

Abstract
STK38 (also known as NDR1) is a Hippo pathway serine/threonine protein kinase with multifarious functions in normal and cancer cells. Using a context‐dependent proximity‐labeling assay, we identify more than 250 partners of STK38 and find that STK38 modulates its partnership depending on the cellular context by increasing its association with cytoplasmic proteins upon nutrient starvation‐induced autophagy and with nuclear ones during ECM detachment. We show that STK38 shuttles between the nucleus and the cytoplasm and that its nuclear exit depends on both XPO1 (aka exportin‐1, CRM1) and STK38 kinase activity. We further uncover that STK38 modulates XPO1 export activity by phosphorylating XPO1 on serine 1055, thus regulating its own nuclear exit. We expand our model to other cellular contexts by discovering that XPO1 phosphorylation by STK38 regulates also the nuclear exit of Beclin1 and YAP1, key regulator of autophagy and transcriptional effector, respectively. Collectively, our results reveal STK38 as an activator of XPO1, behaving as a gatekeeper of nuclear export. These observations establish a novel mechanism of XPO1‐dependent cargo export regulation by phosphorylation of XPO1's C‐terminal auto‐inhibitory domain.
Keywords: APEX2, autophagy, STK38, XPO1, YAP1
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport, Signal Transduction
Introduction
The serine/threonine kinase 38 STK38, also known as NDR1, is important in multifarious and unrelated biological functions, playing roles in cell cycle progression 1, 2, apoptosis 3, and centrosome duplication 4, 5. As a member of the Hippo core signaling, STK38 can directly phosphorylate the Hippo effector YAP1 on S127, resulting in YAP1 inactivation by cytoplasmic sequestration 6. In addition, STK38 has pro‐cancer cell survival functions in stress response and adaptation. STK38 responds to osmotic shock and to the expression of RASSF1A, a Hippo pathway regulator 7. Moreover, STK38 kinase activity is required for autophagy induction in response to starvation by regulating Beclin1 8. Finally, STK38 was also reported to be involved in resistance to anoïkis of cancer cells 9.
This non‐comprehensive inventory illustrates the variety and multiplicity of cellular functions driven by and/or dependent on STK38. How a single kinase can perform these crucial, but clearly distinct, functions? Would it have function‐specific substrates or a yet‐unknown common regulator being permissive for these functions? To address this question, we performed a proximity‐dependent biotinylation to map STK38 partnership in different cellular contexts. This revealed that STK38 interacts mainly with cytoplasmic proteins upon starvation‐induced autophagy, and with nuclear proteins after ECM detachment, suggesting that the subcellular localization of STK38 plays a regulatory role in response to these diverse stimuli. In addition, we confirmed a nuclear/cytoplasmic shuttling of STK38 being under the dependency of its own kinase activity and on XPO1 (exportin‐1, aka CRM1) nuclear export. Moreover, we discovered that STK38 phosphorylates the serine 1055 in the auto‐inhibitory domain of XPO1, thereby triggering the nuclear export of STK38 itself as well as other XPO1 cargoes such as Beclin1 and YAP1. These results suggest that STK38 regulates the subcellular localization and thereby the function of central cellular components by modulating their nuclear export via phosphorylation of XPO1 on its auto‐inhibitory domain.
Results
STK38 interacts with different sets of partners depending on the cellular context
To identify the proteins that interact with STK38 upon nutrient starvation‐induced autophagy, or when cells resist to anoïkis in suspension, we undertook a context‐dependent proteomic approach 10 by establishing cell lines stably expressing APEX2 N‐terminally fused to STK38 (Fig EV1). HeLa cells were used for nutrient starvation‐induced autophagy condition and HEK‐HT‐HRasG12V cells (hereafter referred to as HekRasV12) 11, 12 for anoïkis resistance condition, to match with previous studies of STK38's roles in autophagy 8 and survival upon ECM detachment 9, respectively. A quantitative SILAC proteomic methodology was applied, comparing quantitatively, on the one hand, complete medium incubation versus nutrient starvation‐induced autophagy (see Fig EV1 for this example) and, on the other hand, attached growth versus suspension growth. Validation of autophagy process (Appendix Fig S1A and B) and biotinylation efficiency in both contexts (Appendix Fig S1C and D) was performed before mass spectrometry (MS) identification. Each replicate displayed a good correlation between experiments (Appendix Fig S1E), indicating a high reproducibility between biological triplicate samples.
Figure EV1. STK38 partner identification strategy (related to Fig 1).
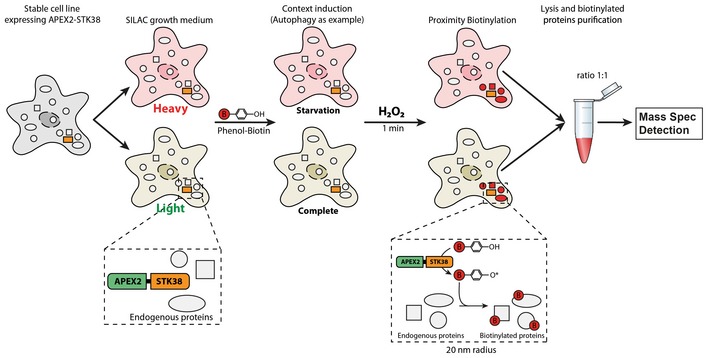
Figure indicating the strategy used to identify STK38 partner association dynamic depending on the context (example is shown for autophagy condition here). Stable cell lines expressing the fusion construct APEX2‐STK38 were generated and then subdivided for amino acid replacement (SILAC). Context was then induced (4 h of EBSS incubation for autophagy induction and suspension growth) as well as a control condition (nutrient‐rich medium incubation and attached growth, respectively). Proximity labeling of STK38 partners was performed as described 56: Briefly, cells were incubated with phenol–biotin for a minimum of 30 min followed by H2O2 incubation for 1 min precisely. Finally, biotinylated proteins (=STK38 partners) were purified from whole‐cell lysates using streptavidin‐coated magnetic beads and subjected to mass spectrometry identification.
Ninety‐seven binding partners of STK38 were identified in the context of autophagy, and 221 partners were identified in the context of suspension growth (Fig 1A) (see Dataset EV1 for the complete list), including several known interactors of STK38 such as MAP4K4 7, HIST2H2AC 13, EWSR1 14, NPM1 15, YWHAZ 16, and MAGOH 17. Among these 97 interactors, 32 displayed an interaction with STK38 that was significantly increased upon nutrient starvation‐induced autophagy as compared to rich medium, while interaction with only one protein was decreased. Upon ECM detachment, 44 proteins displayed an increased interaction with STK38, while for 72 proteins, the interaction with STK38 was decreased. Interestingly, 50 partners were common to both studied conditions, but displayed a differential association status with STK38, depending on the context (Fig 1A). These 50 common partners were classified by unsupervised hierarchical clustering, based on their association status with STK38, resulting in two main clusters as highlighted in orange and purple in Fig 1B. The upper cluster (orange) is composed of 31 proteins mainly increasing their interaction with STK38 upon nutrient starvation‐induced autophagy while decreasing their interaction with STK38 upon ECM detachment. The lower cluster (purple) consists of 19 proteins that increase their interaction with STK38 in suspension but do not see their interaction with STK38 modified upon nutrient starvation‐induced autophagy. Gene Ontology analysis of these two groups of interactors revealed a striking difference (Fig 1C): The most enriched terms upon nutrient starvation‐induced autophagy characterize these proteins as localized in the cytoplasm, while the cluster enriched upon ECM detachment contains mainly nuclear proteins. These results suggest that upon autophagy induction, STK38 associates with cytoplasmic partners while, upon ECM detachment, STK38 appears to preferentially interact with nuclear proteins.
Figure 1. STK38 associates with cytoplasmic interactors upon nutrient starvation and with nuclear interactors upon suspension growth.
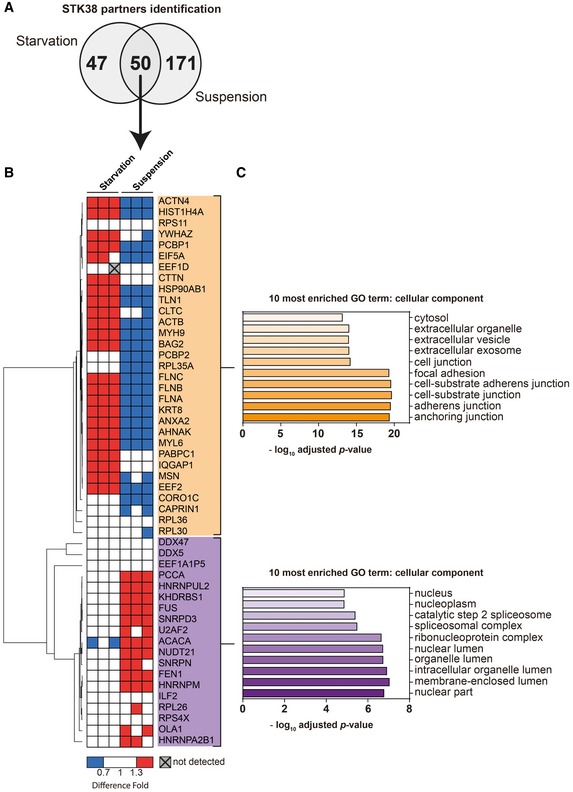
- Venn diagram of STK38 partners identified in both starvation and suspension conditions by proximity biotinylation assay coupled to mass spectrometry identification (see Appendix Table S1 for the complete list of STK38 context‐dependent interactors and Fig EV1 for the context‐dependent protein labeling strategy).
- Heatmap representation of common STK38 interactors identified in both starvation and suspension conditions according to their dynamic of association with STK38 (with a minimal fold increase of 1.38 ± 0.06). Unsupervised hierarchical clustering was generated based on their association fold using Pearson correlation.
- Representation of the 10 most cellular component enriched terms of the two clusters (assessed by using the Gene Ontology (GO) database; http://www.geneontology.org/).
STK38 interacts with XPO1
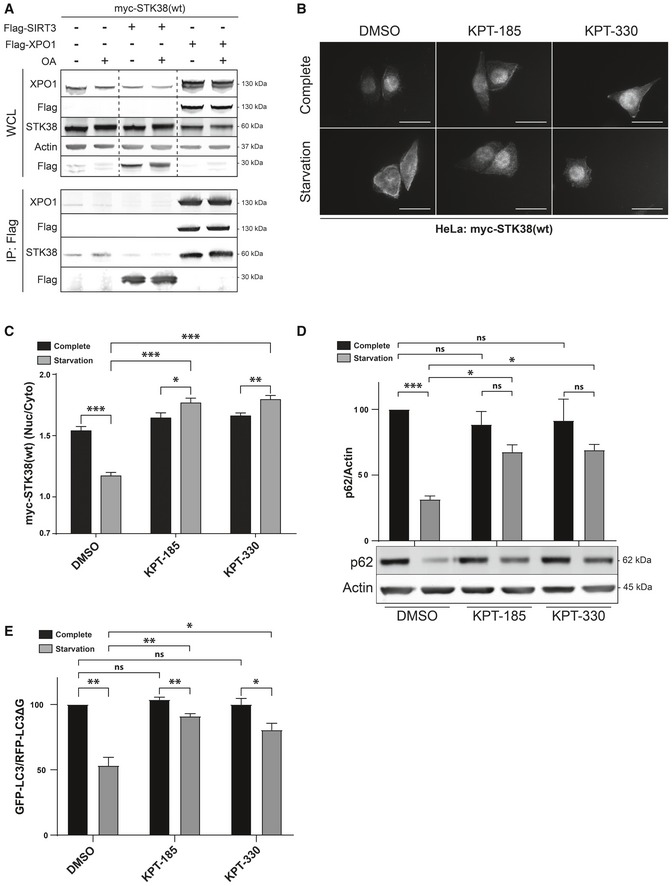
The above observations imply that STK38 shuttles between the nucleus and the cytoplasm. Exportin‐1 (XPO1), also known as the chromosomal region maintenance protein 1 (CRM1), the main nuclear export factor (karyopherin) that transports a wide diversity of proteins from the nucleus to the cytoplasm 18, 19, 20, 21, was identified in our screen as a novel STK38 interactor (Dataset EV1). To investigate whether STK38 interacts with XPO1, we performed a pull‐down experiment. myc‐tagged STK38 was transiently co‐expressed with Flag‐XPO1, or Flag‐Sirt3 as a control. Upon pull‐down using Flag antibody, STK38 co‐immunoprecipitated with XPO1 but not with Flag‐Sirt3 (Fig 2A), suggesting that STK38 physically interacts with XPO1. In addition, inhibiting protein phosphatase type 2A (PP2A) with okadaic acid (OA), which increases STK38 phosphorylation state and activity 22, did not modify the binding of STK38 and XPO1.
Figure 2. XPO1 activity is required for STK38 cytoplasmic accumulation and autophagy upon nutrient starvation.
-
ASTK38 interacts with XPO1. HekRasV12 cells were transiently transfected with myc‐STK38(wt) together with either Flag‐XPO1(wt) plasmid or Flag‐control (ctrl = Sirt3) plasmid or without DNA. Twenty‐four hours later, cells were incubated with okadaic acid (OA) (final concentration = 1 μM) for 1 h or with DMSO. Flag fusions were pulled down, and co‐immunoprecipitated proteins were analyzed by Western blotting (WB). The upper panel displays whole‐cell lysates (WCL), and the lower panel represents immunoprecipitated proteins.
-
B, CSTK38 accumulates in the cytoplasm upon nutrient starvation in a XPO1‐dependent manner. (B) HeLa cells were transfected with myc‐STK38(wt) plasmid. The next day, cells were incubated with DMEM or EBSS in the presence of XPO1 inhibitor KPT‐185 or KPT‐330 as indicated (final concentration = 1 μM) or DMSO for 4 h. Cells were then fixed and stained for myc‐tag. Representative images are shown, and scale bars are 40 μm. (C) Quantification of myc‐STK38(wt) nuclear/cytoplasmic staining (n > 30 cells from 3 independent experiments, mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, Mann–Whitney test).
