

Abstract
Background
Many conventional treatments for uncomplicated malaria are failing because malaria parasites develop resistance to them. One way to combat this resistance is to treat people with a combination of drugs, such as atovaquone‐proguanil.
Objectives
To compare atovaquone‐proguanil with other antimalarial drugs (alone or in combination) for treating children and adults with uncomplicated Plasmodium falciparum malaria.
Search methods
We searched the Cochrane Infectious Diseases Group Specialized Register (June 2005), CENTRAL (The Cochrane Library Issue 2, 2005), MEDLINE (1966 to June 2005), EMBASE (1980 to June 2005), LILACS (1982 to June 2005), reference lists, and conference abstracts. We also contacted relevant pharmaceutical manufacturers and researchers.
Selection criteria
Randomized controlled trials comparing atovaquone‐proguanil with other antimalarial drugs for treating children and adults confirmed to have uncomplicated P. falciparum malaria.
Data collection and analysis
Three authors independently assessed trial eligibility and the risk of bias in the trials, and extracted data for an intention‐to‐treat analysis (where possible). We used risk ratio (RR) and 95% confidence intervals (CI) for dichotomous data. We contacted trial authors for additional information where needed.
Main results
Ten trials, with a total of 2345 participants, met the inclusion criteria. The trials were conducted in four geographical regions and were often small, but they included comparisons across eight drugs. Nine trials were funded by a pharmaceutical company, only three carried out an intention‐to‐treat analysis, and allocation concealment was unclear in seven. Atovaquone‐proguanil had fewer treatment failures by day 28 than chloroquine (RR 0.04, 95% CI 0.00 to 0.57; 27 participants, 1 trial), amodiaquine (RR 0.22, 95% CI 0.13 to 0.36; 342 participants, 2 trials), and mefloquine (RR 0.04, 95% CI 0.00 to 0.73; 158 participants, 1 trial). There were insufficient data to draw a conclusion for this outcome from comparisons with sulfadoxine‐pyrimethamine (172 participants, 2 trials), halofantrine (205 participants, 1 trial), artesunate plus mefloquine (1063 participants, 1 trial), quinine plus tetracycline (154 participants, 1 trial), and dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine (161 participants, 1 trial). Adverse events were mainly common symptoms of malaria and did not differ in frequency between groups.
Authors' conclusions
Data are limited but appear to suggest that atovaquone‐proguanil is more effective than chloroquine, amodiaquine, and mefloquine. There are insufficient data for comparisons against sulfadoxine‐pyrimethamine, halofantrine, artesunate plus mefloquine, quinine plus tetracycline, and dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine in treating malaria. There are not enough data to assess safety, but a number of adverse events were identified with all drugs. Large trials comparing atovaquone‐proguanil with other new combination therapies are needed.
22 March 2019
Update pending
Authors currently updating
The update is due to be published in 2019.
Plain language summary
Atovaquone‐proguanil appears to be more effective than individual drugs for treating uncomplicated malaria, but there are few data comparing atovaquone‐proguanil to other combination therapies
Many conventional treatments for uncomplicated malaria are failing because malaria parasites develop resistance to them. This can be reduced by treating people with combination drugs such as atovaquone‐proguanil. The review found 10 trials, most of low methodological quality and most funded by a single pharmaceutical company. In addition, trials were small and had few participants thus evidence suggesting atovaquone‐proguanil as more effective than a number of single drug treatments at eliminating the Plasmodium falciparum malaria parasite from the blood was limited. There were few good quality data comparing atovaquone‐proguanil with other new combination therapies. There were not enough data to assess adverse events, but all trials recorded some adverse events.
Background
Malaria is a parasitic, mosquito‐borne disease that causes about 300 million clinical cases and more than one million deaths each year (WHO 1999). Young children, pregnant women, and non‐immune people who move into endemic regions are most vulnerable. Over 90% of deaths occur in children under five years of age living in sub‐Saharan Africa (WHO 1999). The associated economic burden of the disease means malaria is a significant impediment to human development in poor countries, where it accounts for 40% of public health expenditure, 30% to 50% of inpatient admissions, and up to 50% of outpatient visits (WHO 2003).
There are four species in the genus Plasmodium that cause malaria in humans: falciparum, malariae, vivax, and ovale. Plasmodium falciparum accounts for 93% of all cases in Africa (RBM 2005). Falciparum malaria can present as uncomplicated or severe forms. Uncomplicated malaria is diagnosed when the plasmodium parasite is seen in the blood and the person has fever (> 37.5 °C) and any of the following symptoms: headache, chills and rigors, general weakness, joint or muscle weakness, abdominal pain, and vomiting (WHO 2001). The disease is said to have progressed to severe malaria when, in addition to the above clinical features, the person also develops a life‐threatening complication such as anaemia (low haemoglobin concentration), convulsions, hypoglycaemia (low blood sugar), or cerebral malaria (malaria with altered level of consciousness or coma) (WHO 2000). Malaria treatment is complicated by the ability of the parasites to develop resistance to antimalarial drugs. Many antimalarial drugs are no longer effective at treating malaria when used alone (monotherapy). For example, 30% of people have chloroquine‐resistant malaria in Uganda (Kamya 2002). One way in which this resistance manifests is through recrudescence, which is the re‐appearance of the same parasite that had been treated earlier. Recrudesced parasites can be distinguished from new ones using a technique called polymerase chain reaction (PCR). Distinguishing new infections from treated infections can help researchers measure the effectiveness of an antimalarial drug (Brockman 1999; Ohrt 1997).
There is a need for safe and effective new therapies to treat malaria. Atovaquone‐proguanil (brand name Malarone) is a combination therapy used to treat multiple‐drug‐resistant malaria (Blanchard 1994; Sabchareon 1998), or prevent it (Overbosch 2001; Sukwa 1999). In combination therapy, two or more drugs with independent modes of action and different biochemical targets in the parasite are used together. This delays the development of resistance to the component drugs thereby prolonging the life span of still effective antimalarial drugs. Combination therapies are recommended for malaria contracted in areas where there is resistance to multiple antimalarial drugs (RBM 2001a).
Atovaquone‐proguanil may be taken as a fixed‐dose combination or as the individual drugs co‐administered together. The fixed‐dose combination (Malarone) contains 250 mg atovaquone and 100 mg proguanil hydrochloride per adult strength tablet and 62.5 mg atovaquone and 25 mg proguanil hydrochloride per child strength tablet. Atovaquone is a synthetic hydroxynaphthoquinone that inhibits mitochondrial electron transport in the parasite (Fry 1992). It is a compound with a high affinity for lipids, and its rate and extent of absorption is increased by dietary fat. It is highly protein bound (> 99%) and is predominantly eliminated unchanged through faeces. The elimination half life is two to three days in adults and one to two days in children (Beerahee 1999). Proguanil is a biguanide derivative that works by inhibiting the parasite's dihydrofolic reductase enzyme via a cyclic triazine metabolite (cycloguanil) (Dollery 1991). Proguanil is partially metabolized and partially excreted in urine. The excretion of its principal metabolite, cycloguanil, is also through urine. Both have elimination half lives of 12 to 15 hours in adults and children, respectively (Beerahee 1999).
The combination of atovaquone‐proguanil appears to act synergistically (Canfield 1995), that is each constituent potentiates the effect of the other against the plasmodium parasite, thereby facilitating its role in the treatment of drug‐resistant malaria. When used alone, either individual agent is associated with high numbers of treatment failures. For example, Looareesuwan 1996 studied 317 people with uncomplicated malaria in Thailand and showed failure rates of 33% when atovaquone was used alone. But when combined with proguanil, treatment failure was less than 3% (Looareesuwan 1996; Sabchareon 1998). However, there are concerns that high failure rates for atovaquone may ultimately affect the useful lifespan of atovaquone‐proguanil if it is used widely (Van Vugt 2002).
Adverse effects associated with atovaquone‐proguanil include abdominal pain, anorexia, nausea, diarrhoea, headache, and cough (Looareesuwan 1999b). Neuropsychiatric effects, such as strange and vivid dreams, insomnia, dizziness and vertigo, anxiety, and depression, have also been reported (Hogh 2000; Overbosch 2001).
Atovaquone‐proguanil is an expensive antimalarial drug, costing US$42 for an adult treatment dose (RBM 2001b). Due to its effectiveness against malaria resistant to first‐line therapies, the manufacturers GlaxoSmithKline decided to donate one million doses of the drug for the management of uncomplicated malaria in poor countries where first‐line therapies have failed (Malarone Donation Programme). The impact and consequences of this programme have been hotly debated (Bloland 1997; Foege 1997; Oyediran 2002; Ringwald 1998; Shretta 2000; Shretta 2001). It was discontinued in September 2001 following the pilot phase, which ran for two and a half years in Kenya and Uganda, due to the conclusion that it would not be an efficient or effective use of malaria resources (Oyediran 2002; Partnerships 2002). This review evaluates the reported evidence on the effectiveness and safety of atovaquone‐proguanil. It also aims to help policy makers make an informed choice of antimalarial for treating uncomplicated P. falciparum malaria.
Objectives
To compare atovaquone‐proguanil with other antimalarial drugs (alone or in combination) for treating children and adults with confirmed uncomplicated P. falciparum malaria.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials.
Types of participants
Children and adults with confirmed uncomplicated P. falciparum malaria.
Uncomplicated malaria is defined as the presence of the plasmodium parasite on blood film in association with fever (temperature of > 37.5°C) and any of the following symptoms: headache, chills and rigors, general weakness, joint or muscle weakness, abdominal pain, vomiting (WHO 2001).
Types of interventions
Intervention
Atovaquone‐proguanil.
Control
Other antimalarial drugs (used alone or in combination).
Types of outcome measures
Primary
Treatment failure* (unadjusted) on or by day 28 (drugs with a half life less or equal to seven days) or day 42 (drugs with a half life greater than seven days), including new infections.
Treatment failure* (adjusted) on or by day 28 (drugs with a half life less or equal to seven days) or day 42 (drugs with a half life greater than seven days), excluding new infections detected by PCR.
Secondary
Treatment failure* on or by day 14.
Parasite clearance time.
Fever clearance time.
Progression to severe malaria.
*Treatment failure is defined as parasitological or clinical evidence of treatment failure between start of treatment and days 14, 28, or 42.
Adverse events
Serious adverse events (fatal, life threatening, or require hospitalization).
Adverse events that result in the discontinuation of treatment.
Other adverse events.
Search methods for identification of studies
We attempted to identify all relevant trials regardless of language or publication status (published, unpublished, in press, and in progress).
Databases
We searched the following databases using the search terms and strategy described in Appendix 1 : Cochrane Infectious Diseases Group Specialized Register (June 2005); Cochrane Central Register of Controlled Trials (CENTRAL), published in The Cochrane Library (Issue 2, 2005); MEDLINE (1966 to June 2005); EMBASE (1980 to June 2005); and LILACS (1982 to June 2005).
Conference proceedings
We searched the following conference proceedings for relevant abstracts: Third European Congress on Tropical Medicine and International Health, Lisbon, Portugal, 8 to 11 September 2002; and The Third Multilateral Initiative on Malaria Pan‐African Conference, Arusha, Tanzania, 18 to 22 November 2002.
Researchers and pharmaceutical companies
We circulated a list of identified studies to individual researchers working in the field and to GlaxoSmithKline to help identify additional trials and provide information on ongoing trials.
Reference lists
We checked the citations of existing reviews and of all trials identified by the above methods.
Data collection and analysis
Selection of studies
The first author screened the results of the search strategy for potentially relevant trials and retrieved the full articles of these trials. Each trial report was scrutinized for multiple publications from the same data set. Using an eligibility form based on the inclusion criteria, the three authors independently assessed the trials for inclusion in the review. We resolved any disagreements through discussion or referred them to Harriet G MacLehose (HGM). We excluded studies that did not meet the inclusion criteria and have stated the reasons for exclusion in the 'Characteristics of excluded studies'.
Data extraction and management
Using a data extraction form, the three authors independently extracted data on the trial characteristics including methods, participants, interventions, and outcomes. We resolved any disagreements by referring to the trial report and through discussion or by consulting HGM. We sought additional information from the authors if data from the trial reports were insufficient or missing.
