

Abstract
Long-read sequencing technologies have contributed greatly to comparative genomics among species and can also be applied to study genomics within a species. In this study, to determine how substantial genomic changes are generated and tolerated within a species, we sequenced a C. elegans strain, CB4856, which is one of the most genetically divergent strains compared to the N2 reference strain. For this comparison, we used the Pacific Biosciences (PacBio) RSII platform (80×, N50 read length 11.8 kb) and generated de novo genome assembly to the level of pseudochromosomes containing 76 contigs (N50 contig = 2.8 Mb). We identified structural variations that affected as many as 2694 genes, most of which are at chromosome arms. Subtelomeric regions contained the most extensive genomic rearrangements, which even created new subtelomeres in some cases. The subtelomere structure of Chromosome VR implies that ancestral telomere damage was repaired by alternative lengthening of telomeres even in the presence of a functional telomerase gene and that a new subtelomere was formed by break-induced replication. Our study demonstrates that substantial genomic changes including structural variations and new subtelomeres can be tolerated within a species, and that these changes may accumulate genetic diversity within a species.
Genetic changes can affect evolution, and different species have accumulated many genetic and phenotypic variations before and after speciation (Schluter 2001; Wu and Ting 2004). Comparison of the genomes of closely related species is one way to understand the mechanisms or consequences of speciation (Alföldi and Lindblad-Toh 2013; Koepfli et al. 2015). The genus Caenorhabditis is a resource for such comparative genomic studies. Species diversity and small genome sizes have made the Caenorhabditis genus a subject for molecular dissection of the genome and of trait evolution (Stein et al. 2003; Slos et al. 2017; Yin et al. 2018). Over 50 species of the Caenorhabditis genus have been collected, and the genomes of 25 of them have been sequenced (Stevens et al. 2018). Although inter-species comparisons have found many genomic differences, which have provided insights into genome evolution, different species have already undergone numerous changes. Little is known about where and how genomic changes within a species have accumulated. To understand genomic changes within a species, we compared the genome of the reference N2 strain with that of CB4856, a highly divergent C. elegans wild strain (Koch et al. 2000; Wicks et al. 2001).
N2 and CB4856 have numerous heritable phenotypic differences. The recombinant inbred lines and the recombinant inbred advanced intercross lines produced by crossing the two strains have revealed several genetic loci that cause phenotypic variations such as aggregation behavior, mating, nictation behavior, pathogen response, and genetic incompatibility (de Bono and Bargmann 1998; Tijsterman et al. 2002; Schulenburg and Müller 2004; Kammenga et al. 2007; Palopoli et al. 2008; Seidel et al. 2008, 2011; Kim et al. 2017; Lee et al. 2017). Attempts have been made to obtain the CB4856 genome that accurately represents these genetic variants, but the currently available CB4856 reference genome has the limitation that it has been assembled from sequences that were obtained using short-read sequencing (Thompson et al. 2015). These sequences may underrepresent genomic rearrangements that are longer than the insert length and may miss insertions and repetitive sequences.
The occurrence of repetitive sequences is generally highest near the ends of chromosomes. A subtelomere is a hypervariable region adjacent to the telomere and has various repeats including segmental duplicated blocks. The repetitive nature of subtelomeric and telomeric regions can impair their assembly by short-read sequencing. For example, in the human genome hg19 version released in 2009, telomeric repeats directly linked to subtelomere sequences appear in only 17 out of 46 chromosome ends (Rudd 2014). In addition, in the C. elegans VC2010 de novo assembly by Nanopore long-read sequencing, telomeric repeats directly linked to subtelomere sequences appear in only six out of 12 chromosome ends (Tyson et al. 2018). Therefore, these regions could be underrepresented in de novo assembled genomes. The high variability of subtelomeres over generations facilitates the emergence of new genes and may help to increase the fitness of organisms. This possibility of the involvement of subtelomeres in chromosome evolution has not been extensively studied because of the difficulty in the genome assembly near subtelomeres.
Telomeres are the ends of linear chromosomes of eukaryotic cells. In most cases, telomeres are composed of specific sequence repeats to form highly ordered structures. Critically shortened telomeres can lead to chromosome dysfunction, so all eukaryotic cells must maintain appropriate telomere length (Harley et al. 1990; O'Sullivan and Karlseder 2010). Organisms that fail to maintain the telomere in germ lines eventually become sterile (Blackburn 1991; Blasco et al. 1997; Meier et al. 2006). The telomere lengthening is mainly fulfilled by using telomerase and telomeric repeats, but in some cases alternative lengthening of telomeres (ALT) can be used to lengthen telomeres without utilizing telomerase (Lundblad and Blackburn 1993; Nakamura et al. 1998).
ALT is defined as telomere lengthening in the absence of functional telomerase activity. ALT occurs in certain cancer cells in humans and in organisms in nature; for example, Drosophila uses retrotransposon rDNA sequences and onions use minisatellite rDNA sequences to maintain telomeres. This ALT process uses sequences other than canonical telomeric repeats (Bryan et al. 1997; Pich and Schubert 1998; Cesare and Reddel 2010; Garavís et al. 2013; Mason et al. 2016). In C. elegans, the telomerase-deficient animals survived telomere attrition by replicating template for ALT (TALT) at the end of every chromosome (Seo et al. 2015; Kim et al. 2016). Break-induced replication (BIR) is another major mechanism to maintain telomeres without the action of telomerase, as reported in human cancer cells and yeasts (Lydeard et al. 2007; Dilley et al. 2016). During BIR, homology templates from either the same chromosome or even a nonallelic region can be used for replication of the templates, up to the size of 200 kb, which can establish new subtelomeres (Costantino et al. 2014; Mason and McEachern 2018).
In this study, we have obtained a nearly completed CB4856 genome by long-read sequencing and report the identification and characterization of structural variations (SVs) within the genome and structural changes in the subtelomeric regions. We also discuss the significance of new subtelomere formation in generating new genetic materials for evolution of new traits.
Results
Long-read sequencing and de novo assembly of the CB4856 genome
To compare the N2 and CB4856 genomes, we used the Pacific Biosciences (PacBio) RSII platform to construct a nearly complete, chromosome-scale, high-quality genome of CB4856. The genome of CB4856 was assembled with Canu (Koren et al. 2017) using 80× coverage raw reads and was composed of 137 contigs of 104 Mb in total length (Supplemental Fig. S1A,B). Elimination of bacterial contamination, followed by base corrections using PacBio and HiSeq raw reads (Chin et al. 2013; Walker et al. 2014), left an assembled genome of 128 contigs, which were assembled to the level of pseudochromosomes by using fosmid, linkage information, and tiling to the N2 genome (Fig. 1A–C; Table 1; Supplemental Figs. S1C–E, S2A,B,E–G; Supplemental Table S1). The final assembled genome of CB4856 was 103 Mb in total, 99.4% identical to the N2 genome, and contained 0.2% SNPs between N2 and CB4856 (Tables 1, 2). BUSCO analysis based on gene content information showed that the completeness of the CB4856 genome was comparable to that of the N2 genome (Supplemental Fig. S2C; Simão et al. 2015). In addition, all of the chromosome ends had assembled telomeres longer than 2 kb; this observation suggests that the genome assembly toward the chromosome ends is of high quality (Supplemental Fig. S2D). Most of the genome regions are covered by PacBio raw reads, an average of 60× (Fig. 1B). To further evaluate the quality of our genome assembly, we measured the quality of alignment among CB4856 HiSeq reads, a reference genome (N2 genome), a CB4856 genome assembly obtained using short reads (Thompson genome) (Thompson et al. 2015), and a CB4856 genome obtained in this study (Kim genome). We aligned the CB4856 HiSeq reads to the genomes (72.2×, 74.6×, 72.5×, respectively) and tried to call SNPs, indels, and heterozygous variants. The CB4856 HiSeq reads were used for alignment, so we expected to get few SNPs, indels, or heterozygous variants from a well-assembled genome of CB4856 and a large number from N2. The number of SNPs and indels found in the Kim genome here was only about 5% of that detected in the Thompson genome (Supplemental Fig. S3). We also found that the numbers of heterozygous variants were 21,432 in the N2 genome, 13,412 in the Thompson genome, and 562 in the Kim genome (Fig. 1D).
Figure 1.

