

Abstract
Over the past few years, the concurrent (1) development of polymer synthesis and (2) introduction of new mathematical models for polymer dynamics have evolved the classical framework for polymer dynamics once established by Doi–Edwards/de Gennes. Although the analysis of supramolecular polymer dynamics based on linear rheology has improved a lot recently, there are a large number of insecurities behind the conclusions, which originate from the complexity of these novel systems. The interdependent effect of supramolecular entities (stickers) and chain dynamics can be overwhelming depending on the type and location of stickers as well as the architecture and chemistry of polymers. This Perspective illustrates these parameters and strives to determine what is still missing and has to be improved in the future works.
Introduction
Supramolecular polymer science has been a promising field of research since it was introduced by Lehn and co-workers.1 Because of the combination of modern synthesis and development of novel theoretical models for describing polymer dynamics, an interdisciplinary research field has arisen in the past decade. Traditionally, supramolecular polymers are defined as polymers that carry one or more supramolecular moieties (sticker) either as the end-group(s), as side groups, or in the main chain. A sticker is a moiety that can either self-associate or heteroassociate by using metal–ligand or ionic interactions or hydrogen bonding.2 Therefore, when incorporated into the polymer, it can alter the properties significantly. A molecule with two or more stickers can act like a monomer and polymerize to form linear polymers or a network, respectively,3,4 or it can stack and/or polymerize into 1-D polymers.5−9 When the stickers are end-groups of a polymer, the corresponding polymer can form longer polymers via binary associations or starlike polymer and gels due to phase separation.10−15
It is important to remember that the presence of a single sticker on the polymer chain end even if it undergoes phase separation or multiple associations cannot form a network or gel, as percolation is needed for the polymer to be called a network or a gel.16 Of course, in exceptional cases where phase-separated domains form a branchlike architecture, a network is plausible, but in this Perspective we neglect this possibility. On the other hand, if the sticker is used as the side group, it can potentially form a transient network exhibiting elastic properties (G′ > G″) in the time scales where a sticker is associated. It is of utmost importance to remember that G′ > G″ does not necessarily correspond to a network formation; there are many cases that lead to an elastic plateau while no cross-linking exists. For instance, the so-called hyperstars can show an elastic plateau which can extend beyond the experimentally accessible frequencies.17−19 On the other hand, microphase-separated block copolymers (BCP) can be considered as colloids in the melt state with G′ > G″.20 Polymers grafted on large cores such as POSS (polyhedral oligomeric silsesquioxane) can also lead to elastic colloidal material due to caging of the individual giant molecules and act as cooperative glasslike clusters.21 Moreover, fibers have been reported to show elastic properties.22,23 In all of these examples, the material is not a cross-linked polymer but exhibits an elastic plateau. This unique behavior should also be kept in mind while analyzing the rheology of supramolecular polymers. For instance, it has been reported that polymers with sticky side groups could form fibers by stacking, and G′ > G″ was seen in the dynamic mechanical analysis. Although the elastic plateau was referred to the transient network formation, no further study proved gel formation, which means the elastic properties can originate from fiber formation and not necessarily from a transient network.24
The exact description of dynamic mechanical properties in associating polymers is therefore not trivial. In this Perspective, the aim is to address the most recent progress in the field of supramolecular polymers via a rheological point of view (with focus on linear rheology) and to introduce the potential of this field in future research. This Perspective focuses on the parameters affecting the rheology of supramolecular polymers, namely, (1) association number per sticker in supramolecular polymers, (2) phase separation, (3) strength of association, and (4) the position of sticker in the polymer (end-group(s), side groups, or in the main chain). Therefore, only recent papers that truly contributed to the linear rheology of supramolecular polymers will be discussed.
Association Number per Sticker
In principle, the number of associations on each sticker depends on the type of sticker. Some can accommodate multiple junction points whereas most of the stickers can provide a one-to-one (binary) association.25,26 Rowan et al. synthesized polymers based on poly(tetrahydrofuran) (PTHF) and poly(ethylene-co-butylene) carrying two ligands as the end-groups. By introducing metals to coordinate with the ligands, depending on the vacancies available on the metals (Eu3+, Zn2+, or La3+), one could obtain a linear or cross-linked polymer. Therefore, in this case the sticker association number can be tuned.27,28 When Zn2+ is incorporated in the polymer, the polymer molecular weight increases as a result of linear association of stickers, and the relaxation time goes to longer time scales beyond the experimental frequency window. With addition of Eu3+ the relaxation time decreases, and a terminal flow could be observed. Although one might suspect that introduction of Eu3+ would lead to a network formation with elastic properties, due to more thermally responsible properties of Eu3+, most of the stickers are dissociated and dangling chains are obtained.28
In case of heteroassociation each sticker can show a different association number depending on the counterpart. For instance, while one cyanuric acid can make strong hydrogen bonding with one Hamilton wedge (HW), it can have two functionalities with weaker hydrogen bonding when 2 equiv of 2,6-diaminotriazine (DAT) is added.29−31
The association number can also be tuned by using different ratios of metal:ligand.32 At low metal content a weakly percolated system can be obtained by many nonassociating stickers available, which leads to a liquidlike behavior. With an increase in the metal ratio to the stoichiometric amount of metal and ligand (1:3 in the case of Fe3+), the maximum cross-linking density is obtained, and an elastic plateau is observed. With further increase in the metal content, metals do not reach their full association potential, and mono- or bis-complexes are formed leading to a decrease in the cross-linking density (Figure 1).
Figure 1.
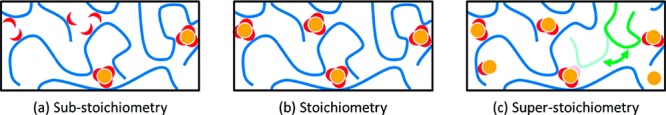
Effect of metal–ligand ratio on the sticker functionality and dynamics of supramolecular polymers: (a) substoichiometry, (b) stoichiometry, and (c) superstoichiometry. Reproduced with permission from ref (32). Copyright 2017 the Society of Rheology.
Moreover, each sticker despite its complementary binary association can form an uncontrolled association as well; this occurs when phase separation play a role. Therefore, if the polymer carries one sticker as the end-group (or center functionalized), either a starlike33,34 or brushlike aggregate can be obtained,23,35−38,34 and if it carries two stickers as end-groups, a cross-linked material can be obtained.13−15,39,40 In particular, the formation of starlike polymers with clusters forming the core leads to an apparent multivalency in stickers (Figure 2).
Figure 2.
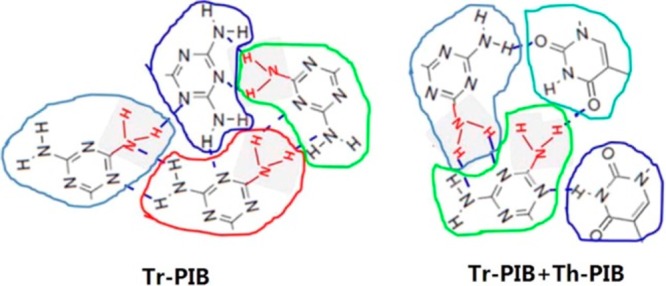
Aggregation of hydrogen-bonding motifs, which leads to an apparent multivalency in the stickers. Adapted with permission from ref (15).
Therefore, in all supramolecular designs it should be carefully evaluated whether such clusters exist or not. This is, however, not always trivial and will be discussed in the following section.
Phase Separation
Despite similarities of controlled (complementary) and uncontrolled associations (aggregates), their melt dynamics differ significantly. The relaxation process in the supramolecular polymers with controlled associations can be considered fundamentally different from the one with uncontrolled associations. For the controlled (binary) associations, Rubinstein and co-workers developed the so-called sticky Rouse model, which predicts that the relaxation time of the chains is proportional to the renormalized bond lifetime τb* (≫τb).41,42 The renormalization is due to the fact that the sticker needs to break a few times before finding another partner to associate with. The sticky reptation model modifies the sticky Rouse model to be applicable to entangled polymers.41 In these models, the stickers have to reach their renormalized lifetime and then, by hopping diffusion, can find a free sticker, and this motion can lead to stress relaxation. In other words, in well-defined binary associations, dissociation of a sticker only leads to a chain relaxation when it is attached to a new partner. This was recently proven by the comprehensive experimental research of Richter and co-workers, concluding that the rheological relaxation time is around 2 orders of magnitude slower than the sticker lifetime and is the time for the sticker to find a new partner.43,44 Alvarez and co-workers also supported this conclusion using 2-ureido-4[1H]-pyrimidinone (UPy)-based polymers, obtaining the effective lifetime of the stickers ∼25 and ∼40 times higher than pure sticker lifetime (for 3% and 8% UPy content, respectively; see Figure 3).45 In both works, no signs of sticker aggregation were observed, and only binary associations were taken into account. In time scales smaller than τb an elastic plateau is formed since the motion of segments between two stickers is hindered due to physical cross-linking.45 In this picture the exchange of partners can occur via different fashions: (1) a concerted mechanism whereby association of one sticker occurs simultaneously with dissociation of another32 or (2) the exchange between partners requires a breaking event that can produce a free site available for a new sticker. In this case, which is inherently more restrictive, the sticker should cover a distance between its initial partner and a newly exposed (opened) sticker in comparison to the former case where the sticker can find any arbitrary complex in its surrounding and perform the partner exchange.32 The presence of a free partner has been shown to be important in particular in the case of low sticker concentrations (high molecular weight precursors with low sticker amount and high sticker lifetime).32 Brassinne and co-workers showed that in such a case the traditional hopping events does not occur (due to low population of the surrounding stickers), and this leads to separation of the sticker lifetime and chain relaxation time. This is because the chain after sticker dissociation should commute a long distance (which takes an intuitively longer time) to find another free partner. Special emphasis is on the free partner because if a guest sticker finds an occupied complex, still no relaxation occurs, and it has to wait until the host complex breaks up. Although this study successfully predicts the relaxation mechanism in the binary associations, it is not always applicable, for instance, to supramolecular systems in which a concerted mechanism is responsible for partner exchange.
Figure 3.
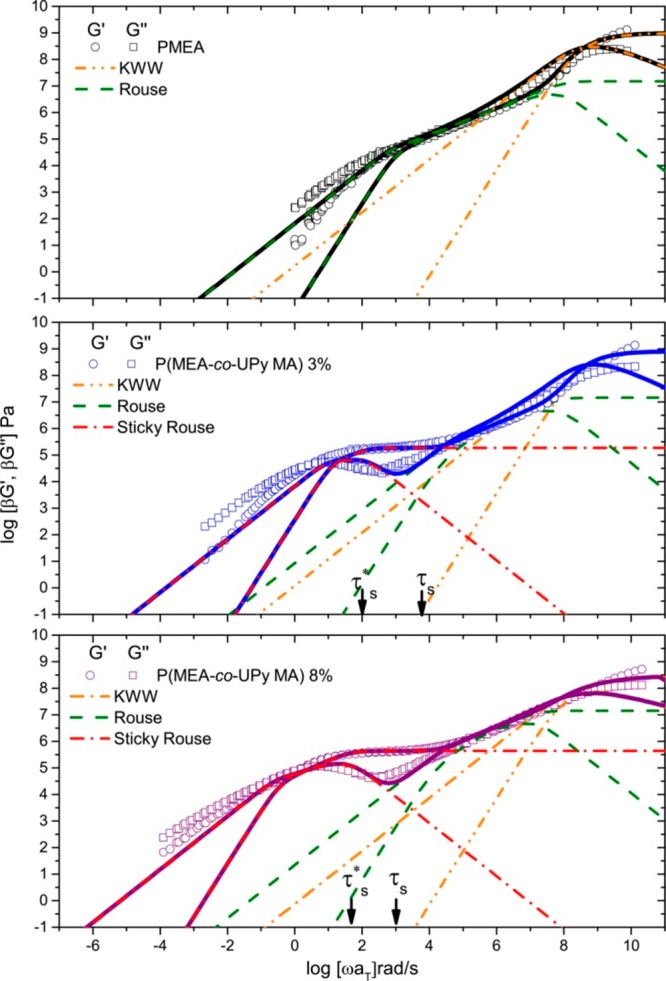
Linear viscoelastic mastercurves of PMEA and P(MEA-co-UPyMA). τs* and τb correspond to the lifetime and normalized lifetime of the stickers. Reproduced with permission from ref (45).
On the other hand, for aggregates, the bond lifetime renormalization is negligible as the aggregates can host an indefinite amount of stickers. When the dissociation energy is within the range of M1/2 < ε/kBT < M4/3 (with M being the aggregation number)46 and as long as the energy change after and before hopping is less than kBT, each sticker can hop, relocate, and contribute to stress relaxation. In the classical sticker hopping picture the stickers can dissociate from the initial cluster and diffuse as open stickers to find another cluster to associate with. Although the overall difference in energy will not change significantly, the dissociation of the sticker from all sticky bonds requires a significant energy barrier and, therefore, energetically unfavorable.47 Therefore, in this case, two small clusters should meet each other and associate, leading to bigger clusters, after which the cluster breaks up to shorter clusters (because the big clusters are entropically unfavorable). This subsequent breakup leads to an exchange of partners, although a few cluster association–dissociation events should occur for all the stickers to change their partner (Figure 4). The size of the cluster also matters, as in bigger clusters there is a higher chance of partner exchange, whereas if an upper cap is considered for cluster size, the terminal relaxation becomes much slower.47
Figure 4.
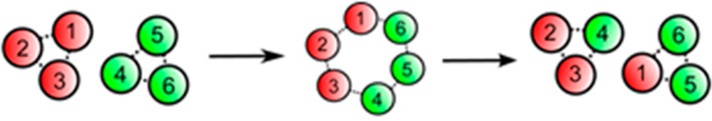
Circles 1–3 and 4–6 show two aggregates consisting of stickers which change their composition via cluster association–dissociation processes. This process leads to a partner exchange event which is followed by stress relaxation. Adapted with permission from ref (47).
A second difference in the controlled (complementary) associations and uncontrolled associations (aggregates) can be the lifetime of the stickers. Usually, the lifetime of the aggregates is reported to be much longer than individual sticker pairs and sometimes exceeds the experimental frequency windows.48−51 van Ruymbeke et al. extensively studied the formation of aggregates in the entangled polymers with sticky side groups.49,51,52Figures 5a and 5b show the model they introduced to describe the relaxation of sticky entangled polymers with aggregates among the chains. Four zones were introduced: (1) A high-frequency Rouse regime, where Rouse motions occur up to the length (time) scales of average spacing between two entanglements and/or stickers. This manifests itself with a slope G′, G″ ∼ ω0.5. (2) Upper plateau zone, wherein the entanglement and the trapped strands contribute to elasticity. In this zone, the dangling chains and starlike aggregates can relax via contour length fluctuations (CLF) and arm retraction mechanism similar to star and comb polymers (see the Supporting Information).53−58 After partial relaxation of the chains via CLF, the tube will be dilated,59 and subsequently in zone (3) the constraint release Rouse (CRR) mechanism leads to equilibration of the trapped strands; once the mobile segments are relaxed, they act like solvents and the trapped segments can undergo Rouse-like motions. Intuitively, this mechanism also shows a scaling G′, G″ ∼ ω0.5. However, care must be taken, as in this picture the aggregates (trapped strands) are not considered to undergo partner exchange events (or hopping) as in this model the number of aggregates per chains is small, and each aggregate is surrounded with polymers instead of other aggregates.51 Although the authors in another study showed that in polymers with low sticker concentrations the relaxation occurs via partner exchange (and not CRR), this can be related to the type of aggregates formed in each system.32 Finally, in the last zone (zone 4) a second plateau appears, which shows the trapped strand contribution to elasticity. Although in this work the emphasis was on the effect of aggregates, in a similar, complementary study, only the effect of binary associations was considered for the same chemistry.60 The authors assigned the third zone with G′, G″ ∼ ω0.5—this time to the hindered fluctuations alongside the blinking of the stickers—as another mechanism of stress relaxation before terminal time. Upon comparison of these two works, it can be concluded that the lifetimes of binary (controlled) associations are much shorter than those of the aggregates (uncontrolled associations).51,60 The same conclusion can be made in another system based on UPy stickers (instead of carboxylic acid groups) where the binary dissociations occur in much shorter times than dissociation of aggregates, some of which never show terminal relaxation.49
Figure 5.

