

Abstract
MAPK inhibitors (MAPKi) show outstanding clinical response rates in melanoma patients harbouring BRAF mutations, but resistance is common. The ability of melanoma cells to switch from melanocytic to mesenchymal phenotypes appears to be associated with therapeutic resistance. High‐throughput, subcellular proteome analyses and RNAseq on two panels of primary melanoma cells that were either sensitive or resistant to MAPKi revealed that only 15 proteins were sufficient to distinguish between these phenotypes. The two proteins with the highest discriminatory power were PTRF and IGFBP7, which were both highly upregulated in the mesenchymal‐resistant cells. Proteomic analysis of CRISPR/Cas‐derived PTRF knockouts revealed targets involved in lysosomal activation, endocytosis, pH regulation, EMT, TGFβ signalling and cell migration and adhesion, as well as a significantly reduced invasive index and ability to form spheres in 3D culture. Overexpression of PTRF led to MAPKi resistance, increased cell adhesion and sphere formation. In addition, immunohistochemistry of patient samples showed that PTRF expression levels were a significant biomarker of poor progression‐free survival, and IGFBP7 levels in patient sera were shown to be higher after relapse.
Keywords: BRAF, mass spectrometry, melanoma, PTRF, resistance
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics
Introduction
BRAF inhibitors (BRAFi) are used routinely for the treatment of metastatic or inoperable melanoma harbouring a BRAFV600E mutation (Chapman et al, 2011; Hauschild et al, 2012). Although BRAFi achieves clinical responses in 50% of patients, acquired drug resistance frequently develops (Wagle et al, 2011; McArthur et al, 2014). The mechanisms of BRAFi resistance have been investigated intensively and have identified MEK reactivation as a critical node. This resulted in the development of BRAFi (i.e. vemurafenib, dabrafenib) and MEKi (i.e. cobimetinib, trametinib) combination therapy, which improved therapeutic response, lowered toxicity, and prolonged progression‐free survival (PFS) and overall survival (OS; Long et al, 2014; McArthur et al, 2014; Robert et al, 2015). However, acquired resistance also develops from BRAFi/MEKi combinatorial therapy and has been shown to be caused by a set of genetic aberrations similar to those responsible for BRAFi resistance (Villanueva et al, 2013; Long et al, 2014; Wagle et al, 2014; Moriceau et al, 2015). In mono‐ or combination therapy, resistance might be due to the plasticity and heterogeneity of melanoma cells (Zipser et al, 2011). Phenotype switching may allow for multiple adaptive mechanisms that enable melanoma cell survival independent of MAPK pathway activation (e.g. EMT like, and differences in proliferation, motility or stem cell‐like characteristics) (Dummer & Flaherty, 2012). Very few proteomic studies have investigated MAPKi resistance phenotype switching in melanoma to get insight into the cellular mechanisms of melanoma drug resistance and to extract clinically relevant features (Koomen & Smalley, 2011; Straussman et al, 2012; Parker et al, 2014, 2015; Fleuren et al, 2016). Proteomics has been used to predict pleiotropic acidophilic protein kinase CK2 (CK2) as a potential drug target, demonstrating that CK2 blockade potentiated the antiproliferative effects of BRAF and MEK inhibition in BRAF‐mutated cancers (Parker et al, 2014). Phosphoproteomics has shown changes in cytoskeletal regulation, GTP/GDP exchange, protein kinase C, insulin growth factor (IGF) signalling and melanosome maturation after transition to a drug‐resistant phenotype (Parker et al, 2015). In a previous feasibility study, we showed in cisplatin‐sensitive versus cisplatin‐resistant melanoma cell lines that proteome profiling facilitates the identification of drug resistance mechanisms (Paulitschke et al, 2013). Recently, we reported that acquired BRAFi resistance is associated with a loss of differentiation, an enhanced expression of the lysosomal compartment, an increased potential for metastasis and epithelial–mesenchymal transition (EMT; Paulitschke et al, 2015). These features are associated with pronounced changes in cellular morphology in line with the model of “phenotype switching” from a melanocytic to a mesenchymal state during progression and resistance (Hoek et al, 2008; Zipser et al, 2011; Paulitschke et al, 2015; Spranger et al, 2015; Hugo et al, 2016), summarized in Paulitschke et al (2016). Mesenchymal melanoma cells are resistant to BRAFi treatment and tend to downregulate lineage‐specific genes (e.g. MelanA, MITF) while upregulating factors known to be involved in drug resistance (e.g. Wnt5a; Weeraratna et al, 2002; Hoek et al, 2008; Eichhoff et al, 2010; Zipser et al, 2011). Chronic exposure of proliferative melanoma cells to TGFβ causes a phenotype switch which involves the activation of PI3K signalling, downregulation of E‐cadherin and the loss of tissue‐specific marker gene expression, which is a process similar to EMT and contributes to melanoma heterogeneity (Schlegel et al, 2015). Recently, proteomic and phosphoproteomic changes of cultured human keratinocytes undergoing EMT and cell cycle arrest in response to stimulation with TGFβ were demonstrated by SMAD‐dependent and SMAD‐independent pathways (D'Souza et al, 2014).
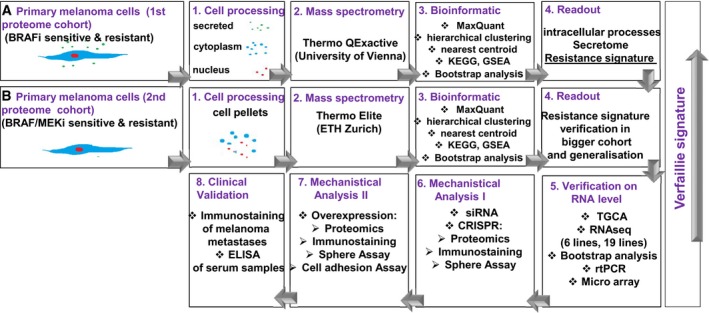
The goal of the present study was to identify a proteomic signature of MAPKi resistance in melanoma cells, to mechanistically dissect the role of genes in that signature and to validate the most informative features on patient biopsies prior to MAPKi therapy. By applying high‐throughput techniques such as subcellular shotgun proteome analyses in two different mass spectrometry (MS) centres and RNAseq, we identified two proteins associated with a mesenchymal phenotype and demonstrated the involvement of PTRF in the invasive phenotype associated with MAPKi resistance in melanoma. Further validation in patient biopsies suggests that PTRF expression is significantly highly expressed in patients who fail to respond to targeted therapy. The study design is highlighted in Fig EV1. To understand the subcellular contribution and to identify possible secreted proteins as serum biomarkers to MAPKi resistance, we first established subcellular fractionation of two sensitive and four resistant cells and obtained a resistance signature by proteome analysis of subcellular compartments (1st proteome cohort). In a next step, we used a larger cohort of seven sensitive and 13 resistant cells and a second MS centre for validation of the signature, generalization of the data by using different MS centres and for additional information for BRAFi and MEKi resistance (2nd proteome cohort). Comparison of RNAseq data (six sensitive and 10 resistant cell lines) to the proteome resistance signature validated our results. The most informative protein PTRF was used for mechanistic analysis and clinical correlation (Fig EV1).
Figure EV1. Overview over experimental and bioinformatic design of the study.
Results
Patient‐derived BRAFi‐resistant short‐term melanoma cultures have an EMT phenotype
Two metastatic primary melanoma cell cultures (S1, S2) sensitive to the BRAFi LGX818 (Encorafenib) and PLX4032 (Vemurafenib), and four BRAFi‐resistant primary cell cultures (R1, R2, R3, R4), were used as short‐term term cultures, characterized (Fig 1A) and processed for shotgun proteomics.
Figure 1. In vivo derived melanoma cultures are resistant to BRAFi treatment and have an EMT phenotype.
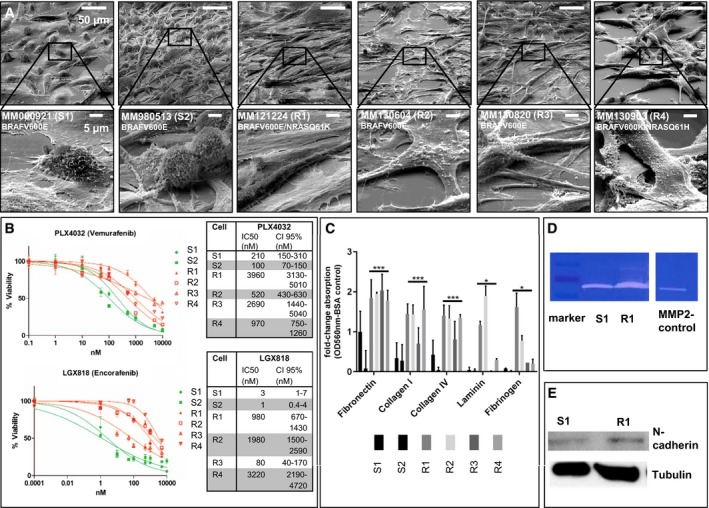
- EM image of S1, S2 and R1–4, Ruler: 50 μM, 5 μM (below).
- IC50 values to PLX4032 and LGX818 of S1, S2 and R1–4. For each cell culture and targeted therapy, we performed the viability analysis in triplicates (n = 3) and depicted the mean values of each concentration (±SD). The normalized data were merged, and the average IC50 was calculated using GraphPad Prism software. We show the 95% confident interval (CI95%) as a measurement of data distribution around the IC50 value of each cell culture.
- Cell adhesion assay of S1, S2 and R1–4, ECM‐mediated cell adhesion was quantified at OD 560 nm after extraction. Each bar graph represents the mean of two independent experiments for each cell culture (±SD). We performed a one‐tailed unpaired Student's t‐test to analyse significance of two groups (sensitive versus resistant cell cultures) (*< 0.05; ***< 0.005).
- Zymography assay of S1, R1, M (marker) and MMP2 C (MMP2‐positive control).
- Western blotting of N‐cadherin of S1 and R1; tubulin serves as a positive control.
The two sensitive cultures, S1 and S2, were established from skin metastases of treatment‐naïve patients. Four resistant cultures, R1‐R4, were taken from different patients who received BRAFi and progressed under treatment.
Resistant melanoma cultures R2 and R3 were established shortly before treatment initiation, suggesting that resistant melanoma cells were already present in these tumours (e.g. intrinsic resistance). The other resistant cultures (R1 and R4) were established from relapsed tumours after an initially successful BRAFi treatment (acquired resistance).
Cell morphology was examined by electron microscopy, revealing sensitive cells that were usually present as smaller cells with a ball‐like structure, whereas resistant cells were larger and flatter with a fibroblast‐like morphology (Fig 1A), consistent with our previous results of melanoma cells with induced resistance (Paulitschke et al, 2015).
All melanoma cells were BRAFV600E mutated, except R3 which has a BRAFV600K mutation. R1 and R4 were shown by single‐cell cloning and following Sanger sequencing to be double mutated for BRAFV600E and NRASQ61K or NRASQ61H, respectively (Raaijmakers et al, 2016). The IC50 to PLX4032 was the lowest in S2, followed by S1, R2, R4, R3 and R1 with the highest IC50. The IC50 to LGX818 was the lowest in S2 followed by S1, R3, R1, R2 and R4 with the highest IC50 (Fig 1B). The IC50 to MEK162 was highest for R1 (Table 1).
Table 1.
Category of primary cells and the IC50 to MAPKi treatment
| IC50 LGX (nM) | IC50 PLX (nM) | IC50 MEK162 (nM) | |
|---|---|---|---|
| Sensitive BRAF mutated | |||
| MM000921 (S1) | 3 | 210 | 50 |
| MM980513 (S2) | 1 | 100 | 10 |
| MM990922 | 10 | 1,032 | 100 |
| MM991104 | 3 | 200 | 100 |
| MM951004 | 7 | 413 | 100 |
| MM990802 | 3 | 176 | 20 |
| MM990706 | 2 | 137 | 10 |
| BRAF mut | |||
| MM130604 (R2) | 1,980 | 520 | 20 |
| MM130820 (R3) | 80 | 2,690 | 20 |
| BRAF/NRAS mut | |||
| MM140307 (DR1) | 200 | > 10,000 | > 1,000 |
| MM121224 (R1)/DMSO | 980 | 3,960 | 100 |
| MM140906 (DR2) | >200 | > 10,000 | > 1,000 |
| MM121224_250 nM MEK162 | 980 | 3,960 | > 10,000 |
| MM121224_500 nM MEK162 | 980 | 3,960 | 1,924 |
| MM130903 (R4) | 3,220 | 970 | 20 |
| MM150423 (DR 3) | 554 | > 10,000 | 1,000 |
| Intrinsic resistant | |||
| MM111031 (IR) | 1670 | 140 | 30 |
| MM150325 | 8*107 | 8*106 | 951.3 |
| MM150405 | 13,321.5 | 36,954 | 5,663 |
As published previously, induced drug‐resistant melanoma cells show an increased adherence to extracellular matrix proteins (Paulitschke et al, 2015). Similarly, the patient‐derived drug‐resistant melanoma cells compared to sensitive ones have a higher adherence in a cell adhesion assay (Fig 1C). Comparing the sensitive (S1) and resistant (R1) melanoma cells by zymography, we detected an increase in MMP2 (matrix metalloproteinase 2) activity in the resistant cells (R1; Fig 1D), a feature associated with invasiveness in EMT (Bae et al, 2013). Consistently, N‐cadherin levels were increased in culture R1 (Fig 1E), which is both in line with the proteome data and our previous publication (Paulitschke et al, 2015).
