

Abstract
Animal behaviours are affected not only by inherited genes but also by environmental experiences. For example, in both rats and humans, stressful early-life events such as being reared by an inattentive mother can leave a lasting trace and affect later stress response in adult life. This is owing to a chemical trace left on the chromatin attributed to so-called epigenetic mechanisms. Such an epigenetic trace often has consequences, sometimes long-lasting, on the functioning of our genes, thereby allowing individuals to rapidly adapt to a new environment. One gene under such epigenetic control is FKBP5, the gene that encodes the protein FKPB51, a crucial regulator of the stress axis and a significant driver of chronic pain states. In this article, we will discuss the possibility that exposure to stress could drive the susceptibly to chronic pain via epigenetic modifications of genes within the stress axis such as FKBP5. The possibility that such modifications, and therefore, the susceptibility to chronic pain, could be transmitted across generations in mammals and whether such mechanisms may be evolutionarily conserved across phyla will also be debated.
This article is part of the Theo Murphy meeting issue ‘Evolution of mechanisms and behaviour important for pain’.
Keywords: epigenetics, stress, FKBP5, chronic pain, vulnerability
1. Stress experiences can leave long-lasting traces onto our chromatin via so-called epigenetic mechanisms
The chromatin is the association of DNA and proteins such as histones that can be found in the nucleus of our cells. The compaction of the chromatin can be modulated in a gene- and a time-dependent manner by epigenetic mechanisms, resulting in the tight regulation of gene expression. This happens through the addition and removal of chemical marks onto the chromatin, such as post-translation modification of histones and methylation of the DNA. While epigenetic marks were once believed to be subject to little alterations in mature systems, it is now well accepted that epigenetic changes are highly dynamic and allow rapid adaptation to the environment within one's lifetime [1–5]. To make a clear distinction between the epigenetic changes occurring during organismal development and that occurring in mature systems, in particular in non-dividing adult neurons, the term neuroepigenetic has been proposed [4]. We now have strong evidence that neuroepigenetic changes can lead to long-lasting modification in neural function implicated in a number of cognitive behaviours [5].
Life experiences can indeed alter the epigenome, the summation of all epigenetic marks on the chromatin, and have a lasting influence upon the way some genes are expressed. Two of the most studied epigenetic modifications are histone acetylation [6–8] and DNA methylation at CpG sites (regions of DNA where a cytosine nucleotide is followed by a guanine nucleotide) [9–11]. Both histone acetylation and decrease in DNA methylation in the promoter of a gene can lead to the localized relaxation of the chromatin and therefore are usually associated with the upregulation of gene expression [12]. Alongside DNA methylation and histone modifications, non-coding RNAs are also crucial modulators of gene expression. Unlike messenger RNAs (mRNAs), non-coding RNAs or regulatory RNAs, that were first thought to possess no coding capacity, regulate gene expression by targeting mRNAs for degradation. They have also been shown to act as guides for the epigenetic machinery to target specific DNA sequence [13–15]. One should note that coding properties have recently been observed from the so-called non-coding RNAs, suggesting that their involvement in cell function regulation is likely to be broader than currently understood [16,17].
So far, the epigenetic changes induced by life experiences have been mostly studied in the context of stress exposure and genes associated with the hypothalamic–pituitary–adrenal (HPA) axis. The first pre-clinical study to elegantly demonstrate the importance of epigenetic mechanisms in the adaptation to our environment was that of Meaney and co-workers [18]. They showed that highly licking and grooming rat mothers were raising pups that became themselves high licking and grooming mothers. This behaviour was associated with an increase in the expression of the glucocorticoid receptor (GR) gene in the hippocampus, and this increased expression was secondary to the low methylation landscape and increased acetylation of histones at the promoter of the NR3C1 gene that encodes the GR protein. Both rodents and humans with high level of GR are more resilient to stress, and studies in human post-mortem brain tissue taken from suicide victims have demonstrated that individuals with a history of childhood trauma are likely to present with increased methylation levels in the NR3C1 gene and therefore reduced GR expression [19–21]. While most work so far has focused on the GR in the HPA axis, more recent studies have also looked at the stress regulator FK506-binding protein 51 (FKBP51).
2. FKBP51: a stress regulator under strong epigenetic regulation
FKBP51, encoded by the gene FKBP5, interacts with a number of molecular partners to impact upon various cellular processes. Most notably, FKBP51 binds to the heat-shock protein 90 (Hsp90) and other co-chaperones of the steroid receptor complex to regulate the stress response [22]. By interacting with the steroid receptor complex, FKBP51 reduces the affinity of the GR to stress hormones, which is particularly important for stress regulation (figure 1). Glucocorticoids are released in response to stress, and activation of the GR usually feeds back to reduce this release, leading to the termination of the stress response [23]. Consequently, changes in levels of FKBP51 perturb the stress response system [22]. The expression of FKBP51 itself is induced by GR activation by stress hormones, producing a direct feedback that regulates GR activity. The result of this feedback is evident in humans expressing FKBP5 variants associated with the heightened induction of FKBP5 mRNA upon stress exposure (known as a risk allele). These variants have been repeatedly associated with anxiety-related disorders, including major depression and post-traumatic stress disorder (PTSD) [22,24]. This is consistent with the finding that inhibition or deletion of the protein reduces anxiety-related behaviour in mice [25,26].
Figure 1.
