopH popH+ | 104.05 ± 5.3 | |
| ΔpopP | 84.96 ± 2.4 | ** |
| ΔpopP popP+ | 93.54 ± 2.5 | |
| ΔpopO | 66.78 ± 8.8 | ** |
| ΔpopO popO+ | 123.29 ± 14.2 | |
| ΔpopS | 60.70 ± 3.5 | *** |
| ΔpopS popS+ | 117.67 ± 11.2 | |
| ΔpopH ΔpopP | 85.59 ± 2.3 | * |
| ΔpopP ΔpepA | 90.90 ± 6.2 | |
| ΔpopS ΔpopP | 40.69 ± 2.8 | *** |
| ΔpopS ΔpopO | 43.37 ± 3.7 | *** |
| ΔpopS ΔpepA | 39.63 ± 6.2 | *** |
| ΔpopO ΔpepA | 60.66 ± 2.4 | ** |
| ΔpopO ΔpopP | 65.20 ± 2.0 | ** |
| ΔpepA pepA::mCherry | 110 ± 1.8 | |
| ΔpopH popH::mCherry | 107 ± 2.0 | |
| ΔpopP popP::mCherry | 111 ± 1.5 | |
| ΔpopO popO::mCherry | 103 ± 2.5 | |
| ΔpopS popS::mCherry | 116 ± 4.8 |
a Represents the average percent germination relative to the wild-type with the standard error of the mean.
b The t-test values represent the following ranges: * = <0.05, ** = <0.01, *** = <0.001.
c The actual wild-type yeast cell formation percentage was 84.3 ± 0.9%.
Aspartyl proteases show specific cellular localization
To begin to address the complexity in the functional relationship of the different pop gene products, carboxyl-terminal mCherry fusion constructs driven by the native promoter were generated and used to transform the respective deletion strains. Phenotypic characterization of these strains suggested that the fusion alleles complemented the mutant phenotype (Table 2). Using live cells grown in vitro, strong fluorescence was evident for strains carrying the popP and popH fusions during yeast growth at 37°C, while strains with the popO, popS and pepA fusions showed no fluorescence under these conditions (Figure 7(a)). No fluorescence was detected from any strains grown in vitro during hyphal growth at 25°C. These results were entirely consistent with the expression profile data. When these strains were used to infect J774 macrophages, none of the five strains showed detectable fluorescence. To increase the sensitivity of the assay, cells growing inside J774 macrophages were fixed and used for immunofluorescence microscopy with an anti-mCherry antibody. Under these conditions, all five strains showed clear expression of the mCherry fusions.
Figure 7.
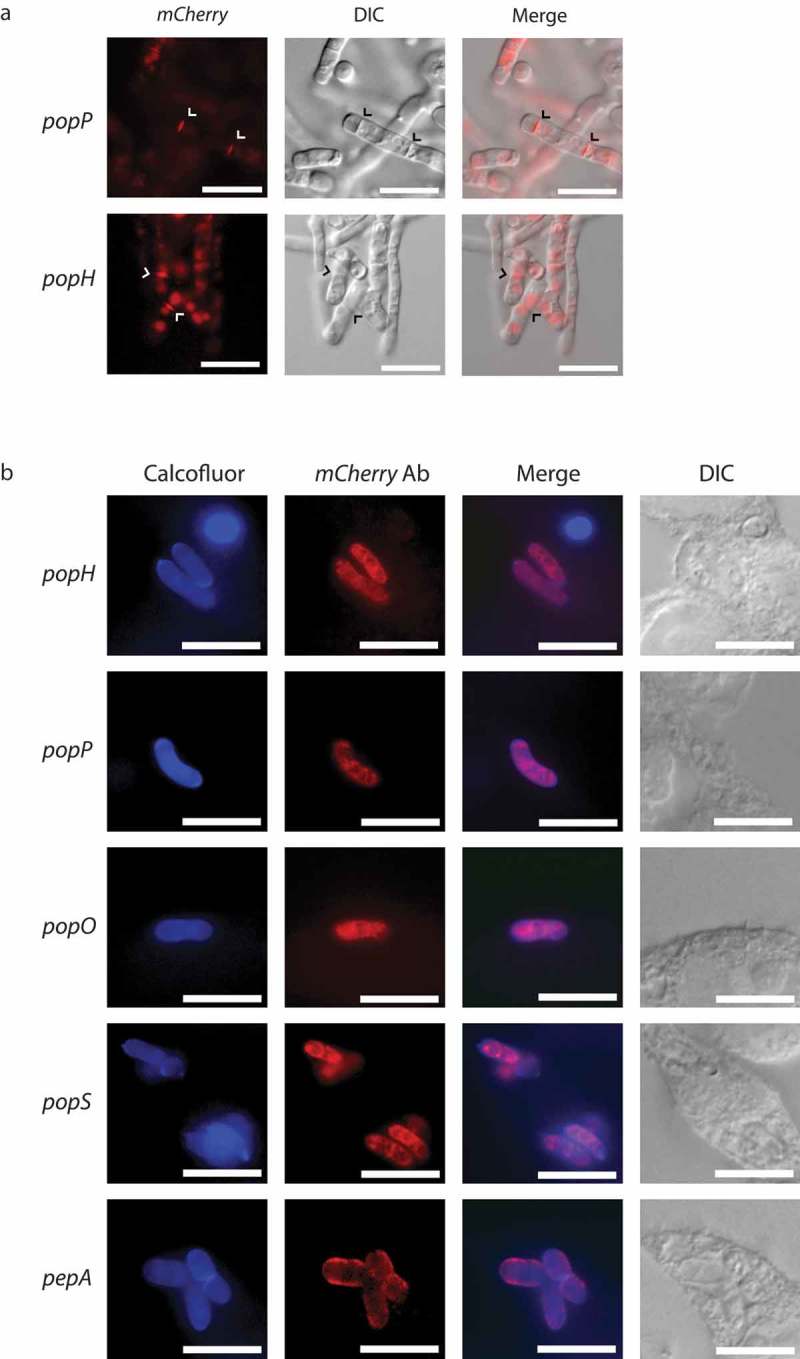
Yeast-specific aspartyl proteases show distinct cellular localization.