Shotgun proteomic analysis of primary MAPKi‐sensitive and MAPKi‐resistant cells
We enriched for subcellular fractions of all cells and performed shotgun proteomic analysis of every fraction. In total, 4,052 proteins in the cytoplasmic, 1,007 proteins in the supernatant and 3,463 proteins in the nuclear fraction were detected. Using Perseus software, we generated heatmaps for every fraction analysed and compared protein expression of sensitive versus resistant cells. In the cytoplasm and nuclei, the functional classes “nucleic acid binding or metabolic process”, “cellular nitrogen compound metabolic process” and “RNA processing” were commonly and significantly upregulated in the sensitive cells, whereas “COPI vesicle coat” and “calcium ion binding” were commonly and significantly upregulated in the resistant cells (Fig EV2).
Figure EV2. Proteome characterization of sensitive (S1,2) and resistant (R1,2,3,4) melanoma cells (cytoplasmic fraction).
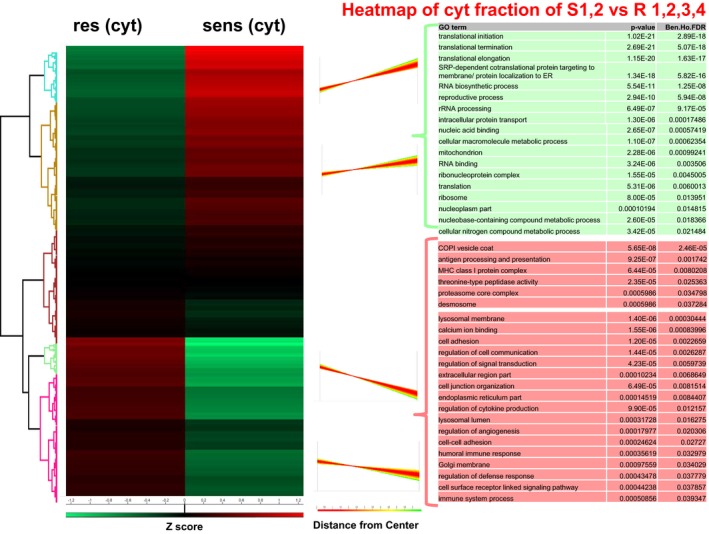
Left: Hierarchical clustering of Z‐scored expression values for significantly changed protein expression, revealing differences between sensitive and resistant cells. Right: Profiles of the four main clusters. Red: upregulated in resistant cells, green: downregulated in sensitive cells.
Groups significantly upregulated in the cytoplasmic fraction of the resistant cultures revealed, e.g. “antigen processing and presentation”, “MHC class I protein complex”, “threonine‐type peptidase activity”, “lysosomal membrane and lumen”, “cell adhesion”, “regulation of cytokine production”, “regulation of angiogenesis” and “immune processes” (Fig EV2).
In the secretome, the identified proteins with significantly different abundance revealed to be upregulated in the resistant cells (Fig EV3). Since many proteins that were upregulated in resistant cells could not be assigned significantly to a specific process, we examined individual proteins that were the most highly differentially expressed. The most highly and significantly upregulated proteins in the resistant secretome were insulin‐like growth factor‐binding protein 7 (IGFBP7), procollagen C‐endopeptidase enhancer 1 (POLCE), nicotinamide phosphoribosyltransferase (NAMPT), nidogen‐1 (NID1), thrombospondin‐2 (THBS2) or interleukin 8 (IL8). Only one pathway was upregulated in the secretome of the sensitive cells: “very‐low‐density lipoprotein particle” (Fig EV3). In the nuclear compartment of the resistant cells, the functional groups “locomotion”, “eukaryotic translation initiation factors”, “proton‐transporting V‐type ATPase”, “clathrin adaptor complex”, “insulin receptor signalling pathway” and “glial cell development” were significantly upregulated (Fig EV4).
Figure EV3. Proteome characterization of sensitive and resistant melanoma cells of secretome.
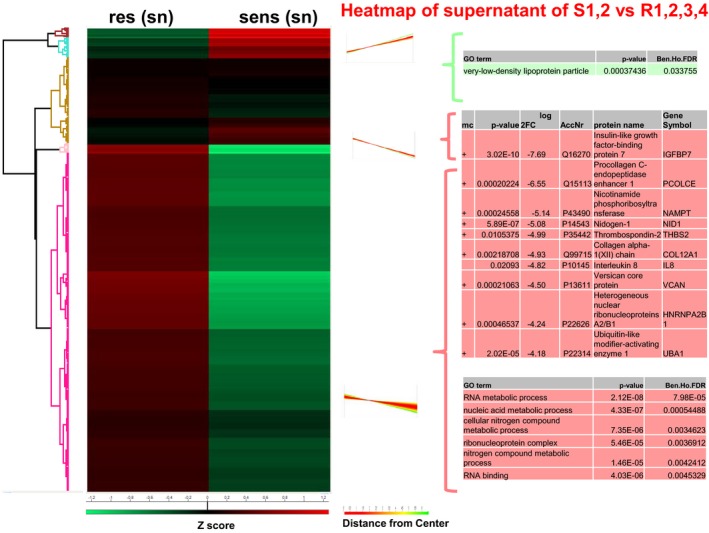
Left: Hierarchical clustering of Z‐scored expression values for significantly changed proteins, revealing differences. Right: Profiles of the one cluster and additional proteins. Red: upregulated in resistant cells, green: downregulated in sensitive cells.
Figure EV4. Proteome characterization of sensitive and resistant melanoma cells of nuclear fraction.
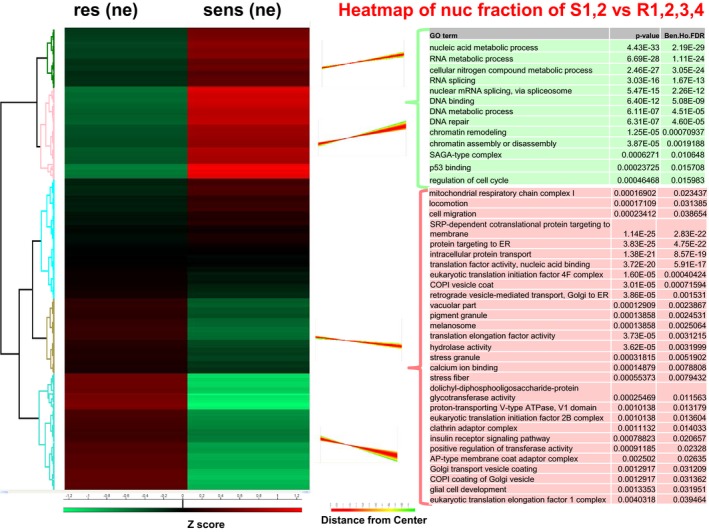
Left: Hierarchical clustering of Z‐scored expression values for significantly changed proteins, revealing differences. Right: Profiles of the four clusters. Red: upregulated in resistant cells, green: downregulated in sensitive cells.
BRAFi and MEKi resistance in KEGG pathway analysis
In a next step, we enhanced the cohort of primary cells by using cell pellets of additional patients of the same cohort to confirm the data, integrating relevant new features such as BRAFi and MEKi double‐resistant primary melanoma cells and intrinsically double‐resistant melanoma cells, in order to analyse MAPKi (BRAFi and MEKi) resistance. This second dataset was comprised of 20 primary melanoma cell cultures with seven MAPKi‐sensitive, four BRAFi‐resistant and five MAPKi‐resistant primary cells including a cell line with increasing in vitro derived MEKi resistance, and three MAPKi intrinsically resistant primary cells. All IC50 values to LGX, PLX and MEK162 and the mutational status of BRAF and NRAS were determined prior to analysis (Table 1). Cell pellets were made, and the MS analysis was conducted at a different institute in order to confirm the earlier findings in a different experimental setting.
To get an insight into the pathways generally upregulated in drug‐resistant cells, a KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis was performed for both proteome cohorts, which are highly comparable. In Fig 3, KEGG pathways of the cytoplasmic fraction (Fig 2A and C) and the 2nd proteome cohort (Fig 2B and D) are exemplified by the pathways “cell adhesion molecules” (Fig 2A and B) and “ECM receptor interaction” (Fig 2C and D), where almost every protein is upregulated (red colour). In addition, gene set enrichment analysis (GSEA) confirmed that these pathways are significantly enriched in both proteome cohorts (Fig 2A’–D’).
Figure 3. Hierarchical clustering of the most informative proteins, correlation with TCGA and RNAseq data.
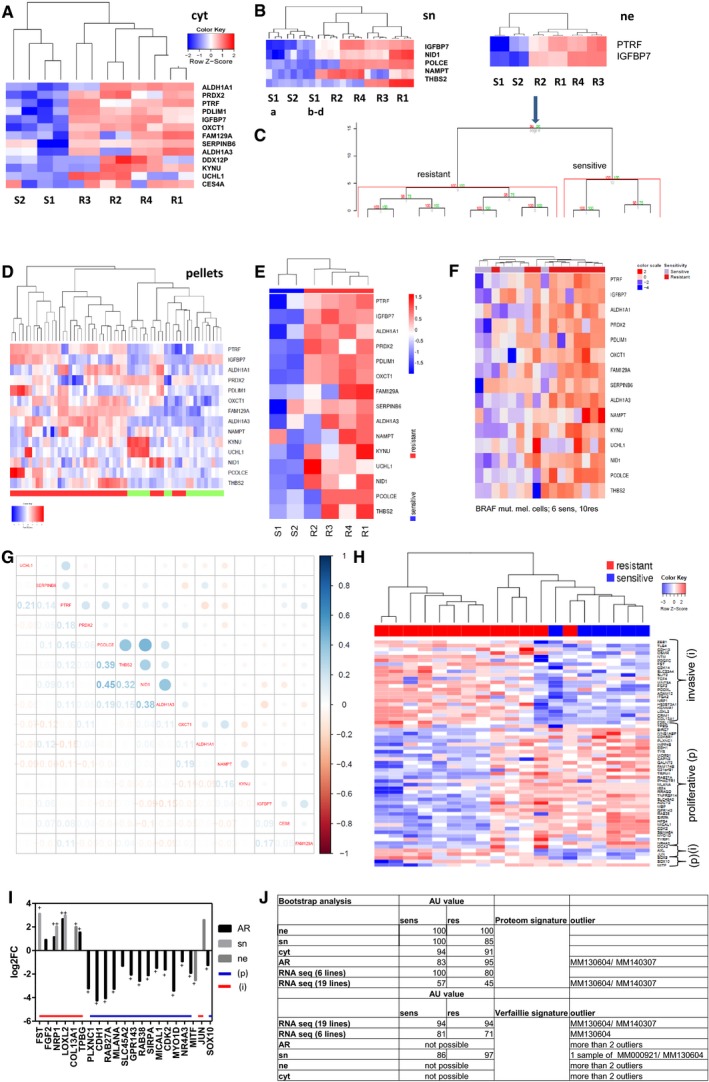
-
A–DProteins needed to distinguish between sensitive and resistant cells in cytoplasm (A), secretome (B) and nucleus with bootstrap analysis (C), of the 15 candidates in cell pellets (D).
-
E, FHeatmap of the 15 candidates with RNAseq data of S1, S2 R1–4 and a second cohort.
-
GCorrelation matrix of the 15 candidates with TCGA, numbers indicate Pearson correlation coefficient.
-
HHeatmap of Verfaillie signature with RNAseq data correlating invasive (i)/proliferative (p) phenotype with resistant/sensitive melanoma cell cultures.
-
IBar graph of 1st cohort (sn, ne) and 2nd cohort (AR; all resistant) correlating the Verfaillie signature of invasive (i) and proliferative (p) genes with the protein expression up or downregulated in resistant cell cultures.
-
JBootstrap analysis with AU value of 1st and 2nd proteome and RNA cohort versus proteome signature (15 proteins) and RNA signature of the Verfaillie publication, + multiparameter correction.
Figure 2. GSEA (gene set enrichment analysis) and KEGG pathway visualization of main regulated pathways.
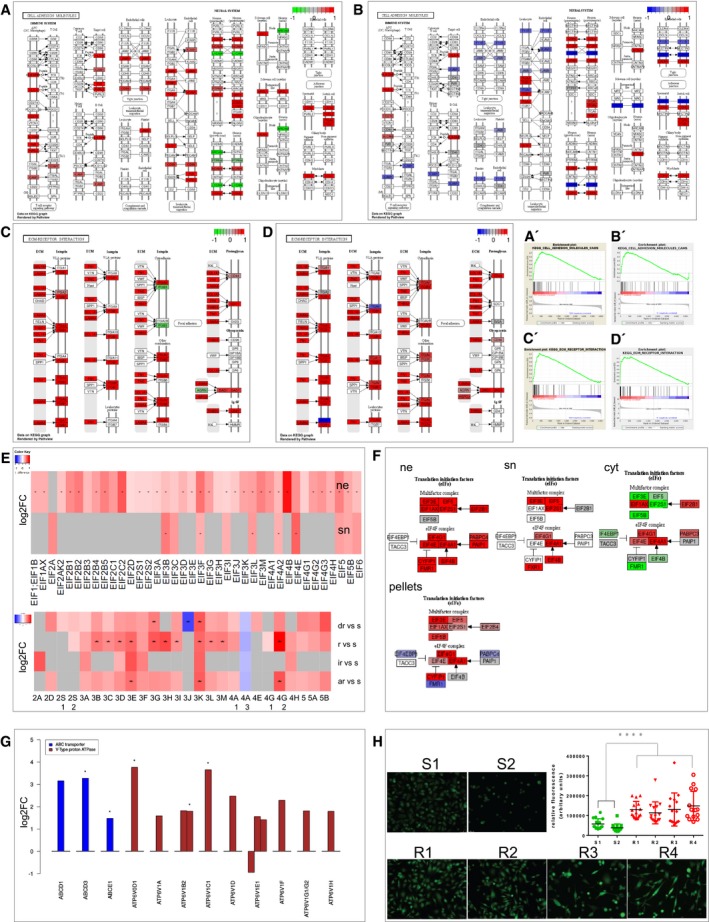
- Cell adhesion molecules 1st proteome cohort.