(a) Schematic representation of supramolecular polymers with sticky side groups, forming starlike aggregates, trapped entanglements, and dangling chains. (b) Four relaxation regimes considered in the model, applicable to linear rheology of entangled supramolecular polymers with aggregate-forming side groups. Reproduced with permission from ref (51). Copyright 2016 the Society of Rheology.
Therefore, phase separations are decisive factors in the dynamics of supramolecular polymers. To this point most of the stickers with few exceptions when incorporated in the polymer matrix can phase separate due to their polarity in comparison to the matrix. For instance, Leibler and Soulié-Ziakovic extensively investigated the self-assembly of polymers end-capped with 2,4-diamino-1,3,5-triazine (DAT)/thymine (THY) and showed that for polymers based on THY or DAT end-functionalized poly(ethylene) (PE) a lamellar morphology could be obtained when THY is solely used as an end-group due to THY crystallization. However, the DAT moiety which does not inherently crystallize still can lead to phase separation.61−63 Similar effects were reported for noncrystalline polymers such as poly(propylene oxide) (PPO) end-capped with DAT and/or THY.64,65 It has to be pointed out that the study of crystallization based on rheology is too optimistic, and therefore, other methods including small- and wide-angle X-ray scattering (SAXS and WAXS) and differential scanning calorimetry (DSC) are necessary.66 For strongly nonpolar polymers such as poly(isobutylene) (PIB) monofunctionalized with DAT, a body-centered-cubic (BCC) morphology was observed, but mesophases were not observed for bifunctional PIB.40 In the case of stronger stickers such as UPy even a lateral aggregation into nanofibers was observed.35,67 Phase separation was also observed in polymers like poly(n-alkyl acrylate)s when no H-bonding exists but rather due to incompatible alkyl side groups (when the alkyl side group has more than four CH2 units). Therefore, the alkyl nanodomains form aggregates with a size of 0.5–2 nm.48,68 But Alvarez et al. attributed this to partial hydrolysis of side groups which turns inevitably (during synthesis) to hydrogen bonding of acrylic acid repeating units (Figure 6a).69
Figure 6.
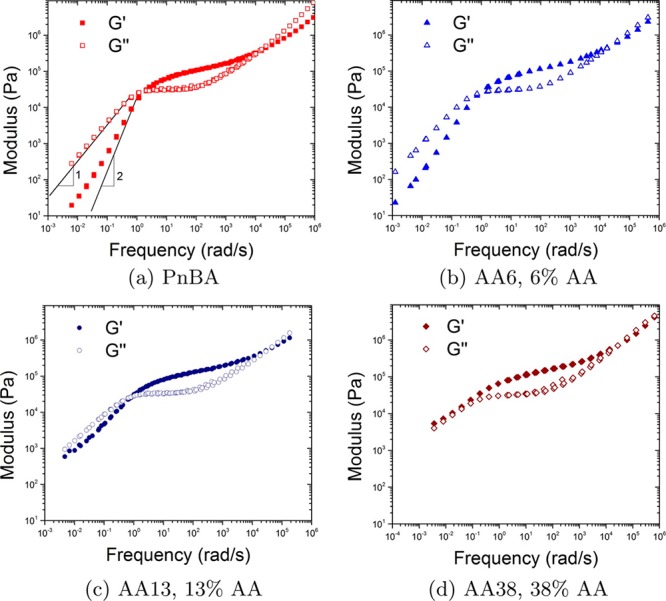
Linear viscoelastic response of (a) PnBA hydrolyzed partially to yield random copolymers with (b) 6%, (c) 13%, and (d) 38% acrylic acid. Even pure PnBA shows diversion from Maxwell-like terminal relaxation, which was attributed to the partial hydrolysis during synthesis. Reproduced with permission from ref (69).
The use of hydrophobic alkyl side groups can also tune the rheology of polymers although the interactions are not strong enough to significantly change the polymer dynamics. Therefore, the side groups rather change the plateau modulus value (Figure 7) or lead to a weak phase separation.70,71 Hydrophobic groups were even used in order to provide self-healing properties by plasticizing effect of alkyl flexible chains.72 On the other hand, strong metal complex based stickers can also make clusters and lead to nanoscale heterogeneity able to enforce the mechanical strength.73 Other studied supramolecular polymers bearing carboxylic acid, UPy, or THY side groups can also show phase separation and change polymer dynamics significantly.24,48,49,51,52 However, their effect can be different depending on the strength of associations, which will be discussed in the following section.
Figure 7.
Mastercurves of the dynamic moduli for the polymers with different size of alkyl side chains. Increase of side chain length leads to a lower plateau modulus. Reproduced with permission from ref (70).
Association Strength
The study of association strength and bond lifetime in supramolecular polymers was originally addressed after the introduction of the UPy moiety by Meijer et al.3,74 Tuning the strength and the bond lifetime of stickers is the key feature in the field of supramolecular polymer materials. At short bond lifetimes (τ < 1 μs), a robust 1D polymeric assembly does not exist, whereas too long a lifetime (τ > 1 min) yields materials without dynamic properties.75 The easy to synthesize UPy moiety was introduced with a dimerization constant of Kdim = 10–7–10–8 M–1 and a lifetime of 0.1–1 s and has been applied in the rheology of supramolecular polymers since then.76 Whereas the measurement of bond lifetime and association constant in solution using nuclear magnetic resonance (NMR) experiments can lead to relatively accurate values, rheology can provide us with better qualitative analysis on the macroscopic scales.
The research on association strength and its effect on polymer dynamics has been predominant for polymers with stickers as side groups, whereas in one study Binder et al. compared the association strength of telechelic polymers with THY and DAT functionalities and found that the association strength can vary depending on the sticker environment: melt or solution. The strength in the melt state was DAT-DAT < THY-THY ≪ THY-DAT, whereas in solution this order was THY-THY < DAT-DAT ≤ THY-DAT. Although, the authors concluded that this is due to different dynamics of hydrogen bonds in different media, from the corresponding polymer melt viscosity, it seems the association is not due to hydrogen bonding and rather to mere phase separation.13 In particular, they showed if THY-DAT hydrogen bonding is blocked by an extra methyl group, the viscosity of the corresponding polymer mixture remains the same as the THY-DAT end-capped polymer mixture (with potential hydrogen bonding).13 The conclusion that in this supramolecular polymer mixture phase separation is more responsible than hydrogen bonding is consistent with the studies by Leibler and Soulié-Ziakovic where they showed THY on its own governs the self-assembly based on crystallization and phase separation without the need for hydrogen bonding.61−63
For polymers with sticky side groups, different types of stickers with different strength have been investigated. Seifert et al. compared solution rheology of polymers with DAT–maleimide, cyanuric acid–Hamilton wedge, and DAT–cyanuric acid hydrogen-bonding interactions (Figure 8). In addition, they used terpyridine and different metal salts for stickers as a comparison.29−31 Performing rheology in two different solvents of different polarities (DMF or a methanol/chloroform mixture), they showed that in the polar solvents, regardless of the sticker type, less association occurs and terminal relaxation was observed. However, in less polar solvents, the terpyridine/Mn2+ complex (Figure 8D) showed the strongest association with G′, G″ ∼ ω0.5, which shows the polymer is close to the gel point.30 The association was slightly weaker when cyanuric acid–Hamilton wedge (Figure 8B) was used, followed by weaker association of DAT–maleimide (Figure 8A). In all the cases where DMF was the solvent of choice, less association was observed, leading to lower cross-linking (although they still exist and contribute to the shallowing of terminal slopes). The authors, however, did not consider the contributions of amide hydrogen bonding in the precursor polymer (p(NIPAAm)) as has been reported in the literature.77 Although the solution rheology of the precursor showed little association,30 the effect can be significant when the amide groups are in the vicinity of stronger stickers in the same polymer. The authors neither considered the effect of aggregation and self-association of the third component (bis-maleimides) added as the cross-linker. Although their interaction would be weak, weak hydrogen bondings have been shown to change the slopes in the frequency sweeps significantly (see Figure 6).48 The terpyridine association got even stronger when Co2+ was used, leading to a plateau reminiscent of a transient network. The formation of transient networks surprisingly occurred in a polar solvent (DMF) not only in the terpyridine system but also when bis-DAT (Figure 8C) was used. This shows that Co2+ has a stronger association strength than Mn2+. But for the bis-DAT system, although the authors justified the plateau by considering higher number of associations per sticker in this system, it is inconsistent with the fact that much stronger stickers (cyanuric acid–Hamilton wedge) were almost totally soluble in DMF. It is also inconsistent with the conclusion of Anthamatten et al. that “a higher concentration of weakly bonding groups is not equivalent to a smaller number of strong bonding groups”.78 The authors compared the effect of stickers (with different hydrogen-bonding strengths) on the rheology of polymers. When comonomers with weaker stickers were used (acrylamidopyridine, acrylic acid, and carboxyethyl acrylate), the corresponding polymer showed similar melt dynamics as the precursor poly(n-butyl acrylate) (PnBA) whereas with UPy as the sticker a plateau was observed, reminiscent of a transient network. For a weak sticker, although a so-called knee-shaped terminal relaxation was observed, by referring to the fast exchange rate of weak stickers, it was concluded that weak stickers do not contribute to the melt rheology. This conclusion is in fact different from our study where weak stickers based on THY could contribute significantly to the polymer dynamics.48 With increasing the amount of stickers along the chain, a sol–gel transition was obtained, but no transient network was formed due to low association constant of THY (Figure 9).48 This shows that weak stickers can indeed affect the dynamics in time scales faster than where phase separation plays a role. In fact, weak binary hydrogen bondings have been shown to change the dynamics in many studies. By use of amide or carboxylic acid groups as side stickers69,77 and hydroxyl, amine, carboxylic acid, or THY as polymer end-groups,10−15 the terminal relaxation time has been shown to be delayed (Figures 5–7 and 9–11). Even if this delay is attributed to the aggregation of side groups, weak stickers play an important role and can even prevent the terminal relaxation in the experimental frequency windows.48,51,52
Figure 8.

Supramolecular polymers based on pNIPAAm and different sticker strengths based on (A) diaminotriazine:maleimides, (B) cyanuric acid:Hamilton wedge, (C) cyanuric acid:diaminotriazine, and (D) terpyridine:metal ions. Reproduced with permission from ref (30). Copyright 2013 the Royal Society of Chemistry.
Figure 9.
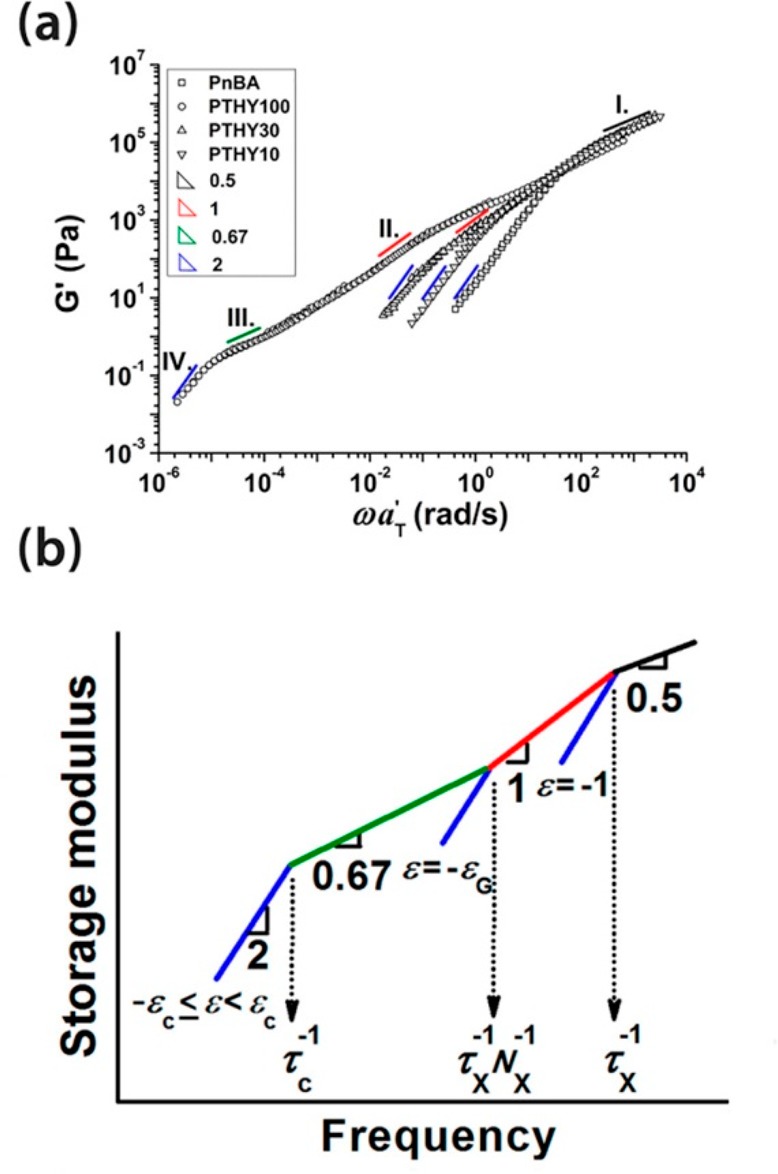
(a) Mastercurves constructed for PnBA and copolymer carrying 10, 30, and 100% THY along the chain. (b) Modified MF percolation theory. Reproduced with permission from ref (48).
Figure 11.
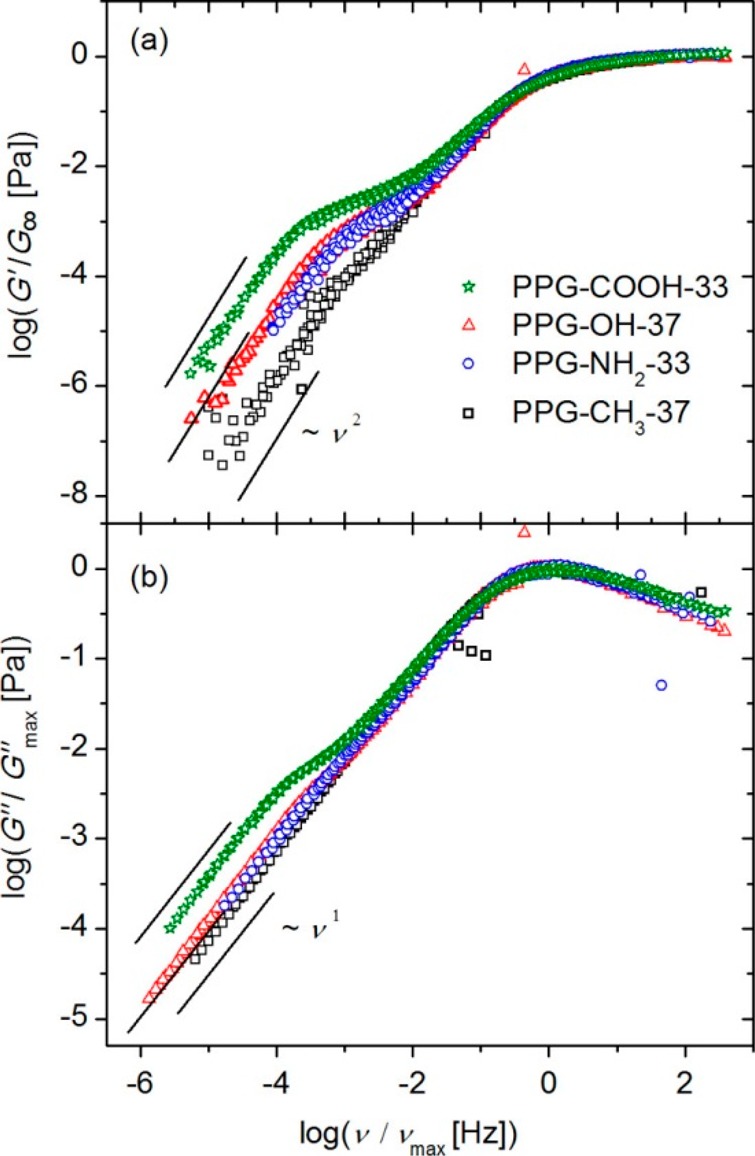
Normalized storage (a) and loss part (b) of the shear modulus mastercurves showing the effect of weak associations on the dynamics of end-capped supramolecular polymers. Reproduced with permission from ref (11).