CB4856 genome assembly and comparison with the N2 genome at a chromosome level. (A) Schematic representation of CB4856 contig lengths mapped to N2 WBcel235 chromosomes. (B) PacBio raw read coverage, mapped on CB4856 chromosomes (100-kb binned). Reads were distributed at average 60× coverage. (C) Schematic of large chromosomal rearrangement between N2 and CB4856 genomes identified using progressiveMauve. The blue box and line indicate inversion; the red box and line, translocation; and the white box indicates the unaligned block. Chr VR has several small rearrangements and unaligned blocks. Chr II: 3,896,126–3,900,949 in N2 was inverted in CB4856 (Chr II: 4,045,653–4,040,823), Chr V: 17,616,880–17,623,484 in N2 was inverted in CB4856 (Chr V: 17,734,209–17,728,873), and Chr V: 19,258,912–19,289,935 in N2 was located at Chr V: 21,193,104–21,237,336 in CB4856. (D) Schematic representation of CB4856 HiSeq reads mapped on the CB4856 genome (blue) or the N2 genome (yellow). Each dot shows the heterozygous base count (100-kb interval) from Chr I to Chr X.
Table 1.
Long-read sequencing-based genome assemblies of CB4856

Table 2.
Comparisons between pairs of N2/Thompson genomes and N2/Kim genomes

To further analyze the two CB4856 genomes, we aligned them to the N2 genome and determined the numbers of SNPs, indels, and SVs larger than 50 bp. The number of SNPs was similar in the Thompson and Kim genomes, but the Kim genome had the largest numbers of indels and SVs (Table 2). The patterns of hypervariable regions in which SNPs are densely distributed was similar in the Thompson and Kim genomes (Supplemental Fig. S4). Taken together, these results indicate that our (Kim) CB4856 genome was of sufficiently high quality.
Long-read sequencing identified new structural variations
With the newly de novo assembled genome of CB4856, we assessed SVs between the N2 reference genome and our CB4856 genome at fine-scale resolution. SVs longer than 50 nucleotides altered more nucleotides than did SNPs. On the chromosomal scale, a 170-kb sequence block from the N2 Chr V: 1,105,418–1,274,268 was located at the CB4856 Chr II: 4,153,071–4,323,030, and a 90-kb sequence block from N2 Chr IV: 9,413,332–9,503,493 was inverted in CB4856 Chr IV: 9,614,155–9,523,953. Furthermore, the Chr V right arm in CB4856 contained numerous small rearrangements that ranged from 10 to 100 kb in size (Fig. 1C; Supplemental Fig. S5). SVs also caused substantial changes in the two genomes (Fig. 2): They included 3349 SVs, which together totaled more than 4.95 Mb (Fig. 2A).
Figure 2.

Structural variations (SVs) between the CB4856 and N2 genomes and their effects on chromosomal contents. (A) SVs between the N2 genome and the short-read-based CB4856 genome, previously reported (left), and between the N2 genome and the long read-based CB4856 genome (right). Repeat expansion, tandem expansion, and insertion SVs are more often detected when using long read-based genome than when using the previous short read-based genome. (B) Tracks representing density at 100-kb intervals; from outside to inside: 1, genomic positions (in Mb) of the six chromosomes based on the N2 genome; 2, density of local recombination rate in CB4856/N2 introgression lines; 3–9, types of SVs identified using Assemblytics: 3, size of SVs; 4, density of repeat-contraction SVs; 5, density of repeat-expansion SVs; 6, density of tandem-contraction SVs; 7, density of tandem-expansion SVs; 8, density of deletion SVs; 9, density of insertion SVs. (C) Tracks representing density at 100-kb intervals; from outside to inside: 1, genomic positions (in Mb) of the six chromosomes based on the N2 genome; 2–4, density of SVs estimated by SnpEff: 2, high-impact SVs; 3, low-impact SVs; 4, modifier SVs. (D) Annotation of SVs. SVs effects were categorized using SnpEff based on their position in the annotated N2 genome. “N2-specific genes” indicates the number of the genes that are completely deleted in CB4856. ‘Genic’ indicates the number of genes whose function is predicted to be affected by the SVs. ‘Intergenic’ indicates the number of SVs in the intergenic region. ‘Upstream’ indicates the number of SVs located within 5 kb upstream of a gene. ‘Downstream’ indicates the number of SVs located within 5 kb downstream from a gene.
We then further analyzed the properties of the SVs that we identified using the Kim genome based on long-read sequencing, compared with those from the Thompson genome. The Kim genome detected an additional 1.6 Mb of SVs, including insertions, tandem expansions, and repeat expansions (Supplemental Fig. S6A). The Kim genome also included ∼4 Mb of unaligned bases that were not present in the Thompson genome (Supplemental Fig. S6B). Unaligned bases occurred in 264,580 regions, which were mostly near the ends of chromosomes (Supplemental Fig. S6C–H). The Kim genome included 467 unaligned regions of >1 kb; of these, 293 regions contained repeat sequences. Over 90% of the SVs found in the Thompson genome were also found in the Kim genome (Supplemental Fig. S6I). We found SVs that had not been found in the short-read-based assembly, so the Kim genome is larger than the Thompson genome.
To examine the consequence of the SVs in the context of the genes affected, we inspected the genome-wide gene annotations of the CB4856 genome based on synteny with N2 or RNA-seq data (Supplemental Fig. S7). The SVs of 2694 genes in CB4856 generated predicted effects on gene function, including start-codon losses, stop-codon losses, frameshifts, or exon losses (Fig. 2B–D; Supplemental Fig. S8). In addition, more than 600 genes are specific to one strain or the other (Supplemental Fig. S7). Half of the completely missing genes were identified in previous CGH data; the other half are identified here for the first time (Supplemental Fig. S9A,B; Maydan et al. 2007, 2010). Among the genes that are missing in CB4856, 31 are reported to cause sterile or lethal phenotypes by RNAi, and six of them, including the incompatibility gene zeel-1, showed sterile or lethal phenotypes when deleted in the N2 background (Supplemental Fig. S9C–E; Supplemental Table S2; Seidel et al. 2008, 2011). High-impact SVs as defined by SnpEff (Cingolani et al. 2012) and strain-specific genes are more concentrated on autosome arms than centers. These results show that chromosomes are changing more rapidly on the arms than at the center (The C. elegans Sequencing Consortium 1998) and show another example of how variants on the chromosome center regions, where recombination frequency is relatively low (Fig. 2B), have been eliminated, together with other deleterious mutations, by background selection (Rockman and Kruglyak 2009; Cutter and Choi 2010; Cutter and Payseur 2013). Our analysis also implies that substantial genetic changes including gene gain or loss have been tolerated during genetic differentiation within these two strains without decreasing brood size (Andersen et al. 2014; Lee et al. 2017).
Long-read sequencing revealed the hypervariable nature of subtelomeres
The subtelomeric regions, which we arbitrarily defined as the 200-kb ends of each chromosome, have many regions without alignment. We used high-coverage long-read sequencing to construct contigs of an average size of 700 kb including telomeres on all chromosomes (Supplemental Fig. S2D). The assembled telomere length of each chromosome end is ∼40% of mean telomere length (Fig. 3C). This information allows a direct comparison of the subtelomeres of the CB4856 genome with those of the reference genome. Only 76% of the sequences from the N2 subtelomeric regions and 74% of those from CB4856 were aligned with those of the other strain. These numbers of aligned nucleotides are relatively small when compared with that of the entire genome, which is 95% of the N2 genome and 93% of the CB4856 genome (Supplemental Fig. S10A).
Figure 3.

New subtelomere formation in CB4856 Chr VR using an alternative lengthening of telomeres (ALT) mechanism. (A) Schematic representation of subtelomere differences between the N2 and CB4856 chromosomes. Yellow bars and blue bars at the end of chromosomes indicate the ratio of unaligned bases of subtelomeres in N2 and CB4856 genome, respectively. (B) Dot plot representing alignment between internal segment (V: 19,377,978–19,606,221) and duplicated segment (V: 21,171,521–21,389,866) of CB4856 Chr VR; 63% of the two regions are aligned, and 91% of the aligned bases are identical. Red: forward strand matches; blue: reverse strand matches. (C) Telomere length of all chromosomes deduced from the long-read CB4856 genome. ‘HiSeq’ data are mean telomere lengths normalized by the telseq software (Ding et al. 2014). The red bar represents the end of N2 (Chr VR internal) in Chr VR of CB4856. Only small portions of the N2 telomere remain in CB4856, followed by a new subtelomere. ‘Chr V terminal’ is from the real end of Chr VR. (D) Schematic representation of Chr V subtelomere in CB4856. Five copies of template for ALT (TALT) (red) are connected to the duplicated segment from the internal segment close to the internal TALT (V: 19,366,148–19,367,611). The bottom shows PacBio raw reads on the tandemly repeated TALT region. Four raw reads almost fully cover this region.