We attempted to extract data to allow an intention‐to‐treat analysis (the analysis was to include all the participants in the groups to which they were originally randomly assigned). We calculated the percentage loss to follow up and reported this in the 'Characteristics of included studies' when there was inconsistency between the number of participants randomized and analysed. For dichotomous data, we recorded the number of participants who experienced the event in each group of the trial. For trials reporting fever and parasite clearance times as mean values, we extracted these values and standard deviations for each group. We also reported the medians and ranges in tables where available.
Assessment of risk of bias in included studies
The three authors independently assessed the risk of bias in each trial. We classified generation of allocation sequence and allocation concealment as adequate, inadequate, or unclear according to Jüni 2001. We described who was blinded, such as the participants, care provider, or outcome assessor. We considered the inclusion of all randomized participants in the analysis (follow up) to be adequate if it was greater than 90%.
Data synthesis
We analysed the data with Review Manager 5 using the fixed‐effect model. We pooled data only where it was appropriate and did not combine results of trials with different comparator drugs. We compared outcome measures using risk ratio (RR) for dichotomous data, mean difference (MD) for continuous data, and 95% confidence intervals (CI).
We assessed heterogeneity between trials by inspecting the forest plots and using the chi‐squared test with a 10% level of statistical significance. We did not detect heterogeneity, but if we do in future updates, and it would still be appropriate to pool the data, we will use the random‐effects model. We intended to explore potential sources of heterogeneity by conducting subgroup analyses by participant age (less or equal to five years old versus greater than five years old), the presence of drug resistance, and drug dose. However, this was not possible due to missing information and lack of replication of drug comparisons.
Once we had included all the trials in the primary analysis, we intended to conduct sensitivity analyses for each of the methodological quality factors. However, this was not possible as there were not enough data for a meaningful analysis.
Results
Description of studies
Trial selection
We identified 11 trials of which 10 fulfilled our inclusion criteria. These are described below and detailed in the 'Characteristics of included studies'. We excluded one trial because the protocol was amended after 40 participants had been recruited so that participants in the comparator group received chloroquine plus sulfadoxine‐pyrimethamine instead of the original chloroquine (Bustos 1999). We also excluded 533 participants from Van Vugt 2002 because they received a combination of atovaquone‐proguanil and artesunate, but this did not upset randomization of the trial.
Trial design
All 10 trials stated that they were randomized controlled trials.
Trial location
Four trials were conducted in Africa − Kenya (Anabwani 1999), Zambia (Mulenga 1999), and Gabon (Borrmann 2003; Radloff 1996), three in South‐East Asia − Thailand (Looareesuwan 1999a; Van Vugt 2002) and Viet Nam (Giao 2004), two in South America − Brazil (De Alencar 1997) and Peru (Llanos‐Cuentas 2001), and one trial in non‐immune participants who had returned from the tropics to France, Europe (Bouchaud 2000).
Participants
There were 2345 participants aged three months to 70 years in the 10 trials. Two trials recruited only children (Anabwani 1999; Borrmann 2003), and three recruited only adults (Bouchaud 2000; De Alencar 1997; Giao 2004).
Interventions
All the drugs were administered orally.
Atovaquone‐proguanil
Atovaquone and proguanil were given in a daily dose of 1000 mg and 400 mg respectively for three days in all except three trials, which recruited children. One trial used a fixed combination containing 62.5 mg atovaquone and 25 mg proguanil hydrochloride (Borrmann 2003). In the other two trials, participants received doses of 15 to 20 mg/kg atovaquone and 8 mg/kg proguanil (Anabwani 1999; Van Vugt 2002).
Control drugs
One trial used chloroquine (Llanos‐Cuentas 2001). This trial's protocol was amended and sulfadoxine‐pyrimethamine replaced chloroquine after 25 participants were recruited because the cure rate was 8% with chloroquine; it was unclear if the amendment was a result of a formal pre‐planned measure. The comparator drugs in the other trials were amodiaquine (Borrmann 2003; Radloff 1996), sulfadoxine‐pyrimethamine (Mulenga 1999), quinine plus tetracycline (De Alencar 1997), halofantrine (Anabwani 1999; Bouchaud 2000), mefloquine (Looareesuwan 1999a), artesunate plus mefloquine (Van Vugt 2002), and dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine (Giao 2004).
Outcomes
All ten trials reported results for treatment failure on or by day 28, parasite clearance and fever clearance times, and adverse events. Two trials used PCR to separate new infections from recrudescence (Borrmann 2003; Van Vugt 2002), but the PCR findings for Borrmann 2003 were not reported in the publication, and consequently we were unable to include them in this review. Giao 2004 reported on recrudescence without genotyping based on the assumption that, due to low transmission rates, recrudescence during the first two weeks was more likely than reinfection. Mulenga 1999 reported the outcome progression to severe disease.
Length of follow up
Eight trials followed up participants to day 28, one to day 35 (Bouchaud 2000), and one to day 42 (Van Vugt 2002). Giao 2004 followed a subset of 92 participants up to 56 days.
Drug resistance
Chloroquine resistance was reported in three study areas (Llanos‐Cuentas 2001; Mulenga 1999; Radloff 1996). Resistance to both chloroquine and sulfadoxine‐pyrimethamine was reported in two trials (De Alencar 1997; Looareesuwan 1999a). De Alencar 1997 also reported some resistance to quinine. Van Vugt 2002 reported multiple‐drug resistance, with resistance to most drugs with the exception of artemisinin derivatives. Anabwani 1999, Borrmann 2003, and Bouchaud 2000 did not clearly describe the drug resistance pattern of their study areas, and it was unclear in Giao 2004.
Source of funding
GlaxoSmithKline, manufacturers of Malarone, supported nine trials by either donating drugs or by providing a grant. The Wellcome Research Laboratories supported and donated drugs to Radloff 1996.
Risk of bias in included studies
See the 'Characteristics of included studies' for details andTable 1for a summary.
1. Risk of bias assessment.
| Trial | Allocation sequence generation | Allocation concealment | Blinding | Inclusiona |
| Anabwani 1999 | Unclear | Unclear | None | Adequate |
| Borrmann 2003 | Adequate | Adequate | None | Inadequate |
| Bouchaud 2000 | Unclear | Unclear | None | Inadequate |
| De Alencar 1997 | Unclear | Unclear | None | Inadequate |
| Giao 2004 | Adequate | Adequate | None | Adequate |
| Llanos‐Cuentas 2001 | Unclear | Unclear | None | Adequate |
| Looareesuwan 1999a | Unclear | Unclear | None | Inadequate |
| Mulenga 1999 | Unclear | Unclear | None | Adequate |
| Radloff 1996 | Adequate | Unclear | None | Inadequate |
| Van Vugt 2002 | Adequate | Adequate | None | Adequate |
aSee the 'Characteristics of included studies' for details. bInclusion of all randomized participants in the final analysis.
Generation of allocation sequence
Four trials reported an adequate method to generate the allocation sequence (Borrmann 2003; Giao 2004; Radloff 1996; Van Vugt 2002). It was unclear how the allocation sequence was generated in the other six trials.
Allocation concealment
Three trials used an adequate method (sealed envelope) to conceal allocation (Borrmann 2003; Giao 2004; Van Vugt 2002). The other trials did not report on this, therefore concealment is unclear.
Blinding
None of the trials used blinding.
Inclusion of all randomized participants
Five trials reported that more than 90% of the randomized participants were included in the analysis (adequate) (Anabwani 1999; Giao 2004; Llanos‐Cuentas 2001; Mulenga 1999; Van Vugt 2002). The other five included 90% or less (inadequate). One hundred and eleven participants across all the trials could not be evaluated for various reasons including withdrawal and loss to follow up.
Effects of interventions
Versus chloroquine
One trial: Llanos‐Cuentas 2001.
Treatment failure on or by day 28
By day 28, parasitaemia prevalence was statistically significantly lower in the atovaquone‐proguanil group (RR 0.04, 95% CI 0.00 to 0.57; 27 participants) (seeAnalysis 1.1 and Appendix 2).
1.1. Analysis.
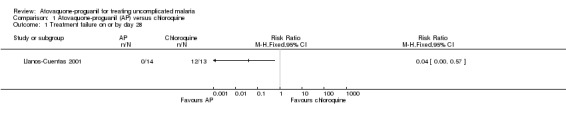
Comparison 1 Atovaquone‐proguanil (AP) versus chloroquine, Outcome 1 Treatment failure on or by day 28.
Parasite and fever clearance times
There was no statistically significant difference in mean parasite clearance time (22 participants; seeAnalysis 1.2) or mean fever clearance time (27 participants; seeAnalysis 1.3) between treatment groups. The median parasite clearance time was statistically shorter in the chloroquine group (seeAppendix 2), however there was no statistically significant difference in the median fever clearance times (seeAppendix 3).
1.2. Analysis.
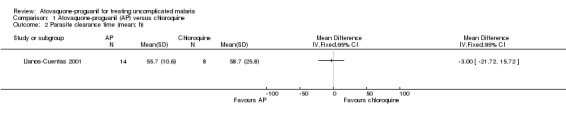
Comparison 1 Atovaquone‐proguanil (AP) versus chloroquine, Outcome 2 Parasite clearance time (mean; h).
1.3. Analysis.
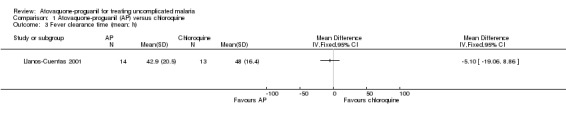
Comparison 1 Atovaquone‐proguanil (AP) versus chloroquine, Outcome 3 Fever clearance time (mean; h).
Adverse events
The adverse events reported were common symptoms of malaria (seeAnalysis 1.4). Most occurred with a similar frequency in both groups, but headache was reported less frequently in the atovaquone‐proguanil group (RR 0.23, 95% CI 0.05 to 0.99; 34 participants). One participant in the atovaquone‐proguanil group had generalized seizures from hyponatraemia that the trialists reported as a "severe adverse event".
1.4. Analysis.
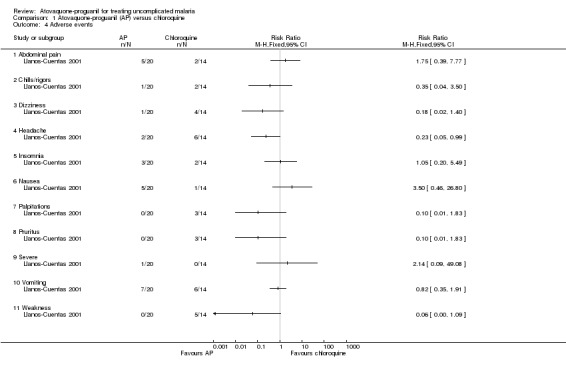
Comparison 1 Atovaquone‐proguanil (AP) versus chloroquine, Outcome 4 Adverse events.
Versus amodiaquine
Two trials: Borrmann 2003 and Radloff 1996.
Treatment failure on or by day 28
A combined estimate of the two trials showed that parasitaemia prevalence by day 28 was statistically significantly lower in the atovaquone‐proguanil group (RR 0.22, 95% CI 0.13 to 0.36; 342 participants; seeAnalysis 2.1 and Appendix 2). We acknowledge that although the sizes of the trials were reasonably comparable, Borrmann 2003 received 90% of the weight due to greater frequency of treatment failures.
2.1. Analysis.
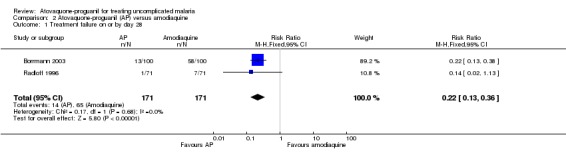
Comparison 2 Atovaquone‐proguanil (AP) versus amodiaquine, Outcome 1 Treatment failure on or by day 28.
Radloff 1996 reported parasitaemia at day 14, but there was no statistically significant difference between treatment groups (142 participants; seeAnalysis 2.2).
2.2. Analysis.
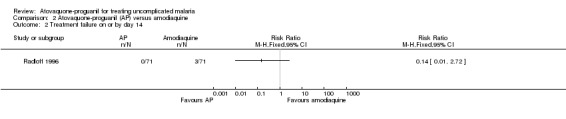
Comparison 2 Atovaquone‐proguanil (AP) versus amodiaquine, Outcome 2 Treatment failure on or by day 14.