Full length, C-terminal fusions of the yeast-specific pepA and pop genes were created with mCherry and introduced into the respective deletion strains. (a) During in vitro growth of yeast cells at 37°C on BHI medium for 6 days, clear mCherry-based fluorescence was evident for the popP and popH fusions only and showed both vesicular and septal localisation (arrowheads). Representative images using DIC and epifluorescence are shown. Scale bar = 10 μm. (b) During growth inside J774 macrophages at 24 hours post-infection, immunofluorescence microscopy using antibodies to mCherry shows that the Pop::mCherry fusion proteins are localised as puncta, often, but not exclusively, perinuclear while the PepA::mCherry fusion protein shows clear cell membrane and/or cell wall localisation. Representative images of cells under DIC and after staining with calcofluor (cell walls) and anti-mCherry antibody (fusions) using epifluorescence are shown. Scale bar = 10 μm.
During in vitro yeast growth, the PopH::mCherry and PopP::mCherry proteins showed localization to the septa of undivided arthroconidial cells as well as to internal vesicles (Figure 7(a)). During ex vivo growth in J774 macrophages, immunofluorescence in the PopH::mCherry and PopP::mCherry strains appeared as a perinuclear ring as well as in diffuse patches within the cytoplasm, which suggests an association with the secretory pathway (Figure 7(b)). Similar localization was evident for the PopO::mCherry and PopS::mCherry strains. In contrast, the PepA::mCherry strain showed strong co-localization with calcofluor staining, indicating localization to the cell membrane and/or cell wall. Despite the prediction of secretion signals for all proteins, no extracellular fluorescence was detected either in vitro or ex vivo.
Aspartyl proteases act cell non-autonomously during macrophage infection
In order to determine if the absence of mCherry fluorescence in the macrophage intracellular environment during infection was due to a lack of secretion or weak signal detection, co-infection experiments with wild-type, ∆popS, and ∆popO mutants were conducted. As single deletion mutant strains possess intact alleles of the remaining pop genes co-infection of macrophages with conidia from two different mutant strains should allow for the production of a full complement of protease gene products in the co-infected macrophage, if the proteases are acting in a cell non-autonomous manner. Macrophages were co-infected with equal numbers of conidia from the ∆popO, ∆popS, and wild-type strains in all pairwise combinations and yeast cell formation rates were determined at 24-h post infection. Infections with the individual wild-type, ∆popS and ∆popO strains were also conducted at the same time. Similar to previous observations the ∆popS and ∆popO mutant strains showed a significant reduction in yeast cell formation (65.9 ± 0.1% and 63.3 ± 2.6%, respectively) compared to wild-type (Table 3). However, the co-infections of ∆popS and ∆popO, wild-type and ∆popS and wild-type and ∆popO were comparable to infections by the wild-type alone with no significant reduction in rates of yeast cell formation (96.90 ± 2.5, 102.22 ± 3.5 and 100.67 ± 2.3, respectively) (Table 3). The data clearly indicate that the activity of these genes is cell non-autonomous.
Table 3.
Germination of pop mutants and wild-type in single and co-infection conditions.
| Strain | Germination (%) a | t-Test b | |
|---|---|---|---|
| Single infections | wild-type | 100 ± 0 | c |
| ΔpopS | 65.89 ± 0.1 | *** | |
| ΔpopO | 63.26 ± 2.6 | * | |
| Co-infections | ΔpopS/ΔpopO | 96.90 ± 2.5 | |
| Wild type/ΔpopS | 102.22 ± 3.5 | ||
| Wild type/ΔpopO | 100.67 ± 2.3 |
a Represents the average percent germination relative to the wild-type with the standard error of the mean.
b The t-test values represent the following ranges: * = <0.05, ** = <0.01, *** = <0.001. No asterisks represent conditions that are not significantly different from wild-type.
c The actual wild-type yeast cell formation percentage was 85.71 ± 2.5%.