- Cell adhesion molecules 2nd proteome cohort.
- ECM receptor interaction in the 1st proteome cohort.
- ECM receptor interaction in the 2nd proteome cohort is highly enriched and upregulated as visualized KEGG pathway visualization and by GSEA (A′–D′) of the two cell culture cohorts.
- Heatmap of significant regulated EIFs in ne (nucleus) and sn (supernatant), pellets. red: upregulated in resistant cells, grey: not identified, * multiparameter correction, s, sensitive; dr, double resistant; r, BRAF resistant; ir, intrinsic resistant; ar, all resistant; vs, versus.
- KEGG pathway visualization of log fold change (FC) regulated EIFs in ne, sn, cyt, cell pellets, red: upregulated, green/blue: downregulated in resistant cells.
- Barplot of proteins involved in drug elimination strategies, regulation is depicted as upregulation in resistant cells by log2FC, order of compartment: cyt, ne, sn, all significant, * multiparameter correction.
- Measurement of the concentration of H+ in the resistant cell cultures and endosomal activity by Molecular Probes® pHrodo® dye in S1, S2 and R1–4. Fluorescence was quantified compared to background using ImageJ software. Five images per cell culture were analysed, and the experiment was performed in triplicates (n = 15). Each measurement was depicted on the dot plot as one single point (mean). One‐tailed unpaired Student's t‐test was performed and revealed statistical significance (P < 0.0001****) of group S against group R.
To take advantage of the different fractions, shifts of proteins assigned to specific functions can be visualized. Interestingly, the ribosomal compartment is enhanced in the nucleus and secretome, whereas the proteasomal compartment is enhanced in the cytoplasm and secretome. This shift in the resistant cells of the ribosomal compartment in the nucleus/ER and the proteasomal compartment in the cytoplasm is also observed to be significant in the GSEA (Fig EV5). In addition, components of both compartments are significantly upregulated in the secretome of the resistant cells (Fig EV5).
Figure EV5. GSEA (gene set enrichment analysis) and KEGG pathway visualization for the differential expression of ribosomal and proteasomal proteins KEGG pathway visualization of logFC regulated ribosomal and proteasomal proteins in cyt, sn and ne, red: upregulated green: downregulated in resistant cells.
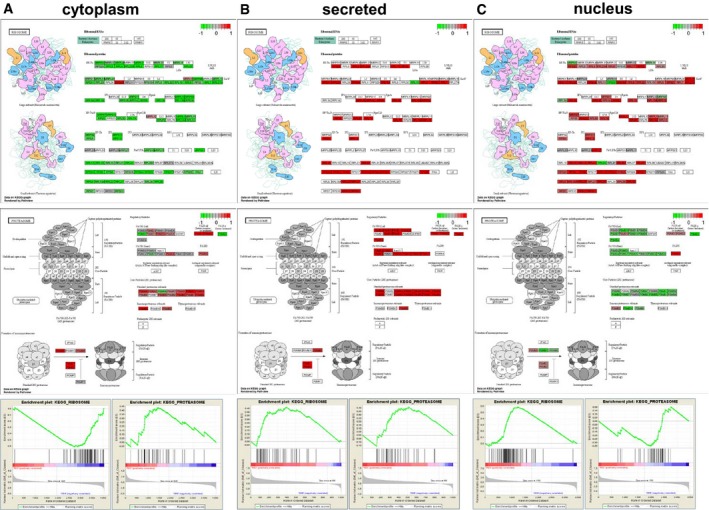
In conclusion, here we provide evidence that BRAFi and MAPKi resistance show common features on the pathway level.
Resistant cells upregulate translation initiation factors and have a significantly lower pH than sensitive cells
Based on a recent publication (Boussemart et al, 2014), the expression of eukaryotic translation initiation factors (EIFs) was also analysed in our cohorts. In the nucleus, all 37 identified EIFs were revealed to be significantly upregulated in resistant cells, and 81% were significant according to multiparameter test correction results (asterisks in Fig 2E). In the secretome, 11 significantly regulated EIFs were identified, and again, all EIFs were upregulated in resistant cells with 45% being significant by multiparameter testing (asterisks in Fig 2E; Slany et al, 2016). This is also depicted in the KEGG analysis using all identified EIFs (Fig 2F). In the cytoplasmic fraction, some EIFs were downregulated (green colour). In the second cohort (cell pellets), the main percentage of the EIFs were upregulated as well (Fig 2E lower part) and consistent with the cytoplasmic fraction; especially, EIF4EBP1 was downregulated (blue colour; Fig 2F lower part).
In addition, the proteome data were analysed for possible different drug elimination strategies such as ABCD transporter and V‐type proton ATPases. In Fig 2G, all corresponding and significantly regulated proteins are listed according to multiparameter testing, as indicated by asterisk. Besides the ABCD transporter, a cluster of proteins of a transporter family (V‐type proton ATPase) were significantly upregulated in the resistant melanoma cells. V‐type proton ATPases transport H+ across membranes, for example from the cytoplasm into endosomes. To verify whether there is a higher concentration of H+ in the resistant cells and higher endosomal activity, a pH‐sensitive fluorescent dye (pHrodo® dye, Molecular Probes®), which fluoresces brightly in acidic environments, was used. Indeed, resistant cell lines showed a significantly stronger fluorescence indicating a lower pH in their endosomes (Fig 2H).
Fifteen proteins have the highest class‐discriminatory power between sensitive and resistant cells
Overall, our shotgun proteomics identified 5,478 proteins over all fractions in the drug‐sensitive and drug‐resistant melanoma cells. To calculate the most class‐discriminating proteins, we used two different bioinformatics strategies: hierarchical clustering and classification to nearest centroids. Initially, proteins with the highest log2FC that was upregulated in the resistant cells were selected in each fraction and added to the heatmap until the dendrogram led to a clear separation of sensitive and resistant melanoma cells (S1,2 versus R1,2,3,4). Only 13 proteins were necessary to distinguish between the cytoplasmic fractions of the drug‐sensitive and drug‐resistant melanoma cells: ALDH1A1, PRDX2, PTRF, PDLIM1, IGFBP7, OXCT1, FAM129A, SERPINB6, ALDH1A3, DDX12P, KYNU, UCHL1 and CES4A (Fig 3A, Table 2). In the secretome, five proteins distinguished between the sensitive and resistant cells (Fig 3B). These are IGFBP7, PCOLCE, NAMPT, NID1 and THBS2. Only IGFBP7 and NID1 were upregulated in all resistant cells and downregulated in all sensitive cells. Most strikingly, only two proteins were necessary to separate the nuclear fractions of drug‐sensitive and drug‐resistant cells: IGFBP7 and PTRF (Fig 3C). Both were among the most strongly differentially expressed proteins of the cytoplasmic fraction. IGFBP7, which is a secreted protein, was also among the candidates that best separated the secretome of drug‐sensitive and drug‐resistant cells (Fig 3B). Using pvclust to estimate the cluster uncertainty showed the highest AU (approximately unbiased) P‐value of 100% for both proteins IGFBP7 and PTRF to cluster between sensitive and resistant cells (Fig 3C). Therefore, these two proteins were informative for separating sensitive from resistant cells in all analysed fractions and were used for further evaluation in clinical samples.
Table 2.
List of most informative proteins using hierarchical clustering in the cytoplasmic (cyt), secreted (sn) and nuclear (nuc) fraction upregulated in the resistant cells
| Fraction | Multiparameter in fractions/in all pellets | P‐value | log2FC | acc‐Nr | Protein name | Gene ID | Role in cancer and resistance |
|---|---|---|---|---|---|---|---|
| cyt | +/+ | 0.00095376 | −4.41 | P00352 | Retinal dehydrogenase 1 | ALDH1A1 | Stemness, disulfiram |
| +/− | 0.00014171 | −5.79 | P32119 | Peroxiredoxin‐2 | PRDX2 | Metastasis in melanoma | |
| +/+ | 8.77E‐05 | −6.19 | Q6NZI2 | Polymerase I and transcript release factor | PTRF | Metastasis, drug resistance | |
| +/− | 0.00038204 | −4.32 | O00151 | PDZ and LIM domain protein 1 | PDLIM1 | Metastasis breast cancer | |
| +/− | 6.06E‐07 | −5.94 | Q16270 | Insulin‐like growth factor‐binding protein 7 | IGFBP7 | EMT | |
| +/+ | 7.29E‐07 | −5.27 | P55809 | Succinyl‐CoA:3‐ketoacid‐coenzyme A transferase 1, mitochondrial | OXCT1 | Prostate cancer progression | |
| −/+ | 0.0121147 | −4.54 | Q9BZQ8 | Protein Niban | FAM129A | Survival mechanisms | |
| −/+ | 0.0189463 | −4.80 | P35237 | Serpin B6 | SERPINB6 | x | |
| −/+ | 0.0456127 | −4.48 | P47895 | Aldehyde dehydrogenase family 1 member A3 | ALDH1A3 | chemoresistance | |
| −/nd | 0.106551 | −4.41 | Q92771;Q96FC9;A8MPP1 | Putative ATP‐dependent RNA helicase DDX12 | DDX12P;DDX11;DDX11L8 | x | |
| −/+ | 0.00951598 | −4.29 | Q16719 | Kynureninase | KYNU | Active in cancer, Tryptophan | |
| −/+ | 0.0419105 | −5.76 | P09936 | Ubiquitin carboxyl‐terminal hydrolase isozyme L1 | UCHL1 | Associated with melanoma cell proliferation, MAPK | |
| 0.126862 | −6.03 | Q5XG92 | Carboxylesterase 4A | CES4A | Drug metabolism | ||
| sn | +/− | 3.02E‐10 | −7.70 | Q16270 | Insulin‐like growth factor‐binding protein 7 | IGFBP7 | |
| +/− | 0.00020224 | −6.55 | Q15113 | Procollagen C‐endopeptidase enhancer 1 | PCOLCE | Interaction with ECM | |
| +/− | 0.00024558 | −5.14 | P43490 | Nicotinamide phosphoribosyltransferase | NAMPT | Higher expression in melanoma, Tryptophan | |
| +/− | 5.89E‐07 | −5.08 | P14543 | Nidogen‐1 | NID1 | Higher expression in cancer, serum marker in breast cancer | |
| +/+ | 0.0105375 | −4.99 | P35442n | Thrombospondin‐2 | THBS2 | Higher expressed in melanoma metastasis | |
| nuc | +/+ | 6.76E‐05 | −7.93 | Q6NZI2 | Polymerase I and transcript release factor | PTRF | |
| +/− | 7.03E‐08 | −7.81 | Q16270 | Insulin‐like growth factor‐binding protein 7 | IGFBP7 |
Interestingly, two of all proteins critical in distinguishing sensitive from resistant cells had not been associated yet with the processes of tumour progression, metastasis or drug resistance, namely SERPINB6 and DDX12P, whereas all other proteins have been described in metastasis, drug resistance, stemness, EMT and ECM remodelling, and are in some cases associated with melanoma and MAPK signalling. To the best of our knowledge, none of the candidates has been previously described to be involved in BRAFi and MEKi resistance (Table 2).
In total, 5,478 proteins from all fractions could be reduced to 15 proteins with the strongest predictive power using two different bioinformatics tools. This 15 protein signature effectively distinguished the sensitive and resistant cell cultures as well in the cohort of cell pellets (2nd proteome cohort; Fig 3D).
In a next step, heatmap separation with increasing numbers of proteins sorted by log2FC was generated. Heatmaps for each fraction with 50 proteins are shown depicting that the separation of sensitive and resistant cells remains true for the cytoplasm and for the nuclear fraction, whereas in the secretome, this separation can only be found with the five proteins depicted in Fig 3B (Appendix Fig S1). Here, the heterogeneity is the highest. In addition, in the cytoplasm and nuclear fractions, up‐ and downregulated proteins were apparently balanced, but the secretome contained the highest proportion of upregulated proteins, as visualized in the GSEA plots and heatmaps of each cellular fraction (Fig EV5). In addition to hierarchical clustering, candidate proteins were selected using classification to nearest centroids and PTRF, IGFBP7, OXCT1, PCOLCE and NID1 were also found to be highly informative (Appendix Fig S2). Therefore, two different bioinformatics analyses led to the confirmation of the signature.
Validation of the signature at the RNA level
The top 15 proteins that distinguished the two phenotypes were also validated using RNAseq data and were highly differentially expressed in sensitive and resistant cells (Fig 3E).
In a next step, a larger cohort of RNAseq data of six BRAFi‐sensitive and MEKi‐sensitive and 10 resistant melanoma cells was analysed independently. A clustering with these 15 candidates between both cohorts could be performed for most of the samples (Fig 3F).
Subsequently, these 15 proteins were compared to publically available melanoma datasets at the TCGA (The Cancer Genome Atlas; https://tcga-data.nci.nih.gov/tcga/) and a strong correlation was found with the secreted proteins POLCE, THBS2, NID 1 and the cytoplasmic protein ALDH1A3 (Fig 3G).
In a next step, we asked whether our proteome signatures represented one of the known phenotype signatures from the literature, such as the publication of Verfaillie et al (2015).