Parasite clearance time
Radloff 1996 did not report a statistically significant difference in mean parasite clearance time between the two groups (142 participants; seeAnalysis 2.3). Borrmann 2003 reported median parasite clearance times (interquartile range), which were 72 hours (10) in the atovaquone‐proguanil group (92 participants) compared with 72 hours (24) in the amodiaquine group (78 participants), P = 0.0002 (seeAppendix 3).
2.3. Analysis.
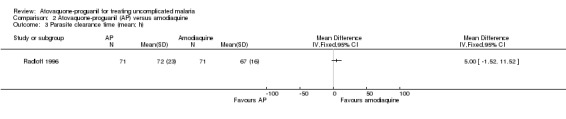
Comparison 2 Atovaquone‐proguanil (AP) versus amodiaquine, Outcome 3 Parasite clearance time (mean; h).
Fever clearance time
This did not differ statistically significantly between the two treatment groups in Radloff 1996 (142 participants; seeAnalysis 2.4). Borrmann 2003 reported median fever clearance times (interquartile range), which were 47 hours (48) in the atovaquone‐proguanil group (92 participants) compared with 46 hours (43) in the amodiaquine group (78 participants), P = 0.85 (Appendix 4).
2.4. Analysis.
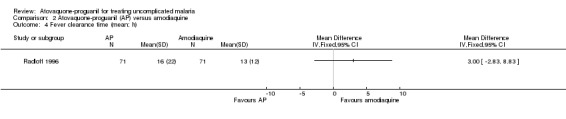
Comparison 2 Atovaquone‐proguanil (AP) versus amodiaquine, Outcome 4 Fever clearance time (mean; h).
Adverse events
Borrmann 2003 reported that most adverse events were mild to moderate in intensity, and that there was no difference in the numbers of adverse events between the two groups. Diarrhoea, cough, and vomiting were the most frequently reported events. Four serious adverse events requiring hospitalization were reported, three (severe anaemia, dystonia, and pneumonia) in the amodiaquine group and one (convulsions) in the atovaquone‐proguanil group. Radloff 1996 reported a statistically significant increase in complaints of abdominal pain (RR 2.80, 95% CI 1.07 to 7.31; 126 participants) and nausea (RR 2.63, 95% CI 1.26 to 5.48; 126 participants) in the atovaquone‐proguanil group, and a decrease in pruritus (RR 0.11, 95% CI 0.04 to 0.35; 126 participants), insomnia (RR 0.29, 95% CI 0.12 to 0.75; 126 participants), and dizziness (RR 0.27, 95% CI 0.08 to 0.93; 126 participants) in the atovaquone‐proguanil group (seeAnalysis 2.5). Both trial authors found weakness was reported less frequently in the atovaquone‐proguanil group (RR 0.12, 95% CI 0.02 to 0.64; 326 participants, 2 trials; seeAnalysis 2.5).
2.5. Analysis.
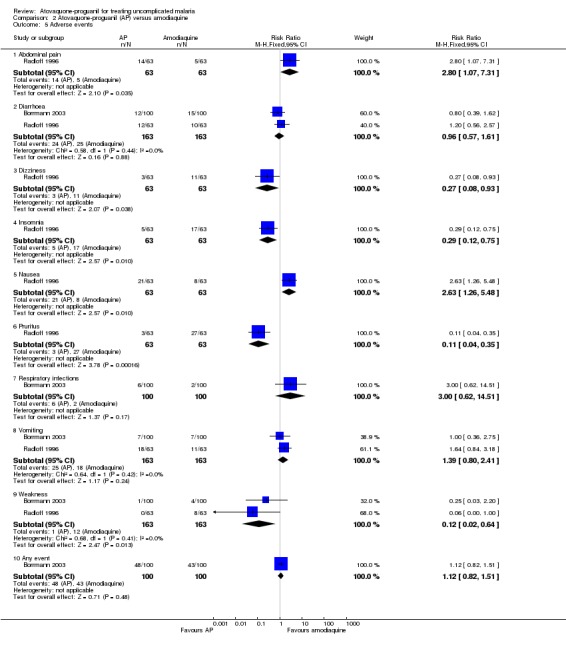
Comparison 2 Atovaquone‐proguanil (AP) versus amodiaquine, Outcome 5 Adverse events.
Versus sulfadoxine‐pyrimethamine
Two trials: Llanos‐Cuentas 2001 and Mulenga 1999
Treatment failure on or by day 28
There was no statistically significant difference between treatment groups (172 participants; seeAnalysis 3.1 and Appendix 2). Llanos‐Cuentas 2001 found no parasites in either treatment group (total of 12 participants), while in Mulenga 1999 one participant (sulfadoxine‐pyrimethamine group) had parasitaemia (total of 160 participants).
3.1. Analysis.
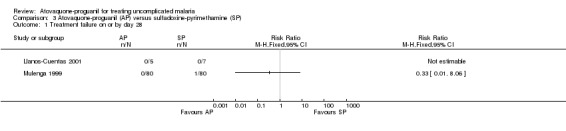
Comparison 3 Atovaquone‐proguanil (AP) versus sulfadoxine‐pyrimethamine (SP), Outcome 1 Treatment failure on or by day 28.
Parasite clearance time
A combined estimate of both trials showed a statistically significantly shorter mean parasite clearance time in the sulfadoxine‐pyrimethamine group (MD 11.23 h, 95% CI 5.43 to 17.03; 174 participants; seeAnalysis 3.2). The median parasite clearance times for both groups were the same in Llanos Cuentas 2001, but Mulenga 1999 found a statistically significantly shorter median parasite clearance time for sulfadoxine‐pyrimethamine (seeAppendix 3).
3.2. Analysis.
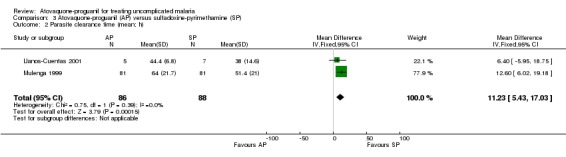
Comparison 3 Atovaquone‐proguanil (AP) versus sulfadoxine‐pyrimethamine (SP), Outcome 2 Parasite clearance time (mean; h).
Fever clearance time
A combined estimate of both trials showed a statistically significantly shorter mean fever clearance time with atovaquone‐proguanil (MD ‐13.73 h, 95% CI ‐21.31 to ‐6.16; 156 participants; seeAnalysis 3.3). Llanos‐Cuentas 2001 reported similar median fever clearance times for both groups, whilst Mulenga 1999 stated a shorter median fever clearance time for atovaquone‐proguanil (seeAppendix 4).
3.3. Analysis.
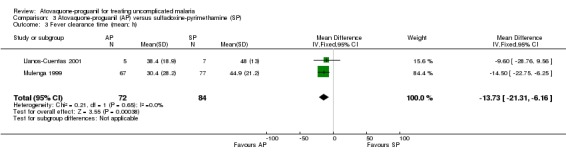
Comparison 3 Atovaquone‐proguanil (AP) versus sulfadoxine‐pyrimethamine (SP), Outcome 3 Fever clearance time (mean; h).
Progression to severe disease
Mulenga 1999 reported that one of 80 in the sulfadoxine‐pyrimethamine group and zero of 80 in the atovaquone‐proguanil group progressed to severe disease (seeAnalysis 3.4).
3.4. Analysis.
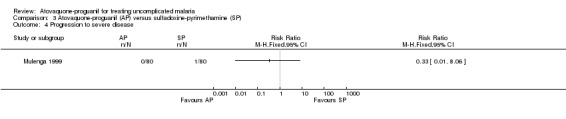
Comparison 3 Atovaquone‐proguanil (AP) versus sulfadoxine‐pyrimethamine (SP), Outcome 4 Progression to severe disease.
Adverse events
The adverse events reported in both trials were said to be typical of malaria symptoms (seeAnalysis 3.5). Most adverse events occurred with a similar frequency in both groups although there were some exceptions. Llanos‐Cuentas 2001 found that nausea occurred more frequently in the atovaquone‐proguanil group, but this was not statistically significant. Mulenga 1999 reported that diarrhoea occurred more frequently in the atovaquone‐proguanil group (RR 43.00, 95% CI 2.65 to 697.91, 160 participants) and orthostatic hypotension occurred less frequently in the atovaquone‐proguanil group (RR 0.03, 95% CI 0.00 to 0.57; 160 participants). In Llanos‐Cuentas 2001, one participant in the atovaquone‐proguanil group had generalized seizures from hyponatraemia, which the trialists reported as a "severe adverse event". This adverse event has been mentioned previously under the comparison with chloroquine.
3.5. Analysis.
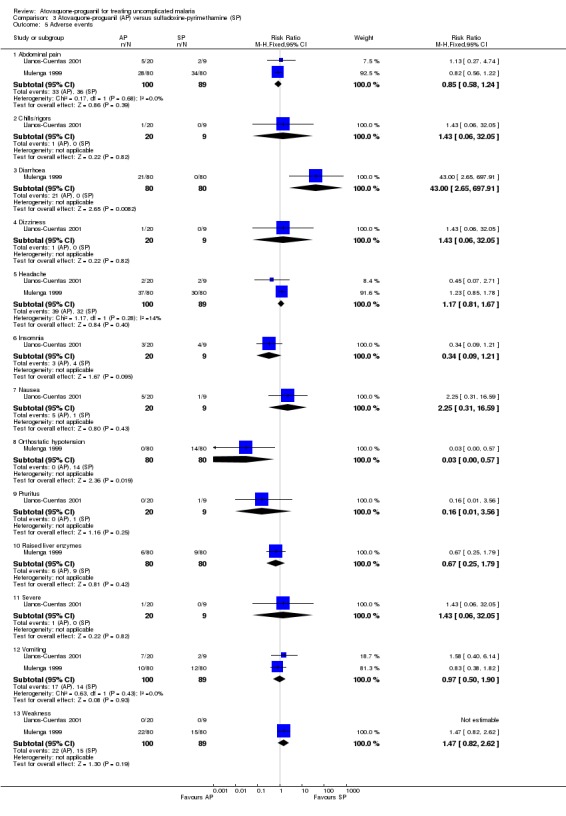
Comparison 3 Atovaquone‐proguanil (AP) versus sulfadoxine‐pyrimethamine (SP), Outcome 5 Adverse events.
Versus quinine plus tetracycline
One trial: De Alencar 1997.
Treatment failure on or by day 28
All participants in both groups were without parasites by day 14, but one of 77 participants in the atovaquone‐proguanil group was parasitaemic on day 21. There was no statistically significant difference between treatment groups at day 28 (seeAnalysis 4.1 and Appendix 2).
4.1. Analysis.
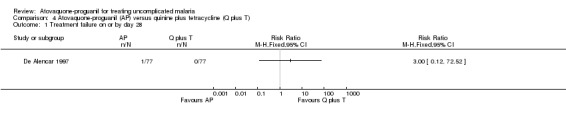
Comparison 4 Atovaquone‐proguanil (AP) versus quinine plus tetracycline (Q plus T), Outcome 1 Treatment failure on or by day 28.
Parasite and fever clearance times
Both the mean parasite clearance times (MD ‐8.40 h, 95% CI ‐14.44 to ‐2.36; 154 participants; seeAnalysis 4.2) and mean fever clearance times (MD ‐9.70 h, 95% CI ‐16.48 to ‐2.92; 119 participants; seeAnalysis 4.3) were statistically significantly shorter in the atovaquone‐proguanil group.
4.2. Analysis.
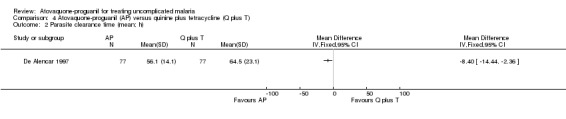
Comparison 4 Atovaquone‐proguanil (AP) versus quinine plus tetracycline (Q plus T), Outcome 2 Parasite clearance time (mean; h).
4.3. Analysis.
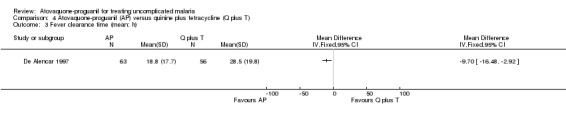
Comparison 4 Atovaquone‐proguanil (AP) versus quinine plus tetracycline (Q plus T), Outcome 3 Fever clearance time (mean; h).
