

Abstract
Catabolic processing of the amyloid precursor protein (APP) is subject to regulatory control by protein kinases. We hypothesized that this regulation involves sequential activation of the enzymes mitogen-activated protein kinase kinase (MEK) and extracellular signal-regulated protein kinase (ERK). In the present investigation, we provide evidence that MEK is critically involved in regulating APP processing by both nerve growth factor and phorbol esters. Western blot analysis of the soluble N-terminal APP derivative APPsdemonstrated that the synthetic MEK inhibitor PD 98059 antagonized nerve growth factor stimulation of both APPs production and ERK activation in PC12 cells. Moreover, PD 98059 inhibited phorbol ester stimulation of APPs production and activation of ERK in both human embryonic kidney cells and cortical neurons. Furthermore, overexpression of a kinase-inactive MEK mutant inhibited phorbol ester stimulation of APP secretion and activation of ERK in human embryonic kidney cell lines. Most important, PD 98059 antagonized phorbol ester-mediated inhibition of Aβ secretion from cells overexpressing human APP695 carrying the “Swedish mutation.” Taken together, these data indicate that MEK and ERK may be critically involved in protein kinase C and nerve growth factor regulation of APP processing. The mitogen-activated protein kinase cascade may provide a novel target for altering catabolic processing of APP.
Keywords: amyloid precursor protein, amyloid β-peptide, protein kinase C, nerve growth factor, mitogen-activated protein kinase, Alzheimer’s disease
Amyloid β-peptide (Aβ), the principle constituent of senile plaques found in Alzheimer’s disease (AD) brain (Hardy, 1997; Selkoe, 1997), is derived by proteolysis of an integral membrane protein known as the amyloid precursor protein (APP). Secretory processing of APP occurs via at least two pathways. One involves activation of an unidentified enzyme known as α-secretase, cleaving APP within the Aβ sequence (Sisodia et al., 1990; Anderson et al., 1991; Wang et al., 1991), precluding Aβ generation and releasing a soluble N-terminal APP fragment (APPs) into the extracellular space. The alternative route involves two unidentified enzymes termed β- and γ-secretase, which cleave APP on the N and C termini of Aβ, respectively. The resultant Aβ is then released into the extracellular space (Haass et al., 1992b; Shoji et al., 1992).
Although catabolism of APP is constitutive, activation of signal transduction pathways can alter the relative amounts of APPs and Aβ produced. Most studies of regulated APP processing have focused on stimulation of protein kinase C (PKC) and receptors linked to phospholipase-C that increase release of APPs while inhibiting the release of soluble Aβ (Buxbaum et al., 1992, 1993; Caporaso et al., 1992; Nitsch et al., 1992; Gabuzda et al., 1993; Hung et al., 1993; Farber et al., 1995; Lee et al., 1995;Wolf et al., 1995; Mills and Reiner, 1996). Other signaling systems shown to stimulate APPs release include calcium (Nitsch et al., 1992; Buxbaum et al., 1994), cAMP (Hu et al., 1996), growth factors (Schubert et al., 1989; Fukuyama et al., 1993), cytokines (Buxbaum et al., 1992, 1994), and estrogen (Jaffe et al., 1994).
The mechanism by which APP catabolism is regulated has not yet been elucidated. Phosphorylation of APP by PKC has been ruled out (Da Cruz et al., 1993; Hung and Selkoe, 1994; Jacobsen et al., 1994). Moreover, PKC-independent regulation of APPs secretion exists (Buxbaum et al., 1994; Nitsch et al., 1996a) and may involve activation of protein–tyrosine kinases (Slack et al., 1995). These observations predict the existence of a pathway activated by multiple first and second messengers capable of regulating APP catabolism in both a PKC-dependent and a PKC-independent manner. These criteria are met by the mitogen-activated protein kinase (MAPK) signal transduction pathway (Cobb and Goldsmith, 1995; Graves et al., 1995; Malarkey et al., 1995;Pelech and Charest, 1996). MAPKs, also known as extracellular signal-regulated protein kinases (ERKs), are the terminal enzymes in a three-level kinase cascade involving the sequential activation ofraf, mitogen-activated protein kinase kinase (MEK), and ERK (Pelech and Charest, 1996). Because MEKs are the only known physiological activators of ERKs (Bardwell and Thorner, 1996), MEKs provide a useful target for manipulating ERK activity. We used both PD 98059, a selective inhibitor of MEK1 (Alessi et al., 1995; Dudley et al., 1995; Lazar et al., 1995), and overexpresssion of a kinase-dead MEK1 mutant (Seger et al., 1994) to test the hypothesis that ERK activation is necessary for regulation of APP processing.
MATERIALS AND METHODS
Cell lines and transfections. Human embryonic kidney (HEK) 293 cells were transiently transfected with pCMV695, an expression vector for APP695 (Selkoe et al., 1988), pCMVβ, an expression vector for bacterial β-galactosidase (Clontech Laboratories), and either pCDNAK97A, an expression vector for kinase-inactive MEK, or the expression vector alone using a high-efficiency calcium phosphate transfection protocol (Chen and Okayama, 1987) as described previously (Raymond et al., 1996). Transfection efficiency was assessed by staining for β-galactosidase and determining the percentage of positively stained cells according to the method of Raymond et al. (1996). HEK 293 cells stably transfected with a construct carrying the Alzheimer’s disease-linked double (“Swedish”) mutation (K695sw), known to secrete elevated levels of both Aβ40 and Aβ42 (Citron et al., 1996), were cultured in DMEM supplemented in 10% fetal calf serum. HEK 293 cells were cultured in MEM supplemented with 10% fetal calf serum as described previously (Raymond et al., 1996). Rat pheochromocytoma (PC12) cells were cultured in DMEM supplemented with 10% horse serum and 5% fetal calf serum. One day before stimulation, HEK 293 cells or PC12 cells were exposed to culture media containing charcoal-inactivated calf serum at the same percentage used previously for cell maintenance. All cell lines were exposed to drugs for 15 min. PC12 cells were exposed to drugs in DMEM according to the method ofBuxbaum et al. (1990). HEK 293 cells were exposed to drugs in MEM supplemented with 1 mg/ml glucose, whereas K695sw cells were exposed to drugs in DMEM.
Cortical cell cultures and drug treatment. Timed pregnant Sprague Dawley rats were anesthetized with halothane at 18 d of gestation, and the cerebral cortex was removed from rat embryos and dissociated using a method described previously (Murphy et al., 1992). Culture maintenance and drug exposure were performed using the method of Fiore et al. (1993) with minor modifications. In brief, before drug treatment, cells were washed once with 1 ml HBSS and preexposed to PD 98059 or drug vehicle for 1 hr. Both PD 98059 and phorbol esters were diluted from 10 mm stocks, made up in dimethylsulfoxide.
Quantification of APPs and Aβ in culture media. After drug exposure, the medium was centrifuged for 10 min at 16,000 × g to remove cellular debris. For APPs detection, the medium was subsequently desalted and concentrated by centrifugation in the presence of protease inhibitors (17 μg/ml phenylmethanesulfonyl fluoride, 2 μg/ml leupeptin, 10 μg/ml aprotinin, and 2 μg/ml pepstatin) according the method ofMills and Reiner (1996). APP was detected by Western blot analysis using an anti-APP N-terminal antibody (anti-PreA4 monoclonal antibody, Boehringer Mannheim, Laval, Quebec, Canada) or WO-2, a monoclonal antibody generated against the first 16 amino acids of the N-terminal region of Aβ (Ida et al., 1996; anti-1–16) as described previously (Mills and Reiner, 1996). All Western blots were probed first with the anti-PreA4 monoclonal antibody (22C11). In some experiments, membranes were subsequently stripped of antibodies and reprobed with the APP-selective antibody WO-2 to prevent detection of secreted APLP (Slunt et al., 1994). For Aβ detection, proteins were precipitated by trichloroacetic acid according to the method of Hames (1981). Aβ was separated by Tris/Tricene SDS-PAGE according to the method of Klafki et al. (1996) and detected by Western blot analysis according to the method of Ida et al. (1996) using the monoclonal antibody WO-2. After densitometric measurements, ANOVA followed by Fisher’s post hoc analysis was used to determine the significance of observed differences. Data are expressed as mean ± SEM and, unless otherwise stated, are representative of three separate trials.
