

Abstract
Background & Aims:
The intestinal microbiome might affect development and severity of nonalcoholic fatty liver disease (NAFLD). We analyzed microbiomes of children with and without NAFLD.
Methods:
We performed a prospective, observational, cross-sectional study of 87 children (8–17 years old) with biopsy-proven NAFLD and 37 children with obesity without NAFLD (controls). Fecal samples were collected and microbiome composition and functions were assessed using 16S rRNA amplicon sequencing and metagenomic shotgun sequencing. Microbial taxa were identified using zero-inflated negative binomial modeling. Genes contributing to bacterial pathways were identified using gene set enrichment analysis.
Results:
Fecal microbiomes of children with NAFLD had lower α-diversity than controls (3.32 vs 3.52; P=.016). Fecal microbiomes from children with nonalcoholic steatohepatitis (NASH) had lowest α-diversity (controls, 3.52; NAFLD, 3.36; borderline NASH, 3.37; NASH 2.97; P=.001). High abundance of Prevotella copri was associated with more severe fibrosis (P=.036). Genes for lipopolysaccharide biosynthesis were enriched in microbiomes from children NASH (P<.001). Classification and regression tree model with level of alanine aminotransferase and relative abundance of the lipopolysaccharide pathway gene encoding 3-deoxy-D-manno-octulosonate 8-phosphate-phosphatase identified patients with NASH with an area under the receiver operating characteristic curve value of 0.92. Genes involved in flagellar assembly were enriched in fecal microbiomes of patients with moderate to severe fibrosis (P<.001). Classification and regression tree models based on level of alanine aminotransferase and abundance of genes encoding flagellar biosynthesis protein had good accuracy for identifying cases with moderate to severe fibrosis (area under the receiver operating characteristic curve, 0.87).
Conclusions:
In an analysis of fecal microbiomes of children with NAFLD, we associated NAFLD and NASH with intestinal dysbiosis. NAFLD and its severity were associated with greater abundance of genes encoding inflammatory bacterial products. Alterations to the intestinal microbiome might contribute to pathogenesis of NAFLD and be used as markers of disease or severity.
Keywords: intestinal microbiota, pediatric, lipopolysaccharide, flagellin
Lay summary:
The intestinal microbiota of children with nonalcoholic liver disease is altered compared to that of children without this chronic liver disease, with increased levels of bacterial proteins that promote inflammation. These microbial features were associated with liver disease severity.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease in American children.1 The diagnosis of NAFLD requires that 5% or more hepatocytes exhibit macrovesicular steatosis, and the exclusion of other identifiable liver diseases and/or clinical conditions, which may cause steatosis. A comprehensive understanding of why certain children develop NAFLD is lacking. Although obesity is a risk factor for NAFLD, most children with obesity do not develop NAFLD. A subset of children with NAFLD has a progressive sub-phenotype known as nonalcoholic steatohepatitis (NASH), characterized by hepatic inflammation and cell injury. Moreover, some children with NASH develop cirrhosis and end-stage liver disease. Thus, NAFLD is not a singular diagnosis, but a broad clinical-pathological spectrum of liver disease.2 Factors that explain the severity of disease are poorly characterized. Emerging evidence suggests that the intestinal microbiome may influence the development and severity of NAFLD.
The intestinal microbiome is a complex ecosystem composed of trillions of microbes, mainly in the large bowel, living in a predominantly symbiotic relationship with the host and broadly impacting host physiology, immune development and function, nutrition, and mucosal protection.3–5 However, it may also impact human pathophysiology, including inflammatory bowel disease, and obesity6 by influencing local and systemic inflammation7,8 and the host capacity for energy harvest.9,10 Additionally, animal models suggest that it may influence several of the putative processes involved in the development and progression of NAFLD,11 including choline metabolism, endotoxemia, obesity, liver inflammation and fibrosis.12–16
While mouse models have implicated intestinal bacteria in the pathophysiology of NAFLD,17 evidence in humans is more limited, especially in children. Although some human studies have evaluated the fecal microbiota in children with NAFLD, they have been small and in some cases, limited by the lack of optimally characterized cases and controls.18,19 Therefore, we evaluated the fecal microbiome of children with NAFLD, and age and Body Mass Index (BMI)-matched control children without NAFLD, characterizing the bacterial composition and functional capacity that discriminate between cases and controls and those that associate with greater disease severity.
METHODS
Study subjects
This was a prospective, observational, cross-sectional ancillary study to the NASH CRN performed at its UC San Diego Clinical Center. Participants were age 8-17 years. Cases were children with NAFLD diagnosed within 90 days using standard clinical history, laboratory, and liver histology criteria.20 Controls were overweight or obese children recruited from primary care clinics without evidence of liver disease based on clinical history, labs and liver Magnetic Resonance Spectroscopy (MRS). Use of probiotics or recent (past 90 days) antibiotics exposure was exclusionary. Institutional Review Boards at each participating center approved the study. Parents provided written informed consent and children provided written assent.
Clinical assessment
Demographic and clinical data for each participant was obtained at a single intake visit at the UCSD Altman Clinical and Translational Research Institute. Height and weight were measured, and fasting blood samples were obtained for glucose, insulin and, liver and lipid panels. Fresh fecal samples were collected.
Liver histology
Liver biopsy specimens were stained with hematoxylin-eosin and Masson’s trichrome stains, and reviewed centrally by the NASH CRN Pathology Committee according to the published NASH CRN histological scoring system scoring system.21 The diagnosis of nonalcoholic steatohepatitis (NASH), borderline NASH, or NAFLD not NASH was made based on the aggregate presence and degree of the individual features of nonalcoholic fatty liver disease. Fibrosis was staged based on the following: stage 0 (no fibrosis), stage 1a (mild perisinusoidal), stage 1b (moderate perisinusoidal), stage 1c (portal fibrosis), stage 2 (perisinusoidal and periportal), stage 3 (bridging fibrosis), and stage 4 (cirrhosis).
Liver MRS
Proton Density Fat Fraction (PDFF) was estimated using MRS by a long-TR, multi-TE stimulated echo acquisition mode (STEAM) sequence using established methodology (see supplement for full details).22,23
Fecal sample collection and processing
Fresh fecal samples were placed in RNAlater™ and rocked overnight at 4°C and stored at −80°C until isolation. Genomic DNA was isolated from fecal samples using the MO BIO PowerFecal DNA Isolation Kit™ (MO BIO) with following modification: after addition of solution C1 and heating at 65°C for 10 minutes, sample was subjected toheating at 95°C for 10 minutes followed by bead beating using the PowerLyzer™ (MO BIO).
16S rRNA gene amplicon sequencing
Polymerase chain reaction (PCR) amplification was performed using primers targeting V1-V3 region of the bacterial 16S rRNA gene. PCR products were purified using 1.8x volume Agencourt AMPure XP™ beads (Beckman Coulter) and quantified using the Qubit® dsDNA BR assay or dsDNA HS assay (Life Technologies). Samples were multiplexed and sequenced on the Illumina MiSeq™ platform to generate 300bp, paired-end reads. (see supplement for full details).
Metagenomic whole genome shotgun (mWGS) sequencing
mWGS libraries were constructed with 100-250 ng of sample DNA, using KAPA HTP library preparation kits™ (KAPA Biosystems), with 8 (for 250 ng) or 10 (for 100 ng) PCR amplification cycles. Library concentrations were determined using the KAPA qPCR kit™ (KAPA Biosystems). Libraries were multiplexed and run on Illumina HiSeq 2000™ platform to generate 100bp paired-end reads.
Bioinformatic Processing of Sequence Data
Paired-end 16S reads were assembled into contiguous sequences and clustered at a 97% sequence similarity threshold using USEARCH 24 to generate set of 1136 Operational Taxonomic Units (OTUs). These were filtered to retain 712 OTUs, which were either present in >5% of samples, or constituted >1% of the reads in one or more sample.