Adverse events
The common adverse events reported were similar to symptoms of malaria (seeAnalysis 4.4). However, there were fewer complaints of tinnitus (RR 0.05, 95% CI 0.02 to 0.17; 154 participants) and dizziness (RR 0.26, 95% CI 0.14 to 0.48; 154 participants) in the atovaquone‐proguanil group. No serious adverse events were reported.
4.4. Analysis.
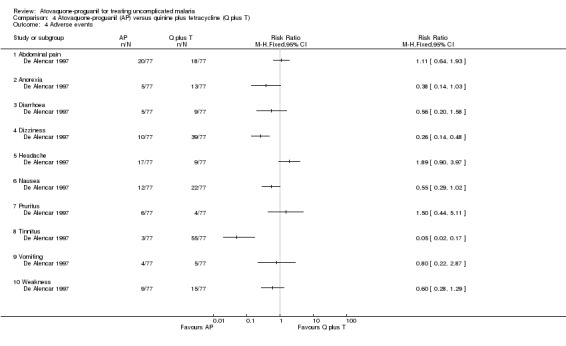
Comparison 4 Atovaquone‐proguanil (AP) versus quinine plus tetracycline (Q plus T), Outcome 4 Adverse events.
Versus halofantrine
Two trials: Anabwani 1999 and Bouchaud 2000
Treatment failure on or by day 28
There was no statistically significant difference in the number of participants with parasitaemia between the atovaquone‐proguanil group (5/81) and the halofantrine group (8/83) by day 28 (Anabwani 1999). No participants in either treatment group were parasitaemic by day 28 in Bouchaud 2000. A combined estimate of these trials shows no statistically significant difference in parasitaemia prevalence between the atovaquone‐proguanil and halofantrine groups at day 28 (205 participants; seeAnalysis 5.1 and Appendix 2).
5.1. Analysis.
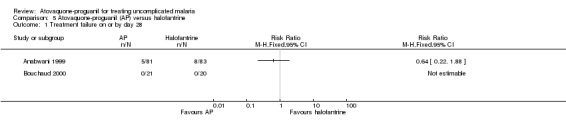
Comparison 5 Atovaquone‐proguanil (AP) versus halofantrine, Outcome 1 Treatment failure on or by day 28.
Parasite clearance time
The mean parasite clearance time was statistically significantly longer for those treated with atovaquone‐proguanil than halofantrine (MD 14.76 h, 95% CI 10.41 to 19.10; 205 participants, 2 trials; seeAnalysis 5.2). The median parasite clearance time as reported by Anabwani 1999 was also statistically longer for the atovaquone‐proguanil group (seeAppendix 3).
5.2. Analysis.
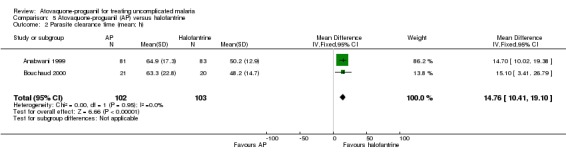
Comparison 5 Atovaquone‐proguanil (AP) versus halofantrine, Outcome 2 Parasite clearance time (mean; h).
Fever clearance time
We did not detect a difference in the mean time to fever clearance between participants receiving atovaquone‐proguanil or halofantrine (205 participants, 2 trials; seeAnalysis 5.3), but the confidence intervals are very wide suggesting that there is not enough power to show any differences. The median fever clearance times reported by Anabwani 1999 showed no statistically significant difference between the two groups (seeAppendix 4).
5.3. Analysis.
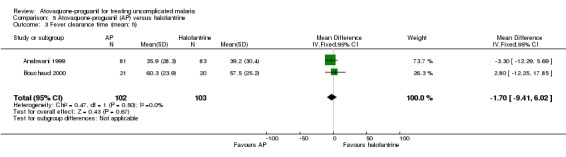
Comparison 5 Atovaquone‐proguanil (AP) versus halofantrine, Outcome 3 Fever clearance time (mean; h).
Adverse events
One hundred and nineteen adverse events were reported in the children who received halofantrine compared with 73 adverse events in those who received atovaquone‐proguanil. Moderate to severe events, as classified by the trial authors, occurred less often in the atovaquone‐proguanil group (6 adverse events) compared with the halofantrine group (16 adverse events) (RR 0.38, 95% CI 0.15 to 0.91; 168 participants, Anabwani 1999; seeAnalysis 5.4). Both trials reported that participants vomited more frequently in the atovaquone‐proguanil group. Three of the 11 participants who vomited after treatment with atovaquone‐proguanil in the Bouchaud 2000 trial were withdrawn from treatment.
5.4. Analysis.
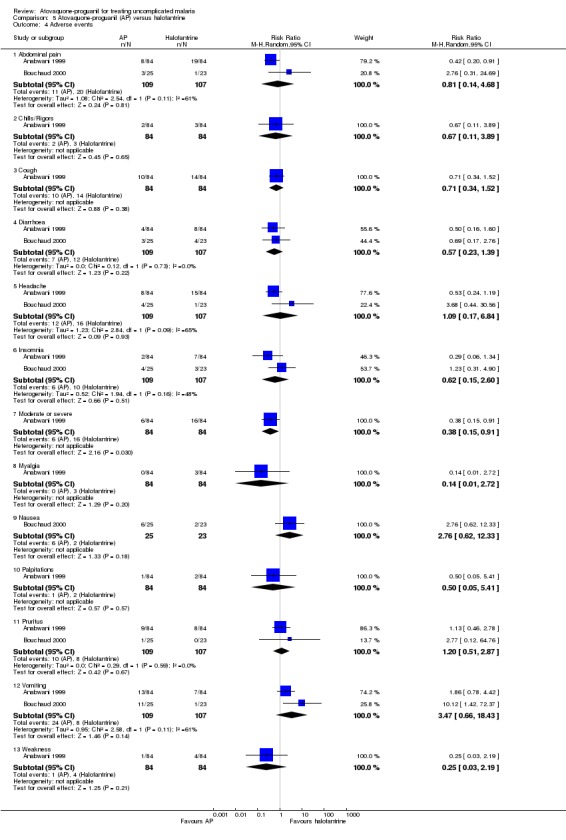
Comparison 5 Atovaquone‐proguanil (AP) versus halofantrine, Outcome 4 Adverse events.
Versus mefloquine
One trial: Looareesuwan 1999a.
Treatment failure on or by day 28
By day 28, statistically significantly less participants in the atovaquone‐proguanil group experienced treatment failure (RR 0.04, 95% CI 0.00 to 0.73; 158 participants; seeAnalysis 6.1 and Appendix 2). None of the 79 participants receiving atovaquone‐proguanil were parasitaemic, while 11 out of 79 receiving mefloquine had parasites on day 28.
6.1. Analysis.
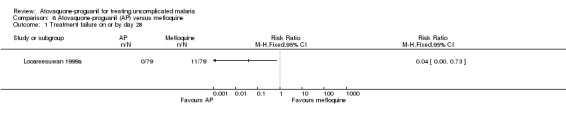
Comparison 6 Atovaquone‐proguanil (AP) versus mefloquine, Outcome 1 Treatment failure on or by day 28.
Parasite clearance time
Mean parasite clearance time was statistically significantly shorter for those receiving atovaquone‐proguanil (MD ‐8.60 h, 95% CI ‐16.08 to ‐1.12; 158 participants; seeAnalysis 6.2). Trialists reported similar median parasite clearance times for atovaquone‐proguanil and mefloquine (seeAppendix 3).
6.2. Analysis.
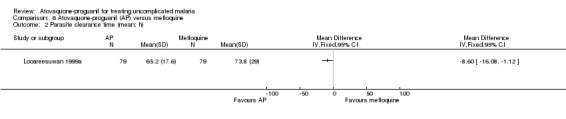
Comparison 6 Atovaquone‐proguanil (AP) versus mefloquine, Outcome 2 Parasite clearance time (mean; h).
Fever clearance time
One trial, including 79 participants, found no statistically significant difference in mean fever clearance time between the two treatment groups (MD 8.00 h, 95% CI ‐2.52 to 18.52; 158 participants; seeAnalysis 6.3); confidence intervals were wide. There was no statistically significant difference in median fever clearance times between both treatment groups (seeAppendix 4).
6.3. Analysis.
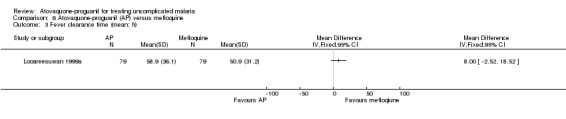
Comparison 6 Atovaquone‐proguanil (AP) versus mefloquine, Outcome 3 Fever clearance time (mean; h).
Adverse events
Adverse events were reported in 36% (33/91) and 35% (32/91) of participants treated with atovaquone‐proguanil and mefloquine, respectively (seeAnalysis 6.4). Vomiting was the most frequent adverse event in the atovaquone‐proguanil group occurring in 10% (9/91) of participants compared with 2% (2/91) of participants in the mefloquine group (RR 4.50, 95% CI 1.00 to 20.26; 182 participants). Raised liver enzymes also occurred more frequently in the atovaquone‐proguanil group (RR 2.50, 95% CI 1.02 to 6.16; 182 participants). The mefloquine group reported sore throat as their most frequent adverse event (8%) (7/91). This event occurred with a similar frequency in the atovaquone‐proguanil group. The trial authors stated that the adverse events reported were typical malaria symptoms.
6.4. Analysis.
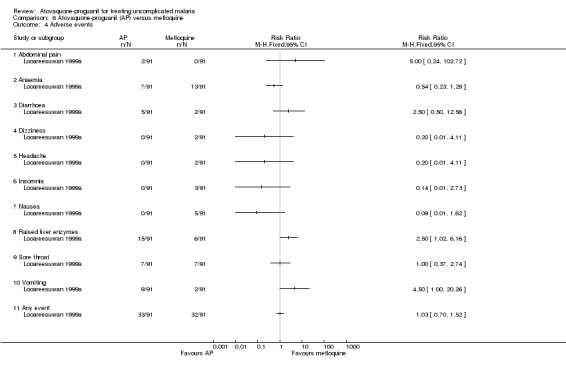
Comparison 6 Atovaquone‐proguanil (AP) versus mefloquine, Outcome 4 Adverse events.
Versus artesunate plus mefloquine
One trial: Van Vugt 2002
Treatment failure on or by day 14 and 28
This trial only reported outcomes adjusted for recrudescence by PCR. It also reported up to day 42 and the results appear similar to day 28. There were wide confidence intervals in the results and no statistically significant difference in parasitaemia at day 14 (1063 participants) or day 28 (1063 participants) (seeAnalysis 7.1 and Analysis 7.2, and Appendix 2); data extrapolated for these days.
7.1. Analysis.
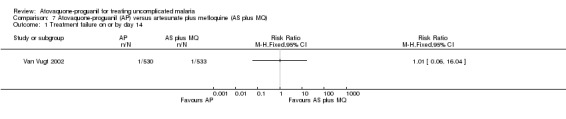
Comparison 7 Atovaquone‐proguanil (AP) versus artesunate plus mefloquine (AS plus MQ), Outcome 1 Treatment failure on or by day 14.
7.2. Analysis.
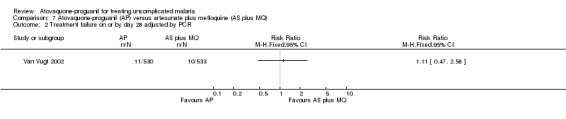
Comparison 7 Atovaquone‐proguanil (AP) versus artesunate plus mefloquine (AS plus MQ), Outcome 2 Treatment failure on or by day 28 adjusted by PCR.
Parasite and fever clearance times
Neither outcome was reported in terms of means or medians. More participants in the atovaquone‐proguanil group (36/530) still had parasites by day three compared with the artesunate plus mefloquine group (2/533). More participants in the atovaquone‐proguanil group (50/530) were still febrile by day two compared with the artesunate plus mefloquine group (6/533).