-
D, EXPO1 activity is required for nutrient starvation‐induced autophagy. (D) Immunoblotting with the indicated antibodies of whole‐cell lysates of cells in (B) and (C) and its graphical representation (n = 3 independent experiments, mean ± SEM; ***P < 0.001, ns, not significant, Student's t‐test). As expected, p62 degradation in starvation conditions was inhibited in the presence of XPO1 inhibitors. (E) HeLa cells stably expressing the GFP‐LC3‐RFP‐LC3ΔG reporter autophagic probe 26 were incubated with DMEM or EBSS in the presence of XPO1 inhibitor KPT‐185 or KPT‐330 (final concentration = 1 μM) or DMSO for 4 h. The GFP‐LC3 (degraded upon autophagy) and RFP‐LC3ΔG (non‐degraded upon autophagy) signals were recorded by FACS analysis and are shown as a ratio recapitulating overall LC3 level (n = 3 independent experiments, mean ± SEM; *P < 0.05, **P < 0.01, ns, not significant, Mann–Whitney test). Here again, incubation with XPO1 inhibitors significantly impaired the autophagy process (see Appendix Fig S2C and D for stable cell line validation).
STK38 accumulates in the cytoplasm upon nutrient starvation‐induced autophagy in a XPO1‐dependent manner
Our proximity‐labeling experiment suggested that STK38 interacts mainly with cytoplasmic proteins upon nutrient starvation, suggesting that in this condition, STK38 is localized to the cytoplasm. Due to the lack of specific and sensitive antibodies to follow endogenous STK38 subcellular localization using immunofluorescence (IF), HeLa cells were transfected with a plasmid expressing myc‐tagged STK38. Immunostaining of STK38 in nutrient‐starved cells (EBSS medium) demonstrated that STK38 localizes mainly in the cytoplasm, while in nutrient‐rich medium (complete DMEM) STK38 is mainly localized in the nucleus (Fig 2B and C). The highly selective XPO1 inhibitors KPT‐185 and KPT‐330 23, 24 inhibited STK38's exit form the nucleus. XPO1 inhibition by KPT‐185 and KPT‐330 was validated in parallel by monitoring the nucleo‐/cytoplasmic localization of a well‐known cargo of XPO1: IκBα (Appendix Fig S2A and B). Together, these results indicate that STK38 shuttles from the nucleus to the cytoplasm upon nutrient starvation in a XPO1‐dependent manner.
To study whether the XPO1‐dependent transport of STK38 contributes to STK38's function in autophagy 8, 25, we monitored the levels of p62/SQSTM1, a well‐known autophagy substrate. As expected, p62 levels decreased upon starvation (Fig 2D). However, the inhibition of XPO1 with the subsequent inhibition of STK38 cytoplasmic localization reduced p62 degradation (Fig 2D), indicating a defect in autophagy flux. As a complementary approach, we generated a stable cell line expressing the autophagic flux probe GFP‐LC3‐RFP‐LC3Δ 26. HeLa GFP‐LC3‐RFP‐LC3Δ cells were validated by silencing autophagy regulators such as ATG5 and Beclin1 followed by measuring the GFP‐LC3/RFP‐LC3Δ ratio by FACS. Similar to the silencing of ATG5 or Beclin1, knockdown of STK38 significantly impaired the reduction of the GFP‐LC3/RFP‐LC3Δ ratio observed in the control condition upon EBSS treatment, indicating a defect in autophagy in these conditions (Appendix Fig S2C and D). This method was used to examine XPO1's contribution to nutrient starvation‐induced autophagy (Fig 2E). Significantly, XPO1 inhibition by both KPT‐185 and KPT‐330 significantly reduced autophagy, as measured by the GFP‐LC3/RFP‐LC3Δ ratios. Taken together, these results indicate that STK38 is exported to the cytoplasm by XPO1 upon starvation‐induced autophagy and that XPO1 has to be functional for the resulting starvation‐induced autophagy process, but are in contrast to a previous report where blockage of XPO1 led to the nuclear accumulation of TFEB and to the induction of autophagy 27. This apparent discrepancy arises in two different settings: with XPO1 being inhibited for 6 h for the transcription‐dependent autophagy induction 27, while in our case, 4 h of inhibition led to a blockage of autophagy. On the other hand, the TFEB‐dependent impact concerned basal autophagy as opposed to our observation related to starvation‐induced autophagy.
STK38 kinase activity is necessary and sufficient to induce its own cytoplasmic relocalization and autophagy
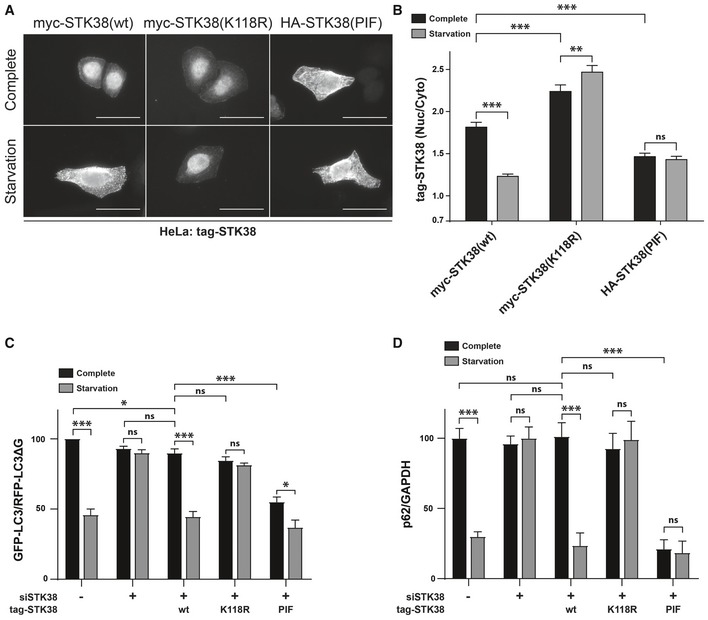
To investigate whether STK38's kinase activity is required for its nuclear/cytoplasmic shuttling, HeLa cells were silenced for endogenous STK38 followed by transient expression of wild‐type STK38 (wt), kinase‐dead (K118R) 3, or constitutively active (PIF) STK38 28. Transfected cells were subsequently submitted to nutrient starvation and stained for STK38 subcellular localization (Fig 3A and B). As expected, STK38(wt) accumulated in the cytoplasm of nutrient‐starved cells. However, kinase‐dead STK38(K118R) remained nuclear upon starvation while a constitutively active version of STK38(PIF) accumulated in the cytoplasm, irrespective of culture conditions (Fig 3A and B). These results indicate that STK38 kinase activity is required and sufficient to induce its export to the cytoplasm, independently of physiological conditions.
Figure 3. STK38 kinase activity is necessary and sufficient to induce its cytoplasmic relocalization and autophagy.
-
A, BSTK38 kinase activity is required and sufficient for its cytoplasmic accumulation upon nutrient starvation. (A) HeLa cells silenced for endogenous STK38 were transfected with myc‐STK38(wt) expressing plasmid, myc‐STK38(K118R) (STK38 kinase‐dead version) plasmid, or HA‐STK38(PIF) (STK38 constitutively active version) plasmid. Twenty‐four hours later, cells were incubated with DMEM or EBSS for 4 h, fixed, and stained for myc‐tag or HA‐tag. Representative images are shown, and scale bars are 40 μm. (B) Graphical representation of tag‐STK38 variant nuclear staining/cytoplasmic staining (n > 30 cells from three independent experiments, mean ± SEM; **P < 0.01, ***P < 0.001, ns, not significant, Mann–Whitney test).
-
C, DSTK38 kinase activity is required and sufficient for nutrient starvation‐induced autophagy. (C) HeLa cells stably expressing the GFP‐LC3‐RFP‐LC3ΔG autophagic probe 26 were transiently transfected with siRNA targeting the 3′UTR region of endogenous STK38 (or with non‐targeting siRNA (siNT)). The next day, cells were transiently transfected with the indicated STK38 mutants expressing plasmids (identical to (A–B)). Twenty‐four hours after plasmid transfection, cells were incubated with DMEM or EBSS for 4 h. The GFP‐LC3 (degraded upon autophagy) and RFP‐LC3ΔG (non‐degraded upon autophagy) signals were recorded by FACS analysis and are shown as a ratio recapitulating overall LC3 level (n = 4 independent experiments, mean ± SEM; *P < 0.05, ***P < 0.001, ns, not significant, Mann–Whitney test). As published 8, depleting STK38 prevents autophagy to take place. This effect was partially reversed by expressing the wt ORF as well as constitutively active STK38, whereas kinase‐dead version failed to reproduce endogenous STK38 effect. Expression of constitutively active STK38 is sufficient to induce a substantial change in the autophagic flux upon nutrient‐rich conditions (see Appendix Fig S3A for STK38 replacement). (D) HeLa cells were transiently transfected with siRNA targeting the 3′UTR region of endogenous STK38 (or with non‐targeting siRNA (siNT)) and then transiently transfected with the indicated STK38 mutants expressing plasmids 48 h after. The next day, the cells were incubated with DMEM or EBSS for 4 h and overall p62 level was quantified for autophagy estimation (see Appendix Fig S3B for blots) (n = 3 independent experiments, mean ± SEM; ***P < 0.001, ns, not significant, Student's t‐test).
To further investigate whether STK38 kinase activity is also involved in nutrient starvation‐induced autophagy, we knocked down endogenous STK38 in HeLa GFP‐LC3‐RFP‐LC3ΔG cells, then complemented these cells with RNAi‐resistant STK38(wt), K118R, or PIF variants (Appendix Fig S3A), and measured the autophagic flux (Fig 3C). As expected 8, STK38 depletion significantly impaired autophagy upon nutrient starvation, which was restored by reintroducing wild‐type STK38 (Fig 3C). In stark contrast, expression of kinase‐dead version of STK38(K118R) failed to restore nutrient starvation‐induced autophagy, while constitutively active STK38(PIF) supported autophagy. Interestingly, expression of constitutively active STK38(PIF) is sufficient to induce a significant decrease in the GFP‐LC3/RFP‐LC3ΔG in nutrient‐rich conditions, indicating an increase in autophagy (Fig 3C). The same effects were observed while using a complementary approach for autophagy measurement: by monitoring the p62 levels (Fig 3D).
In conclusion, these experiments support that the kinase activity of STK38 is important for both its subcellular localization and autophagy and that STK38 is instructive and permissive for autophagy (a situation similar to the one observed in flies 8).
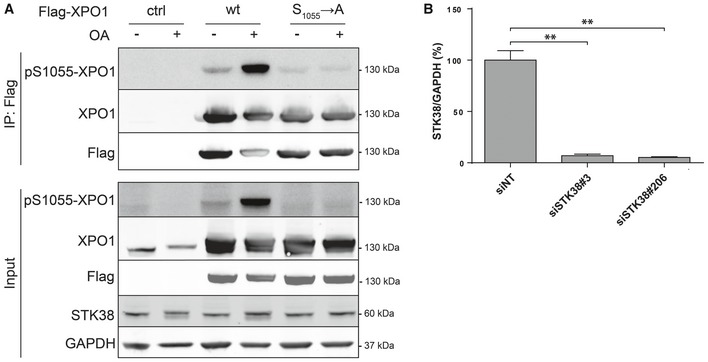
STK38 phosphorylates XPO1 on serine 1055
The presence of a bona fide STK38 HxRxxS/T phosphorylation motif 29 in XPO1 protein sequence offered a patent implication of a potential kinase–substrate relationship between STK38 and XPO1. This motif is centered on serine 1055, reported as being phosphorylated in human cells by an undetermined kinase without any functional relevance 30, 31, 32, 33, and S1055 phosphorylation was required for XPO1 interaction with a 14‐3‐3 34. We generated a phospho‐specific anti‐S1055‐P antibody that was validated using wt or phospho‐acceptor (S1055A) mutant of XPO1 (Fig EV2A). The anti‐XPO1‐S1055‐P antibody exhibited a strong specificity for XPO1(wt) upon OA incubation, a potent inhibitor of protein phosphatase type 2A (PP2A) that boosts most phosphorylation in cells, including STK38 22, leading to XPO1 serine phosphorylation. Concurrently, as expected, the XPO1(S1055A) mutant was not detected with this antibody in both whole‐cell lysates and after pulling down Flag‐XPO1 variants (Fig EV2A).
Figure EV2. Validation of pS1055_XPO1 antibody (related to Fig 4).
- HeLa cells were transiently transfected with the indicated Flag‐XPO1 mutants expressing plasmids (wt or S1055A mutation) or without DNA. The next day, cells were treated with 1 μM OA or with vehicle (DMSO) for 1 h. Flag fusions were immunoprecipitated, and pulled‐down proteins were analyzed by Western blotting. The upper panel displays immunoprecipitated proteins, and the lower panel represents whole‐cell lysates.
- Graphical representation of STK38 protein level for experiment presented in Fig 4A and B (n = 3 independent experiments, mean ± SEM; **P < 0.01, Mann–Whitney test).