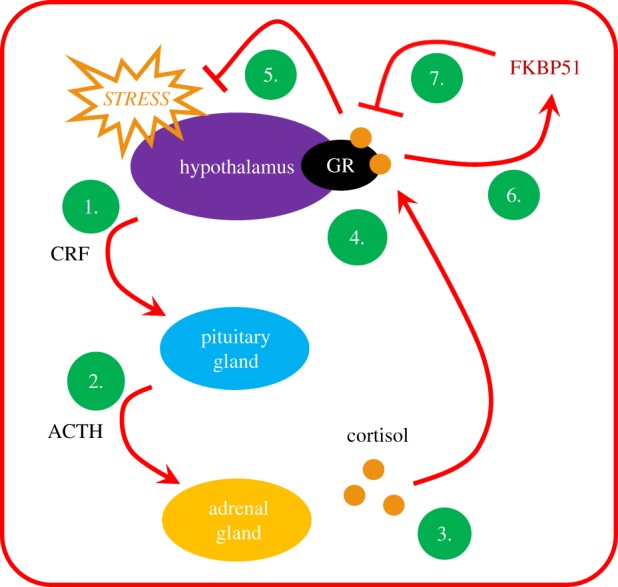
FKBP51 regulates the stress response by interacting with the steroid receptor complex. Stress exposure leads to the release of corticotropin-releasing factor (CRF) from the hypothalamus (1). CRF is transported to the anterior pituitary gland where it stimulates the production of adrenocorticotropic hormone (ACTH) (2). In turns, ACTH stimulates the adrenal glands to produce and release stress hormones (cortisol) into the blood stream (3). When cortisol levels reach a certain level, the binding of cortisol to the glucocorticoid receptor (GR) (4) usually leads to the termination of the stress response (5). FKBP51 expression is induced by GR activation by stress hormones (6) and FKBP51 interacts with the steroid receptor complex and reduces the affinity of GR to stress hormones (7), thereby prolonging the stress response.
A reduction in FKBP5 DNA methylation in intronic regions, which is likely to be associated with an increase in FKBP51 expression, has often been reported following exposure to stress and glucocorticoid stimulation, in both humans and rodents [22,27–31]. Importantly, the reduction in FKBP5 DNA methylation correlates with the trauma intensity in humans [28] and with the degree of exposure to glucocorticoids in rodents [29]. Interestingly, these changes are measured in peripheral blood samples in humans, and in rodents, changes in the brain correlate with those in the blood [31], suggesting that FKBP5 DNA methylation levels in the blood could be used as a biomarker of stress exposure. A seminal study from Binder and co-workers demonstrated that long-lasting, trauma-induced, decrease in intronic regions in FKBP5 DNA methylation measured in human peripheral blood could arise in childhood in individuals with the risk allele. Importantly, this decrease in DNA methylation increased the risk of developing stress-related psychiatric disorders in adulthood [27,32]. More recently, Binder and co-workers [33] have also identified a reduction in DNA methylation at selected enhancer-related FKBP5 sites synergistically modulated by ageing and stress. Individual with stress-related phenotypes show further decrease in ageing-induced DNA demethylation at these specific CG bases and higher FKBP5 mRNA expression in blood samples.
3. FKBP51: a crucial driver of chronic pain
We have recently shown that genetic deletion and pharmacological blockade of FKBP51 alleviated chronic pain states in mice. With these approaches, we were indeed able to reduce the hypersensitivity seen in a number of animal models of persistent pain across sexes: complete Freund's adjuvant-induced joint inflammation [34,35], monoiodoacetate-induced knee inflammation (M. Maiarù, S. M. Géranton 2018, unpublished data), peripheral nerve injury [34,35] and paclitaxel-induced mechanical hypersensitivity [35]. Crucially, models of acute pain, such as IL6- and PGE2-induced inflammation of the hind paw, were insensitive to FKBP51 blockade. Our study also demonstrated that spinal deletion and pharmacological blockade of FKBP51 alleviated established persistent pain as effectively as global deletion, suggesting that FKBP51 could regulate pain independently from its effect on mood, which is likely to be mediated in the brain. While the molecular mechanisms of the regulation of persistent pain by FKBP51 remain to be fully elucidated, our early findings would suggest that this occurs in a glucocorticoid-signalling-dependent mechanism [34,35]. Importantly, a significant body of work from McLean and co-workers [36–38] suggest that genetic variants of FKBP5 alter pain sensitivity after trauma such as car crash and sexual assault, supporting the idea that FKBP51 drives persistent pain states in humans.
In our rodent pre-clinical studies, FKBP51 was upregulated in the dorsal horn after peripheral injury and this upregulation was accompanied at least by two epigenetic changes: the phosphorylation of the methyl CpG binding protein 2 (MeCP2) [39] and the decrease in DNA methylation in the promoter sequence of the FKBP5 gene [34] (figure 2). In these studies, the dorsal horn expression of phosphorylated-MeCP2 and FKBP51 was observed nearly exclusively in neurons [34,39]. However, the changes in DNA methylation were analysed and detected from a mixture of cells, and therefore, while unlikely, we cannot exclude the possibility of a change in FKBP5 DNA methylation in cells other than neurons. All together, these observations support the idea that epigenetic mechanisms are crucial to the development of persistent pain states by promoting the relaxation of the chromatin at the gene FKBP5, leading to the upregulation of FKBP51. A number of rodent studies have since demonstrated that indeed, following injury, epigenetic alterations drive gene expression changes of other contributors to persistent pain states and are therefore crucial to the maintenance of persistent pain. This has been reviewed elsewhere [12,40–43] and will not be discussed further in this manuscript, which focuses on the contribution of epigenetic mechanisms to the susceptibility to chronic pain.
Figure 2.

