

Abstract
Postsynaptic alterations are currently believed to be able to fully account for NMDA-receptor-dependent long-term depression (LTD) and long-term potentiation of synaptic strength, although there is also evidence supporting changes in presynaptic release. Using dualphoton laser scan microscopy of N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide (FM1-43) to directly visualize presynaptic vesicular release at Schaffer collateral–CA1 excitatory synapses in hippocampal slices, we demonstrate reduced vesicular release associated with LTD. Selective loading, by hypertonic shock, of the readily releasable vesicle pool (RRP) showed that LTD of release is a selective modification of release from the RRP. Presynaptic LTD of RRP release required activation of NMDA receptors, production and extracellular diffusion of the intercellular messenger NO, and activation of cGMP-dependent protein kinase.
Keywords: CA1, cGMP, hippocampus, long-term depression, nitric oxide, presynaptic, readily releasable vesicle pool, Schaffer collateral, PKG, transmitter release
Introduction
Activity-dependent, long-term changes in synaptic strength, such as long-term potentiation (LTP) and long-term depression (LTD), are believed to be important for information storage, neural network development, fine-tuning of synaptic connections, learning, and memory (Bailey et al., 1996; Katz and Shatz, 1996; Martin et al., 2000; Braunewell and Manahan-Vaughan, 2001). Despite extensive studies of the mechanisms underlying both LTP and, more recently, LTD, the basic question of whether these forms of synaptic plasticity result from presynaptic and/or postsynaptic changes remains hotly debated (Malenka and Nicoll, 1999; Malinow et al., 2000; Pavlidis et al., 2000). Many lines of evidence support an array of postsynaptic alterations associated with both LTP and LTD, including changes in AMPA-receptor-gated channel conductances (Benke et al., 1998; Lee et al., 1998), insertion and removal of AMPA receptors (Carroll et al., 1999; Shi et al., 1999), and changes in dendritic spine shape (Engert and Bonhoeffer, 1999; Toni et al., 1999). Evidence for presynaptic alterations has been derived largely from quantal analysis studies of pairs of monosynaptically connected neurons (Bekkers and Stevens, 1990; Malinow and Tsien, 1990; Bolshakov and Siegelbaum, 1994), vesicular antibody uptake (Malgaroli et al., 1995), and postsynaptic drug-infusion studies (Reyes and Stanton, 1996; Yeckel et al., 1999; Mellor and Nicoll, 2001). The indirect nature of such evidence, failure to fulfill critical assumptions of quantal analysis, and conflicting data have resulted in continued disagreement.
The styryl fluorescent dye N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide (FM1-43) has been successfully used in isolated neuronal systems to directly visualize presynaptic vesicular release (Betz and Bewick, 1992; Ryan et al., 1993). FM1-43 is taken up into presynaptic vesicles in an activity-dependent manner as a result of endocytosis after transmitter release. Subsequent synaptic stimulation evokes the release of dye, which is visualized as a destaining whose rate is a direct measure of presynaptic release efficacy. Previous studies have documented changes in presynaptic function associated with forms of LTP at synapses between cultured hippocampal neurons based on activity-dependent labeling with antibodies (Malgaroli et al., 1995) or FM1-43 (Ryan et al., 1996; Ma et al., 1999). However, direct evidence for presynaptic changes during LTD at synapses that formed in vivo has been lacking. Recent advances in imaging technology make it possible to monitor vesicle kinetics in acute brain slices directly (Pyle et al., 1999; Stanton et al., 2001; Zakharenko et al., 2001, 2002).
Materials and Methods
Slice preparation and electrophysiology. Wistar rats, 15–22 d of age, were decapitated under deep ether anesthesia, the hippocampi were dissected free, and 300 μm thick transverse slices were cut on a vibratome. Slices were placed in an interface holding chamber at 25°C for at least 1 hr before being transferred to a submerged chamber on the microscope stage, also at 25°C. Slices were perfused with artificial CSF (ACSF; 2 ml/min). ACSF consisted of the following (in mm): 126 NaCl, 5 KCl, 1.25 NaH2PO4, 2 MgCl, 2 CaCl2, 26NaHCO3, and 10 glucose, saturated with 95%O2 and 5%CO2; drugs were bath-applied. Schaffer collateral/commissural axons in stratum radiatum were stimulated at 0.033 Hz, with baseline intensities chosen to evoke half-maximal field EPSPs (fEPSPs) in field CA1. LTD was induced with a single 2 Hz, 10 min train of stimuli of the same intensity.
Loading of the total and readily releasable vesicle pools. After inducing LTD, 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) was bath-applied for the rest of experiments to prevent synaptically driven action potentials from accelerating dye release. Presynaptic boutons were loaded by bath-applying 5 μm FM1-43 (Molecular Probes, Eugene, OR) in 45 mm K + ACSF for 15 min for the total vesicle pool, or in hypertonic ACSF supplemented with sucrose to 800 mOsm for 25 sec for the readily releasable pool (RRP), then returned to normal ACSF. In separate control experiments, neither loading protocol produced long-term changes in amplitude or shape of Schaffer collateral-evoked synaptic fEPSPs, or in pyramidal neuron membrane properties. Stimulus-induced destaining was measured after 30 min in dye-free ACSF, evoked by 5 sec bursts of 10 Hz stimulation, applied once every 30 sec. These short, discontinuous bursts produced a much slower time course of release than does a continuous stimulus train in either brain slices (Stanton et al., 2001; Zakharenko et al., 2001) or cultured neurons (Rosenmund and Stevens, 1996; Pyle et al., 2000), allowing a dynamic range for detecting both increases and decreases in the release rate in response to the induction of long-term synaptic plasticity. At the end of each experiment, complete depolarization-induced destaining was evoked by 85 mm K + ACSF.