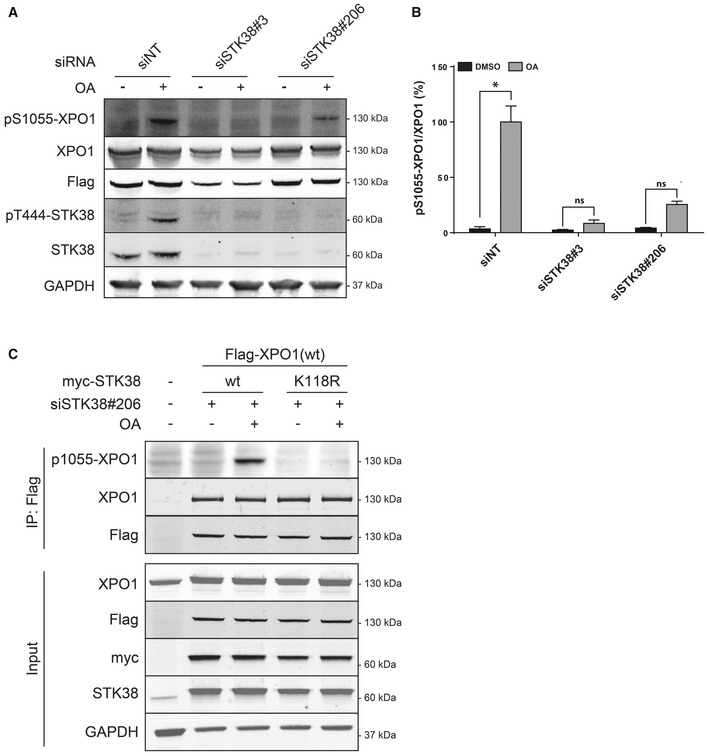
Using this specific anti‐XPO1‐S1055‐P antibody, we investigated whether STK38 can phosphorylate serine 1055 of XPO1. As expected, OA incubation induced both phosphorylation of STK38 on T444, revealing its activated state, and XPO1 on S1055 (Fig 4A and B). On the other hand, knockdown of endogenous STK38 (Fig EV2B) significantly reduced the phosphorylation of XPO1 on S1055 (Fig 4A and B). In addition, in cells where endogenous STK38 was depleted by RNAi, expression of wt STK38, but not kinase‐dead STK38, allowed to detect S1055 phosphorylation of XPO1 (Fig 4C). Finally, the identity of the serine 1055 as the phosphorylation site was confirmed independently by mass spectrometry (Appendix Fig S4). Taken together, these results demonstrate that the serine 1055 of XPO1 appears to be a bona fide phosphorylation residue for STK38.
Figure 4. STK38 is required for phosphorylation of XPO1 on Ser1055.
-
A–CSTK38 is required for XPO1_S1055 phosphorylation. (A) HeLa cells were transiently transfected with the indicated siRNA and subjected to Flag‐XPO1(wt) transient transfection the following day. Forty‐eight hours later, cells were incubated with okadaic acid (OA, final concentration = 1 μM) for 1 h or with DMSO. Immunoblotting was performed on whole‐cell lysates with the indicated antibodies. (B) Graphical representation of the phospho‐S1055‐XPO1 signal on total XPO1 (n = 3 independent experiments, mean ± SEM; *P < 0.05, ns, not significant, Mann–Whitney test). As expected, XPO1 is phosphorylated on its Ser1055 upon OA treatment but not when STK38 is silenced (see Appendix Fig S5 for antibody validation and STK38 silencing quantification). (C) STK38 kinase activity is required for XPO1_S1055 phosphorylation. HekRasV12 cells silenced (or not) for endogenous STK38 were transfected with Flag‐XPO1(wt) expression plasmid in addition to myc‐STK38(wt) or myc‐STK38(K118R) (STK38 kinase‐dead version) plasmids. The next day, cells were treated with 1 μM OA or with vehicle (DMSO) for 1 h. Flag fusions were immunoprecipitated, and pulled‐down proteins were analyzed by Western blotting. The upper panel displays immunoprecipitated proteins, and the lower panel represents whole‐cell lysates.
Phosphorylation of XPO1 on S1055 induces nuclear export of STK38 and autophagy
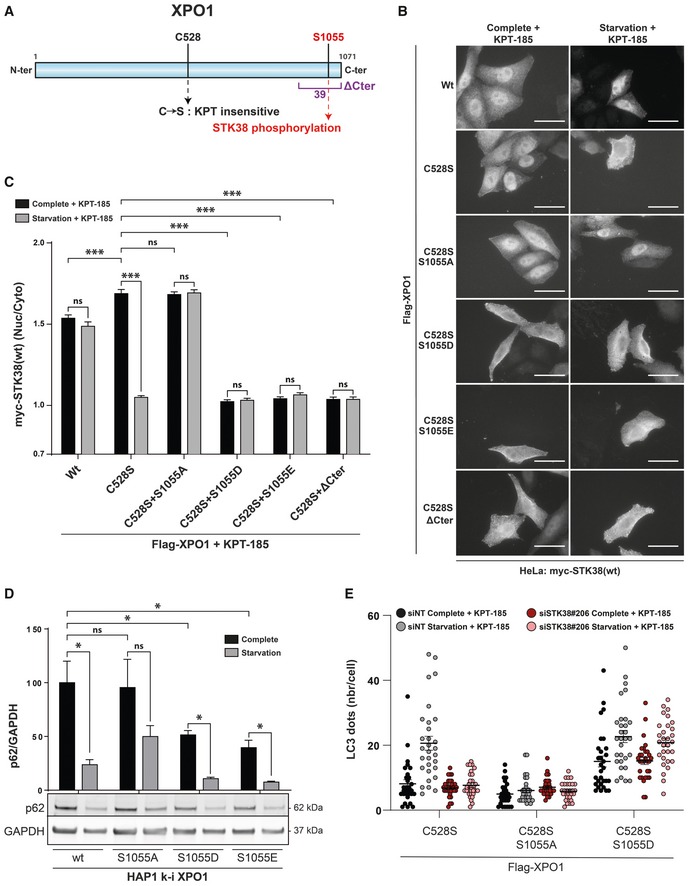
We hypothesized that S1055 phosphorylation of XPO1 is important for its nuclear export function. More precisely, since S1055 is at the tip of the auto‐inhibitory region of XPO1 35, and since STK38 kinase activity is required for its XPO1‐dependent nuclear export, our working model was that this phosphorylation could relieve XPO1 auto‐inhibition and thus enable its export function. To address this point, we analyzed the localization of STK38 in HeLa cells transiently transfected with XPO1 mutants that are either insensitive to phosphorylation (S1055A) or that mimic constitutive phosphorylation (S1055D and S1055E) (Appendix Fig S5A). In order to avoid confusion, we inhibited endogenous XPO1 activity with the inhibitor KPT‐185 while exogenous S1055 mutants of XPO1 were mutated for C528S, which confers XPO1 resistance to both KPT inhibitors without affecting its export function 24 (Fig 5A). Thus, the impact on nuclear export will depend on the exogenously expressed XPO1 only.
Figure 5. XPO1_S1055 phosphorylation is required and sufficient for STK38 nuclear exit and autophagy upon nutrient starvation.
-
AGraphical representation of XPO1 protein. C528 to S amino acid mutation confers XPO1 resistance to both KPT‐185 and KPT‐330 chemical inhibitors 24. S1055 amino acid corresponds to the phosphorylation target of the STK38. Finally, the ΔCter region is also highlighted corresponding to a XPO1 construct lacking the 39 C‐terminal residues 35.
-
B, CXPO1_S1055 phosphorylation is required and sufficient for STK38 nuclear exit upon nutrient starvation. (B) HeLa cells were transiently transfected with myc‐STK38(wt) in addition to the indicated Flag‐XPO1 mutant plasmids. Twenty‐four hours later, cells were incubated with DMEM or EBSS for 4 h, both supplemented with KPT‐185 (final concentration = 1 μM) in order to inhibit endogenous XPO1 activity. Cells were then fixed and stained for Flag and myc tags. Only cells positive for both Flag‐XPO1 and myc‐STK38(wt) were captured. Representative images are shown, and scale bars are 40 μm. (C) Graphical representation of myc‐STK38(wt) nuclear staining/cytoplasmic staining (n > 30 cells from three independent experiments, mean ± SEM; ***P < 0.001, ns, not significant, Mann–Whitney test). Expression of wt XPO1 failed to induce cytoplasmic localization of STK38 upon nutrient starvation in the presence of KPT‐185 where C528S mutant recapitulates results described in Fig 2B and C. Expression of phosphonegative XPO1 (S1055A) failed to induce STK38 cytoplasmic localization upon EBSS treatment while phosphomimetic XPO1 (S1055D and S1055E) was sufficient to promote STK38 cytoplasmic localization without autophagic stimuli. Finally, expression of XPO1 lacking in its 39 C‐terminal residues mimicked phosphomimetic variants.
-
DHAP1 cells carrying genomic XPO1 mutations of S1055 were subjected to IMDM (nutrient‐rich medium) or EBSS (starvation) incubation for 4 h followed by Western blot analysis for p62 level measurement. This result indicates that p62 degradation in starvation conditions was inhibited in XPO1_S1055A HAP1 cells whereas p62 degradation was potentiated in both phosphomimetic variants (S1055D and S1055E) (n = 4 independent experiments, mean ± SEM; *P < 0.05, ns, not significant, Mann–Whitney test; see Fig EV3A for HAP1 genome‐edited cell lines).
-
EHEK‐HT‐iRFP‐LC3 cells were transiently transfected with siRNA targeting endogenous STK38 (or with non‐targeting siRNA (siNT)). After 2 days, cells were transiently transfected with the indicated Flag‐XPO1 mutant plasmids. Twenty‐four hours later, cells were incubated with DMEM or EBSS for 4 h, both supplemented with KPT‐185 (final concentration = 1 μM) in order to inhibit endogenous XPO1 activity and with chloroquine (final concentration = 10 μM). Cells were then fixed, and the number of iRFP‐LC3 dots per cell was recorded only in cells positive for Flag‐XPO1 mutants (see Fig EV3B for representative images and Appendix Fig S5B for STK38 silencing; n = 30 cells from two independent experiments). As expected, silencing of endogenous STK38 prevented the formation of LC3 dots upon starvation where introduction of phosphonegative XPO1 (S1055A) failed also to increase LC3 dots upon starvation. Interestingly, phosphomimetic XPO1 (S1055D) was sufficient to increase the number of LC3 dots even in complete and also STK38‐depleted conditions.
As expected, STK38 remained nuclear in both nutrient‐rich and nutrient‐deprived medium conditions when XPO1(wt) was expressed, since KPT‐185 inhibited both endogenous and exogenous XPO1‐mediated nuclear export (Fig 5B and C). Conversely, STK38 localized in the cytoplasm upon starvation when XPO1(C528S) was expressed (Fig 5B and C), further supporting the findings described in Fig 2. However, upon XPO1(C528S/S1055A) expression, STK38 remained nuclear in starvation condition, suggesting that S1055 phosphorylation of XPO1 is required for the nuclear export of STK38. Reversely, expression of XPO1 carrying phosphomimetic mutations (C528S/S1055D or C528S/S1055E) resulted in a cytoplasmic accumulation of STK38 even in rich medium, further supporting the notion that XPO1 phosphorylation on S1055 is important and instructive for the nuclear export of STK38 (Fig 5B and C).
The localization of the serine 1055 residue within the C‐terminal auto‐inhibitory domain of XPO1 (highlighted in purple in Fig 5A) 35 also prompted us to analyze the effect of a XPO1(C528S) mutant containing a 39‐amino acid C‐terminal deletion. Expression of this C‐terminal deletion mutant indeed resulted in a cytoplasmic accumulation of STK38 irrespective of the culturing conditions (Fig 5B and C). Taken together, these results point toward a mechanism where phosphorylation of S1055 releases XPO1 auto‐inhibitory activity to open up the cargo binding site and allow XPO1 to carry on its nuclear export function.
To independently probe these results, we created S1055 XPO1 mutants in the endogenous genomic locus of haploid HAP1 cells using genome editing (Fig EV3A). HAP1 cells carrying a XPO1S1055A mutant exhibited a reduced autophagy when starved of nutrient, as shown by the limited degradation of the p62 autophagy marker compared to wild‐type cells (Fig 5D). Conversely, both phosphomimetic XPO1 mutants (XPO1S1055D and XPO1S1055E) induced p62 degradation irrespective of culturing conditions (Fig 5D). These results indicate that phosphorylation of XPO1_S1055 is not only required for nutrient starvation‐inducted autophagy, but also sufficient to induce autophagy. Are the phosphorylation and activation of XPO1 linked to the requirement of STK38 for autophagy 8? To address this point, we monitored autophagy by tracking LC3 dots and using HEK‐HT cells stably expressing iRFP‐LC3. We expressed the XPO1 mutants (C528S in addition, or not, of S1055 mutations) while depleting, or not, endogenous STK38 by RNAi, and inhibiting endogenous XPO1 with KPT‐185. Depletion of STK38 (see Appendix Fig S5B for STK38 protein level) did not affect autophagy when phosphomimetic mutant (S1055D) of XPO1 was expressed, as compared to the other conditions by following LC3 dot formation (see Fig 5E for quantification and Fig EV3B for representative images). This result suggests that the main function of STK38 in autophagy is to support XPO1 export activation by phosphorylating XPO1 on S1055.
Figure EV3. CRISPR/Cas9 knock‐in HAP1 cell lines and representative images of LC3 dots (related to Fig 5).
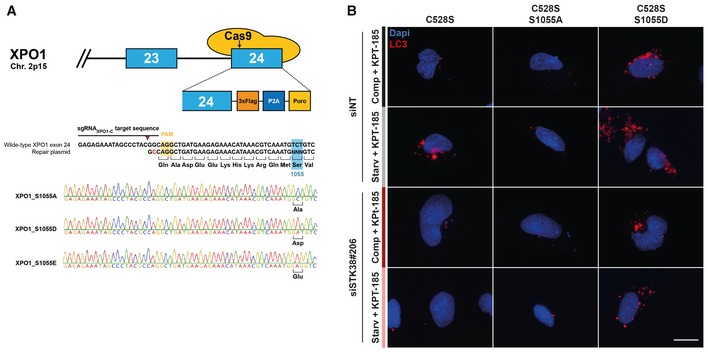
- Schematic overview of CRISPR/Cas9 genome editing of XPO1 in HAP1 cell lines. Exons are shown as blue boxes and introns are visualized as a thick black line, while Cas9 is represented by the yellow oval shapes. The DNA sequence and corresponding amino acid of the region around the Ser1055 residue are enlarged. The Cas9 PAM sequence is indicated in yellow, the sgRNA sequence is highlighted by a black line above the DNA sequence, and the Cas9 cutting site is indicated by the brown arrowhead. The wild‐type sequence is shown together with the sequence of the donor repair plasmid used to generate the S1055 mutations, and the additional silent mutation to prevent recutting of the mutagenized gene is marked in red. Sanger sequencing chromatograms of the generated XPO1 S1055 mutant cell lines are shown with the desired mutations highlighted in bold.