Stickers based on sulfonated styrene and ionic interactions also have different strengths based on the counterion used: Na+ > K+ > Rb+ > Cs+ leading to different terminal relaxation times.79,80 Similarly, the metal complex formation strengths can vary depending on the metal used. Therefore, by use of different metals in bulk of polymers with end-capped ligands, the terminal relaxation time can be delayed with the order81,82 Ni2+ > Co2+ > Cu2+ > Zn2+. This is consistent with the studies in solution rheology, where the order Co2+ > Zn2+ > Mn2+ was obtained for the strengths of association, value of plateau modulus, and terminal relaxation time.73 While these studies reported similar order of strengths, in another study the following order was reported:83 Cu2+ > Zn2+ > Ni2+ > Co2+, which is in fact consistent with their reported association constants in solution (Zn2+ > Co2+ > Mn2+).73 This inconsistency was shown to be due to Co2+ ion aggregation and different metal complex geometries which leads to different association strengths in some studies.73,83 Therefore, the correct choice of ligand, solvent, and polymer matrix is of utmost importance. The same approach was used by Craig et al., whereby use of either of Pd(II) or Pt(II) cross-linkers in a solution of poly(4-vinylpyridine)-based network led to lifetimes ranging from milliseconds to tens of minutes.84−87 Moreover, a similar study was done on poly(butyl acrylate-co-acrylic acid) with varying fractions of ionic groups and the respective Na+, Zn2+, and Co2+ counterions to investigate the role of reversible interactions and network mobility on healing. It was concluded that in the intermediate range of 10 < τb < 100 s ionomers show good scratch healing behavior and good mechanical properties.88
Sticker Position
Polymer dynamics can be tuned based on the location of stickers in the polymer (in the main chain, side groups, or end groups). Using stickers within the main chain can add an internal clock to the polymer so that, in addition to the classical linear polymer behavior, a transient chain with time (temperature)-dependent chain length can be obtained. If the sticker, however, is located as end group(s), a similar behavior can be obtained depending on the polymer chain length. On the other hand, if the sticker is located as side groups of a linear polymer, a transient network can be formed with totally different dynamics. However, in all cases only binary associations are assumed to be present, whereas in reality most of the designs do not follow this assumption and the dynamics diverges from the theory. Therefore, before one designs a supramolecular polymer, it is important to compare the dynamics rising from the position of the stickers.
In the Main Chain
The seminal work of Cates et al. on supramolecular polymers with sticky monomers developed a framework for dynamics of main-chain associating polymers.89 The interplay between the polymer dynamics and sticker association kinetic was studied, and it was shown that depending on the disentanglement time and sticker lifetime, relaxation time can fall into different regimes. In other words, the stress relaxation can proceed by breaking the stickers or reptation, depending on the disentanglement time and sticker lifetime. Watanabe et al. have recently developed new and more robust theories that can explain the dynamics of these polymers.90−94 However, so far fewer studies used stickers as end-groups, in a manner that phase separation does not occur. Using DAT and THY end-capped polymers, one can theoretically form a main-chain supramolecular polymer. The formed supramolecular polymer does not necessarily mean that hydrogen bonding is responsible for the change in rheological properties. The effect of phase separation has been reported to predominate the dynamics in this material; therefore, excluding this effect to form purely main-chain supramolecular polymers based on hydrogen bonding is not quite trivial.13,15,39,40,95 In other words, in order for a polymer chain to be linear and split into shorter chains and associate again, not only highly complementary associations are necessary but also the phase separation should be avoided. For instance, thus far, no study has truly proven the pure binary association of UPy in supramolecular polymers. Indeed, if the amount of sticker is chosen to be as little as possible and the polymer matrix is chosen wisely, the aggregation can be less tangible but still exists. It has been in particular shown that even in high molecular weight polymer matrix (low UPy concentration) the phase separation exists and it contributes to stress relaxation mechanism, so the polymer chains do not behave like linear chains the Cates model hypothesized (Figure 12).96,34 Therefore, a plethora of studies reported the contribution of UPy stacking on polymer dynamics as an inevitable part of this sticker.97,98 This is indeed a big disadvantage when comparing with theories, although in some studies this effect has not been truly addressed.99,100 For instance, in one study by Binder et al. on PnBA end-capped with THY and/or DAT the absence of a plateau in the frequency sweeps was related to the absence of phase separation.13 However, the corresponding slopes in the terminal regime did not show Maxwell behavior, similar to many studies where phase segregation was present and led to smaller terminal slopes instead of plateau formation.101 In fact, the smaller terminal slopes can be explained by the different sizes of aggregates. When the stickers want to undergo hopping motions to relax the applied stress, if there is a distribution of aggregate size, then the partner exchange time differs and leads to a distribution of relaxation times which consequently decreases the terminal slopes. Also, the lack of entanglement plateau in frequency sweeps of telechelic supramolecular polymers does not necessarily mean the absence of associations. For polymers with low Tg obviously the frequency sweeps should be performed at lower temperatures so as to capture all the dynamics; otherwise, the terminal relaxation will be observed depending on the number of associations (and the formed molecular weight). This was recently well explained by van Ruymbeke et al.102
Figure 12.
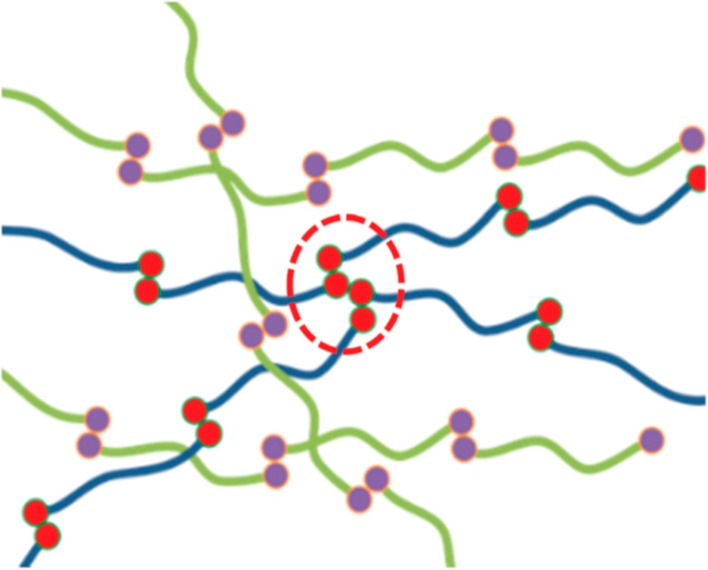
Schematic representation of polymers with end-capped stickers. Some fraction of the stickers (UPy) show aggregation (marked with red dashed line) whereas the rest undergo binary associations. The aggregated end-groups lead to starlike assemblies with different dynamics. Reproduced with permission from ref (96). Copyright 2019 the Royal Society of Chemistry.
The phase separation can therefore change dynamics from linear polymer to transient network dynamics unless only one end-group is carrying stickers whereby starlike assemblies can form rather than a network.
Using metal–ligand interactions, Rowan et al. reported to have obtained longer polymer chains by the presence of stickers as the polymer end-groups.28 Therefore, a rubbery plateau was attributed to the entanglement of elongated chains. This must be, however, a speculative conclusion because due to the lamellar formation (as a result of aggregation of 2,6-bis(N-methylbenzimidazoyl)pyridine (MeBip) stickers), a cross-linked material is obtained, and the corresponding plateau cannot necessarily be due to molecular weight increase. Although the authors showed the addition of different metal ions (Eu3+ instead of Zn2+) can change the rheological properties, the effect of phase separation on polymer dynamics cannot be neglected. In fact, the same group in another work showed no binary associations are actually needed for plateau formation in the very same polymer.103
A few studies could truly investigate the linear supramolecular dynamics without significant contributions from sticker aggregations. By use of tube-based models, in a linear supramolecular polymer with metal–ligand stickers as end-group(s), it has been shown that by adding a 1:2 ratio of metal to ligand (stoichiometry ratio) the polymer chain can be extended 2 (for polymers with one ligand as end-group) to 8 (for polymers with two ligands) times in length without aggregation.102 Interestingly, when the metal content was increased, a rubbery plateau was observed which was attributed to the network formation (not an entanglement plateau). The metal ions act as bridges between PEO chains by complex formation. The attribution of plateau to cross-linking indeed makes sense as linear PEO in order to exhibit terminal relaxation time beyond the experimental frequency window should have a molecular weight beyond 106 g mol–1, which is unrealistic102 (although was considered plausible in other works28). In a similar system with monofunctional PnBA and different metal ions, they showed that depending on the strength of the metal ion, the supramolecular polymer can relax as a mixture of precursor and double-sized polymers, with the latter being more probable for stronger associations (Figure 13). Depending also on the association strength and the terminal relaxation time, the polymers could either relax fully by reptation or break (and then relax via reptation), which is similar to the theory proposed by Cates et al.89 In the case of bifunctional polymers a secondary plateau was observed which was ascribed to the loop formation. In essence, the entanglement between two loops can be released after the sticker dissociation and undergo reptation.82,102 The formation of loops and its effect on the rheological properties have been studied for conventional (covalent) polymeric systems. Covalent polymer networks are generally formed via kinetically controlled processes and, therefore, possess cyclic topological defects. The classical affine and phantom network theories of network are based on acyclic treelike structures, which leads to the overestimation of G′. Therefore, more sophisticated theories must be employed for inclusion of the loops in both supramolecular and conventional polymers.104
Figure 13.

Formation of double-sized PnBAs by using monofunctional precursors and different metal–ligand association strengths. Using (a) linear PnBA-tpy + Ni(II), (b) linear PnBA-tpy + Co(II), (c) linear PnBA-tpy + Cu(II), and (d) linear PnBA-tpy + Zn(II), one can tune the ratio of chains with the size of precursor (PnBA-tpy) and double-sized polymers without aggregation of the end-groups. Reproduced with permission from ref (82).
UPy as Side Groups
The studies on supramolecular polymers with UPy side groups have been so wide that we spend one section only for this type of polymer. The first thorough study on rheology of unentangled polymers with UPy side motifs was published in 2009 by Meijer et al. whereby the polymer dynamics was modeled with the Rouse model.105 Because of weak fitting results, the authors concluded that new models should be developed to describe the melt dynamics in these systems.105 Anthamatten et al. then used the Doi–Edwards theory and considered UPy associations the same as entanglements.78 However, this analogy is quite tricky as the relaxation mechanisms in these two different topological constraints are entirely different; disentanglement occurs via CLF and reptation processes whereas stress relaxation in transient networks with sticky side groups occurs via the partner exchange process. In addition, it has been recently shown that the effect of entanglement on the dynamics is quite different from the presence of stickers.106 Moreover, the molecular weight dependence of terminal relaxation time in entangled polymers is different from transient networks (Figure 14).49 More recently, the relaxation of similar supramolecular polymers was modeled by a modified sticky Rouse model whereby the effect of sticker distribution was taken into account.45 Concurrently, another theory based on modified mean-field percolation theory of Rubinstein and Semenov (see the Supporting Information)42 predicted the linear viscoelastic behavior of the very same polymers.107 The sol–gel transition and the corresponding scaling fairly matched with the theory. In another work, the sticky Rouse model was again modified by considering the effect of sticker distribution, contribution of dangling chains, and the hopping event which occurs after sticker dissociation. Although the result of modeling described the data in the low frequency range pretty well, the middle frequency results were hardly fitting to the model.108 In all these examples, unentangled polymers with UPy sticky side groups were studied in the melt state. Seifert et al. studied the entangled polymer dynamics in block copolymers where one block was hydrophobic and the other block was randomly modified from hydroxyl side groups to UPy side groups with different compositions.109 Surprisingly, no plateau was observed, which was justified by the disruptive effect of remaining hydroxyl groups on UPy association. This is, however, counterintuitive to some degree, as the biggest motivation to use UPy-based stickers is its high association constant, and the 10 mol % hydroxyl group should not dissociate the stickers. This inconsistency can also be seen in two similar works with the same chemistry, where the plateau was actually observed despite the presence of hydroxyl units (compare Figures 14 and 15).49,110 Also, the authors called the corresponding supramolecular polymer a network, whereas no plateau was observed which is also hard to justify.109 The corresponding polymer rather shows a behavior close to branched polymers with dispersity in the molecular weights, which in turn leads to shallowing of the slope. The effect of flexible spacer on the sticker associations was also studied, and in line with our study48 they concluded that a flexible spacer can increase the association.110
Figure 14.
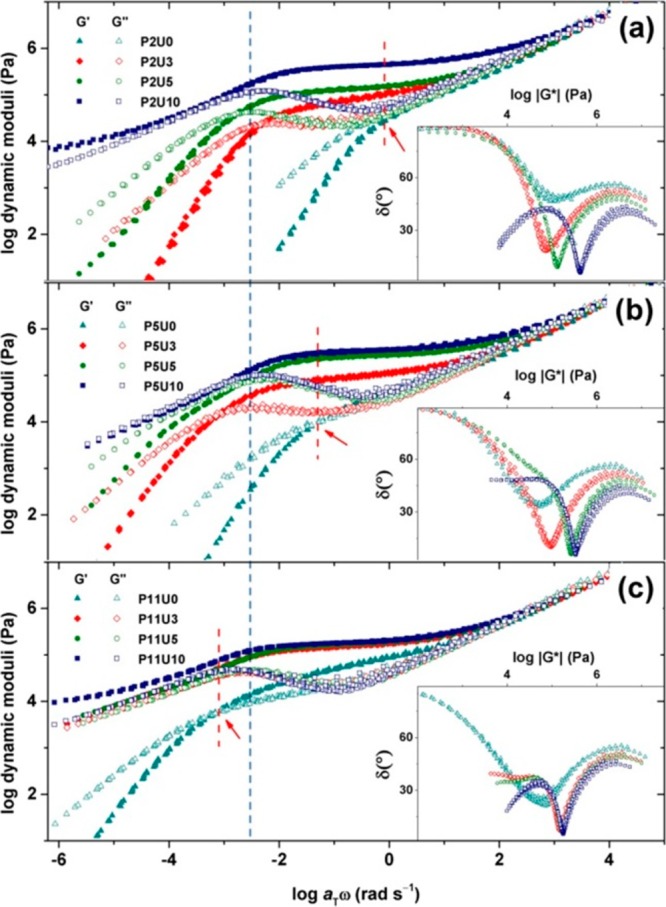
Comparison of linear viscoelasticity of supramolecular polymers with different contents of UPy (3, 5, and 10%) as side groups (sticker) and precursor molecular weights (a) lower (42.5 kg mol–1), (b) close to (76.4 kg mol–1), and (c) above critical molecular weight Mc (165.4 kg mol–1). Reproduced with permission from ref (49).
Figure 15.