The subtelomere sequences show large insertions, deletions, or inversions at more than half of the chromosome ends (Fig. 3A); these changes suggest that half of subtelomeric regions have undergone substantial changes. These subtelomeres showed complex structures composed of sequences with homology to preexisting subtelomeres, sequences with partial homology from internal regions, and sequences with no homology at all (Supplemental Fig. S10B–G).
The structure of Chr VR subtelomere is unique, in consequence of past ALT and BIR events
Among the subtelomeres, Chr VR is unique in that new sequences of more than 200 kb are inserted, and these regions are derived from an internal Chr V region with high homology (71% aligned, 91% identity) (Fig. 3B; Supplemental Fig. S11A). We analyzed the right end of Chr V in more detail to provide an insight into the possible mechanism of new subtelomere formation in the ancestor of CB4856. We found that the right subtelomere of Chr V of CB4856 contained telomere sequences (Fig. 3C; marked as ‘N2 end’ in Fig. 3D) 10-fold shorter than the estimated mean telomere, which were followed by 200 kb of extra sequences (Fig. 3D). This extra region contains five tandemly duplicated copies of the TALT sequence (marked as red bars in Fig. 3D; Supplemental Fig. S11B,C), flanked by telomeric repeats of lengths ranging from 780 to 1182 nt (marked as blue bars in Fig. 3D). The TALT sequence was previously identified and defined as the replication template for ALT in C. elegans animals that survived telomere shortening caused by telomerase deficiency (Seo et al. 2015). These TALT copies were followed by sequences that have 91% identity with an internal 200-kb sequence block next to the internal TALT (Fig. 3D). The real end of the Chr VR in CB4856 contained at least 3-kb-long telomeric repeats. The features of Chr VR are consistent with the hypothesis that the new subtelomere was formed by telomere attrition followed by two sequential telomere damage repair events using ALT and BIR (see Discussion; Fig. 5, below).
Figure 5.
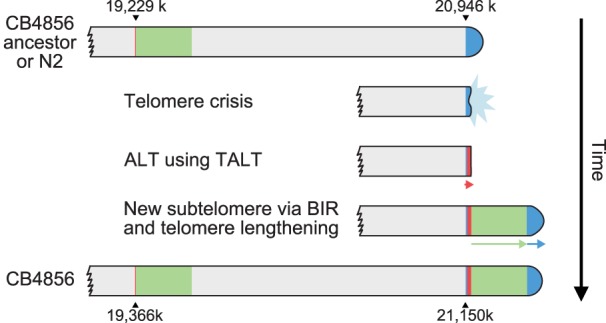
A model of Chr VR subtelomere formation in CB4856. The CB4856 ancestor underwent telomere crisis, and two sequential telomere-damage repair events, one using ALT and the other using BIR, formed new subtelomeres. Finally, the duplicated block end was repaired by telomerase, ending with at least 3-kb-long telomeric repeats.
New genes in the subtelomeric region
The internal region and the newly duplicated subtelomeric regions shared many, but not all, genes (Fig. 4A; Supplemental Fig. S12A). Sixteen common genes are predicted in both regions, and more than 10 genes are predicted to be specific to each region. The duplicated new subtelomere also contains genes copied from different chromosomes. In addition, the analysis of short-read whole-genome sequence data from 151 wild strains (Cook et al. 2016) revealed that seven of them showed a high copy number of TALT sequences and also contained the same unique sequences of the duplicated 200-kb region seen in the CB4856 subtelomere (Fig. 4B,C; Supplemental Table S2). To examine whether the TALT duplication that was observed in the seven strains had arisen independently during evolution, we constructed a phylogenetic tree of the haplotype block that is closely linked to the chromosome arms that bear the TALT duplication. The seven strains that have high TALT copy numbers shared the same TALT-linked haplotype block, and these seven strains are grouped alone into a single cluster (Fig. 4D,E; Supplemental Fig. S12B). Genomic regions that are subject to duplication and changes may act as genetic resources by providing redundant gene sets that can facilitate adaptation to new environments during evolution (Zhang 2003; Leister 2004).
Figure 4.

New subtelomere formation in wild isolates. (A) Internal genes were duplicated to Chr VR subtelomere. The figure shows a putative gene model of the Chr VR subtelomere. Upper panel: internal gene model; lower panel: subtelomeric gene model. (B) TALT copy numbers among wild isolates (Supplemental Table S3). (C) Normalized coverage mapped on the duplicated segment of wild isolates with high TALT copy number (red) strains and low TALT copy number (blue) strains. (D) Haplotype blocks on Chr V of seven strains that have high TALT copy numbers. (E) Phylogenic tree of reference N2 and 151 wild strains whose genomes have been fully sequenced. Strains marked with red color contain several copies of TALT.
Discussion
Since the first collection of C. elegans (Maupas 1901; Nigon and Felix 2017), 330 isotypes comprising more than 750 strains have been collected from all over the world (Cook et al. 2017). Among them, the reference strain N2, collected in the Bristol area of England, and the CB4856 strain, collected in Hawaii, are the best-known and most extensively studied strains. In this study, we constructed a highly contiguous genome of the CB4856 strain by de novo assembly using long-read sequencing. Because of chromosome-scale selective sweeps in C. elegans wild strains, some strains, including CB4856, exhibit distinct polymorphism patterns from most other wild strains (Andersen et al. 2012). For this reason, our completed CB4856 genome will serve as a better reference genome for those wild strains distinct from most other wild strains including N2. In addition, the numerous SVs between N2 and CB4856, identified based on our long-read sequencing, will also help to better understand the effect of SVs on traits by association studies using these strains.
Enrichment of genetic variations in chromosome arms and subtelomeres by background selection and error-prone recombination
Due to background selection, the polymorphism level and the recombination rate are correlated in most species (Kern and Hahn 2018); genetic variations are enriched in chromosome arms, which also show a high recombination rate in many nematodes such as in the genera Pristionchus and Caenorhabditis (Rockman and Kruglyak 2009; Andersen et al. 2012; Rödelsperger et al. 2017; Yin et al. 2018). In particular, repeat sequences are enriched and essential genes are sparsely distributed in chromosome arms (The C. elegans Sequencing Consortium 1998; Kamath et al. 2003). Comparison of Pristionchus species has shown that the more conserved, old genes are present in chromosome centers, whereas newly generated orphan genes are preferentially found in chromosome arms (Prabh et al. 2018; Werner et al. 2018). Similar patterns are shown in C. elegans. Among the C. elegans chromosomes, the largest one, Chr V, contains the fewest essential genes but the highest density of gene families (The C. elegans Sequencing Consortium 1998; Kamath et al. 2003). The right arm of Chr V has the lowest homology gene ratio compared to other closely related species (Stein et al. 2003); it is the region in which mutations accumulate more rapidly than in other chromosome regions, and many deletions are accumulated.
The hypervariable features of the subtelomeric and telomeric regions also contribute to variation enrichment in chromosome arms. Subtelomeres and telomeres are fragile regions that are prone to double-strand breaks (DSBs) during replication, and accurate repair of the DSBs is critical to maintaining genomic integrity (Glover and Stein 1987; Sfeir et al. 2009; Vannier et al. 2012). Most DSBs are repaired by nonhomologous end joining or homologous recombination (Ceccaldi et al. 2016). However, DSBs at subtelomeric and telomeric regions often lead to one-ended DSBs that lose telomeric parts, and thus are repaired by BIR, which finds a homologous sequence instead of missing ends (Bosco and Haber 1998; McEachern and Haber 2006; Kramara et al. 2018). DSBs in telomeres can use remote homologous sequences for repair by executing a searching process (Cho et al. 2014). Repeat sequences are enriched in subtelomeric and telomeric regions, so templates located elsewhere are likely to be used in the homology searching process; their use may increase the variations in the subtelomeric regions. Indeed, each subtelomeric region of CB4856 contains a complex subsequence from a homologous sequence elsewhere in the genome, so they have a new subtelomere that differs from the corresponding one of N2.
