

Abstract
The Burkholderia genus encompasses many Gram-negative bacteria living in the rhizosphere. Some Burkholderia species can cause life-threatening human infections, highlighting the need for clinical interventions targeting specific lipopolysaccharide proteins. Burkholderia cenocepacia O-linked protein glycosylation has been reported, but the chemical structure of the O-glycan and the machinery required for its biosynthesis are unknown and could reveal potential therapeutic targets. Here, using bioinformatics approaches, gene-knockout mutants, purified recombinant proteins, LC-MS–based analyses of O-glycans, and NMR-based structural analyses, we identified a B. cenocepacia O-glycosylation (ogc) gene cluster necessary for synthesis, assembly, and membrane translocation of a lipid-linked O-glycan, as well as its structure, which consists of a β-Gal-(1,3)–α-GalNAc-(1,3)–β-GalNAc trisaccharide. We demonstrate that the ogc cluster is conserved in the Burkholderia genus, and we confirm the production of glycoproteins with similar glycans in the Burkholderia species: B. thailandensis, B. gladioli, and B. pseudomallei. Furthermore, we show that absence of protein O-glycosylation severely affects bacterial fitness and accelerates bacterial clearance in a Galleria mellonella larva infection model. Finally, our experiments revealed that patients infected with B. cenocepacia, Burkholderia multivorans, B. pseudomallei, or Burkholderia mallei develop O-glycan–specific antibodies. Together, these results highlight the importance of general protein O-glycosylation in the biology of the Burkholderia genus and its potential as a target for inhibition or immunotherapy approaches to control Burkholderia infections.
Keywords: cystic fibrosis, glycosylation, nuclear magnetic resonance (NMR), immunogenicity, bacteria, Burkholderia, galleria mellonella, glanders, melioidosis, phenotypic arrays
Introduction
The Burkholderia genus encompasses widespread environmental Gram-negative bacteria adapted to survive in the rhizosphere and freshwater (1, 2). Some Burkholderia species, including those in the Burkholderia cepacia complex, Burkholderia mallei, and Burkholderia pseudomallei, cause life-threatening infections in humans. The B. cepacia complex encompasses over 18 closely-related species typically infecting patients with cystic fibrosis; some of these species, especially Burkholderia cenocepacia, cause the fatal cepacia syndrome (3). B. mallei and B. pseudomallei are potential biological warfare agents (4), which cause glanders and melioidosis, respectively. B. mallei typically infects horses, donkeys, and mules, which are the primary reservoir of this mammalian-adapted pathogen that evolved from the environmental pathogen B. pseudomallei through gene loss (5). Glanders is characterized by pneumonia, often with systemic sepsis, or by skin and lymph node disease (Farcy). B. pseudomallei is endemic to southeast Asia and northern Australia and is being increasingly recognized in the tropics globally, including Africa and the Americas (6–8). Melioidosis can be acquired by inhalation or through skin abrasions or ingestion, causing a wide spectrum of symptoms, including pneumonia and rapidly-progressive systemic sepsis (7). B. pseudomallei can also remain latent and asymptomatic, with subsequent activation and presentation as melioidosis many years after initial exposure (9). Antibiotic treatment of Burkholderia-related infections is difficult due the high-level intrinsic multidrug resistance of these bacteria (10), and there are no clinically approved vaccines for either melioidosis or glanders prevention in humans or animals (11).
Protein glycosylation is a post-translational modification that modulates protein physicochemical properties and functions, and it is common in bacteria (12–14). Glycans may be attached to the amide nitrogen of asparagine residues of a polypeptide (N-glycosylation) or to hydroxyl oxygen of serine or threonine residues (O-glycosylation) (15). In other cases, α-mannopyranose can be linked to tryptophan via a carbon–carbon link (C-mannosylation) (16), and glycans can also be attached to the sulfur of cysteine (S-linked glycosylation) (17). The N-linked general protein glycosylation pathway in bacteria was first elucidated in Campylobacter jejuni (18, 19). This system includes the oligosaccharyltransferase (OTase)5 PglB (20), which is homologous to the eukaryotic Stt3, the catalytic subunit of the OTase complex responsible for N-glycosylation in the lumen of the endoplasmic reticulum.
O-Glycosylation systems have also been noted in many different bacteria (12–15). The O-glycosylation of at least 23 proteins with an uncharacterized predicted trisaccharide glycan, mediated by the OTase PglL, was previously identified in B. cenocepacia (21). However, the genes and gene products for the B. cenocepacia glycosylation machinery and the chemical structure of the glycan moiety were not elucidated. In this study, we report the identification of the Burkholderia O-glycosylation (ogc) cluster, which includes genes in chromosome 1 of B. cenocepacia (BCAL3114 to BCAL3118) encoding the enzymes required for the stepwise assembly of the lipid-linked O-glycan in the cytoplasm and its flipping across the inner membrane. We also discovered these genes are present in all species of Burkholderia and confirm biochemically the presence of O-linked proteins in several species of Burkholderia, suggesting protein O-glycosylation is a conserved feature of this genus. To elucidate the biosynthesis of the O-linked glycan, we generated knockout mutants in the ogc genes characterizing their effect on glycosylation and assessing the function of the epimerase within this cluster. From these studies, we have confirmed the predicted role of the ogc genes and also established the structure of the trisaccharide glycan. We also show that O-glycosylation is required for bacterial fitness in vitro and survival in a larvae infection model. Finally, we demonstrate that individuals previously infected with B. cenocepacia, Burkholderia multivorans, B. pseudomallei, and B. mallei develop anti-O-glycan serum antibodies, suggesting Burkholderia glycoproteins display a common epitope perceived by the human immune system.
Results
Predicted B. cenocepacia O-glycosylation gene cluster is conserved in the Burkholderia genus
Previous work identified PglL (BCAL0960) as the OTase catalyzing the addition of an uncharacterized trisaccharide O-glycan to least 23 B. cenocepacia proteins (21). In OTase-dependent glycosylation pathways, the lipid-linked glycan precursor is assembled on the cytoplasmic side of the membrane, translocated across the membrane, and then transferred onto acceptor proteins at the periplasmic side (22). We therefore hypothesized that at least four genes encoding a phosphoglycosyltransferase (initiating enzyme that mediates the synthesis of a lipid-linked sugar) (23, 24), two additional glycosyltransferases, and a flippase protein would be required for the synthesis and translocation of a B. cenocepacia lipid-linked trisaccharide glycan (22, 24). Chromosome 1 of B. cenocepacia has a putative five-gene operon flanked by the O-antigen synthesis cluster and lipid A-core biosynthesis genes (Fig. 1A) (25), which could be involved in protein glycosylation. These genes were previously annotated, according to their predicted functions, as wecA (BCAL3118, phosphoglycosyltransferase), galE (BCAL3117, UDP-glucose epimerase), wbxA and wbxB (BCAL3116 and BCAL3115, glycosyltransferases, GT2 family forming β-glycosidic bonds and GT4 family forming α-glycosidic bonds, respectively), and wzx (BCAL3114, flippase) (25). This putative operon was herein renamed ogc (for O-glycosylation) (see below). The last ogc gene, BCAL3118, which encodes a predicted UDP–N-acetylhexosamine-1-P transferase homologous to WecA-like initiating enzymes for O-antigen synthesis, was renamed ogcI (for initiating transferase). The immediately upstream gene, BCAL3117, encodes a protein similar to UDP-glucose-4-epimerases of the NAD-dependent epimerase/dehydratase family, and it was renamed ogcE. The two predicted glycosyltransferase-encoding genes, BCAL3115 and BCAL3116, were renamed ogcA and ogcB, respectively. BCAL3114, renamed ogcX, encodes a predicted flippase protein belonging to the Wzx-like polysaccharide transporter family. Therefore, the ogc cluster encodes proteins whose predicted functions fit well with the required enzymatic activities to assemble the lipid-linked trisaccharide glycan for protein glycosylation, according to the model depicted in Fig. 1B.
Figure 1.

Identification of the O-glycosylation cluster (ogc) in B. cenocepacia. A, genetic organization of the ogc cluster (BCAL31140–BCAL3118), placed between the O-antigen and lipid A-core clusters. Names of ORFs indicated above the arrows are according to the original gene annotation (25). Names below are the ones assigned in this study. B, proposed model for the general O-glycosylation assembly pathway in B. cenocepacia; OgcI, initiating enzyme; OgcE, UDP-glucose/galactose epimerase; OgcAB, glycosyltransferases; OgcX, flippase; and PglL, oligosaccharyltransferase. C, synteny of ogc genes in members of the Burkholderia genus. Genes indicated in bold are the ones used to search for synteny in the SyntTax server. The species showing similar arrangements are listed for each group. The synteny scores ranged from 55 to 96 (>30 is considered highly significant conservation) (26). D, relative gel mobility of BCAL2640 and DsbA1 polypeptides in ΔogcX–ogcI and single deletion mutants. Similar results were obtained in three biological repeats. Left panel, Coomassie-stained 14% SDS-polyacrylamide gel of the purified B. cenocepacia BCAL2640 protein expressed in WT K56-2, ΔpglL, and Δogc. Right panel, Western blotting of His-tagged DsbA1 acceptor protein expressed in the parental K56-2 strains and the mutants ΔpglL, ΔogcX, ΔogcX, ΔogcI, ΔogcAB, ΔogcE, and ΔO-antigen cluster (ΔBCAL3119–BCAL3131).
Using the SyntTax web server (26) and the Burkholderia genome database (27), we discovered the ogc cluster is conserved in the majority of Burkholderia and Paraburkholderia species whose genomes have been sequenced (Fig. 1C). Most species retain the five-gene cluster with the genes arranged as in B. cenocepacia. In a reduced number of species, ogcXABE remain together, whereas ogcI appears as a single monocistronic gene separated from the cluster by a variable number of genes depending on the species examined (Fig. 1C). In Paraburkholderia rhizoxinica, ogcB is present as a monocistronic gene, whereas the rest of the genes, ogcXAEI, form a putative operon (Fig. 1C). Therefore, the ogc cluster appears to be a highly-conserved loci encoding the minimal set of gene products to produce the trisaccharide O-linked glycan observed in B. cenocepacia.
Deletion mutant of the ogc cluster cannot glycosylate proteins
To validate the ogc cluster's role in protein glycosylation, we constructed an unmarked deletion mutant (Δogc) in the B. cenocepacia strain K56-2 and a ΔpglL mutant as a control. We also generated the individual unmarked, nonpolar deletion mutants ΔogcX, ΔogcI, ΔogcAB, and ΔogcE. In parallel, we constructed recombinant plasmids expressing His-tagged fusion forms of BCAL2640 and the DsbA1 protein from Neisseria meningitidis. BCAL2640 is a native glycosylated protein in B. cenocepacia, whereas the heterologously expressed DsbA1 is glycosylated by PglL (21) and also by several different OTases (28). Expression of BCAL2640 and DsbA1 was examined in the K56-2 parental strain and its isogenic Δogc and ΔpglL mutants. The purified BCAL2640 polypeptide obtained from Δogc and ΔpglL had increased relative mobility compared with that obtained from the parental strain, suggesting loss of glycosylation (Fig. 1D, left panel). Similarly, examination of DsbA1 mobility by immunoblot with anti-His tag antibodies also revealed DsbA1 polypeptides of lower apparent mass in samples purified from Δogc and ΔpglL, as well as from ΔogcX, ΔogcI, ΔogcAB, and ΔogcE mutants (Fig. 1D, right panel). Conversely, no difference in protein mobility was observed in DsbA1 expressed in an O-antigen synthesis deletion mutant ΔBCAL3119–BCAL3131, ruling out any contribution of the O-antigen genes to protein glycosylation (Fig. 1D, right panel).