Western blots of MAPK, MEK, and cellular APP. Cells were lysed in an extraction buffer containing 1% Nonidet P-40, 1% sodium deoxycholate, 4 mmp-nitrophenylphosphate, and 1 mm sodium vanadate, and the lysate was centrifuged to remove detergent-insoluble material. Twenty-five micrograms of cellular protein were separated by SDS-PAGE on 10% 20 cm gels or 12.5% low-bis (acrylamide/bis ratio of 118.5:1 instead of 37.5:1) mini gels for Western blots of either ERK or MEK. After gel electrophoresis, proteins were transferred electrophoretically to a nitrocellulose membrane and probed using a rabbit polyclonal antibody specific for ERK (Erk1-CT, Upstate Biotechnology, Lake Placid, NY), phosphorylated ERK (phospho-MAPK, New England Biolabs, Mississauga, Ontario, Canada), or MEK (Mek1-NT, Upstate Biotechnology). Five micrograms of cellular protein were separated on 10% mini gels for Western blots of APP, and membranes were probed subsequently with anti-PreA4 monoclonal antibody. Sequential Western blots are representative of three separate trials that may or may not have been taken from the exact same trial.
RESULTS
Pharmacological inhibition of MEK antagonizes nerve growth factor receptor stimulation of APPs secretion and ERK activation
Activation of a wide variety of growth factor receptors having intrinsic or associated tyrosine kinase activity has been shown to stimulate ERK activation (Pelech and Sanghera, 1992; Pelech et al., 1993). Included among these are receptors for nerve growth factor (NGF), epidermal growth factor, and fibroblast growth factor, the stimulation of which has also been shown to increase APPsrelease in cell lines (Refolo et al., 1989; Schubert et al., 1989;Fukuyama et al., 1993). These observations implicate the involvement of ERK in growth factor receptor-mediated regulation of APPsrelease. To determine whether ERK activation is necessary for NGF receptor-dependent stimulation of APPs release, we examined PC12 cells stimulated with NGF in the presence of the MEK1 inhibitor PD 98059. This pharmacological agent has been shown previously to antagonize tyrosine kinase receptor stimulation of ERK1 (Alessi et al., 1995; Dudley et al., 1995; Lazar et al., 1995; Pang et al., 1995) with an IC50 of ∼10 μm (Dudley et al., 1995). APPs production increased significantly when cells were incubated with 100 ng/ml NGF for 15 min, and this increase was antagonized in the presence of 10 μm PD 98059 (2.0 ± 0.3 and 1.0 ± 0.2, respectively; n = 3,p < 0.05) (Fig.1A).
Fig. 1.
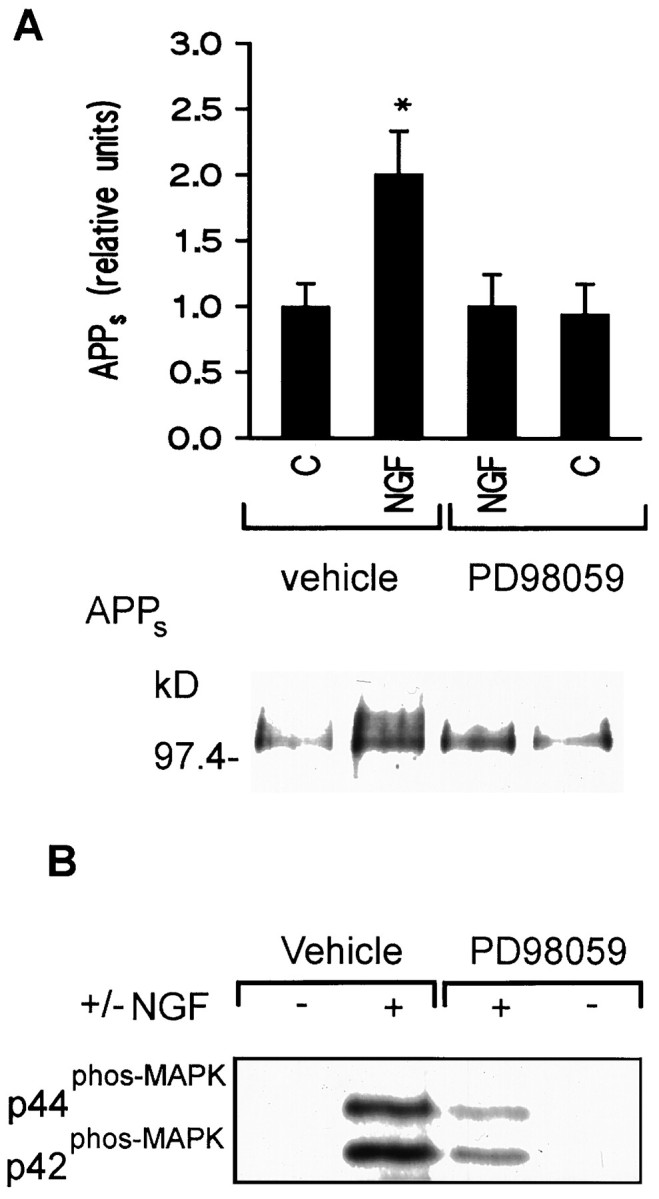
PD 98059 inhibits NGF receptor stimulation of APPs secretion and ERK activation in PC12 cells.A, Top, Densitometric analysis of the effect of NGF (100 ng/ml) on basal APPs release with or without PD 98059 (10 μm). Data are mean ± SEM of three experiments (*p < 0.05, different from all other treatment groups). Bottom, Representative Western blot of APPs fragments released in 15 min by PC12 cells alone or in the presence of NGF with or without PD 98059.B, Representative Western blot of phospho-ERK in PC12 cells after a 15 min drug exposure. The increase in immunoreactivity of the phospho-ERK-specific antibody in the presence of NGF was inhibited by PD 98059.
For ERK to become activated, it first must be phosphorylated by the dual-specificity kinase MEK on both a tyrosine and a threonine residue in the TEY motif (Anderson et al., 1990; Pague et al., 1991). Phosphorylated ERK can be detected either using Western blotting by a gel shift assay in which the electrophoretic mobility of phosphorylated ERK is retarded relative to its nonphosphorylated form (Posada and Cooper, 1992) or using antibodies raised against the phosphorylated TEY consensus sequence. The phosphorylation state of ERK was measured using these methods to ensure 10 μm PD 98059 antagonized NGF receptor stimulation of ERK activation. A 15 min exposure to NGF activated ERK1 and ERK2, and this activation was inhibited by PD 98059 (Fig. 1B).