Two-photon imaging. FM1-43 fluorescence was visualized using a Leica (Nussloch, Germany) DM LFS E upright microscope, two-photon excitation, a water-immersion ultraviolet APO L 40×/0.80 W objective and a Leica multispectral confocal laser scan unit. The light source was a Millennia 5 W diode laser source pumping a Tsunami Ti:sapphire laser (Spectra-Physics, Fremont, CA) that provided ≈130-fsec pulses at 82 MHz, 840 nm center wavelength. Bandpass-filtered epifluorescence was detected with non-descanned photomultiplier tubes behind the objective and a 1.3 numerical aperture oil condenser, optimized for signal over background (540–600 nm) based on spectral analysis with the confocal laser scan head with the pinhole maximally open. Laser intensity was controlled with a variable-beam splitter exploiting polarization of the laser light and neutral density filters. Although there were no signs of photodamage, we always used the lowest intensity necessary for an adequate signal-to-noise ratio. We acquired 512 × 512 pixel images (0.15 μm/pixel in the x- and y-axes). For puncta brightness depth profiles, images were acquired in 4 μm steps in the z-direction. In offline analyses, rectangular regions of interest (ROIs; ∼2–4 μm 2) were selected around centers of bright, punctate fluorescence spots, and 12–16 boutons and three to four background ROIs were measured per slice. If lateral displacement of a bouton beyond the ROI occurred, that data set was discarded. Moreover, in separate experiments to control for z-axis drift, fluorescent beads (0.5 μm) were injected into hippocampal slices, and their movement was monitored in the same slice chamber. The z-axis drift for a typical data set was estimated at not more than 0.15 μm/3 min, which was the period of calculation of initial destaining rates from destaining curves. Only puncta that showed stimulus-dependent unloading were analyzed (∼90% fulfilled this criteria, the nonreleasing sites showed 4% destaining over the entire 20 min time course) (see Fig. 3C). All fields imaged were within the first 100 μm depth, typically 25–60 μm, and 40–60 μm away from stimulating electrode poles. Destaining time courses were generated by normalizing each ROI time course to starting intensity, averaging the background fields to produce a dye bleaching time course (2 hr bleaching, -12.1 ± 1.0%), then dividing each ROI by bleaching at corresponding time points. The half-time of decay of intensity during unloading (t1/2) was calculated for each punctum from single exponential fits to the first five points in destaining curves. Vertical bars are SEMs for the averages of all normalized and corrected boutons across experiments.
Figure 3.

Hypertonic shock (25 sec of 800 mOsm ACSF plus sucrose) selectively loads the RRP, which represents the ∼28% of total vesicle pool brightness with more rapid release kinetics. A, Comparison of the initial 7 min of Schaffer collateral stimulus-evoked FM1-43 release time courses in 45 mm K +-loaded (total vesicle pool) and 25 sec 800 mOsm ACSF-loaded (RRP) slices. The first 10 Hz/5 sec stimulus released ∼12% of the RRP, compared with 2% of the total vesicle pool, confirming that sucrose preferentially loads the RRP in brain slices. B, Comparison of mean ± SEM fluorescence of Schaffer collateral terminals loaded with K + or sucrose, in control (K + = 66; sucrose = 115) versus LTD (K + = 65; sucrose = 86) puncta. *p < 0.05, Student's t test compared with control K +-loaded puncta brightness). C, Comparison of Schaffer collateral stimulus-evoked FM1-43 release time course in puncta that exhibited destaining (CON; n = 115), versus puncta that did not show stimulus-dependent destaining (NONREL; n = 15) in 25 sec 800 mOsm ACSF-loaded (RRP) slices (n = 6 slices in each group). D, Electron micrograph of a Schaffer collateral presynaptic terminal in a hippocampal slice loaded by 25 sec hyperosmotic shock and then fixed 20 min later, showing electron-dense FM1-43 reacted with 3,3′-diaminobenzidine, localized primarily to vesicles closely apposed to the release active zone (arrows).
Photoconversion of FM1-43 for electron microscopy. For electron microscopic (EM) visualization of FM1-43 in individual presynaptic vesicles, we used standard photoconversion methods adapted for use in hippocampal slices (Richards et al., 2000; Harata et al., 2001). In brief, slices were preincubated in 1 mm kynurenic acid, loaded by 25 sec application of hyperosmotic ACSF (800 mOsm) containing 5 μm FM1-43 plus kynurenic acid, and returned to low [Ca 2+] (0.2 mm), high [Mg 2+] (5 mm) ACSF. Either immediately or 20 min after loading, slices were fixed in 2% glutaraldehyde in 50 mm sodium phosphate buffer (PB; resulting solution, 300 mOsm) for 20 min, and washed with 100 mm glycine (in PB) for 1 hr. Slices were then washed for 5 min in 100 mm ammonium chloride (to reduce glutaraldehyde autofluorescence), rinsed quickly in PB, and incubated in DAB (1 mg/ml in PB) for 20 min. To photoprecipitate FM1-43, fluorescence light (480 ± 20 nm) was applied continuously for 20 min in DAB solution, after which slices were washed in ice-cold PB and processed for EM.
Results
Total and readily releasable vesicle pool loading and release
We used two-photon imaging of individual presynaptic terminals loaded with FM1-43 to measure selectively the effects of LTD on release from the total vesicle pool and RRPs at hippocampal CA3–CA1 synapses in vitro. LTD was induced in hippocampal slices 30 min before the loading of presynaptic Schaffer collateral terminals with FM1-43 (experimental protocol) (Fig. 1). After recording the LTD of Schaffer collateral-evoked fEPSPs for 15 min, slices were bathed in the AMPA receptor antagonist CNQX (10 μm) for the rest of the experiment to prevent polysynaptic activity-induced dye release. Either the total vesicle pool or RRP was labeled with FM1-43 by (1) 15 min bath application of FM1-43 (5 μm) plus 45 mm K+ ACSF (Fig. 1A), or (2) 25 sec bath application of FM1-43 (5 μm) in 800 mOsm ACSF plus sucrose (Fig. 1B), respectively. By either loading method, brightly fluorescent spherical clusters of vesicles in individual Schaffer collateral presynaptic terminals (puncta) (Fig. 2) could be imaged by two-photon microscopy (Stanton et al., 2001; Zakharenko et al., 2001) in the apical dendritic field of CA1 stratum radiatum. Fluorescence intensities of these puncta (mean diameter, 1.14 ± 0.03 μm; n = 640) were stable for at least 60 min in the absence of electrical stimulation. During subsequent electrical stimulation, fluorescence intensities of ∼90% of these puncta rapidly diminished, reflecting exocytosis of dye from synaptic vesicles. Figure 2 illustrates two typical CA1 stratum radiatum fields of fluorescent puncta after loading the RRP. Images in the top row are from a control slice, whereas the bottom row illustrates destaining in a slice in which LTD was induced 30 min before loading. In each slice, a 10 Hz/5 sec stimulus burst was applied once each 30 sec, a discontinuous stimulus protocol designed to elicit a slower time course of release capable of detecting increases or decreases in the rate of release. Images are shown 0, 1, 2, 5, and 10 min after the start of destaining stimulation, illustrating a rate of vesicular release that was markedly slower in the slice in which LTD had been previously induced, compared with the unstimulated control.
Figure 1.

Protocols for determining the kinetics of FM1-43 release from presynaptic terminals after the induction of LTD of synaptic strength. Fifteen minutes after inducing LTD, CNQX (10μm) was bath-applied for 15 min, and terminals were labeled by exposure to 5μm FM1-43 during either 45 mm K + application (15 min) to label the total vesicle pool (A), or 800 mOsm ACSF (25 sec) to label the RRP (B). Subsequent 10 Hz/5 sec bursts of electrical stimulation were applied once each 30 sec for a 20 min period to evoke the vesicular release of dye.