To directly monitor the glycosylation status of purified BCAL2640 expressed in strains K56-2, Δogc, and ΔpglL, the purified protein was digested with trypsin and analyzed by LC-MS. The trypsin-derived BCAL2640 peptide 152YAPPPAAVPVAATSGAQGGAAAAAAPAGTKPANAPR187, previously shown to be glycosylated (at the underlined serine residue) (21), was readily observable. Tandem mass spectrometry (MS/MS) and fragmentation by collision-induced dissociation (CID) confirms the peptide is modified with at least two glycans corresponding to HexNAc–HexNAc-262 (Fig. 2A) and HexNAc–HexNAc–Hex (Fig. 2B), whereas higher-energy collisional dissociation (HCD) fragmentation confirmed the peptide sequence (Fig. 2C). The glycosylated form of this peptide was 15-fold more abundant than the unglycosylated form, indicating the majority of BCAL2640 was glycosylated (Fig. 2D). In contrast, this glycopeptide was absent in the purified protein obtained from ΔpglL or Δogc mutants (Fig. 2, E and F). Therefore, deletion of the ogc cluster causes the same defect in protein glycosylation as the loss of the OTase PglL, demonstrating it contains the genes encoding functions for the synthesis and assembly of the protein glycan.
Figure 2.

Identification of PglL and Ogc-dependent glycosylation of BCAL2640. A–C, MS-MS analysis of detected glycopeptide; CID fragmentations confirm that the peptide is modified with at least two glycans corresponding to A, HexNAc–HexNAc-262 (m/z 959.97, +4 corresponding to 3835.868 Da), and B, HexNAc–HexNAc–Hex (m/z 934.97, +4 corresponding to 3735.859 Da). C, HCD fragmentation of the HexNAc–HexNAc–Hex-modified peptide confirming the identity of the peptide sequence. D--F, extracted ion chromatograms of the major charge state, +3, of the glycosylated (modified with HexNAc–HexNAc–Hex) and nonglycosylated forms of 152YAPPPAAVPVAATSGAQGGAAAAAAPAGTKPANAPR187. In K56-2 (WT), the BCAL2640 glycosylated peptide was >15-fold more abundant than the unglycosylated form. This peptide was not glycosylated in ΔpglL or ΔogcX-ogcI. * denotes deglycosylated peptide resulting from in-source fragmentation.
Structural characterization of the O-glycan
To determine the structure of the B. cenocepacia O-glycan, glycopeptides modified with the trisaccharide were generated from glycosylated DsbA1 by digestion with proteinase K and enriched by size-exclusion chromatography. This glycopeptide fraction was identified by nuclear magnetic resonance (NMR) spectroscopy analysis by both homo- and heteronuclear two-dimensional NMR experiments (Fig. 3A and Table S1). The heteronuclear single quantum coherence (HSQC) spectrum (Fig. 3B) showed three main anomeric signals with 1H/13C values at 5.07/94.5, 4.63/101.7, and 4.45/106.3 ppm (Fig. 3A), each labeled with a capital letter. For the A spin system, total correlation spectroscopy (TOCSY) displayed three correlations that connected H-1 up to H-4 (Fig. 3C and Table S1). The attribution of these densities to the right proton in the sequence was inferred by analyzing the homonuclear correlation (COSY) spectrum, whereas the position of H-5 was identified by the H-4/H-5 correlation in the transverse rotating-frame Overhauser enhancement (T-ROESY) spectrum, which in turn defined the position of the two H-6 protons at 3.76 ppm. These results, together with those derived from the HSQC spectrum (Fig. 3B) and the H-2/C-2 values (4.38/51.0 ppm) along with the presence of an acetyl group in the proton spectrum, indicated that A was GalNAc. The 3JH1, H2 value (3.3 Hz) pointed to an α-configuration for the anomeric center. In addition, the downfield displacement of C-3 (79.1 ppm) with respect to the standard value (72.3 ppm) (29) indicated a substitution at this position.
Figure 3.
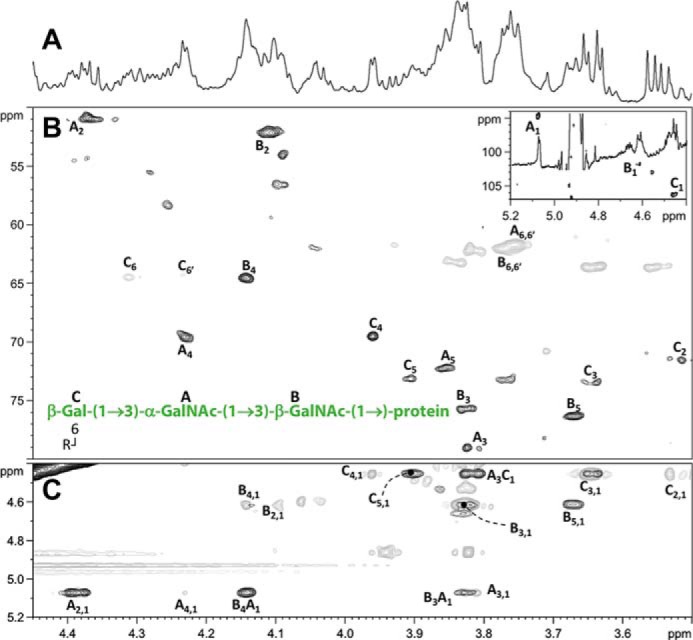
NMR spectra of the glycopeptide. Representative regions of the NMR spectrum acquired at 600 MHz spectrometer, in D2O at 15 °C, for the glycopeptides from DsbA1 after proteinase K digestion and chromatographic purification. A, zoom of 1H NMR spectrum in the ring proton region, B, multiplicity edited 1H-13C HSQC spectrum with signals in gray represent methylene carbon atoms; the inset shows a zoom of the downfield region with the three anomeric resonances of A–C residues. C, superimposition of 1H-1H TOCSY (gray) and 1H-1H T-ROESY (black) spectra. Attribution of most relevant cross-peaks is indicated near the corresponding density, and labels reflect those reported in Table S1. The structure of the glycopeptide's glycan moiety, along with the labels used during spectra attribution, is shown as well B. Unattributed peaks are related to peptides signals.
For B and C residues, their 3JH1, H2 values (8.4 and 8.0 Hz, respectively) disclosed their β-configuration at the anomeric center. Using the same approach as described for residue A, residue B was identified as GalNAc (H-2/C-2 values at 4.11/52.0 ppm) and residue C as galactose (H2/C-2 values at 3.51/71.6 ppm). The comparison of the carbon chemical shifts of these two residues with those taken as in Ref. 29 showed that C-3 of B (75.8 ppm) and C-6 of C (64.4 ppm) were shifted downfield with respect to the standard values (72.3 and 62.0 ppm, respectively), suggesting the presence of a substitution at these positions. The sequence of the three units was inferred by NOE contacts found in the T-ROESY spectrum among H-1 of A and both H-3 and H-4 of B, and H-1 of C with H-3 of A (Fig. 3C). Therefore, combining data from the nuclear Overhauser effects with the 13C chemical shift displacement observed allowed us to build the sequence C-(1→3)–A-(1→3)–B. Importantly, H-1 of B did not correlate with any sugar-related signal but only with protons related to the peptide moiety, for which we conclude the glycan structure is β-Gal-(1→3)–α-GalNAc-(1→3)–β-GalNAc-(1→) directly linked to the protein (Fig. 3B). The T-ROESY spectrum did not contain cross-peaks connecting to any of the two H-6s of C to the substituent at O-6, but the type of chemical displacement of C-6 and C-5, along with the 1H chemical shift of both H-6s, suggested the presence of an acyl substituent that was consistent with a succinylated hexose residue, observed as a mass of 262 Da by MS analysis, but it remains to be structurally identified by NMR.
Dual role of OgcE in protein glycosylation and O-antigen synthesis
The predicted OgcE epimerase (Fig. 1A) could be required for the synthesis of UDP-GalNAc, UDP-Gal, or both, which are the expected precursors for the assembly of the trisaccharide glycan. However, the O-antigen in B. cenocepacia K56-2 is composed of a GalNAc–GalNAc–Rha repeating unit (25), suggesting UDP-GalNAc is a common precursor for both O-antigen and the protein glycan trisaccharide synthesis in this strain. To determine whether gcE is also required for O-antigen synthesis, we examined the lipopolysaccharide (LPS) profile of K56-2, ΔpglL, Δogc, and various deletion mutants in ogc genes. Only the Δogc and ΔogcE mutants produced truncated O-antigen, as indicated by a strong band migrating above the band corresponding to lipid A-core and the loss of bands corresponding to polymeric O-antigen (Fig. 4A). The O-antigen in B. cenocepacia K56-2 is synthesized via the ABC export pathway (25, 30). This requires an adaptor sugar, bound to undecaprenyl-PP, to which the repeating O-antigen units are attached, and this sugar in K56-2 is QuiNAc (31). Therefore, the truncated O-antigen band, in the absence of UDP-GalNAc due to the loss of ogcE (Fig. 4A, asterisk), was interpreted as lipid A-core plus Rha-QuiNAc disaccharide. We have previously shown that a similar band appears in a GalNAc transferase mutant (25). Complementation of Δogc with a plasmid expressing OgcE restored full O-antigen synthesis (Fig. 4A) but did not restore protein glycosylation (Fig. 4B). Therefore, OgcE (BCAL3117) participates in both the synthesis of O-antigen and the protein glycan (Fig. 4, A and B), whereas the remaining ogc genes are not involved in O-antigen synthesis.
Figure 4.

Comparison of effects of single ogc mutants on LPS synthesis and protein glycosylation. Similar results were obtained in three biological repeats. A, proteinase K–treated whole-cell lysates were resolved on 14% Tricine gel and silver staining. Arrowheads indicate a lipid A-core band + truncated O-antigen (see “Results”). B, Western blotting of His-tagged DsbA1 acceptor protein expressed in K56-2 WT, ΔpglL, and Δogc + (OgcE).
OgcE is a UDP-Glc and UDP-GlcNAc epimerase
To elucidate the biochemical function of OgcE, we examined its enzymatic activity by capillary electrophoresis. Purified OgcE showed C4 epimerase activity on UDP-Gal and UDP-Glc (Fig. 5, A and C), reaching a final equilibrium of 79% UDP-Glc and 21% UDP-Gal, irrespective of the starting substrate. OgcE also had C4 epimerase activity on UDP-GalNAc and UDP-GlcNAc (Fig. 5, B and D), reaching a final equilibrium of 68% UDP-GlcNAc and 32% UDP-GalNAc with either substrate. Identical results were obtained with both NAD+ (Fig. 5, A and B) and NADP+ (Fig. 5, C and D), suggesting the enzyme does not use any of these exogenously supplied cofactors. This conclusion is further supported by the unaltered amount of input cofactor after catalysis, the lack of release of reduced NADP+, and because catalysis can also occur in the absence of any cofactor (Fig. S1, top two traces). A small amount of NADH was observed in all enzyme-containing reactions irrespective of the type of cofactor exogenously added, but no NADPH was released. Because the NADH appearance occurs irrespective of the type of added substrate or exogenous cofactor and is not proportional to the amount of catalysis, we surmised that the detected NADH was released by the enzyme. Indeed, release of NADH was also observed in control reactions performed without substrate (Fig. S1, bottom trace). This suggests the OgcE preferentially binds NAD+ over NAPH during overexpression in Escherichia coli and explains why the bound cofactor alleviates the need for any exogenous cofactor. The release may occur due to progressive unfolding of the enzyme during lengthy incubations at 37 °C. To assess which type of substrate is most efficiently catalyzed, reactions were performed with serial dilutions of enzyme on UDP-Gal and UDP-GalNAc. These substrates were selected over UDP-Glc and UDP-GlcNAc because as high as 79 and 67% catalysis can be observed with UDP-Gal and UDP-GalNAc (versus only around 25–30% starting from their glucose counterparts). Enhanced catalysis of UDP-Gal versus UDP-GalNAc was observed at all enzyme dilutions and all time points tested (Figs. S2 and S3). Therefore, we conclude OgcE is an epimerase with dual specificity for the interconversion of UDP-Gal/UDP-Glc and UDP-GlcNAc/UDP-GalNAc, with slightly higher efficiency for nonacetylated substrates.
Figure 5.

OgcE enzymatic activity as UDP-glucose and UDP-GlcNAc epimerase. Capillary electrophoresis electropherograms showing the C4 epimerase activity of OgcE on UDP-Glc (A), UDP-GlcNAc (B), UDP-Gal (C), and UDP-GalNAc (D) and in the presence of NAD+ (A and C) or NADP+ (B and D). + and − indicate the presence or absence of OgcE in the reaction. Reactions were analyzed at equilibrium after 4 h of incubation at 37 °C in the presence of 0.1 mm substrate, 0.1 mm cofactor, and 5 μg of enzyme in a total volume of 10 μl of 200 mm Tris-HCl (pH 8). Two sets of samples were prepared with independent OgcE enzyme fractions and each including one reaction per condition was analyzed with 2 runs per sample. Both sets showed similar data at equilibrium, and only 1 set is shown in the figure. Activity assessment and peak assignments were also based on optimization at different enzyme/substrate ratios and various reaction times, and based on co-injections with standards.