Pharmacological inhibition of MEK antagonizes PKC stimulation of APPs release and ERK activation
PKC stimulation by phorbol esters has been shown to increase dramatically the release of APPs in a wide variety of cell lines (Buxbaum et al., 1992, 1993; Caporaso et al., 1992; Gabuzda et al., 1993; Hung et al., 1993). To determine whether ERKs are necessary for PKC-mediated regulation of APP catabolism, HEK 293 cells were exposed to 0.1 μm phorbol 12-myristate 13-acetate (PMA) with or without 10 μm PD 98059. Stimulation of APPs release by PMA was inhibited by PD 98059 during a 15 min drug exposure as determined using the monoclonal antibody 22C11 (7.7 ± 1.5 and 4.4 ± 1.4, respectively; n = 5, p < 0.05) (Fig.2A) or WO-2 (3.9 ± 0.5 and 2.0 ± 0.6, respectively; n = 3,p < 0.05) (Fig. 2A). To ensure that PD 98059 antagonized PMA-stimulated ERK activation in HEK 293 cells, the phosphorylation state and mobility of ERK were measured in Western blots. The experiments revealed that the PMA-induced electrophoretic shift was antagonized by PD 98059 (Fig. 2B,top; n = 3), as was the increase in phospho-ERK immunoreactivity induced by PMA (Fig. 2B,bottom; n = 3).
Fig. 2.
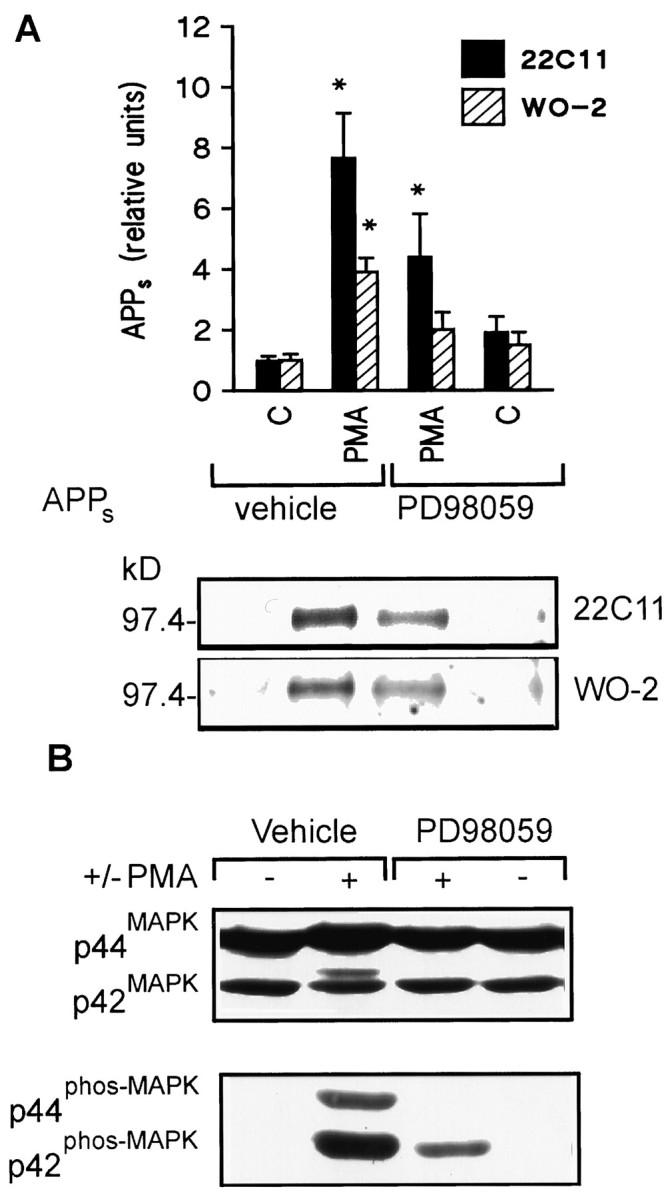
PD 98059 antagonizes phorbol ester stimulation of APPs release in 15 min and ERK activation in HEK 293 cells.A, Top, Densitometric analysis of PMA (0.1 μm) stimulation of APPs secretion with or without PD 98059 (10 μm). Data are mean ± SEM and represent five experiments for 22C11 (filled columns) or three experiments for WO-2 (hatched columns) (*p < 0.05, different from all other treatment groups). Bottom, Representative Western blot of the effect of PMA on basal APPs release alone or in the presence of PD 98059. B, Representative Western blot of ERK isoforms with ERK1 C terminus antibody (top) or phospho-ERK forms (bottom) in HEK 293 cells after a 15 min drug exposure. The PMA-induced “electrophoretic shift” was inhibited by PD 98059. Similarly, the increase in phospho-ERK immunoreactivity in the presence of PMA was antagonized by PD 98059.
PKC-dependent regulation of APPs release has also been observed in primary cultures of hippocampal and cortical neurons (Lee et al., 1995; Mills and Reiner, 1996). To determine whether ERK activation is necessary for PKC-mediated regulation of APPsrelease in neurons, primary cultures of rat cortical neurons were incubated with PDBu (1 μm) with or without PD 98059 (10 μm) for 1 hr. Levels of APPs in the culture media increased significantly in the presence of PDBu, and this increase was antagonized in the presence of PD 98059 (6.52 ± 1.51 and 3.01 ± 0.90, respectively; n = 5,p < 0.05) (Fig.3A). Moreover, phorbol ester stimulation of ERK activation was also suppressed in the presence of PD 98059, as seen by Western blot analysis of ERK mobility (top) or phospho-ERK (bottom) (Fig.3B).
Fig. 3.
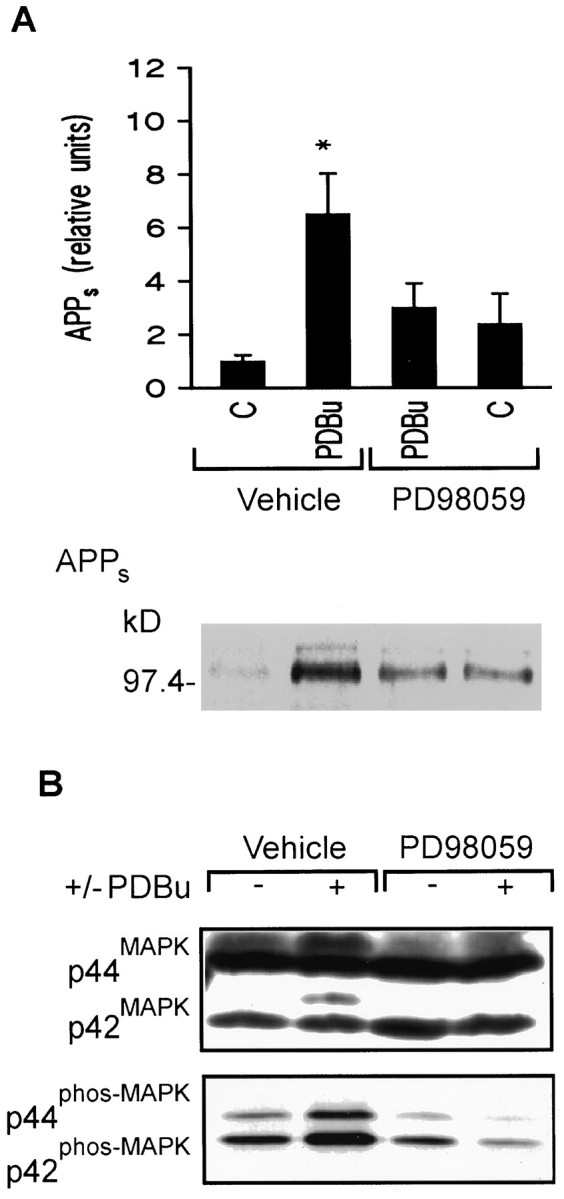
PD 98059 inhibits PKC stimulation of APPs secretion and ERK activation in cortical neurons.A, Top, Densitometric analysis of PDBu (1 μm) stimulation of APPs secretion in rat cortical cultures with or without PD 98059 (10 μm). Data are mean ± SEM of five experiments (*p < 0.05, different from all other treatment groups).Bottom, Representative Western blot of the effect of PD 98059 on PDBu stimulation of APPs release in 15 min.B, Representative Western blot of ERK isoforms with ERK C terminus antibody (top) or phospho-ERK forms (bottom) in cortical cultures after a 1 hr drug exposure: phorbol ester-induced increase in the phosphorylation state of ERK was antagonized by pharmacological inhibition of MEK.