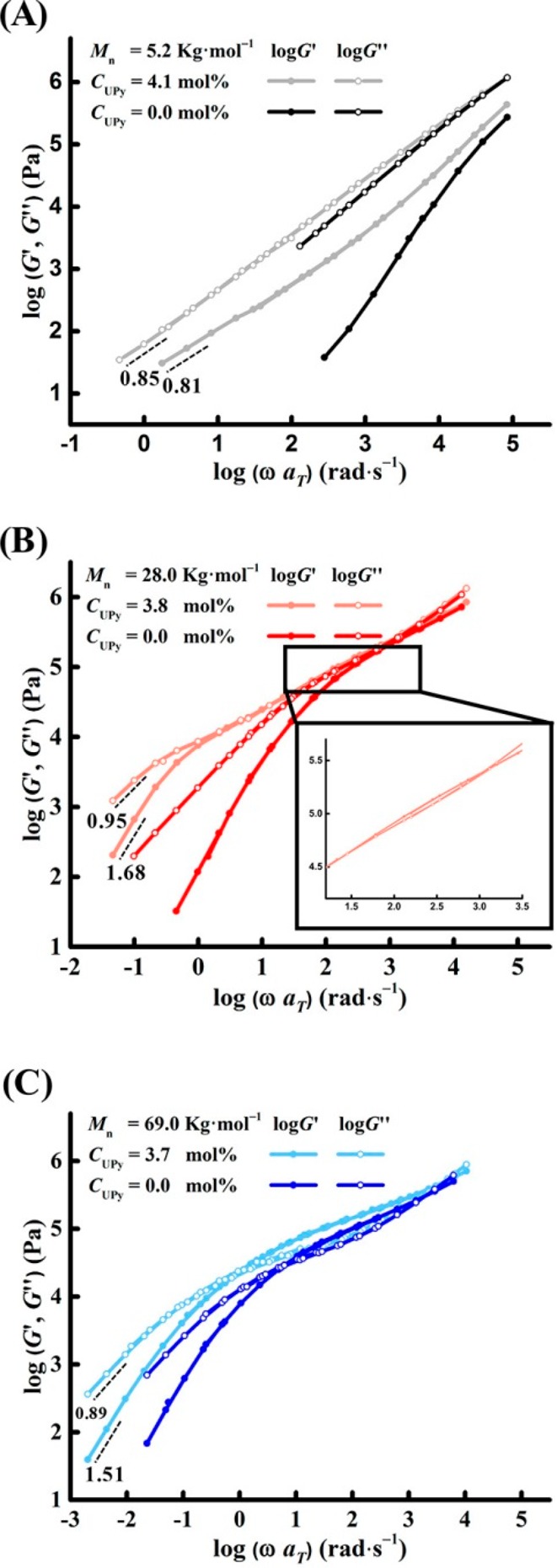
Dynamic moduli (A–C) for precursor and supramolecular polymers with sticker contents of nearly 4% and different polymer molecular weight. Adapted with permission from ref (109).
Another group reported similar work based on PnBA and UPy sticky groups randomly cited along the chain.50 Because of phase separation of UPy stacks, the relaxation of the polymer chains was hindered. Using DSC and optical microscopy, they reported that the phase separation occurs with even 3 mol % sticker. This is, however, inconsistent with the two other studies done (by two independent teams) with the very same polymers (with sticker content close to 3%) whereby the absence of UPy aggregates was proven via SAXS studies. Those works, therefore, assumed only binary associations exist.105,107
Perhaps the most comprehensive theory so far is by van Ruymbeke et al. in which a tube-based model called the time marching algorithm (TMA) (see the Supporting Information) describes the behavior of supramolecular polymers with UPy side units very well.49 One important conclusion was that at longer time scales for large UPy contents a secondary plateau is representative of cluster formation (Figure 14). The intermediate frequency plateau, however, is more tricky to be assigned; with increasing the sticker concentrations in all molecular weights, no increase in the relaxation time was observed. This means the relaxation time belongs to the partner exchange time of UPy units, which does not change with sticker concentration. Moreover, with increasing the molecular weight, the relaxation time did not change, which means the disentanglement process is faster than what a classical sticky reptation model predicts.49 Therefore, disentanglement occurs via delayed CLF processes which takes place nearly at the same time as the sticky Rouse modes, and therefore no additional entanglement plateau can be seen. This assumption makes sense considering the small amount of entanglements.49
Other Side Groups
Other side groups can have ionic origin with potential application as polyelectrolytes and coacervates33,81,82,107−113 or be comprised of metal–ligand interaction,29,30,119,120 weak hydrogen bonding,44,48,51,52,60,69,77,121 or even host–guest interaction.122 Since the seminal work of Craig et al.,123 many works contributed to the metal–ligand type stickers as side groups. In entangled systems, it has been recently shown that the presence of entanglements does not affect the stickers dynamics, but stickers do affect disentanglement by delaying the relaxation process.106 In several studies, the strength of the metal–ligand interaction was studied,119,120 whereas some other research compared the association strength of different types of stickers (based on hydrogen bonding or metal–ligand interaction) (see Figure 8). It was shown that the association strength has a direct influence on the formation of a plateau and delay in the terminal relaxation time.29−31 However, the majority of research on polymers with side stickers focused on weak hydrogen-bonding groups with higher concentration compared to UPy or metal–ligand interactions with less content.44,48,51,52,60,69,77,121 The biggest questions that were addressed in these works were the effect of sticker on the (1) terminal slopes, (2) appearance of one or more plateaus, and (3) terminal relaxation time. Although there have been different theories and experiments to tackle these questions, each of them has its pros and cons and some are inconsistent with each other. This means this field still needs a generalized theory that can match the experimental results.
Following seminal works on scaling in supramolecular polymers,41,42,123 many studies tried to unravel the rationale behind the appearance of various slopes in terminal relaxation of supramolecular polymers. For instance, the shallowing of terminal slopes was attributed to (1) the distribution of sticker, (2) molecular weight distribution, (3) exacerbating effect of stickers on polydispersity, (4) transient branching, and (5) sticker aggregation. Although a distribution of stickers can affect the terminals slope as was suggested in the past,45,49,60,69,108 we have recently shown that using 100% sticker on the chain (one sticker per repeating unit)—a monodisperse distribution of stickers—the shallowing still exists until low frequencies (Figure 9). We concluded that this is due to sol–gel transition, which matched quite well with the newly developed theory of Colby et al.79 The same conclusion was done by other teams using polymers with a distribution of stickers based on either ionic interactions or UPy-based stickers.79,80,107,124−126 Alvarez et al. using polydisperse polymers with randomly distributed stickers concluded that the shallowing of the slope is due to molecular weight and sticker distribution (and not due to aggregation and cluster relaxation; see Figure 3), but they did not test this argument by investigating monodisperse samples to see whether or not they still see the shallowing of slopes.45 In fact, in nearly monodisperse polymers with stickers randomly distributed along the chain the shallowing was still observed.43,44 In another study, on the contrary, with Đ of nearly 2 with each repeating unit carrying a sticker, no shallowing was observed (Figure 10).77 It might be tempting to conclude that the sticker distribution is rather the important parameter, but in another study on monodisperse polymers with randomly located stickers slopes of 1 and 2 were observed.80 Another example is the recent work by Seifert et al. where it was concluded that the shallow slopes due to molecular weight distribution will be exacerbated when stickers are introduced,29,30 whereas the same group reported similar studies wherein a terminal slope of 1.79 and 0.97 for G′ and G″ turned 1.84 and 1.03 upon introduction of stickers (Figure 16).110 Therefore, the conclusion that the presence of stickers can exacerbate the effect of molecular weight distribution is not always correct;29,30 neither is the conclusion that shallow slopes are due to sticker or molecular weight distribution.45,49,60,69,108 Therefore, in line with our studies, we conclude that the shallowing of the slopes is rather due to the formation of clusters of interconnected chains or aggregates of stickers.48,79,80,107,124−126 Aggregates can either influence the terminal relaxation by CRR leading to parallel G′ and G″ (Figure 5b)49,51 (which should not be misinterpreted with weak gel formation as has been seen in the literature69) or introduce a distribution of relaxation times due to size distribution and hopping (partner exchange) time distribution.47 We have shown that the presence of a parallel G′ and G″ and subsequent plateau can even happen with polymers with one sticker (which intuitively cannot lead to a gel formation) due to CRR events.34
Figure 10.

Comparison of the G′ and G″ mastercurves for polymers without (left), with one (middle), and with two (right) amide hydrogen-bonding groups (weak sticker) per repeating unit. Reproduced with permission from ref (77).
Figure 16.
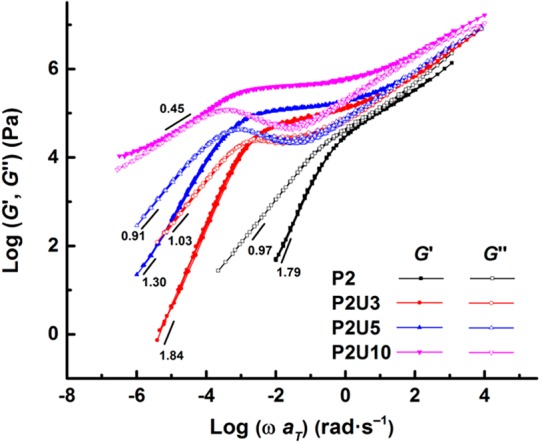
Mastercurve of supramolecular polymers with different contents of sticky side groups (UPy) showing that the shallowing of terminal slopes relative to the precursor only occurs in certain sticker concentrations (not for P2U3). Reproduced with permission from ref (110). Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA.
Miscellaneous
Other types of supramolecular polymers with more sophisticated architecture or dynamics have been studied, too. The majority of works focused on four-armed polymers with sticky end-groups,32,73,81,82,127−131 whereas fewer reports on multiarm (more than four arms)132 or dendronized polymers with sticky ends exist.133,134
On the other hand, perhaps the most interesting supramolecular systems are supramolecular polymer combs and brushes, in which the backbone consists of stickers instead of covalent bonds. The rheological study on comb/brush polymers has been done widely in the literature and can be a fine “database” for comparison with supramolecular systems.135 Staropoli et al. studied the melt dynamics of comb polymers with a few weakly associating arms (Figure 17) and compared their linear rheology with covalent comb polymers.136−138 Although this study was the first of its kind, more research in this field should be done for a general picture in supramolecular graft polymers (with polymeric side chains). For instance, the effect of random presence of arms along the backbone as well as the association strength on the junction point is under investigation by our group.
Figure 17.
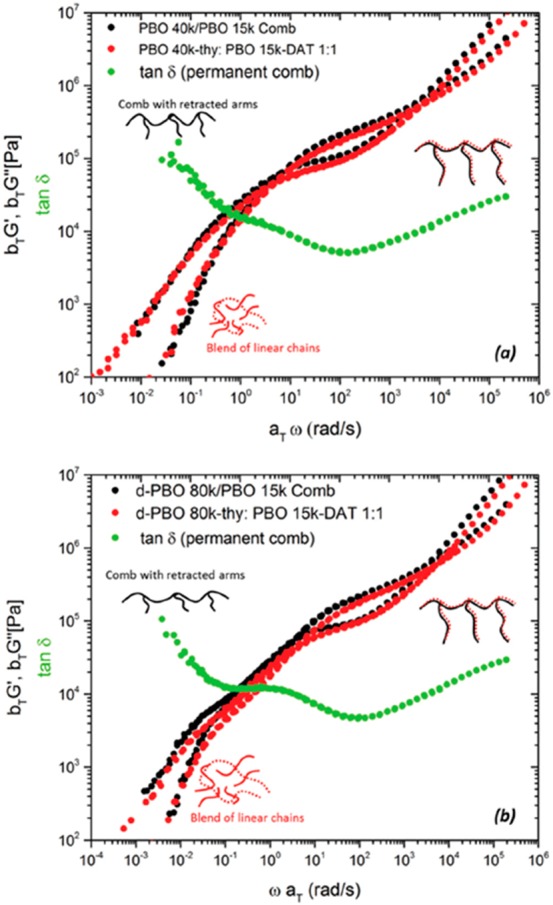
Comparison of rheological mastercurves of covalent (black) and supramolecular comb (red) polymers for two different backbone molecular weights: (a) 40 kg mol–1 and (b) 80 kg mol–1. Reproduced with permission from ref (137). Copyright 2017 the Society of Rheology.
On the other hand, supramolecular brush polymers can show different dynamics in comparison to combs. Bouteiller et al. studied the supramolecular brush dynamics by introducing a sticker in the center of polymer chain, and the corresponding association led to a brush formation.23,33,36,139−142 They explained that different regimes can be found in the rheological mastercurve of these bottlebrush polymers (Figure 18b). For higher concentrations of sticker (shorter arms) at very low frequencies a plateau was observed which was ascribed to gel formation.23 However, assuming that each polymer chain with one central sticker can be considered as monomer, the assembled structure can be a 1D polymer, and the presence of gel cannot be simply explained by the appearance of a plateau. In other words, a gel can be only formed whereby a branched structure increases its size to infinite molecular weight. This system has neither branching (the side chains are dangling arms) nor an infinite molecular weight. However, the presence of a plateau can be due to formation of fibers or due to colloidal jamming. In particular, the magnitude of the plateau modulus is in the range we described as jamming for similar polymers.34 Colloidal jamming is neither a new phenomenon nor limited to supramolecular graft polymers. Hyperstars have been reported to show similar jamming due to impenetrable cores.143 In principle, hyperstars behave as an intermediate between polymers and colloids; after a high-frequency segmental relaxation, a plateau for entangled star arms and then a two-step terminal relaxation can be seen.17−19,144 The two-step process consists of a faster arm retraction and then a structural rearrangement of star cores which is similar to the colloidal materials.143 In the extreme cases of more than 800 short arms, a jamming phenomenon occurs which is due to the excluded volume effects of impenetrable core. This means the center-of-mass motion and terminal relaxation are significantly slowed down, and a plateau is observed instead of liquidlike behavior.143 Hyperstars also do not have any cross-linking but show a plateau very similar to supramolecular brushes (Figure 18a).23,143 Moreover, a Rouse like regime in supramolecular brush polymers was observed in the intermediate frequencies, whose origin was not truly explained (Figure 18b). This regime, however, can be ascribed to CRR relaxation of the arms (see Figure 5).34 We have recently shown that if the supramolecular backbone of the polymer brush imposes steric hindrance, the segments close to the backbone show colloidal jamming. If the molecular weight of the arms is large and mobile enough (high Mn and low Tg), the outermost segments of the arms relax first via arm retraction mechanism. Then, the relaxed parts act as solvent and the backbone can undergo Rouse-like modes (CRR) to partially relax the stress. However, eventually in the time (size) scales close to the backbone of the brush, called the correlation length ξ, each backbone will freeze in the cage of its surrounding counterparts, and a plateau is observed. This plateau is, however, temperature-dependent; at T > Tdiss the stacking will shrink (depolymerize), and a Newtonian behavior is observed. This can be seen schematically in Figure 19. On the other hand, the intermediate frequency plateau was attributed to entanglements (Figure 18b), whereas the arms are far below the critical molecular weight. The authors concluded that the sticker can affect the elasticity of the arms.23 However, similar phenomena have been also observed in the comb polymers without stickers.135,145,146
Figure 18.

Rheological mastercurves. (a) Hyperstars (red) in comparison to the linear analogous (black). Adapted with permission from ref (143). (b) Supramolecular bottlebrush polymers, showing different regimes at different time scales. Reproduced with permission from ref (23).
Figure 19.