Predicted function of the remaining genes in the ogc cluster
To gain clues on the function of the remaining ogc genes, we investigated the glycosylation status of the DsbA1-derived peptide 23VQTSVPADSAPAASAAAAPAGLVEGQNYTVLANPIPQQQAGK64 in the various ogc gene deletion mutants. MS spectra of the DsbA1-derived peptide in K56-2, ΔpglL, Δogc, ΔogcX, ΔogcI, ΔogcAB, and ΔogcE are shown in Fig. S4. Using HCD fragmentation, four glycoforms of this peptide were identified, with all glycopeptides showing nearly identical peptide fragmentation maps (Fig. 6A). The glycan attached to the four glycoforms corresponded to HexNAc–HexNAc–Hex (glycan A), HexNAc–HexNAc-262 (glycan B), QuiNAc-Rha, and a single HexNAc (Fig. 6B). The area under the curve of the extracted ion chromatograms for all observed glycoforms of the peptide enabled us to semi-quantitatively compare each state (Fig. 6C and Table S2). The results show that glycoforms A and B were present in K56-2 with relative abundances of 44 and 52%, respectively, whereas the remaining 4% of the peptide was unmodified. In contrast, 100 and 84% of unmodified peptide was found in preparations obtained from ΔpglL and Δogc, respectively. The peak in Δogc with a relative abundance of 16% corresponded to a peptide modified by a glycan with the predicted mass of a QuiNAc-Rha disaccharide. As shown previously (Fig. 4A), deletion of the ogc cluster results in a truncated O-antigen containing a terminal QuiNAc-Rha attached to the lipid A-core (Fig. 4A, arrowheads). Therefore, we interpreted this result as due to the incorporation of the QuiNAc-Rha disaccharide, which is assembled as Und-PP–QuiNAc-Rha by the O-antigen synthesis machinery and likely transferred to the target protein by PglL. In contrast, the ΔogcI mutant lacked glycosylated peptides, as the glycan assembly is blocked at its initiation step (Fig. 6C, Table S2, and Fig. S4). Both ΔogcX and ΔogcAB showed predominantly unmodified peptide with 90 and 65% relative abundance, respectively. The remaining glycosylated peptide in these two mutants corresponded to an HexNAc glycan form (Fig. 6C, Table S2, and Fig. S4). The low abundance of a modified peptide with a single HexNAc in ΔogcX (9%) suggests the possibility that another flippase in the B. cenocepacia genome could mediate in part the membrane translocation of Und-PP-GalNAc (32). A single ΔogcB mutant also resulted in the production of HexNAc-decorated glycoproteins as in ΔogcAB, supporting the assignment of OgcB as the enzyme responsible for the addition of the second GalNAc (Fig. S5). Multiple attempts to delete ogcA were unsuccessful. It is possible that the absence of this gene may be deleterious for bacterial cell viability due to the accumulation of an Und-PP–linked GalNAc disaccharide that cannot be processed further, suggesting that OgcA is responsible for the addition of the terminal Gal and explaining why Δogc, ΔogcAB, and ΔogcB are well-tolerated.
Figure 6.

Effect of O-glycan biosynthesis gene disruptions on protein glycosylation. A, DsbA1-derived glycopeptide 23VQTSVPADSAPAASAAAAPAGLVEGQNYTVLANPIPQQQAGK64 indicating the sites of HCD fragmentation. B, four glycoforms observed corresponded to glycan A (HexNAc–HexNAc–Hex, m/z 1543.10, +3, 4626.266 Da), glycan B (HexNAc–HexNAc-262, m/z 1576.44, +3), QuiNAc-Rha (m/z 1464.75, +3, 4391.226 Da), and a single HexNAc (m/z 1421.39, +3, 4261.146 Da). C, semi-quantitative comparison of each glycoform in different genetic backgrounds using the area under the curve of the extracted ion chromatograms for all observed glycoforms of the DsbA1 peptide.
O-Glycosylation is common in other Burkholderia species
The gene–by–gene conservation of the ogc cluster in the Burkholderia genus (Fig. 1C) suggests that protein glycosylation with β-Gal-(1→3)–α-GalNAc-(1→3)–β-GalNAc–(1→ is widespread within Burkholderia species. We tested this idea in two ways. First, we showed that glycosylated proteins in lysates from B. cenocepacia, Burkholderia thailandensis, and two different strains of Burkholderia gladioli can be detected using the peanut agglutinin (Fig. S6). This lectin has specificity for Gal-β(1–3)–GalNAc terminal disaccharides. Second, bacterial glycopeptides were investigated by LC-MS in isolates representing several different Burkholderia species. Glycopeptides derived from five proteins were readily detectable from whole-proteome samples of B. thailandensis E264 (supporting Data S1), such as the peptide 159PAAASGAPAPAASGAAAH176 of BTH_I3002ABC, which corresponds to the periplasmic substrate-binding protein of an ABC transporter (Fig. 7) and is modified with the expected HexNAc–HexNAc–Hex. As with B. cenocepacia glycopeptides (21), we also noted the presence of HexNAc–HexNAc-262 and an additional modified glycan corresponding to HexNAc–HexNAc-362 in B. thailandensis (supporting Data S1). Examination of whole-cell lysates from two clinical isolates of B. gladioli revealed at least 14 glycosylated proteins (supporting Data S2 and S3). The analysis of one of the glycosylated peptides, 594AAHPGDIASEAAATGQPR611, of the B. gladioli bifunctional uroporphyrinogen-III synthetase/uroporphyrin-III is shown in Fig. 7. Analysis of eight clinical isolates of B. pseudomallei revealed three modified proteins, one of which was identified across all eight clinical isolates and corresponded to the known virulence factor Ecotin (Fig. 7 and supporting Data S4) (33). As in the other cases, the B. pseudomallei glycopeptides were also modified with either HexNAc–HexNAc–Hex or HexNAc–HexNAc-262. Collectively, our results demonstrate that protein glycosylation occurs in Burkholderia species outside B. cenocepacia and that the addition of the HexNAc–HexNAc–Hex glycan appears to be an invariant feature across members of this genus.
Figure 7.

Glycopeptides detected in B. thailandensis, B. gladioli, and B. pseudomallei are modified by an identical trisaccharide to that in B. cenocepacia. CID fragmentation (left panels) enabled the identification of the expected HexNAc–HexNAc–Hex glycan attached to peptides, whereas HCD fragmentation (right panels) confirmed the identity of the peptide sequences corresponding to the B. thailandensis ABC transporter, periplasmic substrate-binding protein (BTH_I3002, m/z 1007.466, +2, 2012.9161 Da), the B. gladioli bifunctional uroporphyrinogen-III synthetase/uroporphyrin-III C-methyltransferase (bgla_1g31160, m/z 763.359, +3, 2779.3218 Da), and the B. pseudomallei serine protease inhibitor Ecotin (m/z 927.449, +3, 2779.3218 Da).
Loss of O-glycosylation is associated with growth defects under many different carbon sources as well as oxidative and osmotic stress
The conservation of the O-glycosylation pathway in the Burkholderia genus suggested it might be necessary for bacterial cell homeostasis. Therefore, we performed a global comparative analysis of carbon sources utilized by the K56-2 parental strain and ΔpglL using phenotypic microarrays (Biolog). The results show that ΔpglL utilized exogenous carbon sources less effectively than K56-2, especially sugar alcohols such as xylitol, monosaccharides (l-arabinose and d-mannose), deoxy sugars (l-fucose and 2-deoxyribose), and various amino acids (d-alanine, l-alanine, d-serine, l-arginine, l-lysine, and l-ornithine) (Fig. 8A and supporting Data S5). Also, ΔpglL could not utilize putrescine, which is produced from l-ornithine by ornithine decarboxylase (BCAL2641 and BCAM1111) or from l-arginine by arginine decarboxylase (BCAM1112) (34). Furthermore, ΔpglL grew poorly in media with Tweens, formic acid, butyric acid, d-glucosaminic acid, and d-galacturonic acid. A subset of significant phenotypic microarray differences resulting in growth defects relative to the parental strain were independently validated (Fig. 8B). We also examined the sensitivity of ΔpglL toward oxidative stress under a range of H2O2 concentrations. The results showed that the mutant has higher sensitivity toward H2O2 than K56-2 (Fig. S7A). Because in long-term exposure experiments bacteria could adapt to H2O2 stress and because H2O2 is short-lived, we also performed a challenge experiment. In this case, bacteria were challenged with either 300 μm or 1 mm of H2O2 with a bacterial inoculum of OD600 of 0.01 and 0.1, respectively, for 1 h at 37 °C. In both conditions, we found more than a 30% reduction in ΔpglL survival relative to K56-2 (Fig. S7B). The ΔpglL mutant was also more susceptible to osmotic stress induced by high salt (4% NaCl) than K56-2 parental strain (Fig. S7C). We also noted that the ΔpglL mutant was more proteolytically active than the parental strain (Fig. 8, C and D), a phenotype that was complemented by a plasmid carrying a functional pglL gene. Collectively, the results indicate that in the absence of protein glycosylation, B. cenocepacia displays a global defect in utilization of multiple carbon sources, increased proteolytic activity, and reduced tolerance to stress conditions. We therefore conclude that the general protein O-glycosylation pathway may be important for the metabolic fitness of Burkholderia.
Figure 8.

Metabolic alterations in the protein O-glycosylation mutant ΔpglL. A, heat map for carbon source phenotypes as determined by phenotypic microarrays using an Omnilog system (see “Experimental procedures”). Yellow represents WT growth. Blue shading denotes ranges of growth below 80% of the WT growth in the same carbon source, calculated by comparing the area under curve of WT and mutant strain (AUC). The complete results for all carbon sources tested are presented in supporting Data S5. B, selective carbon sources giving differences between K56-2 and ΔpglL were independently validated by generating growth curves measured by the automated bioscreen C using M9 minimal medium with the relevant carbon source under investigation. Statistical differences (mean ± S.D.) of the area under each growth curve were analyzed by multiple t test comparisons with confidence interval of 95%. n = 4 per carbon source. *, p = 0.02; **, p < 0.01; ***, p < 0.001. C, casein proteolytic activity of K56-2, ΔpglL, and the complemented mutant (ΔpglL + PglL) was assayed in casein-containing nutrient agar plates. Plates shown are representatives of at least two experiments done in triplicates. D, quantification of the data in C by measuring the clear halo around the colonies after incubation at 37 °C for 48 h. Values are expressed as mean ± S.D. in millimeters. Statistical differences were analyzed by one-way ANOVA with Tukey's multiple comparisons tests; α = 0.01; ***, p < 0.0001.
Loss of protein glycosylation reduces bacterial growth in the G. mellonella infection model and results in a rapid activation of the larval antimicrobial response
ΔpglL was previously reported to be avirulent in the wax moth G. mellonella and the duckweed Lemna minor, which are widely used insect and plant infection models, respectively, based on end-point experiments (21). However, the mechanisms for the reduced virulence was not investigated. First, we compared the virulence of the O-glycan–defective mutants and the bacterial loads recovered from the G. mellonella hemolymph over 72-h infections. Under our experimental conditions, the parental K56-2 strain caused death of all larvae at 48 h postinfection (Fig. 9A), whereas larvae infected with ΔpglL and Δogc had survival rates of 80 and 98%, respectively. We attributed the reduced virulence of Δogc (compared with ΔpglL) to the cumulative effect caused by the simultaneous loss of protein glycosylation and O-antigen synthesis due to absence of ogcE in Δogc (Fig. 4). Consistent with this, a strain in which Δogc was complemented by OgcE retained the same virulence level as ΔpglL at 48 and 72 h postinfection (Fig. 9A). Second, we examined whether decreased virulence is associated with poor growth of the O-glycosylation–defective mutants in the infected larvae. Analysis of bacterial growth in larval hemolymph showed a 5-log increase in K56-2 (relative to the initial inoculum) at 48 h postinfection (p < 0.01; Fig. 9B). In contrast, ΔpglL and Δogc bacteria only showed 2-log CFU increase at 48 h, and Δogc demonstrated poorer growth overall compared with ΔpglL despite that the differences were not statistically significant.