Kinase-inactive MEK antagonizes PKC stimulation of APPsrelease and ERK activation
Overexpression of mutant proteins has proven to be a powerful tool for studying the role of signaling pathways in various cellular processes. A kinase-inactive MEK mutant, K97A, was generated by mutating lysine 97 to alanine (D. Charest and S. Pelech, unpublished data). This lysine is critical to MEK’s activity because it is found in the ATP-binding site (Seger et al., 1994). Previously, the K97A mutant has been shown to act in a “dominant negative” manner because its overexpression in NIH 3T3 cells inhibited phorbol ester stimulation of endogenous MEK and its downstream substrate ERK (Seger et al., 1994). Stimulation of APPs by 0.1 μmPMA was measured in HEK 293 cells transiently overexpressing human APP695 together with the K97A mutant or vector alone. Densitometric analysis revealed that PMA stimulation of APPs secretion was significantly inhibited in the presence of the kinase-inactive MEK compared with vector alone as determined using 22C11 (3.0 ± 0.3 and 1.9 ± 0.2, respectively;n = 3, p < 0.05) (Fig.4A) or WO-2 (2.3 ± 0.2 and 1.4 ± 0.4, respectively; n = 3,p < 0.05) (Fig. 4A). Moreover, Western blots using the gel shift assay indicate that the PMA-induced increase in ERK phosphorylation was antagonized by expression of the K97A mutant (Fig. 4B). Incomplete antagonism of ERK activation may be attributed in part to transfection efficiency. β-Galactosidase staining indicated that the percentage of transfected cells was 81.4 ± 1.6% (n = 3). Overexpression of the K97A mutant was confirmed using a rabbit polyclonal antibody raised against the N terminus of MEK1 (Upstate Biotechnology) (Fig.4B). Cellular levels of APP695 were not affected by overexpression of the dominant negative MEK (Fig.4B).
Fig. 4.
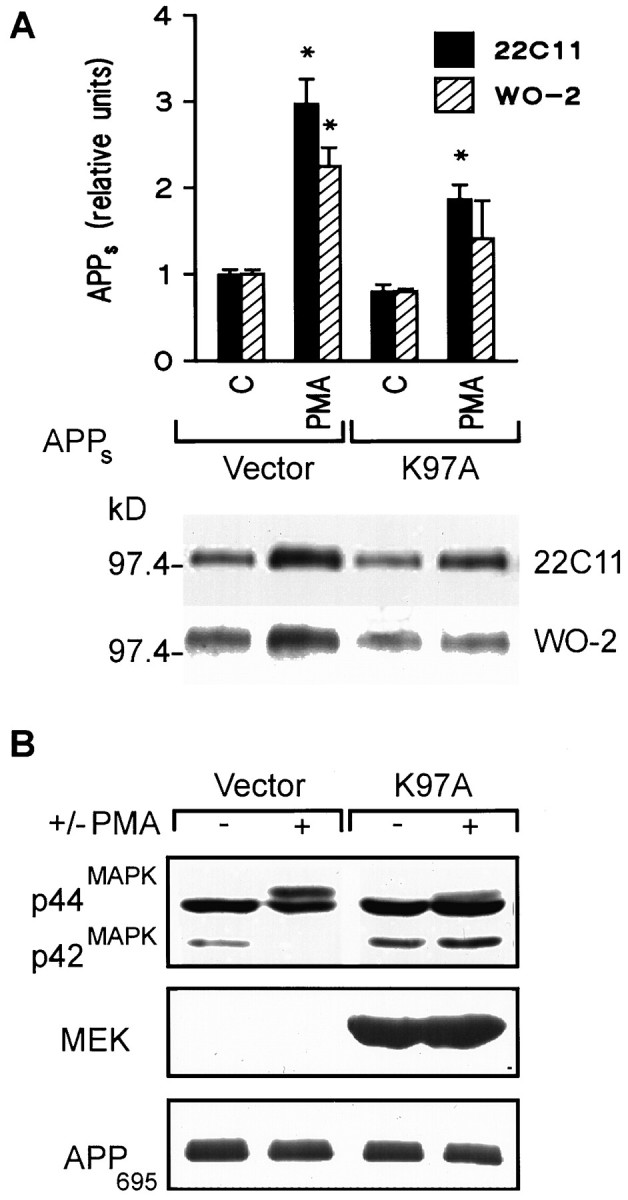
The kinase-dead mutant (K97A) inhibits phorbol ester stimulation of APPs secretion and ERK activation in HEK 293 cells. A, Top, Densitometric analysis of PMA (0.1 μm) stimulation of APPs secretion in cells expressing the MEK mutant (K97A) or vector alone (Vector). Data are mean ± SEM and represent three experiments for both 22C11 (filled columns) and WO-2 (hatched columns) (*p < 0.05, different from all other treatment groups). Bottom, Representative Western blot of the effect of PMA on basal APPs release after transient transfection of vector alone or the K97A mutant.B, Top, Representative Western blot of ERK isoforms with an ERK C terminus antibody in HEK 293 cells transfected with the kinase-dead MEK mutant or vector alone. The “electrophoretic shift” induced by PMA treatment in cells expressing vector alone was inhibited in cells expressing the kinase-dead MEK mutant. Middle, Representative Western blot of MEK1 using a rabbit polyclonal antibody raised against the N terminus of MEK1. Bottom, Representative Western blot of cellular APP using a monoclonal antibody generated against the N terminus of APP.
Pharmacological inhibition of MEK antagonizes PKC regulation of Aβ release
Activation of PKC is also known to regulate Aβ secretion. Specifically, a reduction of Aβ secretion has been observed after phorbol ester treatment (Buxbaum et al., 1993; Gabuzda et al., 1993; Hung et al., 1993; Jacobsen et al., 1994; Querfurth et al., 1994), direct activation of phospholipase C (Buxbaum et al., 1993), and first messengers (Hung et al., 1993) known to activate the PLC/PKC pathway. However, the cellular mechanisms underlying this regulation are poorly understood. To determine whether ERKs are involved in PKC regulation of Aβ secretion, HEK 293 cells overexpressing human APP695 carrying the Swedish mutation were exposed to 1 μm PMA for 15 min with or without 10 μm PD 98059. Densitometric analysis revealed that PMA inhibition of Aβ secretion was antagonized by PD 98059 (0.48 ± 0.05 and 0.83 ± 0.07, respectively; n = 7,p < 0.05) (Fig. 5).
Fig. 5.

PD 98059 antagonizes phorbol ester inhibition of Aβ secretion in K695sw cells. Top, Densitometric analysis of PMA (1 μm) inhibition of Aβ secretion in K695sw cells with or without PD 98059 (10 μm). Data are mean ± SEM of seven experiments [*p < 0.05, different from control (vehicle alone)]. Bottom, Representative Western blot of the effects of the MEK antagonist PD 98059 on PMA inhibition of Aβ release.