- HEK‐HT‐iRFP‐LC3 cells were transiently transfected with siRNA targeting endogenous STK38 (or with non‐targeting siRNA (siNT)). After 2 days, cells were transiently transfected with the indicated Flag‐XPO1 mutant plasmids. Twenty‐four hours later, cells were incubated with DMEM or EBSS for 4 h, both supplemented with KPT‐185 (final concentration = 1 μM) in order to inhibit endogenous XPO1 activity and with chloroquine (final concentration = 10 μM). Cells were then fixed, and the number of iRFP‐LC3 dots per cell was recorded only in cells positive for Flag‐XPO1 mutants. Representative images are shown, and scale bars are 20 μm.
STK38 controls Beclin1 subcellular distribution through XPO1 phosphorylation
In agreement with a previous report 36, we found that Beclin1 shuttles from the nucleus to the cytoplasm of starved HeLa cells in a XPO1‐dependent manner. This shuttling was regulated by STK38, since Beclin1 failed to exit the nucleus when STK38 was silenced (see Appendix Fig S6A for STK38 silencing verification) (Figs 6A and EV4A). This finding was confirmed by immunostaining of endogenous Beclin1 in our genome‐edited HAP1 cell lines. While Beclin1 exited the nucleus from wild‐type HAP1 cell lines upon starvation (Figs 6B and EV4B), Beclin1 remained nuclear in cells containing the mutant XPO1(S1055A). Conversely, in cells expressing the phosphomimetic S1055D or S1055E mutants, the nuclear exit of Beclin1 was promoted even in nutrient‐rich medium (Figs 6B and EV4B). These results suggest that STK38, in addition to regulating its own subcellular fate, also controls the subcellular distribution of Beclin1 through XPO1 phosphorylation, thereby revealing how STK38 through Beclin1 can regulate autophagy. Here again, silencing of endogenous STK38 in cells expressing phosphomimetic XPO1 (S1055D) did not impair Beclin1 nuclear exit upon starvation (Figs 6C and EV4C), providing genetic support to our model.
Figure 6. XPO1_S1055 phosphorylation is required and sufficient for Beclin1 nuclear exit upon nutrient starvation.
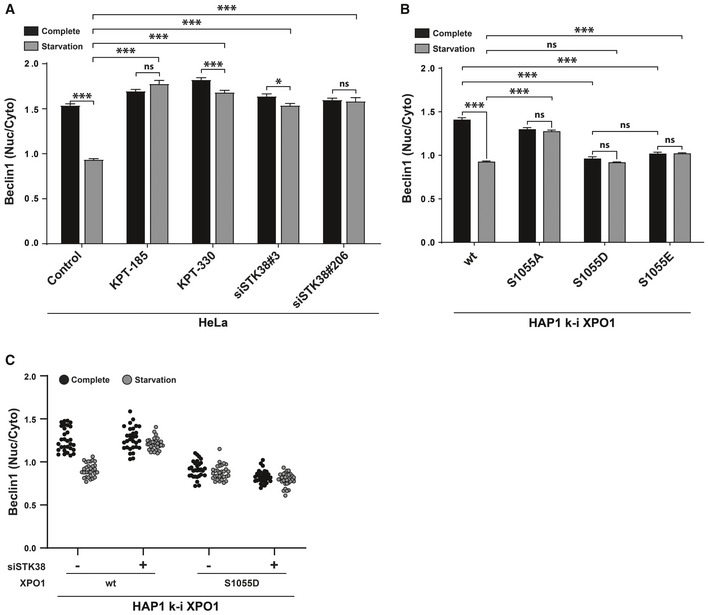
- XPO1 and STK38 are required for Beclin1 cytoplasmic accumulation upon nutrient starvation‐induced autophagy. HeLa cells were transiently transfected with the indicated siRNA (control and KPT conditions = siNT). Seventy‐two hours later, cells were incubated with XPO1 inhibitor KPT‐185 or KPT‐330 as indicated (final concentration = 1 μM) or with DMSO for all other conditions for 2 h prior to incubation with DMEM or EBSS supplemented (or not) with inhibitors for 2 h. Cells were fixed and stained for endogenous Beclin1. Graphical representation of endogenous Beclin1 nuclear staining/cytoplasmic staining (n > 30 cells from three independent experiments, mean ± SEM; *P < 0.05, ***P < 0.001, ns, not significant, Mann–Whitney test). XPO1 activity inhibition by both KPT‐185 and KPT‐330 as well as STK38 silencing failed to induce Beclin1 cytoplasmic accumulation upon nutrient starvation (see Fig EV4A for representative images, Appendix Fig S6A for STK38 silencing, and Appendix Fig S6B for Beclin1 antibody validation).
- XPO1_S1055 phosphorylation is required and sufficient for Beclin1 nuclear exit upon nutrient starvation. Genome‐edited XPO1 mutant HAP1 cells were incubated with IMDM (complete) or EBSS (starvation) for 2 h. Cells were then fixed and stained for endogenous Beclin1. Graphical representation of endogenous Beclin1 nuclear staining/cytoplasmic staining (n > 30 cells from three independent experiments, mean ± SEM; ***P < 0.001, ns, not significant, Mann–Whitney test). Here, phosphonegative XPO1 failed to induce Beclin1 cytoplasmic accumulation upon nutrient starvation, whereas phosphomimetic variants (S1055D and S1055E) potentiated Beclin1 cytoplasmic accumulation (see Fig EV4B for representative images).
- Genome‐edited XPO1 mutant HAP1 cells were transiently transfected with siRNA targeting endogenous STK38 (siSTK38#206 or with non‐targeting siRNA (siNT)). Seventy‐two hours later, cells were incubated with IMDM (complete) or EBSS (starvation) for 2 h. Cells were then fixed and stained for endogenous Beclin1. Graphical representation of endogenous Beclin1 nuclear staining/cytoplasmic staining (n = 30 cells from two independent experiments). Here, phosphomimetic XPO1 (S1055D) potentiated Beclin1 cytoplasmic localization in both complete and starvation condition even in the absence of STK38 as compared to cells carrying wt XPO1 (see Fig EV4C for representative images and Appendix Fig S6C for STK38 silencing).
Figure EV4. Beclin1 subcellular localization (related to Fig 6).
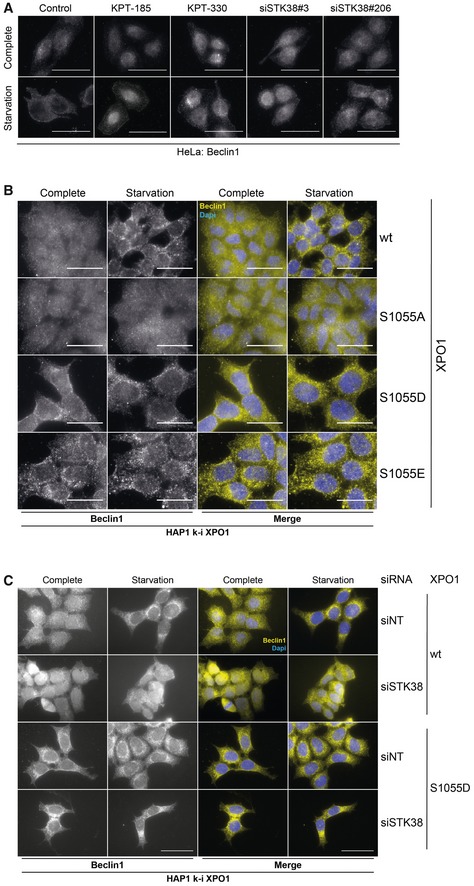
- HeLa cells were transiently transfected with the indicated siRNA (control and KPT conditions = siNT). Seventy‐two hours later, cells were incubated with XPO1 inhibitor KPT‐185 or KPT‐330 as indicated (final concentration = 1 μM) or with DMSO for all other conditions for 2 h prior to incubation with DMEM or EBSS supplemented (or not) with inhibitors for 2 h. Cells were fixed and stained for endogenous Beclin1. Representative images are shown, and scale bars are 40 μm (see Appendix Fig S6A for STK38 silencing).
- Genome‐edited XPO1 mutant HAP1 cells were incubated with IMDM (complete) or EBSS (starvation) for 2 h. Cells were then fixed and stained for endogenous Beclin1. Representative images are shown, and scale bars are 20 μm.
- Genome‐edited XPO1 mutant HAP1 cells were transiently transfected with siRNA targeting endogenous STK38 (siSTK38#206 or with non‐targeting siRNA (siNT)). Seventy‐two hours later, cells were incubated with IMDM (complete) or EBSS (starvation) for 2 h. Cells were then fixed and stained for endogenous Beclin1. Representative images are shown, and scale bars are 20 μm (see Appendix Fig S6C for STK38 silencing).
The subcellular localization of the Hippo pathway terminal effector YAP1 is modulated by STK38‐mediated phosphorylation of XPO1
Can our observations and model be generalized to several, if not all, XPO1 cargoes? We tested this hypothesis on YAP1, a XPO1 cargo 37 but also a STK38 substrate 6, where YAP1 phosphorylation on S127 results in YAP1 nuclear exclusion. In addition, the nuclear/cytoplasmic shuttling of YAP1 has been reported to depend on cell density 38, while its nuclear export has been shown to depend on XPO1 in human cells 39, 40, 41 and in Drosophila 42.
Consistent with reports 43, we found that YAP1 is excluded from the nucleus at high cell density, while total YAP1 protein levels remained unchanged (Appendix Fig S7B), and that XPO1 inhibition prevented YAP1 nuclear exclusion at high cell density (Figs 7A and EV5A). Interestingly, STK38 silencing (Appendix Fig S7A) phenocopied XPO1 inhibition on YAP1 subcellular localization at high cell density (Figs 7A and EV5A), indicating that the nuclear exit of YAP1 is under the control of STK38 and XPO1.
Figure 7. XPO1_S1055 phosphorylation is required and sufficient for YAP nuclear exit at high confluency.
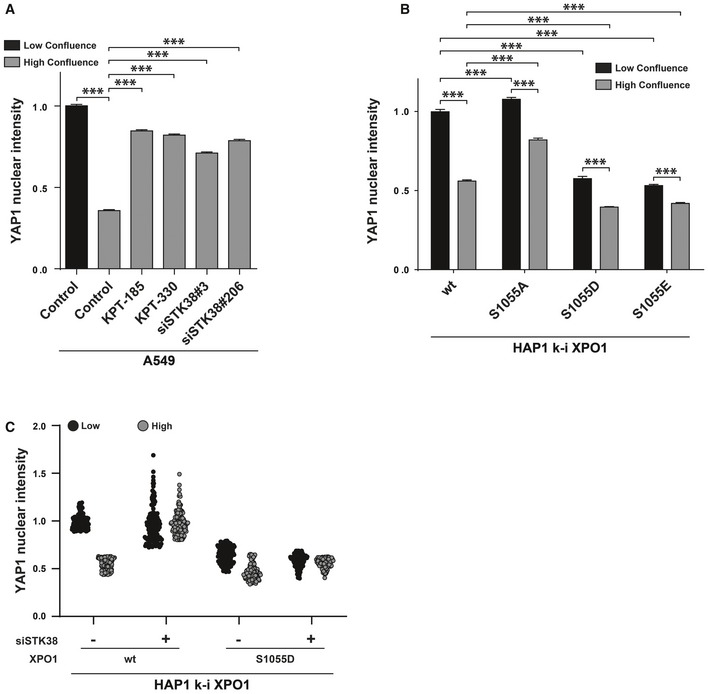
- XPO1 and STK38 are required for YAP1 nuclear export at high confluence. A549 cells were transiently transfected with the indicated siRNA (control and KPT conditions = siNT) at low or high confluency. Forty‐eight hours later, cells were incubated overnight in the presence of XPO1 inhibitors KPT‐185 and KPT‐330 (final concentration = 1 μM) or with DMSO for all other conditions. The next day, cells were fixed and stained for endogenous YAP1. Quantitative representation of YAP1 nuclear fluorescence intensity (n > 300 cells from three independent experiments, mean ± SEM; ***P < 0.001, Mann–Whitney test). XPO1 activity inhibition by both KPT‐185 and KPT‐330 as well as STK38 silencing failed to induce YAP1 nuclear exit at high confluence (see Fig EV5A for representative images and Appendix Fig S7A for STK38 silencing).
- XPO1_S1055 phosphorylation is required and sufficient for YAP nuclear exit. Genome‐edited XPO1 HAP1 cells were cultured for 2 days at low versus high confluency. Cells were then fixed and stained for endogenous YAP1. Quantitative representation of YAP1 nuclear fluorescence intensity (n > 300 cells from 3 independent experiments, mean ± SEM; ***P < 0.001, Mann–Whitney test). Here, phosphonegative XPO1 failed to induce YAP nuclear exit at high confluence, whereas phosphomimetic variants (S1055D and S1055E) potentiated YAP1 nuclear exit (see Fig EV5B for representative images).
- Genome‐edited XPO1 mutant HAP1 cells were transiently transfected with siRNA targeting endogenous STK38 (siSTK38#206 or with non‐targeting siRNA (siNT)) at low or high confluence. Seventy‐two hours later, cells were fixed and stained for endogenous YAP1. Quantitative representation of YAP1 nuclear fluorescence intensity (n = 150 cells from two independent experiments). Here, phosphomimetic XPO1 (S1055D) potentiated YAP1 nuclear exit at both low and high confluences even in the absence of STK38 as compared to cells carrying wt XPO1 (see Fig EV5C for representative images and Appendix Fig S7D for STK38 silencing).
Figure EV5. YAP1 subcellular localization (related to Fig 7).