Figure 9.

Role of O-glycosylation in bacterial proliferation and G. mellonella larval mortality. A, larvae were injected with 8 × 104 CFU of parental and mutant strains, and the percent survival was monitored over 72 h postinfection. Statistical differences (means ± S.D.) were examined by two-way ANOVA with Tukey's multiple comparisons tests; α = 0.05. B, hemolymph from B. cenocepacia–infected larvae (n = 3) was pooled, and CFU were determined by viable count. Results are presented as a superimposed scatter plot, and statistical differences (means ± S.D.) were examined by two-way ANOVA as indicated above. C, transcriptional activation of immune-responsive genes following infection determined by quantitative real-time RT-PCR analysis. The graphs show the normalized fold expression levels of the target transcripts relative to those in PBS-injected animals and represent means ± S.D. of three independent experiments, each with two technical replicas. Expression data were normalized to the expression of the housekeeping actin gene. IMPI, inducible metalloprotease inhibitor. Data were statistically analyzed by two-way ANOVA with Tukey's multiple comparisons tests and α = 0.05. ns, nonsignificant. The calculated p values from individual comparisons are indicated in the figure.
Third, we determined whether the significantly reduced virulence and in larvae growth of the O-glycosylation–defective mutants correlated with rapid killing mediated by the G. mellonella's innate immune system. For this, we determined the expression pattern of innate immune genes in the infected larvae by RT-qPCR analysis. The results showed that the levels of cecropin and galliomycin transcripts were 6–11- and 4-fold higher, respectively, in larvae infected with ΔpglL and Δogc than in those infected with K56-2 at 8 h postinfection (Fig. 9C), while the levels of the galleriomycin transcripts did not differ at this time. By 18 h postinfection, the levels of cecropin and galleriomycin transcripts increased dramatically in larvae infected with the three strains, but the increased expression was more significant in larvae infected with K56-2. Lower levels of cecropin gene expression in Galleria infected with Δogc at 18 h could be related to the rapid death of bacterial cells. For galliomycin, the transcript levels did not increase much, although they were still significantly higher in larvae infected with ΔpglL and Δogc than in those infected with K56-2 (Fig. 9C). As infection in K56-2 was accompanied by increased pigmentation, we inferred the level of melanin formation by measuring the expression of the peptidoglycan recognition protein B (PGRP) gene. PGRP is a microbial pattern recognition molecule that recognizes bacterial cell fragments and mediates host responses toward bacterial infections through activation of the prophenoloxidase cascade, responsible for melanization of pathogens and dead tissues, activation of Tol1 receptor, and phagocytosis induction (35). We found that the level of PGRP gene transcription was similar for larvae infected with the three strains at 8 h postinfection, but at 18 h the increase in PGRP gene expression was significantly higher in larvae infected with the parental strain than in those infected with the O-glycosylation mutants (Fig. 9C). In contrast, transcript levels of the inducible metalloprotease inhibitor gene, which encodes a host inhibitor of bacterial proteases (36), remained similar but significantly higher in larvae infected with both K56-2 and ΔpglL than with Δogc. The expression of the gene encoding the cell wall–degrading enzyme lysozyme (37) remained similar to the larvae infected with K56-2 or both O-glycosylation–defective mutants, although their levels dramatically increased from 8 to 18 h postinfection. Collectively, these findings support the notion that the G. mellonella innate immune system responds early to infection by O-glycosylation–defective bacteria, which compounded by reduced bacterial growth and reduced resistance to oxidative stress could result in rapid and early killing of the mutant bacteria.
Burkholderia-infected patients develop serum antibodies against the Burkholderia O-glycan
Given the previous results and the conservation of the O-glycosylation pathway in Burkholderia species, we examined whether infected patients develop antibodies specifically recognizing Burkholderia O-glycoproteins. For these experiments, we deliberately chose the N. meningitidis DsbA1 as a heterologous glycosylation target expressed in Burkholderia to screen human sera because the assay was designed to reveal antibodies recognizing the Burkholderia glycan independently of the nature of the target protein. The N. meningitidis DsbA1 contains the sequence PAAASAAA with an invariable serine for O-glycosylation, which is similar to the glycosylation motif for the Burkholderia proteins. Convalescent sera collected from 16 patients infected with B. cenocepacia, 14 patients infected with B. multivorans, 20 patients infected with B. pseudomallei, and one patient infected with B. mallei were investigated for the presence of antibodies against purified glycosylated DsbA1 expressed in the K56-2 strain. Fourteen of the 16 samples isolated from B. cenocepacia-infected patients contained antibodies against glycosylated DsbA1 protein, whereas only two serum samples reacted with nonglycosylated DsbA1 (Fig 10A, thick arrows). For B. multivorans-infected patients, 13 of the 14 serum samples tested gave positive reaction with glycosylated DsbA1 (Fig. 10B). Also, 16 of the 20 samples tested for patients with B. pseudomallei reacted with glycosylated DsbA1 and not with the nonglycosylated version (Fig. 10B). This amounts to 86% of the tested serum samples giving positive reaction in the ELISA with glycosylated DsbA1. To confirm that the serum samples contained Burkholderia O-glycan–specific antibodies, positive samples were adsorbed against glycosylated DsbA1 by serial passage on microtiter plates containing bound glycosylated protein, and the adsorbed serum samples were then retested by ELISA. The results showed that all the originally positive samples became negative after adsorption (Fig. 10B). Together, the results reveal that the majority of the convalescent serum samples investigated had antibodies toward the Burkholderia glycan, indicating that either the glycan itself or the glycosylated proteins are antigens recognized upon human infection with Burkholderia species. Also, the results indicate that the antibodies recognize the glycoprotein made in B. cenocepacia irrespective of the type of infection cause, providing additional evidence that the infecting bacteria produce the same O-glycan.
Figure 10.

Immune reaction of Burkholderia O-glycan toward convalescent sera from Burkholderia-infected patients. A, indirect ELISA results using as an antigen purified glycosylated DsbA1 protein produced in B. cenocepacia K56-2 and serum samples collected from patients infected with B. cenocepacia, B. multivorans, B. mallei, and B. pseudomallei. Nonglycosylated heterologously-expressed DsbA1 protein in ΔpglL was used in the experiment. Thin arrows point to the serum samples that reacted with both glycosylated and unglycosylated DsbA1 heterologously expressed in K56-2 and ΔpglL, respectively. Three independent experiments were done, each in duplicate. Thick arrows point to the serum samples that gave negative results with glycosylated DsbA1. B, serum samples were adsorbed by incubation with the glycosylated DsbA1 protein for three successive passages prior to ELISA. Two independent experiments were done. Blue dotted lines in A and B indicate the negative cutoff OD450 value for background color in the ELISA determined by running ELISAs in normal human serum from 30 donors. Error bars represent standard deviation of the mean.
Discussion
In this study, we have uncovered the gene cluster encoding the enzymes responsible for the synthesis and assembly of the lipid-linked glycan precursor for the general protein O-glycosylation pathway in B. cenocepacia and also elucidated the glycan structure as the trisaccharide β-Gal-(1–3)–α-GalNAc-(1–3)–β-GalNAc-peptide. As reported previously (21), we found two forms of the O-glycan, one of which corresponds to a modified trisaccharide. Our data suggest that the trisaccharide core moiety of both glycans derives from the ogc pathway, whereas the modification likely involves nonstoichiometric succinylation at the terminal Gal residue. Succinylation appears as a variable modification in glycans from Rhizobium meliloti (38) and several other Proteobacteria (39). Further studies are required to elucidate the structure of the modified glycan and to identify the gene encoding the modifying enzyme.
Finding the ogc genes allowed us to predict gene functions, most of which were validated by single- or double-deletion mutagenesis followed by LC-MS analyses of the resulting glycopeptides. Thus, the OgcI protein was assigned as a member of the WecA family of phosphoglycosyltransferases, which catalyze the formation of undecaprenyl-diphosphate sugars typically utilizing UDP-GlcNAc or UDP-GalNAc as nucleotide sugar substrates (24). Our results support the assignment of OgcB as the glycosyltransferase adding the second GalNAc residue and predict OgcA is responsible for the addition of the terminal Gal. The inability to generate ΔogcA suggests that absence of this gene may be deleterious for bacterial cell viability. This interpretation is consistent with previous transposon sequencing analysis showing that insertions are unrepresented within the B. cenocepacia ogcA (40) and with a report indicating the B. pseudomallei K96243 ogcA homologue (BPSL2668) is essential for survival (41). It is likely that without the terminal Gal the incomplete O-linked disaccharide glycan cannot be either effectively translocated across the membrane or processed by PglL, leading in both cases to the accumulation of Und-PP that cannot be recycled thus becoming growth-limiting. Further experiments are required to confirm the function of OgcA.
Both the O-glycan and the O-antigen in B. cenocepacia share GalNAc residues (25). Therefore, the epimerization of UDP-Glc to form UDP-Gal would be needed for O-glycan synthesis. Our demonstration that the ogcE gene product catalyzes both reactions explains why the deletion of this gene results both in truncation of O-antigen and loss of protein glycosylation. This scenario is similar to that of C. jejuni GalE, which contributes to the biosynthesis of lipooligosaccharide, capsular polysaccharide, N-linked glycosylation (42, 43), and N. meningitidis GalE, which plays a role in pilin glycosylation and lipooligosaccharide synthesis (44). A closer homologue of OgcE is the UDP-GalNAc–4-epimerase of Yersinia enterocolitica, named Gne, which was reported to be specific for UDP-GalNAc with practically no activity on nonacetylated substrates (45). However, we provide conclusive biochemical evidence that whereas B. cenocepacia OgcE has a slight preference for nonacetylated substrate UDP-Gal it can also interconvert UDP-GalNAc efficiently.
We also demonstrate that the general O-glycosylation pathway is a conserved feature of the Burkholderia genus. This conclusion is supported by multiple evidence. First, the ogc genes are conserved and collinear in most of the Burkholderia species for which genomic sequencing is available, with only few exceptions where some of genes were in different genomic locations. Second, analysis of the O-glycosylation glycoproteome in a subset of B. thailandensis, B. gladioli, and B. pseudomallei isolates confirms the production of a similar trisaccharide structure in these species. Third, B. cenocepacia, B. thailandensis, and B. gladioli protein lysates reveal polypeptides that react with the peanut agglutinin, a lectin that recognizes Gal-β(1–3)–GalNAc terminal disaccharide of the O-glycan moiety.
The conservation of the glycosylation pathway in Burkholderia raises the question of the role of protein glycosylation in the biology of these bacteria, which despite their ability to cause infection in humans and certain domestic animals are also widespread in the environment (1). We provide evidence that loss of protein glycosylation causes global metabolic defects in B. cenocepacia concerning the utilization of several different carbon sources and tolerance to osmotic and oxidative stress. Therefore, general protein O-glycosylation is required for metabolic fitness, possibly because it is involved in the stability of its protein targets, as shown in C. jejuni (46, 47). We also found that not only are glycosylation–defective mutants less virulent in the Galleria mellonella infection model, but also that the pathogen–host interaction at the cellular level is different. Indeed, the level of expression of antimicrobial peptides is higher and temporally faster in larvae infected with the glycosylation–defective mutants. Delayed expression of antimicrobial peptides toward infection with the WT strain could be due to the rapid replication of the bacterium inside the infection model, suggesting that B. cenocepacia may need to reach a certain threshold to cause overwhelming induction of G. mellonella immune response, which leads to larval death. This can also be explained by the higher sensitivity of ΔpglL to oxidative stress, which is also induced upon Galleria infection. Furthermore, a relation between defective transporters and growth in invertebrate cells was demonstrated for Listeria monocytogenes implying that certain nutritional sources are also important substrates for bacterial growth in host cells (48). Potentially, the ΔpglL defect in utilizing several carbon sources suggests that loss of glycosylation affects the function of proteins involved in nutrient transport across the periplasmic space, and it could be another reason for the reduced virulence in G. mellonella. Therefore, loss of the general protein O-glycosylation system would make Burkholderia more susceptible to clearance by the host's innate immune response, suggesting protein O-glycosylation could be considered as a novel target to develop inhibitors for possible clinical applications.