After peripheral noxious stimulation, FKBP51 is upregulated in the dorsal horn following chromatin relaxation. FKBP5 is rapidly epigenetically regulated in the dorsal horn after peripheral noxious stimulation. After complete Freund's adjuvant injection in the ankle joint, we observed an increase in phosphorylation of the transcriptional regulator MeCP2 (1), a decrease in DNA methylation in the promoter sequence of the gene FKBP5 (2) and an increase in FKBP51 protein (3) [34,39]. MeCP2, methyl CpG binding protein 2; HDACs, histone deacetylases; P, phosphorylation. (Online version in colour.)
Early-life trauma in humans can lead to a decrease in FKBP5 DNA methylation, an epigenetic change that primes FKBP5 for hyper-responsiveness and increases the susceptibility to PTSD in adulthood [27]. Could similar processes underlie the vulnerability to chronic pain?
4. Could exposure to environmental stress drive the susceptibly to chronic pain via epigenetic modifications of FKBP5?
To investigate the susceptibility to chronic pain, a number of hyperalgesic priming models have been developed in rodents [44,45]. In these models, animals who have suffered a minor injury remain in a long-lasting latent hyper-responsiveness to an inflammatory or surgical insult. In other words, these models produce a state of sensitization closely resembling clinical situations with an increased risk of developing chronic pain. Rodents that have suffered a minor injury become hyper-responsive to further mild insult that would normally not evoke persistent pain.
It is now known that pain in early life can enhance the duration of the pain response to subsequent injury in animal models, depending on both the nature and timing of the neonatal trauma [46–49]. Various mechanisms have previously been suggested [50], some supporting a role for spinal microglia for the priming of the adult pain responses [44,51]. Interestingly, glial cells have been shown to carry long-term epigenetic changes following peripheral injury [52] and neonatal handling [53] and therefore seem interesting potential drivers of a primed state of hypersensitivity.
While FKBP51 expression in the central nervous system has been mainly reported in neurons in rodents [35], the possibility that FKBP5 could contribute to the susceptibility to chronic pain in adulthood following early-life trauma deserves serious consideration. Indeed, not only early-life injury but also early-life stress exposure, such as sexual assault, could lead to a long-lasting decrease in FKBP5 DNA methylation. Such a reduction in DNA methylation would prime FKBP5 for hyper-responsiveness to injury in later life, enhancing the duration of the pain response and potentially increasing the likelihood of developing chronic pain. The extent, duration and location in the central nervous system of the trauma-induced reduction in DNA methylation remain central to the likelihood that such mechanisms could contribute to the susceptibility to chronic pain. In this context, it is important to note that because pain is a stressor in itself, it is likely that, following injury, changes in FKBP51 expression and FKBP5 DNA methylation occur not only at spinal cord level, as we have reported in mice and rats, but also in brain areas involved with stress, such as the paraventricular nucleus of the hypothalamus [54,55]. Reciprocally, whether traumatic stress experience can lead to changes at the spinal cord level remains a point of discussion.
Could similar mechanisms occur in adult life? A state of latent hypersensitivity can also be induced in adulthood using priming models in rodents [45,50,56] and this latent state of hypersensitivity can last at least for five weeks [50]. Various mechanisms have been suggested including the involvement of dopaminergic descending controls [50], GABAergic signalling at dorsal horn level [57] as well as peripheral mechanisms of nociceptor plasticity [58–60]. However, in the seminal human studies from Binder and co-workers [27], it was reported that trauma in adult life could not lead to long-lasting changes in FKBP5 DNA methylation, as seen in childhood. Mechanisms of epigenetic priming could differ across ages because epigenetic modifications might be differently engaged in the young and the mature nervous system by trauma. Indeed, the epigenome, the complete set of epigenetic modifications on the genetic material of an individual, tends to change as we age; changes in both histone modifications and DNA methylation, specifically gene-specific and global hypermethylation and hypomethylation, have been reported with ageing in various tissue, as well as in the central and peripheral nervous system [61–65]. Some epigenetic actors are also expressed differently in young animals versus adults: e.g. the expression of DNA methyltransferase 1 (DNMT1) and 3a decreases considerably between newborn and middle-age (23–50-year-old) humans [66,67]. Nonetheless, using a gene-specific approach, we did find in our pre-clinical studies that peripheral injury in adult rats induced a reduction in FKBP5 DNA methylation, at least in the superficial dorsal horn [34]. This reduction in DNA methylation lasted at least 7 days. It is, therefore, crucial to characterize precisely the circumstances under which (e.g. trauma intensity, life stages) changes in DNA methylation can occur and can be persistent. This indeed would be necessary for the long-lasting priming of the nociceptive system for hyper-responsiveness to subsequent injury.
5. Epigenetic regulation of the stress axis and the vulnerability to chronic pain: other candidate genes
While this article focused on the gene FKBP5 purposely, other genes from the stress axis are likely to be involved in the susceptibility to chronic pain. For example, the GR itself is known to be involved in spinal mechanisms crucial to the development of persistent pain states through modulation of NMDA receptor expression [68,69]. Because the GR is also under strong epigenetic regulation upon stress exposure [18,20,21], one could assume that stress-induced epigenetic regulation of this receptor could modulate the susceptibility to chronic pain. An important role for the serum and glucocorticoid-regulated kinase 1 (SGK1) in the development of persistent pain states has also been reported at spinal cord level [39,70,71]. The gene that encodes this protein is also known to be sensitive to epigenetic regulation and its expression is modulated by stress in both humans and rodents [72–74]. All together, these observations strongly support the hypothesis that stress exposure can leave long-lasting epigenetic marks onto genes crucial to the development and maintenance of persistent pain states. This suggests that such genes could be primed by stress for injury-induced hyper-responsiveness in later life and therefore could be key to the susceptibility to chronic pain.