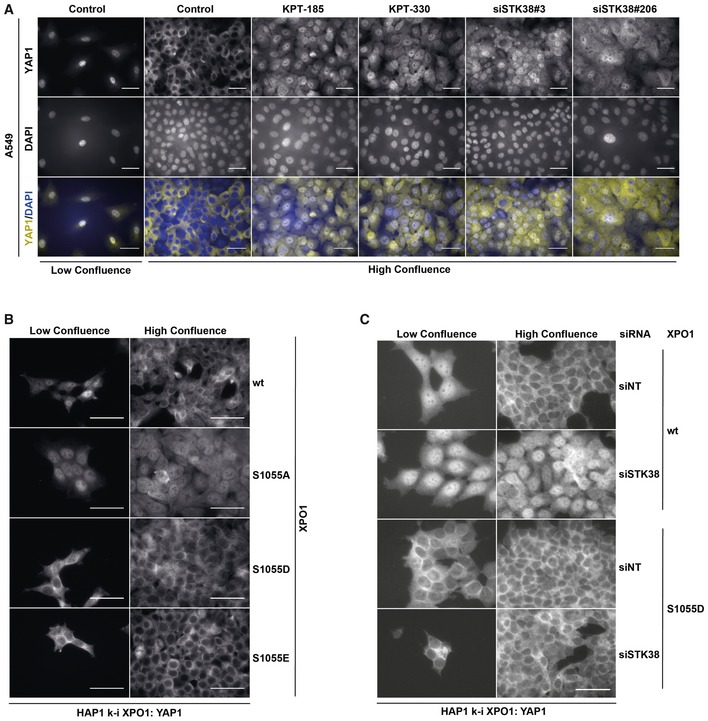
- A549 cells were transiently transfected with the indicated siRNA (control and KPT conditions = siNT) at low or high confluence. Forty‐eight hours later, cells were incubated overnight in the presence of XPO1 inhibitors KPT‐185 and KPT‐330 (final concentration = 1 μM) or with DMSO for all other conditions. The next day, cells were fixed and stained for endogenous YAP1. Representative images are shown, and scale bars are 40 μm (see Appendix Fig S7A for STK38 silencing).
- Genome‐edited XPO1 HAP1 cells were cultured for 2 days at low versus high confluency. Cells were then fixed and stained for endogenous YAP1. Representative images are shown, and scale bars are 40 μm.
- Genome‐edited XPO1 mutant HAP1 cells were transiently transfected with siRNA targeting endogenous STK38 (siSTK38#206 or with non‐targeting siRNA (siNT)) at low or high confluence. Seventy‐two hours later, cells were fixed and stained for endogenous YAP1. Representative images are shown, and scale bars are 40 μm (see Appendix Fig S7D for STK38 silencing).
As an independent approach, we analyzed YAP1 localization in our modified HAP1 cells. YAP1 was excluded from the nucleus in confluent wild‐type HAP1 cells (Figs 7B and EV5B), while YAP1 protein level remained the same between experimental conditions (Appendix Fig S7C). However, cells expressing XPO1(S1055A) failed to induce YAP1 nuclear exclusion at high cell density, whereas cells expressing phosphomimetic S1055 mutants (S1055D or S1055E) induced YAP1 nuclear exclusion, even at low cell density (Figs 7B and EV5B). These results suggest that the subcellular localization of YAP1 can be regulated by phosphorylation of XPO1 by STK38 and somehow support the extension (if not cargo‐wide generalization) of our model to several cargoes. Here again, a phosphomimetic allele of XPO1 (S1055D) suppressed the impact of depletion of STK38 on YAP1 nuclear export (Figs 7C and EV5C). The accumulation of YAP1 in the cytoplasm is dependent on its binding to 14‐3‐3, driven by phosphorylation of YAP1 on S127. Indeed, S127 of YAP1 is phosphorylated in our settings, but this phosphorylation is dependent on LATS1/2 but not on STK38 (Appendix Fig S7E and F). This result suggests that STK38 and LATS1/2 are mobilized for efficient YAP1 nuclear exit: STK38 is needed to activate XPO1 for YAP1 nuclear export per se and LATS1/2 creates an effective 14‐3‐3 binding site that will sequester YAP1 in the cytoplasm.
Discussion
We have shown recently that the kinase STK38 is permissive for nutrient starvation‐induced autophagy 8 and for anoïkis resistance of Ras‐transformed cells 9, adding these functions to a long list of functions where STK38 has been implicated. The STK38 kinase is a core component of the Hippo pathway which controls cellular processes such as stress response 7, cell cycle progression 2, centrosome duplication 4, and NF‐κB activation upon different contexts 44, 45.
For starvation‐induced autophagy and the latter functions, which partner mediates STK38's action remains elusive: We sought to identify these partners with special emphasis on starvation‐induced autophagy and anoïkis resistance. One underlying model would postulate that STK38's diversity of functions is carried by a diversity of partners: function‐specific partners and/or substrates phosphorylated by STK38. Our findings demonstrate that at least for starvation‐induced autophagy, Hippo regulation, centrosome duplication, and NF‐κB activation, one unique substrate of STK38 is the limiting factor of these events, namely the nuclear exportin XPO1.
We found that STK38 phosphorylates XPO1 on its auto‐inhibitory domain and that phosphorylation of XPO1 on S1055 is important in diverse cellular contexts for the nuclear export of crucial intracellular signal transducers such as Beclin1 and YAP1, as well as of Centrin1 (Appendix Fig S8). In this regard, we hypothesize that phosphorylation of S1055 by STK38 induces a change in XPO1 conformation in such a way that the C‐terminal domain, which hinders access to XPO1's NES‐binding pocket in its inactivated state, relocates and frees the cargo binding site, allowing the binding of the cargo to XPO1 for nuclear export (Appendix Fig S9).
The C‐terminal end of XPO1 protein sequence is highly conserved among all chordates (Appendix Fig S10), including the S1055 site. However, the consensus STK38 HxRxxS/T phosphorylation motif appears only in simians but not in all other vertebrates (including non‐simian primates and all the usual model organisms like mouse, xenopus, and zebrafish) which carry a HxLxxS/T motif. The question raised by this observation is whether in these organisms the response to these contexts is regulated by a STK38‐like kinase or another post‐translational modification that would relieve the auto‐inhibition that locks XPO1 in an inactivated state.
The phenomena revealed by this work suggest also that the auto‐inhibition embedded within the structure of XPO1 is not anecdotic but necessary for its proper function and responsiveness to physiological clues. Once XPO1 gets inappropriately activated, it starts an improper behavior disconnected of cell physiology. In rich medium, it triggers early events of autophagy that are supposed to take place only upon starvation. In contrast, in cells with the capacity to proliferate, XPO1 kicks YAP1 out of the nucleus, while nuclear YAP1 is an important pro‐proliferative regulator.
Phosphorylation of XPO1 on S1055 by STK38 is important for the nuclear export of XPO1 cargoes implicated in STK38‐related functions. This allows subtle cellular responses in a context‐dependent manner by modulating the nuclear export of crucial regulators. Although we demonstrated here that Beclin1 and YAP1 are important STK38‐regulated XPO1 cargoes, it remains to be determined how many cargoes are regulated by this mechanism, if it is strictly circumscribed to STK38‐related functions or if this activation mechanism can be generalized.
Pharmacological inhibition of XPO1 is a therapeutic approach for the treatment of cancer 46. Indeed, recently the first‐in‐class XPO1 small‐molecule oral inhibitor selinexor has been approved for the treatment of patients with relapsed refractory multiple myeloma. The role of XPO1 in cancer progression has been evidenced by the identification of recurrent mutations (E571K) in the hydrophobic cargo binding groove of XPO1 in chronic lymphocytic leukemia and other B‐cell malignancies that may affect XPO1 activity 47. Our results suggest that other mutations in the C‐terminal domain of XPO1 could also influence XPO1 activity. Whether such mutations could be found in human tumors remains to be established as their influence on cancer progression.
Materials and Methods
Antibodies
The following primary antibodies were used in the present study: STK38 (H00011329‐M11, Abnova, Mouse monoclonal), XPO1 (sc‐374124, Santa Cruz, Mouse monoclonal), YAP1 (sc‐101199, Santa Cruz, Mouse monoclonal), P‐YAP1(S127) (4911, Cell Signaling, Rabbit polyclonal), Beclin1 (for IF) (sc‐10086, Santa Cruz, Goat polyclonal), Beclin1 (for WB) (3738, Cell Signaling, Rabbit polyclonal), Flag (14793, Cell Signaling, Rabbit polyclonal), myc‐tag (2272, Cell Signaling, Rabbit polyclonal), LC3b (2775, Cell Signaling, Rabbit polyclonal), IκBα (4814, Cell Signaling, Mouse monoclonal), ATG5 (2630, Cell Signaling, Rabbit polyclonal), LATS1 (3477, Cell Signaling, Rabbit monoclonal), LATS2 (5888, Cell Signaling, Rabbit monoclonal), HA (11583816001, Roche, Mouse monoclonal), GAPDH (MAB374, Merck Millipore, Mouse monoclonal), Actin (A2228, Sigma‐Aldrich, Mouse monoclonal), p62/SQSTM1 (M162‐3, MBL, Mouse monoclonal), and P‐STK38(T444) (Rabbit polyclonal) 48. The anti‐pS1055_XPO1 antibody (Davids Biotechnologie) was generated in guinea pigs immunized with specific phosphopeptide mimicking phosphorylated XPO1 S1055. After purification of antibodies with the same phosphopeptide, non‐specific antibodies were eliminated using non‐phosphopeptide.
Chemicals and reagents
Phenol–biotin was purchased from Iris Biotech (LS‐3500 0250). Streptavidin–HRP was purchased from Life Technologies (21126). Both XPO1 inhibitors (KPT‐185 (S7125) and KPT‐330 (S7252)) were purchased from Selleckchem. Okadaic acid was purchased from Santa Cruz (78111). Chloroquine (C6628), H2O2 (H1009), Biotin (B4501), and Flag M2 magnetic beads (M8823) were purchased from Sigma‐Aldrich. Streptavidin magnetic beads (88817) and protein A magnetic beads (10001D) were purchased from Thermo.
Cell culture and drug treatments
Cells were cultured in humidified chambers at 37°C and 5% CO2. HeLa 8, HEK293T (CRL‐3216; ATCC), and A549 (CCL‐185; ATCC) cell lines were grown in DMEM (Gibco) supplemented with 10% FBS (Biosera), 1% penicillin/streptomycin (Gibco), and with 1% l‐glutamine (Gibco). HEK‐HT and HEK‐HT‐HRasG12V (HekRasV12) cells were provided by Christopher Counter 11, 12 and cultured in DMEM supplemented with 100 μg/ml hygromycin and 400 μg/ml geneticin for both of them and an additional 300 μg/μl Zeocin for HekRasV12 cells. Cells stably expressing APEX‐STK38 (pWZL‐APEX2‐STK38; this work) were cultured in appropriate medium supplemented with 10 μg/ml blasticidin. HeLa cells stably expressing the GFP‐LC3‐RFP‐LC3ΔG autophagic flux probe (pMRX‐IP‐GFP‐LC3‐RFP‐LC3ΔG, 84572; Addgene) as well as HEK‐HT‐iRFP‐LC3 cells 49 were cultured in the appropriate medium supplemented with 1 μg/ml puromycin. HAP1 cell lines (C631; Horizon Discovery) were grown in IMDM (Gibco) supplemented with 10% FBS (Biosera), 1% penicillin/streptomycin (Gibco), and with 1% l‐glutamine (Gibco). Knock‐in HAP1 cell lines expressing XPO1 mutants were cultured with the same media described above supplemented with 1 μg/ml puromycin. All antibiotics were from Invivogen.
For okadaic acid (OA) treatments, cells were cultured for 1 h in the appropriate culture media supplemented with 1 μm okadaic acid purchased from Santa Cruz. For EBSS‐induced autophagy, cells were grown in order to reach a maximum of 70% confluency. On the day of the experiment, cells were washed once with PBS followed by incubation with growth medium (GM) or EBSS (24010‐43; Gibco) for 4 h as indicated. For chemical XPO1 inhibition, cells were cultured as indicated in the specific sections with media supplemented with 1 μM of KPT‐185 or KPT‐330 (Selleckchem).
siRNA and DNA transfections
Cells were reversely transfected with siRNAs using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Seventy‐two hours post‐transfection, cells were harvested as defined in the specific sections. For transient DNA transfection, cells were seeded and then transfected the next day with a total amount of 2 μg of DNA using jetPRIME reagent (Polyplus Transfection) according to the manufacturer's instructions. Forty‐eight hours post‐transfection, cells were harvested as defined in the specific sections. If both methods were used, the cells were reversely transfected with siRNA as described above on the first day and then transfected with DNA as described above the next day. Cells were harvested 24 h after DNA transfection as defined in the specific sections. pcDNA3‐Myc‐STK38(wt/K118R) and pcDNA3‐HA‐STK38(PIF) expression plasmids were a gift from Alex Hergovich 48, and pCIG3‐3xFlag‐XPO1(wt/mutants) were generated for this study. siControl ON‐TARGETplus Non‐targeting siRNA (referred to as siNT) was purchased from Dharmacon (D‐001810‐01‐50; Dharmacon). The remaining siRNAs were purchased from Eurogentec:
STK38#3: 5′‐GAGCAGGUUGGCCACAUUCdTT‐3′
STK38#206: 5′‐CGUCGGCCAUAAACAGCUAdTT‐3′
ATG5: 5′‐AACCUUUGGCCUAAGAAGAAAdTT‐3′
Beclin1: 5′‐ACCGACUUGUUCCUUACGGAAdTT‐3′
LATS1: 5′‐AAACUUUGCCGAGGACCCGAAdTT‐3′
LATS2: 5′‐GCCUUUGGAGAAGUGUGCCUUGCUdTT‐3′.