The signature for the heatmap was selected using the listed candidates of Fig 1C of Verfaillie et al where a core subset of invasive and proliferative gene signatures was used [upper part of the heatmap (Fig 3H)] and additional important genes characterized as main players of the phenotype switch and resistance by Verfaillie et al [lower part of the heatmap (Fig 3H)], which we call the Verfaillie signature. With these candidates, we were also able to clearly separate the RNAseq data from sensitive versus resistant cell cultures (Fig 3H), indicating that our data are comparable to those generated by other groups. In a next step, we analysed the two proteome cohorts for the Verfaillie signature and all identified and significant detected proteins of the proteome data were correlated to the phenotypes. The genes correlated to the invasive phenotype (FST, FGF2, NRP1, LOXL2, COL13A1, TGFB, JUN) are significantly upregulated in the resistant cells, whereas the genes correlated to the proliferative phenotype (i.e. CDH1, RAB27A, MLANA, SLC45A2, GPR143, RAB38, SIRPA, MICAL1, CDK2, MYO1D, NR4A3, MITF, SOX10) are significantly downregulated in the resistant cells (Fig 3I). No identified and significantly regulated protein was revealed to be reverse regulated.
To address the question whether there is a difference in signature detection by proteomics and transcriptomics and to quantify cluster uncertainty, we performed bootstrap analysis of the 15 protein signature and the Verfaillie signature on both of our proteome cohorts and the RNAseq datasets. This revealed that we were able to verify signatures on RNA or proteome level, but the proteome signature has the highest AU P‐value for the proteome signature and the RNAseq data for the Verfaillie signature (Fig 3J). Therefore, different signatures can be identified by transcriptomics or proteomics, but both technologies are able to verify known signatures indicating the complementary power of both technologies. In addition, both technologies suggested the same cell cultures were outliers (MM130604, MM140307; Fig 3J).
The EMT‐like transcriptional resistance signature is correlated to PTRF and IGFBP7 expression
Having shown that melanoma cells adapt to drug exposure by adopting EMT features and expressing proteins correlated to the invasive phenotype, we investigated this connection in relation to PTRF and IGFBP7. The expression of both genes was clustered closer to the EMT phenotype in the RNAseq data of 6 BRAFi‐ and MEKi‐sensitive and 10 resistant melanoma cells plus cell states, as characterized by a switch from CDH1 to CDH2 expression (Fig 4A). In addition, BRAFi‐ and MEKi‐sensitive melanoma cell cultures have a lower expression of N‐cadherin but higher expression of E‐cadherin in comparison with the resistant cell cultures.
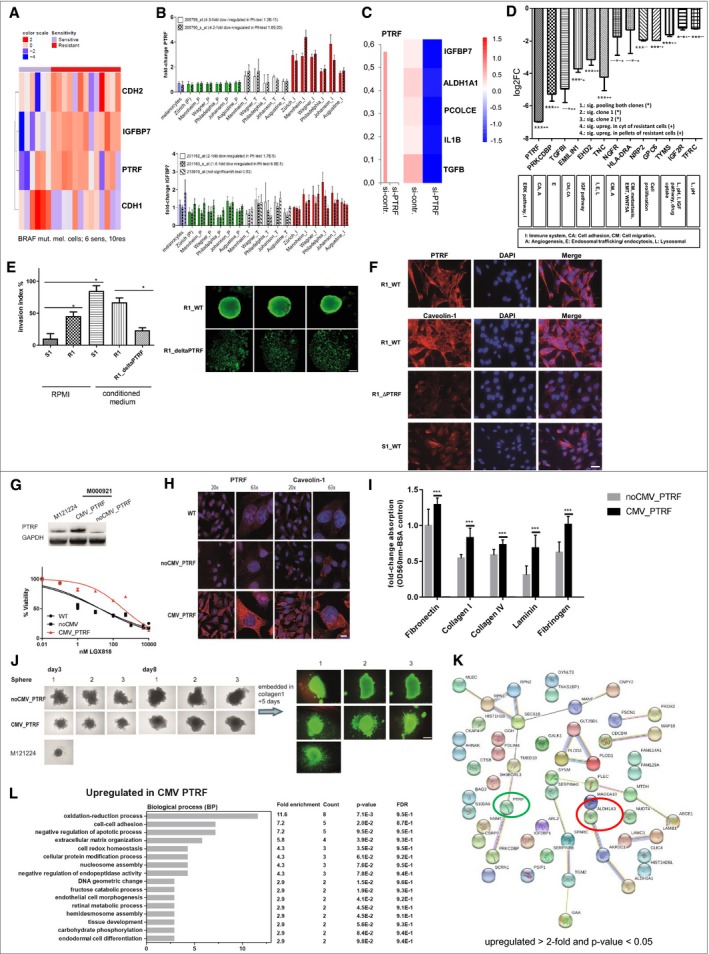
Figure 4. Functional relevance of PTRF .
-
A–DAssociation of PTRF and IGFBP7 with EMT phenotype (RNAseq data) (A) and with the invasive phenotype of melanoma cell cultures in microarray data of six different centres (B). (C) siRNA knockdown of PTRF and involved proteins by PTRF knockdown, Z values of the deltadeltaCT are used for the heatmap. (D) CRISPR of PTRF and the targets.
-
ECell invasion of S1 and R1 with and without supernatant of melanoma‐associated fibroblasts (conditioned medium), and R1 harbouring loss of PTRF by CRISPR/Cas (R1_ΔPTRF). Statistical analysis using a one‐tailed unpaired Student's t‐test to analyse significance of two groups (*< 0.05). Sphere formation of R1 and R1_ΔPTRF showing live fluorescent staining using Calcein AM (Thermo Fisher). Here we show pictures of three independent wells. Ruler: 200 μM.
-
FImmunofluorescence analysis of PTRF and caveolin‐1 in S1, R1 and R1_ΔPTRF. Ruler: 50 μM.
-
GThe BRAF‐mutated and drug‐sensitive melanoma cell culture M000921 (S1) was transduced with a lentivirus containing a PTRF‐overexpressing construct (CMV_PTRF) or a non‐CMV control plasmid (noCMV_PTRF). Expression of PTRF was confirmed by Western blotting. Cells were subjected to dose‐escalating concentrations of the BRAFi LGX818, and cell viability was measured for each concentration in triplicates. Error bars indicate SD. Normalized data were used to calculate the IC50 values by GraphPad Prism software [WT 57 nM (95%CI 32–100 nM); noCMV_PTRF 41 nM (95%CI 26–65 nM) and CMV_PTRF 784 nM (95%CI 592–1,037)].
-
HFluorescent microscopy revealed coregulation of PTRF and caveolin‐1. Ruler: 20×: 30 μM, 63×: 10 μM.
-
ICell adhesion is enhanced in PTRF‐overexpressing cell cultures (S1) (CMV_PTRF) in comparison with control cell cultures (noCMV_PTRF). Each bar graph represents the mean of two independent experiments for each cell culture (±SD). We performed a one‐tailed unpaired Student's t‐test for each adhesion protein to analyse significance between control and PTRF‐overexpressing cells (***< 0.005).
-
JRapid spheroid formation is observed in PTRF‐expressing melanoma cultures which also show higher degree of invasion into a collagen 1 matrix compared to cell cultures lacking PTRF expression. Ruler: 500 μM.
-
KSTRING analysis (red circle) and nearest shrunken centroid (green circle) of proteome data CMV_PTRF versus noCMV_PTRF (upregulated > 2‐fold and P‐value < 0.05).
-
LFunctional annotation categories calculated in DAVID (DAVID Bioinformatics Resources) upregulated in CMV_PTRF. Fold enrichment values, count (genes involved in the term), P‐value and FDR (false discovery rate, calculated using the Benjamini–Hochberg procedure), listed next to the graph, were calculated using DAVID bioinformatics resources.
To exclude bias in cell culture preparation, we performed a meta‐analysis of microarray data of six international centres where data were publicly available and analysed for proliferative and invasive melanoma expression signatures (Widmer et al, 2013). We found PTRF and IGFBP7 to be significantly upregulated in invasive signature melanoma cells across all six analysed datasets (Table 2, Fig 4B).
IGFBP7 expression is downstream of PTRF and PTRF regulates the main features of drug resistance
PTRF plays a role in rRNA transcription and the formation and stabilization of caveolae and was not previously known to be functionally involved in melanoma progression. To determine whether PTRF is functionally connected with IGFBP7 and other proteins that were highly differentially expressed between the sensitive and resistant cells, we treated melanoma cells with siRNA against human PTRF (Yi et al, 2013). Upon PTRF depletion, expression analysis revealed downregulation of three proteins (IGFBP7, ALDH1A3, POLCE), suggesting that these are downstream targets of PTRF (Fig 4C). In addition, PTRF seems to be involved in inflammatory processes, since IL‐1β was also downregulated (Fig 4C).
CRISPR/Cas‐derived knockout of PTRF of two different clones with subsequent shotgun proteomic analysis led to the identification of 12 targets which were significantly and at least twofold downregulated. These proteins are involved in diverse functions such as immune regulatory processes, cell adhesion, cell migration, angiogenesis, endosomal trafficking, endocytosis, the pH regulation and lysosomal function and IGF signalling. Besides downregulation of PTRF itself, PRKCDBP, TGFBI, EMILIN1, EHD2, TNC, NGFR, HLA‐DRA, NRP2, GPC6, TYMS, IGF2R and TFRC are also significantly downregulated. These candidates were checked for their general function, known function in resistance and expression in the cytoplasmic fraction of the resistant cells and the 2nd proteome cohort. PRKCDBP, TGFBI, EHD2, TNC, and TYMS revealed to be upregulated in all analysed BRAFi‐ and MEKi‐resistant cohorts (Fig 4D).
The phenotype‐switching model, as well as the model of EMT in epithelial cancers, postulates that invasiveness is induced by factors released from the microenvironment (Widmer et al, 2015). Tumour‐associated fibroblasts are suspected to be a source for such factors (e.g. TGFβ; Schlegel et al, 2015). To analyse the influence of bystander fibroblasts on melanoma cells, we performed invasion assays using melanoma cells S1 and R1 with and without conditioned media from tumour‐associated fibroblasts. Both melanoma cells invade Matrigel and transmigrate through the chamber membrane, whereby the resistant cells (R1) show this phenotype to a significantly higher extent (Fig 4E). However, when we let the cells invade in tumour‐associated fibroblast conditioned media, both sensitive and resistant cells showed similarly significant enhanced invasive activity. Deletion of PTRF protein expression by CRISPR/Cas (R1_ΔPTRF) led to inhibition of Matrigel invasion of R1 (Fig 4E). In vitro spheroid formation has been associated with stemness in cancer cells. We have reported here that the expression of PTRF is associated with a mesenchymal (stem cell like) phenotype and drug resistance. We have derived 3D spheroid under non‐adhesive culturing conditions. While drug‐resistant cell culture R1 formed compact spheroids in 48 h when grown on agarose‐coated plates, R1_ΔPTRF cells were not able to form spheroid structures at all and cells only rounded up and spread over the well (Fig 4E). Also increasing incubation time did not lead to sphere formation of R1_ΔPTRF cell culture. These data indicate that PTRF is involved in the invasiveness of the resistant melanoma cells as well as cell‐cell contact properties.
Recently, it was reported that PTRF is necessary for caveola formation (Hill et al, 2008). Caveola formation at the cell membrane and trafficking showed also to be part of RTK signalling by internalizing surface receptors like IGFR1 (Salani et al, 2010) as well as lipid regulation and endocytosis.
In drug‐resistant R1 cells, PTRF is localized at the membrane as well as specific spots in the cytoplasm. Caveolin‐1 is observed in a similar staining pattern as PTRF in R1 cells. Deleting PTRF resulted in the downregulation of caveolin‐1, confirming the expression data, as well as its critical role in caveolin‐1 expression in melanoma cells (Fig 4F).
In order to evaluate whether PTRF is critical to resistance against BRAFi, we stably overexpressed PTRF in the drug‐sensitive cell melanoma cell culture M000921 (S1), which lead to resistance against BRAFi (IC50: wild‐type 57 nM; noCMV_PTRF 41 nM and CMV_PTRF 784 nM; Fig 4G).
In line with the observation that the deletion of PTRF in resistant melanoma cells downregulated caveolin‐1 expression, we observed an upregulation of caveolin‐1 together with induced PTRF expression in M000921_CMV_PTRF cells, confirming that PTRF is regulating caveolin‐1 expression. In addition, PTRF induction led to a significant induction of cell adhesion to ECM proteins (Fig 4I).
We again analysed 3D spheroid formation after the induction of PTRF expression and found that spheroids were rapidly formed (after 72 h), which is also observed with the PTRF‐expressing and drug‐resistant cell culture M121224 (Fig 4J). Melanoma culture M000921 harbouring the noCMV_PTRF construct needed 8 days to form a solid sphere, which could be embedded into a Collagen 1 matrix. After 5 days in Collagen 1, only spheroids derived from PTRF‐overexpressing cell cultures spread into the Collagen 1 matrix.
STRING analysis depicts all > 2‐fold upregulated proteins in CMV_PTRF (P‐value < 0.05) of proteome data comparing noCMV_PTRF and CMV_PTRF. PTRF is highly upregulated (green circle) and the ALDH1 A1 and A3 proteins. Performing nearest shrunken centroid revealed that only ALDH1A3 is necessary to separate both cohorts (red circle; Fig 4K). Using DAVID bioinformatic software, i.e. “cell‐cell adhesion” or “regulation of apoptotic process” were revealed to be upregulated in CMV_PTRF (Fig 4L).