Figure2.

LTD of synaptic strength at Schaffer collateral–CA1 synapses produces along-lasting reduction in the evoked vesicular release of FM1-43. Two-photon excitation fluorescent images of RRP puncta in the same field in the stratum radiatum of field CA1 in a control slice (top), versus a slice in which LTD was induced (bottom), at different times after the start of the unloading stimulation protocol (numbers represent the time in minutes; 0′ is the beginning of unloading stimulation). In these slices, presynaptic vesicles in the RRP were selectively loaded with a 25 sec, 800 mOsm hypertonic ACSF plus sucrose.
Hyperosmotic shock has been shown to selectively load the RRP in dissociated cultures (Rosenmund and Stevens, 1996; Goda and Stevens, 1998), but brain slices may present a diffusion barrier to the rapid access of hyperosmotic saline to synapses deep within the tissue. Therefore, we tested the preferential loading of the RRP by hyperosmotic shock in our slices by comparing FM1-43 stimulus-evoked destaining kinetics from terminals loaded hyperosmotically, to the total vesicle pool loaded by prolonged depolarization in 45 mm K+. Figure 3A illustrates the first 10 min release time courses from RRP puncta (open circles) versus puncta in which the total vesicle pool was loaded (filled circles), and then given a 10 Hz/5 sec stimulus burst each 30 sec. The first burst released 12% of the RRP, compared with only 2% of the total pool signal, consistent with our hyperosmotic treatment preferentially loading the RRP. This conclusion is also supported by the brightness of the puncta in each group (Fig. 3B), in which RRP puncta brightness averaged ∼28% of that of the total vesicle pool. During prolonged release from the RRP, slow washout of FM1-43 from the extracellular space could permit the accumulation of sufficient FM1-43 to allow reuptake with rapid re-endocytosis into the RRP. Thus, only early FM1-43 destaining is sure to represent pure exocytosis, whereas later release could be the sum of exocytosis and endocytosis.
Although the vast majority of FM1-43-labeled puncta (∼90%) showed statistically significant destaining on Schaffer collateral stimulation, ∼10% did not. These nonreleasing puncta were removed from the experimental data sets and analyzed separately. Figure 3C shows a comparison of the time courses of destaining of terminals that released after hyperosmotic shock (open circles), which dimmed by ∼47% by the end of the 20 min of stimulation versus the nonreleasing puncta (filled circles), which exhibited only ∼4% total destaining over the same period.
To independently confirm that 25 sec hypertonic shock did preferentially label vesicles in the RRP, we used the photoconversion method of Henkel et al. (1996), modified for hippocampal slices by Harata et al. (2001) (see Materials and Methods), to photoprecipitate FM1-43 for EM visualization. Figure 3D illustrates a typical presynaptic terminal containing dense DAB-positive FM1-43 granules that are primarily concentrated in vesicles closely apposed to the active release zone (arrows). In 58 presynaptic terminals loaded by brief hypertonic shock 20 min before fixation, “docked” vesicles directly apposed to the active zone (13%; 231 of 1782; active zone length, 223 ± 12 nm) contained 5.66 FM1-43 granules per vesicle, compared with undocked vesicles within 200 nm of an active zone (19.6%; 349 of 1782), which contained 2.22 grains per vesicle, and vesicles >200 nm away (67.4%; 1202 of 1782) with only 1.63 grains per vesicle. Vesicles within 200 nm of the active zone showed a bimodal distribution of luminal densities, consisting of 32% FM1-43-positive (mean luminal density, 1.14 ± 0.01), 68% FM1-43-negative (density, 0.66 ± 0.043). In contrast, vesicles >200 nm away from an active zone showed a single Gaussian distribution (density, 0.75 ± 0.015), consistent with unlabeled vesicles. These EM data confirm that RRP vesicles are preferentially labeled after hypertonic loading. They are also consistent with this brief hyperosmotic shock selectively releasing vesicles from the RRP, allowing them to be labeled and then preferentially returned to the RRP (“kiss-and-run” release) (Pyle et al., 2000; Richards et al., 2000).
LTD of total and RRP release
We next compared the effect of the induction of LTD 30–40 min before the measurement of FM1-43 release kinetics from the total, and readily releasable, vesicle pools. Figure 4, A and B, illustrates that the induction of LTD produced a significant slowing in the kinetics of stimulus-evoked FM1-43 release from both the total vesicle pool (Fig. 4A) and the RRP (Fig. 4B). The effect of LTD on release from the RRP was approximately four times the magnitude of that on the total vesicle pool. To estimate release from the reserve vesicle pool, we subtracted the raw FM1-43 release time courses for the RRP from the total vesicle pool, and normalized the resulting time courses, for control and LTD slices (Fig. 4C). There was no difference between control and LTD release kinetics from the reserve pool of vesicles during the first 10 min of stimulation, indicating that the effect of LTD on the total pool was completely accounted for by a selective slowing of release from the RRP.
Figure 4.
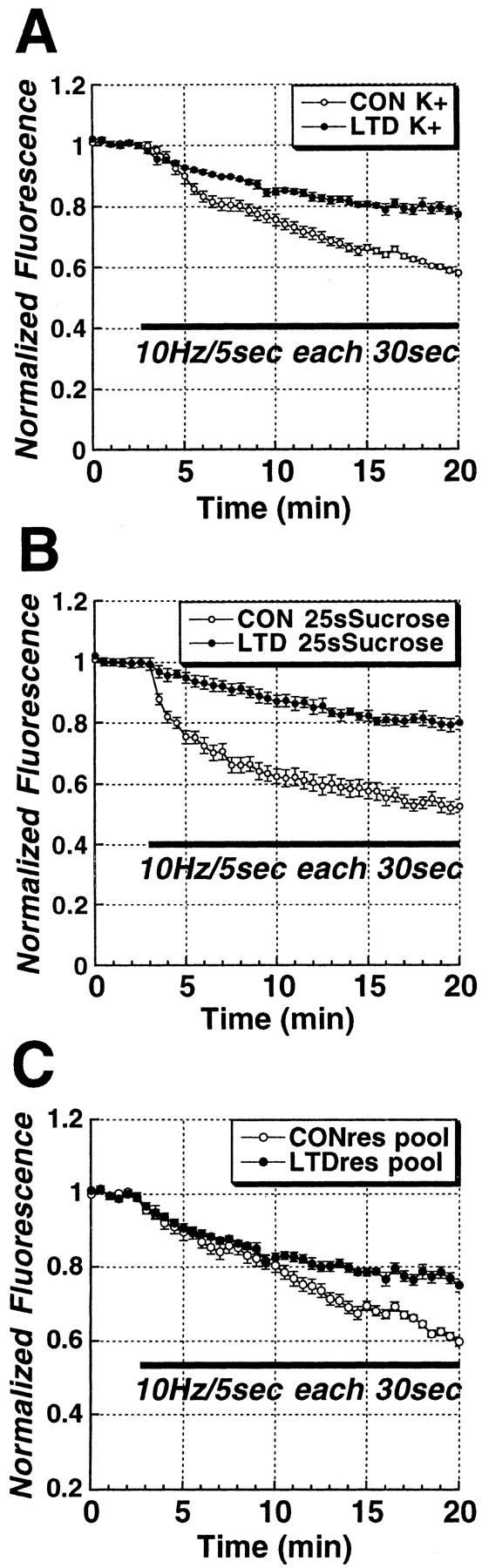
LTD of synaptic strength at Schaffer collateral–CA1 synapses is associated with a selective long-lasting reduction in evoked release from the RRP. A, Time courses of Schaffer collateral stimulus-evoked (solid bar;10 Hz/5 sec bursts each 30 sec) FM1-43 destaining from the total vesicle pool in control (n = 5) versus LTD (n = 5) slices. B, Time courses of Schaffer collateral stimulus-evoked (solid bar; 10 Hz/5 sec bursts each 30 sec) FM1-43 destaining from the RRP in control (n = 7) versus LTD (n = 6) slices, illustrating the preferential reduction in release from the RRP. C, Time courses of Schaffer collateral stimulus-evoked (solid bar; 10 Hz/5 sec each 30 sec) FM1-43 release from the reserve vesicle pool in control versus LTD slices, calculated by subtracting RRP from total vesicle pool time courses. During the first 10 min, release from the reserve pool was not altered by previous induction of LTD.