To examine the secretion of these proteases by T. marneffei during macrophage infection, western blot analysis was used to identify the presence of these proteins in fractions of T. marneffei infected macrophages (see Materials and Methods). J774 macrophages were infected with conidia from each of the pepA::mCherry, popP::mCherry, popH::mCherry, popS::mCherry and popO::mCherry strains and protein was extracted from purified yeast cells isolated from within macrophages and the remaining macrophage lysate lacking T. marneffei yeast. A strain carrying a histone H1 mCherry fusion (H1::mCherry) allele was used as a control. Western blot analysis revealed the presence of the H1::mCherry protein in the T. marneffei yeast extract but not in the macrophage fraction (Supplementary Figure 4). In contrast, the Pop::mCherry fusion proteins were detected in both T. marneffei yeast and macrophage factions. This was most clearly evident for the popP::mCherry strain (Supplementary Figure 4), consistent with the expression profiling data that showed the popP gene has the highest expression of all the pop family members. A higher level of PopP::mCherry fusion protein is observed in the T. marneffei yeast cell faction. Coupled with the co-infection results, these data strongly suggest that at least a number of the proteases encoded by the pop gene family are secreted and function outside of T. marneffei cells.
Discussion
The evolution of traits that permit exploitation of new environments can proceed by the repurposing of genes with or without loss of the starting function, or the evolution or acquisition of new genes. While the former is constrained if there is selection acting to maintain the ancestral function of a gene, this is not so for the latter. The evolution or acquisition of new genes can occur by horizontal gene transfer or by gene amplification followed by neofunctionalisation. Gene amplification as a mechanism for evolving new traits provides redundancy for maintaining the original function, relaxes selection on individual genes and increases the level of the gene product, which may in itself have selective advantages.
Here we have described the identification of a novel expansion of aspartyl protease-encoding genes in T. marneffei that may have been critical in the evolution of pathogenicity. T. marneffei is the only identified animal pathogenic species in the Talaromyces genus and the only species in the Eurotiomycetes order that exhibits dimorphic switching; an essential trait for intracellular growth. This study showed that the genes in this expansion form one monophyletic clade, with only single orthologues of the proteases present in other species. In most cases, these genes are associated with subtelomeric regions and surrounded by repetitive sequences, including transposable elements, suggesting mechanisms for the genesis of the expansion. Such mechanisms may have included transposon-driven duplication or unequal crossing-over between repetitive sequences. The role of transposons in the evolution of this gene family is further supported by the presence of a sub-clade of pop genes that lack introns, and which are likely to have arisen by retrotransposition. Furthermore, these genes vary in copy number amongst T. marneffei isolates, indicating an ongoing dynamic process in the evolution of this family. Given these observations, we hypothesized that this family may have evolved early in the acquisition of pathogenicity in T. marneffei.
The activity of genes within expanded gene families is associated with virulence in a number of protozoan and fungal species. These include the VAR genes of Plasmodium falciparum, the VSG genes of Trypanosoma bruscei and the TLO family in Candida species [23–25]. In some of these examples, the gene products have diverged from the ancestral form and the expression of individual genes can vary significantly within and between cellular populations. For cell surface-localized gene products, like the VAR and VSG gene families, this leads to antigenic variation in a population, enabling evasion of the host immune system. In T. marneffei, variation in pop protein sequence showed clear positive selection and was primarily observed in residues exposed to the host. Given it is predicted that pop gene products are secreted, this is consistent with the hypothesis that this may provide a source of antigenic variation for T. marneffei to allow escape from detection or help modulate immune interactions during infection.
For the T. marneffei pop genes, it is not clear if the expression of each gene shows significant cell to cell variation, as would be expected if they were functioning similarly to the VAR and VSG genes, where switching expression from one member of the gene family to another permits immune evasion and affects infection outcome. Expression profiling suggests that this may not be a significant factor in the action of these genes as each gene showed distinct cell type and growth condition dependent patterns. Fluorescent tagging of members of the Pop family confirmed the expression profiles and revealed novel localization for PopH and PopP. Collectively, these data suggest that at least some pop genes in this family are more likely to require the enzymatic function of the gene product to assist the growth of T. marneffei under various environmental conditions. This does not preclude some or all of these genes also serving a more structural antigenic variation role.