6. Epigenetic transgenerational inheritance
The possibility of inheritance of acquired characteristics was first proposed by the French naturalist Jean-Baptiste Lamarck at the end of the eighteenth century. Lamarck suggested that the environment could make lasting and potentially heritable alterations in gene function. This idea has remained controversial and the possibility that acquired traits could be passed on through generation remains an intense area of investigation [75,76].
While meiotic epigenetic inheritance is strongly debated, experience-dependent transgenerational transmission has gathered significant support. Meaney and co-workers’ [18] work was the first to clearly demonstrate in pre-clinical models that a mothering style of grooming and nursing could be passed on to the next generation through an epigenetic mechanism. The female rats that were resilient to stress brought up offspring that were also resilient to stress in adulthood and both mothers and progenies had a lower level of DNA methylation and higher expression of the GR gene in the hippocampus than stress-vulnerable controls [18]. More recent human data also support the idea that the effects of stress on the epigenome could be inherited through social interactions. In particular, the intergenerational effects of trauma have received a lot of attention and have been widely observed clinically. Parental PTSD, for example, in holocaust survivors, has been linked with an increased risk for psychopathology in offspring which has been associated with epigenetic programming of glucocorticoid functions [77–79]. DNA methylation patterns of the gene NR3C1, in particular, were shown to be related to parental PTSD, with maternal and paternal PTSD having different effects [78]. Similar findings have been extended to less extreme forms of stress [80]. For example, there was a positive association between the methylation status of the NR3C1 promoter in offspring of mothers stressed during pregnancy (reviewed in [80]).
It is interesting to note that changes in DNA methylation in the gene FKBP5 were reported in both holocaust survivors and their offspring, but that parents and offspring exhibited inverse methylation changes [28]. These results suggested that, if the changes observed in the parents reflected exposure to extreme stress, the opposite change in the offspring could be a sign of resilience. This would suggest that epigenetic transgenerational transmission could sometimes be not only the transfer of the negative consequences of stress exposure but also a mechanism to enhance environmental adaptation [80]. Whether similar mechanisms could apply to the transmission of an increased vulnerability or resilience to chronic pain remains to be demonstrated.
The transgenerational transfer of potentially acquired traits through gametes remains much debated. While considerable efforts have been made in identifying the contribution of epigenetic modifications to the heritability of complex and stress-related diseases, this has been extremely difficult to elucidate owing to the lack of a clear mechanism. This is because, during mammalian development, the embryo goes through epigenetic reprogramming, when epigenetic marks are erased and remodelled, making it unlikely for a mark to be transferred from parents to offspring. Nonetheless, a number of genes, including imprinted genes, bypass epigenetic reprogramming and can therefore carry epigenetic marks across generations [81]. Occurrences of such transmission have already been reported including transmission across generations of exposure to pesticides and experiences, such as metabolic deprivation, increased fat intake and fear [82]. However, evidence for this type of heritability in humans remains very limited [83].
Nevertheless, a study in mice was able to demonstrate that odour fear conditioning could be transmitted to offspring via changes in methylation in the germ line that were shown to affect the expression of the relevant olfactory receptor gene [84,85]. Importantly, up to two generations of offspring showed increased fear of the smell that had been used to condition the parents and strong evidence was provided to demonstrate that the transmission occurred through the germ line and not behaviourally. In another study, traumatic stress in early life could influence the expression of small non-coding RNAs which are also known to mediate the effect of the environment on our genome [86,87]. The changes in behaviour and small non-coding RNAs could be seen in the serum and hippocampus of offspring for at least two generations and injection of sperm RNAs from traumatized males into fertilized oocytes reproduced the same alterations in the offspring, demonstrating a non-social transmission of the traits [86]. In both studies reported here, the assimilation of environmentally induced phenotypes could act as a driver of evolutionary change and could even be seen as contributing to the rapid transformation of a learned behaviour into an instinct [88]. This would undeniably be extremely beneficial to offspring to be able to react to a threat from birth. However, one could also argue that, while adapting to one's environment before birth should help with survival, these changes could have serious consequences if the environment at birth does not match the environment one was prepared for.
7. Epigenetic inheritance in other phyla
While our knowledge of epigenetic mechanisms in animals studied in medical research is rapidly expanding, it is not always the case for other species. Current understanding suggests that, while the patterns of DNA methylation are well preserved across species of vertebrates, there are large differences in terms of the timing and nature of reprogramming and genomic imprinting. For example, fishes and frogs do not undergo global DNA methylation remodelling during embryogenesis [89], while in zebrafish the maternal methylome is reconfigured to match the paternal methylome pattern [90,91]. In the case of invertebrates, some genomes have no DNA methylation, while others could be as methylated as vertebrates. Importantly, DNA methylation may have different functions in invertebrates such as alternative splicing [89]. More relevant to the study of pain mechanisms are observations made in one of the most studied invertebrate, Aplysia, used by Kandel and co-workers [92,93] in pioneering studies in the field of learning and memory. While the DNA methyltransferase 1 (DNMT1) was only recently discovered in Aplysia, Kandel and co-workers [94] demonstrated that DNA methylation was necessary for 5HT-dependent long-term facilitation. These findings were supported by more recent studies indicating that DNA methylation was a crucial mechanism for both the consolidation and maintenance of long-term memory in Aplysia [95]. Overall, these observations would suggest that epigenetic mechanisms are likely to contribute to the development of persistent sensitized states across phyla. However, the possibility of inheritance of some kind of vulnerable state through epigenetic mechanisms in invertebrates is still unclear and has been discussed elsewhere [89].