New subtelomere formation by ALT and BIR
Among the newly formed subtelomeres, Chr VR shows a unique feature that is reminiscent of telomere damage, ALT, and BIR. Our hypothesis for the Chr VR subtelomere formation in the ancestor of CB4856 is that the telomere underwent attrition followed by two sequential telomere-damage repair events, one using ALT and the other using BIR (Fig. 5).
The presence of short telomeric repeats within the subtelomeric region of CB4856 Chr VR implies that telomere attrition and repair had occurred. The multiple copies of TALT sequences next to the telomeric sequences suggests that the repair of telomere attrition was not performed by the canonical telomerase-mediated lengthening mechanism but by an ALT mechanism, even in the presence of the telomerase gene. TALT copies were not the end of the Chr VR: TALT copies were followed by sequences very similar to the region next to the internal TALT, probably by segmental duplication of a 200-kb internal sequence block. The last TALT sequences may have acted as a homology template for BIR in this process. The chromosome ends with a few TALT copies may have been recognized as a breakage, which in turn could induce the BIR mechanism. Searching for homologous sequences with that of the TALT homology template must have found the internal TALT, resulting in the duplication of sequences next to the internal TALT up to 200 kb via BIR. We postulate that harsh environmental stimuli or stresses, yet to be identified, may have induced Chr VR–specific DSBs in CB4856 ancestors and that these stimuli activated the intrinsic subtelomeric recombination mechanisms by which a new subtelomere was formed by ALT and BIR. We did not fail to notice that telomerase also had an important, though limited, function in the new subtelomere formation. Short traces of telomeric repeats between the tandem TALT copies suggest that telomerase was briefly activated on each end of TALT but was not enough to produce long telomeric repeats. In addition, the duplicated block end was repaired by the action of telomerase, as the real end of the Chr VR contains at least 3-kb-long telomeric repeats.
Our analysis of the genomic feature in the CB4856 subtelomere of Chr VR shows that an ALT template can repair telomere attrition even when the telomerase gene is intact. Consistent with this inference, mouse embryonic stem cells or mouse somatic cells may have ALT features when telomerase is present, and ALT and telomerase coexist to perform their unique functions in cells (Zalzman et al. 2010; Neumann et al. 2013). Currently, little is known about the normal function of ALT, and our analysis of the genomic features of CB4856 shows for the first time that ALT activity may be present in the germline to repair abrupt telomere attrition of an individual that already has telomerase activity. Our analysis also shows that BIR can induce subtelomere evolution by replicating internal genetic materials. Subtelomeres are enriched with ‘contingency genes,’ which are critical for adaptation to novel or stressful environments, and the gene families located in subtelomeres tend to expand rapidly (Barry et al. 2003; Brown et al. 2010). By this process, the subtelomere and telomere DSB-induced BIR can operate as a mechanism in the evolutionary process. To summarize, our findings suggest that a species can tolerate substantial structural changes in the genome without losing integrity as the same species and that new subtelomeres, and eventually new chromosomal contents, can evolve by the ALT and BIR mechanisms.
Methods
C. elegans culture
Worms were cultured at 20°C under standard culture conditions.
gDNA extraction and PacBio sequencing
Mixed stage worms were collected and washed 5× in M9 buffer. Worms were lysed in lysis buffer for 8 h (100 µg mL−1 Proteinase K, 50 mM KCl, 10 mM Tris (pH 8.3), 2.5 mM MgCl2, 0.45% NP-40, 0.45% Tween 20, and 1% beta-mercaptoethanol). DNA was extracted using phenol-chloroform extraction and ethanol precipitation. To minimize DNA shearing, we used phase-lock gel and minimized pipetting. DNA in TE buffer was treated with RNase (10 µg mL−1) for 2 h and re-extracted, before being dissolved in TE buffer. Macrogen performed library preparation and sequencing using the PacBio Single Molecule, Real-Time (SMRT) DNA sequencing technology (platform: PacBio RS II; chemistry: P6-C4).
Total RNA extraction and RNA sequencing
Mixed stage worms were harvested in the M9 buffer and TRIzol. To disrupt worms, we performed flash-freeze/thaw cycles 10×. RNA was extracted using chloroform and isopropanol precipitation. Macrogen performed library preparation and sequencing using HiSeq 4000 (Illumina) with 101-bp paired-end reads. Technical duplicate samples were sequenced in this study.
Genome assembly and polishing
De novo genome assembly was generated with 80× coverage PacBio reads using Canu (Koren et al. 2017) (version 1.6; canu minReadLength=1000 correctedErrorRate=0.040 genomeSize=100 m -pacbio-raw *.pacbio.subreads.fastq.gz). To increase base quality, the assembly was corrected using PacBio raw reads with Quiver (Chin et al. 2013) and HiSeq raw reads with Pilon (Walker et al. 2014). First, we converted PacBio raw reads to BAM files using bax2bam (version 0.0.8; bax2bam --subread --pulsefeatures=DeletionQV,DeletionTag,InsertionQV,IPD,MergeQV,SubstitutionQV,PulseWidth,SubstitutionTag), aligned PacBio raw reads to the Canu-only assembly using pbalign (version 0.3.1; default option), merged BAM files using BamTools (version 2.4.1; bamtools merge), and polished it using Quiver (version 2.2.1; variantCaller --algorithm quiver). Quiver, bax2bam, BamTools, and pbalign were from the GenomicConsensus package (https://github.com/PacificBiosciences/GenomicConsensus). We repeated this process with the Quiver-polished assembly instead of the Canu-only one. Next, to remove bacterial sequence contamination, we aligned the contigs with 3000 bacterial genomes downloaded at European Nucleotide Archive (ENA) (on March 30, 2018) from ftp://ftp.ebi.ac.uk/pub/databases/fastafiles/embl_genomes/genomes/Bacteria using BLAST+ (Camacho et al. 2009) (version 2.7.1; makeblastdb -input_type fasta -dbtype nucl and blastn -task megablast -evalue 1e-06 -outfmt 6 -perc_ identity 50). Nine contigs were excluded that contain bacterial homology sequences longer than 50% in contig length using a custom Python script (Supplemental Code). Lastly, homopolymers were corrected with mapping CB4856 short reads downloaded from NCBI (accession numbers: SRR3440952, SRR3441150, SRR3441428, and SRR3441550; 73× coverage) (Cook et al. 2017) to 128 contigs using BWA-MEM (Li 2013) (version 0.7.17) and Pilon (version 1.22). The following rounds of Pilon polishing were performed with the same parameters except using the previous round Pilon-polished contigs as a reference. We repeated the polishing using Pilon 4× in total.
Scaffolding contigs
To determine a subset of CB4856 genome assembly that aligned syntenically onto the N2 genome, we used NUCmer and show-tiling from the MUMmer package (Kurtz et al. 2004; Marçais et al. 2018) (version 4.0.0 beta). The final 128 polished contigs were aligned onto the N2 genome (Ensembl WBcel235/ce11) using NUCmer (nucmer --mum -l 100 -c 300). The most well-aligned 74 contigs were obtained using show-tiling (show-tiling -l 1 -g -1 -i 80.0 -v 1.0 -V 0.0). The right end contigs of Chr I and Chr V had no telomeric repeats, so we manually selected telomere-containing contigs among not placed ones. We judged whether these contigs showed similarity to either end using NUCmer (nucmer --mum -l 100 -c 300), then assessed linkage data from recombinant inbred lines between N2 and CB4856. First, reads aligned onto the N2 genome were extracted using Picard (http://broadinstitute.github.io/picard/) (version 2.18.6; picard SamToFastq), realigned to the CB4856 genome using BWA-MEM, and sorted using SAMtools (Li et al. 2009) (version 1.6; samtools sort). Duplicated reads were removed using picard MarkDuplicatesWithMateCigar, REMOVE_ DUPLICATES=true, read groups were added using picard AddOrReplaceReadGroups, and indexed using samtools index. Variants were called using GATK (Poplin et al. 2017) (version 4.0.5.1; Haplotype caller -ERC GVCF --use-new-qual-calculator, GenomicsDBImport, and Genotype GVCFs --founder-id ‘CB4856’ --use-new-qual-calculator --max-alternate-alleles 2). We further analyzed whether leftover telomere-containing contigs have linkage with the ends of Chr I and Chr V, then placed the remaining right end contigs for Chr I and Chr V (Supplemental Code). Lastly, the initial version of mitochondrial contig was aligned to the N2 mitochondrial genome using progressiveMauve (Darling et al. 2010). The CB4856 mitochondrial contig was repeated twice as compared with the N2's, so the ends were trimmed to make a linearized-circular genome. Placed and not-placed contigs were compared for their length, lower-quality nucleotide ratio based on Quiver, and repetitive element ratio using RepeatMasker (Smit et al. 2016) (version open-4.0.7; http://www.repeatmasker.org). Scatterplots were created using an excel template (Weissgerber et al. 2015). All gaps between contigs were filled with 1000 Ns to generate a chromosome-level assembly. Assembly statistics were measured using nucmer --maxmatch -l 100 -c 300 and dnadiff, and the numbers of SNPs were counted using show-snps -C. Fosmids were also used to scaffold contigs. We used 15,360 fosmids, removed <500 bp, and mapped them using BWA-MEM. Only fosmids that had both ends mapped were used to check mapping regions, and two contigs were scaffolded if they had at least the same mapped fosmid. Unless otherwise specified, this assembly was used for all following analyses.