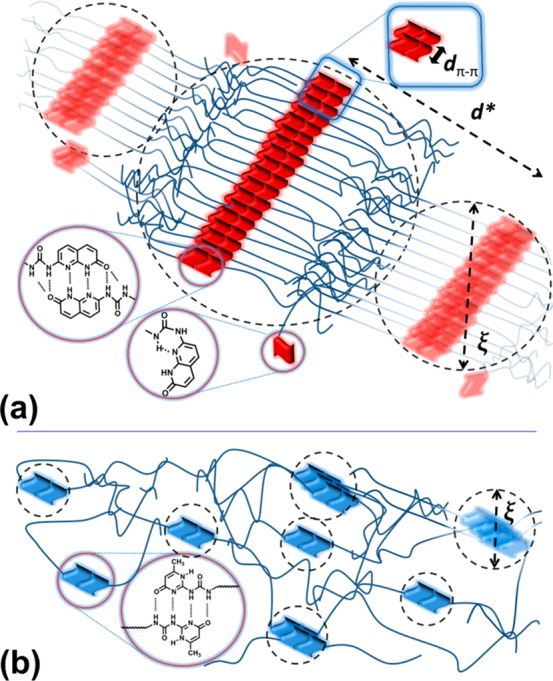
Tentative representation of (a) supramolecular bottlebrush polymers (assuming the interaction between the cores of the supramolecular brushes is responsible for the elastic behavior) in comparison with (b) polymers with small assemblies composed of a few chains, without long-range interaction. d*, dπ–π, and ξ are the lamellar periodicity, the stacking distance, and the correlation length, respectively. Reproduced with permission from ref (34).
We have recently found that the supramolecular polymer brushes can show a transition from colloidal to polymeric materials by changing the arm molecular weight similar to (hyper)star polymers. Therefore, increase of arm molecular weight decreases the stacking (due to decrease in sticker concentration) and increases the polymeric composition of the material (thereby leading to polymeric behavior). Although the supramolecular polymer brush can also be considered as a kind of block copolymer by the sticker being a block and arms the second block, the rheological properties and dynamics are quite different. For one, a block copolymer above TODT relaxes as two polymers attached to each other on the junction point and each block has its own dynamics, but a supramolecular polymer brush can have three regimes: stacked whereby colloidal jamming is present, binary associated (mixed with starlike aggregates) where binary polymers relax similar to linear but double-sized polymers, and single polymers in a melt with no significant effect from the sticker.
New approaches
involving block copolymers in one way or another
lead to materials capable of self-healing.147,148 However, their dynamics are still not clear. In one study by Binder
et al. on dendritic supramolecular copolymers based on the PnBA-Ba/PI-HW
mixture, the authors observed a plateau and a crossover between G′ and G″ and assigned the
corresponding frequency to bond lifetime, whereas the presence of
a plateau can be related to the entanglements and the crossover the
disentanglement time of entangled PI. This could have been more clear
by examination of the molecular dependency of the crossover.149 On the other hand, pure PI-HW also showed a
plateau similar to the PnBA-Ba/PI-HW mixture, and although the authors
referred this discrepancy to the HW aggregation, it seems the supramolecular
association in PnBA-Ba/PI-HW is actually not formed (not the other
way around). In fact, the PnBA used in this study was far below Me and could have already relaxed, and therefore,
no significant change in the dynamics of the PnBA-Ba/PI-HW (in comparison
to pure PI-HW) was observed. Therefore, the assignment of plateau
or moduli crossover is not always trivial. In another study, a triblock
copolymer with outer blocks carrying stickers was synthesized, and
the corresponding mastercurve showed a plateau. The authors used the
plateau modulus, and by considering 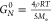 , they calculated the “entanglement
molecular weight”.150 However, the
plateau can have different origins, such as cross-linking of stickers
or long-range phase separation of blocks.20,151 The correct assignment of the plateau was done accurately in a similar
work by another group, whereby a block copolymer with one block carrying
UPy moieties and the other block being PnBA was synthesized. The presence
of a plateau was therefore attributed to the percolated network.152
, they calculated the “entanglement
molecular weight”.150 However, the
plateau can have different origins, such as cross-linking of stickers
or long-range phase separation of blocks.20,151 The correct assignment of the plateau was done accurately in a similar
work by another group, whereby a block copolymer with one block carrying
UPy moieties and the other block being PnBA was synthesized. The presence
of a plateau was therefore attributed to the percolated network.152
The application of hydrophobic blocks as stickers has been reported as well.153−155 By using one type of sticker (metal–ligand interaction) at one end and a hydrophobic block at the other end, one could observe two plateaus with two sticker lifetimes.
The
last important parameter is the calculation of entanglement
molecular weight, which is not always trivial. For instance, in the
synthesis of supramolecular polymers, especially with side stickers,
the precursor which usually has a well-known Me is converted partially to a supramolecular derivative by
changing the side groups. It has been shown that the magnitude of
the plateau modulus GN, terminal relaxation
time τd, and Me depend
on the packing length.69,156 This means if the precursor
has modified side groups, the rheological parameters are not comparable
anymore. Recently, one study compared the rheological properties of
three different polymers with one (P1AP), two (P2AP), or without stickers
(P1EP) on each repeating unit.77 It was
found that the polymers with two stickers per monomer, instead of
having longest terminal relaxation time, in comparison to no or one
sticker, had the shortest terminal relaxation time (Figure 10). The authors justified this
counterintuitive result as the opposing effect of side-chain flexibility
and hydrogen bonding. The authors used the formula τ ∼ Mw3.4 to compare the effect of molecular
weight on terminal relaxation time. However, the three polymers studied,
have different packing density, can be considered as three different
polymers with different Me; considering
τ = KMw3.4, K depends on Me and chemistry of polymers.
In other words, τrep ∼ 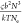 , which can be simplified to τ ∼ Mw3.4 for the same polymers (with
the same Me), but the polymers studied
were quite different in Me.157 Also, the calculated Ge for P1AP was 4 times higher than P2AP. It was concluded that
by using Me =
, which can be simplified to τ ∼ Mw3.4 for the same polymers (with
the same Me), but the polymers studied
were quite different in Me.157 Also, the calculated Ge for P1AP was 4 times higher than P2AP. It was concluded that
by using Me = 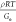 , P1AP has lower Me and therefore higher concentration of physical cross-linking
(associated stickers) than P2AP.77 However,
the lower Me can be related to the lower
packing length of P1AP in comparison to P2AP and therefore more entanglements
(not necessarily more sticker associations). In another study, the
precursor copolymers based on PnBA and PHEA (poly(hydroxylethyl acrylate)
were modified to copolymers with UPy groups instead of hydroxyl moieties
of PHEA. The number of entanglements was calculated based on PnBA Me (= 16.8 g mol–1).109 The authors did not include the effect of packing
length on the final polymers as both the Ge and Me would be different upon side-group
modification.69 In order to have a better
understanding of how much packing length and copolymer composition
can influence the Me, one can compare Me in different compositions of poly(styrene-ran-(methyl methacrylate) (SMMA). For poly(styrene) (PS)
with Me of ∼21 kg mol–1, addition of 23% MMA can lead to a copolymer Me of around 19 kg mol–1.163 This effect is even stronger when comparing the bulky UPy
sticker in comparison to a hydroxyl group in the literature,77,109 whereas styrene and MMA are less different in bulkiness.
, P1AP has lower Me and therefore higher concentration of physical cross-linking
(associated stickers) than P2AP.77 However,
the lower Me can be related to the lower
packing length of P1AP in comparison to P2AP and therefore more entanglements
(not necessarily more sticker associations). In another study, the
precursor copolymers based on PnBA and PHEA (poly(hydroxylethyl acrylate)
were modified to copolymers with UPy groups instead of hydroxyl moieties
of PHEA. The number of entanglements was calculated based on PnBA Me (= 16.8 g mol–1).109 The authors did not include the effect of packing
length on the final polymers as both the Ge and Me would be different upon side-group
modification.69 In order to have a better
understanding of how much packing length and copolymer composition
can influence the Me, one can compare Me in different compositions of poly(styrene-ran-(methyl methacrylate) (SMMA). For poly(styrene) (PS)
with Me of ∼21 kg mol–1, addition of 23% MMA can lead to a copolymer Me of around 19 kg mol–1.163 This effect is even stronger when comparing the bulky UPy
sticker in comparison to a hydroxyl group in the literature,77,109 whereas styrene and MMA are less different in bulkiness.
Outlook
With a deep insight into the rheology of supramolecular polymers, the next step is to manufacture new materials with state-of-the-art technologies like 3D printing. The 3D printing technique or additive manufacturing has evolved recently, combing precision and cost effectiveness to fabricate new materials on demand.158,159 Supramolecular polymers specifically have been manufactured by using this technique to form biomedical scaffolds for regenerative medicine.160,162
Another promising and still developing field of research depending on rheology of supramolecular polymers is self-healing.4,39,98,122,128,147,161 In one type of these materials using noncovalent interactions, one can repair material, and because of highly complementary associations, the mechanical properties stay the same even after a few cycles of damage and repair.
Therefore, in order to apply self-healing characteristic to the new self-associative materials or using 3D printing techniques to fabricate them, one needs to master the rheology of supramolecular polymers.
Concluding Remarks
The research in the field of supramolecular polymers can be considered as “forever evolving” as concurrent development in supramolecular polymer synthesis opens a pathway for new supramolecular polymer dynamics. This Perspective is intended to critically compare the methods developed recently for the linear rheology of supramolecular polymers. Although this research field has gained some degree of maturity, there is still inconsistency in many details within the literature which needs to be addressed in the future. Combining different techniques such as piezo-rheometry, solid-state NMR, and/or dielectric spectroscopy can be beneficial in unraveling the polymer dynamics in complex systems. Moreover, a general and more comprehensive theory is necessary to include the effects of chemistry and phase separation on polymer dynamics. Nevertheless, thus far linear rheology has shown to be a strong and promising tool to reflect the dynamics at the macroscopic level, and it is predicted that by more advanced synthetic techniques soon a new generation of polymers with unprecedented dynamics will arise.
Acknowledgments
The authors are grateful to Evelyne van Ruymbeke for fruitful discussions and Dina Maniar for designing the graphics. The research was supported by a NWO-VICI innovational research grant.
Biographies

Milad Golkaram was born in Tehran, Iran. In 2013 he completed his B.Sc. degree in chemical engineering with a major in polymer chemistry from Arak University. Afterward, he moved to Germany (2013–2015) to attend a master’s program in polymer material science at Martin-Luther University Halle Wittenberg, Halle (Saale), whereby he received a M.Sc. Degree (magna cum laude). During his master’s, he obtained several scholarships such as DAAD and the Young Polymer Scientist Scholarship. He defended his thesis under the supervision of Prof. W. H. Binder on self-healing polymers and moved to The Netherlands to do his PhD at the University of Groningen. Under the supervision of Prof. K. Loos, his PhD involved the synthesis of new supramolecular polymers and study of their melt self-assembly.

Katja Loos is Professor at the Zernike Institute for Advanced Materials of the University of Groningen, The Netherlands, holding the chair of Macromolecular Chemistry and New Polymeric Materials. She specialized in Organic Chemistry and Polymer Chemistry during her university studies at the Johannes Gutenberg Universität in Mainz, Germany, and the University of Massachusetts in Amherst, USA. She moved into the field of Enzymatic Polymerizations during her doctoral research at the University of Bayreuth, Germany, and the Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil. After a postdoctoral research stay at Polytechnic University in Brooklyn, NY, USA, she started an independent research group at the University of Groningen. Already early in her career, Katja Loos was awarded two travel scholarships of the German Academic Exchange Service (DAAD), and she received the very prestigious Feodor Lynen Fellowship award of the Alexander von Humboldt Foundation to conduct her postdoctoral research. During her independent research career, on the basis of her unique approach in combining modern polymer synthesis, (macro) molecular self-assembly, and utilization of the arising structures, The Netherlands Organisation for Scientific Research (NWO) has awarded her the prestigious VIDI grant in 2009 and VICI grant in 2014, the German Research Council (DFG) the Eleonore Trefftz guest professorship within the scope of its excellency initiative and the Alexander von Humboldt Foundation a Friedrich Wilhelm Bessel Research Award in 2019. Katja Loos is a Fellow of the Dutch Polymer Institute (DPI) and the Royal Society of Chemistry (RSC).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.macromol.9b02085.