8. Conclusion
Epigenetic mechanisms have been highlighted as key players of the development and maintenance of persistent pain states and are expected to contribute to the susceptibility to chronic pain, at least partly through the modulation of the stress axis and in particular the gene FKBP5. However, it is likely that epigenetic regulation and genetic pre-dispositions are working together to drive vulnerability or resilience, as seen with the gene FKBP5 in the context of mood disorders. While epigenetic modifications can be transmitted across generations, the transmission seems more likely to occur via social interactions than biological inheritance. Indeed, considering the fast dynamics of epigenetic mechanisms that allow the adaptation to the environment within a lifetime, one could question the advantage of a biological transmission of epigenetic traits (for humans at least).
Acknowledgements
The author would like to thank S.P. Hunt (UCL), S. Koch (UCL) and M. Maiarù (UCL) for their helpful conversation during the preparation of this manuscript.
Data accessibility
This article has no additional data.
Competing interests
The author has no competing interests.
Funding
This work was supported by the UK Medical Research Council grant no. G1100577, a NIH Fast Track Grant, and a grant from the Pain Relief Foundation.
References
- 1.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. 2008. Decoding the epigenetic language of neuronal plasticity. Neuron 60, 961–974. ( 10.1016/j.neuron.2008.10.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renthal W, Nestler EJ. 2008. Epigenetic mechanisms in drug addiction. Trends Mol. Med. 14, 341–350. ( 10.1016/j.molmed.2008.06.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Day JJ, Sweatt JD. 2010. DNA methylation and memory formation. Nat. Neurosci. 13, 1319–1323. ( 10.1038/nn.2666) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sweatt JD. 2013. The emerging field of neuroepigenetics. Neuron 80, 624–632. ( 10.1016/j.neuron.2013.10.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Day JJ, Sweatt JD. 2011. Epigenetic mechanisms in cognition. Neuron 70, 813–829. ( 10.1016/j.neuron.2011.05.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gräff J, Tsai L-H. 2013. Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111. ( 10.1038/nrn3427) [DOI] [PubMed] [Google Scholar]
- 7.Peixoto L, Abel T. 2013. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology 38, 62–76. ( 10.1038/npp.2012.86) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. 2004. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 279, 40 545–40 559. ( 10.1074/jbc.M402229200) [DOI] [PubMed] [Google Scholar]
- 9.Lax E, Szyf M. 2018. The role of DNA methylation in drug addiction: implications for diagnostic and therapeutics. Prog. Mol. Biol. Transl. Sci. 157, 93–104. ( 10.1016/bs.pmbts.2018.01.003) [DOI] [PubMed] [Google Scholar]
- 10.Szyf M. 2011. The early life social environment and DNA methylation: DNA methylation mediating the long-term impact of social environments early in life. Epigenetics 6, 971–978. ( 10.4161/epi.6.8.16793) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szyf M. 2013. DNA methylation, behavior and early life adversity. J. Genet. Genomics 40, 331–338. ( 10.1016/j.jgg.2013.06.004) [DOI] [PubMed] [Google Scholar]
- 12.Géranton SM, Tochiki KK. 2015. Regulation of gene expression and pain states by epigenetic mechanisms. Prog. Mol. Biol. Transl. Sci. 131, 147–183. ( 10.1016/bs.pmbts.2014.11.012) [DOI] [PubMed] [Google Scholar]
- 13.Di Ruscio A, et al. 2013. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 503, 371–376. ( 10.1038/nature12598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghildiyal M, Zamore PD. 2009. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 10, 94–108. ( 10.1038/nrg2504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dykes IM, Emanueli C. 2017. Transcriptional and post-transcriptional gene regulation by long non-coding RNA. Genomics Proteomics Bioinformatics 15, 177–186. ( 10.1016/j.gpb.2016.12.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubé F, Francastel C. 2018. Coding and non-coding RNAs, the frontier has never been so blurred. Front. Genet. 9, 140 ( 10.3389/fgene.2018.00140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pang Y, Mao C, Liu S. 2018. Encoding activities of non-coding RNAs. Theranostics 8, 2496–2507. ( 10.7150/thno.24677) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weaver ICG, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. 2004. Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854. ( 10.1038/nn1276) [DOI] [PubMed] [Google Scholar]
- 19.Suderman M, McGowan PO, Sasaki A, Huang TCT, Hallett MT, Meaney MJ, Turecki G, Szyf M. 2012. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc. Natl Acad. Sci. USA. 109(Suppl. 2), 17 266–17 272. ( 10.1073/pnas.1121260109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGowan PO, Suderman M, Sasaki A, Huang TCT, Hallett M, Meaney MJ, Szyf M, Sirigu A. 2011. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS ONE 6, e14739 ( 10.1371/journal.pone.0014739) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonté B, Szyf M, Turecki G, Meaney MJ. 2009. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12, 342–348. ( 10.1038/nn.2270) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zannas AS, Wiechmann T, Gassen NC, Binder EB. 2016. Gene–stress–epigenetic regulation of FKBP5: clinical and translational implications. Neuropsychopharmacology 41, 261–274. ( 10.1038/npp.2015.235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herman JP, McKlveen JM, Solomon MB, Carvalho-Netto E, Myers B. et al. 2012. Neural regulation of the stress response: glucocorticoid feedback mechanisms. Braz. J. Med. Biol. Res. 45, 292–298. ( 10.1590/S0100-879X2012007500041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binder EB, et al. 2004. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat. Genet. 36, 1319–1325. ( 10.1038/ng1479) [DOI] [PubMed] [Google Scholar]