Genome quality assessment
BUSCO (Simão et al. 2015) and BWA-MEM were used to verify the completeness of the CB4856 genome. First, the N2 and CB4856 genomes were assessed using BUSCO OrthoDB v9 (-l eukaryota_odb9 -m geno -sp caenorhabditis). Next, PacBio raw reads were aligned to the Kim genome by using pbalign from the GenomicConsensus package, and its average coverage was calculated by SAMtools depth. Finally, CB4856 HiSeq reads were aligned to two genomes, variants were called using BCFtools (Li 2011) (version 1.6; bcftools mpileup -Ou -f | bcftools call -vmO z -o and bcftools filter -O v -o -s LOWQUAL -i'%QUAL>10′), and positions with allele frequency of 40%–60% were extracted to visualize them.
Gene annotation transfer and gene prediction
The EMBL-formatted gene annotation (Ensembl 91) was transferred to the CB4856 genome using the Rapid Annotation Transfer Tool (Otto et al. 2011) (RATT; version 24-Dec-2011). We optimized parameters of start.ratt.sh (Strain, -c 400 -l 20 -g 500, and -o 75), and reformatted the resulting EMBL file to the GFF format. N2-specific genes were defined as genes whose exons were not transferred at all using RATT. According to the canonical gene set of WS266 version downloaded from WormBase (c_elegans.PRJNA13758.WS266.canonical_geneset.gtf), the annotations of 45,457 genes in N2 46,742 genes (including 1655 genes of total 1891 N2 pseudogenes) were transferred to the CB4856 genome. We also confirmed that 19,355 of the total of 20,039 N2 protein-coding genes were transferred into the CB4856 genome. N2-specific genes were defined as genes from which exons were not transferred at all using RATT. To further confirm that 684 N2-specific genes (including 661 protein-coding genes) are not found in the CB4856 genome, we searched the sequence of those genes in the CB4856 genome using BLAST+ (blastn -outfmt 7 -html -perc_identity 95.0 -qcov_hsp_perc 95). We identified five genes with copy-number changes only. We repeated this same procedure for the Thompson genome and finally identified 619 genes that are specific to N2.
We then used the MAKER annotation pipeline (Cantarel et al. 2008) (version 2.31.9) to further annotate the CB4856 genome and generated ab initio gene prediction with several tools, including AUGUSTUS (Stanke et al. 2006) (version 3.2.3), SNAP (Korf 2004) (version 2006-07-28), and BUSCO, referred to the pipeline posted on a GitHub website (https://gist.github.com/darencard/bb1001ac1532dd4225b030cf0cd61ce2). Data analyzed in the MAKER pipeline included (1) de novo assembled transcripts from CB4856 RNA-seq data with two biological replicates, (2) N2 strain proteome sequences for protein homology evidence (Caenorhabditis_elegans.WBcel235.pep.all.fa; download from the WBcel235 release of WormBase), (3) trained ab initio prediction data set from the SNAP gene prediction tool, and (4) another trained ab initio AUGUSTUS data set optimized by BUSCO. De novo assembled transcripts of CB4856 RNA-seq data were generated using STAR (Dobin et al. 2013) (version 020201; STAR --readFilesIn --readFilesCommand gzip -cd) and Trinity (Haas et al. 2013) (version 2.6.6; Trinity --genome_guided_bam --genome_ guided_max_intron 100920). Before running the first-round MAKER, we masked repeat sequences in the CB4856 genome using RepeatMasker (RepeatMasker --engine ncbi -lib celrep.Repbase.ref -pa 60) and Repbase data (Bao et al. 2015) (https://www.girinst.org/repbase/). Complex repeats were isolated and reformatted using a custom Perl script (Supplemental Code). Taken together, the gene annotation using MAKER was guided by hints from de novo assembled transcript, known protein sequences, and complex repeat and transposable element protein sequences bundled in RepeatMasker (maker -base cb4856_rnd1_RM_trinity_mixed_ published round1_maker_opts.Repbase.repeat.trinity.mixed.published. ctl maker_bopts.ctl maker_exe.ctl; est2genome=1, protein2genome=1 in the maker_exe.ctl file). We combined the resulting FASTA files and GFF files using fasta-merge and gffmerge in the MAKER package. We then predicted genes in the CB4856 genome with ab initio gene prediction tools to improve our gene annotation. For training AUGUSTUS, we used nematode-specific BUSCO gene models (nematode_odb9) and the sequence with mRNA annotations based on the initial MAKER result containing 1 kb on each side. At the end, we refined training parameters for AUGUSTUS using BUSCO (BUSCO.py -i maker.all.maker.transcripts1000.fasta -o rnd1_maker -l nematode_ odb9/ -m genome -c 8 --long -sp worm -z --augustus_parameters=“--progress=true”). For training SNAP, we used maker2zff, fathom, forge, and hmm-assembler in the MAKER package to filter the initial MAKER result (maker2zff -x0.25 -l 50) and extracted the annotation and sequences containing 1 kb on each side for the training (fathom -gene-stats; fathom -validate; fathom -categorize 1000; fathom -export 1000 -plus). Based on this information, we generated training parameters for SNAP (forge; hmm-assembler.pl -params). Then, the second round of MAKER was run to predict genes with the AUGUSTUS and SNAP training data set (maker -base cb4856_RM_trinity_mixed_published_ rnd2round2_maker_opts.Repbase.repeat.trinity.mixed.published.ctlmaker_ bopts.ctl maker_exe.ctl). Parameters were changed for ab initio gene prediction (est2genome=0, protein2genome=0).
After running two rounds of the MAKER ab initio gene prediction pipeline, we filtered out less reliable genes by using the following criteria (Stanley et al. 2018): (1) Discard MAKER gene models that overlap regions that are covered by genes annotated in RATT gene-transfer pipeline; (2) discard genes that encode proteins of shorter than 30 amino acids (as 90 bp); (3) if two or more different MAKER gene models overlap in their coding sequence, discard the model that has the lower eAED score. After these steps, we predicted 781 MAKER gene models and integrated them into the previous gene lists to make the complete set of 46,238 genes. The resulting FASTA files and GFF files were merged using fasta-merge and gffmerge in the MAKER package as well (Supplemental Material).
Structural variations and GO analysis
We used NUCmer (nucmer --maxmatch -l 100 -c 500) to align the final Quiver-Pilon-polished 128 contigs to the N2 genome, or to a CB4856 genome that had been assembled from short reads (Thompson et al. 2015), then called SVs by using the NUCmer output file and Assemblytics (Nattestad and Schatz 2016) (http://assemblytics.com/). Large rearrangements on the chromosome scale were analyzed using progressiveMauve. To assess the effects of genetic variations, we reformatted the Assemblytics result using a custom Python script (Supplemental Code) and annotated effects of SVs using SnpEff (Cingolani et al. 2012) (version 4.3t; java -jar snpEff.jar). The SnpEff result was summarized based on size and impact categories (modifier, low, moderate, and high) on genes and visualized using Circos version 0.69-6 (Krzywinski et al. 2009) (http://circos.ca/software/download/circos). To evaluate the functional effects of high-impact SVs on genes, we further identified genes which have “lethal” or “sterile” phenotypic evidence reported by RNAi depletion experiment or allelic deletion mutation experiments using the SimpleMine web tool (Lee et al. 2018) (https://www.wormbase.org/tools/mine/simplemine.cgi) and also predicted Gene Ontology (GO) terms for gene functions with the gene set enrichment analysis web tool (Angeles-Albores et al. 2016) (https://www.wormbase.org/tools/enrichment/tea/tea.cgi). N2/CB4856 local recombination data were obtained from https://github.com/AndersenLab/linkagemapping.