To quantify relative gene abundance in the intestinal metagenome, mWGS sequences were mapped to curated reference gene database using HUMAnN2.25 The function of detected genes was determined by aligning them to the Kyoto Encyclopedia of Genes and Genomes (KEGG) reference database (see supplement for full details).26
Data Analysis
Patients’ characteristics were summarized as median and interquartile range or n (%). A non-parametric Mann-Whitney-Wilcoxon test was used to compare cases and controls while a Kruskal-Wallis test was used to compare Prevotella copri low, medium, and high abundance (see supplementary methods) in continuous variables. A Chi-square or Fisher’s exact test was performed for categorical variables. To compare continuous variables between groups, a t test or Analysis of Variance (ANOVA) was performed. If parametric assumptions were not met even after transformation (typically log), then a non-parametric method was used. Individuals were classified as low, medium, or high based on relative abundance of P. copri (see results and supplementary methods), and distribution of these categories by fibrosis, NASH, or steatosis was examined using a linear-by-linear association exact test. Fibrosis stage was dichotomized into none to mild (stages 0 to 1) or moderate to severe.
Due to the sparseness of the microbiome data, a zero-inflated negative binomial model 27 was applied to test the differential effect of NAFLD or fibrosis on the relative abundance of bacteria at different taxonomic levels. For those that did not converge, a negative binomial or Poisson model was used instead. To account for the potential confounding factor of ethnicity, ethnicity (Hispanic vs Non-Hispanic) was included as a covariate for those genera or OTUs that were significant at p<0.05.
Comparisons of microbiome community between individuals (β-diversity) were based on a Bray-Curtis distance matrix of normalized OTU counts and evaluated using principal coordinates analysis in conjunction with analysis of similarity (ANOSIM) tests.28
Classification and regression trees (CART) were used to explore relationships between fibrosis, α-diversity and microbial taxon abundance. Fibrosis (absent/mild vs moderate/severe) was treated as the target variable and the following were predictors: Bacteroides vulgatus abundance, P. copri abundance, and Shannon α-diversity index. The options for parent node and terminal node were 10 and 5, respectively. Ten percent leave-out samples were used for cross-validation. Optimization was performed using a Gini function.
Differential abundance of microbial gene pathways was tested by gene set enrichment analysis (GSEA). Microbial genes were represented as KEGG Orthologs (KOs, see supplementary methods), and a p-value was generated for each KO using a gamma model or a mixture model. KOs were ranked by p-value and the relative enrichment of KEGG pathways within KO rankings was calculated using the R package piano.29 Direction of the change in relative abundance of each KO was determined based on the direction (positive vs. negative) of the normalized Wilcoxon signed-rank test statistic. Significantly enriched pathways were identified at p<0.05 following False Discovery Rate (FDR) correction. In comparisons of none/mild vs. moderate/severe fibrosis GSEA was carried out separately for high and low P. copri individuals. Pathways were reported as significant if they had an FDR-adjusted p-value<0.05 for both groups (for justification of this approach see supplementary methods). CART were also performed to determine important factors in predicting case vs control, NASH vs NAFLD but not NASH, Moderate-to-severe fibrosis vs. absent-to-mild fibrosis using ALT and abundance of KOs from the LPS biosynthetic pathway or the flagellar assembly pathway as predictor variables. Area Under the Receiver Operating Characteristics curve (AUROC) was calculated to evaluate the performance.
The relative contribution of different bacterial genera to microbial gene pathways of interest was assessed using indicator values,30 which were generated and tested using the R package labdsv (see supplementary methods).31
Unless otherwise stated, statistical significance was assigned at p<0.05. FDR-adjusted p<0.2 was considered significant for comparisons of taxa at different levels. Statistical analyses were performed using SAS 9.4 (SAS Institute, Cary, NC), StatXact (Cytel Studio version 8.0.0. Cytel Inc.), R (version 3.3.2 or 3.4.2, Vienna, Austria), and Salford Predictive Modeler (SPM) software suite CART (Salford Systems, San Diego, CA). R package phyloseq 32 was used and microbial community analysis was performed using the R package Vegan.33
RESULTS
Study population
There were 124 children studied, with a median age of 12 years. Demographics and clinical features are shown in Table 1 (Supplementary Fig. 1). Cases (n=87) and controls (n=37) did not differ by age or race. Children with NAFLD were more likely to be male, 71% versus 46% (p=0.0073). Cases and controls were well-matched for BMI and percent body fat.
Table 1.
Patient Characteristics: Case vs Control
| Case (n=87) | Control (n=37) | P value | |
|---|---|---|---|
| Gender | |||
| Male | 62(71) | 17(46) | 0.0073a |
| Ethnic | |||
| Hispanic | 77(89) | 28(76) | 0.070 |
| Race | |||
| White | 39(45) | 13(35) | 0.32a |
| Age | 12(10,14) | 13(11,14) | 0.20b |
| Height (cm) | 160(150,167) | 157(152,165) | 0.62b |
| Weight (kg) | 74(61,93) | 71(62,84) | 0.41b |
| BMI | 29(26,34) | 29(25,31) | 0.35b |
| BMI percentilec | 99(98,99) | 98(95,99) | 0.022b |
| BMI Z scorec | 2(2,2) | 2(1,2) | 0.022b |
| ALT | 57(36,100) | 16(12,20) | <.0001b |
| AST | 40(27,56) | 20(18,25) | <.0001b |
| GGT | 30(22,45) | 18(15,21) | <.0001b |
| Insulin | 28(18,41) | 23(14,39) | 0.21b |
| Glucose | 85(79,89) | 85(82,89) | 0.41b |
| MRS Liver Fat | 19(12,24) | 4(2,5) | <.0001b |
| Body Fat | 42(39,46) | 41(37,46) | 0.34b |
| Triglyceride | 126(90,196) | 86(77,121) | 0.0073b |
| Total cholesterol | 164(141,186) | 154(135,172) | 0.11b |
| HDL cholesterol | 42(36,49) | 44(39,50) | 0.17b |
| LDL cholesterol | 94(76,111) | 83(72,95) | 0.11b |
| Steatosis | |||
| 5-33% | 28(32) | ||
| 34-66% | 25(29) | ||
| >66% | 34(39) | ||
| NASH Diagnosis | |||
| NAFLD, not NASH | 38(44) | ||
| Borderline NASH | 37(43) | ||
| Definite NASH | 11(13) | ||
| Fibrosis | |||
| Stage 0 | 39(45) | ||
| Stage 1a | 4(5) | ||
| Stage 1b | 2(2) | ||
| Stage 1c | 25(29) | ||
| Stage 2 | 10(11) | ||
| Stage 3 | 7(8) |
Data are presented as median (interquartile range) or n (%).
Chi-Square test;
Mann-Whitney-Wilcoxon test;
Calculated based on the 2000 Centers for Disease Control and Prevention (CDC) growth charts
Taxonomic composition of the intestinal microbiome (full study cohort)
Using 16S rRNA gene sequencing, an average of 67,852 gene sequences were generated per individual and 712 discrete bacterial taxa (OTUs) identified. At the phylum level, dominant taxa consisted of the Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria. At the genus level, dominant taxa were Bacteroides, Faecalibacterium, and Blautia, as well as a number of unclassified members of the family Lachnospiraceae (Supplementary Fig. 2).
Differences in the composition of the intestinal microbiome between cases and controls
The phyla Bacteroidetes and Proteobacteria were higher in cases (zero-inflated negative binomial model: fold change (FC): 1.34, FDR-adjusted p=0.051 and FC: 1.57, FDR-adjusted p=0.025, respectively), whereas the phylum Firmicutes was higher in controls (FC: 0.84, FDR-adjusted p=0.025). Genus-level comparisons revealed statistically significant differences in the relative abundance of multiple sparse genera, but few differences in dominant taxa (Fig. 1A, Supplementary Table 1). After adjusting for ethnicity, the majority (14/19) of the genera remained significant. In some cases multiple OTUs assigned to the same genus showed opposite trends of increased/decreased relative abundance between cases and controls (Supplementary Fig. 3, Supplementary Table 1), indicating the potential for differences between distinct taxa belonging to a single genus.
Figure 1. Taxonomic and community-level changes between NAFLD versus controls.