DISCUSSION
The major finding of the present study is that activation of the MAPK pathway is necessary for regulation of the secretory processing of APP. Antagonism of MEK inhibits phorbol ester and NGF receptor stimulation of APPs release as well as phorbol ester-mediated inhibition of Aβ release. The strength of the current study derives from the use of two distinct approaches for inhibiting the MAPK cascade, the pharmacological agent PD 98059 and gene transfer with a kinase-dead MEK mutant, both of which provided mutually supportive results. Moreover, the effects that we have observed are manifest in several different cell lines including neurons, suggesting that they are likely to be general rather than cell-specific.
These data also have broader implications for the function of ERKs. Correlative evidence has suggested that secretory stimuli activate ERKs in a variety of cells (Stratton et al., 1991; Frodin et al., 1995; Cox et al., 1996), but evidence demonstrating a requirement for ERK activation in secretion has not been obtained. Moreover, it has been shown that activation of the MAPK pathway is not required in some instances (Khoo and Cobb, 1997). The present experiments clearly implicate ERKs in regulation of APP secretory processing and, therefore, provide the first direct evidence for the necessity of the MAPK pathway in secretory events.
These results are relevant to our understanding of the molecular mechanisms by which APP catabolism is regulated in cells. A strong case has been made for the role of PKC activation in the regulation of APP catabolism (Nitsch and Growdon, 1994). PKC regulation of APP processing has been characterized extensively and has been shown to occur in a wide variety of cell lines (Buxbaum et al., 1990; Caporaso et al., 1992; Buxbaum et al., 1993; Gabuzda et al., 1993) and in central neurons (Lee et al., 1995; Mills and Reiner, 1996). However, the downstream effectors remain unknown. Our studies using both pharmacological and gene transfer approaches imply that MEK and/or ERK are necessary effectors for PKC-mediated stimulation of APPs release in both cell lines and neurons. Antagonism of PKC-mediated inhibition of Aβ secretion with the MEK inhibitor PD 98059 indicates that the MAPK pathway is also downstream of PKC regulation of Aβ production and that activation of MEK and/or ERK may reduce Aβ secretion.
The best characterized means of stimulating the MAPK pathway is by activation of receptor tyrosine kinases (Pelech and Sanghera, 1992;Cobb and Goldsmith, 1995). After ligand binding, these receptors autophosphorylate, promoting the association of ras with GTP leading to the sequential activation of raf1, MEK, and ERK. Autophosphorylation also promotes interaction of the receptor with a number of alternative target proteins including PLC-γ (Meisenhelder et al., 1989; Ronnstrand et al., 1992; Middlemas et al., 1994; Eriksson et al., 1995). Because of the abundant evidence that PKC activation regulates APP catabolism (Nitsch and Growdon, 1994), it is tempting to hypothesize that regulation of APP catabolism via receptor tyrosine kinases might be mediated by activation of PLC-γ. However, it is equally plausible that the “direct route” of ERK activation by receptor tyrosine kinases may be sufficient for regulation of APP catabolism by growth factor receptors. Regardless of the detailed molecular circuitry involved, our data demonstrate that ERK activation is necessary for growth factor stimulation of APPssecretion.
The mechanism by which the MAPK pathway regulates APP catabolism is unknown. Because of the time course involved in the present experiments, ERKs are unlikely to increase APPs secretion by increasing overall expression of cellular APP. Rather, it seems likely that ERKs are acting to phosphorylate one or more targets within the cell to modify APP catabolism. Direct phosphorylation of the APP holoprotein is unlikely because activated ERK does not phosphorylate the cytoplasmic domain of APP under conditions in which it is able to hyperphosphorylate tau (Alplin et al., 1996). Alternatively, ERKs may regulate APP processing indirectly by phosphorylating proteins involved in intracellular trafficking. For example, like PKC, ERK may increase APPs secretion by phosphorylating a tightly associatedtrans-Golgi network protein, thereby altering the formation of constitutive secretory vesicles containing mature APP (Xu et al., 1995). Also, presenilin-1, another protein thought to alter APP processing via its effects on protein trafficking (Borchelt et al., 1996; Lemere et al., 1996; Citron et al., 1997; Weidemann et al., 1997), has a consensus sequence for ERK-dependent phosphorylation and has been shown recently to be a substrate for PKC (Seeger et al., 1997;Walter et al., 1997a). Of course, the yet to be identified secretases that cleave APP remain potential candidates for phosphorylation by the MAPK cascade, either directly or indirectly.
A number of structurally unrelated membrane proteins undergo cleavage and subsequent release of their ectodomains into the extracellular medium, much like APP (Echlers and Riordon, 1991; Mattson et al., 1997); many of these share a common mechanism of regulation (Arribas and Massague, 1995). In addition to APP, PKC regulation of membrane protein processing has been observed for proTGF-α (Pandiella and Massague, 1991) colony-stimulating factor-1 (Stein and Rettenmier, 1991), colony-stimulating factor-1 receptor (Downing et al., 1989), and LAR transmembrane protein tyrosine phosphatase (Mullberg et al., 1992). Our data implicating the MAPK cascade in regulation of APP catabolism suggest that this mechanism of regulation may also be relevant to these membrane proteins.
Juxtamembrane cleavage serves to liberate APPs, which may act as a paracrine signaling factor. For example, APPs has been shown to stimulate a cGMP-dependent protein kinase (Furukawa et al., 1996) as well as ERKs (Greenberg et al., 1994,1995), and this function may be altered by phosphorylation of the ectodomain (Walter et al., 1997b). ERK activation by APPsis intriguing in light of the present findings, because it suggests that there may be a positive-feedback pathway whereby activation of ERK stimulates APPs release, which in turn activates the MAPK pathway.
Cell surface receptors known to regulate APP processing include heterotrimeric G-protein-coupled receptors and tyrosine kinase-coupled receptors (for review, see Beyreuther et al., 1996; Buxbaum and Greengard, 1996; Nitsch et al., 1996b). The effector system responsible can be regulated in either a PKC-dependent or a PKC-independent manner (Buxbaum et al., 1994; Slack et al., 1995; Nitsch et al., 1996a) and may involve activation of tyrosine kinases (Slack et al., 1995). All of these criteria are met by the MAPK signal transduction pathway. Our data for the first time implicate MEK and/or ERK in both PKC and tyrosine kinase receptor regulation of APP catabolism. Indeed, preliminary evidence from our laboratory suggests that MEK and/or ERK are critically involved in NMDA receptor stimulation of APPs secretion (J. Mills and P. Reiner, unpublished data), suggesting that the MAPK pathway may be critical for regulation of APP catabolism by a number of first messengers.
It is widely hypothesized that production and deposition of amyloid are early events in AD and may be the key pathological event that triggers the disease process (Hardy, 1997; Selkoe, 1997). As such, any manipulation that diminishes the production of Aβ is of potential therapeutic utility. The results presented in this study suggest that strategies aimed at activating the MAPK cascade may be a viable approach in this respect.
Footnotes
J.M. and F.L. were supported by studentships from the Alzheimer Society of Canada, D.L.C. was supported by a studentship from the Medical Research Council (MRC), and P.B.R. and S.L.P. are MRC Scientists. We acknowledge financial support provided by the Alzheimer Society of Canada, the British Columbia Health Research Foundation, and the MRC of Canada. We thank Rouzbeh Shooshtarian and Andy Laycock for technical assistance and Dennis Selkoe for his generous provision of the APP695 expression vector and K695sw cells.
Correspondence should be addressed to Peter B. Reiner, Kinsmen Laboratory of Neurological Research, Department of Psychiatry, University of British Columbia, 2255 Wesbrook Mall, Vancouver, British Columbia, Canada V6T 1Z3.