Generation of stable cell lines
HeLa and HekRasV12 cell lines stably expressing the APEX2‐STK38 N‐terminal fusion protein as well as the HeLa cells stably expressing the GFP‐LC3‐RFP‐LC3ΔG autophagic flux reporter probe 26 were generated by retroviral transduction. For retroviral production, HEK293T packaging cells were transfected with the retroviral vector pWZL‐APEX2‐STK38 or with the pMRX‐IP‐GFP‐LC3‐RFP‐LC3ΔG retroviral vector (84572; Addgene) and the packaging plasmid pcl10A1 (NBP2‐29542; Novus) using FuGENE 6 (E2691; Promega) according to the manufacturer's instructions. Forty‐eight hours after transfection, supernatants were harvested, filtered through a 0.45‐μm filter, and added to the recipient cells in the presence of 4 μg/ml polybrene (Sigma). Twenty‐four hours post‐transduction, cells were harvested and seeded in fresh culture dishes and subsequently selected with appropriate antibiotics for 1 week.
Following antibiotic selection, HeLa cells stably expressing the GFP‐LC3‐RFP‐LC3ΔG autophagic probe were subjected to clonal selection. Briefly, cells were washed twice with PBS and incubated with TrypLE Express (Gibco) according to the manufacturer's instructions and then resuspended in PBS supplemented with 25 mM HEPES (Gibco), 1 mM EDTA (Invitrogen), and 1% BSA (Euromedex) previously filtered through a 0.45‐μm filter. Cells positive for both GFP and RFP were then sorted individually on a BD FACSAria III in a 96‐well plate and incubated for several weeks with DMEM supplemented with antibiotic selection (1 μg/ml puromycin). One clone was chosen according to GFP and RFP expression and cultured under antibiotic selection in order to use it for experiments.
CRISPR/Cas9 genome editing, DNA extraction, and sequencing
Genome‐edited HAP1 cell lines were generated using the CRISPaint principle 50 and similarly as described before 51. Briefly, cells were transfected using TurboFectin 8.0 (Origene) according to the manufacturer's instructions with a plasmid encoding an sgRNA targeting the C‐terminus of XPO1 (5′‐GAGAGAAATAGCCCTACGGC‐3′), a plasmid encoding SpCas9 and an sgRNA targeting the donor plasmid (5′‐GCCAGTACCCAAAAAGCGCC‐3′), and a repair donor plasmid. This repair plasmid contains the targeting site, the sequence to restore the C‐terminus of XPO1 with a silent mutation upstream of the endogenous C‐terminal PAM sequence, the desired S1055 mutation, and 3xFLAG followed by a P2A‐coupled puromycin resistance gene to stably integrate the mutation in the last exon of Xpo1 at its endogenous locus. Following transfection, cells were incubated for 2 days and then selected over a period of 1 week with 1 μg/ml puromycin. Cells were harvested and plated at a density of 0.5 cells/well in 96‐well plates in 20% FBS‐containing medium to obtain single‐cell‐derived colonies. Colonies were grown for 2–4 weeks and were regularly screened.
When single‐cell‐derived colonies were sufficiently grown, cells were washed and then lysed in Bradley lysis buffer at 56°C (10 mM Tris–HCl (pH 7.5), 10 mM EDTA, 0.5% SDS, 10 mM NaCl, and 1 μg/ml proteinase K). The genomic DNA was extracted from the lysate using ethanol–salt precipitation and centrifugation. The C‐terminus of XPO1 was amplified by in–out PCR with the following primers: fwd: 5′‐CTCAAGTAAAGCTCTTTGTGACAGGGC‐3′; rv: 5′‐CAGCCATTCTCGGGCCGATC‐3′. The PCR product was sequenced with the reverse primer 5′‐GGAACGTCGTCTCTTGTAGC‐3′ by Sanger sequencing for correct integration of the mutation (Macrogen).
SILAC
HeLa and HekRasV12 cell lines stably expressing the APEX2‐STK38 construct were metabolically labeled with light or heavy isotopes of arginine and lysine. Early passage of each cell was split on day 0 into two T25 flasks (Falcon). One flask was cultured in light SILAC media consisting of DMEM deficient in l‐lysine and l‐arginine (#89985; Thermo) that we supplemented with l‐lysine (Lys0) and l‐arginine (Arg0) (L8662 and A8094, respectively; Sigma) at 146 and 84 mg/l, respectively; 10% FBS (Biosera); 1% Pen/Strep (Gibco); and 1% glutamine (Gibco). The other flask was cultured in heavy SILAC media as above except Lys0 and Arg0 that were replaced by l‐lysine‐13C6,15N2 (Lys8) and L‐arginine‐13C6,15N4 (Arg10) (88209 and 89990, respectively; Thermo). Every 4 days, before cells reached confluency, the heavy and light SILAC cultures were split into fresh SILAC heavy and light media, respectively. After 8 passages, the heavy and light SILAC cultures were expanded in 10‐cm petri dishes and cultured for two more passages. When heavy and light SILAC HeLa and HekRasV12 cells reached passage number 10, a fraction of each condition was sent to mass spectrometry analysis to confirm that amino acids have been well substituted by the heavy or light ones.
Context‐dependent proximity biotinylation assay
For the context‐dependent proximity biotinylation assay (PBA), we first induced the context in both heavy and light isotopic cell lines stably expressing the APEX2‐STK38 construct (autophagy for Hela cells and suspension for HekRasV12 cells), then to proceed to the proximity labeling and finally to purify biotinylated proteins.
Light and heavy SILAC HeLa APEX2‐STK38 cells were seeded in 10‐cm petri dishes in order to obtain 80% confluence. On the day of the experiment, cells were washed twice with PBS (Gibco) and then incubated in the presence of DMEM for the light SILAC condition or in the presence of EBSS (24010‐043, Life Technologies) for the heavy SILAC condition during 4 h. Light and heavy SILAC HekRasV12 APEX2‐STK38 cells were seeded to obtain 80% confluence the next day in attached (normal 10‐cm petri dish growth condition) for the light SILAC condition or in suspension (see below) for the heavy SILAC condition and cultured overnight. For suspension culture, cells were grown overnight in Ultra‐Low Attachment Surface 6‐well plates or 10‐cm dishes (Greiner) that were previously incubated in 0.02% pluronic acid‐coated for 1 h at 37°C followed by two washes with PBS.
On the day of the PBA, cells were pre‐incubated for 30 min with phenol–biotin (LS‐3500 0250, Iris Biotech) at a concentration of 500 μM at 37°C before the addition of 1 mM of H2O2 (H1009; Sigma) during 1 min at room temperature. The cells were immediately quenched by two following washes of PBS supplemented with 10 mM sodium azide (S2002; Sigma), 10 mM sodium ascorbate (A7631; Sigma), and 5 mM Trolox (238813; Sigma) followed by two washes of PBS (10 ml at each time) allowing the removal of the phenol–biotin/H2O2 solution. Attached cells were then collected using TrypLE Express (Gibco) as indicated by the manufacturer's instructions followed by centrifugation at 300 g during 5 min. Cell pellets were then lysed on ice by adding 1 ml of freshly prepared RIPA lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X‐100, 1× cOmplete EDTA‐free protease inhibitor cocktail (05892791001, Roche)) supplemented with 1 mM PMSF, 10 mM sodium azide, 10 mM sodium ascorbate, and 5 mM Trolox. Lysates were then incubated on tube rotator for 20 min at 4°C followed by centrifugation at 14,000 g for 10 min at 4°C. Supernatants were separated from pellets and then directly flash‐frozen in liquid nitrogen before being stored at −80°C. Protein concentration was measured using the Pierce BCA protein assay kit according to the manufacturer's instructions (23225; Thermo) with bovine serum albumin as standard, and the absorbance at 262 nm was recorded on a FLUOstar OPTIMA plate reader (BMG Labtech).
Samples from the lysed SILAC cells were combined in a 1:1 ratio (2 mg of total protein) as indicated in Fig EV1 for the autophagy condition and incubated for 1 h at 4°C in the presence of protein A magnetic beads (10001D, Thermo) on a tube rotor in order to perform a pre‐clear. Non‐bound fractions were then incubated in the presence of 500 μl of streptavidin magnetic beads (88817; Thermo) previously washed as for the protein A magnetic beads and then incubated for 1 h at 4°C on a tube rotor. Streptavidin beads were then washed two times with 1 ml of the same lysis buffer described above, one time with 1 ml of KCl 1 M, one time with 1 ml of Na2CO3 0.1 M, one time with 1 ml of 2 M urea in 10 mM Tris–HCl pH 8.0, and then two times with lysis buffer. Biotinylated proteins were then eluted by incubating the beads with 60 μl of 1× Laemmli sample buffer (1610747; Bio‐Rad) supplemented with 50 mM DTT and 2 mM biotin (B4501; Sigma) and by heating them at 95°C for 5 min.
Sample preparation and digestion
Immunoprecipitates were prepared as described above. After the final elution in 1× reducing Laemmli buffer, the samples were proceeded for digestion using the filter‐assisted sample preparation (FASP) method, performed essentially as described 52, 53. Briefly, protein extracts were applied to 30‐kDa MWCO centrifugal filter units (Microcon; Millipore), mixed with UA buffer (8M urea, 100 mM Tris–HCl pH 8.9), and centrifuged. Alkylation was carried out by incubation for 20 min in the dark with UA buffer containing 50 mM iodoacetamide. Filters were then washed twice with UA buffer followed by two washes with ABC buffer (50 mM ammonium bicarbonate). Finally, 1 μg of trypsin (Promega, France) was added and digestion was achieved by overnight incubation at 37°C.
After recovery, peptides were subjected to the detergent removal procedure using HiPPR (High Protein and Peptide Recovery) Detergent Removal Resin from Thermo Fisher Scientific, as recommended by the manufacturer. Briefly, the spin columns containing detergent removal resin were centrifuged at 1,500 g for 1 min to remove the storage buffer. The resin was washed three times by adding ABC buffer and centrifuging at 1,500 g for 1 min. Samples were then added to resin in a vol:vol ratio and incubated for 10 min at room temperature. The spin columns were placed in a collection tube and centrifuged at 1,500 g for 2 min to collect the detergent‐free sample. Peptides were vacuum‐dried and resuspended in 10% acetonitrile and 0.1% formic acid for LC‐MS/MS.
Immunoprecipitates for the phosphoprotein analysis were run on a precast 4–15% 1D SDS–PAGE gel. Gel bands at 120 kDa were excised, washed, and dehydrated with 100% acetonitrile. Proteins inside the gel were reduced, alkylated, and finally digested with trypsin for 16 h at 37°C. Peptides were extracted with 50% acetonitrile and 1% trifluoroacetic acid followed by 100% acetonitrile. Peptides were vacuum‐dried and resuspended in 20 μl of 10% acetonitrile and 0.1% trifluoroacetic acid for titanium dioxide purification. Phosphopeptide enrichments by titanium dioxide were carried out using Titansphere TiO2 Spin tips (3 mg/200 μl, Titansphere PHOS‐TiO; GL Sciences Inc., Japan) according to the manufacturer's protocol. After phosphopeptide elution from the TiO2 Spin tips, GC Spin Tips (GL‐Tip, Titansphere; GL Sciences Inc., Japan) were used to purify the phosphopeptides according to the manufacturer's instructions. Finally, phosphopeptides were eluted with 70 μl of 80% acetonitrile and 0.1% TFA (1,000 g for 5 min) and vacuum‐dried.
Mass spectrometry
For each run, 1 μl was injected in a nanoRSLC‐Q Exactive PLUS (Dionex RSLC Ultimate 3000; Thermo Scientific, Waltham, MA, USA). Extracted peptides were resuspended in 0.1% (v/v) trifluoroacetic acid and 10% acetonitrile and were loaded onto a μ‐precolumn (Acclaim PepMap 100 C18, cartridge, 300 μm i.d. × 5 mm, 5 μm; Dionex), followed by separation on the analytical 50‐cm nanocolumn (0.075 mm ID, Acclaim PepMap 100, C18, 2 μm; Dionex). Chromatography solvents were (A) 0.1% formic acid in water and (B) 80% acetonitrile and 0.08% formic acid. Peptides were eluted from the column using a gradient from 5 to 40% B over 38 min and were analyzed by data‐dependent MS/MS, using the top‐10 acquisition method. Briefly, the instrument settings were as follows: Resolution was set to 70,000 for MS scans and 17,500 for the data‐dependent MS/MS scans in order to increase the speed. The MS AGC target was set to 3.106 counts with a maximum injection time of 200 ms, while MS/MS AGC target was set to 1.105 with a maximum injection time of 120 ms. Dynamic exclusion was set to 30 s. Each sample was analyzed in three to five biological replicates. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 54 partner repository with the dataset identifier PXD011968.
Peptide mixtures were also analyzed with a nanoElute UHPLC (Bruker) coupled to a timsTOF Pro mass spectrometer (Bruker). Peptides were first washed using a C18 PepMap 100 precolumn (5 mm, 300 μm i.d., 100 Å, 5 μm; Thermo) with mobile phase A (ACN 2%/FA 0.1%) before being separated on an analytical column RP‐C18 Odyssey (25 cm, 75 μm i.d., 120 Å, 1.6 μm; IonOpticks) at a flow rate of 400 nl/min, at 50°C, with mobile phases A (ACN 2%/FA 0.1%) and B (ACN 99.9%/FA 0.1%). Elution gradient was run from 2 to 35% B in 15 min and then from 35 to 95% B in 8 min.
MS acquisition was run in DDA mode with PASEF. Accumulation time was set to 100 ms in the TIMS tunnel. Capillary voltage was set to 1.4 kV, and mass ranged from 100 to 1,700 m/z in MS and MS/MS. A collision energy stepping was applied during each TIMS‐MS/MS separation event: 51 ev for the first 19 ms, 47 ev from 19 to 38 ms, then 42 ev between 38 and 57 ms, further lowered to 37 eV between 57 and 76 ms, and raised back to 42 ev between 76 and 95 ms. The quadrupole isolation width was 2 Th below 800 m/z, and 3 Th otherwise. Dynamic exclusion was activated for ions within 0.015 m/z and 0.015 V.s/cm2 and released after 0.4 min. Exclusion was reconsidered if precursor ion intensity was 4 times superior. Low‐abundance precursors below the target value of 20,000 a.u. and intensity of 2,500 a.u. were selected several times for PASEF‐MS/MS until the target value was reached. Parent ion selection was achieved by using a two‐dimensional m/z and 1/k0 selection area filter allowing the exclusion of singly charged ions. Total cycle time was 1.15 s with 10 PASEF cycles.