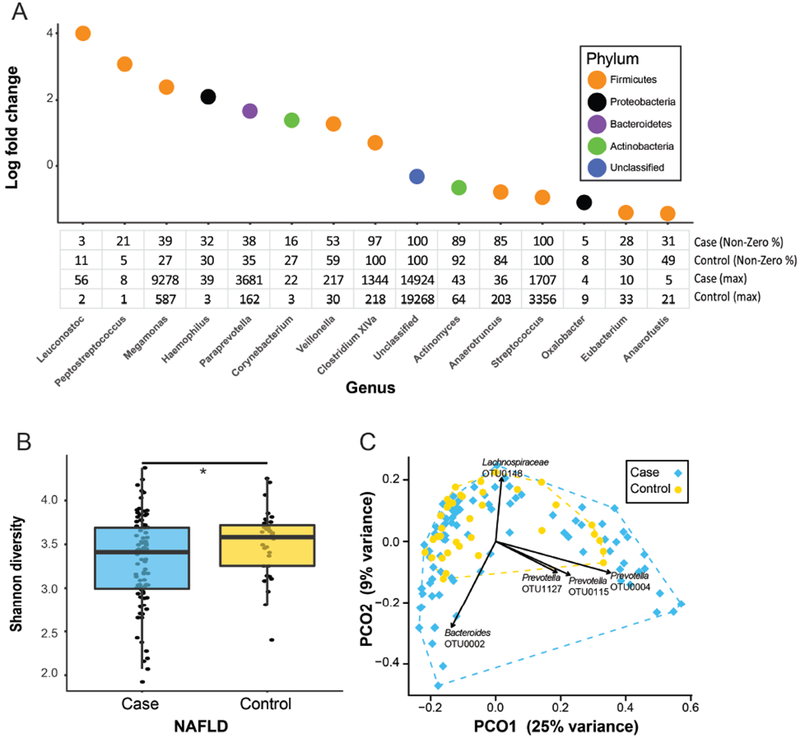
(A) Bacterial genera identified as being differentially abundant when comparing cases to controls and accounting for ethnicity. The table shows percentage of individuals with non-zero read counts for each genus and maximum number of reads detected in any single individual following normalization. (B) Difference in α-diversity between cases and controls (*p<0.05). (C) Principal coordinates analysis (PCoA) plot showing variation in β-diversity between cases and controls. Each point represents a single patient. Overlaid vectors indicate the direction of change in abundance for top five OTUs correlating with distribution of individuals within the plot.
At the community level, cases had significantly lower α-diversity than controls (3.32 vs 3.52, t-test: p=0.016, Fig. 1B). Microbiome composition (β-diversity) did not differ between cases and controls (ANOSIM test, p=0.71); however, principal coordinates analysis (PCoA) demonstrated greater person-to-person microbiome variability among cases (Fig. 1C, multivariate homogeneity groups dispersion test: p=0.061). Person-to-person variability in microbiome composition was correlated with relative abundance of OTUs for two genera - Bacteroides and Prevotella. Species represented by these OTUs were inferred by reconstructing their sequence-based phylogeny (Supplementary Fig. 4, Supplementary Methods) indicating that dominant OTUs likely represented the species P. copri and B. vulgatus.
Individuals clustered into three discrete groups based on relative abundance of P. copri (Supplementary Fig. 5, Supplementary Methods), but did not cluster by B. vulgatus relative abundance. The dominance of B. vulgatus or P. copri within the microbiome appeared mutually exclusive (Supplementary Fig. 5C); however, this trend may not indicate competitive exclusion as suggested previously,19 but may reflect that with finite sequencing abundance estimates are relative, not absolute. Similarly, greater relative abundance of both B. vulgatus and P. copri correlated with lower α-diversity (Supplementary Fig. 6). In addition, high P. copri abundance was significantly associated with Hispanic ethnicity (p=0.025, Supplementary Table 2).
Association of microbiome composition with disease severity
The phyla Proteobacteria and TM7 were higher in patients with NASH (FC: 1.97, p=0.027), while the phyla Fusobacteria, Verrucomicrobia, and Lentisphaerae were higher in patients with NAFLD but not NASH (FC: 0.0006, FDR-adjusted p=0.0033, FC: 0.013, FDR-adjusted p<0.001, FC: 0.084, and FDR-adjusted p=0.15, respectively). At genus level, Lactobacillus and Oribacterium were higher in patients with NASH, while Oscillibacter, Lactonifactor, Akkermansia, and Enterococcus were higher in patients with NAFLD but not NASH (FDR-adjusted p<0.2 Fig. 2A). The genera Akkermansia, Lactobacillus, Oscillibacter, and Parabacteroides remained significant after adjusting for ethnicity. At OTU level, multiple taxa belonging to the same genus showed opposite trends in abundance (Supplementary Fig. 7).
Figure 2. Taxonomic and community-level changes between NAFLD vs. NASH.
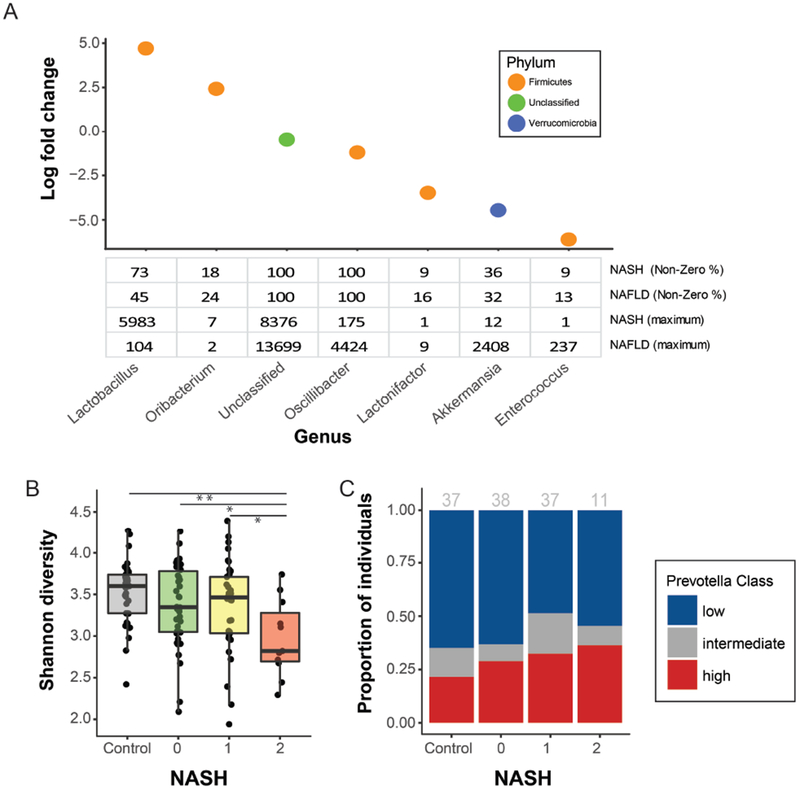
(A) Bacterial genera identified as differentially abundant comparing cases with NAFLD but not NASH vs. definite NASH when accounting for ethnicity. The table shows percentage of individuals with non-zero read counts for each genus and the maximum reads detected in any single individual following normalization. (B) Difference in α-diversity between controls and cases separated by severity (control, 0 = NAFLD but not NASH, 1 = borderline NASH, 2 = definite NASH). (C) Distribution of Prevotella abundance categories (low, intermediate, high,) by severity (control, 0 = NAFLD but not NASH, 1 = borderline NASH, 2 = definite NASH); number per group shown in gray.low, intermediate, or high Prevotella intestinal microbiome by severity (control, 0 = NAFLD but not NASH, 1 = borderline NASH, 2 = definite NASH). Gray numbers above each bar indicate the number of individuals per group.
Community-level changes accompanying NASH were characterized by a significant difference in α-diversity across the spectrum of NAFLD (ANOVA test: p=0.012, Fig. 2B). Patients with NASH had lower α-diversity compared to controls (2.97 vs 3.52, p=0.001), patients with NAFLD but not NASH (2.97 vs 3.36, p=0.017), or patients with borderline NASH (2.97 vs 3.37, p=0.016).
When classified by fibrosis stage (≤1 vs ≥2), the phyla Bacteroidetes (FC: 1.43, FDR-adjusted p=0.091), Proteobacteria (FC: 1.72, FDR-adjusted p=0.062) and TM7 (FC: 3.41, FDR-adjusted p=0.062) were more abundant in patients with moderate-to-severe fibrosis. Fusobacteria (FC: 0.0007, FDR-adjusted p=0.0022), Verrucomicrobia (FC: 0.027, FDR-adjusted p<0.0001), Firmicutes (FC: 0.85, FDR-adjusted p=0.16) were more abundant in patients with absent-to-mild fibrosis. At genus level, the most abundant taxa identified as differentially abundant were Lactobacillus (higher in moderate-to-severe fibrosis), Akkermansia (higher in absent-to-mild fibrosis, Fig. 3A). Slackia, Akkermansia, Gemella, Granulicatella, Lactobacillus, and Turicibacter remained significant after adjusting ethnicity. Also, in several cases multiple OTUs assigned to the same genus showed conflicting trends with fibrosis severity (Supplementary Fig. 8).