Appendices for a brief description of physics behind the time marching algorithm (TMA) and hindered fluctuations, arm retraction, and modified mean-field percolation theory of Rubinstein and Semenov (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fouquey C.; Lehn J. M.; Levelut A. M. Molecular Recognition Directed Self-Assembly of Supramolecular Liquid Crystalline Polymers from Complementary Chiral Components. Adv. Mater. 1990, 2, 254–257. 10.1002/adma.19900020506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. L.; Xiao T.; Lin C.; Wang L. Advanced Supramolecular Polymers Constructed by Orthogonal Self-Assembly. Chem. Soc. Rev. 2012, 41, 5950–5968. 10.1039/c2cs35099h. [DOI] [PubMed] [Google Scholar]
- Sijbesma R. P.; Beijer F. H.; Brunsveld L.; Folmer B. J. B.; Hirschberg J. H. K. K.; Lange R. F. M.; Lowe J. K. L.; Meijer E. W. Reversible Polymers Formed from Self-Complementary Monomers Using Quadruple Hydrogen Bonding. Science 1997, 278, 1601–1604. 10.1126/science.278.5343.1601. [DOI] [PubMed] [Google Scholar]
- Cordier P.; Tournilhac F.; Soulié-Ziakovic C.; Leibler L. Self-Healing and Thermoreversible Rubber from Supramolecular Assembly. Nature 2008, 451, 977–980. 10.1038/nature06669. [DOI] [PubMed] [Google Scholar]
- Obert E.; Bellot M.; Bouteiller L.; Andrioletti F.; Lehen-Ferrenbach C.; Boué F. Both Water- and Organo-Soluble Supramolecular Polymer Stabilized by Hydrogen-Bonding and Hydrophobic Interactions. J. Am. Chem. Soc. 2007, 129, 15601–15605. 10.1021/ja074296l. [DOI] [PubMed] [Google Scholar]
- Peterca M.; Percec V.; Imam M. R.; Leowanawat P.; Morimitsu K.; Heiney P. A. Molecular Structure of Helical Supramolecular Dendrimers. J. Am. Chem. Soc. 2008, 130, 14840–14852. 10.1021/ja806524m. [DOI] [PubMed] [Google Scholar]
- Hill J. P.; Jin W.; Kosaka A.; Fukushima T.; Ichihara H.; Shimomura T.; Ito K.; Hashizume T.; Ishii N.; Aida T. Self-Assembled Hexa-peri-hexabenzocoronene Graphitic Nanotube. Science 2004, 304, 1481–1483. 10.1126/science.1097789. [DOI] [PubMed] [Google Scholar]
- Arnaud A.; Belleney J.; Boué F.; Bouteiller L.; Carrot G.; Wintgens V. Aqueous Supramolecular Polymer Formed from an Amphiphilic Perylene Derivative. Angew. Chem., Int. Ed. 2004, 43, 1718–1721. 10.1002/anie.200353434. [DOI] [PubMed] [Google Scholar]
- Alvarenga B. G.; Raynal M.; Bouteiller L.; Sabadini E. Unexpected Solvent Influence on the Rheology of Supramolecular Polymers. Macromolecules 2017, 50, 6631–6636. 10.1021/acs.macromol.7b00786. [DOI] [Google Scholar]
- Krutyeva M.; Brás A. R.; Antonius W.; Hövelmann C. H.; Poulos A. S.; Allgaier J.; Radulescu A.; Lindner P.; Pyckhout-Hintzen W.; Wischnewski A.; Richter D. Association Behavior, Diffusion, and Viscosity of End-Functionalized Supramolecular Poly(Ethylene Glycol) in the Melt State. Macromolecules 2015, 48, 8933–8946. 10.1021/acs.macromol.5b02060. [DOI] [Google Scholar]
- Xing K.; Tress M.; Cao P.-F.; Fan F.; Cheng S.; Saito T.; Sokolov A. P. The Role of Chain-End Association Lifetime in Segmental and Chain Dynamics of Telechelic Polymers. Macromolecules 2018, 51, 8561–8573. 10.1021/acs.macromol.8b01210. [DOI] [Google Scholar]
- Xing K.; Chatterjee S.; Saito T.; Gainaru C.; Sokolov A. P. Impact of Hydrogen Bonding on Dynamics of Hydroxyl-Terminated Polydimethylsiloxane. Macromolecules 2016, 49, 3138–3147. 10.1021/acs.macromol.6b00262. [DOI] [Google Scholar]
- Herbst F.; Binder W. H. Comparing Solution and Melt-State Association of Hydrogen Bonds in Supramolecular Polymers. Polym. Chem. 2013, 4, 3602–3609. 10.1039/c3py00362k. [DOI] [Google Scholar]
- Yan T.; Schröter K.; Herbst F.; Binder W. H.; Thurn-Albrecht T. What Controls the Structure and the Linear and Nonlinear Rheological Properties of Dense, Dynamic Supramolecular Polymer Networks?. Macromolecules 2017, 50, 2973–2985. 10.1021/acs.macromol.6b02507. [DOI] [Google Scholar]
- Yan T.; Schröter K.; Herbst F.; Binder W. H.; Thurn-Albrecht T. Nanostructure and Rheology of Hydrogen-Bonding Telechelic Polymers in the Melt: From Micellar Liquids and Solids to Supramolecular Gels. Macromolecules 2014, 47, 2122–2130. 10.1021/ma402007f. [DOI] [Google Scholar]
- Bobade S. L.; Malmgren T.; Baskaran D. Micellar-Cluster Association of Ureidopyrimidone Functionalized Monochelic Polybutadiene. Polym. Chem. 2014, 5, 910–920. 10.1039/C3PY01002C. [DOI] [Google Scholar]
- Pakula T.; Vlassopoulos D.; Fytas G.; Roovers J. Structure and Dynamics of Melts of Multiarm Polymer Stars. Macromolecules 1998, 31, 8931–8940. 10.1021/ma981043r. [DOI] [Google Scholar]
- Roovers J.; Zhou L. L.; Toporowski P. M.; van der Zwan M.; Iatrou H.; Hadjichristidis N. Regular Star Polymers with 64 and 128 Arms. Models for Polymeric Micelles. Macromolecules 1993, 26, 4324–4331. 10.1021/ma00068a039. [DOI] [Google Scholar]
- Kapnistos M.; Semenov A. N.; Vlassopoulos D.; Roovers J. Viscoelastic Response of Hyperstar Polymers in the Linear Regime. J. Chem. Phys. 1999, 111, 1753–1759. 10.1063/1.479436. [DOI] [Google Scholar]
- Kossuth M. B.; Morse D. C.; Bates F. S. Viscoelastic Behavior of Cubic Phases in Block Copolymer Melts. J. Rheol. 1999, 43, 167–196. 10.1122/1.550981. [DOI] [Google Scholar]
- Liu G.; Feng X.; Lang K.; Zhang R.; Guo D.; Yang S.; Cheng S. Z. D. Dynamics of Shape-Persistent Giant Molecules: Zimm-like Melt, Elastic Plateau, and Cooperative Glass-Like. Macromolecules 2017, 50, 6637–6646. 10.1021/acs.macromol.7b01058. [DOI] [Google Scholar]
- Li J.; Wang Z.; Wen L.; Nie J.; Yang S.; Xu J.; Cheng S. Z. D. Highly Elastic Fibers Made from Hydrogen-Bonded Polymer Complex. ACS Macro Lett. 2016, 5, 814–818. 10.1021/acsmacrolett.6b00346. [DOI] [PubMed] [Google Scholar]
- Callies X.; Véchambre C.; Fonteneau C.; Pensec S.; Chenal J.-M.; Chazeau L.; Bouteiller L.; Ducouret G.; Creton C. Linear Rheology of Supramolecular Polymers Center-Functionalized with Strong Stickers. Macromolecules 2015, 48, 7320–7326. 10.1021/acs.macromol.5b01583. [DOI] [Google Scholar]
- Cheng S.; Zhang M.; Dixit N.; Moore R. B.; Long T. E. Nucleobase Self-Assembly in Supramolecular Adhesives. Macromolecules 2012, 45, 805–812. 10.1021/ma202122r. [DOI] [Google Scholar]
- Zhukhovitskiy A. V.; Zhong M.; Keeler E. G.; Michaelis V. K.; Sun J. E. P.; Hore M. J. A.; Pochan D. J.; Griffin R. G.; Willard A. P.; Johnson J. A. Highly Branched and Loop-Rich Gels via Formation of Metal–Organic Cages Linked by Polymers. Nat. Chem. 2016, 8, 33–41. 10.1038/nchem.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing B.; Xu D.; Wang X.; Zhu Y. Multiresponsive, Critical Gel Behaviors of Polyzwitterion–Polyoxometalate Coacervate Complexes. Macromolecules 2018, 51, 9405–9411. 10.1021/acs.macromol.8b01759. [DOI] [Google Scholar]
- Weng W.; Beck J. B.; Jamieson A. M.; Rowan S. J. Understanding the Mechanism of Gelation and Stimuli-Responsive Nature of a Class of Metallo-Supramolecular Gels. J. Am. Chem. Soc. 2006, 128, 11663–11672. 10.1021/ja063408q. [DOI] [PubMed] [Google Scholar]
- Kumpfer J. R.; Wie J. J.; Swanson J. P.; Beyer F. L.; Mackay M. E.; Rowan S. J. Influence of Metal Ion and Polymer Core on the Melt Rheology of Metallosupramolecular Films. Macromolecules 2012, 45, 473–480. 10.1021/ma201659d. [DOI] [Google Scholar]
- Seiffert S. Effect of Supramolecular Interchain Sticking on the Low-Frequency Relaxation of Transient Polymer Networks. Macromol. Rapid Commun. 2016, 37, 257–264. 10.1002/marc.201500605. [DOI] [PubMed] [Google Scholar]
- Rossow T.; Hackelbusch S.; van Assenbergh P.; Seiffert S. A Modular Construction Kit for Supramolecular Polymer Gels. Polym. Chem. 2013, 4, 2515–2527. 10.1039/c3py00104k. [DOI] [Google Scholar]
- Hackelbusch S.; Rossow T.; van Assenbergh P.; Seiffert S. Chain Dynamics in Supramolecular Polymer Networks. Macromolecules 2013, 46, 6273–6286. 10.1021/ma4003648. [DOI] [Google Scholar]
- Brassinne J.; Cadix A.; Wilson J.; van Ruymbeke E. Dissociating Sticker Dynamics from Chain Relaxation in Supramolecular Polymer Networks—The Importance of Free Partner!. J. Rheol. 2017, 61, 1123–1134. 10.1122/1.4997594. [DOI] [Google Scholar]
- Callies X.; Fonteneau C.; Véchambre C.; Pensec S.; Chenal J.-M.; Chazeau L.; Bouteiller L.; Ducouret G.; Creton C. Linear Rheology of Bis-Urea Functionalized Supramolecular Poly(Butylacrylate)s: Part I – Weak Stickers. Polymer 2015, 69, 233–240. 10.1016/j.polymer.2014.12.053. [DOI] [Google Scholar]
- Golkaram M.; Boetje L.; Dong J.; Suarez L. E. A.; Fodor C.; Maniar D.; van Ruymbeke E.; Faraji S.; Portale G.; Loos K. Supramolecular Mimic for Bottlebrush Polymers in Bulk. ACS Omega 2019, 4, 16481–16492. 10.1021/acsomega.9b02126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel W. P. J.; Portale G.; Wisse E.; Dankers P. Y. W.; Meijer E. W. Aggregation of Ureido-Pyrimidinone Supramolecular Thermoplastic Elastomers into Nanofibers: A Kinetic Analysis. Macromolecules 2011, 44, 6776–6784. 10.1021/ma201303s. [DOI] [Google Scholar]
- Véchambre C.; Callies X.; Fonteneau C.; Ducouret G.; Pensec S.; Bouteiller L.; Creton C.; Chenal J.-M.; Chazeau L. Microstructure and Self-Assembly of Supramolecular Polymers Center-Functionalized with Strong Stickers. Macromolecules 2015, 48, 8232–8239. 10.1021/acs.macromol.5b01584. [DOI] [Google Scholar]
- Callies X.; Herscher O.; Fonteneau C.; Robert A.; Pensec S.; Bouteiller L.; Ducouret G.; Creton C. Combined Effect of Chain Extension and Supramolecular Interactions on Rheological and Adhesive Properties of Acrylic Pressure-Sensitive Adhesives. ACS Appl. Mater. Interfaces 2016, 8, 33307–33315. 10.1021/acsami.6b11045. [DOI] [PubMed] [Google Scholar]
- Callies X.; Ressouche E.; Fonteneau C.; Ducouret G.; Pensec S.; Bouteiller L.; Creton C. Effect of the Strength of Stickers on Rheology and Adhesion of Supramolecular Center-Functionalized Polyisobutenes. Langmuir 2018, 34, 12625–12634. 10.1021/acs.langmuir.8b02533. [DOI] [PubMed] [Google Scholar]
- Yan T.; Schröter K.; Herbst F.; Binder W. H.; Thurn-Albrecht T. Unveiling the Molecular Mechanism of Self-Healing in a Telechelic, Supramolecular Polymer Network. Sci. Rep. 2016, 6, 32356. 10.1038/srep32356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst F.; Schröter K.; Gunkel I.; Gröger S.; Thurn-Albrecht T.; Balbach J.; Binder W. Aggregation and Chain Dynamics in Supramolecular Polymers by Dynamic Rheology: Cluster Formation and Self-Aggregation. Macromolecules 2010, 43, 10006–10016. 10.1021/ma101962y. [DOI] [Google Scholar]
- Rubinstein M.; Semenov A. N. Dynamics of Entangled Solutions of Associating Polymers. Macromolecules 2001, 34, 1058–1068. 10.1021/ma0013049. [DOI] [Google Scholar]
- Rubinstein M.; Semenov A. N. Thermoreversible Gelation in Solutions of Associating Polymers. 2. Linear Dynamics. Macromolecules 1998, 31, 1386–1397. 10.1021/ma970617+. [DOI] [Google Scholar]
- Gold B. J.; Hövelmann C. H.; Lühmann N.; Pyckhout-Hintzen W.; Wischnewski A.; Richter D. The Microscopic Origin of the Rheology in Supramolecular Entangled Polymer Networks. J. Rheol. 2017, 61, 1211–1226. 10.1122/1.4998159. [DOI] [Google Scholar]
- Gold B. J.; Hövelmann C. H.; Lühmann N.; Székely N. K.; Pyckhout-Hintzen W.; Wischnewski A.; Richter D. Importance of Compact Random Walks for the Rheology of Transient Networks. ACS Macro Lett. 2017, 6, 73–77. 10.1021/acsmacrolett.6b00880. [DOI] [PubMed] [Google Scholar]
- Shabbir A.; Javakhishvili I.; Cerveny S.; Hvilsted S.; Skov A. L.; Hassager O.; Alvarez N. J. Linear Viscoelastic and Dielectric Relaxation Response of Unentangled UPy-Based Supramolecular Networks. Macromolecules 2016, 49, 3899–3910. 10.1021/acs.macromol.6b00122. [DOI] [Google Scholar]
- Semenov A. N.; Rubinstein M. Dynamics of Entangled Associating Polymers with Large Aggregates. Macromolecules 2002, 35, 4821–4837. 10.1021/ma0117965. [DOI] [Google Scholar]
- Amin D.; Likhtman A. E.; Wang Z. Dynamics in Supramolecular Polymer Networks Formed by Associating Telechelic Chains. Macromolecules 2016, 49, 7510–7524. 10.1021/acs.macromol.6b00561. [DOI] [Google Scholar]
- Golkaram M.; Fodor C.; van Ruymbeke E.; Loos K. Linear Viscoelasticity of Weakly Hydrogen-Bonded Polymers near and below the Sol–Gel Transition. Macromolecules 2018, 51, 4910–4916. 10.1021/acs.macromol.8b00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadi M.; Jangizehi A.; van Ruymbeke E.; Seiffert S. Deconvolution of the Effects of Binary Associations and Collective Assemblies on the Rheological Properties of Entangled Side-Chain Supramolecular Polymer Networks. Macromolecules 2019, 52, 5255–5267. 10.1021/acs.macromol.9b00323. [DOI] [Google Scholar]
- Jangizehi A.; Ghaffarian R.; Ahmadi M. Dynamics of Entangled Supramolecular Polymer Networks in Presence of High-Order Associations of Strong Hydrogen Bonding Groups. Polym. Adv. Technol. 2018, 29, 726–735. 10.1002/pat.4178. [DOI] [Google Scholar]
- Hawke L. G. D.; Ahmadi M.; Goldansaz H.; van Ruymbeke E. Viscoelastic Properties of Linear Associating Poly(n-Butyl Acrylate) Chains. J. Rheol. 2016, 60, 297–310. 10.1122/1.4942231. [DOI] [Google Scholar]
- Goldansaz H.; Fustin C.-A.; Wübbenhorst M.; van Ruymbeke E. How Supramolecular Assemblies Control Dynamics of Associative Polymers: Toward a General Picture. Macromolecules 2016, 49, 1890–1902. 10.1021/acs.macromol.5b01535. [DOI] [Google Scholar]
- Milner S. T.; Mcleish T. Parameter-Free Theory for Stress Relaxation in Star Polymer Melts. Macromolecules 1997, 30, 2159–2166. 10.1021/ma961559f. [DOI] [Google Scholar]