Adverse events
The trialists compared adverse events seen in the artesunate plus mefloquine arm with pooled data for the atovaquone‐proguanil and atovaquone‐proguanil plus artesunate arms (seeAnalysis 7.3). The number of adverse events was reported separately for days one to two and days three to seven. Those that received atovaquone‐proguanil had a less frequent occurrence of dizziness (RR 0.64, 95% CI 0.53 to 0.78; 1596 participants), palpitations (RR 0.62, 95% CI 0.47 to 0.83; 1596 participants), tremor (RR 0.39, 95% CI 0.17 to 0.87; 1596 participants), vomiting (RR 0.28, 95% CI 0.09 to 0.83; 1596 participants), nausea (RR 0.29, 95% CI 0.12 to 0.68; 1596 participants), and sleep disorders (RR 0.43, 95% CI 0.25 to 0.74; 1596 participants). No serious adverse events were reported.
7.3. Analysis.
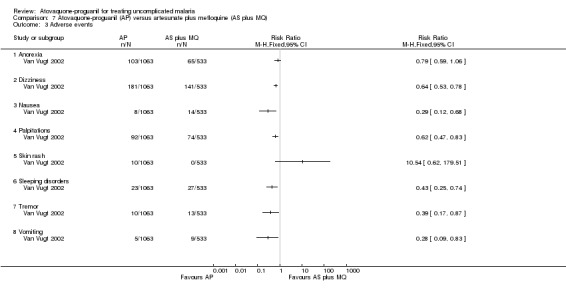
Comparison 7 Atovaquone‐proguanil (AP) versus artesunate plus mefloquine (AS plus MQ), Outcome 3 Adverse events.
Versus dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine
One trial: Giao 2004.
Treatment failure on or by day 28
There was no statistically significant difference between the treatment groups for treatment failures on or by day 28 (161 participants; seeAnalysis 8.1 and Appendix 2).
8.1. Analysis.
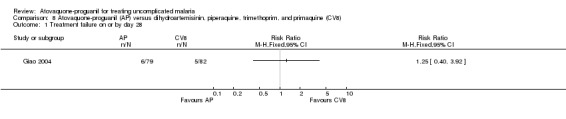
Comparison 8 Atovaquone‐proguanil (AP) versus dihydroartemisinin, piperaquine, trimethoprim, and primaquine (CV8), Outcome 1 Treatment failure on or by day 28.
Parasite and fever clearance time
There was no statistically significant difference in the mean parasite clearance time (161 participants; seeAnalysis 8.2) or mean fever clearance time (161 participants; seeAnalysis 8.3) between the two treatment groups. We derived the standard deviations for these clearance times from the confidence interval stated by the trialists. The trial article did not report on median parasite and fever clearance times.
8.2. Analysis.
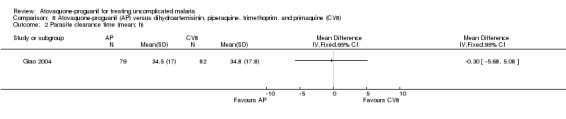
Comparison 8 Atovaquone‐proguanil (AP) versus dihydroartemisinin, piperaquine, trimethoprim, and primaquine (CV8), Outcome 2 Parasite clearance time (mean; h).
8.3. Analysis.
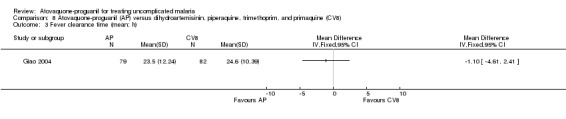
Comparison 8 Atovaquone‐proguanil (AP) versus dihydroartemisinin, piperaquine, trimethoprim, and primaquine (CV8), Outcome 3 Fever clearance time (mean; h).
Adverse events
One participant in each of the treatment groups complained of dermal itch. The atovaquone‐proguanil group was reported to have had one complaint of diarrhoea and two of vomiting, while one participant in the dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine group complained of dry mouth (same one who had an itch) and another of headache. See Analysis 8.4 for details.
8.4. Analysis.
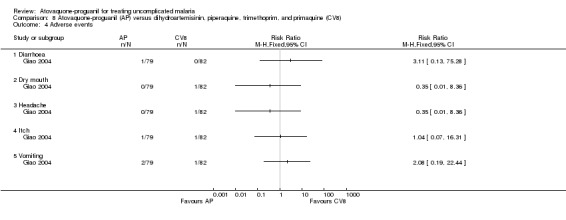
Comparison 8 Atovaquone‐proguanil (AP) versus dihydroartemisinin, piperaquine, trimethoprim, and primaquine (CV8), Outcome 4 Adverse events.
Discussion
This review set out to assess the effectiveness and safety of atovaquone‐proguanil compared with other antimalarial drugs (used alone or in combination) for treating children and adults with confirmed uncomplicated P. falciparum malaria. Many conventional therapies for malaria are failing because malaria parasites are developing resistance to them. The main interest of this review was to determine whether the combination atovaquone‐proguanil may be an effective and safe replacement.
Although over 2300 participants are included in the review, the comparisons are across eight different drugs and four geographical regions, thus statistical power to detect differences is low and generalizations are not possible. All the included trials followed up participants to day 28. However, it is now thought that patients receiving drugs with a half life of more than seven days, such as chloroquine (10 days) and mefloquine (10 to 40 days) should be followed up to day 42 (Stepniewska 2004). The trial using artesunate plus mefloquine followed up participants to day 42, but the trial using chloroquine followed up participants to day 28. Few trials used PCR to determine cure rates and re‐infection. Of the 10 included trials, seven had unclear methods of concealment, which allows potential for bias. Most of the trials reported data only for the participants deemed "evaluable" as per the protocol, usually those who completed the scheduled study period of 28 days. Only three trials carried out an intention‐to‐treat analysis, thus results may be subject to attrition bias.
Within the limitations described above, atovaquone‐proguanil performed better than chloroquine, amodiaquine, and mefloquine in terms of treatment failure on or by day 28. However, data for only 25 participants treated with chloroquine were available. All other treatment comparisons, including those with other combination therapies (sulfadoxine‐pyrimethamine, halofantrine, artesunate plus mefloquine, quinine plus tetracycline, and dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine), found no difference in treatment failure on or by day 28. This may be partly due to the small size of the trials. In addition, one of the two halofantrine trials was conducted on non‐immune participants who had contracted malaria from various endemic countries and their varying immune responses may be a contributory factor.
Atovaquone‐proguanil cleared parasites faster than quinine plus tetracycline and mefloquine. However, sulfadoxine‐pyrimethamine and halofantrine worked faster than atovaquone‐proguanil at clearing parasites. These latter trials recruited fewer participants. No difference was found between atovaquone‐proguanil and either chloroquine, amodiaquine, or dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine in clearing parasites peripherally. It is worth noting that one of the two trials that used amodiaquine as a comparator reported parasite clearance time and fever clearance times as medians with ranges and thus did not form part of the meta‐analysis.
The fever clearance times exhibit a skewed distribution, thus some caution is required in the interpretation of the pooled mean differences as they do not form a good representation of the data. However, atovaquone‐proguanil reduced fever quicker in comparison to sulfadoxine‐pyrimethamine and quinine plus tetracycline; 22% (35/154) of the randomized participants were not included in the calculation of fever clearance time, which could be a potential source of bias. There was no difference in fever clearance time between atovaquone‐proguanil and either chloroquine, amodiaquine, halofantrine, mefloquine, or dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine. Clearance times for artesunate plus mefloquine have not been discussed since the data are not explicit; we are awaiting clarification from trialists and will include these data in a future update.
One participant treated with sulfadoxine‐pyrimethamine progressed from uncomplicated malaria to severe malaria with renal insufficiency that required dialysis. None of the recipients of atovaquone‐proguanil developed this complication. Other adverse events were reported to be mostly typical of symptoms of malaria. Most of the trials reported the number of participants experiencing a particular event as a fraction of the total number of participants in that group.
Apart from the effectiveness of a drug, there are a number of other factors that should be taken into account when deciding which treatment to use. There are concerns that malaria parasites may quickly develop resistance to atovaquone‐proguanil because resistance to atovaquone is already high (Van Vugt 2002). Consequently, this combination should be used with due consideration, and the emergence of any resistance should be closely monitored. Malaria burdens the developing world, which can least afford the therapies to combat the disease. Thus, the cost of malaria treatment is a crucial factor in adopting a therapy. Atovaquone‐proguanil is expensive, and an objective cost‐benefit analysis could assist clinicians to judge which antimalarial would most benefit each patient.
Authors' conclusions
Implications for practice.
Although the data are limited, in terms of treatment failure on or by day 28, atovaquone‐proguanil performed better than chloroquine, amodiaquine, and mefloquine. There is insufficient data for comparisons against sulfadoxine‐pyrimethamine, halofantrine, artesunate plus mefloquine, quinine plus tetracycline, and dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine. Hence knowledge about the performance of this drug in areas with multiple‐drug resistance is limited.
Atovaquone‐proguanil clears parasites quicker than mefloquine and quinine plus tetracycline, is comparable to chloroquine, amodiaquine, and dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine, but slower than sulfadoxine‐pyrimethamine and halofantrine.
There are not enough data to assess safety, but a number of adverse effects of all drugs were identified.
Implications for research.
We recommend that trialists conduct large randomized controlled trials comparing atovaquone‐proguanil with new combination therapies, and follow participants for at least 28 days (if the drug half life is less than seven days) or 42 days (if the drug half life is more than seven days). Allocation concealment should be achieved through reliable means, such as sealed envelopes. We stress that data at all time points should be kept on any randomized person (irrespective of whether they are withdrawn) to allow an intention‐to‐treat analysis. Vulnerable groups such as children under five years of age should also be considered for inclusion.
Trials should assess the safety and effectiveness of atovaquone‐proguanil compared with existing best regimens for malaria, such as artemisinin‐based combination therapies.
What's new
| Date | Event | Description |
|---|---|---|
| 5 August 2008 | Amended | Converted to new review format with minor editing. |
Acknowledgements
The protocol for this Cochrane Review was developed during the Mentorship Programme organized by the Cochrane Infectious Diseases Group, July 2002. The UK Department for International Development (DFID) supports this Programme through the Effective Health Care Alliance Programme (EHCAP) at the Liverpool School of Tropical Medicine. Alex Osei‐Akoto and Shirley Owusu‐Ofori received funding from DFID through EHCAP to travel to the Liverpool School of Tropical Medicine to complete the review. Harriet G MacLehose (Assistant Editor, Cochrane Infectious Disease Group) resolved disagreements between authors about trial selection and data extraction.
This document is an output from a project funded by DFID for the benefit of developing countries. The views expressed are not necessarily those of DFID.
Appendices
Appendix 1. Search methods: detailed search strategies
| Search set | CIDG SRa | CENTRAL | MEDLINEb | EMBASEb | LILACSb |
| 1 | atovaquone | atovaquone | malaria | malaria | malaria |
| 2 | proguanil | proguanil | Exp MALARIA | Exp MALARIA | proguanil |
| 3 | Malarone | Malarone | 1 or 2 | 1 or 2 | atovaquone |
| 4 | malaria | malaria | atovaquone | atovaquone | Malarone |
| 5 | — | — | proguanil | proguanil | — |
| 6 | — | — | atovaquone‐proguanil | atovaquone‐proguanil | — |
| 7 | — | — | chloriguane | chloriguane | — |
| 8 | — | — | Chlorguanid* | cycloguanil | — |
| 9 | — | — | cycloguanil | 7 or 8 | — |
| 10 | — | — | 7 or 8 or 9 | 5 or 9 | — |
| 11 | — | — | 5 or 10 | 4 and 10 | — |
| 12 | — | — | 4 and 10 | 6 or 11 | — |
| 13 | — | — | 6 or 12 | Malarone | — |
| 14 | — | — | Malarone | 12 or 13 | — |
| 15 | — | — | 13 or 14 | 3 and 14 | — |
| 16 | — | — | 3 and 15 | — | — |
| 17 | — | — | — | — | — |
aCochrane Infectious Diseases Group Specialized Register. bSearch terms used in combination with the search strategy for retrieving trials developed by The Cochrane Collaboration (Higgins 2005); upper case: MeSH or EMTREE heading; lower case: free text term.