PTRF and IGFBP7 expressions are significant biomarkers for BRAFi therapy
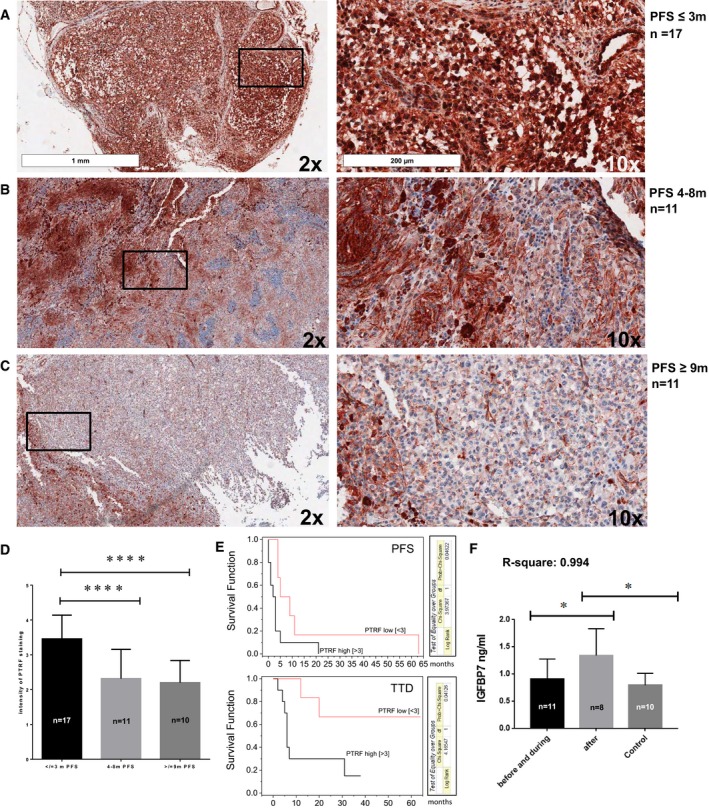
To evaluate the clinical relevance of PTRF expression, staining of melanoma biopsies from patients with 3‐month PFS, 6‐month PFS and > 9‐month PFS and of melanoma metastasis prior and after BRAFi therapy was performed. A total of 39 tissue stainings were evaluated, each from one patient. The highest PTRF expression was seen in the patient cohort with the lowest PFS (Fig 5A–C).
Figure 5. Clinical relevance of PTRF and IGFBP7 and drug activity.
-
A–D(A–C) Immunohistochemical staining for PTRF and (D) statistical analysis by one‐way ANOVA based on PFS. *P‐value < 0.05; **< 0.01, ***< 0.001.
-
EKaplan–Meier curves with hazard ratio correlating PFS and TTD (time to death) with PTRF expression.
-
FIGFBP7 analysis by one‐way ANOVA in the serum samples of patients with MAPKi therapy by ELISA. *P‐value < 0.05.
There was a significant difference in PTRF expression in patients with low PFS compared to patients with intermediate and high PFS (Fig 5D). In addition, PTRF was expressed more strongly in the nucleus of patient biopsies with the 3‐month PFS compared to the other groups (Fig 5A–C).
Kaplan–Meier curves with hazard ratios correlating PFS and TTD (time to death) with PTRF expression revealed a significant correlation to both clinical parameters (Fig 5E).
In addition to PTRF, IGFBP7 was the only other protein needed in the nucleus to predict MAPKi response and was the most differentially expressed protein. Since it is a secreted protein, we performed an ELISA of 19 serum samples of patients receiving MAPKi therapy, and serum samples from healthy donors as controls. We could confirm significantly higher IGFBP7 levels in the blood of patients after relapse (Fig 5F). The patient data are summarized in Table 3.
Table 3.
Patient data including histological number, treatment, gender, age and PFS
| Histonumber | Treatment | Gender | Age | PFS | Staining |
|---|---|---|---|---|---|
| H 2012.13197 | Encorafenib | Male | 52 | 0 | |
| H 2012.21212 | |||||
| H 2012.22814 | |||||
| H 2011.23411 | Vemurafenib | Male | 33 | 0 | |
| H 2012.565 | |||||
| H 2012.3743 | Vemurafenib | Male | 44 | 1 | |
| H 2012.26647 | |||||
| H 2012.26648 | |||||
| H 2012.15227 | Encorafenib | Male | 37 | 1 | |
| H 2012.22589 | |||||
| H 2012.28351 | |||||
| H 2013.11851 | Vemurafenib | Male | 71 | 2 | |
| H 2014.3532 | Vemurafenib | Female | 48 | 3 | |
| H 2014.7225 | |||||
| H 2014.17199 | |||||
| H 2012.13943 | Vemurafenib | Male | 71 | 3 | |
| H 2012.15651 | |||||
| H 2012.21917‐19 | Vemurafenib | Male | 51 | 4 | |
| H 2014.9111 | Vemurafenib | Female | 53 | 4 | |
| H 2012.27219 | |||||
| HS2013.147 | |||||
| H 2012.7765 | Vemurafenib | Female | 73 | 4 | |
| H 2012.19800 | |||||
| H 2012.27486 | |||||
| H 2012.15676 | Vemurafenib | Female | 53 | 5 | |
| H 2013.13544 | |||||
| H 2012.6191 | Vemurafenib | Female | 61 | 5 | |
| H 2012.6826 | |||||
| H 2012.15122 | Vemurafenib | Female | 50 | 9 | |
| H 2013.13614 | |||||
| HS2014.34 | |||||
| H 2012.17690 | Vemurafenib | Male | 55 | 11 | |
| H 2013.18481 | |||||
| H 2010.25483 | Vemurafenib | Male | 59 | 21 | |
| H 2012.21406 | |||||
| H 2014.2708 | |||||
| H 2009.17175 | Vemurafenib | Male | 44 | Stable | |
| H 2009.21860 |
| Histonumber | Treatment | Gender | Age | PFS | ELISA |
|---|---|---|---|---|---|
| HG12.1108 | Encorafenib | Male | 37 | 1 | |
| HG14.189 | Vemurafenib | Male | 71 | 1 | |
| HG14.281 | |||||
| HG14.358 | |||||
| HG13.1620 | Vemurafenib | Female | 53 | 4 | |
| HG14.06 | |||||
| HG14.525 | |||||
| HG14.82 | |||||
| HG14.1020 | Encorafenib | Male | 34 | 4 | |
| HG14.620 | |||||
| HG14.1096 | Vemurafenib | Female | 53 | 5 | |
| HG14.1117 | |||||
| HG13.1399 | |||||
| HG13.1622 | Encorafenib | Male | 60 | 14 | |
| HG14.1131 | |||||
| HG14.23 | |||||
| HG15.06 | |||||
| HG14.1069 | Encorafenib | Female | 30 | Stable | |
| HG15.06 |
Discussion
In this study, we applied proteomic screens to differentiate MAPKi‐resistant and MAPKi‐sensitive melanoma cells in order to identify resistance mechanisms to MAPKi and novel biomarkers indicating resistance. We discovered a signature that reliably distinguishes resistant from sensitive cells. We assume that the discovered proteins reflect pathways closely related to the mechanisms of resistance. This signature was validated by transcriptome data, by functional analyses and by the evaluation of patient samples.
Pathways characteristic of MAPKi resistance
Previously, we identified in an acquired resistance model seven characteristics that were associated with vemurafenib resistance (Paulitschke et al, 2015). We were also able to confirm these data in the primary cells tested in this study. Therefore, different types of resistant mechanisms might be active due to the heterogeneity in melanoma.
Recently, it was shown that the inactivation of the immune system and the influence of the tumour microenvironment contribute to MAPKi resistance (Hugo et al, 2015). The observed upregulation of the V‐type ATPases leads to an acidic environment and to inactivation of T cells (Conboy et al, 1999; Sun‐Wada & Wada, 2013). These transporters might also be involved in drug efflux and inactivation and are part of the lysosomal compartment. The role of these V‐type ATPases maintaining multi‐drug resistance (MDR) by acidification of the ECM is nicely demonstrated by others (Daniel et al, 2013). Our data suggest a similar mechanism in the context of MAPKi resistance.
EIFs were shown to be involved in the different hallmarks of cancer (Hanahan & Weinberg, 2011; Spilka et al, 2013). Very recently, eIF4E was suggested to promote melanoma cell invasion and metastasis and as a possible prognostic marker (Khosravi et al, 2015) and the eIF4F complex was shown to be a nexus of resistance to anti‐BRAF and anti‐MEK cancer therapies (Boussemart et al, 2014). Here we confirm these data on the protein level using different BRAFi as well as BRAFi‐ and MEKi‐sensitive and BRAFi‐ and MEKi‐resistant melanoma primary cell cohorts and measuring at two different MS centres. We provide evidence that BRAFi and MAPKi resistance mechanisms provide comparable features.
Protein signature characteristic for MAPKi resistance
We identified a 15 protein signature to discriminate in two different cohorts of BRAF‐sensitive and BRAF‐resistant primary cells and another cohort of MAPKi sensitive and resistant cells.
One of the most strongly upregulated proteins associated with BRAFi resistance was polymerase I and transcript release factor (PTRF), also termed cavin‐1, which has been implicated in the endocytosis pathway caveolae formation, which contributes to an enhanced extracellular matrix remodelling, cell migration and cell adhesion and lysosomal function (Hill et al, 2008; Parton & del Pozo, 2013). The endolysosomal pathway has recently been shown by gene set enrichment analysis (GSEA) to be specifically prominent in melanoma compared to more than 35 other cancer types (Alonso‐Curbelo et al, 2014).
Validation of the resistance signature by transcriptomics and clinical validation
The 15 candidates were verified at the RNA level using two different cohorts of RNAseq data. We were also able to use the Verfaillie signature to discriminate between sensitive and resistant cells using the RNAseq data.
In a next step, we analysed the two proteome cohorts, which could be correlated with the Verfaillie signature. The master genes of the invasive/mesenchymal phenotype such as TGFβ or JUN are upregulated in the resistant cells, and in contrast to this, the genes associated with the proliferative phenotype such as CDH1, MLANA, MITF and SOX10 are downregulated in the resistant cells.
To address the question whether there is a difference in signature detection by proteomics and transcriptomics and to quantify cluster uncertainty, we performed bootstrap analysis of the 15 protein signature and the Verfaillie signature on the 1st and 2nd RNA and proteome cohort. This revealed that the proteome signature had the highest AU P‐value for the proteome signature and the RNAseq data for the Verfaillie signature. Therefore, different signatures can be obtained by transcriptomics or proteomics, but both technologies are able to verify the significant signatures regardless of the platform, suggesting the complementary power of both technologies. Proteomics led to a new link of resistance by identifying PTRF and IGFBP7 as the two most differentially expressed proteins with AU P‐value of 100%. Knockdown of PTRF demonstrated that PTRF regulates TGFβ. TGFβ has been described as one of the main players to induce EMT and drug resistance (Bordelon & Grichnik, 2015; Schlegel et al, 2015). IGFBP7, a TGFß target, is already known to be involved in tumour progression, cell migration, angiogenesis, decreased patient survival and EMT, and PCOLCE is involved in cell adhesion and extracellular matrix remodelling and ALDH1A3, which is already described to be involved in chemoresistance, seems to be downstream targets of PTRF (Rupp et al, 2015). PTRF could potentially indirectly regulate IGFBP7 through TGFβ which is in line with recent data, demonstrating that PTRF is required for the function of insulin‐like growth factor‐I receptor (Salani et al, 2010). Based on our data, this IGFBP7‐PTRF link seems to be important in BRAFi resistance, which might be regulated by the JUN pathway (Fig 3I). c‐Jun was shown to mediate phenotype switching and BRAFi resistance in melanoma by transcriptomics (Ramsdale et al, 2015). In label‐free quantitative proteomic analysis of lipid raft proteins in multi‐drug‐resistant MCF‐7 breast cancer cells, it could be demonstrated that among other proteins PTRF was upregulated and conferred drug resistance and an EMT cell phenotype (Yi et al, 2013).
PTRF together with caveolin‐1 (CAV‐1) is known to be involved in caveolae biogenesis and function. Caveolae are abundant in mammalian cells and reported to be involved in lipid regulation, endocytosis and insulin signalling by regulating the internalization of insulin‐like growth factor‐I receptor (IGF‐IR; Salani et al, 2010; Cheng & Nichols, 2016). Insulin was shown to lead to translocation of PTRF to the cytoplasm (Aboulaich et al, 2006), and cytoplasmic PTRF was shown to be required for caveolae formation (Hill et al, 2008). Knockout of PTRF led to the identification of novel targets of PTRF, 12 targets were significant, and at least twofold downregulated by knockout of PTRF in both clones. These proteins are involved in diverse functions such as immune regulatory processes (e.g. PRKCDBP, HLA‐DRA2; McMahon et al, 2009; Whitmarsh, 2013), cell adhesion (e.g. Tenascin, Glypican‐6; Shao et al, 2015), cell migration (e.g. Neuropilin 2, Tenascin, Glypican‐6; Shao et al, 2015), angiogenesis (e.g. Neuropilin 2), endosomal trafficking (e.g. EDH2, PRKCDBP), endocytosis (e.g. IGF2R, TFRC; Shi et al, 2015), pH regulation (e.g. IGF2R, TFRC; Shi et al, 2015) and lysosomal function (e.g. HLA‐DRA2, IGF2R) and IGF signalling (e.g. NGFR, IGF2R). Therefore, these processes have been partly described to be effects of PTRF or involved in the process of resistance as well in our analysed cell systems. In line with siRNA results, we identified a protein involved in TGFβ signalling (TGFB), which is known to be involved in cell adhesion, and EMT and tumour progression (uniprot) is significantly upregulated in BRAFi‐resistant cells. We correlated PTRF to an invasive phenotype and important features of resistance such as migration and invasion as demonstrated by CRISPR PTRF cells by the loss of sphere formation and invasive index.
Overexpression of PTRF led to MAPKi resistance and increased cell adhesion and sphere formation.
We validated the clinical relevance of PTRF and IGFBP7 on tumour biopsies, and most strikingly, we demonstrated that lower expression of PTRF before treatment initiation was correlated with longer PFS in melanoma patients receiving BRAFi therapy (Fig 4A–D).