LTD of stimulus-evoked FM1-43 uptake
A comparison of the brightness of total vesicle pool puncta in LTD and control slices before the unloading stimulus (Fig. 3B) showed that the induction of LTD led to less loading of presynaptic terminals by 45 mm K+, consistent with a reduced release probability. However, puncta were measured at different depths from slice to slice, and the strong depolarization used is one that maximally loads the total vesicle pool. To test the effect of plasticity on fusion probability with more physiologically relevant action-potential-driven release, we collected a z-series profile through the top 80 μm of slices in 4 μm steps from a set of control slices, and slices in which either LTD had been induced 45 min earlier, and then vesicles were loaded with repeated bursts of Schaffer collateral stimulation (10 Hz/5 sec bursts every 30 sec for 20 min) in 5 μm FM1-43. Figure 5 is a plot of these depth profiles of mean puncta brightness, showing that LTD (filled circles) was indeed associated with less stimulus-evoked uptake of FM1-43 compared with controls (open circles), throughout the depth profile. Bath application of the NMDA receptor antagonist d-AP-5 (20 μm) during low-frequency stimulation, followed by a 30 min drug washout, completely blocked this long-term effect on stimulus-evoked FM1-43 uptake.
Figure 5.

LTD produces a long-lasting decrease in stimulus-evoked uptake of FM1-43 that is NMDA-receptor dependent. Comparison of z-axis depth profiles of mean ± SEM puncta fluorescence induced to take up FM1-43 by Schaffer collateral stimulation (10 Hz/5 sec each 30 sec for 20 min) in control slices (n = 4), versus slices in which LTD (n = 4) had been induced previously (2 Hz/10 min stimulus train), and slices in which the LTD stimulus train was applied in the presence of the NMDA receptor antagonist d,l-AP-5 (20 μm; n = 4). Independent of depth, LTD was associated with decreased FM1-43 endocytosis. This decrease in FM1-43 endocytosis was completely prevented by NMDA receptor blockade.
RRP release before and after induction of LTD in the same slices
Although our previous data indicate that the induction of LTD produces a decrease in release from RRP vesicles, those experiments were performed on separate groups of slices. To confirm this conclusion, we designed a protocol to examine the kinetics of FM1-43 release from the RRP in the same slices before and after the induction of LTD (Fig. 6A). We used the reversible, nonselective glutamate receptor antagonist kynurenic acid (KYN; 10 mm) to reversibly block synaptic transmission during hypertonic loading of the RRP. The first 20 min of the plot in Figure 6B (Pre-LTD) illustrates the stimulus-evoked (10 Hz/5 sec each 30 sec; first bar) RRP release kinetics before the induction of LTD. After this first stimulus, 45 mm K+ was used to complete the unloading, and then KYN was washed out to recover synaptic transmission so that LTD could be induced (2 Hz/10 min). After LTD was recorded for 15 min, KYN was reperfused for 15 min, and the same slices were loaded with FM1-43 using an identical hypertonic shock (800 mOsm, 25 sec), and release was tested a second time. The second identical stimulus now evoked significantly slower FM1-43 release (third bar), consistent with the previously observed effect of LTD on RRP release kinetics. In control experiments in which no LTD was induced, the second release profile was identical to the first (n = 3; data not shown).
Figure 6.