Comparison of SVs and the determination of coverage of specific SVs
Each set of SVs was further analyzed using a custom Python script (nucmer --maxmatch -l 100 -c 500, Assemblytics SV minimum length: 50 bp) (Supplemental Code). First, we extracted the coordinations of SVs on each chromosome from the SV files by using NUCmer and Assemblytics. On each chromosome of the Thompson genome or the Kim genome, we collected the SV region and the additional left side 500 bp (start position −500) and right side 500 bp (end position +500 bp) of the coordinates. The widening of the region was done to prevent mistakes that may occur due to trivial coordination errors. We determined genome-specific SVs and their corresponding genomic positions as a BED file by using a custom Python script (Supplemental Code). Finally, from the BAM file that aligned the Canu corrected reads to the genomes using pbalign, we extracted the depth information using mosdepth (mosdepth 0.2.4; mosdepth --by v1.novel.snps.v1_coordination.bed cb4856.v1.only.sv.pbalign. depth v1.correctedReads.pbalign.sorted.bam mosdepth --by v2.cb4856_ contig_scaffold_novel_sv_v2.v2_coord.bed cb4856.v2.only.sv.pbalign. depth v2.correctedReads.pbalign.sorted.bam) (Pedersen and Quinlan 2018).
SNP and indel calling by use of GATK
The calling was performed using the FASTQ files downloaded from NCBI (accession numbers: SRR3440952, SRR3441150, SRR3441428, and SRR3441550) (Cook et al. 2017). The FASTQ files are aligned to the reference genome by BWA-MEM (bwa mem -M -R). Aligned SAM files were processed with Picard SortSam and MarkDuplicates to remove PCR duplicates and were converted to BAM files (picard SortSam SORT_ORDER=coordinate picard MarkDuplicates). Four BAM files were used for SNP and indel calling with GATK (McKenna et al. 2010) HaplotypeCaller (Poplin et al. 2017) (GenomeAnalysisTK -T HaplotypeCaller) against the reference genome. We then distinguished SNPs (GenomeAnalysisTK SelectVariants -selectType SNP) and indels by using GATK SelectVariants (GenomeAnalysisTK SelectVariants -selectType INDEL). We then filtered SNPs and indels using GATK VariantFiltration with a standard filter option (GenomeAnalysisTK -T VariantFiltration –filterExpression ‘QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0 || SOR > 4.0′’, GenomeAnalysisTK -T VariantFiltration –filterExpression ‘QD < 2.0 || FS > 200.0 || ReadPosRankSum < -20.0 || SOR > 10.0’). Base calibration was done for each BAM file using the first round SNPs and indels, with GATK BaseRecalibrator (GenomeAnalysisTK -T BaseRecalibrator -BQSR recal_data.table). Correction was performed with GATK PrintReads (GenomeAnalysisTK -T PrintReads -BQSR recal_data.table). Finally, we integrated each BAM file into a single file by using Picard MergeSamFile (Picard MergeSamFiles), and the second round of calling for SNPs and indels was done with GATK HaplotypeCaller (GenomeAnalysisTK -T HaplotypeCaller). GATK SelectVariants was again used to distinguish SNPs and INDELs (GenomeAnalysisTK SelectVariants -selectType SNP and GenomeAnalysisTK SelectVariants -selectType INDEL). SNP results from the second round were processed with corresponding filters, and only the SNPs common to all four short-read sequencing data (accession numbers: SRR3440952, SRR3441150, SRR3441428, and SRR3441550) were collected with GATK SelectVariants (GenomeAnalysisTK -T VariantFiltration –filterExpression ‘QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0 || SOR > 4.0’) and maxNOCALLnumber (GenomeAnalysisTK -T SelectVariants -ef –maxNOCALLnumber 0).
Subtelomere analysis
The subtelomere was defined as the 200-kb end of each chromosome. All subtelomere pairs of N2 and CB4856 strains were aligned using NUCmer and progressiveMauve, and unaligned regions were obtained. These regions were searched using BLAST+ (blastn -task megablast -evalue 1e-06 -outfmt 6 -perc_identity 50) to identify any homology in the N2 genome. To analyze the extreme difference of Chr VR, internal and duplicated sequences were extracted and aligned to each other using nucmer --maxmatch, and the alignment was visualized using mummerplot. Lastly, short reads of 14 strains were aligned to the CB4856 genome using BWA-MEM, and the positional depth of the last contig was parsed using samtools depth -a -r. The short reads were downloaded from NCBI (accession numbers: CB4856: SRR3440952, SRR3441150, SRR3441428, SRR3441550; CX11262: SRR3441573, SRR3441359; CX11264: SRR3452248, SRR3452255, SRR3441549; CX11314: SRR3441488, SRR3441191, SRR3440991; CX11315: SRR3441659, SRR3441435, SRR3441151; DL226: SRR3441461, SRR3441168, SRR3440967; DL238: SRR3452231, SRR3452104, SRR3452184; LKC34: SRR3452180, SRR3441481, SRR3441206; MY16: SRR3452112, SRR3441454, SRR3441180; MY23: SRR3452187, SRR3452234, SRR3441433; N2: SRR3441391, SRR3452263, SRR3441113; QX1791: SRR3452145, SRR3452136, SRR3441468; QX1794: SRR3441473, SRR3441189, SRR3440987; QX1793: SRR3452168, SRR3452175, SRR3441470) (Cook et al. 2017). This depth was normalized by the average whole genome depth of each strain.
TALT copy number estimation and phylogenetic analysis
TALT copy number was estimated by calculating the normalized coverage of putative TALT regions. Normalized coverage was calculated by dividing the depth of coverage within TALT regions by the mean depth of coverage of the nuclear genome. Depth of coverage calculations were performed using VCF-kit (Cook and Andersen 2017) across sequence-alignment files for 150 wild isolates. Variant data for dendrogram comparisons were assembled by constructing a FASTA file with the genome-wide variant positions across all strains and subsetting by regions as described (Cook et al. 2016). MUSCLE (Edgar 2004) (version v3.8.31) was used to construct neighbor-joining trees. The R packages APE (Paradis et al. 2004) (version 3.4) and phyloseq (McMurdie and Holmes 2013) (version 1.12.2) were used for data processing and plotting. Haplotype block analysis was conducted as previously described (Lee et al. 2019).
Data access
All sequencing reads and assembly from this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA523481. All custom scripts generated in this study are available as Supplemental Code.
Supplementary Material
Acknowledgments
We thank B. Seo and Macrogen for initial analyses of the CB4856 genome and D. Lee for haplotype block discussion. CB4856 was kindly provided by the Caenorhabditis Genetics Center. This work was supported by the Samsung Science and Technology Foundation under Project Number SSTF-BA1501-04. C.K. was supported by the BK21 program. J.K. was supported by a POSCO Science Fellowship from the POSCO TJ Park Foundation. This work was partly supported by a National Institutes of Health R01 subcontract to E.C.A. (GM107227), the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust, and an American Cancer Society Research Scholar grant to E.C.A. (127313-RSG-15-135-01-DD), along with support from the National Science Foundation Graduate Research Fellowship (DGE-1324585) to D.E.C. Additionally, we thank WormBase without which most genomic analyses would not be possible.
Author contributions: C.K., J.K., and J.L. designed the experiments and analyzed and interpreted the data. S.K. analyzed SVs and gene annotation. C.K., J.K., and J.L. wrote the manuscript. D.E.C. and K.S.E. counted TALT copy numbers across the wild isolates, and E.C.A. analyzed the phylogenetic tree of the wild isolates.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.246082.118.
Freely available online through the Genome Research Open Access option.