- 25.Hartmann J, et al. 2012. The involvement of FK506-binding protein 51 (FKBP5) in the behavioral and neuroendocrine effects of chronic social defeat stress. Neuropharmacology 62, 332–339. ( 10.1016/j.neuropharm.2011.07.041) [DOI] [PubMed] [Google Scholar]
- 26.Hartmann J, et al. 2015. Pharmacological inhibition of the psychiatric risk factor FKBP51 has anxiolytic properties. J. Neurosci. 35, 9007–9016. ( 10.1523/jneurosci.4024-14.2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klengel T, et al. 2013. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 16, 33–41. ( 10.1038/nn.3275) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yehuda R, Daskalakis NP, Bierer LM, Bader HN, Klengel T, Holsboer F, Binder EB. 2015. Holocaust exposure induced intergenerational effects on FKBP5 methylation. Biol. Psychiatry 80, 372–380. ( 10.1016/j.biopsych.2015.08.005) [DOI] [PubMed] [Google Scholar]
- 29.Lee RS, Tamashiro KLK, Yang X, Purcell RH, Huo Y, Rongione M, Potash JB, Wand GS. 2011. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology (Berl.) 218, 303–312. ( 10.1007/s00213-011-2307-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee RS, et al. 2010. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology 151, 4332–4343. ( 10.1210/en.2010-0225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ewald ER, Wand GS, Seifuddin F, Yang X, Tamashiro KL, Potash JB. et al. 2014. Alterations in DNA methylation of Fkbp5 as a determinant of blood–brain correlation of glucocorticoid exposure. Psychoneuroendocrinology 44, 112–122. ( 10.1016/j.psyneuen.2014.03.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klengel T, Binder EB. 2015. FKBP5 allele-specific epigenetic modification in gene by environment interaction. Neuropsychopharmacology 40, 244–246. ( 10.1038/npp.2014.208) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zannas AS, et al. 2019. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-κB-driven inflammation and cardiovascular risk. Proc. Natl Acad. Sci. USA 116, 11 370–11 379. ( 10.1073/pnas.1816847116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maiarù M, Tochiki KK, Cox MB, Annan LV, Bell CG, Feng X, Hausch F, Géranton SM. 2016. The stress regulator FKBP51 drives chronic pain by modulating spinal glucocorticoid signaling. Sci. Transl. Med. 8, 325ra19 ( 10.1126/scitranslmed.aab3376) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maiarù M, et al. 2018. The stress regulator FKBP51: a novel and promising druggable target for the treatment of persistent pain states across sexes. Pain 159, 1224–1234. ( 10.1097/j.pain.0000000000001204) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Auvergne L, et al. 2016. Association of epidemiologic factors and genetic variants influencing hypothalamic-pituitary-adrenocortical axis function with postconcussive symptoms after minor motor vehicle collision. Psychosom. Med. 78, 68–78. ( 10.1097/PSY.0000000000000253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bortsov AV, et al. 2013. Polymorphisms in the glucocorticoid receptor co-chaperone FKBP5 predict persistent musculoskeletal pain after traumatic stress exposure. Pain 154, 1419–1426. ( 10.1016/j.pain.2013.04.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ulirsch JC, et al. 2014. No man is an island: living in a disadvantaged neighborhood influences chronic pain development after motor vehicle collision. Pain 155, 2116–2123. ( 10.1016/j.pain.2014.07.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Géranton SM, Morenilla-Palao C, Hunt SP. 2007. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J. Neurosci. 27, 6163–6173. ( 10.1523/jneurosci.1306-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denk F, McMahon SB. 2012. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron 73, 435–444. ( 10.1016/j.neuron.2012.01.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Géranton SM, Tochiki KK. 2015. Could targeting epigenetic processes relieve chronic pain states? Curr. Opin. Support. Palliat. Care 9, 138–146. ( 10.1097/SPC.0000000000000127) [DOI] [PubMed] [Google Scholar]
- 42.Odell DW. 2018. Epigenetics of pain mediators. Curr. Opin. Anaesthesiol. 31, 402–406. ( 10.1097/ACO.0000000000000613) [DOI] [PubMed] [Google Scholar]
- 43.Niederberger E, Resch E, Parnham MJ, Geisslinger G. 2017. Drugging the pain epigenome. Nat. Rev. Neurol. 13, 434–447. ( 10.1038/nrneurol.2017.68) [DOI] [PubMed] [Google Scholar]
- 44.Beggs S, Currie G, Salter MW, Fitzgerald M, Walker SM. 2012. Priming of adult pain responses by neonatal pain experience: maintenance by central neuroimmune activity. Brain 135(Pt 2), 404–417. ( 10.1093/brain/awr288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reichling DB, Levine JD. 2009. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 32, 611–618. ( 10.1016/j.tins.2009.07.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fitzgerald M, Walker SM. 2009. Infant pain management: a developmental neurobiological approach. Nat. Clin. Pract. Neurol. 5, 35–50. ( 10.1038/ncpneuro0984) [DOI] [PubMed] [Google Scholar]
- 47.Walker SM, Tochiki KK, Fitzgerald M. 2009. Hindpaw incision in early life increases the hyperalgesic response to repeat surgical injury: critical period and dependence on initial afferent activity. Pain 147, 99–106. ( 10.1016/j.pain.2009.08.017) [DOI] [PubMed] [Google Scholar]