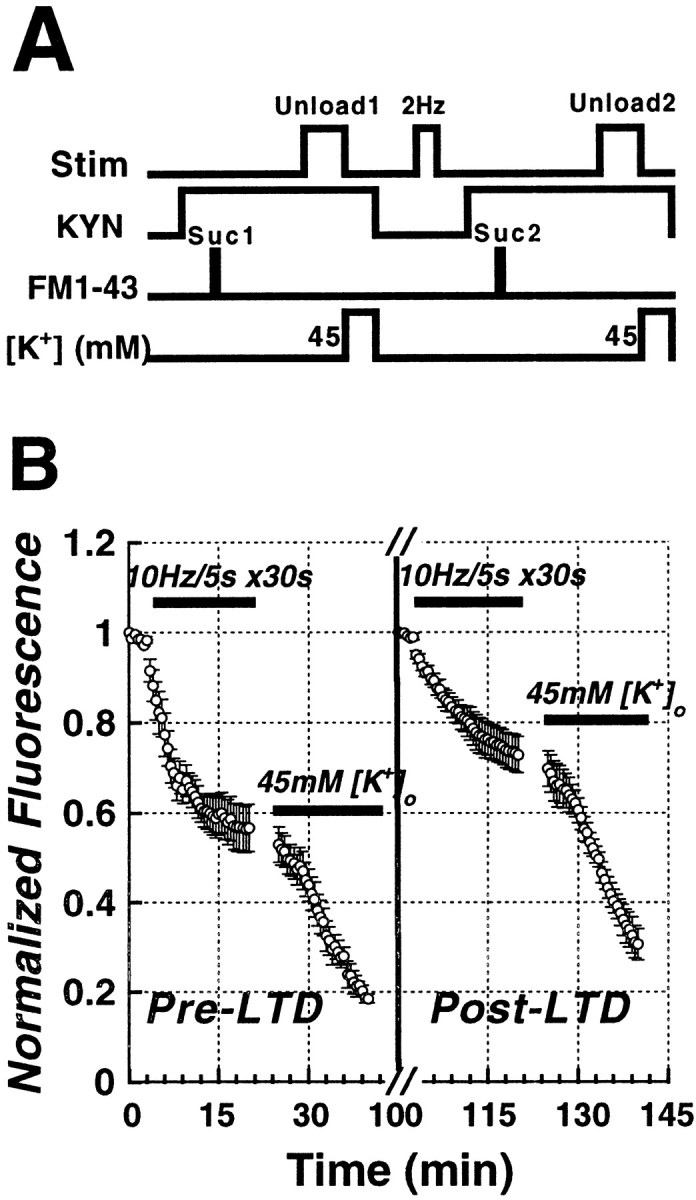
The time course of Schaffer collateral stimulus-evoked (solid bars;10 Hz/5 sec bursts each 30 sec) FM1-43 release from the RRP before and after inducing LTD in the same set of slices. A, Protocol for determining the kinetics of FM1-43 release from presynaptic terminals before and during LTD in the same slices. First, kynurenic acid was bath-applied (10 mm KYN, 15 min), then terminals were loaded with 5 μm FM1-43 in 800 mOsm sucrose ACSF for 25 sec. Release was tested 30 min later with 10 Hz/5 sec bursts of electrical stimulation each 30 sec. After residual dye was released with 45 mm K +, kynurenate was washed out and LTD induced (1 Hz/10 min). The identical release protocol was then repeated. B, After evoking FM1-43 release from the pre-LTD RRP (first solid bar; 10 Hz/5 sec bursts each 30 sec), 45 mm K + was applied to release all dye, LTD was induced, and the RRP was reloaded with FM1-43. Afterward, post-LTDFM1-43 release was evoked by a second, identical stimulus (third solid bar; 10 Hz/5 sec bursts each 30 sec). After LTD, release t1/2 was significantly smaller than release before LTD (p < 0.05; paired t test; n = 5).
NMDA receptor, NO, and cGMP-dependent protein kinase dependence of LTD of RRP release
Both LTD and LTP of synaptic strength have been reported to consist of multiple forms mediated by different receptor and messenger pathways. In particular, there are NMDA-receptor-dependent and NMDA-receptor-independent forms of LTD and LTP. Therefore, we tested whether LTD-induced changes in RRP release kinetics depend on NMDA receptor activation. Bath application of the NMDA receptor antagonist dl-2-amino-5-phosphonopentanoic acid (AP-5, 20 μm) before the induction of LTD completely blocked the induction of LTD of RRP release (Fig. 7, open circles). We and others have reported previously evidence that the gaseous intercellular messenger NO, cGMP, and cGMP-dependent protein kinase (PKG) activity, are all required for the induction of a presynaptic form of LTD (Izumi and Zorumski, 1993; Gage et al., 1997; Wu et al., 1998; Reyes-Harde et al., 1999; Stanton et al., 2001). Therefore, we performed studies to determine whether this cascade might be involved in plasticity-associated changes in RRP release. Bath application of the selective PKG inhibitor KT5823 (1 μm) markedly reduced the induction of LTD of synaptic transmission (see Fig. 10D), and completely prevented LTD of FM1-43 release from the RRP (Fig. 8, open circles). Bath application of the competitive nitric oxide (NO) synthase (NOS) inhibitor l-nitroarginine (l-NA; 100 μm) partially blocked (∼50%) the induction of LTD of synaptic transmission (see Fig. 10D). As with KT5823, l-NA completely prevented the effect of LTD on RRP release (Fig. 9A, open circles). These data suggest that there are components, in addition to changes in presynaptic release, that also contribute to LTD and LTP. Presynaptic alterations evoked by postsynaptic NMDA receptor activation must depend on a retrograde messenger, such as NO. Consistent with this hypothesis, when we examined the ability of the extracellular NO scavenger hemoglobin (100 μm)to impair LTD, it produced a partial blockade of both LTD (Fig. 10D) and of the effects of LTD on RRP release mechanisms (Fig. 9B, open circles). The residual presynaptic LTD component could be attributable to either other retrograde messengers or incomplete scavenging of NO.
Figure 7.

LTD of vesicular release from the RRP is NMDA-receptor dependent. The time course of Schaffer collateral stimulus-evoked release from the RRP (solid bar;10 Hz/5 sec bursts each 30 sec) 45 min after the induction of LTD. Application of the NMDA receptor antagonist AP-5 (20μm) 30 min before inducing LTD (n = 5) completely blocked the reduction of release, compared with control slices (n = 5).
Figure 10.

Distributions of RRP puncta unloading rates (1/t1/2) in different groups of slices. A, Histogram of unloading rates (1/t1/2) of individual puncta in control slices (solid bars), versus slices in which LTD had been induced (graybars). B, Histogram of unloading rates (1/t1/2) of individual puncta in slices pretreated with the NMDA antagonist d,l-AP-5 (20μm) 30 min before the application of the LTD stimulus. C, Mean ± SEM 1/t1/2(s -1) in control (solid bar), LTD (gray bar), and slices pretreated with 20 μm d,l-AP-5 (coarsely hatched bar, left), 10 μm KT5823 (finely hatched bar, left), 10 μm l-NA (coarsely hatched bar, right), or 100 μm Hb (finely hatched bar, right) before the application of the LTD stimulus. *p < 0.05; Student's t test compared with control 1/t1/2. D, Left, Mean ± SEM percentage change in fEPSP amplitude 15 min after the application of LTD-inducing stimuli (2 Hz/10 min) in the same control and drug-treated slices in which FM1-43 release was measured (*p < 0.05; Student's t test compared with control LTD); right, percentage change in 1/t1/2(sec -1) in the same drug conditions.
Figure 8.

LTD of vesicular release from the RRP is PKG dependent. The time course of Schaffer collateral stimulus-evoked release from the RRP (solid bar; 10 Hz/5 sec bursts each 30 sec) in slices treated with the PKG inhibitor KT5823 (10 μm; n = 5) 30 min before induction of LTD, compared with control slices (n = 5).
Figure 9.

LTD of vesicular release from the RRP is NO dependent. A, Time course of Schaffer collateral stimulus-evoked release from the RRP (solid bar; 10 Hz/5 sec bursts each 30 sec) in slices treated with the NOS inhibitorl-NA)(10μm;n=4)comparedwithcontrolslices(n=5). l-NA completely blocked LTD of release from the RRP. B, The NO scavenger hemoglobin (Hb; 10 μm; n = 4) partially blocked the reduction in RRP release seen in LTD slices compared with control LTD (n = 5) and unstimulated slices (n = 6).