Data processing following LC‐MS/MS acquisition
Raw MS files were processed with the MaxQuant software version 1.5.3.30 and searched with Andromeda search engine against the Homo Sapiens UniProtKB/Swiss‐Prot v.06/2016. To search parent mass and fragment ions, we set an initial mass deviation of 4.5 ppm and 20 ppm, respectively. The minimum peptide length was set to 7 amino acids and strict specificity for trypsin cleavage was required, allowing up to two missed cleavage sites. Carbamidomethylation (Cys) was set as fixed modification, whereas oxidation (Met) and N‐term acetylation were set as variable modifications. The false discovery rates (FDRs) at the protein and peptide levels were set to 1%. Scores were calculated in MaxQuant as described previously 55. The reverse and common contaminant hits were removed from MaxQuant output. Proteins were quantified on parent ions, selecting multiplicity “2” for standard quantification in SILAC; the heavy label was Arg10 and Lys8, while the light label corresponded to non‐labeled Arg and Lys. The maximum value of labeled amino acids per peptide was set to 3. Protein quantification was obtained using at least 2 peptides ration counts per protein.
Three biological replicates were analyzed for each condition studied. Statistical analysis was performed with Perseus software (version 1.5.5.3) freely available at www.perseus-framework.org. The matrix was filtered to keep proteins quantified at least three times in at least one condition. Statistical analysis was performed on each condition (group of three biological replicates) by one‐sample test against the value 1, and we retained the proteins significative according to t‐test (S0 = 1, P‐value < 0.05). Subsequently, we applied a personalized priority scoring in order to highlight the proteins that were changing the most and more significant priority score = (1∕P‐value × (<ratio > −1)2).
Mascot search engine (version 2.2.07; Matrix Science) was used to identify the phosphorylated XPO1. Default parameters used were as follows: Fixed modification (Carbamidomethyl (C)) and variable modification (Oxidation (M)) were allowed as well as Phospho (STY) and Gln‐>pyro‐Glu (N‐term Q) with one missed cleavage. Enzyme was trypsin, monoisotopic peptide mass tolerance was ± 5 ppm (after linear recalibration), and fragment mass tolerance was ± 0.05 Da.
Western blot
After the indicated treatments, cells were washed with ice‐cold PBS and lysed at 4°C in freshly made lysis buffer (20 mM Tris–HCl pH = 8, 150 mM NaCl, 10% glycerol, 1% Triton X‐100, 1 mM EDTA, 1 mM PMSF, 50 mM NaF, 1 mM DTT, 1 mM Na3VO4, protease inhibitor cocktail). Lysates were incubated for 20 min at 4°C on a rotary wheel and then subjected to centrifugation at 13,000 g for 10 min at 4°C. Sample supernatants were resuspended in Laemmli buffer (2% SDS, 60 mM Tris–HCl pH = 6.8, 10% glycerol, 100 mM DTT, and 0.005% bromophenol blue) followed by boiling at 95°C for 5 min. An equal amount of protein was run on NuPAGE precast gradient SDS–PAGE gels (Life technologies). After protein separation by electrophoresis, proteins were transferred on a 0.2‐μm nitrocellulose transfer membrane (GE Healthcare). Non‐specific sites were blocked for 1 h in TBS–Tween (TBST) 0.2% with 5% BSA at room temperature. Membranes were then incubated overnight at 4°C under gentle agitation with appropriate primary antibodies diluted in TBST–BSA 5%. The next day, membranes were washed 3 times with TBST at room temperature (RT) and incubated with appropriate secondary antibodies for 1 h at RT under gentle agitation prior to washes as described before. Luminescent signal was visualized and recorded with the enhanced chemiluminescence method (Western Lightning Plus‐ECL; PerkinElmer) using horseradish peroxidase (HRP)‐conjugated secondary antibodies (Jackson ImmunoResearch) followed by image acquisition using a ChemiDoc MP imaging system (Bio‐Rad) or with the LI‐COR Odyssey Infrared Imaging System (LI‐COR Biosciences) using IRDye‐conjugated secondary antibodies.
Immunoprecipitations
For immunoprecipitation experiments, cells were grown in 10‐cm petri dishes in order to reach 80% confluency on the day of the experiment. After the indicated treatments, cells were washed with ice‐cold PBS and lysed at 4°C using the same lysis buffer as described in the Western Blot section. Lysates were incubated for 20 min at 4°C on a rotary wheel and then subjected to centrifugation at 13,000 g for 10 min at 4°C. Supernatant was then incubated for 1 h at 4°C in the presence of protein A magnetic beads (10001D; Thermo) on a tube rotor in order to perform a pre‐clear. Non‐bound fractions were then incubated in the presence of anti‐Flag M2 magnetic beads (M8823; Sigma) for 1 h at 4°C on a tube rotor. Magnetic beads were then washed two times with the same lysis buffer, one time with high‐salt lysis buffer (300 mM NaCl) and one time with normal lysis buffer. Magnetic beads were then resuspended in Laemmli buffer and then heated at 95°C for 5 min for elution.
Flow cytometry
To determine the autophagic flux in HeLa GFP‐LC3‐RFP‐LC3ΔG cells, cells were seeded in 96‐well view plate (PerkinElmer) in order to reach 80% confluence on the day of acquisition. Cells were subjected to treatment as indicated and described above. Cells were resuspended using phenol red lacking trypsin–EDTA (59418C; Sigma) prior to being quenched using Defined Trypsin Inhibitor (R007100; Thermo). All data were acquired on a CytoFLEX (Beckman Coulter) and analyzed using FlowJo (LLC).
Immunofluorescence and image analysis
Cells were grown on coverslips, washed twice in PBS, fixed with 4% paraformaldehyde for 5 min at room temperature (RT) followed by three washes in PBS, and then quenched with 50 mM NH4Cl in PBS for 15 min at RT. After three PBS washes, if labeling using primary antibody was performed, fixed cells were permeabilized in 0.1% Triton X‐100 in PBS for 10 min at RT followed by three washes in PBS and then blocked in 5% BSA and 10% FBS in PBS for 30 min at RT. Cells were subsequently incubated for 1 h at RT with primary antibodies diluted in blocking buffer. After three washes in PBS, cells were incubated with secondary antibodies diluted in blocking buffer for 1 h at RT followed by three washes in PBS. Coverslips were dried and then mounted with ProLong™ Gold Antifade Mountant with DAPI (P36941; Thermo). Images were acquired on a Zeiss Axioplan 2 microscope through a CoolSNAP HQ2 camera (Photometrics) under the control of MetaMorph software (Universal Imaging). Cells were selected for acquisition if they were positive for all staining.
Image processing was performed using ImageJ software (NIH). For nucleo‐/cytoplasmic ratio, the fluorescent signal was quantified in the nuclear and cytosolic compartments. The nucleus was identified by DAPI staining and the cell area by thresholding the background. The cytosolic area was determined by subtracting the nuclear compartment to the cell compartment. The ratio was obtained by dividing the mean of fluorescence in the cytosolic area by the mean of fluorescence in the nuclear one. For LC3 dot assay, dots were recorded using the iRFP channel. After background subtraction, the number of iRFP‐LC3 dots per cell was calculated. For YAP1 nuclear intensity, the mean of fluorescence intensity was calculated only in the nuclear area identified by the DAPI staining.
Snapshots of live cells: centrosomes
For centrosome experiment, HeLa cells stably expressing GFP‐Centrin were cultured on glass‐bottom 6‐well plates (MatTek) and incubated with the siRNAs or drugs as indicated in the figure legend. On the day of the experiment, the GFP signal was recorded using a Zeiss AxioObserver Z1 microscope through a Hamamatsu Flash4 camera under the control of MetaMorph software (Universal Imaging). The number of centrosomes was counted manually on Z‐stack images by counting the number of centrosomes (Centrin “spots”) without differentiating unique separated centrioles in the G1 phase from separated centrosomes (harboring 2 centrioles each) in the S/G2 phase. Image processing was performed using ImageJ software (NIH).
Bioinformatics and statistical analysis
Heatmap of STK38 interactor association fold was generated using the online tool Morpheus from the Broad Institute (https://software.broadinstitute.org/morpheus). Rows and columns were clustered using the hierarchal clustering tool in Morpheus by the one‐minus‐Pearson correlation matrix and the average linking method. Enrichment analysis was performed using Gene Ontology (David Bioinformatics) website (http://www.geneontology.org/) for enriched terms as cellular components in Homo sapiens. The 10 most enriched terms according to their adjusted P‐value were plotted. Adjusted P‐value was calculated using the Bonferroni correction for multiple testing. Correlogram was generated using the “corrplot” package in R.
Statistical significance was quantified by P‐values using GraphPad Prism v5.0 software. Student's t‐test was used if data followed normal distribution; otherwise, the Mann–Whitney test was used. All tests performed were two‐sided. For all tests, differences were considered statistically significant when P‐values were below 0.05 (*), 0.01 (**), or 0.001 (***). In the figures, P‐values are indicated as follows: *P < 0.05; **P < 0.01; ***P < 0.001; and ns, not significant. Graphs represent mean ± standard error of the mean (SEM).
Protein sequence alignment
Protein sequences were retrieved from UniProt (https://www.uniprot.org/) as FASTA files, aligned in msa R package {https://doi.org/10.1093/bioinformatics/btv494} using the Clustal Omega method {https://doi.org/10.1038/msb.2011.75} with default parameters. The following sequences were used: Human: O14980 (Homo sapiens); Chimpanzee: H2R0K9 (Pan troglodytes); Gibbon: G1RF15 (Nomascus leucogenys); Cercocebus: A0A2K5LXU0 (Cercocebus atys); Drill: A0A2K5YJR1 (Mandrillus leucophaeus); Colobus: A0A2K5I3H1 (Colobus angolensis); Capuchin: A0A2K5SFU2 (Cebus capucinus); Saimiri: A0A2K6UYF0 (Saimiri boliviensis); Tarsier: A0A1U7TKR2 (Tarsius syrichta); Sifaka: A0A2K6G794 (Propithecus coquereli); Galago: H0WFU2 (Otolemur garnettii); Rabbit: G1SMY6 (Oryctolagus cuniculus); Beaver: A0A250Y6Q4 (Castor canadensis); Guinea_pig: A0A286XB55 (Cavia porcellus); Mole_rat: A0A0P6K7E8 (Heterocephalus glaber); Hamster: A0A1U7QPJ1 (Mesocricetus auratus); Mouse: Q6P5F9 (Mus musculus); Rat: Q80U96 (Rattus norvegicus); Ground_squirrel: A0A287CSQ9 (Ictidomys tridecemlineatus); Tupaia: L9KQ84 (Tupaia chinensis); Panda: D2HZX2 (Ailuropoda melanoleuca); Dog: E2R9K4 (Canis lupus); Horse: F6S8L9 (Equus caballus); Sheep: W5QG19 (Ovis aries); Pig: A0A218PI30 (Sus scrofa); Bat: G1PGH6 (Myotis lucifugus); Hedgehog: A0A1S3ARI2 (Erinaceus europaeus); Manatee: A0A2Y9RGB7 (Trichechus manatus); Elephant: G3TDG6 (Loxodonta africana); Tasman_devil: G3WCI2 (Sarcophilus harrisii); Opossum: F7EIW1 (Monodelphis domestica); Platypus: F7DTN5 (Ornithorhynchus anatinus); Chicken: A0A1D5P8H7 (Gallus gallus); Owl: A0A093FS26 (Tyto alba); Alligator: A0A1U7RM64 (Alligator sinensis); Green_turtle: M7B3V0 (Chelonia mydas); Copperhead: A0A1W7RHI2 (Agkistrodon contortrix); Anole_lizard: H9G6E3 (Anolis carolinensis); Xenopus: A0A1L8G775 (Xenopus laevis); Salmon: A0A1S3PKN6 (Salmo salar); Zebrafish: E7FBU7 (Danio rerio); Gar: W5NGS6 (Lepisosteus oculatus); Catfish: W5UJN6 (Ictalurus punctatus); Latimeria: H3BFQ8 (Latimeria chalumnae); Ghostshark: V9K8G4 (Callorhinchus milii); Lamprey: S4R883 (Petromyzon marinus); Ascidia: A0A1W3JKJ9 (Ciona intestinalis); Octopus: A0A0L8GCW5 (Octopus bimaculoides); Sea_cucumber: A0A2G8LJC7 (Stichopus japonicus); Lingula: A0A1S3IRI2 (Lingula unguis); Slug: A0A0B7BCQ8 (Arion vulgaris); Leech: T1FP79 (Helobdella robusta); C_elegans: Q23089 (Caenorhabditis elegans); Flour_beetle: D6X0Q6 (Tribolium castaneum); Black_ant: A0A0J7KFR1 (Lasius niger); Honeybee: A0A087ZMS1 (Apis mellifera); Drosophila: A0A0S0WNN6 (Drosophila melanogaster); Crab: A0A0P4WHK7 (Scylla olivacea); Ixodes: V5I1V1 (Ixodes ricinus); Dictyostelium: Q54EV7 (Dictyostelium discoideum); Arabidopsis: Q9SMV6 (Arabidopsis thaliana); Bakers_yeast: P30822 (Saccharomyces cerevisiae); and Fission_yeast: P14068 (Schizosaccharomyces pombe).