REFERENCES
- 1.Arribas J, Massague J. Transforming growth factor-α and β-amyloid precursor protein share a secretory mechanism. J Cell Biol. 1995;128:433–441. doi: 10.1083/jcb.128.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 3.Anderson JP, Esch FS, Keim PS, Sambamurti K, Lieberburg I, Robakis NK. Exact cleavage site of Alzheimer amyloid precursor in neuronal PC-12 cells. Neurosci Lett. 1991;128:126–128. doi: 10.1016/0304-3940(91)90775-o. [DOI] [PubMed] [Google Scholar]
- 4.Anderson NG, Maller JL, Tonks NK, Sturgill TW. Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature. 1990;343:651–653. doi: 10.1038/343651a0. [DOI] [PubMed] [Google Scholar]
- 5.Aplin AE, Gibb GM, Jacobsen S, Gallo J-M, Anderton BH. In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3β. J Neurochem. 1996;76:699–707. doi: 10.1046/j.1471-4159.1996.67020699.x. [DOI] [PubMed] [Google Scholar]
- 6.Bardwell L, Thorner J. The conserved motif at the amino termini of MEKs might mediate high-affinity interaction with the cognate MAPKs. Trends Biochem Sci. 1996;21:373–374. [PubMed] [Google Scholar]
- 7.Beyreuther K, Multhaup G, Monning U, Sandbrink R, Beher D, Hesse L, Small DH, Masters CL. Regulation of APP expression, biogenesis and metabolism by extracellular matrix and cytokines. Ann NY Acad Sci. 1996;777:74–75. doi: 10.1111/j.1749-6632.1996.tb34403.x. [DOI] [PubMed] [Google Scholar]
- 8.Borchelt DR, Thinakaren G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada C-M, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 9.Buxbaum JD, Greengard P. Regulation of APP processing by intra- and intercellular signals. Ann NY Acad Sci. 1996;777:327–331. doi: 10.1111/j.1749-6632.1996.tb34441.x. [DOI] [PubMed] [Google Scholar]
- 10.Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P. Processing of Alzheimer β/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci USA. 1990;87:6003–6006. doi: 10.1073/pnas.87.15.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buxbaum JD, Oishi M, Chen HI, Pinkas-Kramarski R, Jaffe EA, Gandy SE, Greengard P. Cholinergic agonists and interleukin 1 regulate processing and release of the Alzheimer β/A4 amyloid protein precursor. Proc Natl Acad Sci USA. 1992;89:10075–10078. doi: 10.1073/pnas.89.21.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buxbaum JD, Koo EH, Greengard P. Protein phosphorylation inhibits production of Alzheimer amyloid β/A4 peptide. Proc Natl Acad Sci USA. 1993;90:9195–9198. doi: 10.1073/pnas.90.19.9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buxbaum JD, Ruefli AA, Parker CA, Cypess AA, Greengard P. Calcium regulates processing of the Alzheimer amyloid protein precursor in a protein kinase C-independent manner. Proc Natl Acad Sci USA. 1994;91:4489–4493. doi: 10.1073/pnas.91.10.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caporaso GL, Gandy SE, Buxbaum JD, Ramabhadran TV, Greengard P. Protein phosphorylation regulates secretion of Alzheimer β/A4 amyloid precursor protein. Proc Natl Acad Sci USA. 1992;89:3055–3059. doi: 10.1073/pnas.89.7.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2744–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Citron M, Diehl TS, Gordon G, Biere AL, Seubert P, Selkoe DJ. Evidence that the 42- and 40-amino acid forms of amyloid β protein are generated from the β-amyloid precursor protein by different protease activities. Proc Natl Acad Sci USA. 1996;93:13170–13175. doi: 10.1073/pnas.93.23.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St. George-Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 18.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 19.Cox ME, Ely CM, Catling AD, Weber MJ, Parsons SJ. Tyrosine kinases are required for catecholamine secretion and mitogen-activated protein kinase activation in bovine adrenal chormaffin cells. J Neurochem. 1996;66:1103–1112. doi: 10.1046/j.1471-4159.1996.66031103.x. [DOI] [PubMed] [Google Scholar]
- 20.Da Cruz E, Silva OAB, Iverfeldt K, Oltersdorf T, Sinha S, Lieberburg I, Ramabhadran TV, Suzuke T, Sisodia SS, Gandy S, Greenbard P. Regulated cleavage of Alzheimer β-amyloid precursor protein in the absence of the cytoplasmic tail. Neuroscience. 1993;57:873–877. doi: 10.1016/0306-4522(93)90031-a. [DOI] [PubMed] [Google Scholar]
- 21.Downing JR, Roussel MF, Sherr CJ. Ligand and protein kinase C downmodulate the colony-stimulating factor 1 receptor by independent mechanisms. Mol Cell Biol. 1989;9:2890–2896. doi: 10.1128/mcb.9.7.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Echlers MRW, Riordon JF. Membrane proteins with soluble counterparts: role of proteolysis in the release of transmembrane proteins. Biochemistry. 1991;30:10065–10074. doi: 10.1021/bi00106a001. [DOI] [PubMed] [Google Scholar]
- 24.Eriksson A, Nanberg E, Ronnstrand L, Engstrom U, Hellman U, Rupp E, Carpenter G, Heldin CH, Claesson-Welsh L. Demonstration of functionally different interactions between phospholipase C-γ and the two types of platelet-derived growth factor receptors. J Biol Chem. 1995;270:7773–77781. doi: 10.1074/jbc.270.13.7773. [DOI] [PubMed] [Google Scholar]
- 25.Farber SA, Nitsch RM, Schulz JG, Wurtman RJ. Regulated Secretion of β-amyloid precursor protein in rat brain. J Neurosci. 1995;15:7442–7451. doi: 10.1523/JNEUROSCI.15-11-07442.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiore RS, Murphy TH, Sanghera JS, Pelech SL, Baraban JM. Activation of p42 mitogen-activated protein kinase by glutamate receptor stimulation in rat primary cortical cultures. J Neurochem. 1993;61:1626–1633. doi: 10.1111/j.1471-4159.1993.tb09796.x. [DOI] [PubMed] [Google Scholar]
- 27.Frodin M, Sekine N, Roche E, Filloux C, Prentki M, Wollheim CB, Van Obberghen E. Glucose, other secretagogues, and nerve growth factor stimulate mitogen-activated protein kinase in the insulin-secreting beta-cell line INS-1. J Biol Chem. 1995;270:7882–7889. doi: 10.1074/jbc.270.14.7882. [DOI] [PubMed] [Google Scholar]
- 28.Fukuyama R, Chandrasekaran K, Rapoport SI. Nerve growth factor induced neuronal differentiation is accompanied by differentiated induction and localization of the amyloid precursor protein (APP) in PC12 cells and variant PC12S cells. Mol Brain Res. 1993;17:17–22. doi: 10.1016/0169-328x(93)90067-y. [DOI] [PubMed] [Google Scholar]
- 29.Furukawa K, Sopher B, Rydel RE, Begley JG, Martin GM, Mattson MP. Increase activity-regulating and neuroprotective efficacy of α-secretase-derived secreted APP is conferred by a C-terminal heparin-binding domain. J Neurochem. 1996;67:1882–1896. doi: 10.1046/j.1471-4159.1996.67051882.x. [DOI] [PubMed] [Google Scholar]