References
- Alföldi J, Lindblad-Toh K. 2013. Comparative genomics as a tool to understand evolution and disease. Genome Res 23: 1063–1068. 10.1101/gr.157503.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen EC, Gerke JP, Shapiro JA, Crissman JR, Ghosh R, Bloom JS, Félix M-A, Kruglyak L. 2012. Chromosome-scale selective sweeps shape Caenorhabditis elegans genomic diversity. Nat Genet 44: 285–290. 10.1038/ng.1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen EC, Bloom JS, Gerke JP, Kruglyak L. 2014. A variant in the neuropeptide receptor npr-1 is a major determinant of Caenorhabditis elegans growth and physiology. PLoS Genet 10: e1004156 10.1371/journal.pgen.1004156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeles-Albores D, Lee RYN, Chan J, Sternberg PW. 2016. Tissue enrichment analysis for C. elegans genomics. BMC Bioinformatics 17: 366 10.1186/s12859-016-1229-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O. 2015. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6: 11 10.1186/s13100-015-0041-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry J, Ginger ML, Burton P, McCulloch R. 2003. Why are parasite contingency genes often associated with telomeres? Int J Parasitol 33: 29–45. 10.1016/S0020-7519(02)00247-3 [DOI] [PubMed] [Google Scholar]
- Blackburn EH. 1991. Structure and function of telomeres. Nature 350: 569–573. 10.1038/350569a0 [DOI] [PubMed] [Google Scholar]
- Blasco MA, Lee H-W, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. 1997. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91: 25–34. 10.1016/S0092-8674(01)80006-4 [DOI] [PubMed] [Google Scholar]
- Bosco G, Haber JE. 1998. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 150: 1037–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CA, Murray AW, Verstrepen KJ. 2010. Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr Biol 20: 895–903. 10.1016/j.cub.2010.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR. 1997. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med 3: 1271–1274. 10.1038/nm1197-1271 [DOI] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium. 1998. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282: 2012–2018. 10.1126/science.282.5396.2012 [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10: 421 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sanchez Alvarado A, Yandell M. 2008. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18: 188–196. 10.1101/gr.6743907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Rondinelli B, D'Andrea AD. 2016. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 26: 52–64. 10.1016/j.tcb.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare AJ, Reddel RR. 2010. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 11: 319–330. 10.1038/nrg2763 [DOI] [PubMed] [Google Scholar]
- Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. 10.1038/nmeth.2474 [DOI] [PubMed] [Google Scholar]
- Cho Nam W, Dilley Robert L, Lampson Michael A, Greenberg Roger A. 2014. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159: 108–121. 10.1016/j.cell.2014.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6: 80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DE, Andersen EC. 2017. VCF-kit: assorted utilities for the variant call format. Bioinformatics 33: 1581–1582. 10.1093/bioinformatics/btx011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DE, Zdraljevic S, Tanny RE, Seo B, Riccardi DD, Noble LM, Rockman MV, Alkema MJ, Braendle C, Kammenga JE. 2016. The genetic basis of natural variation in Caenorhabditis elegans telomere length. Genetics 204: 371–383. 10.1534/genetics.116.191148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DE, Zdraljevic S, Roberts JP, Andersen EC. 2017. CeNDR, the Caenorhabditis elegans natural diversity resource. Nucleic Acids Res 45: D650–D657. 10.1093/nar/gkw893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD. 2014. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343: 88–91. 10.1126/science.1243211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Choi JY. 2010. Natural selection shapes nucleotide polymorphism across the genome of the nematode Caenorhabditis briggsae. Genome Res 20: 1103–1111. 10.1101/gr.104331.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Payseur BA. 2013. Genomic signatures of selection at linked sites: unifying the disparity among species. Nat Rev Genet 14: 262–274. 10.1038/nrg3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5: e11147 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono M, Bargmann CI. 1998. Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell 94: 679–689. 10.1016/S0092-8674(00)81609-8 [DOI] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA. 2016. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539: 54–58. 10.1038/nature20099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Mangino M, Aviv A, UK10K Consortium, Spector T, Durbin R. 2014. Estimating telomere length from whole genome sequence data. Nucleic Acids Res 42: e75 10.1093/nar/gku181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5: 113 10.1186/1471-2105-5-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavís M, González C, Villasante A. 2013. On the origin of the eukaryotic chromosome: the role of noncanonical DNA structures in telomere evolution. Genome Biol Evol 5: 1142–1150. 10.1093/gbe/evt079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover T, Stein C. 1987. Induction of sister chromatid exchanges at common fragile sites. Am J Hum Genet 41: 882–890. [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8: 1494–1512. 10.1038/nprot.2013.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. 1990. Telomeres shorten during ageing of human fibroblasts. Nature 345: 458–460. 10.1038/345458a0 [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M. 2003. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237. 10.1038/nature01278 [DOI] [PubMed] [Google Scholar]
- Kammenga JE, Doroszuk A, Riksen JA, Hazendonk E, Spiridon L, Petrescu A-J, Tijsterman M, Plasterk RH, Bakker J. 2007. A Caenorhabditis elegans wild type defies the temperature–size rule owing to a single nucleotide polymorphism in tra-3. PLoS Genet 3: e34 10.1371/journal.pgen.0030034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern AD, Hahn MW. 2018. The neutral theory in light of natural selection. Mol Biol Evol 35: 1366–1371. 10.1093/molbev/msy092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Sung S, Lee J. 2016. Internal genomic regions mobilized for telomere maintenance in C. elegans. Worm 5: e1146856 10.1080/21624054.2016.1146856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee D, Lee J. 2017. A quantitative trait locus for nictation behavior on chromosome V. microPublication Biology 10.17912/W23D39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch R, van Luenen HG, van der Horst M, Thijssen KL, Plasterk RH. 2000. Single nucleotide polymorphisms in wild isolates of Caenorhabditis elegans. Genome Res 10: 1690–1696. 10.1101/gr.GR-1471R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepfli K-P, Paten B, Genome 10K Community of Scientists, O'Brien SJ. 2015. The Genome 10K Project: a way forward. Annu Rev Anim Biosci 3: 57–111. 10.1146/annurev-animal-090414-014900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27: 722–736. 10.1101/gr.215087.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5: 59 10.1186/1471-2105-5-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramara J, Osia B, Malkova A. 2018. Break-induced replication: the where, the why, and the how. Trends Genet 34: 518–531. 10.1016/j.tig.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Yang H, Kim J, Brady S, Zdraljevic S, Zamanian M, Kim H, Paik Y-K, Kruglyak L, Andersen EC, et al. 2017. The genetic basis of natural variation in a phoretic behavior. Nat Commun 8: 273 10.1038/s41467-017-00386-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RYN, Howe KL, Harris TW, Arnaboldi V, Cain S, Chan J, Chen WJ, Davis P, Gao S, Grove C, et al. 2018. WormBase 2017: molting into a new stage. Nucleic Acids Res 46: D869–D874. 10.1093/nar/gkx998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Zdraljevic S, Cook DE, Frézal L, Hsu J-C, Sterken MG, Riksen JAG, Wang J, Kammenga JE, Braendle C, et al. 2019. Selection and gene flow shape niche-associated copy-number variation of pheromone receptor genes. bioRxiv 10.1101/580803 [DOI] [PMC free article] [PubMed]
- Leister D. 2004. Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance genes. Trends Genet 20: 116–122. 10.1016/j.tig.2004.01.007 [DOI] [PubMed] [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27: 2987–2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN].