Figure 3. Taxonomic and community-level changes between absent-to-mild vs. moderate-to-severe-fibrosis.
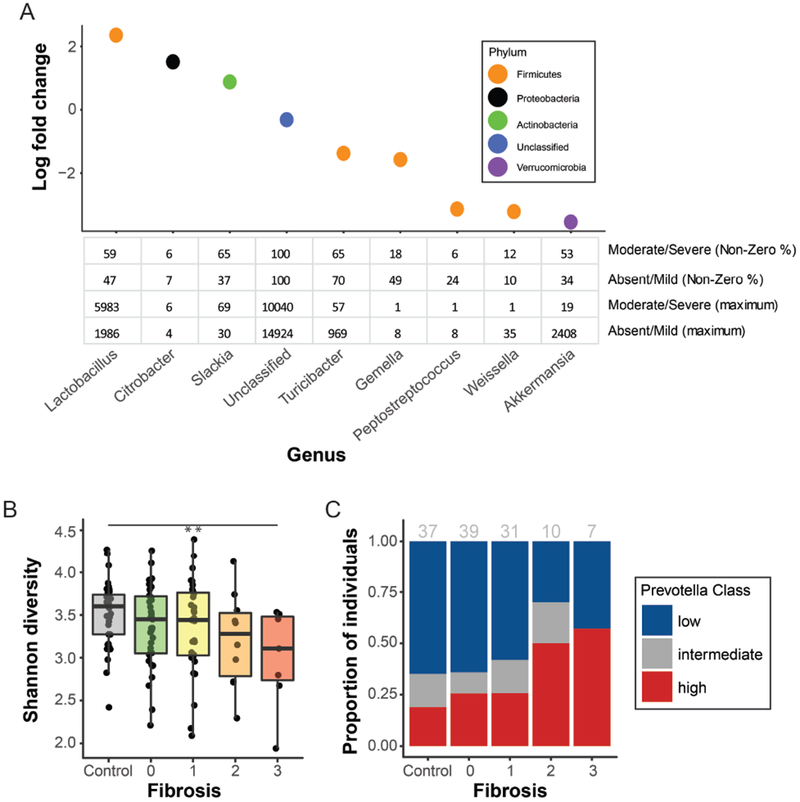
(A) Bacterial genera differentially abundant by fibrosis stage ( ≤1 vs ≥2) when accounting for ethnicity. Table shows percentage of individuals with non-zero read counts for each genus and maximum reads detected in any single individual following normalization. (B) Difference in α-diversity across fibrosis stages (control, NAFLD with stages 1,2,3) (C) Distribution of Prevotella abundance categories (low, intermediate, high) across across fibrosis stages (control, NAFLD with stages 1,2,3); number per group shown in gray.
Comparison across the range from control to NAFLD without or with severe fibrosis showed lower α-diversity (p=0.009, Fig. 3B) and higher frequency of high P. copri abundance (p=0.036, Fig. 3C) among those with greater fibrosis. Because P. copri abundance was associated with both lower α-diversity and Hispanic ethnicity, CART analysis was performed and showed that BMI and P. copri abundance were the best predictors of fibrosis severity (Supplementary Fig. 9) and that ethnicity did not add to the models ability to identify fibrosis severity.
Association of microbiome gene pathways with case status and severity
Differences in the composition of the microbiome (Fig. 1C) correlated strongly with functional differences, as reflected by variation in microbial gene diversity (Mantel R=0.86, p (perm) <0.001, Supplementary Fig. 10A). However, the total number of genes in the metagenome did not vary with presence of NAFLD, or disease severity (Supplementary Figs. 10–11).
Using GSEA, 30 pathways were differentially abundant in one or more pairwise comparisons (Table 2). No evidence was found for variation in abundance of gene pathways associated with alcohol metabolism and this negative result was subsequently corroborated using serum alcohol assays (see supplementary methods). There was significantly greater prevalence of pathways associated with carbohydrate metabolism in the milder disease state for each pairwise comparison (Case vs. control, NASH vs NAFLD but not NASH, Moderate-to-severe fibrosis vs. absent-to-mild fibrosis), while there was significantly greater prevalence of pathways whose products stimulate host inflammation in the more severe disease state for each pairwise comparison.
Table 2. Metabolic pathways enriched in different stages of NAFLD.
Gene set enrichment analysis (GSEA) results showing metabolic pathways that are enriched in comparisons of i) cases (NAFLD+NASH) vs. obese controls, ii) NAFLD (steatosis 0) vs. NASH (steatosis 2), iii) non-severe (fibrosis <2) vs. severe (fibrosis 2+) fibrosis.
| Enriched in | Pathway Description | KEGG ID | KOs In Pathway | KOs Detected In Study | GSEA Statistic | p-value | q-value | KOs Up In Rank | KOs Down In Rank |
|---|---|---|---|---|---|---|---|---|---|
| Controls | |||||||||
| Glycolysis / Gluconeogenesis | ko00010 | 94 | 62 | 0.43754 | 1.00E-04 | 0.00355 | 37 | 25 | |
| Pentose phosphate pathway | ko00030 | 75 | 53 | 0.47768 | 0.0041 | 0.041586 | 30 | 23 | |
| Starch and sucrose metabolism | ko00500 | 101 | 64 | 0.32807 | 0 | 0 | 48 | 16 | |
| Amino sugar and nucleotide sugar metabolism | ko00520 | 144 | 94 | 0.46333 | 1.00E-04 | 0.00355 | 57 | 37 | |
| Glucagon signaling pathway | ko04922 | 55 | 7 | 0.13289 | 6.00E-04 | 0.00852 | 7 | 0 | |
| Lysine degradation | ko00310 | 71 | 22 | 0.37371 | 0.0019 | 0.022483 | 15 | 7 | |
| D-Alanine metabolism | ko00473 | 5 | 5 | 0.12772 | 0.0021 | 0.022938 | 5 | 0 | |
| Peptidoglycan biosynthesis | ko00550 | 40 | 34 | 0.39466 | 6.00E-04 | 0.00852 | 21 | 13 | |
| Tetracycline biosynthesis | ko00253 | 20 | 6 | 0.15449 | 0.0014 | 0.018073 | 6 | 0 | |
| beta-Lactam resistance | ko01501 | 99 | 37 | 0.41112 | 6.00E-04 | 0.00852 | 23 | 14 | |
| Vancomycin resistance | ko01502 | 21 | 17 | 0.27211 | 2.00E-04 | 0.0047333 | 13 | 4 | |
| ABC transporters | ko02010 | 451 | 321 | 0.55299 | 5.00E-04 | 0.00852 | 148 | 173 | |
| Phosphotransferase system (PTS) | ko02060 | 83 | 67 | 0.38192 | 0 | 0 | 47 | 20 | |
| Aminoacyl-tRNA biosynthesis | ko00970 | 64 | 35 | 0.3799 | 2.00E-04 | 0.0047333 | 21 | 14 | |
| Cases | |||||||||
| Lipopolysaccharide biosynthesis | ko00540 | 38 | 37 | 0.10385 | 0 | 0 | 4 | 33 | |
| Riboflavin metabolism | ko00740 | 26 | 16 | 0.055318 | 1.00E-04 | 0.00355 | 0 | 16 | |
| Bacterial chemotaxis | ko02030 | 26 | 26 | 0.087381 | 0 | 0 | 1 | 25 | |
| Flagellar assembly | ko02040 | 41 | 38 | 0.053467 | 0 | 0 | 1 | 37 | |
| NAFLD | |||||||||
| Methane Metabolism | ko00680 | 158 | 103 | 0.26365 | 1.00E-04 | 0.0044996 | 27 | 76 | |
| Carbon Metabolism | ko01200 | 342 | 203 | 0.43357 | 1.00E-04 | 0.0044996 | 89 | 114 | |
| Ribosome biogenesis in eukaryotes | ko03008 | 82 | 11 | 0.18249 | 0.00049995 | 0.016873 | 2 | 9 | |
| RNA Transport | ko03013 | 138 | 12 | 0.10798 | 1.00E-04 | 0.0044996 | 1 | 11 | |
| RNA Polymerase | ko03020 | 51 | 15 | 0.24024 | 0.0009999 | 0.026997 | 4 | 11 | |
| NASH | |||||||||
| Phenylalanine metabolism | ko00360 | 75 | 38 | 0.25417 | 0.00019998 | 0.0089991 | 30 | 8 | |