Appendix 2. Treatment failure on or by day 28: summary
| Atovaquone‐proguanil vs | No. trials | No. participants | Risk ratio | 95% confidence interval | Location | Trial |
| Chloroquine | 1 | 27 | 0.04 | 0.00 to 0.57 | Peru | Llanos‐Cuentas 2001 |
| Amodiaquine | 2 | 342 | 0.22 | 0.13 to 0.36 | Gabon | Borrmann 2003, Radloff 1996 |
| Sulfadoxine‐pyrimethamine | 2 | 172 | 0.33 | 0.01 to 8.06 | Peru, Zambia | Llanos‐Cuentas 2001, Mulenga 1999 |
| Quinine plus tetracycline | 1 | 154 | 3.00 | 0.12 to 72.52 | Brazil | De Alencar 1997 |
| Halofantrine | 2 | 205 | 0.64 | 0.22 to 1.88 | Kenya, France | Anabwani 1999, Bouchaud 2000 |
| Mefloquine | 1 | 158 | 0.04 | 0.00 to 0.73 | Thailand | Looareesuwan 1999a |
| Artesunate plus mefloquine | 1 | 1063 | 1.11 | 0.47 to 2.58 | Thailand | Van Vugt 2002 |
| Dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine | 1 | 161 | 1.25 | 0.40 to 3.92 | Vietnam | Giao 2004 |
aData from graphs.
Appendix 3. Parasite clearance time: median and range
| Trial | Intervention | No. participants | Median (h) | Range (h) | 95% CI | P‐value |
| Llanos‐Cuentas 2001 | Atovaquone‐proguanil | 14 | 57 | — | — | < 0.0001 |
| Chloroquine | 13 | 48 | — | — | — | |
| Borrmann 2003 | Atovaquone‐proguanil | 92 | 72 | 10 (IQR) | — | 0.0002 |
| Amodiaquine | 78 | 70 | 24 (IQR) | — | — | |
| Llanos‐Cuentas 2001 | Atovaquone‐proguanil | 5 | 42 | — | — | — |
| Sulfadoxine‐pyrimethamine | 7 | 42 | — | — | — | |
| Mulenga 1999 | Atovaquone‐proguanil | 81 | 72 | 12 to 102 | 12 to 24 | < 0.05 |
| Sulfadoxine‐pyrimethamine | 81 | 48 | 6 to 114 | — | — | |
| Anabwani 1999 | Atovaquone‐proguanil | 81 | 64.5 | 15 to 103 | — | — |
| Halofantrine | 83 | 50.4 | 17 to 78 | 11.5 to 20.8 | < 0.001 | |
| Looareesuwan 1999a | Atovaquone‐proguanil | 79 | 66.5 | 24 to 127 | — | < 0.05 |
| Mefloquine | 79 | 65.0 | 24 to 167 | — | — |
CI: confidence interval; IQR: interquartile range.
Appendix 4. Fever clearance time: median and range
| Trial | Intervention | No. participants | Median (h) | Range (h) | 95% CI | P‐value |
| Llanos‐Cuentas 2001 | Atovaquone‐proguanil | 14 | 46 | 8 to 92 | — | — |
| Chloroquine | 13 | 48 | 20 to 72 | — | — | |
| Borrmann 2003 | Atovaquone‐proguanil | 92 | 47 | 48 (IQR) | — | 0.85 |
| Amodiaquine | 78 | 46 | 43 (IQR) | — | — | |
| Llanos‐Cuentas 2001 | Atovaquone‐proguanil | 5 | 40 | 20 to 26 | — | — |
| Sulfadoxine‐pyrimethamine | 7 | 44 | 28 to 72 | — | — | |
| Mulenga 1999 | Atovaquone‐proguanil | 67 | 23 | 1 to 160.5 | — | — |
| Sulfadoxine‐pyrimethamine | 77 | 48 | 6 to 101 | — | — | |
| Anabwani 1999 | Atovaquone‐proguanil | 81 | 29.8 | 0.5 to 99 | ‐11 to 6.4 | 0.60 |
| Halofantrine | 83 | 35.3 | 0.5 to 123 | — | — | |
| Looareesuwan 1999a | Atovaquone‐proguanil | 84 | 53.5 | 3 to 152 | — | — |
| Mefloquine | 88 | 50.0 | 4 to 147 | — | — |
CI: confidence interval; IQR: interquartile range.
Data and analyses
Comparison 1. Atovaquone‐proguanil (AP) versus chloroquine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment failure on or by day 28 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Parasite clearance time (mean; h) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3 Fever clearance time (mean; h) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Abdominal pain | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Chills/rigors | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Dizziness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 Headache | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 Insomnia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 Nausea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 Palpitations | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.8 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.9 Severe | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.10 Vomiting | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.11 Weakness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2. Atovaquone‐proguanil (AP) versus amodiaquine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment failure on or by day 28 | 2 | 342 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.13, 0.36] |
| 2 Treatment failure on or by day 14 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Parasite clearance time (mean; h) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4 Fever clearance time (mean; h) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5 Adverse events | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Abdominal pain | 1 | 126 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.8 [1.07, 7.31] |
| 5.2 Diarrhoea | 2 | 326 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.57, 1.61] |
| 5.3 Dizziness | 1 | 126 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.08, 0.93] |
| 5.4 Insomnia | 1 | 126 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.12, 0.75] |
| 5.5 Nausea | 1 | 126 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.63 [1.26, 5.48] |
| 5.6 Pruritus | 1 | 126 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.04, 0.35] |
| 5.7 Respiratory infections | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.62, 14.51] |
| 5.8 Vomiting | 2 | 326 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.80, 2.41] |
| 5.9 Weakness | 2 | 326 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.02, 0.64] |
| 5.10 Any event | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.82, 1.51] |
Comparison 3. Atovaquone‐proguanil (AP) versus sulfadoxine‐pyrimethamine (SP).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment failure on or by day 28 | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Parasite clearance time (mean; h) | 2 | 174 | Mean Difference (IV, Fixed, 95% CI) | 11.23 [5.43, 17.03] |
| 3 Fever clearance time (mean; h) | 2 | 156 | Mean Difference (IV, Fixed, 95% CI) | ‐13.73 [‐21.31, ‐6.16] |
| 4 Progression to severe disease | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5 Adverse events | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Abdominal pain | 2 | 189 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.58, 1.24] |
| 5.2 Chills/rigors | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.06, 32.05] |
| 5.3 Diarrhoea | 1 | 160 | Risk Ratio (M‐H, Fixed, 95% CI) | 43.0 [2.65, 697.91] |
| 5.4 Dizziness | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.06, 32.05] |
| 5.5 Headache | 2 | 189 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.81, 1.67] |
| 5.6 Insomnia | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.09, 1.21] |
| 5.7 Nausea | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.25 [0.31, 16.59] |
| 5.8 Orthostatic hypotension | 1 | 160 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.03 [0.00, 0.57] |
| 5.9 Pruritus | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.16 [0.01, 3.56] |
| 5.10 Raised liver enzymes | 1 | 160 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.25, 1.79] |
| 5.11 Severe | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.06, 32.05] |
| 5.12 Vomiting | 2 | 189 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.50, 1.90] |
| 5.13 Weakness | 2 | 189 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [0.82, 2.62] |
Comparison 4. Atovaquone‐proguanil (AP) versus quinine plus tetracycline (Q plus T).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment failure on or by day 28 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Parasite clearance time (mean; h) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3 Fever clearance time (mean; h) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Abdominal pain | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Anorexia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Diarrhoea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 Dizziness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 Headache | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 Nausea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.8 Tinnitus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.9 Vomiting | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.10 Weakness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 5. Atovaquone‐proguanil (AP) versus halofantrine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment failure on or by day 28 | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Parasite clearance time (mean; h) | 2 | 205 | Mean Difference (IV, Fixed, 95% CI) | 14.76 [10.41, 19.10] |
| 3 Fever clearance time (mean; h) | 2 | 205 | Mean Difference (IV, Fixed, 95% CI) | ‐1.70 [‐9.41, 6.02] |
| 4 Adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Abdominal pain | 2 | 216 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.14, 4.68] |
| 4.2 Chills/Rigors | 1 | 168 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.11, 3.89] |
| 4.3 Cough | 1 | 168 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.34, 1.52] |
| 4.4 Diarrhoea | 2 | 216 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.23, 1.39] |
| 4.5 Headache | 2 | 216 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.17, 6.84] |
| 4.6 Insomnia | 2 | 216 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.15, 2.60] |
| 4.7 Moderate or severe | 1 | 168 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.15, 0.91] |
| 4.8 Myalgia | 1 | 168 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.72] |
| 4.9 Nausea | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 2.76 [0.62, 12.33] |
| 4.10 Palpitations | 1 | 168 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.05, 5.41] |
| 4.11 Pruritus | 2 | 216 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.51, 2.87] |
| 4.12 Vomiting | 2 | 216 | Risk Ratio (M‐H, Random, 95% CI) | 3.47 [0.66, 18.43] |
| 4.13 Weakness | 1 | 168 | Risk Ratio (M‐H, Random, 95% CI) | 0.25 [0.03, 2.19] |
Comparison 6. Atovaquone‐proguanil (AP) versus mefloquine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment failure on or by day 28 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Parasite clearance time (mean; h) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3 Fever clearance time (mean; h) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Abdominal pain | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Anaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Diarrhoea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 Dizziness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 Headache | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 Insomnia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 Nausea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.8 Raised liver enzymes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.9 Sore throat | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.10 Vomiting | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.11 Any event | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 7. Atovaquone‐proguanil (AP) versus artesunate plus mefloquine (AS plus MQ).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment failure on or by day 14 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Treatment failure on or by day 28 adjusted by PCR | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Anorexia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Dizziness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Nausea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 Palpitations | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Skin rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Sleeping disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.7 Tremor | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.8 Vomiting | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 8. Atovaquone‐proguanil (AP) versus dihydroartemisinin, piperaquine, trimethoprim, and primaquine (CV8).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment failure on or by day 28 | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Parasite clearance time (mean; h) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3 Fever clearance time (mean; h) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Diarrhoea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Dry mouth | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Headache | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 Itch | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 Vomiting | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Anabwani 1999.
| Methods | Randomized controlled trial Length of follow up: 28 d Generation of allocation sequence: random assignment of study number Allocation concealment: unclear Blinding: none Inclusion of all randomized participants in the analysis: 164 analysed/168 randomized (97.6%) |
|
| Participants | Number: 168 enrolled; 164 analysed; 4 discontinued intervention and were excluded Age range: 3 to 12 years Gender: male and female Inclusion criteria: age 3 to 12 years; uncomplicated malaria; fever; parasitaemia between 1000 to 200,000 parasites/µL; ability to tolerate oral therapy; weight > 10 kg; written informed consent given by parent or guardian Exclusion criteria: severe or cerebral malaria; prolonged QTc interval (above 0.44 s); mixed infections with other Plasmodium species; concomitant disease |
|
| Interventions | 1. Atovaquone‐proguanil (60 mg/kg atovaquone and 24 mg/kg proguanil over 3 d) 2. Halofantrine (24 mg/kg over 12 h) | |
| Outcomes | 1. 28‐d cure rate 2. Parasite clearance time 3. Fever clearance time 4. Adverse events | |
| Notes | Location: Kenya Drug resistance: not stated |
|
Borrmann 2003.
| Methods | Randomized controlled trial Length of follow up: 28 d Generation of allocation sequence: blocks of 10 and sequentially assigned to groups Allocation concealment: sealed envelopes Blinding: none Inclusion of all randomized participants in the final analysis: 170 analysed/200 randomized (85%) |
|
| Participants | Number: 200 enrolled; 170 analysed, 92 for the atovaquone‐proguanil group and 78 for amodiaquine group Gender: male and female Age range: 3 to 43 months Inclusion criteria: documented uncomplicated falciparum malaria with parasitaemia between 1000 and 200,000 parasites/µL; weight between 5 kg and 11 kg; written or verbal informed consent by parent or guardian Exclusion criteria: administration of antimalarials or medications with antimalarials or haemolytic effects with previous 7 d; underlying severe diseases or concomitant infections causing fever; hypersensitivity to atovaquone, proguanil, or amodiaquine; predefined abnormal laboratory values at screening; symptoms and signs of severe malaria |
|
| Interventions | 1. Atovaquone‐proguanil (fixed dose combination containing 62.5 mg atovaquone and 25 mg proguanil for 3 d) 2. Amodiaquine (10 mg/kg of a 1% suspension of amodiaquine chlorohydrate once daily for 3 d) | |
| Outcomes | 1. 28‐d cure rate 2. Parasite clearance time 3. Fever clearance time 4. Adverse events | |
| Notes | Location: Gabon Drug resistance: not stated |
|
Bouchaud 2000.