- 30.Gabuzda D, Busciglio J, Yanker BA. Inhibition of β-amyloid production by activation of protein kinase C. J Neurochem. 1993;61:2326–2329. doi: 10.1111/j.1471-4159.1993.tb07479.x. [DOI] [PubMed] [Google Scholar]
- 31.Graves JD, Campbell JS, Krebs EG. Protein serine/threonine kinases of the MAPK cascade. Ann NY Acad Sci. 1995;766:320–343. doi: 10.1111/j.1749-6632.1995.tb26684.x. [DOI] [PubMed] [Google Scholar]
- 32.Greenberg SM, Koo EH, Selkoe DJ, Qiu WQ, Kosik KS. Secreted β-amyloid precursor protein stimulates mitogen-activated protein kinase and enhances τ phosphorylation. Proc Natl Acad Sci USA. 1994;91:7104–7108. doi: 10.1073/pnas.91.15.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenberg SM, Qiu WQ, Selkoe DJ, Ben-Itzhak A, Kosik KS. Amino-terminal region of the β-amyloid protein activates mitogen-activated protein kinase. Neurosci Lett. 1995;198:52–56. doi: 10.1016/0304-3940(95)11944-r. [DOI] [PubMed] [Google Scholar]
- 34.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 35.Hames BD. In gel electrophoresis of proteins. A Practical approach. IRL; London: 1991. [Google Scholar]
- 36.Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 37.Hu H, Sweeney D, Greengard P, Gandy S. Metabolism of Alzheimer β-amyloid precursor protein: regulation by protein kinase A in intact cells and in a cell-free system. Proc Natl Acad Sci USA. 1996;93:4081–4084. doi: 10.1073/pnas.93.9.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hung AY, Selkoe DJ. Selective ectodomain phosphorylation and regulated cleavage of β-amyloid precursor protein. EMBO J. 1994;13:534–542. doi: 10.1002/j.1460-2075.1994.tb06291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hung AY, Haass C, Nitsch RM, Qiu WQ, Citron M, Wurtman RJ, Growdon JH, Selkoe D. Activation of protein kinase C inhibits cellular production of the amyloid β-protein. J Biol Chem. 1993;268:22959–22962. [PubMed] [Google Scholar]
- 40.Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, Sandbrink R, Masters CL, Beyreuther K. Analysis of heterogeneous βA4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- 41.Jacobsen JS, Spruyt MA, Brown AM, Sahasrabubhe SR, Blume AJ, Vitek MP, Muenkel HA, Sonnenberg-Reines J. The release of Alzheimer’s disease β-amyloid peptide is reduced by phorbol treatment. J Biol Chem. 1994;269:8376–8382. [PubMed] [Google Scholar]
- 42.Jaffe AB, Toran-Allerand CD, Greengard P, Gandy SE. Estrogen regulates metabolism of Alzheimer amyloid β precursor protein. J Biol Chem. 1994;269:13065–13068. [PubMed] [Google Scholar]
- 43.Khoo S, Cobb MH. Activation of mitogen-activating protein kinase by glucose is not required for insulin secretion. Proc Natl Acad Sci USA. 1997;94:5599–5604. doi: 10.1073/pnas.94.11.5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klafki H-W, Wiltfang J, Staufenbiel M. Electrophoretic separation of βA4 peptides (1–40) and (1–42). Anal Biochem. 1996;237:24–29. doi: 10.1006/abio.1996.0195. [DOI] [PubMed] [Google Scholar]
- 45.Lazar DF, Wiese RJ, Brady MJ, Mastick CC, Waters SB, Yamauchi K, Pessin JE, Cuatrecasas P, Saltiel AR. Mitogen-activated protein kinase kinase inhibition does not block the stimulation of glucose utilization by insulin. J Biol Chem. 1995;270:20801–20807. doi: 10.1074/jbc.270.35.20801. [DOI] [PubMed] [Google Scholar]
- 46.Lee RK, Wurtman RJ, Cox AJ, Nitsch RM. Amyloid precursor protein processing is stimulated by metabotropic glutamate receptors. Proc Natl Acad Sci USA. 1995;92:8083–8087. doi: 10.1073/pnas.92.17.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, Yamaguchi H, Ruiz A, Maritnez A, Madrigal L, Hincapie L, Arango JC, Anthony DC, Koo EH, Goate AM, Selkoe DJ, Arango JC. The E280A presenilin 1 Alzheimer mutation produces increased Aβ42 deposition and severe cerebellar pathology. Nat Med. 1996;2:1146–1150. doi: 10.1038/nm1096-1146. [DOI] [PubMed] [Google Scholar]
- 48.Malarkey K, Belham CM, Paul A, Graham A, McLees A, Scott PH, Plevin R. The regulation of tyrosine kinase signalling pathways by growth factor and G-protein-coupled receptors. Biochem J. 1995;309:361–375. doi: 10.1042/bj3090361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mattson MP, Barger SW, Furukawa K, Bruce AJ, Wyss-Coray T, Mark RJ, Mucke L. Cellular signaling roles of TGFβ, TNFα and βAPP in brain injury responses and Alzheimer’s disease. Brain Res Rev. 1997;23:47–61. doi: 10.1016/s0165-0173(96)00014-8. [DOI] [PubMed] [Google Scholar]
- 50.Meisenhelder J, Suh P-G, Rhee SG, Hunter T. Phospholipase C-γ is a substrate for the PDGF and EGF receptor protein-tyrosine kinases in vivo and in vitro. Cell. 1989;57:1109–1122. doi: 10.1016/0092-8674(89)90048-2. [DOI] [PubMed] [Google Scholar]
- 51.Middlemas DS, Meisenhelder J, Hunter T. Identification of TrkB autophosphorylation sites and evidence that phospholipase C-γ 1 is a substrate of the Trk B receptor. J Biol Chem. 1994;269:5458–5466. [PubMed] [Google Scholar]
- 52.Mills J, Reiner PB. Phorbol esters but not cholinergic agonist oxotremorine-M and carbachol increase release of the amyloid precursor protein in cultured rat cortical neurons. J Neurochem. 1996;67:1511–1518. doi: 10.1046/j.1471-4159.1996.67041511.x. [DOI] [PubMed] [Google Scholar]
- 53.Mullberg J, Schooltink H, Stoyan T, Heinrich PC, Rose-John S. Protein kinase C activity is rate limiting for shedding of the interleukin-6 receptor. Biochem Biophys Res Commun. 1992;189:794–800. doi: 10.1016/0006-291x(92)92272-y. [DOI] [PubMed] [Google Scholar]
- 54.Murphy TH, Wright DD, Baraban JM. Phosphoinositide turnover associated with glutamatergic synaptic transmission. J Neurochem. 1992;59:2336–2339. doi: 10.1111/j.1471-4159.1992.tb10130.x. [DOI] [PubMed] [Google Scholar]
- 55.Nitsch RM, Growdon JH. Roles of neurotransmission in the regulation of amyloid β-protein precursor processing. Biochem Pharmacol. 1994;47:1275–1284. doi: 10.1016/0006-2952(94)90325-5. [DOI] [PubMed] [Google Scholar]
- 56.Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- 57.Nitsch RM, Deng M, Growdon JH, Wurtman RJ. Serotonin 5-HT2a and 5-HT2c receptors stimulate amyloid precursor protein ectodomain secretion. J Biol Chem. 1996a;271:4188–4194. doi: 10.1074/jbc.271.8.4188. [DOI] [PubMed] [Google Scholar]
- 58.Nitsch RM, Wurtman RJ, Growdon JH. Regulation of APP processing: potential for the therapeutical reduction of brain amyloid burden. Ann NY Acad Sci. 1996b;777:175–182. doi: 10.1111/j.1749-6632.1996.tb34416.x. [DOI] [PubMed] [Google Scholar]