Another significant question arising from the conservation of the Burkholderia glycosylation pathway is whether Burkholderia glycoproteins are recognized by the human immune system. Burkholderia human infections represent a health risk to susceptible patient groups such as those with cystic fibrosis, as well as patients in endemic areas where B. pseudomallei prevails. Being multidrug-resistant pathogens compounds this problem because Burkholderia infections are also difficult to treat (49). We show that the vast majority of serum samples from cystic fibrosis patients infected with B. cenocepacia and B. multivorans, as well as from patients with melioidosis and glanders, have antibodies that react with the O-glycosylated DsbA1 protein. This suggests that either the Burkholderia glycan or the combination of the glycan and the target protein at the glycosylation site are epitopes specifically recognized by the immune system, paving the way to future research to determine whether these antibodies afford protection against infection.
In summary, this study has comprehensively characterized a conserved general protein O-glycosylation pathway in the Burkholderia genus demonstrating the relevance of protein glycosylation in the biology of B. cenocepacia and by extension in this group of bacteria widespread in multiple environmental niches and also responsible for opportunistic infections.
Experimental procedures
Strains and growth conditions
Strains and plasmids used in this study are listed in Table S3. Bacteria were grown at 37 °C in Luria-Bertani (LB) medium. Antibiotics were used at the following final concentrations: 50 μg of trimethoprim ml−1 for E. coli and 100 μg ml−1 for B. cenocepacia; 30 μg of tetracycline ml−1 for E. coli and 100 μg ml−1 for B. cenocepacia; and 40 μg of kanamycin ml−1 for E. coli. Ampicillin at 200 μg ml−1 and polymyxin at 20 μg ml−1 were used to select against donor and helper E. coli strains in triparental mating. Antibiotics and chemicals were purchased from Sigma-Aldrich (UK).
Recombinant DNA methods and deletion mutagenesis
The primers used are listed in Table S4. DNA ligations, restriction endonuclease digestions, and agarose gel electrophoresis were performed according to standard techniques (50) or by Gibson assembly (51). Restriction enzymes, Antarctic phosphatase, and T4 DNA ligase were purchased from New England Biolabs (Ipswich, MA) and used as recommended by the manufacturer. E. coli GT115 and DH5α cells were transformed by the calcium chloride method (50). PCR amplifications were carried out using the HotStar HiFidelity polymerase (Qiagen). Colony-PCR was performed with Taq polymerase (Qiagen). Amplifications reactions were optimized for each primer pair. DNA sequencing was performed at the sequencing facility in GATC Biotech (London, UK). Unmarked, nonpolar gene deletion mutants were constructed as described previously (52) and verified by DNA sequencing of PCR amplicons spanning the deletion end points.
Purification of BCAL2640
A plasmid constitutively expressing the BCAL2640 protein was constructed by amplifying the gene with primers Q539 and Q540. The resulting amplicon was digested with NdeI and XbaI and ligated into a similarly digested pDA12 resulting in pDA12-BCAL2640, which was introduced into K56-2 WT, ΔpglL, and Δogc by triparental mating (52). Exconjugants were isolated by plating on LB agar plates supplemented with 100 μg of tetracycline ml−1, 200 μg of ampicillin ml−1, 20 μg of polymyxin ml−1. Cultures of pDA12-BCAL2640 expressed in WT, ΔpglL, or Δogc were grown overnight at 37 °C. Bacteria were harvested and lysed using a cell disrupter (Constant Systems Ltd., Northants, UK) at 18,000 p.s.i. Supernatants containing soluble BCAL2640 protein were incubated with 0.2 m Ni2+-coated Sepharose beads (GE Healthcare Life Sciences, UK) overnight at 4 °C with mixing. The beads were washed, and the His-tagged BCAL2640 was eluted twice with 250 mm imidazole. Purified BCAL2640 was run on 14% SDS-PAGE and stained by PageBlue Protein Staining Solution (Thermo Fisher Scientific, UK). Gel bands were excised and prepared for MS analysis.
Purification of DsbA1
Plasmid pMF22 (pMLBAD-DsbA1) was introduced into WT B. cenocepacia K56-2, ΔpglL, Δogc, ΔogcX, ΔogcI, ΔogcAB, ΔogcE, and ΔBCAL3119–3131 by triparental mating (52). Exconjugants were selected on LB agar plates supplemented with 100 μg of trimethoprim ml−1. For the purification of the lipoprotein DsbA1, stationary phase bacterial cultures were harvested at 12,000 × g for 15 min at 4 °C and resuspended in buffer A (1 μg ml−1 DNase I, complete EDTA-free protease inhibitor mixture, 2% Triton X-114 in PBS). Bacteria were lysed with the cell disrupter, as described above. The supernatant was incubated at 37 °C until phase separation was complete. The aqueous phase was removed after centrifugation at 10,000 × g for 10 min at 30 °C. The remaining detergent layer containing the glycosylated lipoproteins was diluted to the original solution volume with buffer A without detergent for 1 h. Ten millimolar imidazole and 300 mm NaCl were added to the detergent phase prior to the addition of an appropriate volume of Ni2+-coated Sepharose beads equilibrated in buffer B (2% Triton X-114 and 30 mm NaCl in PBS). This solution was allowed to mix overnight at 4 °C. The beads were packed and then washed three times with buffer B containing 50 mm imidazole. The selected His-tagged DsbA1 was eluted twice, with 2 column volumes, with buffer B containing 300 mm imidazole. Purified DsbA1 was resolved on 14% SDS-PAGE and stained by PageBlue protein-staining dye for MS analysis. Purified DsbA1 was either concentrated by 10% TCA precipitation overnight or passed on detergent removal spin column (Thermo Fisher Scientific) prior to ELISA and NMR analysis, respectively.
Protein manipulation and immunoblotting
Whole-cell lysates were prepared from overnight cultures of K56-2 and all mutants containing pAMF22 and induced by 0.2% arabinose overnight at 37 °C in the presence of 100 μg of trimethoprim ml−1. Two hundred μl of cells with OD600 of 1 were pelleted, then resuspended in 1× sample buffer with 5% β-mercaptoethanol, and boiled for 10 min. Protein separation was performed by 14% SDS-PAGE. His-tagged DsbA1 was revealed by immunoblotting using a 1:10,000 dilution of the mouse anti-His mAb (GE Healthcare Life Sciences, UK). Proteins were visualized using a Licor IR Imaging System with Odyssey software. These experiments were replicated three times.
In-gel digestion of proteins
Gel-separated proteins were processed as described previously (53) with minor modifications. Briefly, gel bands of interest were excised and destained in a 50:50 solution of 50 mm NH4HCO3 (pH 8.0), 100% ethanol for 20 min at room temperature with shaking at 750 rpm, and destained samples were then washed with 100% ethanol and vacuum-dried to dryness. Dried samples were then rehydrated in 10 mm DTT in 50 mm NH4HCO3 and reduced for 60 min at 56 °C with shaking. Following reduction, samples were washed twice in 100% ethanol for 10 min to ensure the complete removal of DTT and vacuum-dried to dryness. Reduced samples were rehydrated in 55 mm iodoacetamide in 50 mm NH4HCO3 in the dark for 45 min at room temperature. Following alkylation, samples were washed twice with 100% ethanol and vacuum-dried. Dried alkylated samples were then rehydrated with 12 ng/μl trypsin (Promega, Madison WI) in 40 mm NH4HCO3 at 4 °C for 1 h. Following rehydration, excess trypsin was removed, and gel pieces were covered in 40 mm NH4HCO3 and incubated overnight at 37 °C. Peptide samples were extracted from the gel sample twice using 4 gel volumes of 30% ethanol, 3% acetic acid followed by 4 gel volumes of 100% ethanol with the supernatant from each extraction pooled. The resulting peptide mixtures were dried down, desalted using C18 stage tips (54), and stored on tips at 4 °C. Peptides were eluted in buffer B (80% acetonitrile, 0.1% formic acid) and dried down before analysis by LC-MS.
Generation of whole-cell lysates for proteome analysis
Burkholderia strains of interest were grown overnight on confluent LB plates. Plates were flooded with 5 ml of pre-chilled sterile PBS, and colonies were removed with a cell scraper. Cells were washed three times in PBS and collected by centrifugation at 10,000 × g at 4 °C and then snap-frozen. Frozen whole-cell samples were resuspended in 4% SDS, 100 mm Tris (pH 8.0), 20 mm DTT, and boiled at 95 °C at 2000 rpm for 10 min. Samples were clarified by centrifugation at 17,000 × g for 10 min; supernatant was collected, and protein concentration was determined by bicinchoninic acid assay (Thermo Fisher Scientific Pierce). 200 μg of protein from each sample was acetone-precipitated by mixing 4 volumes of ice-cold acetone with 1 volume of sample. Samples were precipitated overnight at −20 °C and then spun down at 16,000 × g for 10 min at 0 °C. The precipitated protein pellets were resuspended with 80% ice-cold acetone and precipitated for an additional 4 h at −20 °C. Samples were centrifuged at 17,000 × g for 10 min at 0 °C, and the supernatant was discarded, and excess acetone was driven off at 65 °C for 5 min.
Digestion of complex protein lysates
Dried protein pellets were resuspended in 6 m urea, 2 m thiourea, 40 mm NH4HCO3 and reduced/alkylated prior to digestion with Lys-C (1:200 w/w) and trypsin (1:50 w/w) overnight (55). Digested samples were acidified to a final concentration of 0.5% formic acid and desalted with homemade high-capacity StageTips composed of 5 mg of EmporeTM C18 material (3M, Maplewood, Minnesota) and 5 mg of OLIGO R3 reverse-phase resin (Thermo Fisher Scientific) according to the protocols of Ishihama et al. (56) and Rappsilber et al. (57). Bound peptides were eluted with buffer B, dried, and stored at −20 °C.
Liquid chromatography and mass spectrometry analysis
Prior to LC-MS analysis, samples were resuspended in 15 μl of buffer A (2% acetonitrile, 0.1% formic acid). LC-MS was performed on either an Agilent 1290 Series HPLC (Agilent Technologies, Mississauga, Ontario, Canada) coupled to LTQ-Orbitrap Velos (Thermo Fisher Scientific, San Jose CA), a Dionex Ultimate 3000 UPLC (Thermo Fisher Scientific) coupled to an Orbitrap Elite, or an EASY-nLC1000 system coupled to a Q-Exactive. For the LTQ-Orbitrap Velos and Q-Exactive, LC-MS was accomplished using a two-column system in which samples were concentrated prior to separation onto a 2-cm-long, 100-μm inner diameter fused silica trap column containing 5.0-μm Aqua C-18 beads (Phenomenex) and then separated using an in-house packed C18 analytical 75-μm inner diameter × 360-μm outer diameter column composed of 35 cm of ReproSil-Pur C18 AQ 1.9bμm (Dr. Maisch, Ammerbuch-Entringen, Germany) column for the EASY-nLC1000 system or a 20-cm ReproSil-Pur C18 AQ 3.0 μm for the Agilent 1290 Series HPLC. Samples were concentrated onto the trap for 10 min using 100% buffer A at 5 μl/min after which the gradient was altered from 100% phase A to 40% buffer B over 90 min at 250 nl/min with the eluting peptides infused directly into the mass spectrometers via nano-electrospray ionization. For the Orbitrap Elite, LC-MS was accomplished using a two-column chromatography setup comprising a PepMap100 C18 20 mm × 75-μm trap and a PepMap C18 500 mm × 75-μm analytical column (Thermo Fisher Scientific). Samples were concentrated onto the trap column at 5 μl/min for 5 min and infused into an Orbitrap Elite at 300 nl/min via the analytical column. One hundred and 80-min gradients were run altering the buffer composition from 1% buffer B to 28% B over 145 min, then from 28% B to 40% B over 10 min, then from 40% B to 100% B over 2 min, and the composition was held at 100% B for 3 min and then dropped to 3% B over 5 min and held at 3% B for another 15 min. All instruments were operated in a data-dependent manner using Xcalibur version 2.2 (Thermo Fisher Scientific). For samples analyzed on the LTQ-Velos, one full precursor scan in the Orbitrap (resolution 60,000; 500–2000 Th, AGC target of 1 × 106) was followed by the selection of the top most intense and multiply charged ions above 5000 counts for CID (normalized collision energy 35, AGC of 4 × 104) followed by HCD (resolution 7500, normalized collision energy 40, AGC of 2 × 105) with 30 s of dynamic exclusion enabled as described previously (55). For samples analyzed on the Orbitrap Elite, one full precursor scan in the Orbitrap (resolution 60,000; 500–2,000 Th, AGC target of 1 × 106) was followed by the selection of the five most intense and multiply charged ions above 10,000 counts for CID (normalized collision energy 35, AGC of 4 × 104) followed by HCD (resolution 15,000, normalized collision energy 40, AGC of 2 × 105) with 45 s of dynamic exclusion enabled. For samples analyzed on the Q-Exactive, one full precursor scan (resolution 70,000; 350–2000 m/z, AGC target of 3 × 106) was followed by 10 data-dependent HCD MS-MS events (resolution 35,000, normalized collision energy of 25 with 50% stepping, AGC target of 1 × 106) with 25 s of dynamic exclusion enabled. Area of the most intense charge state of each species was used to assess the relative levels/distribution of observed peptides.