- 48.Lidow MS, Song ZM, Ren K. 2001. Long-term effects of short-lasting early local inflammatory insult. Neuroreport 12, 399–403. ( 10.1097/00001756-200102120-00042) [DOI] [PubMed] [Google Scholar]
- 49.Schwaller F, Fitzgerald M. 2014. The consequences of pain in early life: injury-induced plasticity in developing pain pathways. Eur. J. Neurosci. 39, 344–352. ( 10.1111/ejn.12414) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim J-YV, et al. 2015. Spinal dopaminergic projections control the transition to pathological pain plasticity via a D1/D5-mediated mechanism. J. Neurosci. 35, 6307–6317. ( 10.1523/jneurosci.3481-14.2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moriarty O, Tu Y, Sengar AS, Salter MW, Beggs S, Walker SM. 2019. Priming of adult incision response by early life injury: neonatal microglial inhibition has persistent but sexually dimorphic effects in adult rats. J. Neurosci. 39, 3081–3093. ( 10.1523/jneurosci.1786-18.2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Denk F, Crow M, Didangelos A, Lopes DM, McMahon SB. 2016. Persistent alterations in microglial enhancers in a model of chronic pain. Cell Rep. 15, 1771–1781. ( 10.1016/j.celrep.2016.04.063) [DOI] [PubMed] [Google Scholar]
- 53.Schwarz JM, Hutchinson MR, Bilbo SD. 2011. Early-life experience decreases drug-induced reinstatement of morphine CPP in adulthood via microglial-specific epigenetic programming of anti-inflammatory IL-10 expression. J. Neurosci. 31, 17 835–17 847. ( 10.1523/jneurosci.3297-11.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herman J, Flak J, Jankord R. 2008. Chronic stress plasticity in the hypothalamic paraventricular nucleus. In Progress in brain research (eds Waxman S, Stein DG, Swaab D, Fields H), pp. 353–364. Amsterdam, The Netherlands: Elsevier; See http://linkinghub.elsevier.com/retrieve/pii/S0079612308004299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibbison B, Angelini GD, Lightman SL. 2013. Dynamic output and control of the hypothalamic–pituitary–adrenal axis in critical illness and major surgery. Br. J. Anaesth. 111, 347–360. ( 10.1093/bja/aet077) [DOI] [PubMed] [Google Scholar]
- 56.Megat S, Shiers S, Moy JK, Barragan-Iglesias P, Pradhan G, Seal RP, Dussor G, Price TJ. 2018. A critical role for dopamine D5 receptors in pain chronicity in male mice. J. Neurosci. 38, 379–397. ( 10.1523/JNEUROSCI.2110-17.2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim J-YV, Megat S, Moy JK, Asiedu MN, Mejia GL, Vagner J, et al. 2016. Neuroligin 2 regulates spinal GABAergic plasticity in hyperalgesic priming, a model of the transition from acute to chronic pain. Pain 157, 1314–1324. ( 10.1097/j.pain.0000000000000513) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moy JK, Khoutorsky A, Asiedu MN, Dussor G, Price TJ. 2018. eIF4E phosphorylation influences Bdnf mRNA translation in mouse dorsal root ganglion neurons. Front. Cell Neurosci. 12, 29 ( 10.3389/fncel.2018.00029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferrari LF, Araldi D, Levine JD. 2015. Distinct terminal and cell body mechanisms in the nociceptor mediate hyperalgesic priming. J. Neurosci. 35, 6107–6116. ( 10.1523/jneurosci.5085-14.2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Araldi D, Khomula EV, Ferrari LF, Levine JD. 2018. Fentanyl induces rapid onset hyperalgesic priming: type I at peripheral and Type II at central nociceptor terminals. J. Neurosci. 38, 2226–2245. ( 10.1523/jneurosci.3476-17.2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.D'Aquila P, Rose G, Bellizzi D, Passarino G. 2013. Epigenetics and aging. Maturitas 74, 130–136. ( 10.1016/j.maturitas.2012.11.005) [DOI] [PubMed] [Google Scholar]
- 62.Tammen SA, Friso S, Choi S-W. 2013. Epigenetics: the link between nature and nurture. Mol. Aspects Med. 34, 753–764. ( 10.1016/j.mam.2012.07.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rakyan VK, et al. 2010. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 20, 434–439. ( 10.1101/gr.103101.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garriga J, Laumet G, Chen S-R, Zhang Y, Madzo J, Issa J-PJ, Pan H-L, Jelinek J. 2018. Nerve injury-induced chronic pain is associated with persistent DNA methylation reprogramming in dorsal root ganglion. J. Neurosci. 38, 6090–6101. ( 10.1523/JNEUROSCI.2616-17.2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Issa J-P. 2014. Aging and epigenetic drift: a vicious cycle. J. Clin. Invest. 124, 24–29. ( 10.1172/JCI69735) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oliveira AMM, Hemstedt TJ, Bading H. 2012. Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat. Neurosci. 15, 1111–1113. ( 10.1038/nn.3151) [DOI] [PubMed] [Google Scholar]
- 67.Zhang Z, Deng C, Lu Q, Richardson B. 2002. Age-dependent DNA methylation changes in the ITGAL (CD11a) promoter. Mech. Ageing Dev. 123, 1257–1268. ( 10.1016/S0047-6374(02)00014-3) [DOI] [PubMed] [Google Scholar]