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad V, Blackburn EH. 1993. An alternative pathway for yeast telomere maintenance rescues est1− senescence. Cell 73: 347–360. 10.1016/0092-8674(93)90234-H [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, Haber JE. 2007. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448: 820–823. 10.1038/nature06047 [DOI] [PubMed] [Google Scholar]
- Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. 2018. MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol 14: e1005944 10.1371/journal.pcbi.1005944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JMO, McEachern MJ. 2018. Mild telomere dysfunction as a force for altering the adaptive potential of subtelomeric genes. Genetics 208: 537–548. 10.1534/genetics.117.300607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JM, Randall TA, Capkova Frydrychova R. 2016. Telomerase lost? Chromosoma 125: 65–73. 10.1007/s00412-015-0528-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maupas E. 1901. Modes et formes de reproduction des nematodes. Arch Zool Exp Gén 8: 463–624. [Google Scholar]
- Maydan JS, Flibotte S, Edgley ML, Lau J, Selzer RR, Richmond TA, Pofahl NJ, Thomas JH, Moerman DG. 2007. Efficient high-resolution deletion discovery in Caenorhabditis elegans by array comparative genomic hybridization. Genome Res 17: 337–347. 10.1101/gr.5690307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maydan JS, Lorch A, Edgley ML, Flibotte S, Moerman DG. 2010. Copy number variation in the genomes of twelve natural isolates of Caenorhabditis elegans. BMC Genomics 11: 62 10.1186/1471-2164-11-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEachern MJ, Haber JE. 2006. Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75: 111–135. 10.1146/annurev.biochem.74.082803.133234 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8: e61217 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier B, Clejan I, Liu Y, Lowden M, Gartner A, Hodgkin J, Ahmed S. 2006. trt-1 is the Caenorhabditis elegans catalytic subunit of telomerase. PLoS Genet 2: e18 10.1371/journal.pgen.0020018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TM, Cooper JP, Cech TR. 1998. Two modes of survival of fission yeast without telomerase. Science 282: 493–496. 10.1126/science.282.5388.493 [DOI] [PubMed] [Google Scholar]
- Nattestad M, Schatz MC. 2016. Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32: 3021–3023. 10.1093/bioinformatics/btw369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann AA, Watson CM, Noble JR, Pickett HA, Tam PP, Reddel RR. 2013. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev 27: 18–23. 10.1101/gad.205062.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigon VM, Felix MA. 2017. History of research on C. elegans and other free-living nematodes as model organisms. WormBook 2017: 1–84. 10.1895/wormbook.1.181.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan RJ, Karlseder J. 2010. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 11: 171–181. 10.1038/nrm2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto TD, Dillon GP, Degrave WS, Berriman M. 2011. RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res 39: e57 10.1093/nar/gkq1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palopoli MF, Rockman MV, TinMaung A, Ramsay C, Curwen S, Aduna A, Laurita J, Kruglyak L. 2008. Molecular basis of the copulatory plug polymorphism in Caenorhabditis elegans. Nature 454: 1019–1022. 10.1038/nature07171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. 2004. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20: 289–290. 10.1093/bioinformatics/btg412 [DOI] [PubMed] [Google Scholar]
- Pedersen BS, Quinlan AR. 2018. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics 34: 867–868. 10.1093/bioinformatics/btx699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pich U, Schubert I. 1998. Terminal heterochromatin and alternative telometric sequences in Allium cepa. Chromosome Res 6: 315–321. 10.1023/A:1009227009121 [DOI] [PubMed] [Google Scholar]
- Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D, et al. 2017. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 10.1101/201178 [DOI]
- Prabh N, Roeseler W, Witte H, Eberhardt G, Sommer RJ, Rödelsperger C. 2018. Deep taxon sampling reveals the evolutionary dynamics of novel gene families in Pristionchus nematodes. Genome Res 28: 1664–1674. 10.1101/gr.234971.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L. 2009. Recombinational landscape and population genomics of Caenorhabditis elegans. PLoS Genet 5: e1000419 10.1371/journal.pgen.1000419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rödelsperger C, Meyer JM, Prabh N, Lanz C, Bemm F, Sommer RJ. 2017. Single-molecule sequencing reveals the chromosome-scale genomic architecture of the nematode model organism Pristionchus pacificus. Cell Rep 21: 834–844. 10.1016/j.celrep.2017.09.077 [DOI] [PubMed] [Google Scholar]
- Rudd MK. 2014. Human and primate subtelomeres. In Subtelomeres (ed. Louis EJ, Becker MM), pp. 153–164. Springer, New York. [Google Scholar]
- Schluter D. 2001. Ecology and the origin of species. Trends Ecol Evol 16: 372–380. 10.1016/S0169-5347(01)02198-X [DOI] [PubMed] [Google Scholar]
- Schulenburg H, Müller S. 2004. Natural variation in the response of Caenorhabditis elegans towards Bacillus thuringiensis. Parasitology 128: 433–443. 10.1017/S003118200300461X [DOI] [PubMed] [Google Scholar]
- Seidel HS, Rockman MV, Kruglyak L. 2008. Widespread genetic incompatibility in C. elegans maintained by balancing selection. Science 319: 589–594. 10.1126/science.1151107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel HS, Ailion M, Li J, van Oudenaarden A, Rockman MV, Kruglyak L. 2011. A novel sperm-delivered toxin causes late-stage embryo lethality and transmission ratio distortion in C. elegans. PLoS Biol 9: e1001115 10.1371/journal.pbio.1001115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo B, Kim C, Hills M, Sung S, Kim H, Kim E, Lim DS, Oh H-S, Choi RMJ, Chun J, et al. 2015. Telomere maintenance through recruitment of internal genomic regions. Nat Commun 6: 8189 10.1038/ncomms9189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. 2009. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138: 90–103. 10.1016/j.cell.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Slos D, Sudhaus W, Stevens L, Bert W, Blaxter M. 2017. Caenorhabditis monodelphis sp. n.: defining the stem morphology and genomics of the genus Caenorhabditis. BMC Zool 2: 4 10.1186/s40850-017-0013-2 [DOI] [Google Scholar]
- Smit A, Hubley R, Green P. 2016. RepeatMasker Open-4.0. 2015. http://www.repeatmasker.org/.
- Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. 2006. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34: W435–W439. 10.1093/nar/gkl200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley E, Coghlan A, Berriman M. 2018. A MAKER pipeline for prediction of protein-coding genes in parasitic worm genomes. Protoc Exch 10.1038/protex.2018.056 [DOI] [Google Scholar]
- Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, et al. 2003. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol 1: E45 10.1371/journal.pbio.0000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens L, Félix MA, Beltran T, Braendle C, Caurcel C, Fausett S, Fitch D, Frézal L, Kaur T, Kiontke K, et al. 2018. Comparative genomics of ten new Caenorhabditis species. bioRxiv 10.1101/398446 [DOI] [PMC free article] [PubMed]
- Thompson OA, Snoek LB, Nijveen H, Sterken MG, Volkers RJM, Brenchley R, Van't Hof A, Bevers RPJ, Cossins AR, Yanai I, et al. 2015. Remarkably divergent regions punctuate the genome assembly of the Caenorhabditis elegans Hawaiian strain CB4856. Genetics 200: 975–989. 10.1534/genetics.115.175950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijsterman M, Okihara KL, Thijssen K, Plasterk RH. 2002. PPW-1, a PAZ/PIWI protein required for efficient germline RNAi, is defective in a natural isolate of C. elegans. Curr Biol 12: 1535–1540. 10.1016/S0960-9822(02)01110-7 [DOI] [PubMed] [Google Scholar]
- Tyson JR, O'Neil NJ, Jain M, Olsen HE, Hieter P, Snutch TP. 2018. MinION-based long-read sequencing and assembly extends the Caenorhabditis elegans reference genome. Genome Res 28: 266–274. 10.1101/gr.221184.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannier J-B, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. 2012. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149: 795–806. 10.1016/j.cell.2012.03.030 [DOI] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissgerber TL, Milic NM, Winham SJ, Garovic VD. 2015. Beyond bar and line graphs: time for a new data presentation paradigm. PLoS Biol 13: e1002128 10.1371/journal.pbio.1002128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner MS, Sieriebriennikov B, Prabh N, Loschko T, Lanz C, Sommer RJ. 2018. Young genes have distinct gene structure, epigenetic profiles, and transcriptional regulation. Genome Res 28: 1675–1687. 10.1101/gr.234872.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicks SR, Yeh RT, Gish WR, Waterston RH, Plasterk RH. 2001. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat Genet 28: 160–164. 10.1038/88878 [DOI] [PubMed] [Google Scholar]
- Wu C-I, Ting C-T. 2004. Genes and speciation. Nat Rev Genet 5: 114–122. 10.1038/nrg1269 [DOI] [PubMed] [Google Scholar]
- Yin D, Schwarz EM, Thomas CG, Felde RL, Korf IF, Cutter AD, Schartner CM, Ralston EJ, Meyer BJ, Haag ES. 2018. Rapid genome shrinkage in a self-fertile nematode reveals sperm competition proteins. Science 359: 55–61. 10.1126/science.aao0827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalzman M, Falco G, Sharova LV, Nishiyama A, Thomas M, Lee S-L, Stagg CA, Hoang HG, Yang H-T, Indig FE. 2010. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature 464: 858–863. 10.1038/nature08882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. 2003. Evolution by gene duplication: an update. Trends Ecol Evol 18: 292–298. 10.1016/S0169-5347(03)00033-8 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.