| Benzoate degradation | ko00362 | 90 | 29 | 0.26513 | 0.00069993 | 0.015748 | 23 | 6 | |
| Lipopolysaccharide biosynthesis | ko00540 | 38 | 37 | 0.27594 | 0.00049995 | 0.013499 | 31 | 6 | |
| Folate biosynthesis | ko00790 | 44 | 27 | 0.27806 | 0.0017998 | 0.034711 | 22 | 5 | |
| Degradation of aromatic compounds | ko01220 | 198 | 38 | 0.17672 | 1.00E-04 | 0.0067493 | 34 | 4 | |
| Ribosome | ko03010 | 143 | 73 | 0.33263 | 0.00039996 | 0.013499 | 52 | 21 | |
| Staphylococcus aureus infection | ko05150 | 66 | 7 | 0.014195 | 1.00E-04 | 0.0067493 | 7 | 0 | |
| Absent/Mild Fibrosis | |||||||||
| Phosphotransferase system (PTS) | ko02060 | 83 | 64 | 0.31372 | 0 | 0 | 16 | 48 | |
| 65 | 0.34107 | 0 | 0 | 18 | 47 | ||||
| Fructose and mannose metabolism | ko00051 | 96 | 70 | 0.42412 | 5.00E-04 | 0.0095714 | 27 | 43 | |
| 66 | 0.40166 | 2.00E-04 | 0.0132 | 27 | 39 | ||||
| Moderate/Severe Fibrosis | |||||||||
| Flagellar assembly | ko02040 | 41 | 37 | 0.21208 | 0 | 0 | 31 | 6 | |
| 37 | 0.25479 | 0.0004 | 0.0132 | 33 | 4 | ||||
| Ribosome | ko03010 | 143 | 84 | 0.15512 | 0 | 0 | 81 | 3 | |
| 60 | 0.14784 | 0 | 0 | 54 | 6 |
Lipopolysaccharide (LPS) biosynthesis was significantly enriched in cases (FDR-adjusted p<0.0001, Table 2, Fig. 4A) and further enriched in those with NASH (FDR-adjusted p=0.0135, Table 2, Fig. 4E). CART models using ALT and the abundance of KOs within the LPS biosynthesis pathway as predictors had good diagnostic accuracy for discriminating NAFLD cases (AUROC=0.95) and for discriminating cases with NAFLD but not NASH from those with definite NASH (AUROC=0.92). Measuring the abundance of a single KO from the LPS pathway was sufficient in each CART model: the relative abundance of genes encoding for 3-deoxy-D-manno-octulosonate 8-phosphate phosphatase (kdsC) provided the best prediction for NAFLD (Fig. 4B,C); while the relative abundance of genes encoding for UDP-3-O-acyl-N-acetylglucosamine deacetylase (lpxC) provided the best prediction for NASH (Fig. 4F,G). Examination of the taxonomic origin of shotgun-sequenced reads indicated that 32 bacterial genera contributed to LPS biosynthesis. Indicator values were used as a concise metric to represent the extent to which the contribution of different genera to this pathway was characteristic of either cases vs. controls, or NAFLD vs. NASH (Fig. 4D,H).
Figure 4. Lipopolysaccharide biosynthesis (LPS) pathway association with NAFLD and NASH.
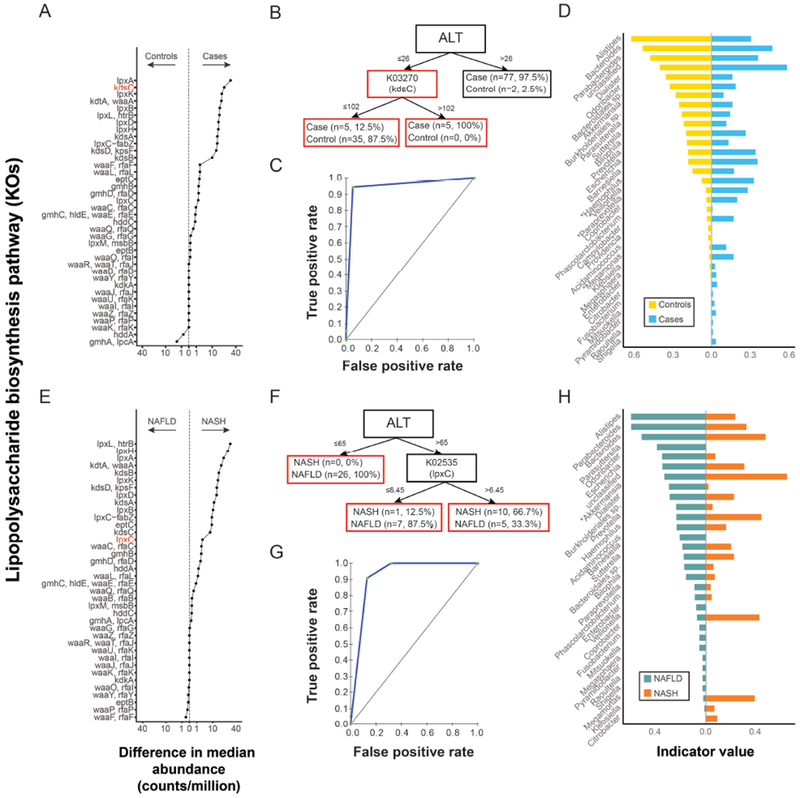
(A) KEGG Orthologs (KOs) in LPS pathway shown on y-axis and the absolute difference (controls vs cases) in median relative abundance of each KO shown on the x-axis. Values to the left and right of the dashed line indicate KOs found at greater relative abundance in controls and cases, respectively. KOs in red are those appearing in panel B. (B) Classification and regression tree (CART) model for NAFLD based on ALT and the abundance of KOs from the LPS biosynthesis pathway . The corresponding ROC is shown in panel (C). (D) Bacterial genera contributing to LPS assembly in controls and cases are shown on the y-axis. The x-axis depicts indicator values. A large indicator value indicates a genus with both high fidelity to a condition (i.e. the genus was detected as contributing to this pathway in a large proportion of individuals with that condition) and high specificity to a condition (i.e. the genus was found to contribute more to the condition in question than to the alternative condition in the contrast). Asterisks indicate genera present in Fig. 1A. (E) Difference in the median number of reads mapping to the LPS pathway in patients with NAFLD vs NASH. The x and y axis are as described for A. (F) CART model for detecting NASH based on ALT and the abundance of KOs from the LPS pathway as predictor variables. The corresponding ROC is shown in panel (G). (H) Bacterial genera contributing to LPS assembly in patients with NAFLD and patients with NASH. The x and y axis are as described for D. Asterisks indicate genera present in Fig. 2A.
Flagellar assembly was also enriched in cases (FDR-adjusted p<0.0001, Table 2, Fig. 5A), and in those with moderate-to-severe fibrosis (FDR-adjusted p<0.0001, Fig. 5A, E). CART models based on ALT and the abundance of KOs within the flagellar assembly pathway had good diagnostic accuracy for discriminating NAFLD cases (Fig. 5B,C, AUROC=0.97) and for discriminating cases with absent-to-mild from those with moderate-to-severe fibrosis (Fig. 5F,G, AUROC=0.87). KOs selected for inclusion in CART models predicting NAFLD represented genes encoding motility protein B (MotB), flagellar hook-basal body complex protein (FliE), flagellar basal body P-ring formation protein (FlgA, Fig. 5A,B). The single KO included in CART models for fibrosis represented genes encoding Flagellar biosynthesis protein (FlhA, Fig. 5E,F). There were 16 bacterial genera that contributed to flagellar assembly and the relative contribution of specific taxa to this pathway varied across conditions (Fig. 5D, H). In particular, the genus Lactobacillus made a significantly greater contribution to flagellar assembly in cases vs control (FDR-adjusted p=0.187) and in moderate/severe fibrosis (FDR-adjusted p=0.03).