Until now, few predictive biomarkers have been available for targeted therapy. PTRF has been associated with multi‐drug resistance in glioblastoma (Wang et al, 2014) and breast cancer (Yi et al, 2013). There is conflicting literature regarding the role of PTRF in cancer progression, which might be also due to the cellular localization of the protein, the caveolin‐dependent or caveolin‐independent activity, and the association with caveolin‐1 (Hill et al, 2012). Having predictive markers for MAPKi resistance is of high clinical relevance, since the decision for combinatorial targeted treatment versus immune treatment in BRAF‐mutated melanoma patients is a major clinical challenge. For instance, markers depicting intrinsic MAPKi resistance would help guide patient stratification towards immune therapy.
In addition to PTRF, we have identified the highly upregulated factor IGFBP7 in the secretome of resistant cells. IGFBP7 was shown to exhibit no influence in BRAFV600E senescence (Wajapeyee et al, 2010), but little is known about the role in resistance to BRAFi or to other targeted therapies in melanoma. Here we provide evidence that IGFBP7 can serve as a marker in patients receiving therapy with MAPKi, as there was a significant correlation with disease progression in the cohort tested (Fig 4E). Two of the most discriminating proteins are involved in the kynurenine pathway (NAMPT and KYNU). Nicotinamide phosphoribosyltransferase (NAMPT) was shown to be overexpressed in melanoma lesions (Maldi et al, 2013), decreased serum tryptophan concentrations were demonstrated to predict a poor prognosis in melanoma patients (Weinlich et al, 2007), and the inhibition of NAMPT was described to serve as a therapeutic target in cancer therapy (Sampath et al, 2015).
Conclusion
Predictive markers which help to stratify melanoma patients into either targeted or immune therapy are urgently needed in the clinic. Here, a proteomic screen identified two markers (PTRF and IGFBP7) that could be validated on clinical samples and that could play a role in resistance to MAPKi.
Materials and Methods
Cell lines and chemicals
Surplus tumour material was obtained after surgical removal of melanoma metastases from patients after written informed consent approved by the local institutional research board (EK647 and EK800). The experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report. Clinical diagnosis of the tumour material was confirmed by histology and immunohistochemistry. Primary melanoma cells were established from patient biopsies using the selective adherence method (Raaijmakers et al, 2015) and included in the URPP biobank, University Hospital Zürich, Department of Dermatology. Cells were grown in RPMI 1640 (Sigma Life Science, USA) supplemented with 10% foetal bovine serum (Gibco, Life Technologies, USA), 2 mM glutamine (Biochrom, Germany) and sodium pyruvate (Sigma Life Science, USA). The BRAFi Vemurafenib (PLX4032), Encorafenib (LGX818) and MEKi Binimetinib (MEK162) were purchased at Selleckchem, USA. The ERK1/2 inhibitor SCH772984 was purchased at Chemical probes. All used primary cells, and the IC50 are listed in Table 1.
Cell viability assay and IC50 determination
Melanoma cells were seeded to a density of 2.5 × 103 cells in each well of a 96‐well plate and challenged with dose‐escalating concentrations of BRAFi for 72 h. Cell viability was estimated using a colorimetric (MTT) assay. Briefly, on the day of the assay the medium was changed to medium containing 10% MTT‐stock solution (stock solution 10 mg/ml MTT in PBS, Sigma‐Aldrich, Switzerland). Plates were incubated at 37°C for 2–4 h depending on the cell line, medium was removed, and formazan crystals were solubilized in 100 μl dimethyl sulfoxide (Sigma‐Aldrich, Switzerland). Absorbance was measured at 595 nm (reference 620 nm) using a microplate reader (Tecan infinite M200 Pro). The half‐maximal inhibitory concentration (IC50 value) was calculated using GraphPad Prism software (USA).
Matrigel invasion chamber assay
Melanoma cells (S1 and R1) were pre‐starved with RPMI containing reduced FCS concentration (3%) for 48 h. On the day of experiment, 2 × 104 melanoma‐associated fibroblast were seeded in 800 μl in 24‐well plates and let to adhere. Empty and Matrigel‐coated invasion inserts (8 μm pore size, BD Biosciences, Bedford, MA) were re‐hydrated in RPMI without FCS for 2 h. Pre‐starved melanoma cells were trypsinized and collected into RPMI without FCS. Cell titre was adjusted to 1 × 105 cells/ml. Empty and Matrigel‐coated, re‐hydrated inserts were put into the wells of a 24‐well plate with and without fibroblast seeded on the bottom. 500 μl of cell suspension was seeded on top of the inserts. We used 800 μl RPMI containing 10% FCS as a chemoattractant. After 16 h, cells on the top of the insert were removed by cotton swap. Membranes were cut out and mounted on microscopy slides in mounting medium Prolong Gold anti‐fade reagent containing DAPI staining (Promega). Slides were imaged using a Leica CTR 6000 fluorescent microscope. A total of eight face fields were imaged per insert. All experiments were carried out in duplicates, and two sets of experiments were performed on two different days. Images were analysed with ImageJ. The invasion index was calculated by the per cent of cells invading through Matrigel in relation to motile cells moving through the membrane.
CytoSelect™ 48‐well cell adhesion assay (ECM Array, Colorimetric Format)
The ECM Array was performed as described previously (Paulitschke et al, 2013). Briefly, melanoma cells were seeded in a density of 1 × 105 cells/well in ECM proteins pre‐coated 24‐well plates and incubated for 1 h at 37°C. Unbound cells were washed away, and the adherent cells were fixed in 70% ethanol and stained with 0.2% crystal blue staining solution and quantified colorimetrically.
Spheroid formation assay and invasion assay
In order to create non‐adhesive conditions, a 96‐well plate was coated with 100 μl of 1.5% Agarose dissolved in RPMI containing no supplements or FCS and kept under UV light in the laminar flow until agarose polymerized. 5,000 cells per well were seeded on top of the agarose solution in 100 μl melanoma cell culture medium. Spheroid formation was followed over several days using a Zeiss Primovert light microscope.
Spheroids were also embedded in Collagen 1 (from rat tails, BD, USA). Here, spheres were harvested and kept on ice. Residual cell culture medium was removed, and sphere was mixed with 100 μl Collagen 1 solution containing 10% FCS, 10% DMEM, 4 mM L‐glutamine and 0.4% sodium bicarbonate and transferred into a 96‐well plate coated with 1.5% Agarose. The collagen solution was left to polymerize in the cell culture incubator for 1 h. Collagen was topped up to 100 μl of melanoma medium. After 5 days, Calcein‐Green 1:50 (stock 4 nM, Sigma‐Aldrich, USA) and Ethidium homodimer 1:50 (stock 2 nM, Sigma‐Aldrich, USA) were added to each well containing the spheroids and incubated for 1–2 h at 37°C. Pictures of spheroids were taken with a fluorescence microscope Leica DMi8.
Immunohistochemical staining of PTRF, IGFBP7, MHC I, CD3 and CD99
Blocks of paraffin‐embedded, formalin‐fixed tissues corresponding to melanoma patients who received targeted therapy were used for immunohistochemical analysis, which was performed as described previously (Belloni et al, 2012). The following primary antibodies were used: rabbit polyclonal Anti‐PTRF antibody (ab48824, Abcam; 1:200), monoclonal mouse Anti‐CD3 (DAKO M7254; 1:50), mouse monoclonal anti‐CD99 (ab17083, Abcam; 1:10) and rabbit monoclonal anti‐MHC class I antibody (ABIN650045). Blinded evaluation was performed independently by two trained dermatologists (EG and VP). Kaplan–Meier estimates were used to describe the survival experiences of patients with low (< 3) compared to high (> 3) expression of the PTRF protein on melanoma cells, 1 was ranked for < 50% staining, 2 for up to 75%, 3 for up to 100% and 4 for positive nuclear staining. Images were performed with Aperio ImageScope Slide Scanner.
Western blots
Cells were frozen in liquid nitrogen and lysed with lysis buffer containing phosphatase and protease inhibitors as described previously (Hoeller et al, 2005; Paulitschke et al, 2012). Membranes were incubated with the following primary antibodies: rabbit polyclonal anti‐PTRF antibody (ab48824).
Zymography assay
The zymography assay was performed as described previously (Paulitschke et al, 2012). The supernatants of the primary cells were collected. The SDS gel contained gelatine (1 mg/ml) was stained in Coomassie solution for 30 min and stripped with an isopropanol–acetic acid solution (BioTeZ Berlin‐Buch GmbH, Berlin, Germany).
Proteome analysis
Proteome analysis was performed as described previously (Paulitschke et al, 2012, 2013).
Subcellular fractionation and sample preparation
All fractionation steps were performed on ice. To obtain the cytoplasmic fraction, the primary melanoma cells were lysed in isotonic lysis buffer (10 mM HEPES/NaOH, pH 7.4, 0.25 M sucrose, 10 mM NaCl, 3 mM MgCl2, 0.5% Triton X‐100) supplemented with protease inhibitors (pepstatin, leupeptin and aprotinin, each at 1 μg/ml; 1 mM PMSF) and mechanical shear stress. In addition, we performed nuclear fractionation. By centrifugation at 2,300 × g at 4°C, the cytoplasmic proteins were separated from the nuclei and precipitated overnight with ice‐cold ethanol at −20°C. In addition, cell pellets were used for the second cohort. Supernatant was collected after 6‐h incubation in serum‐free medium and sterile filtered through a 0.2‐μm filter.
After precipitation, all protein samples were dissolved in sample buffer (7.5 M urea, 1.5 M thiourea, 4% CHAPS, 0.05% SDS, 100 mM DDT) and protein concentrations were determined by means of a Bradford assay (Bio‐Rad‐Laboratories, Germany).
For proteomic analyses, in‐solution digests were prepared, as previously described (Bileck et al, 2014; Slany et al, 2016). Briefly, 20 μg of each protein sample was concentrated onto a 3kD MWCO filter (Pall Austria Filter GmbH) pre‐washed with LC‐MS grade water (Millipore GesmbH); proteins were reduced with DTT and alkylated with iodoacetamide (IAA). After centrifugation at 14,000 g for 10 min, proteins were washed with 50 mM ammonium bicarbonate buffer. Trypsin (Roche Diagnostics, Germany) was then added, and incubation was performed at 37°C overnight. After tryptic digestion, peptide samples were cleaned up with C‐18 spin columns (Pierce, Thermo Fisher Scientific) and eluted two times with 50% acetonitrile (ACN), 0.1% trifluoroacetic acid (TFA) and once with 80% ACN, 0.1% TFA. The peptides were dried and stored at −20°C until LC‐MS analysis.
Liquid chromatography/tandem mass spectrometry analysis
QExactive
Peptides of the cellular fractions were separated using nanoflow UHPLC (Dionex Ultimate 3000RLSC) using a 2 cm × 75 μm C18 PepMap100 pre‐column for pre‐concentration of peptides using mobile phase A (98% H2O, 2% ACN, 0.2% FA) at a flow rate of 10 μl/min and a 50 cm × 75 μm Pepmap100 analytical column (Thermo Fisher Scientific, Austria). Peptide separation was achieved applying a multi‐step gradient of 8–40% mobile phase B (80% ACN, 20% H2O, 0.1% FA) over 235 min at a flow rate of 300 nl/min. Peptides were analysed in positive ionization mode at a resolution of 70,000 (at m/z = 200) in the range from m/z 400 to 1,400 and fragmented using HCD in a QExactive MS (Thermo) in a data‐dependent mode upon fragmentation at 30% normalized collision energy (isolation width of Δm/z = 2, 150‐ms collection time, resolution 17,500 at m/z = 200, top 10 method). All samples were analysed by LC‐MS/MS as duplicates.
Orbitrap elite
Peptides of the cellular fractions were separated using nanoflow UHPLC (Thermo Easy‐nLC 1000) using a 15 cm × 75 μm PepMap RSLC C18 analytical column (2 μm particle size, Thermo Fisher Scientific). Peptide separation was achieved applying a linear gradient of 5–30% mobile phase B (98% ACN, 2% H2O, 0.15% FA; mobile phase A contained 98% H2O, 2% ACN, 0.15% FA) over 180 min at a flow rate of 300 nl/min. Peptides were analysed in positive ionization mode using ion trap CID in an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) in data‐dependent acquisition mode. MS1 resolution was set to 120,000 (at m/z = 400), and the 15 most abundant precursors (m/z 350–1,600, charge state + 2 and above) were selected per cycle. Fragmentation was performed with an isolation width of 2 m/z and a normalized collision energy of 35%. Fragment ions were detected in the linear ion trap in normal resolution mode. All samples were analysed by LC‐MS/MS as duplicates in randomized order.
Data processing
Carbamidomethylation of cysteines was set as fixed modification, whereas methionine oxidation as well as N‐terminal protein acetylation was set as variable modifications.
As described in Bileck et al (2014), label‐free quantitative (LFQ) data analysis was supported by MaxQuant 1.3.0.5 including the Andromeda search engine and the integrated statistical analysis package Perseus (Cox & Mann, 2008, 2012; Tyanova et al, 2016). Protein identification was done by searching against the SwissProt Database (version 012013 with 20,264 entries) allowing a mass tolerance of 5 ppm for MS spectra and 20 ppm for MS/MS spectra. Furthermore, search criteria included a maximum of two missed cleavages, a minimum of two peptide identifications per protein, at least one of them unique and an FDR less than 0.01 for both peptide identification and protein identification. Again carbamidomethylation of cysteine residues was set as a fixed modification and oxidation of methionine residues and N‐terminal protein acetylation as variable modifications.