| Methods | Randomized controlled trial Length of follow up: 35 d Generation of allocation sequence: unclear Allocation concealment: unclear Blinding: none Inclusion of all randomized participants in the final analysis: 41 analysed/48 randomized (85%) |
|
| Participants | Number enrolled: 48 Age range: 15 to 65 years Gender: male and female Inclusion criteria: > 16 years old; had malaria from a short stay in an endemic country; non‐immune individual; parasitaemia between 1000 and 100,000 parasites/µL Exclusion criteria: severe malaria; prolonged QTc interval (above 0.44 s); presence of mixed infections with other Plasmodium species; presence of concomitant disease; inability to take oral treatment; history of syncope; pregnancy; breastfeeding mother; weighed < 40 kg; resided in an endemic area for the previous year |
|
| Interventions | 1. Atovaquone‐proguanil (4 x 250 mg atovaquone and 4 x 100 mg proguanil as single daily dose for 3 d) 2. Halofantrine (total of 1500 mg in 3 doses of 500 mg 6 h apart) | |
| Outcomes | 1. 28‐d cure rate 2. Parasite clearance time 3. Fever clearance | |
| Notes | Location: France Drug resistance: difficult to say as malaria was imported |
|
De Alencar 1997.
| Methods | Randomized controlled trial Length of follow up: 28 d Generation of allocation sequence: unclear Allocation concealment: unclear Blinding: none Inclusion of all randomized participants in the final analysis: 154 analysed/175 randomized (88%) |
|
| Participants | Number enrolled: 175 Age range: 18 to 65 years Gender: male Inclusion criteria: adult men; age 18 to 68 years; smear positive falciparum malaria; general good health; parasitaemia between 1000 and 100,000 parasites/µL Exclusion criteria: grossly abnormal laboratory results; refusal to be hospitalized for 28 d; inability to tolerate study medication; missing study medication |
|
| Interventions | 1. Atovaquone plus proguanil (atovaquone 1 g and proguanil 400 mg daily for 3 d) 2. Quinine plus tetracycline (600 mg quinine 3 times a day and 250 mg tetracycline 4 times a day for 7 d) | |
| Outcomes | 1. 28‐d cure rate 2. Parasite clearance time 3. Fever clearance time 4. Adverse events | |
| Notes | Location: Brazil Drug resistance: high for chloroquine, sulfadoxine‐pyrimethamine, and quinine to some extent |
|
Giao 2004.
| Methods | Randomized controlled trial Length of follow up: all followed up for 28 d; 92 participants followed up for 56 d Generation of allocation sequence: codes were allocated in randomized blocks of 10 Allocation concealment: sealed envelope Blinding: none Inclusion of all randomized participants in the final analysis: 161 analysed/165 randomized (98%) |
|
| Participants | Number enrolled: 165; 161 analysed; 4 excluded for P. vivax infection Age range: 17 to 64 years for atovaquone‐proguanil group; 16 to 73 years for dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine group Gender: male and female; though slight difference in ratio Inclusion criteria: uncomplicated falciparum malaria with parasitaemia > 1000 parasites/µL; age > 16 years Exclusion criteria: pregnancy; lactation; complicated malaria; inability to take oral medication; known allergy to study drugs; verbal confirmation of taking artemisinin within 24 h, mefloquine/tetracycline/doxycycline in 7 days and quinine in previous 12 h |
|
| Interventions | 1. Atovaquone‐proguanil (4 x 250 mg atovaquone and 4 x 100 mg proguanil) as single dose daily for 3 d 2. Dihydroartemisinin‐piperaquine‐trimethoprim‐primaquine (2 x 32 mg dihydroartemisinin + 320 mg piperaquine phosphate + 90 mg trimethoprim + 5 mg primaquine phosphate at time 0, 8 h, 24 h, and 48 h | |
| Outcomes | 1. Radical cure at d 28 2. Recrudescence (early and late) 3. Parasite clearance time 4. Fever clearance time 5. Adverse events | |
| Notes | Location: Binh Thuan, south Viet Nam Drug resistance: unclear |
|
Llanos‐Cuentas 2001.
| Methods | Randomized controlled trial Length of follow up: 28 d Generation of allocation sequence: unclear Allocation concealment: unclear Blinding: none Inclusion of all randomized participants in the final analysis: 39 analysed/43 randomized (91%) |
|
| Participants | Number enrolled: 43 Age range: 15 to 65 years Gender: male and female Inclusion criteria: age 12 to 65 years; presence of acute uncomplicated falciparum malaria; lifelong residents of the study area; parasitaemia between 1000 and 200,000 parasites/µL Exclusion criteria: severe malaria; presence of mixed infections with other Plasmodium species; presence of concomitant disease; inability to take oral treatment; pregnancy; breastfeeding mother |
|
| Interventions | 1. Atovaquone plus proguanil (1000 mg atovaquone, 400 mg proguanil over 3 d) 2. Chloroquine (1500 mg over 3 d) 3. Sulfadoxine‐pyrimethamine (1500 mg sulfadoxine and 75 mg pyrimethamine) | |
| Outcomes | 1. 28‐d cure rate 2. Parasite clearance time 3. Fever clearance time 4. Adverse events | |
| Notes | Location: Peru Drug resistance: high for chloroquine |
|
Looareesuwan 1999a.
| Methods | Randomized controlled trial Length of follow up: 28 d Generation of allocation sequence: unclear Allocation concealment: unclear Blinding: none Inclusion of all randomized participants in the final analysis: 158 analysed/182 randomized (87%) |
|
| Participants | Number enrolled: 182 Age range: 15 to 63 years Gender: male and female Inclusion criteria: age 16 to 65 years; presence of acute uncomplicated falciparum malaria; parasitaemia between 1000 and 200,000/µL; weight 40 kg and above Exclusion criteria: presence of mixed infections with other Plasmodium species; presence of concomitant disease (intercurrent febrile infections); inability to take oral treatment (persistent vomiting); pregnancy; breastfeeding mother |
|
| Interventions | 1. Atovaquone plus proguanil (1000 mg atovaquone and 400 mg proguanil daily over 3 d) 2. Mefloquine (1250 mg over 6 h) | |
| Outcomes | 1. 28‐d cure rate 2. Parasite clearance time 3. Fever clearance | |
| Notes | Location: Thailand Drug resistance: high for chloroquine and sulfadoxine‐pyrimethamine |
|
Mulenga 1999.
| Methods | Randomized controlled trial Length of follow up: 28 d Generation of allocation sequence: unclear Allocation concealment: unclear Blinding: none Inclusion of all randomized participants in the final analysis: 160 analysed/163 randomized (98%) |
|
| Participants | Number randomized: 163 Age range: 14 to 54 years Gender: male and female (mainly male) Inclusion criteria: age 12 to 65 years; presence of acute uncomplicated falciparum malaria; parasitaemia between 1000 and 200,000/µL; weight 40 kg and above; no underlying disease Exclusion criteria: presence of mixed infections with other Plasmodium species; presence of concomitant disease (intercurrent febrile infections); inability to take oral treatment (persistent vomiting); pregnancy breastfeeding mother |
|
| Interventions | 1. Atovaquone plus proguanil (1000 mg atovaquone and 400 mg proguanil daily over 3 d) 2. Sulfadoxine‐pyrimethamine (1500 mg sulfadoxine and 75 mg pyrimethamine as single dose) | |
| Outcomes | 1. 28‐d cure rate 2. Parasite clearance time 3. Fever clearance time | |
| Notes | Location: Zambia Drug resistance: high for chloroquine |
|
Radloff 1996.
| Methods | Randomized controlled trial Length of follow up: 28 d Generation of allocation sequence: participants given a sequential study number, which was randomly assigned to treatment option Allocation concealment: unclear Blinding: none Inclusion of all randomized participants in the final analysis: 126 analysed/142 randomized (89%) |
|
| Participants | Number enrolled: 142 Age range: 15 to 65 years Gender: male and female (mainly male) Inclusion criteria: age 15 to 65 years; presence of acute uncomplicated falciparum malaria; parasitaemia between 200 and 100,000 parasites/µL; weight 40 kg and above; urine test negative for chloroquine or sulphonamides Exclusion criteria: severe malaria; presence of mixed infections with other Plasmodium species; presence of concomitant disease (intercurrent febrile infections); 2‐week history of antimalarial administration; pregnancy; breastfeeding mother |
|
| Interventions | 1. Atovaquone plus proguanil (1000 mg atovaquone and 400 mg proguanil daily over 3 d) 2. Amodiaquine (1500 mg over 3 d: 600 mg on admission, 600 mg 24 h later, and 300 mg after a further 24 h) | |
| Outcomes | 1. 28‐d cure rate 2. Parasite clearance time 3. Fever clearance time | |
| Notes | Location: Gabon Drug resistance: high for chloroquine |
|
Van Vugt 2002.
| Methods | Randomized controlled trial Length of follow up: 42 d Generation of allocation sequence: block randomization Allocation concealment: sealed envelopes Blinding: none Inclusion of all randomized participants in the final analysis: 1063 analysed/1063 randomized (100%) |
|
| Participants | Number enrolled:1063 Age range: 2 to 70 years Gender: male and female Inclusion criteria: age 2 to 70 years; slide confirmed acute uncomplicated falciparum malaria; weight > 10 kg; written informed consent by patient or guardian; not pregnant; not received mefloquine in the previous 63 d; not obtunded; not vomiting; no other clinical or laboratory signs of severe illness Exclusion criteria: severe malaria; presence of mixed infections with other Plasmodium species; presence of concomitant disease (intercurrent febrile infections); 2‐week history of antimalarial administration; pregnancy; breastfeeding mother |
|
| Interventions | 1. Atovaquone plus proguanil (atovaquone 15 mg/kg/d, proguanil 8 mg/kg/d for 3 d)
2. Artesunate plus mefloquine (artesunate 4 mg/kg/d for 3 d; mefloquine 15 mg/kg on day 1 and 10 mg/kg on day 2) Participants with axillary temperature > 38 °C given antipyretics and cooled by tepid sponging before drug administration |
|
| Outcomes | 1. Incidence of microscopically and genetically confirmed recrudescent infections 2. Parasite clearance time 3. Fever clearance time 4. Adverse events 5. Degree of anaemia | |
| Notes | Location: Thailand Drug resistance: multiple‐drug resistance except artemisinin derivatives |
|
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bustos 1999 | Trial protocol initially randomized participants to receive chloroquine or atovaquone‐proguanil. However, after 40 participants had been recruited, the cure rate for chloroquine was found to be < 35%, thus subsequent participants in the chloroquine arm were given sulfadoxine‐pyrimethamine in addition. This ultimately resulted in a 3‐arm study. Participants in the atovaquone‐proguanil arm at the time of the protocol amendment should have been separated from those who were recruited after the amendment in order to make the comparisons |
Differences between protocol and review
Issue 4, 2005 (first review version): In line with the Cochrane Infectious Diseases Group (CIDG) new guidelines we no longer include quasi‐randomized controlled trials, modified the primary outcome measure (protocol: "Parasitaemia by days 14 and 28"), removed the secondary outcome measure "Parasitaemia by day 28, adjusted to exclude new infections using PCR", added "Treatment failure* on or by day 14" as a secondary outcome measure, and modified the method to assess blinding in trials.
Contributions of authors
Alex Osei‐Akoto and Shirley Owusu‐Ofori drafted the review, and Lois Orton commented on the review. All three authors completed the review.
Sources of support
Internal sources
No sources of support supplied
External sources
Department for International Development, UK.
Declarations of interest
None known.