Aspartyl protease expansions are found in a small number of other pathogenic fungi, where they are proposed to influence pathogenicity and infection. In species of the Candida genus, the number of secreted aspartyl protease (SAP) genes differ, with larger numbers hypothesized to confer greater pathogenicity [26]. In Candida albicans, SAPs are associated with distinct stages of pathogenesis. Mutations in SAP 1–3 show virulence defects in murine vaginitis models while SAP 4–6 are implicated in peritonitis [27,28]. Deletions of several of the genes in T. marneffei that are expressed during ex vivo macrophage growth (pepA, popS, and popO) led to reductions in yeast cell formation under these conditions, and combinatorial deletions of these genes showed more severe or synthetic phenotypes. This suggests that these genes play a role in virulence and the strong conservation of the functional domains in these gene products is consistent with the idea that their enzymatic activity is important. Moreover, the strong positive selection signature that was restricted to the pop expansion, the partially redundant function for some of these genes and the ongoing variation in copy number amongst T. marneffei strains suggests that the biological roles(s) of these genes may be a recent development in the evolution of pathogenicity of T. marneffei and that these genes may act as a source of new effectors to allow T. marneffei to respond to new environments. Some genes such as popP and popH showed minor or no phenotypes when deleted despite strong expression and may therefore be functionally redundant with one or more other pop genes.
The pop gene products have conserved pre-pro peptide sequences suggesting that they are secreted proteins. Localization of PopH and PopP in vitro was to the septa of arthroconidial filaments, which go on to produce yeast cells, and dividing yeast cells, suggestive of a role in the division of yeast cells by fission, a process that is unique to T. marneffei amongst the Eurotiomycetes. However, single and double mutants of these two genes showed no defects in this process, indicating that their role is non-essential or redundant. Localization of PopP, PopS, PopH and PopO ex vivo was perinuclear and vesicular, typically representing secretory machinery such as the endoplasmic reticulum and Golgi. While none of the gene products were detected extracellularly by fluorescence microscopy, we showed that some of these gene products could be detected in macrophage lysates after infection with T. marneffei, even after T. marneffei had been removed from the infected cells, and that complementation of the mutant phenotypes could be achieved by coinfection of macrophages with two mutant strains. These data strongly support the prediction that the pop gene products are secreted and that the lack of detection extracellularly by fluorescence microscopy indicates a sensitivity issue due to either diffusion or degradation of these proteins. This is consistent with the PepA data, which showed localization immediately adjacent to the cell wall, thus likely outside the cell membrane. In C. albicans SAP9 is localized to the cell wall and mediates interactions with phagocytic cells of the immune system [29]. Similarly, pep1, the orthologue of pepA in A. fumigatus is classified as an allergen and as such interacts with the host immune system [30]. The pepA gene may also mediate intracellular interactions between T. marneffei and host immune cells such as macrophages.
Another possible role for PepA is in an activation pathway for secreted pop proteins, which need to be proteolytically cleaved for function (Figure 6(c)). Some support for this hypothesis comes from the phenotypes of double deletion mutants. Combinations with popS show that it acts independently of other pop genes in yeast cell formation. Conversely, pepA or popP deletions combined with popO show epistatic effects suggesting that they act redundantly in one stage of a pathway that also includes popO. If this hypothesis is correct, it is more complex because the popP;pepA double deletion did not mimic the popO single deletion. Given that only five pop genes were characterized in depth in this study it is entirely possible that one or more of the other pop genes may also contribute to this pathway, thereby compensating for the loss of activity in popP and pepA.
Overall the findings clearly show that the pop gene family contributes to T. marneffei intracellular growth, a key aspect to initiation of infection. Despite the large number of genes comprising this family and their potential for functional overlap, the contributions of specific members to the germination of T. marneffei inside macrophages appears to be distinct, underscoring the importance of this expanded group. Recent studies have identified other expanded gene families as significant contributors to T. marneffei pathogenicity [31] (unpublished data). Further analysis of all members of the expansion is necessary to understand fully the role these proteases play in the pathogenicity of T. marneffei. Interestingly, anti-retroviral therapies used in HIV treatment that target the HIV aspartyl protease can inhibit aspartyl proteases of C. albicans and have been shown to influence the virulence of this species in vivo [32]. If these compounds also inhibit pop proteins in T. marneffei they may open new avenues of treatment, especially in HIV infected patients where these medications may already be in use.
Materials and methods
Bioinformatic analysis
Protein alignments were performed using ClustalW in the MEGA7 software package [33,34]. A relatedness tree was inferred from the alignment using the neighbor joining method [35] with 500 bootstrap replicates [36], branches with less than 50% support were collapsed. Secretion signals and likelihood of secretion were determined using signalP 4.1 [37] with Eukaryotes organism group and default settings. Repetitive sequences were identified using a custom python script, which counted the occurrence of each 30mer in the genome and then scored the repetitiveness of each position in the genome. Transposable elements were classified using RepeatMasker 4.0.5 with RepBase Update 20,140,131 with the species group set to fungi. Read depth analysis was performed using custom python scripts briefly, normalized depth of read coverage over a given gene was used to estimate the number of copies of that gene in an isolate. ClustalW was used to align coding sequences of pop genes in order to carry out positive selection analysis in MEGA7. This analysis consisted of a codon-based z-test for positive selection [38]. The dN-dS test statistic was used in the final table to allow examination of both positive and negative selection. Alignments for Supplementary Figure 1 were performed using Clustal Omega [39]. These alignments were then used to calculate and map conservation scores onto the Aspergillus oryzae pepA protein structure [20] with AL2CO [40]. Protein structure visualization was performed in MacPyMol v1.8 [41]. Surface exposure (solvent accessibility) was calculated with ASAView [42].