Glycopeptide identification
Raw data files were processed with Proteome Discover version 1.4 (Thermo Fisher Scientific) to generate mgf files. The resulting mgf files were searched using MASCOT version 2.4 (Matrix Science, provided by the BC Proteomics Network). Searches were carried out using semi-trypsin specificity, carbamidomethylation of cysteine as a fixed modification, and oxidation (M) as a variable modification. A precursor and product tolerance of 20 ppm was used, and B. cenocepacia data were searched against the B. cenocepacia K56-2Valvano proteome (http://www.uniprot.org/taxonomy/985076, downloaded from NCBI February 15, 2013),6 whereas the non-B. cenocepacia species were searched against the NCBI proteome database (downloaded September 1, 2016) with the taxonomy restricted to “Other Proteobacteria.” Scan events that did not result in peptide identification were manually inspected and identified as possible glycopeptides based on the presence of the diagnostic oxonium ion 204.09 m/z of HexNAc or 188.09 m/z in the case of QuiNAc-modified peptides. To facilitate glycopeptide assignments from HCD scans, the ions below the mass of the predicted deglycosylated peptides were extracted with Xcalibur version 2.2 using the Spectrum list function. Ions with a deconvoluted mass above that of the deglycosylated peptide and ions corresponding to known carbohydrate oxoniums were removed in a similar approach to post-spectral processing of electron-transfer dissociation data (58) and searched as above with Mascot. All spectra were searched with the decoy option enabled with all peptides passing a 1% false discovery rate. Identified glycopeptide spectra were manually inspected and spectra annotated according to the nomenclature of Roepstorff and Fohlman (59) (supporting Data S1–S4). Glycopeptides derived from clinical Burkholderia strains were matched to either the B. gladioli reference strain BSR3 or the B. pseudomallei reference strain K96243.
Structural characterization of the protein glycan moiety
DsbA1, heterologously expressed in B. cenocepacia and obtained from TCA precipitation, was dissolved in water (1 ml), and 2 aliquots of proteinase K (0.3 mg each, Sigma P-2308) were added at 37 °C, at 8 h intervals, with last addition left overnight. The solution was directly loaded on a column packed with Bio-Gel P10 (d = 1.5 cm, h = 118 cm) using water as eluent (flow = 16 ml/h) and monitoring the eluate with a refractive index detector (Knauer K-2301). Fractions were pooled and monitored by 1H NMR, and the fraction was enriched in the glycopeptide was eluted after ∼40% of the total column volume. NMR analyses were performed on a Bruker 600 MHz equipped with a cryo-probe. Spectra were recorded at 15 °C using acetone as internal standard (1H 2.225 ppm, 13C 31.45 ppm). 2D spectra (DQF-COSY, TOCSY, T-ROESY, and gHSQC) were recorded using Bruker software (TopSpin 2.1). Homonuclear experiments were recorded using 512 FIDs of 2048 complex points and 40 scans per FID, and for TOCSY and T-ROESY spectra a mixing time of 100 and 300 ms was applied, respectively. HSQC spectrum was acquired with 512 FIDs of 2048 complex point and 60 scans per FID, and spectra were processed and analyzed using Bruker TopSpin 3 program.
Extraction and analysis of B. cenocepacia lipopolysaccharide
Proteinase K–treated whole-cell lysates were resolved by SDS-PAGE (16% v/v polyacrylamide) and visualized following silver staining (60). Complementation of O-antigen synthesis in Δogc was done by conjugating pXO23 (pAP20::ogcE) into the mutant by triparental mating, and exconjugants were counterselected on 100 μg of chloramphenicol ml−1, 200 μg of ampicillin ml−1, and 20 μg of polymyxin ml−1.
OgcE enzymatic assay
BCAL3117 was amplified by primers Q729 and Q728, cut by restriction enzymes NdeI and EcoRI, and cloned in pET28 vector containing an N-terminal His6-tag that had been cut with the same restriction enzymes. Expression was induced by 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 3 h at 37 °C for a 300-ml culture. The cells were harvested by centrifugation and lysed by passage in a cell disruptor after resuspension in buffer A: 50 mm Tris-HCl, 100 mm NaCl. OgcE was purified using a 3-ml Fast Flow chelating Sepharose column loaded with Ni2+ and equilibrated in buffer A. After extensive washing with buffer A, OgcE was eluted with increasing concentrations of imidazole (100–400 mm). The purified enzyme was preserved by addition of glycerol (25% final concentration) and frozen at −20 °C. Reactions were set on four substrates: UDP-Gal, UDP-Glc, UDP-GalNAc, and UDP-GlcNAc each with NADP+ or NAD+. Reactions were done in 200 mm Tris (pH 8), with 0.1 mm substrate and 0.1 mm cofactor in a volume of 10 μl, with 7.3 μl of enzyme fraction (comprising ∼5 μg of enzyme) and incubated for 4 h at 37 °C unless stated otherwise in the legends to the figures. The reactions were analyzed by capillary electrophoresis (PACE MDQ, Beckman) using a bare silica capillary, 200 mm Borax buffer (pH 9), and UV detection at 254 nm (61). The % of conversion was obtained by integrating the substrate and product peak surface areas using the 32Karat software.
Oxidative and osmolar susceptibility testing
Sensitivity toward different concentrations of hydrogen peroxide (Sigma) was assessed using the bioscreen. Bacterial cultures were diluted to an OD600 of 0.01 in LB and dispensed in 100-well plates in 270-μl volumes. 30-ml aliquots of hydrogen peroxide in different concentrations were added to the bacterial cultures. Plates were incubated in a Bioscreen C automated growth curve reader for 16 h with OD600 readings taken every 1 h. The same procedure was employed to determine the susceptibility toward high concentrations of NaCl using glucose as a carbon source.
Survival assay was also made after challenging bacterial cultures in LB with hydrogen peroxide at concentrations of either 300 μm with bacterial inoculum of 8 × 106 CFU/ml or 1 mm with bacterial inoculum of 8 × 107 for 1 h at 37 °C with shaking. Samples were withdrawn and serially diluted in PBS. Then, 10-μl portions were dropped onto the surface of LB agar plates. The plates were incubated at 37 °C for 24 h; the resulting colonies were counted, and the % of survival was determined relative to parallel-untreated control samples.
Phenotypic microarray experiments
The metabolic phenome of B. cenocepacia K56-2 and ΔpglL was assessed using pre-configure Biolog Phenotype MicroArrays (PMs) from Biolog Inc., Hayward, CA. We used PM plates 1 and 2, which contain 190 of the most common carbon sources in 96-well arrangements composed of one negative control well and 95 wells prefilled with a given carbon source in a dried state. PM experiments were performed following the standard protocol (62). Briefly, B. cenocepacia strains were grown on LB agar plates for 36 h at 37 °C. A cell suspension of 95% transmittance in Biolog solution IF-0 was prepared by resuspending bacteria grown on LB agar plates. Dye A was then added, and then 100 μl/well were transferred to each of the 96-well PM microplates (a set of 95 substrates and one blank well). The plates were incubated at 37 °C for 48 h. The optical density (OD600) values were then measured using a microtiter plate reader. Each experiment was performed as biological duplicates and growth differences were established by comparing the area under the growth curve (AUC). Growth in a given carbon source was considered positive when median AUC was more than 40% higher than negative control AUC. A cutoff value of 80% was used to determine a significant growth reduction of the mutant compared with WT. Hits ranged from reduction in growth from 80% up to less than 60% (Fig. 8A). Positive results were subsequently validated by the performing growth studies using the automated Bioscreen C in M9 minimal media to test the utilization of selected carbon sources compared with PM1 and PM2 plates. Bacterial cultures were diluted to approximately an OD600 of 0.01 in M9 minimal media deprived of carbon sources and dispensed in 100-well plates in 270-μl volumes. Each required carbon source was tested individually by adding 30 μl of a stock solution to each to the 100-well Bioscreen plate to give final concentrations of 0.2% (w/v) in the wells. Plates were incubated in a Bioscreen C automated growth curve analyzer for 48 h, and bacterial growth was assessed turbidimetrically at 600 nm every 2 h. Comparisons were also made by determining the AUC obtained over four biological replicas in triplicate.
Proteolytic activity
To determine protease activity, overnight cultures grown in LB were diluted to an OD600 of 1, and 3 μl of this culture was spotted onto a nutrient agar plate containing 1.5% skim milk (63). The plates were incubated for 48 h at 37 °C, after which the radii of the cleared zones surrounding the colonies were measured.
G. mellonella larvae infection
G. mellonella wax moth larvae were acquired from UK Waxworms Ltd., and stored in wood shavings in the dark at 16 °C prior to infection. Larvae with approximate weight of 250–350 mg were used. Bacteria were grown in 5 ml of LB, harvested during exponential phase, resuspended in sterile PBS, and serially diluted. The surface of the larvae was disinfected by ethanol (70%), and then the larvae were injected with 10 μl of bacterial suspension, containing ∼8 × 104 CFU/ml of either K56-2 or the tested mutants, into the last right proleg by use of a Hamilton syringe with a 26-gauge needle. A group of 10 control larvae were injected with 10 μl of PBS in parallel. Larvae were kept at 37 °C in the dark. Larval survival was monitored at 24-h intervals over a period of 72 h and was judged based on visual appearance and lack of movement in response to stimuli. Three independent experiments were performed.
Determination of in vivo bacterial loads in G. mellonella
Larvae were infected with ∼8 × 104 CFU of K56-2 or ΔpglL or Δogc. Groups of three insects were collected at 24 and 48 h, and hemolymph samples from three larvae (∼100 μl) were collected in microcentrifuge tubes containing 10 μl of a saturated solution of N-phenylthiourea (Sigma-Aldrich, UK). Serial dilutions of the homogenate in PBS were plated on LB agar supplemented with 100 μg ml−1 spectinomycin, and colonies were counted after incubation at 37 °C for 24 h. Three independent experiments were performed. No CFU were recovered from noninfected larvae in LB agar supplemented with spectinomycin.
RNA extraction and RT-PCR
Larvae were infected with ∼1 × 104 CFU of exponentially growing bacterial cultures. At 8 and 18 h post-infection, three larvae from either the control PBS group or the infected group were homogenized on ice with 1 ml of TRI-reagent (Ambion), using Stuart homogenizer SHM2 (Bibby Scientific Ltd., Staffordshire, UK). Total RNA was purified by a standard chloroform/isopropyl alcohol protocol, and the obtained RNA was further purified using a Nucleospin RNAII kit (Macherey-Nagel) that included one step of on-column DNase treatment, following the manufacturer's instructions. The quantification of purified RNA samples was performed with a nanodrop (Nanovue Plus, GE Healthcare). cDNA was obtained from 1 μg of total RNA by using a commercial Moloney murine leukemia virus reverse transcriptase (Invitrogen, UK). Real-time PCR (RT-PCR) analyses were performed with an Mx3005p qPCR system (Agilent Technologies, UK). One hundred nanograms of cDNA were used as the template in a 20-μl reaction mixture containing KapaSYBR Fast qPCR mix (Kapa Biosystems) and primer mix. Actin and 18S rRNA genes were amplified as housekeeping genes. The primers used are listed in Table S1. The thermocycling protocol was as follows: 95 °C for 3 min for hot-start polymerase activation, followed by 45 cycles of denaturation at 95 °C for 15 s and annealing at 60 °C for 30 s. SYBR Green dye fluorescence was measured at 521 nm during the annealing phase. Fold changes in gene expression were calculated using the Livak method (ΔΔCT) (64) with normalization to the actin gene (supporting Data S6).