Unchanged
References
References to studies included in this review
Anabwani 1999 {published data only}
- Anabwani G, Canfield CJ, Hutchinson DB. Combination atovquone and proguanil hydrochloride vs. halofantrine for treatment of acute Plasmodium falciparum malaria in children. Pediatric Infectious Disease Journal 1999;18(5):456‐61. [DOI] [PubMed] [Google Scholar]
Borrmann 2003 {published data only}
- Borrmann S, Faucher J‐F, Bagaphou T, Missinou MA, Binder RK, Pabisch S, et al. Atovaquone and proguanil versus amodiaquine for the treatment of Plasmodium falciparum malaria in African infants and young children. Clinical Infectious Diseases 2003;37(11):1441‐7. [DOI] [PubMed] [Google Scholar]
Bouchaud 2000 {published data only}
- Bouchaud O, Monlun E, Muanza K, Fontanet A, Scott T, Goetschel A, et al. Atovaquone plus proguanil versus halofantrine for the treatment of imported acute uncomplicated Plasmodium falciparum malaria in non‐immune adults: a randomized comparative trial. American Journal of Tropical Medicine and Hygiene 2000;63(5‐6):274‐9. [PubMed] [Google Scholar]
De Alencar 1997 {published data only}
- Alencar FE, Cerutti C Jr, Durlacher RR, Boulos M, Alves FP, Milhous W, et al. Atovaquone and proguanil for the treatment of malaria in Brazil. Journal of Infectious Diseases 1997;175(6):1544‐7. [DOI] [PubMed] [Google Scholar]
Giao 2004 {published data only}
- Giao PT, Vries PJ, Hung le Q, Binh TQ, Nam NV, Kager PA. CV8, a new combination of dihydroartemisinin, piperaquine, trimethoprim and primaquine, compared with atovaquone‐proguanil against falciparum malaria in Vietnam. Tropical Medicine and International Health 2004;9(2):209‐16. [DOI] [PubMed] [Google Scholar]
Llanos‐Cuentas 2001 {published data only}
- Llanos‐Cuentas A, Campos P, Clendenes M, Canfield CJ, Hutchinson DB. Atovaquone and proguanil hydrochloride compared with chloroquine or pyrimethamine/sulfadoxine for treatment of acute Plasmodium falciparum malaria in Peru. Brazilian Journal of Infectious Diseases 2001;5(2):67‐72. [DOI] [PubMed] [Google Scholar]
Looareesuwan 1999a {published data only}
- Looareesuwan S, Wilairatana P, Chalermarut K, Rattanapong Y, Canfield CJ, Hutchinson DB. Efficacy and safety of atovaquone/proguanil compared with mefloquine for treatment of acute Plasmodium falciparum malaria in Thailand. American Journal of Tropical Medicine and Hygiene 1999;60(4):526‐32. [DOI] [PubMed] [Google Scholar]
Mulenga 1999 {published data only}
- Mulenga M, Sukwa TY, Canfield CJ, Hutchinson DB. Atovaquone and proguanil versus pyrimethamine/sulfadoxine for the treatment of acute falciparum malaria in Zambia. Clinical Therapeutics 1999;21(5):841‐52. [DOI] [PubMed] [Google Scholar]
Radloff 1996 {published data only}
- Radloff PD, Philipps J, Nkeyi M, Hutchinson D, Kremsner PG. Atovaquone and proguanil for Plasmodium falciparum malaria. Lancet 1996;347(9014):1511‐4. [DOI] [PubMed] [Google Scholar]
Van Vugt 2002 {published data only}
- Vugt M, Leonardi E, Phaipun L, Slight T, Thway KL, McGready R, et al. Treatment of uncomplicated multidrug‐resistant falciparum malaria with artesunate‐atovaquone‐proguanil. Clinical Infectious Diseases 2002;35(12):1498‐504. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Bustos 1999 {published data only}
- Bustos DG, Canfield CJ, Canete‐Miguel E, Hutchinson DB. Atovaquone‐proguanil compared with chloroquine‐sulfadoxine‐pyrimethamine for treatment of acute Plasmodium falciparum malaria in the Philippines. The Journal of Infectious Diseases 1999;179(6):1587‐90. [DOI] [PubMed] [Google Scholar]
Additional references
Beerahee 1999
- Beerahee M. Clinical pharmacology of atovaquone and proguanil hydrochloride (http://www.malarone.com/hcp/clinical.html). Journal of Travel Medicine 1999; Vol. 6 Suppl 1. [PubMed]
Blanchard 1994
- Blanchard TJ, Mabey DC, Hunt‐Cooke A, Edwards G, Hutchinson DB, Benjamin S, et al. Multiresistant falciparum malaria cured using atovaquone and proguanil. Transactions of the Royal Society of Tropical Medicine and Hygiene 1994;88(6):693. [DOI] [PubMed] [Google Scholar]
Bloland 1997
- Bloland PB, Kazembe PN, Watkins WM, Doumbo OK, Nwanyanwu OC, Ruebush TK 2nd. Malarone‐donation programme in Africa. Lancet 1997;350(9091):1624‐5. [DOI] [PubMed] [Google Scholar]
Brockman 1999
- Brockman A, Paul RE, Anderson TJ, Hackford I, Phaiphun L, Looareesuwan S, et al. Application of genetic markers to the identification of recrudescent Plasmodium falciparum infections on the northwestern border of Thailand. American Journal of Tropical Medicine and Hygiene 1999;60(1):14‐21. [DOI] [PubMed] [Google Scholar]
Canfield 1995
- Canfield CJ, Pudney M, Gutteridge WE. Interactions of atovaquone with other antimalarial drugs agaist Plasmodium falciparum in vitro. Experimental Parasitology 1995;80(3):373‐81. [DOI] [PubMed] [Google Scholar]
Dollery 1991
- Dollery C, editor. Poguanil hydrochloride. Therapeutic Drugs. 2nd Edition. Vol. 2, New York: Churchill Livingstone, 1991:247‐51. [Google Scholar]
Foege 1997
- Foege WH. Malarone‐donation programme. The Lancet 1997;350:1628‐9. [DOI] [PubMed] [Google Scholar]
Fry 1992
- Fry M, Pudney M. Site of action of the antimalarial hydroxynaphttoquinone, 2‐[trans‐4‐(4'‐chlophenyl) cyclohexyl]‐3‐hydroxy‐1, 4 naphthoquinone (566C80). Biochemical Pharmarmacology 1992;43(7):1545‐53. [DOI] [PubMed] [Google Scholar]
Higgins 2005
- Higgins J, Green S, editors. Highly sensitive search strategies for identifying reports of randomized controlled trials in MEDLINE. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]; Appendix 5b. http://www.cochrane.org/resources/handbook/index.htm (accessed 10 August 2005).
Hogh 2000
- Hogh B, Clarke PD, Camus D, Nothdurft HD, Overbosch D, Gunther M, et al. Malarone International Study Team. Atovaquone‐proguanil versus chloroquine‐proguanil for malaria prophylaxis in non‐immune travellers: a randomised, double‐blind study. Lancet 2000;356(9245):1888‐94. [DOI] [PubMed] [Google Scholar]
Jüni 2001
- Jüni P, Altman DG, Egger M. Systematic reviews in health care: Assessing the quality of controlled clinical trials. BMJ 2001;323(7303):42‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kamya 2002
- Kamya MR, Bakyaita NN, Talisuna AO, Were WM, Staedke SG. Increasing antimalarial drug resistance in Uganda and revision of the national drug policy. Tropical Medicine and International Health 2002;7(12):1031‐41. [DOI] [PubMed] [Google Scholar]
Looareesuwan 1996
- Looareesuwan S, Viravan C, Webster HK, Kyle DE, Hutchinson DB, Canfield CJ. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. American Journal of Tropical Medicine and Hygiene 1996;54(1):62‐6. [DOI] [PubMed] [Google Scholar]
Looareesuwan 1999b
- Looareesuwan S, Chulay JD, Canfield CJ, Hutchinson DB. Malarone (atovaquone and proguanil hydrochloride): a review of its clinical development for treatment of malaria. Malarone Clinical Trials Study Group. American Journal of Tropical Medicine and Hygiene 1999;60(4):533‐41. [DOI] [PubMed] [Google Scholar]
Ohrt 1997
- Ohrt C, Mirabelli‐Primdahl L, Karnasuta C, Chantakulkij S, Kain KC. Distinguishing Plasmodium falciparum treatment failures from reinfections by restriction fragment length polymorphism and polymerase chain reaction genotyping. American Journal of Tropical Medicine and Hygiene 1997;57(4):430‐7. [DOI] [PubMed] [Google Scholar]
Overbosch 2001
- Overbosch D, Schilthuis H, Bienzle U, Behrens RH, Kain KC, Clarke PD, et al. Atovaquone‐proguanil versus mefloquine for malaria prophylaxis in nonimmune travelers: results from a randomized, double‐blind study. Clinical Infectious Diseases 2001;33(7):1015‐21. [DOI] [PubMed] [Google Scholar]
Oyediran 2002
- Oyediran AB, Ddumba EM, Ochola SA, Lucas AO, Koporc K, Dowdle WR. A public‐private partnership for malaria control: lessons from the Malarone Donation Programme. Bulletin of the World Health Organization 2002;80(10):817‐21. [PMC free article] [PubMed] [Google Scholar]
Partnerships 2002
- Initiative on Public‐Private Partnerships for Health. Malarone Donation Program (Historical). www.ippph.org/data/abstract.cfm?id=28 (accessed 16 July 2002).
RBM 2001a
- Global Partnership to Roll Back Malaria. The use of antimalarial drugs: report of an WHO informal consultation, 13‐17 November 2000. Geneva: World Health Organization, 2001:17. [Google Scholar]
RBM 2001b
- Global Partnership to Roll Back Malaria. The use of antimalarial drugs: report of an WHO informal consultation, 13‐17 November 2000. Geneva: World Health Organization, 2001:32. [Google Scholar]
RBM 2005
- Global Partnership to Roll Back Malaria. World malaria report: 2005. Geneva: World Health Organization, 2005. [Google Scholar]
Review Manager 5 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Ringwald 1998
- Ringwald P, Basco LK. Malarone‐donation programme in Africa. Lancet 1998;351(9103):673‐4. [DOI] [PubMed] [Google Scholar]
Sabchareon 1998
- Sabchareon A, Attanath P, Phanuaksook P, Chanthavanich P, Poonpanich Y, Mookmanee D, et al. Efficacy and pharmacokinetics of atovaquone and proguanil in children with multidrug‐resistant Plasmodium falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 1998;92(2):201‐6. [DOI] [PubMed] [Google Scholar]
Shretta 2000
- Shretta R, Brugha R, Robb A, Snow RW. Sustainability, affordability, and equity of corporate drug donations: the case of Malarone. Lancet 2000;355(9216):1718‐20. [DOI] [PubMed] [Google Scholar]
Shretta 2001
- Shretta R, Walt G, Brugha R, Snow RW. A political analysis of corporate drug donations: the example of Malarone in Kenya. Health Policy and Planning 2001;16(2):161‐70. [DOI] [PubMed] [Google Scholar]
Stepniewska 2004
- Stepniewska K, Taylor WR, Mayxay M, Price R, Smithuis F, Guthmann JP, et al. In vivo assessment of drug efficacy against Plasmodium falciparum malaria: duration of follow‐up. Antimicrobial Agents and Chemotherapy 2004;48(11):4271‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sukwa 1999
- Sukwa TY, Mulenga M, Chisdaka N, Roskell NS, Scott TR. A randomised, double‐blind, placebo‐controlled field trial to determine the efficacy and safety of Malarone (atovaquone/proguanil) for prophylaxis of malaria in Zambia. American Journal of Tropical Medicine and Hygiene 1999;60(4):521‐5. [DOI] [PubMed] [Google Scholar]
WHO 1999
- World Health Organization. The world health report: 1999: Making a difference. Geneva: World Health Organization, 1999:1. [PMC free article] [PubMed] [Google Scholar]
WHO 2000
- World Health Organization. Severe falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 2000;94(1):1‐10. [PubMed] [Google Scholar]
WHO 2001
- World Health Organization. Training manual for management of malaria at district level, facilitator's guide. Harare: World Health Organization (Regional Office for Africa), 2001:4‐6. [Google Scholar]
WHO 2003
- World Health Organization. Malaria Control Unit. The Africa malaria report 2003. Geneva: World Health Organization, 2003:19. [Google Scholar]