- 68.Wang S, Lim G, Zeng Q, Sung B, Yang L, Mao J. 2005. Central glucocorticoid receptors modulate the expression and function of spinal NMDA receptors after peripheral nerve injury. J. Neurosci. 25, 488–495. ( 10.1523/jneurosci.4127-04.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang S, Lim G, Zeng Q, Sung B, Ai Y, Guo G, Yang L, Mao J. 2004. Expression of central glucocorticoid receptors after peripheral nerve injury contributes to neuropathic pain behaviors in rats. J. Neurosci. 24, 8595–8605. ( 10.1523/jneurosci.3058-04.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peng H-Y, Chen G-D, Lai C-Y, Hsieh M-C, Lin T-B. 2013. Spinal serum-inducible and glucocorticoid-inducible kinase 1 mediates neuropathic pain via kalirin and downstream PSD-95-dependent NR2B phosphorylation in rats. J. Neurosci. 33, 5227–5240. ( 10.1523/jneurosci.4452-12.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng H-Y, Chen G-D, Hsieh M-C, Lai C-Y, Huang Y-P, Lin T-B. 2012. Spinal SGK1/GRASP-1/Rab4 is involved in complete Freund's adjuvant-induced inflammatory pain via regulating dorsal horn GluR1-containing AMPA receptor trafficking in rats. Pain 153, 2380–2392. ( 10.1016/j.pain.2012.08.004) [DOI] [PubMed] [Google Scholar]
- 72.Cattaneo A, Riva MA. 2016. Stress-induced mechanisms in mental illness: a role for glucocorticoid signalling. J. Steroid Biochem. Mol. Biol. 160, 169–174. ( 10.1016/j.jsbmb.2015.07.021) [DOI] [PubMed] [Google Scholar]
- 73.Iurato S, et al. 2017. DNA methylation signatures in panic disorder. Transl. Psychiatry 7, 1287 ( 10.1038/s41398-017-0026-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anacker C, et al. 2013. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc. Natl Acad. Sci. USA 110, 8708–8713. ( 10.1073/pnas.1300886110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Szyf M. 2015. Nongenetic inheritance and transgenerational epigenetics. Trends Mol. Med. 21, 134–144. ( 10.1016/j.molmed.2014.12.004) [DOI] [PubMed] [Google Scholar]
- 76.Horsthemke B. 2018. A critical view on transgenerational epigenetic inheritance in humans. Nat. Commun. 9, Article number: 2973 ( 10.1038/s41467-018-05445-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lehrner A, et al. 2014. Maternal PTSD associates with greater glucocorticoid sensitivity in offspring of Holocaust survivors. Psychoneuroendocrinology 40, 213–220. ( 10.1016/j.psyneuen.2013.11.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yehuda Ret al. 2014. Influences of maternal and paternal PTSD on epigenetic regulation of the glucocorticoid receptor gene in Holocaust survivor offspring. Am. J. Psychiatry 171, 872–880. ( 10.1176/appi.ajp.2014.13121571) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bader HN, Bierer LM, Lehrner A, Makotkine I, Daskalakis NP, Yehuda R. 2014. Maternal age at holocaust exposure and maternal PTSD independently influence urinary cortisol levels in adult offspring. Front. Endocrinol. (Lausanne) 5, 103 ( 10.3389/fendo.2014.00103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bowers ME, Yehuda R. 2016. Intergenerational transmission of stress in humans. Neuropsychopharmacology 41, 232–244. ( 10.1038/npp.2015.247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iurlaro M, von Meyenn F, Reik W. 2017. DNA methylation homeostasis in human and mouse development. Curr. Opin. Genet. Dev. 43, 101–109. ( 10.1016/j.gde.2017.02.003) [DOI] [PubMed] [Google Scholar]
- 82.Grossniklaus U, Kelly WG, Kelly B, Ferguson-Smith AC, Pembrey M, Lindquist S. 2013. Transgenerational epigenetic inheritance: how important is it? Nat. Rev. Genet. 14, 228–235. ( 10.1038/nrg3435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Skvortsova K, Iovino N, Bogdanović O. 2018. Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol. 19, 774–790. ( 10.1038/s41580-018-0074-2) [DOI] [PubMed] [Google Scholar]
- 84.Szyf M. 2014. Lamarck revisited: epigenetic inheritance of ancestral odor fear conditioning. Nat. Neurosci. 17, 2–4. ( 10.1038/nn.3603) [DOI] [PubMed] [Google Scholar]
- 85.Dias BG, Ressler KJ. 2014. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat. Neurosci. 17, 89–96. ( 10.1038/nn.3594) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gapp K, Jawaid A, Sarkies P, Bohacek J, Pelczar P, Prados J, Farinelli L, Miska E, Mansuy IM. 2014. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat. Neurosci. 17, 667–669. ( 10.1038/nn.3695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gapp K, Bohacek J. 2018. Epigenetic germline inheritance in mammals: looking to the past to understand the future. Genes Brain Behav. 17, e12407 ( 10.1111/gbb.12407) [DOI] [PubMed] [Google Scholar]
- 88.Robinson GE, Barron AB. 2017. Epigenetics and the evolution of instincts. Science 356, 26–27. ( 10.1126/science.aam6142) [DOI] [PubMed] [Google Scholar]
- 89.Head JA. 2014. Patterns of DNA methylation in animals: an ecotoxicological perspective. Integr. Comp. Biol. 54, 77–86. ( 10.1093/icb/icu025) [DOI] [PubMed] [Google Scholar]
- 90.Potok ME, Nix DA, Parnell TJ, Cairns BR. 2013. Reprogramming the maternal zebrafish genome after fertilization to match the paternal methylation pattern. Cell 153, 759–772. ( 10.1016/j.cell.2013.04.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang L, et al. 2013. Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell 153, 773–784. ( 10.1016/j.cell.2013.04.041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bailey CH, Bartsch D, Kandel ER. 1996. Toward a molecular definition of long-term memory storage. Proc. Natl Acad. Sci. USA 93, 13 445–13 452. ( 10.1073/pnas.93.24.13445) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hawkins RD, Kandel ER, Bailey CH. 2006. Molecular mechanisms of memory storage in Aplysia. Biol. Bull. 210, 174–191. ( 10.2307/4134556) [DOI] [PubMed] [Google Scholar]
- 94.Rajasethupathy P, et al. 2012. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 149, 693–707. ( 10.1016/j.cell.2012.02.057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pearce K, Cai D, Roberts AC, Glanzman DL. 2017. Role of protein synthesis and DNA methylation in the consolidation and maintenance of long-term memory in Aplysia. Elife 6, e18299 ( 10.7554/eLife.18299) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.