ELISA
Antibodies against glycosylated and unglycosylated DsbA1 were detected by indirect ELISA. Ninety six-well Nunc MaxiSorp plates were coated with 50 μl of purified DsbA1 protein expressed in K56-2 WT (glycosylated) or in ΔpglL (nonglycosylated) and diluted in coating buffer (100 mm carbonate/bicarbonate, pH 9.6) to reach a final concentration of 2–4 μg/ml. Control wells contained only coating buffer. The plates were covered by plastic adhesive film and incubated at 4 °C overnight. Coating solution was removed, and plates were washed with 300 μl of PBS/Tween 20 (0.05%). Additional blocking was achieved by adding 300 μl of blocking buffer (5% BSA). Plates were covered and incubated at room temperature for 1 h and then washed three times with PBS/Tween 20. Fifty microliters of serum samples, diluted in half-strength blocking buffer, were added to the wells and incubated for 90 min at room temperature. Normal human serum from noninfected individuals was used as a negative control. Plates were washed four times with PBS/Tween 20. Fifty microliters of biotinylated rabbit anti-human IgG secondary antibody diluted to 1:20,000 (1 μl in 20 ml PBS) were added and incubated for 1 h at room temperature. Plates were washed five times with PBS/Tween 20. Fifty microliters of streptavidin/horseradish peroxidase diluted 1:300 in PBS were then added to the wells and incubated for 1 h in the dark at room temperature. After washing four times with PBS/Tween 20, 50 μl of the substrate solution 3,3′,5,5′-tetramethylbenzidine were added per well and incubated in the dark at room temperature. After sufficient color development, 30 μl of stop solution (3 m HCl) were added, and the absorbance of each well was read with POLARstar Omega microplate reader (BMG LABTECH, Ortenberg, Germany) at 450 nm. Three independent repeats were made for every sample, each done in duplicate. A positive ELISA test was defined by an absorbance reading exceeding the cutoff value computed by two standard deviations above the mean of the negative control. We also performed a confirmatory reverse ELISA was done, in which the glycan-specific antibodies from positive serum samples were adsorbed using the glycosylated DsbA1 protein by incubating the samples with the glycosylated protein at a concentration of 100 μg/ml for 2 h at room temperature and overnight at 4 °C for three successive passages. Serum samples were then collected and used in a normal ELISA procedure as described above in parallel to the same samples without adsorption. Results are presented in ELISA units in which a cutoff of 2× standard deviation of the mean of the negative control was used to decide whether a serum sample is reactive toward the glycan or not.
Ethics
Patients with a confirmed diagnosis of CF were recruited between 2010 and 2014 during routine outpatient appointments at the adult CF Centre in the Belfast Health and Social Care Trust (Belfast City Hospital) and provided written informed consent for the provision of serum samples. The study was approved by the Office for Research Ethics Committees Northern Ireland (10/NIR01/41) and co-sponsored by the Belfast Health and Social Care Trust and Queen's University Belfast (10067SE-OPMS). Samples from patients attending the Manchester Adult CF Clinic were collected under the framework of the Manchester Respiratory and Allergy Biobank (ManRAB) (full ethical approval was provided by the ManRAB ethics committee; REC reference: 10/H1010/7). Blood samples were collected from adult patients with melioidosis who had received written information before signing the consent form, approved by the Khon Kaen University Ethics Committee for Human Research No. HE561234. The convalescent human glanders serum was obtained under USAMRIID protocol FY00-15. The investigators adhered to the policies regarding the protection of human subjects as prescribed by 45 CFR 46 and 32 CFR 219 (Protection of Human Subjects). Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the United States Army or any of the granting agencies that supported this study. In all of the above cases obtaining serum samples, proper handling and anonymity were conducted based on the Declaration of Helsinki principles.
Author contributions
Y. F. M., N. E. S., C. C., X. O., and M. A. V. formal analysis; Y. F. M., N. E. S., R. I., and M. A. V. validation; Y. F. M., N. E. S., C. C., X. O., R. I., C. D. C., and M. A. V. investigation; Y. F. M., A. M., and C. D. C. visualization; Y. F. M., N. E. S., A. M., C. C., X. O., D. D., B. J. C., L. J. F., R. I., and C. D. C. methodology; Y. F. M., N. E. S., and C. D. C. writing-original draft; Y. F. M., N. E. S., C. C., X. O., G. L., M. M. T., H. G., A. M. J., D. D., B. J. C., L. J. F., R. I., C. D. C., and M. A. V. writing-review and editing; N. E. S., G. L., M. M. T., H. G., A. M. J., D. D., and B. J. C. resources; N. E. S., A. M., and C. D. C. software; H. G. data curation; B. J. C., L. J. F., R. I., and M. A. V. supervision; M. A. V. conceptualization; M. A. V. funding acquisition; M. A. V. project administration.
Supplementary Material
Acknowledgments
We thank M. Feldman for the plasmid pAMF22; J. Torres Bustos for technical assistance with the Omnilog; B. Oickle and K. Patel for OgcE expression and purification; T. Stinear, B. Howden, and S. Pidot for providing clinical B. gladioli strains; and M. Mayo, V. Theobald, and M. Barnes for isolate selection, curation, and sample preparation of genetically diverse clinical B. pseudomallei strains.
This work was supported in part by a grant from the Canadian Institutes of Health Research (to M. A. V. and C. C.) and from the UK Medical Research Council Confidence in Concept Project CD1617-CIC04 (to M. A. V.). The authors declare that they have no conflicts of interest with the contents of this article.
This paper is dedicated to the memory of Professor Mary Jane Osborn, deceased on January 17, 2019, for her pioneering work in microbial glycobiology.
This article contains Figs. S1–S7, Tables S1–S4, and supporting Data S1–S6.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- OTase
- oligosaccharyltransferase
- HCD
- higher-energy collisional dissociation
- PGRP
- peptidoglycan recognition protein B
- CID
- collision-induced dissociation
- qPCR
- quantitative PCR
- HSQC
- heteronuclear single quantum coherence
- TOCSY
- total correlation spectroscopy
- T-ROESY
- transverse rotating-frame Overhauser enhancement spectroscopy
- LPS
- lipopolysaccharide
- AGC
- automatic gain control
- Th
- thomson
- PM
- Phenotype MicroArray
- AUC
- area under growth curve
- CFU
- colony-forming unit
- CF
- cystic fibrosis
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- Und-PP
- undecaprenyl-PP
- FIF
- free induction decay.
References
- 1. Coenye T., and Vandamme P. (2003) Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ. Microbiol. 5, 719–729 10.1046/j.1462-2920.2003.00471.x [DOI] [PubMed] [Google Scholar]
- 2. Eberl L., and Vandamme P. (2016) Members of the genus Burkholderia: good and bad guys. F1000Res. 5, F1000 Faculty Rev–1007 10.12688/f1000research.8221.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scoffone V. C., Chiarelli L. R., Trespidi G., Mentasti M., Riccardi G., and Buroni S. (2017) Burkholderia cenocepacia infections in cystic fibrosis patients: drug resistance and therapeutic approaches. Front. Microbiol. 8, 1592 10.3389/fmicb.2017.01592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lipsitz R., Garges S., Aurigemma R., Baccam P., Blaney D. D., Cheng A. C., Currie B. J., Dance D., Gee J. E., Larsen J., Limmathurotsakul D., Morrow M. G., Norton R., O'Mara E., Peacock S. J., et al. (2012) Workshop on treatment of and postexposure prophylaxis for Burkholderia pseudomallei and B. mallei infection, 2010. Emerg. Infect. Dis. 18, e2 10.3201/eid1812.120638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Losada L., Ronning C. M., DeShazer D., Woods D., Fedorova N., Kim H. S., Shabalina S. A., Pearson T. R., Brinkac L., Tan P., Nandi T., Crabtree J., Badger J., Beckstrom-Sternberg S., Saqib M., et al. (2010) Continuing evolution of Burkholderia mallei through genome reduction and large-scale rearrangements. Genome Biol. Evol. 2, 102–116 10.1093/gbe/evq003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Limmathurotsakul D., Golding N., Dance D. A. B., Messina J. P., Pigott D. M., Moyes C. L., Rolim D. B., Bertherat E., Day N. P. J., Peacock S. P., and Hay S. I. (2016) Predicted global distribution of Burkholderia pseudomallei and burden of melioidosis. Nat. Microbiol. 1, 15008 10.1038/nmicrobiol.2015.8 [DOI] [PubMed] [Google Scholar]
- 7. Wiersinga W. J., Currie B. J., and Peacock S. J. (2012) Melioidosis. N. Engl. J. Med. 367, 1035–1044 10.1056/NEJMra1204699 [DOI] [PubMed] [Google Scholar]
- 8. Wiersinga W. J., Virk H. S., Torres A. G., Currie B. J., Peacock S. J., Dance D. A. B., and Limmathurotsakul D. (2018) Melioidosis. Nat. Rev. Dis. Primers 4, 17107 10.1038/nrdp.2017.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Currie B. J., Ward L., and Cheng A. C. (2010) The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl. Trop. Dis. 4, e900 10.1371/journal.pntd.0000900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rhodes K. A., and Schweizer H. P. (2016) Antibiotic resistance in Burkholderia species. Drug Resist. Updat. 28, 82–90 10.1016/j.drup.2016.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Limmathurotsakul D., Funnell S. G., Torres A. G., Morici L. A., Brett P. J., Dunachie S., Atkins T., Altmann D. M., Bancroft G., Peacock S. J., and Steering Group on Melioidosis Vaccine, D. (2015) Consensus on the development of vaccines against naturally acquired melioidosis. Emerg. Infect. Dis. 2015 21, 10.3201/eid2106.141480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eichler J., and Koomey M. (2017) Sweet new roles for protein glycosylation in prokaryotes. Trends Microbiol. 25, 662–672 10.1016/j.tim.2017.03.001 [DOI] [PubMed] [Google Scholar]
- 13. Schäffer C., and Messner P. (2017) Emerging facets of prokaryotic glycosylation. FEMS Microbiol. Rev. 41, 49–91 10.1093/femsre/fuw036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Valguarnera E., Kinsella R. L., and Feldman M. F. (2016) Sugar and spice make bacteria not nice: protein glycosylation and its influence in pathogenesis. J. Mol. Biol. 428, 3206–3220 10.1016/j.jmb.2016.04.013 [DOI] [PubMed] [Google Scholar]
- 15. Nothaft H., and Szymanski C. M. (2010) Protein glycosylation in bacteria: sweeter than ever. Nat. Rev. Microbiol. 8, 765–778 10.1038/nrmicro2383 [DOI] [PubMed] [Google Scholar]
- 16. Spiro R. G. (2002) Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12, 43R–56R 10.1093/glycob/12.4.43R [DOI] [PubMed] [Google Scholar]
- 17. Stepper J., Shastri S., Loo T. S., Preston J. C., Novak P., Man P., Moore C. H., Havlíček V., Patchett M. L., and Norris G. E. (2011) Cysteine S-glycosylation, a new post-translational modification found in glycopeptide bacteriocins. FEBS Lett. 585, 645–650 10.1016/j.febslet.2011.01.023 [DOI] [PubMed] [Google Scholar]
- 18. Szymanski C. M., Yao R., Ewing C. P., Trust T. J., and Guerry P. (1999) Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol. Microbiol. 32, 1022–1030 10.1046/j.1365-2958.1999.01415.x [DOI] [PubMed] [Google Scholar]
- 19. Wacker M., Linton D., Hitchen P. G., Nita-Lazar M., Haslam S. M., North S. J., Panico M., Morris H. R., Dell A., Wren B. W., and Aebi M. (2002) N-Linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 298, 1790–1793 10.1126/science.298.5599.1790 [DOI] [PubMed] [Google Scholar]