Figure 5. Flagellar assembly pathway associations with NAFLD and with fibrosis severity.
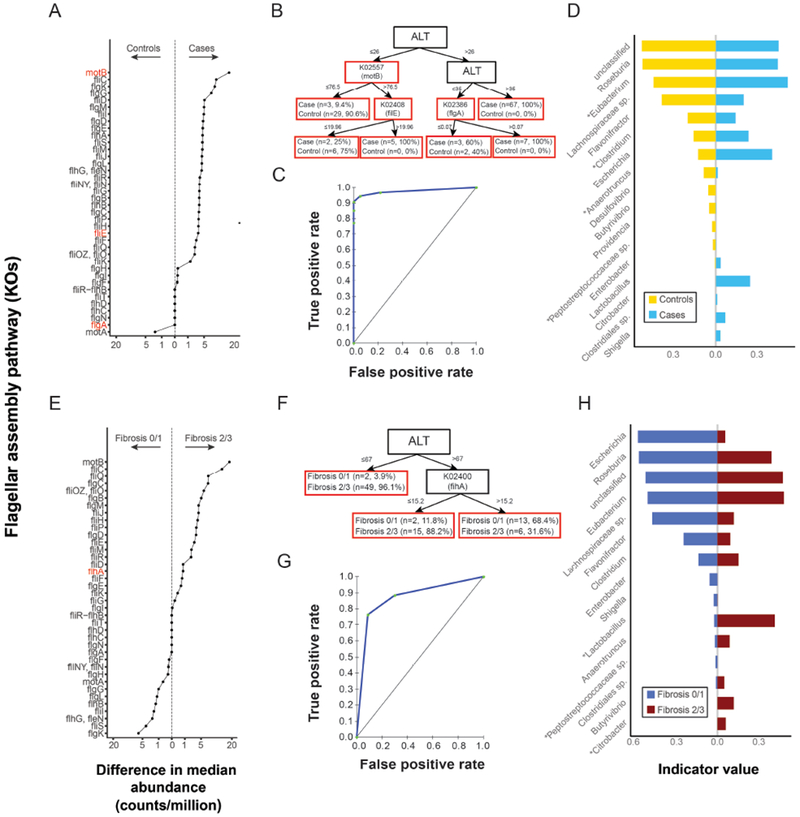
(A) KEGG Orthologs (KOs) in the flagellar assembly pathway shown on y-axis and the absolute difference (controls vs cases) in median relative abundance of each KO shown on the x-axis. Values to the left and right of the dashed line indicate KOs found at greater relative abundance in controls and cases, respectively. KOs in red are those appearing in panel B. (B) CART model for NAFLD based on ALT and the abundance of KOs from the flagellar assembly pathway as predictor variables. The corresponding ROC is shown in panel (C). (D) Bacterial taxa genera contributing to flagellar assembly in controls and cases are shown on the y-axis. The x-axis depicts indicator values. Asterisks indicate genera present in Fig. 1A. (E) difference in the median number of reads mapping to the flagellar assembly pathway in patients by fibrosis stage ( ≤1 vs ≥2) . The x and y axis are as described for A. (F) CART model for moderate-to-severe fibrosis based on ALT and abundance of KOs from the the flagellar assembly pathway as predictor variables. The corresponding ROC is shown in panel (G). (H) Bacterial genera contributing to flagellar assembly in patients by fibrosis stage ( ≤1 vs ≥2). The x and y axis are as described for D. Asterisks indicate genera present in Fig. 3A.
Examination of the genera contributing to pro-inflammatory gene pathways was additionally carried out on an individual-by-individual basis (Supplementary Figs 12–15). This indicated that a substantial contribution from the genus Lactobacillus to flagellar assembly was only observed in a subset of individuals (Supplementary Figs. 14–15). Furthermore, other bacterial taxa appeared to make substantial contributions to these pathways in only one, or few, individuals. This indicated that, while the overall abundance of these pathways associated with the presence and severity of NAFLD, the taxa responsible for each pathway was likely to vary between individuals.
DISCUSSION
We studied the association of intestinal microbiota with NAFLD in well-characterized children with obesity with and without NAFLD. Lower α-diversity of the intestinal microbiome was associated with both the condition of having NAFLD and the severity of NAFLD. High P. copri abundance was associated with lower α-diversity and greater fibrosis. Genes for lipopolysaccharide biosynthesis were enriched with NASH and genes for flagellar assembly were enriched with moderate-to-severe fibrosis. Microbial genes were associated with disease severity and provided for prediction models for NASH and for moderate-to-severe fibrosis with good accuracy.
NAFLD is associated with dysbiosis of the intestinal microbiome
In this study, we demonstrated that pediatric NAFLD was associated with two community-level changes in the intestinal microbiome. The first of these was a decline in α-diversity, which may reflect a reduction in the number of different microbial taxa (i.e. richness), and a bias towards overrepresentation of a smaller number of these taxa. Interestingly, declines in α-diversity were not associated with fewer microbial genes detected, meaning there was no evidence of lower metabolic potential despite lower diversity. This differs from previous studies of adult obesity where loss of diversity6 was linked to a reduction in metabolic potential.34
The second community-level change associated with NAFLD was greater person-to-person variability in composition of the microbiome. While this trend was relatively minor, a reanalysis of microbiome data originating from obese vs. lean twins showed similar increases in variability within obese individuals.35 This trend is also consistent with recent observations that increased heterogeneity is associated with a variety of stressors in many microbiome-related disorders.35,36 In contrast to α-diversity, person-to-person variability correlated strongly with the composition of the intestinal metagenome, suggesting that greater heterogeneity within cases was accompanied by differences in microbial metabolic potential.
The influence of Bacteroides, Prevotella, and diversity on NAFLD
The genera Bacteroides and Prevotella are dominant members of the healthy human intestinal microbiome.4 We classified patients into discrete groups based on a bimodal distribution in the relative abundance of P. copri, which was the dominant species belonging to the genus Prevotella.
When considering the relative abundance of Bacteroides and Prevotella, previous studies have found conflicting results. Zhu et al.37 reported no difference in Bacteroides and a significant difference in Prevotella relative abundance when comparing children with healthy-weight, obesity, or NASH from Buffalo, NY; Prevotella was more abundant in children with NASH or obesity and less abundant in healthy controls.38 However, this trend was driven by greater Prevotella relative abundance between healthy-weight vs. obese/NASH individuals. In a study of 61 Italian children with obesity or NAFLD matched to 54 controls, there was no association between liver disease and Bacteroides or the Prevotella genus.18 However, an examination of publically available data suggests that few children with NAFLD in Italy would have been classified as having high Prevotella relative abundance. In adults in France, Boursier et al. reported significantly higher Bacteroides relative abundance, and lower Prevotella relative abundance in patients with NASH.19 These differences may be attributed in part to both diet and geography, with high Prevotella abundance linked to fiber-rich diets and high Bacteroides abundance linked to a high protein, Western diet.39–41 Perhaps varying conclusions in studies from different geographic regions are due to host factors in that race/ethnicity, genetics, and diet may play a larger role in the relationship between disease and microbiome than is currently known.
A common theme is that lower microbial α-diversity is associated with NAFLD.18,37,42 As microbial abundance estimates are relative,43 changes in α-diversity are closely linked to the relative abundance of all taxa. Accordingly, we show that the relative abundance of dominant OTUs from both Bacteroides and Prevotella correlated inversely with α-diversity. Given this observation, we hypothesize that previously reported increases in the relative abundance of either taxon may be explained by a loss of diversity accompanying NAFLD-associated dysbiosis. The specific dominant taxon showing increased relative abundance because of such dysbiosis may thus depend on whether Bacteroides or Prevotella dominate the background of the individual in question. Under this hypothesis, it also follows that it may be the loss of diversity, not the increased relative abundance of dominant taxa that is affecting NAFLD.