- Kapnistos M.; Vlassopoulos D.; Roovers J.; Leal L. Linear Rheology of Architecturally Complex Macromolecules: Comb Polymers with Linear Backbones. Macromolecules 2005, 38, 7852–7862. 10.1021/ma050644x. [DOI] [Google Scholar]
- Milner S. T.; McLeish T. C. B. Arm-Length Dependence of Stress Relaxation in Star Polymer Melts. Macromolecules 1998, 31, 7479–7482. 10.1021/ma980060d. [DOI] [Google Scholar]
- Inkson N. J.; Graham R. S.; McLeish T. C. B.; Groves D. J.; Fernyhough C. M. Viscoelasticity of Monodisperse Comb Polymer Melts. Macromolecules 2006, 39, 4217–4227. 10.1021/ma060018f. [DOI] [Google Scholar]
- Mcleish T. Molecular Rheology of H-Polymers. Macromolecules 1988, 21, 1062–1070. 10.1021/ma00182a037. [DOI] [Google Scholar]
- Zhu J.; Likhtman A.; Wang Z. Arm Retraction Dynamics of Entangled Star Polymers: A Forward Flux Sampling Method Study. J. Chem. Phys. 2017, 147, 044907. 10.1063/1.4995422. [DOI] [PubMed] [Google Scholar]
- van Ruymbeke E.; Masubuchi Y.; Watanabe H. Effective Value of the Dynamic Dilution Exponent in Bidisperse Linear Polymers: From 1 to 4/3. Macromolecules 2012, 45, 2085–2098. 10.1021/ma202167q. [DOI] [Google Scholar]
- Ahmadi M.; Hawke L. G. D.; Goldansaz H.; van Ruymbeke E. Dynamics of Entangled Linear Supramolecular Chains with Sticky Side Groups: Influence of Hindered Fluctuations. Macromolecules 2015, 48, 7300–7310. 10.1021/acs.macromol.5b00733. [DOI] [Google Scholar]
- German I.; D’Agosto F.; Boisson C.; Tencé-Girault S.; Soulié-Ziakovic C. Microphase Separation and Crystallization in H-Bonding End-Functionalized Polyethylenes. Macromolecules 2015, 48, 3257–3268. 10.1021/ma502304k. [DOI] [Google Scholar]
- Lacombe J.; Soulié-Ziakovic C. Lamellar Mesoscopic Organization of Supramolecular Polymers: A Necessary Pre-Ordering Secondary Structure. Polym. Chem. 2017, 8, 5954–5961. 10.1039/C7PY01219E. [DOI] [Google Scholar]
- Lacombe J.; Pearson S.; Pirolt F.; Norsic S.; D’Agosto F.; Boisson C.; Soulié-Ziakovic C. Structural and Mechanical Properties of Supramolecular Polyethylenes. Macromolecules 2018, 51, 2630–2640. 10.1021/acs.macromol.8b00270. [DOI] [Google Scholar]
- Cortese J.; Soulié-Ziakovic C.; Cloitre M.; Tencé-Girault S.; Leibler L. Order-Disorder Transition in Supramolecular Polymers. J. Am. Chem. Soc. 2011, 133, 19672–19675. 10.1021/ja209126a. [DOI] [PubMed] [Google Scholar]
- Cortese J.; Soulié-Ziakovic C.; Tencé-Girault S.; Leibler L. Suppression of Mesoscopic Order by Complementary Interactions in Supramolecular Polymers. J. Am. Chem. Soc. 2012, 134, 3671–3674. 10.1021/ja2119496. [DOI] [PubMed] [Google Scholar]
- Chen S.; Binder W. H. Dynamic Ordering and Phase Segregation in Hydrogen-Bonded Polymers. Acc. Chem. Res. 2016, 49, 1409–1420. 10.1021/acs.accounts.6b00174. [DOI] [PubMed] [Google Scholar]
- Cheng C. C.; Chang F. C.; Wang J. H.; Chu Y.; Wang Y. S.; Lee D. J.; Chuang W. T.; Xin Z. Large-Scale Production of Ureido-Cytosine Based Supramolecular Polymers with Well-Controlled Hierarchical Nanostructures. RSC Adv. 2015, 5, 76451–76457. 10.1039/C5RA15849D. [DOI] [Google Scholar]
- Beiner M.; Huth H. Nanophase Separation and Hindered Glass Transition in Side-Chain Polymers. Nat. Mater. 2003, 2, 595–599. 10.1038/nmat966. [DOI] [PubMed] [Google Scholar]
- Shabbir A.; Goldansaz H.; Hassager O.; van Ruymbeke E.; Alvarez N. J. Effect of Hydrogen Bonding on Linear and Nonlinear Rheology of Entangled Polymer Melts. Macromolecules 2015, 48, 5988–5996. 10.1021/acs.macromol.5b00757. [DOI] [Google Scholar]
- López-Barrón C. R.; Tsou A. H.; Hagadorn J. R.; Throckmorton J. A. Highly Entangled α-Olefin Molecular Bottlebrushes: Melt Structure, Linear Rheology, and Interchain Friction Mechanism. Macromolecules 2018, 51, 6958–6966. 10.1021/acs.macromol.8b01431. [DOI] [Google Scholar]
- Qian Z.; Koh Y. P.; Pallaka M. R.; Chang A. B.; Lin T.-P.; Guzmán P. E.; Grubbs R. H.; Simon S. L.; McKenna G. B. Linear Rheology of a Series of Second-Generation Dendronized Wedge Polymers. Macromolecules 2019, 52, 2063–2074. 10.1021/acs.macromol.8b02122. [DOI] [Google Scholar]
- Susa A.; Mordvinkin A.; Saalwächter K.; van der Zwaag S.; Garcia S. J. Identifying the Role of Primary and Secondary Interactions on the Mechanical Properties and Healing of Densely Branched Polyimides. Macromolecules 2018, 51, 8333–8345. 10.1021/acs.macromol.8b01396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossow T.; Seiffert S. Supramolecular Polymer Gels with Potential Model-Network Structure. Polym. Chem. 2014, 5, 3018–3029. 10.1039/c3py01692g. [DOI] [Google Scholar]
- Söntjens S. H. M.; Sijbesma R. P.; van Genderen M. H. P.; Meijer E. W. Stability and Lifetime of Quadruply Hydrogen Bonded 2-Ureido-4[1H]-Pyrimidinone Dimers. J. Am. Chem. Soc. 2000, 122, 7487–7493. 10.1021/ja000435m. [DOI] [Google Scholar]
- Aida T.; Meijer E. W.; Stupp S. I. Functional Supramolecular Polymers. Science 2012, 335, 813–817. 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goor O. J. G. M.; Hendrikse S. I. S.; Dankers P. Y. W.; Meijer E. W. From Supramolecular Polymers to Multi-Component Biomaterials. Chem. Soc. Rev. 2017, 46, 6621–6637. 10.1039/C7CS00564D. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Wang C.; Guo Y.; Peng C.; Narayanan A.; Kaur S.; Xu Y.; Weiss R. A.; Joy A. Opposing Effects of Side-Chain Flexibility and Hydrogen Bonding on the Thermal, Mechanical, and Rheological Properties of Supramolecularly Cross-Linked Polyesters. Macromolecules 2018, 51, 9294–9305. 10.1021/acs.macromol.8b01781. [DOI] [Google Scholar]
- Lewis C. L.; Stewart K.; Anthamatten M. The Influence of Hydrogen Bonding Side-Groups on Viscoelastic Behavior of Linear and Network Polymers. Macromolecules 2014, 47, 729–740. 10.1021/ma402368s. [DOI] [Google Scholar]
- Chen Q.; Huang C.; Weiss R. A.; Colby R. H. Viscoelasticity of Reversible Gelation for Ionomers. Macromolecules 2015, 48, 1221–1230. 10.1021/ma502280g. [DOI] [Google Scholar]
- Zhang Z.; Huang C.; Weiss R. A.; Chen Q. Association Energy in Strongly Associative Polymers. J. Rheol. (Melville, NY, U. S.) 2017, 61, 1199–1207. 10.1122/1.4997586. [DOI] [Google Scholar]
- Zhuge F.; Hawke L.; Fustin C.-A.; Gohy J.-F.; van Ruymbeke E. Decoding the Linear Viscoelastic Properties of Model Telechelic Metallo-Supramolecular Polymers. J. Rheol. (Melville, NY, U. S.) 2017, 61, 1245–1262. 10.1122/1.4997593. [DOI] [Google Scholar]
- Zhuge F.; Brassinne J.; Fustin C.-A.; van Ruymbeke E.; Gohy J.-F. Synthesis and Rheology of Bulk Metallo-Supramolecular Polymers from Telechelic Entangled Precursors. Macromolecules 2017, 50, 5165–5175. 10.1021/acs.macromol.7b00646. [DOI] [Google Scholar]
- Henderson K. J.; Zhou T. C.; Otim K. J.; Shull K. R. Ionically Cross-Linked Triblock Copolymer Hydrogels with High Strength. Macromolecules 2010, 43, 6193–6201. 10.1021/ma100963m. [DOI] [Google Scholar]
- Loveless D. M.; Jeon S. L.; Craig S. L. Rational Control of Viscoelastic Properties in Multicomponent Associative Polymer Networks. Macromolecules 2005, 38, 10171–10177. 10.1021/ma0518611. [DOI] [Google Scholar]
- Yount W. C.; Loveless D. M.; Craig S. L. Small-Molecule Dynamics and Mechanisms Underlying the Macroscopic Mechanical Properties of Coordinatively Cross-Linked Polymer Networks. J. Am. Chem. Soc. 2005, 127, 14488–14496. 10.1021/ja054298a. [DOI] [PubMed] [Google Scholar]
- Yount W. C.; Loveless D. M.; Craig S. L. Strong Means Slow: Dynamic Contributions to the Bulk Mechanical Properties of Supramolecular Networks. Angew. Chem., Int. Ed. 2005, 44, 2746–2748. 10.1002/anie.200500026. [DOI] [PubMed] [Google Scholar]
- Serpe M. J.; Craig S. L. Physical Organic Chemistry of Supramolecular Polymers. Langmuir 2007, 23, 1626–1634. 10.1021/la0621416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R. K.; Hohlbein N.; Garcia S. J.; Schmidt A. M.; van der Zwaag S. Connecting Supramolecular Bond Lifetime and Network Mobility for Scratch Healing in Poly(Butyl Acrylate) Ionomers Containing Sodium, Zinc and Cobalt. Phys. Chem. Chem. Phys. 2015, 17, 1697–1704. 10.1039/C4CP04015E. [DOI] [PubMed] [Google Scholar]
- Cates M. E. Reptation of Living Polymers: Dynamics of Entangled Polymers in the Presence of Reversible Chain-Scission Reactions. Macromolecules 1987, 20, 2289–2296. 10.1021/ma00175a038. [DOI] [Google Scholar]
- Watanabe H.; Matsumiya Y.; Masubuchi Y.; Urakawa O.; Inoue T. Viscoelastic Relaxation of Rouse Chains Undergoing Head-to-Head Association and Dissociation: Motional Coupling through Chemical Equilibrium. Macromolecules 2015, 48, 3014–3030. 10.1021/acs.macromol.5b00409. [DOI] [Google Scholar]
- Kwon Y.; Matsumiya Y.; Watanabe H. Viscoelastic and Orientational Relaxation of Linear and Ring Rouse Chains Undergoing Reversible End-Association and Dissociation. Macromolecules 2016, 49, 3593–3607. 10.1021/acs.macromol.6b00424. [DOI] [Google Scholar]
- Watanabe H.; Matsumiya Y.; Kwon Y. Viscoelastic and Dielectric Relaxation of Reptating Type-A Chains Affected by Reversible Head-to-Head Association and Dissociation. Macromolecules 2018, 51, 6476–6496. 10.1021/acs.macromol.8b00691. [DOI] [Google Scholar]
- Matsumiya Y.; Watanabe H.; Urakawa O.; Inoue T. Experimental Test for Viscoelastic Relaxation of Polyisoprene Undergoing Monofunctional Head-to-Head Association and Dissociation. Macromolecules 2016, 49, 7088–7095. 10.1021/acs.macromol.6b01313. [DOI] [Google Scholar]
- Watanabe H.; Matsumiya Y.; Kwon Y. Dynamics of Rouse Chains Undergoing Head-to-Head Association and Dissociation: Difference between Dielectric and Viscoelastic Relaxation. J. Rheol. 2017, 61, 1151–1170. 10.1122/1.4997579. [DOI] [Google Scholar]
- Kieltyka R. E.; Pape A. C. H.; Albertazzi L.; Nakano Y.; Bastings M. M. C.; Voets I. K.; Dankers P. Y. W.; Meijer E. W. Mesoscale Modulation of Supramolecular Ureidopyrimidinone-Based Poly(Ethylene Glycol) Transient Networks in Water. J. Am. Chem. Soc. 2013, 135, 11159–11164. 10.1021/ja403745w. [DOI] [PubMed] [Google Scholar]
- Boothroyd S. C.; Hoyle D. M.; McLeish T. C. B.; Munch E.; Schach R.; Smith A. J.; Thompson R. L. Association and Relaxation of Supra-Macromolecular Polymers. Soft Matter 2019, 15, 5296–5307. 10.1039/C8SM02580K. [DOI] [PubMed] [Google Scholar]
- Kan L.; Zhang P.; Jiang H.; Zhang S.; Liu Z.; Zhang X.; Ma N.; Qiu D.; Wei H. Microphase Separation of a Quadruple Hydrogen Bonding Supramolecular Polymer: Effect of the Steric Hindrance of the Ureido-Pyrimidone on Their Viscoelasticity. RSC Adv. 2019, 9, 8905–8911. 10.1039/C8RA08861F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston K. R.; Jackson A.-M. S.; Yost R. W.; Carman H. S.; Sheares Ashby V. Supramolecular Engineering Polyesters: Endgroup Functionalization of Glycol Modified PET with Ureidopyrimidinone. Polym. Chem. 2016, 7, 6744–6751. 10.1039/C6PY01421F. [DOI] [Google Scholar]
- Dankers P. Y. W.; van Leeuwen E. N. M.; van Gemert G. M. L.; Spiering A. J. H.; Harmsen M. C.; Brouwer L. A.; Janssen H. M.; Bosman A. W.; van Luyn M. J. A.; Meijer E. W. Chemical and Biological Properties of Supramolecular Polymer Systems Based on Oligocaprolactones. Biomaterials 2006, 27, 5490–5501. 10.1016/j.biomaterials.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Lou N.; Wang Y.; Li X.; Li H.; Wang P.; Wesdemiotis C.; Sokolov A. P.; Xiong H. Dielectric Relaxation and Rheological Behavior of Supramolecular Polymeric Liquid. Macromolecules 2013, 46, 3160–3166. 10.1021/ma400088w. [DOI] [Google Scholar]
- van Beek D. J. M.; Spiering A. J. H.; Peters G. W. M.; te Nijenhuis K.; Sijbesma R. P. Unidirectional Dimerization and Stacking of Ureidopyrimidinone End Groups in Polycaprolactone Supramolecular Polymers. Macromolecules 2007, 40, 8464–8475. 10.1021/ma0712394. [DOI] [Google Scholar]
- Goldansaz H.; Voleppe Q.; Piogé S.; Fustin C. A.; Gohy J. F.; Brassinne J.; Auhl D.; van Ruymbeke E. Controlling the Melt Rheology of Linear Entangled Metallo-Supramolecular Polymers. Soft Matter 2015, 11, 762–774. 10.1039/C4SM02319F. [DOI] [PubMed] [Google Scholar]
- Sivakova S.; Bohnsack D. A.; Mackay M. E.; Suwanmala P.; Rowan S. J. Utilization of a Combination of Weak Hydrogen-Bonding Interactions and Phase Segregation to Yield Highly Thermosensitive Supramolecular Polymers. J. Am. Chem. Soc. 2005, 127, 18202–18211. 10.1021/ja055245w. [DOI] [PubMed] [Google Scholar]
- Zhong M.; Wang R.; Kawamoto K.; Olsen B. D.; Johnson J. A. Quantifying the Impact of Molecular Defects on Polymer Network Elasticity. Science 2016, 353, 1264–1268. 10.1126/science.aag0184. [DOI] [PubMed] [Google Scholar]
- Feldman K. E.; Kade M. J.; Meijer E. W.; Hawker C. J.; Kramer E. J. Model Transient Networks from Strongly Hydrogen-Bonded Polymers. Macromolecules 2009, 42, 9072–9081. 10.1021/ma901668w. [DOI] [Google Scholar]
- Shivokhin M.; Narita T.; Talini L.; Habicht A. K. T.; Seiffert S.; Indei T.; Schieber J. Interplay of Entanglement and Association Effects on the Dynamics of Semidilute Solutions of Multisticker Polymer Chains. J. Rheol. (Melville, NY, U. S.) 2017, 61, 1231–1241. 10.1122/1.4997740. [DOI] [Google Scholar]
- Zhang Z.; Liu C.; Cao X.; Gao L.; Chen Q. Linear Viscoelastic and Dielectric Properties of Strongly Hydrogen-Bonded Polymers near the Sol–Gel Transition. Macromolecules 2016, 49, 9192–9202. 10.1021/acs.macromol.6b02017. [DOI] [Google Scholar]