Author contributions
APJM and JHC designed the study and experiments. APJM conducted experiments with the help of BM and NC and analyzed the data. JL and CC prepared the proteomic samples and performed mass spectrometry. ICG performed initial analysis on the proteomic data. APJM and MKS designed ImageJ macro for nuclear/cytoplasmic IF quantifications. MJ and DD performed the CRISPR/Cas9 genome editing and provided expertise on XPO1. VNA performed the protein sequence alignment. MP, PC, AH, and GZ assisted expertly centrosome, autophagy, STK38, and YAP1 data, respectively. MCP supervised biochemical experiments. APJM designed the figures. APJM and JHC, with the assistance of AH and DD, wrote the manuscript with input from all authors. All authors approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Acknowledgements
A.PJ.M. was supported by funding from Paris Sciences et Lettres Research University and Institut Curie. M.K.S. was supported by fellowship from Institut national de la Santé et de la Recherche médicale (INSERM ITMO Plan Cancer 2014‐2018, PC201530 to MCP, fellowship to M.K.S.). This work was supported by institutional funding by INSERM and Institut Curie, Association pour la Recherche sur le Cancer (Grant Number 29928 to J.H.C.), Ligue Contre le Cancer (Grant Number 24098 to J.H.C.), Association Christelle Bouillot, and the Fonds Wetenschappelijk Onderzoek (FWO to D.D.). We thank the staff of the Flow Cytometry Core Facility of Institut Curie for their expert support and advice. We thank Alice Y. Ting (MIT) for her generous advices about APEX2 labeling system. We would like to thank Cedric Pionneau (Plateforme P3S, Paris) for the help in the phosphoprotein analysis.
EMBO Reports (2019) 20: e48150
Data availability
The SILAC and APEX2 mass spectrometry proteomic data from this study have been deposited to the ProteomeXchange‐PRIDE repository (https://www.ebi.ac.uk/pride/archive/) and assigned the dataset number PXD011968.
References
- 1. Cornils H, Kohler RS, Hergovich A, Hemmings BA (2011) Downstream of human NDR kinases: impacting on c‐myc and p21 protein stability to control cell cycle progression. Cell Cycle 10: 1897–1904 [DOI] [PubMed] [Google Scholar]
- 2. Cornils H, Kohler RS, Hergovich A, Hemmings BA (2011) Human NDR kinases control G(1)/S cell cycle transition by directly regulating p21 stability. Mol Cell Biol 31: 1382–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vichalkovski A, Gresko E, Cornils H, Hergovich A, Schmitz D, Hemmings BA (2008) NDR kinase is activated by RASSF1A/MST1 in response to FAS receptor stimulation and promotes apoptosis. Curr Biol 18: 1889–1895 [DOI] [PubMed] [Google Scholar]
- 4. Hergovich A, Lamla S, Nigg EA, Hemmings BA (2007) Centrosome‐associated NDR kinase regulates centrosome duplication. Mol Cell 25: 625–634 [DOI] [PubMed] [Google Scholar]
- 5. Hergovich A, Kohler RS, Schmitz D, Vichalkovski A, Cornils H, Hemmings BA (2009) The MST1 and hMOB1 tumor suppressors control human centrosome duplication by regulating NDR kinase phosphorylation. Curr Biol 19: 1692–1702 [DOI] [PubMed] [Google Scholar]
- 6. Zhang L, Tang F, Terracciano L, Hynx D, Kohler R, Bichet S, Hess D, Cron P, Hemmings BA, Hergovich A et al (2015) NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr Biol 25: 296–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Selimoglu R, Bettoun A, Joffre C, Meunier B, Parrini MC, Fesquet D, Formstecher E, Cascone I, Hergovich A, Camonis JH (2014) RalA GTPase and MAP4K4 function through NDR1 activation in stress response and apoptotic signaling. HSOA J Cell Biol Cell Metab 1: 1–11 [Google Scholar]
- 8. Joffre C, Dupont N, Hoa L, Gomez V, Pardo R, Gonçalves‐Pimentel C, Achard P, Bettoun A, Meunier B, Bauvy C et al (2015) The pro‐apoptotic STK38 kinase is a new Beclin1 partner positively regulating autophagy. Curr Biol 25: 2479–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bettoun A, Joffre C, Parrini MC, Gundogdu R, Ahmad AD, Gomez M, Cascone I, Meunier B, White MA, Codogno P et al (2016) Mitochondrial clearance by the STK38 kinase supports oncogenic Ras‐induced cell transformation. Oncotarget 7: 44142–44160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rhee H‐W, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY (2013) Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339: 1328–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Counter CM, Hahn WC, Wei W, Caddle SD, Beijersbergen RL, Lansdorp PM, Sedivy JM, Weinberg RA (1998) Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci USA 95: 14723–14728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA (1999) Creation of human tumour cells with defined genetic elements. Nature 400: 464–468 [DOI] [PubMed] [Google Scholar]
- 13. Campos EI, Smits AH, Kang Y‐H, Landry S, Escobar TM, Nayak S, Ueberheide BM, Durocher D, Vermeulen M, Hurwitz J et al (2015) Analysis of the histone H3.1 interactome: a suitable chaperone for the right event. Mol Cell 60: 697–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Elzi DJ, Song M, Hakala K, Weintraub ST, Shiio Y (2014) Proteomic analysis of the EWS‐Fli‐1 interactome reveals the role of the lysosome in EWS‐Fli‐1 turnover. J Proteome Res 13: 3783–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen R, Wang Y, Liu Y, Zhang Q, Zhang X, Zhang F, Shieh C‐HP, Yang D, Zhang N (2013) Quantitative study of the interactome of PKCζ involved in the EGF‐induced tumor cell chemotaxis. J Proteome Res 12: 1478–1486 [DOI] [PubMed] [Google Scholar]
- 16. Jin J, Smith FD, Stark C, Wells CD, Fawcett JP, Kulkarni S, Metalnikov P, O'Donnell P, Taylor P, Taylor L et al (2004) Proteomic, functional, and domain‐based analysis of in vivo 14‐3‐3 binding proteins involved in cytoskeletal regulation and cellular organization. Curr Biol 14: 1436–1450 [DOI] [PubMed] [Google Scholar]
- 17. Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ (2012) The cellular EJC interactome reveals higher‐order mRNP structure and an EJC‐SR protein nexus. Cell 151: 750–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishida E, Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M (1997) CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 390: 308–311 [DOI] [PubMed] [Google Scholar]
- 19. Ossareh‐Nazari B, Bachelerie F, Dargemont C (1997) Evidence for a role of CRM1 in signal‐mediated nuclear protein export. Science 278: 141–144 [DOI] [PubMed] [Google Scholar]
- 20. Stade K, Ford CS, Guthrie C, Weis K (1997) Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 90: 1041–1050 [DOI] [PubMed] [Google Scholar]
- 21. Fornerod M, Ohno M, Yoshida M, Mattaj IW (1997) CRM1 is an export receptor for leucine‐rich nuclear export signals. Cell 90: 1051–1060 [DOI] [PubMed] [Google Scholar]
- 22. Millward TA, Hess D, Hemmings BA (1999) Ndr protein kinase is regulated by phosphorylation on two conserved sequence motifs. J Biol Chem 274: 33847–33850 [DOI] [PubMed] [Google Scholar]
- 23. Lapalombella R, Sun Q, Williams K, Tangeman L, Jha S, Zhong Y, Goettl V, Mahoney E, Berglund C, Gupta S et al (2012) Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 120: 4621–4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neggers JE, Vercruysse T, Jacquemyn M, Vanstreels E, Baloglu E, Shacham S, Crochiere M, Landesman Y, Daelemans D (2015) Identifying drug‐target selectivity of small‐molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem Biol 22: 107–116 [DOI] [PubMed] [Google Scholar]
- 25. Joffre C, Codogno P, Fanto M, Hergovich A, Camonis J (2016) STK38 at the crossroad between autophagy and apoptosis. Autophagy 8627: 1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaizuka T, Morishita H, Hama Y, Tsukamoto S, Matsui T, Toyota Y, Kodama A, Ishihara T, Mizushima T, Mizushima N (2016) An autophagic flux probe that releases an internal control. Mol Cell 64: 835–849 [DOI] [PubMed] [Google Scholar]
- 27. Silvestrini MJ, Johnson JR, Kumar AV, Thakurta TG, Blais K, Neill ZA, Marion SW, St. Amand V, Reenan RA, Lapierre LR (2018) Nuclear export inhibition enhances HLH‐30/TFEB activity, autophagy, and lifespan. Cell Rep 23: 1915–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cook D, Hoa LY, Gomez V, Gomez M, Hergovich A (2014) Constitutively active NDR1‐PIF kinase functions independent of MST1 and hMOB1 signalling. Cell Signal 26: 1657–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hergovich A (2016) The roles of NDR protein kinases in hippo signalling. Genes 7: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP (2004) Large‐scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci USA 101: 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F et al (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534: 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, Clauser KR, Qiao JW, Gritsenko MA, Moore RJ et al (2014) Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics 13: 1690–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharma K, D'Souza RCJ, Tyanova S, Schaab C, Wiśniewski JR, Cox J, Mann M (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr‐based signaling. Cell Rep 8: 1583–1594 [DOI] [PubMed] [Google Scholar]
- 34. Kang SS, Shin SH (2013) Phosphorylation of human chromosome maintenance 1 mediates association with 14‐3‐3 proteins. Animal Cells Syst 17: 186–195 [Google Scholar]
- 35. Dian C, Bernaudat F, Langer K, Oliva MF, Fornerod M, Schoehn G, Müller CW, Petosa C (2013) Structure of a truncation mutant of the nuclear export factor CRM1 provides insights into the auto‐inhibitory role of its C‐terminal helix. Structure 21: 1338–1349 [DOI] [PubMed] [Google Scholar]
- 36. Liang XH, Yu J, Brown K, Levine B (2001) Beclin 1 contains a leucine‐rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res 61: 3443–3449 [PubMed] [Google Scholar]
- 37. Kirli K, Karaca S, Dehne HJ, Samwer M, Pan KT, Lenz C, Urlaub H, Gorlich D (2015) A deep proteomics perspective on CRM1‐mediated nuclear export and nucleocytoplasmic partitioning. Elife 4: 1–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao B, Zhao B, Wei X, Wei X, Li W, Li W, Udan RS, Udan RS, Yang Q, Yang Q et al (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S et al (2011) Role of YAP/TAZ in mechanotransduction. Nature 474: 179–184 [DOI] [PubMed] [Google Scholar]
- 40. Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, Chen AC, Sah RL, Taylor SS, Engler AJ et al (2015) Matrix stiffness drives epithelial‐mesenchymal transition and tumour metastasis through a TWIST1‐G3BP2 mechanotransduction pathway. Nat Cell Biol 17: 678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ege N, Dowbaj AM, Jiang M, Howell M, Hooper S, Foster C, Jenkins RP, Sahai E (2018) Quantitative analysis reveals that actin and Src‐family kinases regulate nuclear YAP1 and its export. Cell Syst 6: 692–708 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ren F, Zhang L, Jiang J (2010) Hippo signaling regulates Yorkie nuclear localization and activity through 14‐3‐3 dependent and independent mechanisms. Dev Biol 337: 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Das A, Fischer RS, Pan D, Waterman CM (2016) YAP nuclear localization in the absence of cell‐cell contact is mediated by a filamentous actin‐dependent, myosin II‐ and phospho‐YAP‐independent pathway during extracellular matrix mechanosensing. J Biol Chem 291: 6096–6110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Paul I, Batth TS, Iglesias‐gato D, Al‐araimi A, Al‐ I, Alkharusi A, Norstedt G, Olsen JV, Zadjali F (2017) The ubiquitin ligase Cullin5 SOCS2 regulates NDR1/STK38 stability and NF‐κB transactivation. Sci Rep 7: 42800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shi DD, Shi H, Lu D, Li R, Zhang Y, Zhang J (2012) NDR1/STK38 potentiates NF‐κB activation by its kinase activity. Cell Biochem Funct 30: 664–670 [DOI] [PubMed] [Google Scholar]
- 46. Parikh K, Cang S, Sekhri A, Liu D (2014) Selective inhibitors of nuclear export (SINE)– a novel class of anti‐cancer agents. J Hematol Oncol 7: 78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, Escaramis G, Jares P, Beà S, González‐Díaz M et al (2011) Whole‐genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 475: 101–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hergovich A, Bichsel SJ, Hemmings BA (2005) Human NDR kinases are rapidly activated by MOB proteins through recruitment to the plasma membrane and phosphorylation. Mol Cell Biol 25: 8259–8272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Singh MK, Martin APJ, Joffre C, Zago G, Camonis J, Coppey M, Parrini MC (2019) Localization of RalB signaling at endomembrane compartments and its modulation by autophagy. Sci Rep 9: 11797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schmid‐Burgk JL, Höning K, Ebert TS, Hornung V (2016) CRISPaint allows modular base‐specific gene tagging using a ligase‐4‐dependent mechanism. Nat Commun 7: 12338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vercruysse T, De Bie J, Neggers JE, Jacquemyn M, Vanstreels E, Schmid‐Burgk JL, Hornung V, Baloglu E, Landesman Y, Senapedis W et al (2017) The second‐generation exportin‐1 inhibitor KPT‐8602 demonstrates potent activity against acute lymphoblastic leukemia. Clin Cancer Res 23: 2528–2541 [DOI] [PubMed] [Google Scholar]
- 52. Lipecka J, Chhuon C, Bourderioux M, Bessard M‐A, van Endert P, Edelman A, Guerrera IC (2016) Sensitivity of mass spectrometry analysis depends on the shape of the filtration unit used for filter aided sample preparation (FASP). Proteomics 16: 1852–1857 [DOI] [PubMed] [Google Scholar]
- 53. Wiśniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362 [DOI] [PubMed] [Google Scholar]
- 54. Vizcaíno JA, Csordas A, del‐Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez‐Riverol Y, Reisinger F, Ternent T et al (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- 56. Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, Pedram K, Carr SA, Ting AY (2016) Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc 11: 456–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Data Availability Statement
The SILAC and APEX2 mass spectrometry proteomic data from this study have been deposited to the ProteomeXchange‐PRIDE repository (https://www.ebi.ac.uk/pride/archive/) and assigned the dataset number PXD011968.