- 20. Lizak C., Gerber S., Numao S., Aebi M., and Locher K. P. (2011) X-ray structure of a bacterial oligosaccharyltransferase. Nature 474, 350–355 10.1038/nature10151 [DOI] [PubMed] [Google Scholar]
- 21. Lithgow K. V., Scott N. E., Iwashkiw J. A., Thomson E. L., Foster L. J., Feldman M. F., and Dennis J. J. (2014) A general protein O-glycosylation system within the Burkholderia cepacia complex is involved in motility and virulence. Mol. Microbiol. 92, 116–137 10.1111/mmi.12540 [DOI] [PubMed] [Google Scholar]
- 22. Hug I., and Feldman M. F. (2011) Analogies and homologies in lipopolysaccharide and glycoprotein biosynthesis in bacteria. Glycobiology 21, 138–151 10.1093/glycob/cwq148 [DOI] [PubMed] [Google Scholar]
- 23. Lukose V., Walvoort M. T. C., and Imperiali B. (2017) Bacterial phosphoglycosyl transferases: initiators of glycan biosynthesis at the membrane interface. Glycobiology 27, 820–833 10.1093/glycob/cwx064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Valvano M. A. (2011) Common themes in glycoconjugate assembly using the biogenesis of O-antigen lipopolysaccharide as a model system. Biochemistry 76, 729–735 10.1134/S0006297911070029 [DOI] [PubMed] [Google Scholar]
- 25. Ortega X., Hunt T. A., Loutet S., Vinion-Dubiel A. D., Datta A., Choudhury B., Goldberg J. B., Carlson R., and Valvano M. A. (2005) Reconstitution of O-specific lipopolysaccharide expression in the Burkholderia cenocepacia strain J2315 that is associated with transmissible infections in patients with cystic fibrosis. J. Bacteriol. 187, 1324–1333 10.1128/JB.187.4.1324-1333.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oberto J. (2013) SyntTax: a web server linking synteny to prokaryotic taxonomy. BMC Bioinformatics 14, 4 10.1186/1471-2105-14-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winsor G. L., Khaira B., Van Rossum T., Lo R., Whiteside M. D., and Brinkman F. S. (2008) The Burkholderia genome database: facilitating flexible queries and comparative analyses. Bioinformatics 24, 2803–2804 10.1093/bioinformatics/btn524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gebhart C., Ielmini M. V., Reiz B., Price N. L., Aas F. E., Koomey M., and Feldman M. F. (2012) Characterization of exogenous bacterial oligosaccharyltransferases in Escherichia coli reveals the potential for O-linked protein glycosylation in Vibrio cholerae and Burkholderia thailandensis. Glycobiology 22, 962–974 10.1093/glycob/cws059 [DOI] [PubMed] [Google Scholar]
- 29. Bock K., and Pedersen C. (1983) Carbon-13 nuclear magnetic resonance spectroscopy of monosaccharides. Adv. Carbohydr. Chem. Biochem. 41, 27–66 10.1016/S0065-2318(08)60055-4 [DOI] [Google Scholar]
- 30. Cuthbertson L., Kos V., and Whitfield C. (2010) ABC transporters involved in export of cell surface glycoconjugates. Microbiol. Mol. Biol. Rev. 74, 341–362 10.1128/MMBR.00009-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ortega X., Silipo A., Saldías M. S., Bates C. C., Molinaro A., and Valvano M. A. (2009) Biosynthesis and structure of the Burkholderia cenocepacia K56-2 lipopolysaccharide core oligosaccharide: truncation of the core oligosaccharide leads to increased binding and sensitivity to polymyxin B. J. Biol. Chem. 284, 21738–21751 10.1074/jbc.M109.008532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Feldman M. F., Marolda C. L., Monteiro M. A., Perry M. B., Parodi A. J., and Valvano M. A. (1999) The activity of a putative polyisoprenol-linked sugar translocase(Wzx) involved in Escherichia coli O-antigen assembly is independent of the chemical structure of the O repeat. J. Biol. Chem. 274, 35129–35138 10.1074/jbc.274.49.35129 [DOI] [PubMed] [Google Scholar]
- 33. Ireland P. M., Marshall L., Norville I., and Sarkar-Tyson M. (2014) The serine protease inhibitor Ecotin is required for full virulence of Burkholderia pseudomallei. Microb. Pathog. 67, 55–58 10.1016/j.micpath.2014.01.001 [DOI] [PubMed] [Google Scholar]
- 34. El-Halfawy O. M., and Valvano M. A. (2014) Putrescine reduces antibiotic-induced oxidative stress as a mechanism of modulation of antibiotic resistance in Burkholderia cenocepacia. Antimicrob. Agents Chemother. 58, 4162–4171 10.1128/AAC.02649-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Royet J., Gupta D., and Dziarski R. (2011) Peptidoglycan recognition proteins: modulators of the microbiome and inflammation. Nat. Rev. Immunol. 11, 837–851 10.1038/nri3089 [DOI] [PubMed] [Google Scholar]
- 36. Vertyporokh L., and Wojda I. (2017) Expression of the insect metalloproteinase inhibitor IMPI in the fat body of Galleria mellonella exposed to infection with Beauveria bassiana. Acta Biochim. Pol. 64, 273–278 10.18388/abp.2016_1376 [DOI] [PubMed] [Google Scholar]
- 37. Andrejko M., Mizerska-Dudka M., and Jakubowicz T. (2008) Changes in Galleria mellonella lysozyme level and activity during Pseudomonas aeruginosa infection. Folia Microbiol. 53, 147–151 10.1007/s12223-008-0021-2 [DOI] [PubMed] [Google Scholar]
- 38. York G. M., and Walker G. C. (1998) The succinyl and acetyl modifications of succinoglycan influence susceptibility of succinoglycan to cleavage by the Rhizobium meliloti glycanases ExoK and ExsH. J. Bacteriol. 180, 4184–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bohin J. P. (2000) Osmoregulated periplasmic glucans in Proteobacteria. FEMS Microbiol. Lett. 186, 11–19 10.1111/j.1574-6968.2000.tb09075.x [DOI] [PubMed] [Google Scholar]
- 40. Gislason A. S., Turner K., Domaratzki M., and Cardona S. T. (2017) Comparative analysis of the Burkholderia cenocepacia K56-2 essential genome reveals cell envelope functions that are uniquely required for survival in species of the genus Burkholderia. Microb. Genom 3, 10.1099/mgen.0.000140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moule M. G., Hemsley C. M., Seet Q., Guerra-Assunção J. A., Lim J., Sarkar-Tyson M., Clark T. G., Tan P. B., Titball R. W., Cuccui J., and Wren B. W. (2014) Genome-wide saturation mutagenesis of Burkholderia pseudomallei K96243 predicts essential genes and novel targets for antimicrobial development. MBio 5, e00926–13 10.1128/mBio.00926-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bernatchez S., Szymanski C. M., Ishiyama N., Li J., Jarrell H. C., Lau P. C., Berghuis A. M., Young N. M., and Wakarchuk W. W. (2005) A single bifunctional UDP-GlcNAc/Glc 4-epimerase supports the synthesis of three cell surface glycoconjugates in Campylobacter jejuni. J. Biol. Chem. 280, 4792–4802 10.1074/jbc.M407767200 [DOI] [PubMed] [Google Scholar]
- 43. Fry B. N., Feng S., Chen Y. Y., Newell D. G., Coloe P. J., and Korolik V. (2000) The galE gene of Campylobacter jejuni is involved in lipopolysaccharide synthesis and virulence. Infect. Immun. 68, 2594–2601 10.1128/IAI.68.5.2594-2601.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stimson E., Virji M., Makepeace K., Dell A., Morris H. R., Payne G., Saunders J. R., Jennings M. P., Barker S., and Panico M. (1995) Meningococcal pilin: a glycoprotein substituted with digalactosyl 2,4-diacetamido-2,4,6-trideoxyhexose. Mol. Microbiol. 17, 1201–1214 10.1111/j.1365-2958.1995.mmi_17061201.x [DOI] [PubMed] [Google Scholar]
- 45. Bengoechea J. A., Pinta E., Salminen T., Oertelt C., Holst O., Radziejewska-Lebrecht J., Piotrowska-Seget Z., Venho R., and Skurnik M. (2002) Functional characterization of Gne (UDP-N-acetylglucosamine-4-epimerase), Wzz (chain length determinant), and Wzy (O-antigen polymerase) of Yersinia enterocolitica serotype O:8. J. Bacteriol. 184, 4277–4287 10.1128/JB.184.15.4277-4287.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Larsen J. C., Szymanski C., and Guerry P. (2004) N-Linked protein glycosylation is required for full competence in Campylobacter jejuni 81-176. J. Bacteriol. 186, 6508–6514 10.1128/JB.186.19.6508-6514.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cain J. A., Dale A. L., Niewold P., Klare W. P., Man L., White M. Y., Scott N. E., and Cordwell S. J. (2019) Proteomics reveals multiple phenotypes associated with N-linked glycosylation in Campylobacter jejuni. Mol. Cell. Proteomics 18, 715–734 10.1074/mcp.RA118.001199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mukherjee K., Altincicek B., Hain T., Domann E., Vilcinskas A., and Chakraborty T. (2010) Galleria mellonella as a model system for studying Listeria pathogenesis. Appl. Environ. Microbiol. 76, 310–317 10.1128/AEM.01301-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Regan K. H., and Bhatt J. (2016) Eradication therapy for Burkholderia cepacia complex in people with cystic fibrosis. Cochrane Database Syst. Rev. 11, CD009876 10.1002/14651858.CD009876.pub3 [DOI] [PubMed] [Google Scholar]
- 50. Sambrook J., Fritsch E. F., and Maniatis T. (1990) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 51. Gibson D. G., Young L., Chuan R.-Y., Venter J. C., Hutchinson C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 52. Aubert D. F., Hamad M. A., and Valvano M. A. (2014) in Host–Bacteria Interactions: Methods and Protocols (Vergunst A., and Callaghan D. O., eds) pp. 311–327, Springer Science+Business Media, New York [Google Scholar]
- 53. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 10.1038/nprot.2006.468 [DOI] [PubMed] [Google Scholar]
- 54. Rappsilber J., Ishihama Y., and Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 10.1021/ac026117i [DOI] [PubMed] [Google Scholar]
- 55. Scott N. E., Parker B. L., Connolly A. M., Paulech J., Edwards A. V., Crossett B., Falconer L., Kolarich D., Djordjevic S. P., Højrup P., Packer N. H., Larsen M. R., and Cordwell S. J. (2011) Simultaneous glycan–peptide characterization using hydrophilic interaction chromatography and parallel fragmentation by CID, higher energy collisional dissociation, and electron transfer dissociation MS applied to the N-linked glycoproteome of Campylobacter jejuni. Mol. Cell. Proteomics 10, M000031–MCP201 10.1074/mcp.M000031-MCP201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ishihama Y., Rappsilber J., and Mann M. (2006) Modular stop and go extraction tips with stacked disks for parallel and multidimensional peptide fractionation in proteomics. J. Proteome Res. 5, 988–994 10.1021/pr050385q [DOI] [PubMed] [Google Scholar]
- 57. Rappsilber J., Mann M., and Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 10.1038/nprot.2007.261 [DOI] [PubMed] [Google Scholar]
- 58. Good D. M., Wenger C. D., and Coon J. J. (2010) The effect of interfering ions on search algorithm performance for electron-transfer dissociation data. Proteomics 10, 164–167 10.1002/pmic.200900570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roepstorff P., and Fohlman J. (1984) Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601 10.1002/bms.1200111109 [DOI] [PubMed] [Google Scholar]
- 60. Marolda C. L., Lahiry P., Vinés E., Saldías S., and Valvano M. A. (2006) Micromethods for the characterization of lipid A-core and O-antigen lipopolysaccharide. Methods Mol. Biol. 347, 237–252 10.1385/1-59745-167-3:237 [DOI] [PubMed] [Google Scholar]
- 61. Butty F. D., Aucoin M., Morrison L., Ho N., Shaw G., and Creuzenet C. (2009) Elucidating the formation of 6-deoxyheptose: biochemical characterization of the GDP-B1dB0-glycero-B1dB0-manno-heptose C6 dehydratase, DmhA, and its associated C4 reductase, DmhB. Biochemistry 48, 7764–7775 10.1021/bi901065t [DOI] [PubMed] [Google Scholar]
- 62. Bochner B. R. (2009) Global phenotypic characterization of bacteria. FEMS Microbiol. Rev. 33, 191–205 10.1111/j.1574-6976.2008.00149.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kazanas N. (1968) Proteolytic activity of microorganisms isolated from freshwater fish. Appl. Microbiol. 16, 128–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.