Distribution histograms of individual release sites before and after LTD
The previous time courses consist of mean intensities derived from averaging multiple puncta (12–16 per slice) over all slices, which might obscure significant individual differences in rates of destaining or distinctly different populations of release sites. To determine whether there were different populations of release sites, we calculated, for each punctum, the time required for fluorescence intensity to decay to half of its initial value during unloading stimulation (t1/2), by fitting the first 3 min of the decay time course of each punctum with a monoexponential. Histograms comparing the distribution of individual time constants in the different groups are shown in Figure 10. In Figure 10A, the distribution of 1/t1/2(sec-1) in control slices was shifted significantly to the left in slices in which LTD had been induced. Blockade of NMDA receptor activation with 20 μm d,l-AP-5 during the low-frequency stimulus train prevented the effects of LTD (Fig. 10B) on 1/t1/2 (sec-1) distributions compared with controls. In all cases, t1/2 distributions were well fitted by single Gaussians, giving no sign of multiple populations of releasing puncta. Comparison of mean 1/t1/2(sec-1) in all LTD groups (Fig. 10C) confirms that previous induction of LTD produced a 64% reduction compared with control slices. This effect of LTD was completely prevented by blocking NMDA receptor activation, PKG, or NOS activity, whereas the extracellular NO scavenger partially reduced the effect of LTD. The percentage change in 1/t1/2(sec-1) in all LTD groups (Fig. 10D, right) mimicked the percentage changes in the strength of synaptic transmission (Fig. 10D, left).
Discussion
The results presented here demonstrate, for the first time, selective labeling of the RRP and direct imaging of RRP release kinetics from presynaptic terminals in acute brain slices. In particular, although high K+ appears to load the entire releasable vesicle population, a rapid hypertonic shock loads a smaller pool of vesicles whose size (∼25% of the total) and more rapid release kinetics are consistent with the RRP in dissociated cell cultures characterized electrophysiologically by similar loading and release methods (Rosenmund and Stevens, 1996; Goda and Stevens, 1998). We also demonstrate that two-photon laser scanning microscopy can visualize the kinetics of stimulus-evoked release of FM1-43 from individual presynaptic terminals, without additional compounds that might affect synaptic physiology, such as sulforhodamine or cyclodextrins.
In comparing Schaffer collateral stimulus-evoked release from these pools, measured by the rate of dye loss, we found that the induction of LTD of synaptic transmission evoked a long-lasting decrease in FM1-43 release from presynaptic boutons. These data are consistent with a previous report of LTD associated with a decrease in RRP size at synapses between cultured hippocampal neurons (Goda and Stevens, 1998). A comparison of the effect of LTD on release kinetics from the total (K+-loaded) vesicle pool versus the RRP showed that the action of LTD was completely confined to the RRP. When we subtracted the RRP release time course from the total vesicle pool before normalization, to estimate release from the reserve pool, there was no difference in LTD and control release during the first 10 min of stimulation. Interestingly, there was a divergence of LTD and control time courses during the final 10 min of stimulation. If the rate of movement from the reserve pool into the RRP was not changed by LTD, subtracting time courses should have accounted for the entire effect of LTD. The fact that it did not suggests that entry from the reserve pool into the RRP might also be slowed by LTD, but that the exchange rate between these pools is significantly slower than release from the RRP.
It is worth noting that, although it is generally agreed that a brief hypertonic shock selectively releases transmitter from the RRP, our data are consistent with vesicles from this pool, once released, being preferentially recycled back into the RRP. That is, when we loaded using a stimulus that selectively releases the RRP, this labeled vesicle pool exhibited brightness and release kinetics appropriate for the RRP when released a second time, consistent with studies on synapses in dissociated neuron cultures, suggesting that vesicles in the RRP are reclaimed via a rapidly recycling route that returns them preferentially to the RRP (Pyle et al., 2000; Richards et al., 2000). In multiple dye studies of the recycling times of vesicle pools in hippocampal neuronal cultures, Pyle et al. (2000) found that RRP vesicles undergo rapid exocytosis and endocytosis (τ ≅ 1 sec), whereas vesicles recruited from the reserve pool recycle much more slowly (τ ≅ 30 sec).
EM ultrastructural characterization of the distribution of FM1-43 in presynaptic vesicles loaded by brief hypertonic shock confirmed the selective loading of the RRP. Moreover, it also confirmed that the exchange rate between the RRP and reserve vesicle pools was sufficiently slow that there was still twofold to threefold more FM1-43 in vesicles either docked or within 200 nm of the active zone 20 min after loading. The sum of the percentage of vesicles that appeared to be directly linked to the active zone (docked; 13%), plus those within 200 nm (19.6%), is consistent with physiological estimates of the size of the RRP (Rosenmund and Stevens, 1996; Goda and Stevens, 1998; Schneggenburger et al., 1999; Pyle et al., 2000; Richards et al., 2000), as well as the relative brightness of RRP puncta in this study (28%). The retention of FM1-43 in the RRP is probably the result of repeated recycling of RRP vesicles back into the RRP (Pyle et al., 2000; Richards et al., 2000).
Recent findings of Zakharenko et al. (2002), using similar FM1-43 techniques to image the total vesicle pool of Schaffer collateral–CA1 terminals in hippocampal slices from 2- to 3-d-old rats, show both interesting parallels and differences. Although they also observed LTD-induced reduction in presynaptic vesicular release probability, slices at this early developmental stage do not yet express significant amounts of NMDA receptors or LTP, and LTD depends instead on the activation of metabotropic glutamate receptors (mGluRs) (Bolshakov and Siegelbaum, 1994). Moreover, their observations suggest that mGluR-dependent LTD can turn release sites completely off, although it is unclear whether the NMDA-receptor-dependent LTD we studied later in development can produce a similar inactivation. Combining our data indicates that multiple forms of activity-dependent LTD can produce long-term changes in presynaptic transmitter release via different receptors, and that different forms of LTD may be expressed at different stages in development.
It is also noteworthy that a previous study by Zakharenko et al. (2001) examining the release of FM1-43 at Schaffer collateral–CA1 terminals in slices from 14- to 30-d-old mice, found that the LTP of release associated with a strong LTP-inducing stimulus required both NMDA-receptor- and voltage-dependent calcium channel activation. When combined with our data, this indicates that both NMDA-receptor-dependent LTP and NMDA- receptor-dependent LTD can be associated with long-lasting changes in presynaptic transmitter release, although it remains to be determined whether voltage-dependent calcium channel activation on either or both sides of the synapse are selectively involved in cooperatively evoking LTP of release.