Alternatively, the setting of decreased diversity may increase the potential for the dominant organisms themselves to impart negative consequences. In our study we report evidence for an association between the genus Prevotella and aspects of the NAFLD phenotype. In particular we observed that individuals classified as high P. copri were more frequently cases with moderate-to-severe fibrosis. We also demonstrated that it was P. copri relative abundance, not α-diversity, which best predicted liver fibrosis. These findings along with the known biology of P. copri suggest a potential link between high levels of P. copri and fibrosis progression. Previous studies have reported that increased P. copri relative abundance may exacerbate arthritis,44 possibly via TH17 cell-mediated inflammation.45 Moreover, in contrast to other bacteria present in the intestinal microbiome (e.g. Bacteroides), P. copri produces superoxide reductase and phosphoadenosine phosphosulphate reductase. Together, these enzymes grant P. copri a higher tolerance to host-derived reactive oxygen species through the conversion of highly toxic superoxide to less toxic hydrogen peroxide, and the production of thioredoxin, thus providing P. copri a competitive advantage by allowing it not only to thrive in a proinflammatory environment, but also perhaps increase intestinal inflammation for its own advantage.46 Such pro-inflammatory effects may therefore exacerbate liver damage during NAFLD progression and are worthy of further investigation.
Therefore, we hypothesize both that a loss in diversity and an increase in the abundance of P. copri may be causal factors influencing NAFLD. While these hypotheses are inevitably confounded, they need not be mutually exclusive. Indeed they may each independently be exerting influence on different aspects of the NAFLD disease spectrum.
Pro-inflammatory pathways associated with disease severity
In addition to taxonomic differences, we identified two pro-inflammatory pathways, containing genes encoding for LPS biosynthesis and flagellar assembly, which were present at greater relative abundance in the NAFLD metagenome. LPS biosynthesis genes had greater relative abundance in the metagenome of cases vs. controls and individuals with NASH vs. NAFLD, while flagellar assembly genes had greater relative abundance in the metagenome of cases vs. controls and individuals with moderate-to-severe fibrosis vs. absent-to-mild fibrosis.
LPS is a potent activator of innate immunity via toll-like receptor TLR4 stimulation in macrophages, neutrophils, and dendritic cells.47 It is a component of the cell walls of gramnegative bacteria, including the phylum Proteobacteria as well as the genera Bacteroides and Prevotella. Critically, TLR4 activation has also been shown to stimulate the production of Th17 cells from naïve CD4 T cells,47 and a Th17 driven pro-inflammatory state is involved in NAFLD pathogenesis. Th17 subsets are elevated in patients with NAFLD and obesity and the Th17 response has been linked with progression to NASH.48 TLR4 signaling plays a critical role in NAFLD progression as well. TLR4 expression is increased in wild-type mice with steatohepatitis in response to high-fat diet,15 and TLR4-mutant mice are resistant to NAFLD.49,50
Bacterial flagellin is also a potent activator of the innate immune system, both via the toll-like receptor TLR551 and via NOD-like receptor-mediated inflammasome activation.52 TLR5 is expressed in the intestinal epithelium where it plays an important role in exacerbating the symptoms of inflammatory bowel disease. While LPS is a ubiquitous feature of gram-negative bacteria, only a subset of bacteria express flagella. Furthermore, only certain taxa are known to stimulate TLR5.51 The taxonomic origin of genes detected in this pathway is therefore of interest. However, we caution that overlap between genes encoding for flagellar assembly and type III secretion systems mean further investigation is warranted in order to validate the true function of genes identified as contributing to this pathway.
Multiple bacteria were found to contribute to these pro-inflammatory pathways. Most notably, the genus Lactobacillus made a significantly greater relative contribution to flagellar assembly in cases and in individuals with moderate-to-severe fibrosis. This genus also showed the largest increase in relative abundance of any taxon identified as significant in comparisons of both NAFLD vs. NASH and absent-to-mild vs. moderate-to-severe fibrosis. Motility in Lactobacillus appears restricted to a small number of species.53 While some of these species possess adaptations that limit their pro-inflammatory effects,54 broad conservation of the TLR5 interaction motif suggests that many are capable of triggering a TLR5-mediated immune response.53
Implication of Lactobacillus via both 16S and shotgun approaches makes this genus of particular interest. However, another prominent feature of our analysis was that a variety of taxa made substantial contributions to pro-inflammatory pathways in a small number of individuals. These taxa may therefore also be relevant to the etiology of NAFLD; however, their taxonomic diversity, sparse occurrence, and low global abundance of may have prevented their detection in this, and other, 16S based studies. Many of these taxa belong to the phylum Proteobacteria, which we observed at greater abundance in NAFLD cases. Similarly, Zhu et al. reported greater abundance of Proteobacteria in patients with NASH.37 This phylum may be particularly permissive for opportunistic pathogens that can influence NAFLD.
There have been recent advances using bacterial taxonomy to predict NAFLD disease states55 however evidence of functional redundancy between different microbial taxa linked to pro-inflammatory pathways suggests that genes, rather than taxa are likely to prove better biomarkers for the identification and characterization of NAFLD. Accordingly, we found that genus abundance measurements were inadequate for predicting disease using CART. In contrast, accounting for the abundance of genes within the LPS biosynthesis and flagellar assembly pathways resulted in a significant improvement over predictive models based on ALT alone.
The current study was notable for the large sample size of well-characterized children with NAFLD including liver histology evaluated in a standardized fashion by expert liver pathologists blinded to the clinical and laboratory details. In addition, the study design maximized the ability of the controls to serve as a proper comparison group. These children were recruited from the same local communities as the cases and absence of liver disease was proven with history, physical, labs, and MRS-PDFF. However, factors that can influence the microbiome such as diet were not evaluated. Notably, our study population was predominantly of Hispanic ethnicity and this was controlled for in our analyses. We encourage further elucidation of the relationship between ethnicity and the microbiome in the context of NAFLD and this will require studies that consider both cultural and biological factors. Finally, the cross-sectional design allowed for association but not causation. Mechanistic links could be evaluated through longitudinal studies in patients, and in targeted translational studies in gnotobiotic and/or genetically modified animal models.
Conclusions
We examined the intestinal microbiome in a well-characterized pediatric cohort with and without NAFLD. We identified significant differences in the NAFLD intestinal microbial community characterized by lower α-diversity and greater variation in β-diversity. Collectively, we refer to these changes as NAFLD-associated dysbiosis. We further conclude that NAFLD-associated dysbiosis is characterized by changes in the functional capacity of the intestinal microbiome, including greater abundance of genes encoding for the pro-inflammatory bacterial products LPS and flagellin. CART models using microbial gene abundance as predictors support the potential for the microbiome to be a clinically useful biomarker. Multiple bacteria were found to contribute to these pro-inflammatory pathways, with distinct groups of contributing taxa in each individual. These findings may explain the conflicting findings of prior studies, while suggesting essential biological roles for the microbiome in NAFLD pathophysiology.
Supplementary Material
What you need to know:
BACKGROUND AND CONTEXT:
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in children in the United States. Alterations to the intestinal microbiome might affect development or severity of NAFLD.
NEW FINDINGS:
We found fecal microbiomes of children with NAFLD to be disrupted compared to children without NAFLD. Increased levels of bacterial genes that regulate synthesis of lipopolysaccharide assembly of flagella were associated with disease severity and identified children with steatohepatitis or moderate to severe fibrosis.
LIMITATIONS:
This was a cross-sectional study of associations; the findings should be replicated in other cohorts. The mechanisms proposed should be assessed in animal models.
IMPACT:
Characterization of fecal microbiomes and genes expressed by intestinal microbes might provide insights into the pathogenesis of NAFLD in children and lead to discovery of therapeutic targets and diagnosis markers.