- Cui G.; Boudara V. A. H.; Huang Q.; Baeza G. P.; Wilson A. J.; Hassager O.; Read D. J.; Mattsson J. Linear Shear and Nonlinear Extensional Rheology of Unentangled Supramolecular Side-Chain Polymers. J. Rheol. (Melville, NY, U. S.) 2018, 62, 1155–1174. 10.1122/1.5012349. [DOI] [Google Scholar]
- Jangizehi A.; Ghaffarian S. R.; Schmolke W.; Seiffert S. Dominance of Chain Entanglement over Transient Sticking on Chain Dynamics in Hydrogen-Bonded Supramolecular Polymer Networks in the Melt. Macromolecules 2018, 51, 2859–2871. 10.1021/acs.macromol.7b02180. [DOI] [Google Scholar]
- Jangizehi A.; Ahmadi M.; Seiffert S. Dynamics of Supramolecular Associative Polymer Networks at the Interplay of Chain Entanglement, Transient Chain Association, and Chain-Sticker Clustering. J. Polym. Sci., Part B: Polym. Phys. 2019, 57, 1209–1223. 10.1002/polb.24782. [DOI] [Google Scholar]
- Wu S.; Cao X.; Zhang Z.; Chen Q.; Matsumiya Y.; Watanabe H. Molecular Design of Highly Stretchable Ionomers. Macromolecules 2018, 51, 4735–4746. 10.1021/acs.macromol.8b00617. [DOI] [Google Scholar]
- Wu S.; Liu S.; Zhang Z.; Chen Q. Dynamics of Telechelic Ionomers with Distribution of Number of Ionic Stickers at Chain Ends. Macromolecules 2019, 52, 2265–2276. 10.1021/acs.macromol.8b01776. [DOI] [Google Scholar]
- Chen Q.; Zhang Z.; Colby R. H. Viscoelasticity of Entangled Random Polystyrene Ionomers. J. Rheol. (Melville, NY, U. S.) 2016, 60, 1031–1040. 10.1122/1.4955432. [DOI] [Google Scholar]
- Lopez C. G. Entanglement Properties of Polyelectrolytes in Salt-Free and Excess-Salt Solutions. ACS Macro Lett. 2019, 8, 979–983. 10.1021/acsmacrolett.9b00161. [DOI] [PubMed] [Google Scholar]
- Rubinstein M.; Liao Q.; Panyukov S. Structure of Liquid Coacervates Formed by Oppositely Charged Polyelectrolytes. Macromolecules 2018, 51, 9572–9588. 10.1021/acs.macromol.8b02059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreev M.; Prabhu V. M.; Douglas J. F.; Tirrell M.; de Pablo J. J. Complex Coacervation in Polyelectrolytes from a Coarse-Grained Model. Macromolecules 2018, 51, 6717–6723. 10.1021/acs.macromol.8b00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamad F. G.; Chen Q.; Colby R. H. Linear Viscoelasticity and Swelling of Polyelectrolyte Complex Coacervates. Macromolecules 2018, 51, 5547–5555. 10.1021/acs.macromol.8b00401. [DOI] [Google Scholar]
- Chen Q.; Bao N.; Wang J.-H. H.; Tunic T.; Liang S.; Colby R. H. Linear Viscoelasticity and Dielectric Spectroscopy of Ionomer/Plasticizer Mixtures: A Transition from Ionomer to Polyelectrolyte. Macromolecules 2015, 48, 8240–8252. 10.1021/acs.macromol.5b01958. [DOI] [Google Scholar]
- Rao Y. L.; Feig V.; Gu X.; Nathan Wang G. J.; Bao Z. The Effects of Counter Anions on the Dynamic Mechanical Response in Polymer Networks Crosslinked by Metal-Ligand Coordination. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 3110–3116. 10.1002/pola.28675. [DOI] [Google Scholar]
- Brassinne J.; Jochum D. F.; Fustin C.-A.; Gohy J.-F. Revealing the Supramolecular Nature of Side-Chain Terpyridine-Functionalized Polymer Networks. Int. J. Mol. Sci. 2015, 16, 990–1007. 10.3390/ijms16010990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M.; Noro A.; Matsushita Y. Viscoelastic Properties of Supramolecular Soft Materials with Transient Polymer Network. J. Polym. Sci., Part B: Polym. Phys. 2014, 52, 755–764. 10.1002/polb.23479. [DOI] [Google Scholar]
- Tan C. S. Y.; Agmon G.; Liu J.; Hoogland D.; Janeček E.-R.; Appel E. A.; Scherman O. A. Distinguishing Relaxation Dynamics in Transiently Crosslinked Polymeric Networks. Polym. Chem. 2017, 8, 5336–5343. 10.1039/C7PY00574A. [DOI] [Google Scholar]
- Xu D.; Craig S. L. Scaling Laws in Supramolecular Polymer Networks. Macromolecules 2011, 44, 5465–5472. 10.1021/ma200096s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Wang C.; Chen Q.; Colby R. H.; Weiss R. A. Reversible Gelation Model Predictions of the Linear Viscoelasticity of Oligomeric Sulfonated Polystyrene Ionomer Blends. Macromolecules 2016, 49, 3936–3947. 10.1021/acs.macromol.6b00620. [DOI] [Google Scholar]
- Chen Q.; Colby R. H. Linear Viscoelasticity of Sulfonated Styrene Oligomers near the Sol-Gel Transition. Korea-Australia Rheol. J. 2014, 26, 257–261. 10.1007/s13367-014-0030-4. [DOI] [Google Scholar]
- Zhang Z.; Chen Q.; Colby R. H. Dynamics of Associative Polymers. Soft Matter 2018, 14, 2961–2977. 10.1039/C8SM00044A. [DOI] [PubMed] [Google Scholar]
- Tang S.; Habicht A.; Li S.; Seiffert S.; Olsen B. D. Self-Diffusion of Associating Star-Shaped Polymers. Macromolecules 2016, 49, 5599–5608. 10.1021/acs.macromol.6b00959. [DOI] [Google Scholar]
- Stukalin E. B.; Cai L.-H.; Kumar N. A.; Leibler L.; Rubinstein M. Self-Healing of Unentangled Polymer Networks with Reversible Bonds. Macromolecules 2013, 46, 7525–7541. 10.1021/ma401111n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wietor J.-L.; van Beek D. J. M.; Peters G. W.; Mendes E.; Sijbesma R. P. Effects of Branching and Crystallization on Rheology of Polycaprolactone Supramolecular Polymers with Ureidopyrimidinone End Groups. Macromolecules 2011, 44, 1211–1219. 10.1021/ma1026065. [DOI] [Google Scholar]
- Rossow T.; Habicht A. K. T.; Seiffert S. Relaxation and Dynamics in Transient Polymer Model Networks. Macromolecules 2014, 47, 6473–6482. 10.1021/ma5013144. [DOI] [Google Scholar]
- Habicht A. K. T.; Czarnecki S.; Rossow T.; Seiffert S. Connectivity Defects Enhance Chain Dynamics in Supramolecular Polymer Model-network Gels. J. Polym. Sci., Part B: Polym. Phys. 2017, 55, 19–29. 10.1002/polb.24250. [DOI] [Google Scholar]
- Metri V.; Louhichi A.; Yan J.; Baeza G. P.; Matyjaszewski K.; Vlassopoulos D.; Briels W. J. Physical Networks from Multifunctional Telechelic Star Polymers: A Rheological Study by Experiments and Simulations. Macromolecules 2018, 51, 2872–2886. 10.1021/acs.macromol.7b02613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz L. F.; Costanzo S.; Huang Q.; Schlüter A. D.; Vlassopoulos D. Dendronized Polymers with Ureidopyrimidinone Groups: An Efficient Strategy To Tailor Intermolecular Interactions, Rheology, and Fracture. Macromolecules 2017, 50, 5176–5187. 10.1021/acs.macromol.7b00747. [DOI] [Google Scholar]
- Costanzo S.; Scherz L. F.; Schweizer T.; Kröger M.; Floudas G.; Schlüter A. D.; Vlassopoulos D. Rheology and Packing of Dendronized Polymers. Macromolecules 2016, 49, 7054–7068. 10.1021/acs.macromol.6b01311. [DOI] [Google Scholar]
- Hu M.; Xia Y.; Mckenna G.; Kornfield J.; Grubbs R. Linear Rheological Response of a Series of Densely Branched Brush Polymers. Macromolecules 2011, 44, 6935–6943. 10.1021/ma2009673. [DOI] [Google Scholar]
- Allgaier J.; Hövelmann C. H.; Wei Z.; Staropoli M.; Pyckhout-Hintzen W.; Lühmann N.; Willbold S. Synthesis and Rheological Behavior of Poly(1,2-Butylene Oxide) Based Supramolecular Architectures. RSC Adv. 2016, 6, 6093–6106. 10.1039/C5RA24547H. [DOI] [Google Scholar]
- Staropoli M.; Raba A.; Hovelmann C. H.; Appavou M.-S.; Allgaier J.; Krutyeva M.; Pyckhout-Hintzen W.; Wischnewski A.; Richter D. Melt Dynamics of Supramolecular Comb Polymers: Viscoelastic and Dielectric Response. J. Rheol. (Melville, NY, U. S.) 2017, 61, 1185–1196. 10.1122/1.5001059. [DOI] [Google Scholar]
- Staropoli M.; Raba A.; Hövelmann C. H.; Krutyeva M.; Allgaier J.; Appavou M.-S.; Keiderling U.; Stadler F. J.; Pyckhout-Hintzen W.; Wischnewski A.; et al. Hydrogen Bonding in a Reversible Comb Polymer Architecture: A Microscopic and Macroscopic Investigation. Macromolecules 2016, 49, 5692–5703. 10.1021/acs.macromol.6b00978. [DOI] [Google Scholar]
- Louhichi A.; Jacob A. R.; Bouteiller L.; Vlassopoulos D. Humidity Affects the Viscoelastic Properties of Supramolecular Living Polymers. J. Rheol. (Melville, NY, U. S.) 2017, 61, 1173–1182. 10.1122/1.4997600. [DOI] [Google Scholar]
- Pensec S.; Nouvel N.; Guilleman A.; Creton C.; Boué F.; Bouteiller L. Self-Assembly in Solution of a Reversible Comb-Shaped Supramolecular Polymer. Macromolecules 2010, 43, 2529–2534. 10.1021/ma901709e. [DOI] [Google Scholar]
- Catrouillet S.; Fonteneau C.; Bouteiller L.; Delorme N.; Nicol E.; Nicolai T.; Pensec S.; Colombani O. Competition Between Steric Hindrance and Hydrogen Bonding in the Formation of Supramolecular Bottle Brush Polymers. Macromolecules 2013, 46, 7911–7919. 10.1021/ma401167n. [DOI] [Google Scholar]
- Catrouillet S.; Bouteiller L.; Nicol E.; Nicolai T.; Pensec S.; Jacquette B.; Le Bohec M.; Colombani O. Self-Assembly and Critical Solubility Temperature of Supramolecular Polystyrene Bottle-Brushes in Cyclohexane. Macromolecules 2015, 48, 1364–1370. 10.1021/ma5024022. [DOI] [Google Scholar]
- Gury L.; Gauthier M.; Cloitre M.; Vlassopoulos D. Colloidal Jamming in Multiarm Star Polymer Melts. Macromolecules 2019, 52, 4617–4623. 10.1021/acs.macromol.9b00674. [DOI] [Google Scholar]
- Johnson K. J.; Glynos E.; Sakellariou G.; Green P. Dynamics of Star-Shaped Polystyrene Molecules: From Arm Retraction to Cooperativity. Macromolecules 2016, 49, 5669–5676. 10.1021/acs.macromol.6b00456. [DOI] [Google Scholar]
- Kempf M.; Ahirwal D.; Cziep M.; Wilhelm M. Synthesis and Linear and Nonlinear Melt Rheology of Well-Defined Comb Architectures of PS and PpMS with a Low and Controlled Degree of Long-Chain Branching. Macromolecules 2013, 46, 4978–4994. 10.1021/ma302033g. [DOI] [Google Scholar]
- Tsukahara Y.; Namba S.; Iwasa J.; Nakano Y.; Kaeriyama K.; Takahashi M. Bulk Properties of Poly(Macromonomer)s of Increased Backbone and Branch Lengths. Macromolecules 2001, 34, 2624–2629. 10.1021/ma0016937. [DOI] [Google Scholar]
- Chen S.; Mahmood N.; Beiner M.; Binder W. H. Self-Healing Materials from V- and H-Shaped Supramolecular Architectures. Angew. Chem., Int. Ed. 2015, 54, 10188–10192. 10.1002/anie.201504136. [DOI] [PubMed] [Google Scholar]
- Chen S.; Yan T.; Fischer M.; Mordvinkin A.; Saalwächter K.; Thurn-Albrecht T.; Binder W. H. Opposing Phase-Segregation and Hydrogen-Bonding Forces in Supramolecular Polymers. Angew. Chem., Int. Ed. 2017, 56, 13016–13020. 10.1002/anie.201707363. [DOI] [PubMed] [Google Scholar]
- Chen S.; Döhler D.; Binder W. H. Rheology of Hydrogen-Bonded Dendritic Supramolecular Polymer Networks in the Melt State. Polymer 2016, 107, 466–473. 10.1016/j.polymer.2016.08.046. [DOI] [Google Scholar]
- Rahmstorf E.; Abetz V. Supramolecular Networks from Block Copolymers Based on Styrene and Isoprene Using Hydrogen Bonding Motifs—Part 1: Synthesis and Characterization. Materials 2018, 11, 1608. 10.3390/ma11091608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzette A. V.; Leibler L. Block Copolymers in Tomorrow’s Plastics. Nat. Mater. 2005, 4, 19–31. 10.1038/nmat1295. [DOI] [PubMed] [Google Scholar]
- Goldansaz H.; Jangizehi A.; Nikravan G.; Verkinderen O.; Goderis B.; Ghaffarian S. R.; van Ruymbeke E.; Ahmadi M. Rheological Modifiers Based on Supramolecular Block Copolymers: From Weak Associations to Interconnected Micelles. React. Funct. Polym. 2019, 137, 27–37. 10.1016/j.reactfunctpolym.2019.01.008. [DOI] [Google Scholar]
- Zinn T.; Willner L.; Lund R. Telechelic Polymer Hydrogels: Relation between the Microscopic Dynamics and Macroscopic Viscoelastic Response. ACS Macro Lett. 2016, 5, 1353–1356. 10.1021/acsmacrolett.6b00824. [DOI] [PubMed] [Google Scholar]
- Brassinne J.; Gohy J.-F.; Fustin C.-A. Orthogonal Control of the Dynamics of Supramolecular Gels from Heterotelechelic Associating Polymers. ACS Macro Lett. 2016, 5, 1364–1368. 10.1021/acsmacrolett.6b00831. [DOI] [PubMed] [Google Scholar]
- Brassinne J.; Fustin C.-A.; Gohy J.-F. Control over the Assembly and Rheology of Supramolecular Networks via Multi-Responsive Double Hydrophilic Copolymers. Polym. Chem. 2017, 8, 1527–1539. 10.1039/C6PY02143C. [DOI] [Google Scholar]
- Fetters L.; Lohse D.; Colby R.. Physical Properties of Polymers; Springer: New York, 2007. [Google Scholar]
- Rubinstein M.; Colby R. H.. Polymer Physics; Oxford University Press: New York, 2003. [Google Scholar]
- Jungst T.; Smolan W.; Schacht K.; Scheibel T.; Groll J. Strategies and Molecular Design Criteria for 3D Printable Hydrogels. Chem. Rev. 2016, 116, 1496–1539. 10.1021/acs.chemrev.5b00303. [DOI] [PubMed] [Google Scholar]
- Voet V. S. D.; Strating T.; Schnelting G. H. M.; Dijkstra P.; Tietema M.; Xu J.; Woortman A. J. J.; Loos K.; Jager J.; Folkersma R. Biobased Acrylate Photocurable Resin Formulation for Stereolithography 3D Printing. ACS Omega 2018, 3, 1403–1408. 10.1021/acsomega.7b01648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart L. R.; Li S.; Sturgess C.; Wildman R.; Jones J. R.; Hayes W. 3D Printing of Biocompatible Supramolecular Polymers and Their Composites. ACS Appl. Mater. Interfaces 2016, 8, 3115–3122. 10.1021/acsami.5b10471. [DOI] [PubMed] [Google Scholar]
- Campanella A.; Döhler D.; Binder W. H. Self-Healing in Supramolecular Polymers. Macromol. Rapid Commun. 2018, 39, 1700739. 10.1002/marc.201700739. [DOI] [PubMed] [Google Scholar]
- Rupp H.; Döhler D.; Hilgeroth P.; Mahmood N.; Beiner M.; Binder W. H. 3D printing of supramolecular polymers: Impact of nanoparticles and phase separation on printability. Macromol. Rapid Commun. 2019, 1900467. 10.1002/marc.201900467. [DOI] [PubMed] [Google Scholar]
- Lomellini P.; Rossi A. G. Effect of Composition on the Entanglement Density in Random Copolymers. Makromol. Chem. 1990, 191, 1729–1737. 10.1002/macp.1990.021910723. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.