Perseus software and hierarchical clustering
Protein identifications were further analysed using Perseus (version 1.3.0.4). Therefore, proteins were filtered for reversed sequences and contaminants as well as a minimum of three independent identifications per protein. Annotation enrichment analysis was performed based on gene ontology cellular compartment, molecular function and biological process terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways according to Geiger et al (2010).
GSEA (gene set enrichment analysis) and KEGG pathway visualization
Expression of proteins was analysed for significantly enriched protein sets using gene set enrichment analysis (GSEA).
Shrunken Centroids for calculation of key players in resistance and Bootstrap analysis
To identify a potential biomarker profile, a classifier based on shrunken centroids was constructed using ClaNC and R (Dabney, 2006). Priors were chosen according to the number of samples. The performance of the classifier was tested by leaving‐one‐out cross validation.
Cluster uncertainty was estimated using pvclust, which is an implementation of multiscale bootstrap resampling for assessing the uncertainty in hierarchical cluster analysis.
Construction of heatmaps
Heatmaps were constructed using standard R procedures, hierarchical clustering and the Pearson correlation coefficient with average linkage as distance metrics. Different sizes of heatmaps were performed.
siRNA knockdown
Silencing RNA (siRNA) transfection of melanoma cells was carried out using INTERFERin transfection solution according to the manufacturer's protocol (Polyplus‐transfection, France). Cells were transfected with 20 nM of siRNA (Qiagen) for 72 h before RNA or protein was extracted. For the knockdown of PTRF, mRNA with the following anti‐sense sequence was used: 5′GGAAAGAUUGAAUCCUAAAdTdT3′. Non‐human transcript targeting anti‐sense RNA was used as a negative control (AllStars negative control, Qiagen, The Netherlands).
CRISPR
CRISPRed cell cultures of MM121224 were generated using the MuLE (Multiple Lentiviral Expression) System according to Albers et al (2015).
sgRNAs used in this study were designed according to the CRISPR Design Tool (https://chopchop.rc.fas.harvard.edu/) as standard primers (Microsynth). The PAM sequence depicted in blue was not included in the cloned sgRNA. A 5′ ACCG was added to the 5′ end of the forward direction oligo and a 5′ AAAC to the 5′ end of the reverse complement oligo. Oligos were first annealed to generate double‐stranded DNA fragments and then ligated into the BfuAI‐digested Entry vector (pMuLE ENTR U6 stuffer sgRNA scaffold L1‐R5 Plasmid #62127, Addgene). The sequence of the resulting Entry vectors was verified by Sanger sequencing.
Ptrf Exon1/Target sequence 5′‐sgRNA‐PAM‐3′: GCTCGACAATATAGAGCGTGGGG
The Entry vector (Plasmid #62127, Addgene) containing the gRNA, the Entry vector containing Cas9 (pMuLE ENTR SV40‐hCas9 L5‐L2 Plasmid #62134, Addgene) and a lentiviral destination vector (expressing EGFP) were recombined using the Gateway LR Clonase II Plus Enzyme mix (Life Technologies #12538‐120).
Lentivirus production, cell transduction and generation of CRISPR‐Clones
Lentivirus was prepared as described recently (Albers et al, 2015). HEK293T cell supernatant was centrifuged at 1,200 g for 5 min and then applied on subconfluent melanoma cells.
Cells positive for GFP were sorted by FACS using an ARIA3 device. Bulk culture was amplified followed by single‐cell sorting also using the ARIA3 device. Cell clones were tested for the absence of the target protein by Western blot and in case of PTRF were further analysed by proteomics.
Cloning of PTRF expression construct, generation of lentivirus particles and transduction of melanoma cells
PTRF lentiviral expressing constructs were generated using the multiple lentiviral expression system (MuLE) as described by Albers et al (2015) (Albers and Ian Frew, University Zürich). Shortly, PTRF was cloned from a commercially available vector (Origene SC101318) into either the ENTRY vector pMuLE ENTR CMV L5‐L2 (Addgene 62091) or as control into the ENTRY vector pMuLE ENTR MCS L1‐R5 (Addgene 61084). After MultiSite gateway recombination using the two above entry vectors (PTRF cloned in one or the other, respectively) and a GFP destination vector (Addgene 62175), the following constructs were received: CMV_PTRF_eGFP and noCMV_PTRF_CMV_MCS_eGFP (denoted shortly as noCMV_PTRF). GFP‐positive cells were sorted with an Aria III 4L cell sorter.
Lentivirus particles were prepared via transfection of subconfluent HEK293T cells cultured in DMEM with 10% FCS. For a 10‐cm dish, 8 μg of total vector DNA containing CMV_PTRF_eGFP or noCMV_PTRF_eGFP vector and the lentiviral packaging plasmids psPAX2 (Addgene, no 12260) and pMD2.G (Addgene, no 12259) were mixed in a ratio 4:2:1 in 1 ml of serum‐free DMEM. 24 μl PEI (1 mg/ml) was added and incubated for 15 min at RT and mixed with 9 ml DMEM with 10% FCS before mixture was added to HEK cells. Supernatant containing lentivirus particles were harvested twice in 96 h, spin at 1,500 g for 10 min, and melanoma cells were incubated with supernatant for 72 h before sorted for GFP expression with an Aria III 4L cell sorter.
RNAseq
To perform RNAseq, one microgram of total RNA from melanoma cells was processed by the standard Illumina RNAseq Stranded mRNA protocol and sequenced on the Illumina HiSeq2500 paired‐end 125 bp. RNAseq data were aligned using the R implementation of subread with Hg19 as a reference genome (Liao et al, 2013). Mapped reads were summarized on the gene level.
Immunofluorescence imaging, SEM (Scanning electron microscopy) and pH staining
Cells were seeded to a density of 5 × 104 to 8 × 104 cells per well into a four‐well chamber slide (BD, USA) and incubated at 37°C and 5% CO2 overnight. Cells were washed in PBS and fixed in acetone at −20°C for 5–10 min. After two additional washes, unspecific binding was blocked with sterile‐filtered blocking buffer as described recently (Eichhoff et al, 2010; PCMR). Primary antibodies (PTRF (1:100; ab48824, Abcam) and caveolin‐1 (1:500; ab17052, Abcam) were diluted in blocking buffer and incubated for 1 h at room temperature. After three additional washing steps with PBS, the cells were incubated 30 min with blocking buffer containing secondary antibodies diluted 1:2,000 [anti‐mouse Alexa Fluor‐568, anti‐mouse Alexa Fluor‐488 and anti‐rabbit Alexa Fluor‐488 (Invitrogen)]. Cells were washed extensively for at least another three times with PBS, chambers were removed from glass slides, and cells were mounted into Prolong Gold anti‐fade reagent containing DAPI staining (Promega). Slides were imaged using a Leica DiM 6 fluorescent microscope or a Leica Sp8 confocal microscope.
SEM with S1, S2, R1‐R4 was performed as described previously (Bray et al, 1993; Paulitschke et al, 2015). The SEM images were taken at a magnification between 500 and 3,000 using a Leo DSM 982 field emission scanning electron microscope at 4 kV. For pH‐sensitive staining, cells were seeded as above, stained according to manufacturer's instruction (pHrodo® dye, Molecular Probes®) and imaged using identical exposure and acquisition conditions. Relative fluorescence of single cells in each image was quantified using ImageJ software.
Statistical analysis
Patient data
The log‐rank test was used to test for the equal distribution of survival times between the two groups as described previously (Kleffel et al, 2015). The corresponding hazard ratio was estimated with the use of a Cox proportional hazards model. A P‐value of less than 0.05 was considered to indicate statistical significance. All analyses were conducted with the use of the R programming language and Origin Pro 9.1G Software.
Proteome data
Data were analysed using Proteome Discoverer 1.4 equipped with Mascot 2.2 searching against the UniProt database for human proteins (version 102014 with 20,195 entries, restricted to reviewed entries only). Search criteria included a minimum of two peptide identifications per protein, at least one of them unique as well as a maximum of 5 ppm initial mass deviation for precursor ion spectra and 20 ppm for MS/MS spectra and only peptides with a false discovery rate (FDR) less than 0.01 (validation based on q‐value). Regarding the LFQ‐values, as these are Log2 values, the difference directly corresponds to logarithmic fold‐control values. Changes in protein abundance values between sensitive and resistant cells were determined by a two‐sided t‐test with P < 0.05 and a minimum of a twofold abundance difference. In addition, to emphasize the most robust regulatory effects observed within one kind of cell, we determined significantly regulated proteins with a global FDR < 0.05 as determined by a permutation‐based method, referring to Cox et al (2014) and Tusher et al (2001). Significantly up‐ and downregulated proteins were determined by applying a two‐sided t‐test with a significance level of P < 0.05 (permutation‐based correction). In addition, hierarchical clustering was achieved using Euclidean distance and average linkage clustering of Z‐scored expression values. UniProt accession numbers were mapped to gene symbols, and GSEA was carried out on KEGG, BIOCARTA and REACTOME pathways available at the C2 curated dataset available at the molecular signature database as well as the Gene Ontology Biological Process using the GSEA software with the parameters set to ranking according to log fold change, minimum gene set size 15, maximum gene set size 500 (Subramanian et al, 2005). An FDR of equal or below 0.25 was considered to be significant. KEGG pathways were visualized using the Pathview package of R (Luo & Brouwer, 2013).
It provides AU (approximately unbiased) P‐value as well as BP (bootstrap probability) value for each cluster in a dendrogram. AU P‐value, which is computed by multiscale bootstrap resampling, is a better approximation to unbiased P‐value than BP value computed by normal bootstrap resampling, and therefore, this value is used. http://www.sigmath.es.osaka-u.ac.jp/shimo-lab/prog/pvclust/
RNAseq data
Genes with less than 5 reads over all samples were filtered previous to analysis. Differential expression was estimated using DEGSeq2 (Love et al, 2014). For visualization in heatmaps, the z‐score of the moderated logarithm of normalized counts was used.
Author contributions
Conceptualization: VP, RD, MPL. Methodology: VP, CG, PP, OE, EG, TM, AL, RA, MPL. Investigation: VP, PP, MPL, OE, PFC, CG, SNF, EG, IS, TM, AB, AL, RA, T‐PC, AI, NZ, JS, JM, BK. Writing Original Draft: VP. Writing Review and Editing: MPL, OE, PFC, RD, RK, CG, HP, AI, NZ, TM, SNF, EG, RA. Funding Acquisition: VP, RD, MPL, EG, RK, HP, CG. Resources: VP, RD, MPL, RA, RK, HP. Supervision: MPL, RD.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
Elisabeth Hofstaetter, Alice Langer, Melanie Maudrich, Daniel Hug, Ines Kleiber‐Schaaf, Mirka Schmid and Tabea Koch are acknowledged for technical assistance and biobank work. Luxi Zhang and Jan Käsler are acknowledged for technical support. We also thank the University Research Priority Program (URPP) in translational cancer research for the primary cell cultures. This study was supported by the following grants: Promedica (R.A., V.P.), MUW mobility grant (V.P.), Comprehensive Cancer Center Forschungsförderung der Initiative Krebsforschung, MedUni Wien (V.P.), Forschungskredit of the University of Zurich (FK‐14‐032 to E.G.), URPP “Translational Cancer Research” grant (R.D., V.P.), Louis Widmer grant (V.P.), Bürgermeistergrant (V.P.), Verein Hautkrebsforschung, Swiss National Science Foundation (PMPDP3_151326 to E.G. and SNSF; 31003A_166435 to R.A.), Verein für Hautkrebsforschung (M.L.), the Hochspezialisierte Medizin Schwerpunkt Immunologie (HSM‐2‐Immunologie) Schweiz and Krebsliga Schweiz (KLS 3151‐02‐2013), and Prof. Bruno Bloch foundation (L.F.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. No additional external funding was received for this study. Data are patented (EP17192493).
The EMBO Journal (2019) 38: e95874
Data availability
Proteome data are available via Proteome exchange with identifiers PXD005432, PXD005433. RNAseq data are available via https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128915.