Molecular methods and plasmid construction
T. marneffei genomic DNA was isolated as previously described [43]. Southern and blotting was performed with Amersham Hybond XL membrane using [α-32P] dATP labeled probe hybridization using standard methods [44]. Sequences of primers are provided in Supplementary Table 2. Deletion constructs were created using the modified GatewayTM method [45]. The deletion constructs of the following three genes were created using pHW7771 containing the pDONR-pyrGBlaster cassette. Wild-type PCR product of pepA (PMAA_090410) was generated with primers QQ87 and QQ88 and cloned into pBluescript II SK+ to generate pMP7880. To make the deletion construct pMP7886, PCR primers QQ89 and QQ90 were used to generate an inverse PCR product (which removes from -25 to +1409 of pepA) for GatewayTM reaction. Wild-type PCR product of popH (PMAA_058850) was generated with primers QQ81 and QQ82 and cloned into pBluescript II SK+ to generate pMP7876. To make the deletion construct pMP7881, PCR primers QQ83 and QQ84 were used to generate an inverse PCR product (which removes from -110 to +1328 of popH) for GatewayTM reaction. Wild-type PCR products of popO (PMAA_062510) was generated with primers QQ98 and QQ99 and cloned into pBluescript II SK+ to generate pHW7877. To make the deletion construct pHW7882, PCR primers QQ100 and RR1 were used to generate an inverse PCR product (which removes from -59 to +1212 of popO) for GatewayTM reaction. The deletion constructs of the following gene were created using pHW7772 containing the pDONR-bar cassette. Wild-type PCR products of popS (PMAA_089390) was generated with primers QQ75 and QQ76 and cloned into pBluescript II SK++ to generate pHW7879. To make the deletion construct pHW7884, PCR primers QQ83 and QQ84 were used to generate an inverse PCR product (which removes from -121 to +1225 of popS) for GatewayTM reaction. The deletion constructs of the following gene were created using pHW7773 containing the pDONR-riboB cassette. Wild-type PCR products of popP (PMAA_065480) was generated with primers QQ93 and QQ94 and cloned into pBluescript II SK+ to generate pMP7878. To make the deletion construct, pMP7883, PCR primers QQ95 and QQ96 were used to generate an inverse PCR product (which removes from -144 to +1979 of popP) for GatewayTM reaction. Complementation constructs were generated by ligating the wild-type PCR products of each gene into EcoCRI digested pHB7615 (niaD targeting vector) to generate pMP7891 (pepA), pMP7887 (popH), pHW7888 (popO), pHW7890 (popS) and pMP7889 (popP).
The mCherry tagged constructs were generated by inserting promoter and open reading frame sequences of each gene into SpeI digested pHW7911 (mCherry::niaD targeting vector). These sequences excluded the stop codon and native terminator sequence of each gene. For the pepA::mCherry fusion construct, PCR product generated using QQ87 and XX20 was cut with XbaI and ligated into SpeI digested pHW7911 to generate pMP8062 For the popH::mCherry fusion construct, PCR product generated using QQ81 and XX19 was cut with SpeI and ligated into SpeI digested pHW7911 to generate pMP8058 For the popO::mCherry fusion construct, PCR product generated using QQ98 and XX17 was cut with XbaI and ligated into SpeI digested pHW7911 to generate pMP8059 For the popS::mCherry fusion construct, PCR product generated using QQ75 and XX18 was cut with XbaI and ligated into SpeI digested pHW7911 to generate pMP8061 For the popP::mCherry fusion construct, PCR product generated using QQ93 and XX18 was cut with SpeI and ligated into SpeI digested pHW7911 to generate pMP8060.
Fungal strains and media
At 25°C strains were grown on A. nidulans minimal medium (ANM) supplemented with 10 mM ammonium sulfate ((NH4)2SO4) as a sole nitrogen source. At 37°C strains were grown on Brain Heart Infusion (BHI) medium (3.7% brain heart infusion).