Mechanisms that might underlie the decreased rate of FM1-43 release during LTD, and a converse increased rate associated with strong LTP, have been discussed previously (Zakharenko et al., 2001). First, there could be a decrease (or increase) in the probability of vesicle fusion at the active zone in response to terminal depolarization and Ca2+ influx. Second, there could be a change in the rate of fusion pore formation kinetics, leading to altered peak transmitter concentration in the synaptic cleft. Finally, there could be a decrease (or increase) in the number of active zones present within a presynaptic bouton. The fact that we observed selective plasticity of release from the RRP does not definitively differentiate between these possibilities, but it is easier to explain our data by a selective change in the efficacy of existing active zone coupling to RRP vesicles, because a decrease in active zone numbers without any change in release efficacy should decrease RRP and total pool release equally.
Previous studies have come down on both sides of this issue. cAMP-induced potentiation in dissociated hippocampal cell cultures has been shown to increase the number of FM1-43-labeled (Ma et al., 1999) and synaptophysin-labeled (Bozdagi et al., 2000) puncta, and induction of LTP in hippocampal slice cultures has been shown to increase the number of multiple spine synapses (Toni et al., 1999). However, an FM1-43 study of LTP in acute hippocampal slices found no evidence for an increase in the number of functional release sites (Zakharenko et al., 2001). Zakharenko et al. (2002) concluded that mGluR-dependent LTD in slices from 2- to 3-d-old rats is associated with an increase in the number of FM1-43-labeled puncta that do not release at all in response to electrical stimulation. Thus, LTD may involve both the modification and deactivation of existing release sites, whereas LTP may involve the activation of silent or the formation of new sites, a question that needs additional investigation.
With respect to modulation of existing active zone function, there are a number of rates that could be modified (Fig. 11). These include the rate of transfer from the reserve pool to the RRP (1), priming and release from the RRP (2), the preferential recycling of vesicles back into the RRP, perhaps after kiss-and-run release (3), and the return of vesicles to the reserve pool for later conversion back into the RRP (4). Of these, any of the first three could result in a selective decrease in release kinetics from the RRP, although the presence of an effect on release during the first stimulus burst favors an action on priming and/or release probability (pr; 2). In contrast, an effect on rate (1) should produce an additional component of LTD of release from the K+-loaded vesicle pools not accounted for by sucrose loading the RRP. The late divergence of the difference curves between RRP and reserve pool release (Fig. 4C) may indicate an effect on transfer from the reserve pool to the RRP, but this rate is too slow to account for initial release effects. The rate of refilling of the RRP has been shown to be dependent on firing frequency (Wang and Kaczmarek, 1998), whereas RRP size can be altered at mossy fiber–CA3 synapses by seizure activity (Goussakov et al., 2000), suggesting that RRP recycling rates are a likely target for the longterm modifications underlying activity-dependent long-term plasticity.
Figure 11.

Potential sites of modifications underlying the presynaptic LTD of release selectively targeting the RRP. Vesicular transmitter release targets before (Pre-LTD) and during (LTD) expression of LTD: (1) Transfer from the reserve vesicle pool to the RRP. (2) Priming and release of docked vesicles. (3) Kiss-and-run recycling of vesicles preferentially into the RRP. (4) Recycling of vesicles into the reserve pool. Although a reduction in the rates of any of these steps could produce presynaptic LTD, our observation that LTD preferentially reduces release from the RRP, without altering reserve pool size or early release kinetics suggests that the rates of RRP priming and pr (2), and/or re-entry (3), are reduced during LTD.
Our data also support a particular second-messenger cascade underlying LTD of release. NMDA-receptor-dependent LTD of release appears to be mediated by retrograde diffusion of the intercellular messenger NO, because inhibition of NOS completely blocked LTD of RRP release. However, the observation that the NO scavenger hemoglobin significantly reduced, but did not completely prevent, LTD of release could either mean that Hb could not completely scavenge extracellular NO before it found its way to the terminals, or that presynaptic NOS contributes some intraterminal NO.
LTD of release throughout the 20 min stimulus time course was also dependent on PKG activity, consistent with previous indirect studies indicating that NO, cGMP, and PKG are crucial for a presynaptic form of LTD (Izumi and Zorumski, 1993; Gage et al., 1997; Wu et al., 1998). Previous studies have also implicated NO, cGMP, and PKG in the induction of LTP, including data from dissociated neuronal cultures (Arancio et al., 1995; 2001) and hippocampal slices (Lu et al., 1999; Bon and Garthwaite, 2001), but there are contradictory studies in hippocampal slices (Williams et al., 1993; Cummings et al., 1994), and a report of a lack of impairment of LTP in a double type I and II PKG knockout mouse (Kleppisch et al., 1999). It is of particular interest that this last study, which argued strongly against a role for PKG in LTP, also reported that LTP was susceptible to the inhibition of NOS, suggesting that NO may have actions crucial for LTP apart from the activation of guanylyl cyclase. Our data strongly support a crucial role for the NO–cGMP–PKG pathway in a presynaptic form of LTD. It has been suggested (Arancio et al., 2001) that complicated, and often contradictory, reports may reflect the presence of dual roles in LTD and LTP, with both cGMP-dependent and cGMP-independent forms of each complicating comparison of long-term plasticity induced by differing stimulus protocols. The NO–cGMP–PKG pathway might be necessary for the induction of both LTD and LTP of presynaptic release. In that case, other, as yet unidentified, mechanisms must then control the switch from one to the other effect dominating the net change.
Footnotes
This work was supported by National Institutes of Health Grant NS44421, the Alexander von Humboldt Stiftung and the Whitehall Foundation (P.K.S.), the Grass and Onassis Foundations (A.K.), and Deutsche Forschungsgemeinschaft (W.M.). We thank D. Hall for invaluable assistance with electron microscopy, H. Glasser for expert technical assistance, and R. Carroll, D. Faber, S. Nawy, and S. Siegelbaum for insightful comments.