Acknowledgments
Grant support
The project described was partially supported by the National Institutes of Health, NIDDK Grant No. R01 DK088831(NHS, GW), Grant UL1TR000100 of CTSA funding and Grant UL1TR001442 of CTSA funding. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations
- NAFLD
Nonalcoholic Fatty Liver Disease
- NASH
Nonalcoholic Steatohepatitis
- NASH CRN
NASH Clinical Research Network
- BMI
Body Mass Index
- MRI
Magnetic Resonance Imaging
- MRS
Magnetic Resonance Spectroscopy
- HDL
High Density Lipoprotein
- ALT
Alanine Aminotransferase
- AST
Aspartate Aminotransferase
- GGT
Gamma Glutamyl Transpeptidase
- STEAM
Stimulated Echo Acquisition Mode
- PDFF
Proton Density Fat Fraction
- AMARES
Advanced Method for Accurate, Robust and Efficient Spectral
- PCR
Polymerase Chain Reaction
- ANOVA
Analysis of Variance
- ANOSIM
Analysis of Similarity
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KO
KEGG Orthologs
- OTU
Operational Taxonomic Unit
- FDR
False Discovery Rate
- CART
Classification and Regression Tree
- PCoA
Principal Coordinate Analysis
- GSEA
Gene Set Enrichment Analysis
- LPS
Lipopolysaccharide
- TLR
Toll-Like Receptor
- mWGS
Whole Metagenome Shotgun
- UniProt
Universal Protein Resource
- SPM
Salford Predictive Modeler
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Additional authors are listed alphabetically
There is no conflict of interest to disclose
REFERENCES
- 1.Schwimmer JB, Deutsch R, Kahen T, et al. Prevalence of fatty liver in children and adolescents. Pediatrics 2006; 118:1388–1393. [DOI] [PubMed] [Google Scholar]
- 2.Schwimmer JB, Behling C, Newbury R, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005;42:641–649. [DOI] [PubMed] [Google Scholar]
- 3.Turnbaugh PJ, Ley RE, Hamady M, et al. The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature 2007;449:804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The Human Microbiome Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012;486:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Round JL, Mazmanian SK. The gut microbiome shapes intestinal immune responses during health and disease. Nat Rev Immunol 2009;9:313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ley RE. Obesity and the human microbiome. Curr Opin Gastroenterol 2010;26:5–11. [DOI] [PubMed] [Google Scholar]
- 7.Caesar R, Tremaroli V, Kovatcheva-Datchary P, et al. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab 2015;22:658–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chassaing B, Gewirtz AT. Gut microbiota, low-grade inflammation, and metabolic syndrome. Toxicol Pathol 2014;42:49–53. [DOI] [PubMed] [Google Scholar]
- 9.Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027–1031. [DOI] [PubMed] [Google Scholar]
- 10.Sonnenburg JL, Bäckhed F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016;535:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chassaing B, Etienne-Mesmin L, Gewirtz AT. Microbiota-liver axis in hepatic disease. Hepatology 2014;59:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dumas M-E, Barton RH, Toye A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci 2006;103:12511–12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henao-Mejia J, Elinav E, Strowig T, et al. Inflammasomes: Far beyond inflammation. Nat Immunol 2012;13:321–324. [DOI] [PubMed] [Google Scholar]
- 14.Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic diet–induced obesity and diabetes in mice. Diabetes 2008;57:1470–81. [DOI] [PubMed] [Google Scholar]
- 15.Rivera C, Adegboyega P, Rooijen N van, et al. Toll-like recepter-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepa 2007;47:571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seki E, Minicis S De, Österreicher CH, et al. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med 2007;13:1324–1332. [DOI] [PubMed] [Google Scholar]
- 17.Abu-Shanab A, Quigley E. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2010;7:697–701. [DOI] [PubMed] [Google Scholar]
- 18.Chierico F Del, Nobili V, Vernocchi P, et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017;65:451–464. [DOI] [PubMed] [Google Scholar]
- 19.Boursier J, Mueller O, Barret M, et al. The severity of NAFLD is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016;63:764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newton KP, Hou J, Crimmins NA, et al. Prevalence of Prediabetes and Type 2 Diabetes in Children With Nonalcoholic Fatty Liver Disease. 2017;92123:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleiner DE, Brunt EM, Natta M Van, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313–1321. [DOI] [PubMed] [Google Scholar]
- 22.Hamilton G, Yokoo T, Bydder M, et al. ln vivo characterization of the liver fat 1H MR spectrum. NMR Biomed 2011. ;24:784–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bydder M, Hamilton G, Yokoo T, et al. Optimal phased-array combination for spectroscopy. Magn Reson Imaging 2006;26:847–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgar RC. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat Methods 2013;10:996–998. [DOI] [PubMed] [Google Scholar]
- 25.Abubucker S, Segata N, Goll J, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 2012;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S, Furumichi M, et al. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 2010;38:355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ridout M, Hinde J, Demétrio CGB, et al. A Score Test for Testing a Zero-Inflated Poisson Regression Model against Zero-Inflated Negative Binomial Alternatives. Biometrics 2001;57:219–223. [DOI] [PubMed] [Google Scholar]
- 28.Clarke KR. Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 1993;18:117–143. [Google Scholar]
- 29.Väremo L, Nielsen J, Nookaew I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res 2013;41:4378–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dufrene M, Legendre P. Species assemblages and indicator species: The need for a flexible asymmetrical approach. Ecol Monogr 1997;67:345–66. [Google Scholar]
- 31.Roberts D labdsv: Ordination and Multivariate Analysis for Ecology. 2016. [Google Scholar]
- 32.McMurdie PJ, Holmes S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One 2013;8(4):e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahmud S, Lou W, Johnston N. A probit- log- skew-normal mixture model for repeated measures data with excess zeros, with application to a cohort study of paediatric respiratory symptoms. BMC Med Res Methodol 2010;10:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chatelier E Le, Nielsen T, Qin J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013:500(7464):541–6. [DOI] [PubMed] [Google Scholar]
- 35.Holmes I, Harris K, Quince C. Dirichlet multinomial mixtures: Generative models for microbial metagenomics. PLoS One 2012;7:e30126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zaneveld JR, McMinds R, Thurber RV. Stress and stability: Applying the Anna Karenina principle to animal microbiomes. Nat Microbiol 2017: (2) :17121. [DOI] [PubMed] [Google Scholar]
- 37.Zhu L, Baker SS, Gill C, et al. Characterization of Gut Microbiomes in Nonalcoholic Steatohepatitis (NASH) Patients: A Connection Between Endogenous Alcohol and NASH. Hepatology 2013:601–609. [DOI] [PubMed] [Google Scholar]
- 38.Zhu L, Baker R, Zhu R, et al. Sequencing the gut metagenome as a noninvasive diagnosis for advanced nonalcoholic steatohepatitis. Hepatology 2017;66:2080–2083. [DOI] [PubMed] [Google Scholar]
- 39.De Filippo C, Cavalieri D, Di M, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci 2010;107:14691–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature 2012. 2012;486:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014;505:559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen F, Zheng RD, Sun XQ, et al. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat Dis Int 2017;16:375–381. [DOI] [PubMed] [Google Scholar]
- 43.Gloor GB, Macklaim JM, Pawlowsky-Glahn V, et al. Microbiome Datasets Are Compositional: And This Is Not Optional. Front Microbiol 2017;8:2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013:2:e01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maeda Y, Kurokawa T, Umemoto E, et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol. 2016;68:2646–2661. [DOI] [PubMed] [Google Scholar]
- 46.Hofer U Microbiome: Pro-inflammatory Prevotella? Nat Rev Microbiol 2014;12:5. [DOI] [PubMed] [Google Scholar]
- 47.Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol 2014;5:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chackelevicius CM, Gambaro SE, Tiribelli C, et al. Th17 involvement in nonalcoholic fatty liver disease progression to non-Alcoholic steatohepatitis. World J Gastroenterol 2016;22:9096–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Csak T, Velayudham A, Hritz I, et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. AJP Gastrointest Liver Physiol 2011. ;300:G433–G441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poggi M, Bastelica D, Gual P, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 2007;50:1267–1276. [DOI] [PubMed] [Google Scholar]
- 51.Vijay-Kumar M, Gewirtz AT. Guardians of the Gut: Newly Appreciated Role of Epithelial Toll-Like Receptors in Protecting the Intestine. Gastroenterology 2008;135:347–351. [DOI] [PubMed] [Google Scholar]
- 52.Miao EA, Andersen-Nissen E, Warren SE, et al. TLR5 and Ipaf: Dual sensors of bacterial flagellin in the innate immune system. Semin Immunopathol 2007;29:275–288. [DOI] [PubMed] [Google Scholar]
- 53.Cousin FJ, Lynch SM, Harris HMB, et al. Detection and genomic characterization of motility in Lactobacillus curvatus: Confirmation of motility in a species outside the Lactobacillus salivarius clade. Appl Environ Microbiol 2015: 81(4):1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kajikawa A, Midorikawa E, Masuda K, et al. Characterization of flagellins isolated from a highly motile strain of Lactobacillus agilis. BMC Microbiol 2016; 16:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caussy C, Tripathi A, Humphrey G, et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat Commun 2019;10:1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.