Transformation was performed previously described using PEG-mediated protoplast fusion [43]. Full details of strains used in this study are listed in Supplementary Table 3. The ∆pepA (G1004), ∆popH (G994) and ∆popO (G997) strains were generated by transforming strain G816 (∆ligD, pyrG1) [46] with linearised deletion constructs pHW7886 (pepA), pHW7881 (popH), pHW7882 (popO) and selecting for uracil prototrophy. The ∆popS (G1002) strain was generated by transforming strain G809 (∆ligD::pyrG, pyrG1) with linearised deletion construct pHW7884 (popS) and selecting for glufosinate resistance. The ∆popP (G999) strain were generated by transforming strain G829 (∆ligD, riboB::pyrG) [42] with linearized deletion constructs pHW7883 (popP) and selecting for riboflavin prototrophy. The ∆pepA, ∆popH, ∆popO, ∆popS and ∆popP mutations were complemented by transforming the respective strains with pMP7891 (pepA), pMP7887 (popH), pHW7888 (popO), pHW7890 (popS) and pMP7889 (popP) to generate ∆pepA pepA+ (G1005), ∆popH popH+ (G995), ∆popO popO+ (G998), ∆popS popS+ (G1003) and ∆popP popP+ (G1000) strains. To generate ∆popP ∆popH (G1007), ∆popP ∆pepA (G1011), ∆popS ∆popP (G1013), ∆popS ∆popO (G1009), ∆popS ∆pepA (G1012), ∆popO ∆pepA (G1010) and ∆popO ∆popP (G1008) double mutants, ∆pepA, ∆popH, ∆popO, ∆popS and ∆popP single mutant strains were grown on 1 mg/ml 5-FOA supplemented with 10 mM ((NH4)2SO4) and 5 mM uracil to select for the loss of the pyrG marker. These were then transformed with the respective linearised deletion constructs corresponding to the combination required for the strain. The mCherry tagged strains were generated by transforming the ∆pepA+, ∆popH, ∆popO, ∆popS+ and ∆popO strains with circular pepA::mCherry (pMP8062), popH::mCherry (pMP8058), popO::mCherry (pHW8059), popP::mCherry (pHW8060) and popS::mCherry (pMP8061) constructs into the respective strains, to generate ∆pepA pepA::mCherry (G1006), ∆popH popH::mCherry (G996), ∆popO popO::mCherry (G1061), ∆popS popS::mCherry (G1060) and ∆popO popO::mCherry (G1001) strains.
Macrophage infection assay
J774 and THP-1 macrophage infection assay was performed as per the previously described protocol [47]. Briefly, macrophages were seeded at a concentration of 1 × 105 cells/m into a 6 well microtitre tray containing sterile coverslips and 2 mL of DMEM (J774) or RPMI (THP-1) medium and incubated at 37°C for 24 h. Macrophages were activated with 0.1 μg/mL lipopolysaccharide (LPS)(J774) or differentiated by the addition of 32 μM phorbol 12-myristate 13-acetate (PMA)(THP-1) for 24 h at 37°C. Cells were washed with PBS and 2 mL of complete DMEM or RPMI medium containing 1 × 106 conidia was added. A control lacking conidia was also performed. Macrophages were incubated for 2 h at 37°C to allow conidia to be engulfed, washed once in PBS to remove non-phagocytosed conidia and incubated a further 24 or 48 h at 37°C. Macrophages were fixed in 4% paraformaldehyde and stained with 1 mg/mL fluorescent brightener 28 (calcofluor, CAL) to observe fungal cell walls. Mounted coverslips were examined using differential interference contrast (DIC) and epifluorescence optics for cell wall staining and viewed on a Reichart Jung Polyvar II microscope. Images were captured using a SPOT CCD camera (Diagnostic Instruments Inc) and processed in Adobe Photoshop. The numbers of ungerminated conidia, germlings or yeast cells were recorded in a population of approximately 100 macrophages in three independent experiments.
Protein extraction and western blot analysis
The macrophage infection assay was performed as previously described (46). Briefly, J774 murine macrophages were seeded at 1 × 105 cells/mL in a flask containing Dulbecco’s modified Eagle medium (DMEM) and grown for 2 days. Macrophages were activated with 0.1 μg/mL lipopolysaccharide (LPS), incubated for 24 h and infected with 3 × 108 conidia of T. marneffei. After 2 h the unphagocytosed conidia were washed off, fresh DMEM added and incubated for an additional 24 h. The media was removed and RIPA buffer (10mM Tris, pH7.4, 100mM NaCl, 1mM EDTA, 1mM EGTA, 1% Triton X-100, 10% glycerol, 0.1% SDS and 0.5% deoxycholate) containing cOmpleteTM Protease Inhibitor (Roche) and 1mM PMSF, was added to lyse the macrophages. The cell lysate was centrifuged at 2000xg and the pellet predominantly consisting of T. marneffei yeast cells was separated from the J774 macrophage protein supernatant. T. marneffei cells were homogenized in a FastPrep®-24. Approximately 15 μg of each protein extract, as determined by Bradford assay, was used for SDS-PAGE. Western blot transfer was performed using Immobilon-P polyvinylidene difluoride (PVDF) filters (Merck Millipore Ltd). The mCherry-tagged protein was detected using an anti-mCherry monoclonal rat primary antibody (ThermoFisher Scientific) and an anti-rat IgG horseradish peroxidase (HRP)-linked secondary antibody (Cell Signaling Technology). HRP activity was detected using ClarityTM Western enhanced chemiluminescence (ECL) substrate (Bio-Rad) on a Bio-Rad ChemiDocTM MP imaging system.