References
- Aboulaich N, Ortegren U, Vener AV, Stralfors P (2006) Association and insulin regulated translocation of hormone‐sensitive lipase with PTRF. Biochem Biophys Res Comm 350: 657–661 [DOI] [PubMed] [Google Scholar]
- Albers J, Danzer C, Rechsteiner M, Lehmann H, Brandt LP, Hejhal T, Catalano A, Busenhart P, Goncalves AF, Brandt S et al (2015) A versatile modular vector system for rapid combinatorial mammalian genetics. J Clin Invest 125: 1603–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso‐Curbelo D, Riveiro‐Falkenbach E, Perez‐Guijarro E, Cifdaloz M, Karras P, Osterloh L, Megias D, Canon E, Calvo TG, Olmeda D et al (2014) RAB7 controls melanoma progression by exploiting a lineage‐specific wiring of the endolysosomal pathway. Cancer Cell 26: 61–76 [DOI] [PubMed] [Google Scholar]
- Bae GY, Choi SJ, Lee JS, Jo J, Lee J, Kim J, Cha HJ (2013) Loss of E‐cadherin activates EGFR‐MEK/ERK signaling, which promotes invasion via the ZEB1/MMP2 axis in non‐small cell lung cancer. Oncotarget 4: 2512–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloni B, Schonewolf N, Rozati S, Goldinger SM, Dummer R (2012) Cutaneous drug eruptions associated with the use of new oncological drugs. Chem Immunol Allergy 97: 191–202 [DOI] [PubMed] [Google Scholar]
- Bileck A, Kreutz D, Muqaku B, Slany A, Gerner C (2014) Comprehensive assessment of proteins regulated by dexamethasone reveals novel effects in primary human peripheral blood mononuclear cells. J Proteome Res 13: 5989–6000 [DOI] [PubMed] [Google Scholar]
- Bordelon JR, Grichnik JM (2015) TGF‐beta may control the switch between tumorigenic growth and “stem cell/mesenchymal” potentially drug‐resistant states. Dermatol Ther 28: 177–178 [DOI] [PubMed] [Google Scholar]
- Boussemart L, Malka‐Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, Thomas M, Basmadjian C, Ribeiro N, Thuaud F et al (2014) eIF4F is a nexus of resistance to anti‐BRAF and anti‐MEK cancer therapies. Nature 513: 105–109 [DOI] [PubMed] [Google Scholar]
- Bray DF, Bagu J, Koegler P (1993) Comparison of hexamethyldisilazane (HMDS), Peldri II, and critical‐point drying methods for scanning electron microscopy of biological specimens. Microsc Res Tech 26: 489–495 [DOI] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364: 2507–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JP, Nichols BJ (2016) Caveolae: one function or many? Trends Cell Biol 26: 177–189 [DOI] [PubMed] [Google Scholar]
- Conboy IM, Manoli D, Mhaiskar V, Jones PP (1999) Calcineurin and vacuolar‐type H+‐ATPase modulate macrophage effector functions. Proc Natl Acad Sci USA 96: 6324–6329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M (2012) 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high‐throughput data. BMC Bioinformatics 13(Suppl 16): S12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney AR (2006) ClaNC: point‐and‐click software for classifying microarrays to nearest centroids. Bioinformatics 22: 122–123 [DOI] [PubMed] [Google Scholar]
- Daniel C, Bell C, Burton C, Harguindey S, Reshkin SJ, Rauch C (2013) The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochem Biophys Acta 1832: 606–617 [DOI] [PubMed] [Google Scholar]
- D'Souza RC, Knittle AM, Nagaraj N, van Dinther M, Choudhary C, ten Dijke P, Mann M, Sharma K (2014) Time‐resolved dissection of early phosphoproteome and ensuing proteome changes in response to TGF‐beta. Sci Signal 7: rs5 [DOI] [PubMed] [Google Scholar]
- Dummer R, Flaherty KT (2012) Resistance patterns with tyrosine kinase inhibitors in melanoma: new insights. Curr Opin Oncol 24: 150–154 [DOI] [PubMed] [Google Scholar]
- Eichhoff OM, Zipser MC, Xu M, Weeraratna AT, Mihic D, Dummer R, Hoek KS (2010) The immunohistochemistry of invasive and proliferative phenotype switching in melanoma: a case report. Melanoma Res 20: 349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleuren ED, Zhang L, Wu J, Daly RJ (2016) The kinome ‘at large’ in cancer. Nat Rev Cancer 16: 83–98 [DOI] [PubMed] [Google Scholar]
- Geiger T, Cox J, Mann M (2010) Proteomic changes resulting from gene copy number variations in cancer cells. PLoS Genet 6: e1001090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E et al (2012) Dabrafenib in BRAF‐mutated metastatic melanoma: a multicentre, open‐label, phase 3 randomised controlled trial. Lancet 380: 358–365 [DOI] [PubMed] [Google Scholar]
- Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, Walser P, Abankwa D, Oorschot VM, Martin S et al (2008) PTRF‐Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 132: 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MM, Daud NH, Aung CS, Loo D, Martin S, Murphy S, Black DM, Barry R, Simpson F, Liu L et al (2012) Co‐regulation of cell polarization and migration by caveolar proteins PTRF/Cavin‐1 and caveolin‐1. PLoS One 7: e43041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KS, Eichhoff OM, Schlegel NC, Dobbeling U, Kobert N, Schaerer L, Hemmi S, Dummer R (2008) In vivo switching of human melanoma cells between proliferative and invasive states. Can Res 68: 650–656 [DOI] [PubMed] [Google Scholar]
- Hoeller C, Thallinger C, Pratscher B, Bister MD, Schicher N, Loewe R, Heere‐Ress E, Roka F, Sexl V, Pehamberger H (2005) The non‐receptor‐associated tyrosine kinase Syk is a regulator of metastatic behavior in human melanoma cells. J Invest Dermatol 124: 1293–1299 [DOI] [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB et al (2015) Non‐genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 162: 1271–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu‐Lieskovan S, Berent‐Maoz B, Pang J, Chmielowski B, Cherry G et al (2016) Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 165: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi S, Tam KJ, Ardekani GS, Martinka M, McElwee KJ, Ong CJ (2015) eIF4E is an adverse prognostic marker of melanoma patient survival by increasing melanoma cell invasion. J Invest Dermatol 135: 1358–1367 [DOI] [PubMed] [Google Scholar]
- Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q et al (2015) Melanoma cell‐intrinsic PD‐1 receptor functions promote tumor growth. Cell 162: 1242–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koomen JM, Smalley KS (2011) Using quantitative proteomic analysis to understand genotype specific intrinsic drug resistance in melanoma. Oncotarget 2: 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2013) The Subread aligner: fast, accurate and scalable read mapping by seed‐and‐vote. Nucleic Acids Res 41: e108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ et al (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371: 1877–1888 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Brouwer C (2013) Pathview: an R/Bioconductor package for pathway‐based data integration and visualization. Bioinformatics 29: 1830–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldi E, Travelli C, Caldarelli A, Agazzone N, Cintura S, Galli U, Scatolini M, Ostano P, Miglino B, Chiorino G et al (2013) Nicotinamide phosphoribosyltransferase (NAMPT) is over‐expressed in melanoma lesions. Pigment Cell Melanoma Res 26: 144–146 [DOI] [PubMed] [Google Scholar]
- McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA et al (2014) Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation‐positive melanoma (BRIM‐3): extended follow‐up of a phase 3, randomised, open‐label study. Lancet Oncol 15: 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon KA, Zajicek H, Li WP, Peyton MJ, Minna JD, Hernandez VJ, Luby‐Phelps K, Anderson RG (2009) SRBC/cavin‐3 is a caveolin adapter protein that regulates caveolae function. EMBO J 28: 1001–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, Koya RC, Samatar AA, Khanlou N, Braun J et al (2015) Tunable‐combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 27: 240–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Clifton‐Bligh R, Molloy MP (2014) Phosphoproteomics of MAPK inhibition in BRAF‐mutated cells and a role for the lethal synergism of dual BRAF and CK2 inhibition. Mol Cancer Ther 13: 1894–1906 [DOI] [PubMed] [Google Scholar]
- Parker R, Vella LJ, Xavier D, Amirkhani A, Parker J, Cebon J, Molloy MP (2015) Phosphoproteomic analysis of cell‐based resistance to BRAF inhibitor therapy in melanoma. Front Oncol 5: 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, del Pozo MA (2013) Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol 14: 98–112 [DOI] [PubMed] [Google Scholar]
- Paulitschke V, Gruber S, Hofstatter E, Haudek‐Prinz V, Klepeisz P, Schicher N, Jonak C, Petzelbauer P, Pehamberger H, Gerner C et al (2012) Proteome analysis identified the PPARgamma ligand 15d‐PGJ2 as a novel drug inhibiting melanoma progression and interfering with tumor‐stroma interaction. PLoS One 7: e46103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulitschke V, Haudek‐Prinz V, Griss J, Berger W, Mohr T, Pehamberger H, Kunstfeld R, Gerner C (2013) Functional classification of cellular proteome profiles support the identification of drug resistance signatures in melanoma cells. J Proteome Res 12: 3264–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulitschke V, Berger W, Paulitschke P, Hofstatter E, Knapp B, Dingelmaier‐Hovorka R, Fodinger D, Jager W, Szekeres T, Meshcheryakova A et al (2015) Vemurafenib resistance signature by proteome analysis offers new strategies and rational therapeutic concepts. Mol Cancer Ther 14: 757–768 [DOI] [PubMed] [Google Scholar]
- Paulitschke V, Eichhoff O, Cheng PF, Levesque MP, Holler C (2016) Proteomics approaches to understanding mitogen‐activated protein kinase inhibitor resistance in melanoma. Curr Opin Oncol 28: 172–179 [DOI] [PubMed] [Google Scholar]
- Raaijmakers MI, Widmer DS, Maudrich M, Koch T, Langer A, Flace A, Schnyder C, Dummer R, Levesque MP (2015) A new live‐cell biobank workflow efficiently recovers heterogeneous melanoma cells from native biopsies. Exp Dermatol 24: 377–380 [DOI] [PubMed] [Google Scholar]
- Raaijmakers MI, Widmer DS, Narechania A, Eichhoff O, Freiberger SN, Wenzina J, Cheng PF, Mihic‐Probst D, Desalle R, Dummer R et al (2016) Co‐existence of BRAF and NRAS driver mutations in the same melanoma cells results in heterogeneity of targeted therapy resistance. Oncotarget 7: 77163–77174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsdale R, Jorissen RN, Li FZ, Al‐Obaidi S, Ward T, Sheppard KE, Bukczynska PE, Young RJ, Boyle SE, Shackleton M et al (2015) The transcription cofactor c‐JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal 8: ra82 [DOI] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L et al (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372: 30–39 [DOI] [PubMed] [Google Scholar]
- Rupp C, Scherzer M, Rudisch A, Unger C, Haslinger C, Schweifer N, Artaker M, Nivarthi H, Moriggl R, Hengstschlager M et al (2015) IGFBP7, a novel tumor stroma marker, with growth‐promoting effects in colon cancer through a paracrine tumor‐stroma interaction. Oncogene 34: 815–825 [DOI] [PubMed] [Google Scholar]
- Salani B, Passalacqua M, Maffioli S, Briatore L, Hamoudane M, Contini P, Cordera R, Maggi D (2010) IGF‐IR internalizes with Caveolin‐1 and PTRF/Cavin in HaCat cells. PLoS One 5: e14157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath D, Zabka TS, Misner DL, O'Brien T, Dragovich PS (2015) Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol Ther 151: 16–31 [DOI] [PubMed] [Google Scholar]
- Schlegel NC, von Planta A, Widmer DS, Dummer R, Christofori G (2015) PI3K signalling is required for a TGFbeta‐induced epithelial‐mesenchymal‐like transition (EMT‐like) in human melanoma cells. Exp Dermatol 24: 22–28 [DOI] [PubMed] [Google Scholar]
- Shao H, Kirkwood JM, Wells A (2015) Tenascin‐C signaling in melanoma. Cell Adh Migr 9: 125–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Tan SH, Ng S, Zhou J, Yang ND, Koo GB, McMahon KA, Parton RG, Hill MM, Del Pozo MA et al (2015) Critical role of CAV1/caveolin‐1 in cell stress responses in human breast cancer cells via modulation of lysosomal function and autophagy. Autophagy 11: 769–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slany A, Bileck A, Kreutz D, Mayer RL, Muqaku B, Gerner C (2016) Contribution of human fibroblasts and endothelial cells to the Hallmarks of Inflammation as determined by proteome profiling. Mol Cell Proteomics 15: 1982–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilka R, Ernst C, Mehta AK, Haybaeck J (2013) Eukaryotic translation initiation factors in cancer development and progression. Cancer Lett 340: 9–21 [DOI] [PubMed] [Google Scholar]
- Spranger S, Bao R, Gajewski TF (2015) Melanoma‐intrinsic beta‐catenin signalling prevents anti‐tumour immunity. Nature 523: 231–235 [DOI] [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily‐Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT et al (2012) Tumour micro‐environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487: 500–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun‐Wada GH, Wada Y (2013) Vacuolar‐type proton pump ATPases: acidification and pathological relationships. Histol Histopathol 28: 805–815 [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L et al (2015) Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun 6: 6683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Infante JR, Krepler C, Reyes‐Uribe P, Samanta M, Chen HY, Li B, Swoboda RK, Wilson M, Vultur A et al (2013) Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep 4: 1090–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC et al (2011) Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol 29: 3085–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor‐Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN et al (2014) MAP kinase pathway alterations in BRAF‐mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov 4: 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR (2010) Role for IGFBP7 in senescence induction by BRAF. Cell 141: 746–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Liu T, Bai Y, Liao H, Qiu S, Chang Z, Liu Y, Yan X, Guo H (2014) Polymerase I and transcript release factor acts as an essential modulator of glioblastoma chemoresistance. PLoS One 9: e93439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM (2002) Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 1: 279–288 [DOI] [PubMed] [Google Scholar]
- Weinlich G, Murr C, Richardsen L, Winkler C, Fuchs D (2007) Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology 214: 8–14 [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ (2013) A new regulator of caveolae signalling. Elife 2: e01428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer DS, Hoek KS, Cheng PF, Eichhoff OM, Biedermann T, Raaijmakers MI, Hemmi S, Dummer R, Levesque MP (2013) Hypoxia contributes to melanoma heterogeneity by triggering HIF1alpha‐dependent phenotype switching. J Invest Dermatol 133: 2436–2443 [DOI] [PubMed] [Google Scholar]
- Widmer DS, Eichhoff OM, Dummer R, Levesque MP (2015) Melanoma's next top model, it is in the air. Exp Dermatol 24: 659–660 [DOI] [PubMed] [Google Scholar]
- Yi JS, Mun DG, Lee H, Park JS, Lee JW, Lee JS, Kim SJ, Cho BR, Lee SW, Ko YG (2013) PTRF/cavin‐1 is essential for multidrug resistance in cancer cells. J Proteome Res 12: 605–614 [DOI] [PubMed] [Google Scholar]
- Zipser MC, Eichhoff OM, Widmer DS, Schlegel NC, Schoenewolf NL, Stuart D, Liu W, Gardner H, Smith PD, Nuciforo P et al (2011) A proliferative melanoma cell phenotype is responsive to RAF/MEK inhibition independent of BRAF mutation status. Pigment Cell Melanoma Res 24: 326–333 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
Proteome data are available via Proteome exchange with identifiers PXD005432, PXD005433. RNAseq data are available via https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128915.