Correspondence should be addressed to Dr. Patric K. Stanton, Departments of Neuroscience and Neurology, Kennedy Center Room B21, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461-1602. E-mail: stanton@aecom.yu.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235936-09$15.00/0
References
- Arancio O, Antonova I, Gambaryan S, Lohmann SM, Wood JS, Lawrence DS, Hawkins RD ( 2001) Presynaptic role of cGMP-dependent protein kinase during long-lasting potentiation. J Neurosci 21: 143-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio O, Kandel ER, Hawkins RD ( 1995) Activity-dependent long-term enhancement of transmitter release by presynaptic 3′-5′-cGMP in cultured hippocampal neurons. Nature 376: 74-80. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Bartsch D, Kandel ER ( 1996) Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA 93: 13445-13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benke TA, Luthi A, Isaac JT, Collingridge GL ( 1998) Modulation of AMPA receptor unitary conductance by synaptic activity. Nature 393: 793-797. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF ( 1990) Presynaptic mechanism for long-term potentiation in the hippocampus. Nature 346: 724-729. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS ( 1992) Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science 255: 200-203. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA ( 1994) Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science 264: 1148-1152. [DOI] [PubMed] [Google Scholar]
- Bon CL, Garthwaite J ( 2001) Exogenous nitric oxide causes potentiation of hippocampal synaptic transmission during low-frequency stimulation via the endogenous nitric oxide-cGMP pathway. Eur J Neurosci 14: 585-594. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW ( 2000) Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron 28: 245-259. [DOI] [PubMed] [Google Scholar]
- Braunewell KH, Manahan-Vaughan D ( 2001) Long-term depression: a cellular basis for learning? Rev Neurosci 12: 121-140. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, von Zastrow M, Nicoll RA, Malenka RC ( 1999) Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci 2: 454-460. [DOI] [PubMed] [Google Scholar]
- Cummings JA, Nicola SM, Malenka RC ( 1994) Induction in the rat hippocampus of long-term potentiation (LTP) and long-term depression (LTD) in the presence of a nitric oxide synthase inhibitor. Neurosci Lett 176: 110-114. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T ( 1999) Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399: 66-70. [DOI] [PubMed] [Google Scholar]
- Gage AT, Reyes M, Stanton PK ( 1997) Nitric oxide-guanylyl cyclase-dependent and -independent components of multiple forms of long-term synaptic depression. Hippocampus 7: 286-295. [DOI] [PubMed] [Google Scholar]
- Goda Y, Stevens CF ( 1998) Readily releasable pool size changes associated with long term depression. Proc Natl Acad Sci USA 95: 1283-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goussakov IV, Fink K, Elger CE, Beck H ( 2000) Metaplasticity of mossy fiber synaptic transmission involves altered release probability. J Neurosci 20: 3434-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harata N, Ryan TA, Smith SJ, Buchanan J, Tsien RW ( 2001) Visualizing recycling synaptic vesicles in hippocampal neurons by FM1-43 photoconversion. Proc Natl Acad Sci USA 98: 12748-12753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel AW, Lubke J, Betz WJ ( 1996) FM1-43 dye ultrastructural localization in and release from frog motor terminals. Proc Natl Acad Sci USA 93: 1918-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF ( 1993) Nitric oxide and long-term synaptic depression in the rat hippocampus. Neuro Report 4: 1131-1134. [PubMed] [Google Scholar]
- Katz LC, Shatz CJ ( 1996) Synaptic activity and the construction of cortical circuits. Science 274: 1133-1138. [DOI] [PubMed] [Google Scholar]
- Kleppisch T, Pfeifer A, Klatt P, Ruth P, Montkowski A, Fassler R, Hofmann F ( 1999) Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J Neurosci 19: 48-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF ( 1998) NMDA induces longterm synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21: 1151-1162. [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD ( 1999) Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci 19: 10250-10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Zablow L, Kandel ER, Siegelbaum SA ( 1999) Cyclic AMP induces functional presynaptic boutons in hippocampal CA3-CA1 neuronal cultures. Nat Neurosci 2: 24-30. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA ( 1999) Long-term potentiation: a decade of progress? Science 285: 1870-1874. [DOI] [PubMed] [Google Scholar]
- Malgaroli A, Ting AE, Wendland B, Bergamaschi A, Villa A, Tsien RW, Scheller RH ( 1995) Presynaptic component of long-term potentiation visualized at individual hippocampal synapses. Science 268: 1624-1628. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW ( 1990) Presynaptic enhancement shown by wholecell recordings of long-term potentiation in hippocampal slices. Nature 346: 177-180. [DOI] [PubMed] [Google Scholar]
- Malinow R, Mainen ZF, Hayashi Y ( 2000) LTP mechanisms: from silence to four-lane traffic. Curr Opin Neurobiol 10: 352-357. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG ( 2000) Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23: 649-711. [DOI] [PubMed] [Google Scholar]
- Mellor J, Nicoll RA ( 2001) Hippocampal mossy fiber LTP is independent of postsynaptic calcium. Nat Neurosci 4: 125-126. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Montgomery J, Madison DV ( 2000) Presynaptic protein kinase activity supports long-term potentiation at synapses between individual hippocampal neurons. J Neurosci 20: 4497-4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Choi S, Tsien RW ( 1999) Visualization of synaptic activity in hippocampal slices with FM1-43 enabled by fluorescence quenching. Neuron 24: 803-808. [DOI] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Piedras-Renteria ES, Tsien RW ( 2000) Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron 28: 221-231. [DOI] [PubMed] [Google Scholar]
- Reyes M, Stanton PK ( 1996) Induction of hippocampal long-term depression requires release of Ca 2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci 16: 5951-5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Harde M, Potter BVL, Galione A, Stanton PK ( 1999) Induction of hippocampal LTD requires nitric-oxide-stimulated PKG activity and Ca 2+ release from cyclic ADP-ribose-sensitive stores. J Neurophysiol 82: 1569-1576. [DOI] [PubMed] [Google Scholar]
- Richards DA, Guatimosim C, Betz WJ ( 2000) Two endocytic recycling routes selectively fill two vesicle pools in frog motor nerve terminals. Neuron 27: 551-559. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF ( 1996) Definition of the readily-releasable pool of vesicles at hippocampal synapses. Neuron 16: 1197-1207. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ ( 1993) The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron 11: 713-724. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Ziv NE, Smith SJ ( 1996) Potentiation of evoked vesicle turnover at individually resolved synaptic boutons. Neuron 17: 125-134. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E ( 1999) Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23: 399-409. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia R, Zaman S, Wenthold R, Svoboda K, Malinow R ( 1999) Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 284: 1811-1816. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Heinemann U, Müller W ( 2001) FM1-43 imaging reveals cGMP-dependent long-term depression of presynaptic transmitter release. J Neurosci 21: RC167(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D ( 1999) LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402: 421-425. [DOI] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK ( 1998) High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394: 384-388. [DOI] [PubMed] [Google Scholar]
- Williams JH, Li YG, Nayak A, Errington ML, Murphy KP, Bliss TVP ( 1993) The suppression of long-term potentiation in rat hippocampus by inhibitors of nitric oxide synthase is temperature and age dependent. Neuron 11: 877-884. [DOI] [PubMed] [Google Scholar]
- Wu J, Wang Y, Rowan MJ, Anwyl R ( 1998) Evidence for involvement of the cGMP-protein kinase G signaling system in the induction of long-term depression, but not long-term potentiation, in the dentate gyrus in vitro J Neurosci 18: 3589-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeckel MF, Kapur A, Johnston D ( 1999) Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci 2: 625-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA ( 2001) Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat. Neurosci 4: 711-717. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA ( 2002) mGluR-dependent LTD alters mode of presynaptic exocytosis. Neuron 35: 1099-1110. [DOI] [PubMed] [Google Scholar]