Microscopy
Immunofluorescence localization of the mCherry-tagged strains was performed with an anti-mCherry rat monoclonal primary (Life Technologies) and a goat anti-rat ALEXA 488 secondary antibody (Molecular Probes) using standard protocols [48]. Immunofluorescence microscopy controls using only primary or secondary antibodies as well as an untagged strain were performed to confirm the specificity of the antibodies.
Funding Statement
This work was supported by the National Health and Medical Research Council [1126384].
Acknowledgments
We would like to thank Prof. M. J. Hynes for critical reading of the manuscript and other members of the group for their helpful suggestions and comments during the progress of this work. The research was supported by a National Health and Medical Research grant to AA.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Kyes SA, Kraemer SM, Smith JD.. Antigenic variation in Plasmodium falciparum: gene organization and regulation of the var multigene family. Eukaryot Cell. 2007;6:1511–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Davies DR. The structure and function of the aspartic proteinases. Ann Rev Biophys Biophys Chem. 1990;19:189–215. [DOI] [PubMed] [Google Scholar]
- [3].Silva NC, Nery JM, Dias ALT. Aspartic proteinases of Candida spp.: role in pathogenicity and antifungal resistance. Mycoses. 2014;57:1–11. [DOI] [PubMed] [Google Scholar]
- [4].Joneson S, Stajich JE, Shiu S-H, et al. Genomic transition to pathogenicity in chytrid fungi. PLOS Pathog. 2011;7:e1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gao Q, Jin K, Ying S-H, et al. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLOS Genet. 2011;7:e1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Naglik JR, Challacombe SJ, Hube B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev. 2003;67:400–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gropp K, Schild L, Schindler S, et al. The yeast Candida albicans evades human complement attack by secretion of aspartic proteases. Mol Immunol. 2009;47:465–475. [DOI] [PubMed] [Google Scholar]
- [8].Wu H, Downs D, Ghosh K, et al. Candida albicans secreted aspartic proteases 4–6 induce apoptosis of epithelial cells by a novel Trojan horse mechanism. FASEB J. 2013;27:2132–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Correia A, Lermann U, Teixeira L, et al. Limited role of secreted aspartyl proteinases Sap1 to Sap6 in Candida albicans virulence and host immune response in murine hematogenously disseminated candidiasis. Infect Immun. 2010;78:4839–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kumar R, Saraswat D, Tati S, et al. Novel aggregation properties of Candida albicans secreted aspartyl proteinase Sap6 mediates virulence in oral candidiasis. Infect Immun. 2015;83:2614–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pericolini E, Gabrielli E, Amacker M, et al. Secretory aspartyl proteinases cause vaginitis and can mediate vaginitis caused by Candida albicans in mice. MBio. 2015;6:e00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Winter MB, Salcedo EC, Lohse MB, et al. Global identification of biofilm-specific proteolysis in Candida albicans. MBio. 2016;7:e01514–01516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dagdeviren M, Cerikcioglu N, Karavus M. Acid proteinase, phospholipase and adherence properties of Candida parapsilosis strains isolated from clinical specimens of hospitalised patients. Mycoses. 2005;48:321–326. [DOI] [PubMed] [Google Scholar]
- [14].Silva S, Henriques M, Oliveira R, et al. Characterization of Candida parapsilosis infection of an in vitro reconstituted human oral epithelium. Eur J Oral Sci. 2009;117:669–675. [DOI] [PubMed] [Google Scholar]
- [15].Monod M, Capoccia S, Léchenne B, et al. Secreted proteases from pathogenic fungi. Int J Med Microbiol. 2002;292:405–419. [DOI] [PubMed] [Google Scholar]
- [16].Lee JD, Kolattukudy PE. Molecular cloning of the cDNA and gene for an elastinolytic aspartic proteinase from Aspergillus fumigatus and evidence of its secretion by the fungus during invasion of the host lung. Infect Immun. 1995;63:3796–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Reichard U, Monod M, Odds F, et al. Virulence of an aspergillopepsin-deficient mutant of Aspergillus fumigatus and evidence for another aspartic proteinase linked to the fungal cell wall. J Med Vet Mycol. 1997;35:189–196. [DOI] [PubMed] [Google Scholar]
- [18].Ustianowski AP, Sieu TPM, Day JN. Penicillium marneffei infection in HIV. Curr Opin Infect Dis. 2008;21:31–36. [DOI] [PubMed] [Google Scholar]
- [19].Andrianopoulos A. Control of morphogenesis in the human fungal pathogen Penicillium marneffei. Int J Med Microbiol. 2002;292:331–347. [DOI] [PubMed] [Google Scholar]
- [20].Kamitori S, Ohtaki A, Ino H, et al. Crystal structures of Aspergillus oryzae aspartic proteinase and its complex with an inhibitor pepstatin at 1.9Å resolution. J Mol Biol. 2003;326:1503–1511. [DOI] [PubMed] [Google Scholar]