- 59.Pague DH, Rossomondo AJ, Martino P, Erickson AK, Her JH, Shabanowitz J, Hunt DF, Weber MJ, Sturgill TW. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 1991;10:885–892. doi: 10.1002/j.1460-2075.1991.tb08021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pandiella A, Massague J. Multiple signals activate cleavage of membrane transforming growth factor-α precursor. J Biol Chem. 1991;266:5769–5773. [PubMed] [Google Scholar]
- 61.Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 62.Pelech SL, Charest DL. Cell cycle control in progress. In: Meijer L, Guidet S, Lim Tung HY, editors. Cell cycle research, Vol 1. Plenum; New York: 1996. pp. 33–52. [DOI] [PubMed] [Google Scholar]
- 63.Pelech SL, Sanghera JS. MAP kinases: charting the regulatory pathways. Science. 1992;257:1355–1356. doi: 10.1126/science.1382311. [DOI] [PubMed] [Google Scholar]
- 64.Pelech SL, Charest DL, Mordret GP, Siow YL, Palaty C, Campbell D, Charlton L, Samiei M, Sanghera JS. Networking with mitogen-activated kinases. Mol Cell Biochem. 1993;127:157–169. doi: 10.1007/BF01076767. [DOI] [PubMed] [Google Scholar]
- 65.Posada J, Cooper JA. Requirement for phosphorylation of MAP kinase during meiosis in Xenopus oocytes. Science. 1992;255:182–184. doi: 10.1126/science.1313186. [DOI] [PubMed] [Google Scholar]
- 66.Querfurth HW, Selkoe DJ. Calcium ionophore increases amyloid β peptide production by cultured cells. Biochemistry. 1994;33:4550–4561. doi: 10.1021/bi00181a016. [DOI] [PubMed] [Google Scholar]
- 67.Raymond LM, Moshaver A, Tingley WG, Shalaby I, Huganir RL. Glutamate receptor ion channel properties predict vulnerability to cytotoxicity in a transfected nonneuronal cell line. Mol Cell Neurosci. 1996;7:102–115. doi: 10.1006/mcne.1996.0008. [DOI] [PubMed] [Google Scholar]
- 68.Refolo LM, Salton SR, Anderson JP, Mehta P, Robakis NK. Nerve and epidermal growth factors induce the release of the Alzheimer amyloid precursor from PC12 cell cultures. Biochem Biophys Res Commun. 1989;164:664–670. doi: 10.1016/0006-291x(89)91511-8. [DOI] [PubMed] [Google Scholar]
- 69.Ronnstrand L, Mori S, Arridsson A-K, Eriksson A, Wernstedt C, Hellman U, Claesson-Welsh L, Heldin C-H. Identification of two C-terminal autophosphorylation sites in the PDGF β-receptor: involvement in the interaction with phospholipase C-γ. EMBO J. 1992;11:3911–3919. doi: 10.1002/j.1460-2075.1992.tb05484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schubet D, Jin L-W, Saitoh T, Cole G. The regulation of amyloid β protein precursor secretion and its modulatory role in cell adhesion. Neuron. 1989;3:689–694. doi: 10.1016/0896-6273(89)90237-7. [DOI] [PubMed] [Google Scholar]
- 71.Seeger M, Nordstedt C, Petanceska S, Kovac DM, Gouras GK, Hahne S, Fraser P, Levesque L, Czernik AJ, St. George-Hyslop P, Sisodia SS, Thinkakaran G, Tanzi RE, Greengard P, Gandy S. Evidence for phosphorylation and oligomeric assembly of presenilin 1. Proc Natl Acad Sci USA. 1997;94:5090–5094. doi: 10.1073/pnas.94.10.5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seger R, Seger D, Reszka AA, Munar ES, Eldar-Finkelman H, Dobrowolska G, Jensen AM, Campbell JS, Fischer EH, Krebs EG. Overexpression of mitogen-activated protein kinase kinase (MAPKK) and its mutants in NIH 3T3 cells. J Biol Chem. 1994;269:25699–25709. [PubMed] [Google Scholar]
- 73.Selkoe DJ. Alzheimer’s disease: genotypes, phenotype and treatment. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 74.Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz L, Oltersdorf T. β-Amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci USA. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shoji M, Golde TE, Cheung TT, Ghiso J, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, Younkin SG. Production of the Alzheimer amyloid β protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 76.Sisodia SS, Koo EH, Beyreuther K, Unterbeck A, Price DL. Evidence that β-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science. 1990;248:492–495. doi: 10.1126/science.1691865. [DOI] [PubMed] [Google Scholar]
- 77.Slack BE, Breu J, Petryniak MA, Srivastava K, Wurtman RJ. Tyrosine phosphorylation of amyloid precursor protein secretion by the M3 muscarinic acetylcholine receptor. J Biol Chem. 1995;270:8337–8344. doi: 10.1074/jbc.270.14.8337. [DOI] [PubMed] [Google Scholar]
- 78.Slunt HH, Thinakaran G, Van Koch C, Lo ACY, Tanzi RE, Sisodia SS. Expression of a ubiquitous, cross-reactive homologue of the mouse β-amyloid precursor protein (APP). J Biol Chem. 1994;269:2637–2644. [PubMed] [Google Scholar]
- 79.Stein J, Rettenmier CW. Proteolytic processing of a plasma membrane-bound precursor to human macrophage colony-stimulating factor (CSF-1) is accelerated by phorbol ester. Oncogene. 1991;6:601–605. [PubMed] [Google Scholar]
- 80.Stratton KR, Worley PF, Litz JS, Parsons SJ, Huganir RL, Baraban JM. Electroconvulsive treatment induces a rapid and transient increase in tyrosine phosphorylation of a 40-kilodalton protein associated with microtubule-associated protein 2 kinase activity. J Neurochem. 1991;56:147–152. doi: 10.1111/j.1471-4159.1991.tb02574.x. [DOI] [PubMed] [Google Scholar]
- 81.Walter J, Grunberg J, Capell A, Pesold B, Schindzielorz A, Citron M, Mendla K, St. George-Hyslop P, Multhaup G, Selkoe DJ, Haass C. Proteolytic processing of the Alzheimer disease-associated presenilin-1 generates an in vivo substrate for protein kinase C. Proc Natl Acad Sci USA. 1997a;94:5349–5354. doi: 10.1073/pnas.94.10.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walter J, Capell A, Hung AY, Langen H, Schnölzer M, Thinakaran G, Sisodia SS, Selkoe DJ, Haass C. Ectodomain phosphorylation of β-amyloid precursor protein at two distinct cellular locations. J Biol Chem. 1997b;272:1896–1903. doi: 10.1074/jbc.272.3.1896. [DOI] [PubMed] [Google Scholar]
- 83.Wang R, Meschia JF, Cotter RJ, Sisodia SS. Release of the β/A4 amyloid precursor protein: identification of a cleavage site in cultured mammalian cells. J Biol Chem. 1991;266:16960–16964. [PubMed] [Google Scholar]
- 84.Weidemann A, Paliga K, Durrwang U, Czech C, Evin G, Masters CL, Beyreuther K. Formation of stable complexes between two Alzheimer’s disease gene products: presenilin-2 and β-amyloid precursor protein. Nat Med. 1997;3:328–332. doi: 10.1038/nm0397-328. [DOI] [PubMed] [Google Scholar]
- 85.Wolf BA, Wertkin AM, Jolly YC, Yasuda RP, Wolfe BB, Konrad RJ, Manning D, Ravi S, Williamson JR, Lee VM-Y. Muscarinic regulation of Alzheimer’s disease amyloid precursor protein secretion and amyloid β-protein production in human neuronal NT2N cells. J Biol Chem. 1995;270:4916–4922. doi: 10.1074/jbc.270.9.4916. [DOI] [PubMed] [Google Scholar]