- [21].Szecsi PB. The aspartic proteases. Scand J Clin Lab Invest. 1992;52:5–22. [PubMed] [Google Scholar]
- [22].Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1-4):462-7. [DOI] [PubMed] [Google Scholar]
- [23].Bachmann A, Predehl S, May J, et al. Highly co-ordinated var gene expression and switching in clinical Plasmodium falciparum isolates from non-immune malaria patients. Cell Microbiol. 2011;13(9):1397–1409. [DOI] [PubMed] [Google Scholar]
- [24].McCulloch R, Field MC. Quantitative sequencing confirms VSG diversity as central to immune evasion by Trypanosoma brucei. Trends Parasitol. 2015;31(8):346–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].van Het Hoog M, Rast TJ, Martchenko M, et al. Assembly of the Candida albicans genome into sixteen supercontigs aligned on the eight chromosomes. Genome Biol. 2007;8(4):R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Parra-Ortega B, Cruz-Torres H, Villa-Tanaca L, et al. Phylogeny and evolution of the aspartyl protease family from clinically relevant Candida species. Mem Inst Oswaldo Cruz. 2009;104:505–512. [DOI] [PubMed] [Google Scholar]
- [27].De Bernardis F, Arancia S, Morelli L, et al. Evidence that members of the secretory aspartyl proteinase gene family, in particular SAP2, are virulence factors for Candida vaginitis. J Infect Dis. 1999;179:201–208. [DOI] [PubMed] [Google Scholar]
- [28].Kretschmar M, Hube B, Bertsch T, et al. Germ tubes and proteinase activity contribute to virulence of Candida albicans in murine peritonitis. Infect Immun. 1999;67:6637–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hornbach A, Heyken A, Schild L, et al. The glycosylphosphatidylinositol-anchored protease Sap9 modulates the interaction of Candida albicans with human neutrophils. Infect Immun. 2009;77:5216–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Reichard U, Eiffert H, Rüchel R. Purification and characterization of an extracellular aspartic proteinase from Aspergillus fumigatus. Med Mycol. 1994;32:427–436. [DOI] [PubMed] [Google Scholar]
- [31].Yang E, Chow W-N, Wang G, et al. Signature gene expression reveals novel clues to the molecular mechanisms of dimorphic transition in Penicillium marneffei. PLOS Genet. 2014;10:e1004662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].De Bernardis F, Tacconelli E, Mondello F, et al. Anti-retroviral therapy with protease inhibitors decreases virulence enzyme expression in vivo by Candida albicans without selection of avirulent fungus strains or decreasing their anti-mycotic susceptibility. FEMS Immunol Med Microbiol. 2004;41:27–34. [DOI] [PubMed] [Google Scholar]
- [33].Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. [DOI] [PubMed] [Google Scholar]
- [34].Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. [DOI] [PubMed] [Google Scholar]
- [36].Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. [DOI] [PubMed] [Google Scholar]
- [37].Petersen TN, Brunak S, von Heijne G, et al. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Method. 2011;8:785–786. [DOI] [PubMed] [Google Scholar]
- [38].Zhang J, Rosenberg HF, Nei M. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci USA. 1998;95:3708–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using clustal omega. Mol Syst Biol. 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pei J, Grishin NV. AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics. 2001;17:700–712. [DOI] [PubMed] [Google Scholar]
- [41].The PyMOL Molecular Graphics System Version 1.8. Schrödinger, LLC; 2015. Available from: https://www.schrodinger.com/pymol
- [42].Ahmad S, Gromiha M, Fawareh H, et al. ASAView: database and tool for solvent accessibility representation in proteins. BMC Bioinf. 2004;5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Borneman AR, Hynes MJ, Andrianopoulos A. The abaA homologue of Penicillium marneffei participates in two developmental programmes: conidiation and dimorphic growth. Mol Microbiol. 2000;38:1034–1047. [DOI] [PubMed] [Google Scholar]
- [44].Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. New York: Cold Spring Laboratory Press; 1989. [Google Scholar]
- [45].Boyce KJ, Bugeja HE, Weerasinghe H. Strategies for the molecular genetic manipulation and visualization of the human fungal pathogen Penicillium marneffei. Fungal Genet Rep. 2012;59:1–12. [DOI] [PubMed] [Google Scholar]
- [46].Bugeja HE, Hynes MJ, Andrianopoulos A. AreA controls nitrogen source utilisation during both growth programs of the dimorphic fungus Penicillium marneffei. Fungal Biol. 2012;116:145–154. [DOI] [PubMed] [Google Scholar]
- [47].Boyce K, Andrianopoulos A. Macrophage infection by fungi. Bio-Protocol. 2(14):e236. [Google Scholar]
- [48].Fischer R, Timberlake WE. Aspergillus nidulans apsA (anucleate primary sterigmata) encodes a coiled-coil protein required for nuclear positioning and completion of asexual development. J Cell Biol. 1995;128:485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.