

Abstract
Purpose:
Small molecule inhibitors have revolutionized treatment of certain genomically-defined solid cancers. Despite breakthroughs in treating systemic disease, central nervous system (CNS) metastatic progression is common, and advancements in treating CNS malignancies remain sparse. By improving drug penetration across a variably permeable blood-brain barrier and diffusion across intratumoral compartments, more uniform delivery and distribution can be achieved to enhance efficacy.
Experimental Design:
Ultrasmall fluorescent core-shell silica nanoparticles, Cornell prime dots (C’ dots), were functionalized with αv integrin-binding (cRGDY), or non-targeting (cRADY) peptides, and PET labels (124I, 89Zr), to investigate the utility of dual-modality cRGD-C’ dots for enhancing accumulation, distribution, and retention (ADR) in a genetically engineered mouse model of glioblastoma (mGBM). mGBMs were systemically treated with 124I-cRGD- or 124I-cRAD-C’ dots and sacrificed at 3 and 96 hours, with concurrent intravital injections of FITC-dextran for mapping blood-brain barrier breakdown and the nuclear stain Hoechst. We further assessed target inhibition and ADR following attachment of dasatinib, creating nanoparticle-drug conjugates (Das-NDCs). Imaging findings were confirmed with ex-vivo autoradiography, fluorescence microscopy, and p-S6RP IHC.
Results:
Improvements in brain tumor delivery and penetration, as well as enhancement in the ADR were observed following administration of integrin-targeted C’ dots, as compared with a non-targeted control. Furthermore, attachment of the small molecule inhibitor, dasatinib, led to its successful drug delivery throughout mGBM, demonstrated by downstream pathway inhibition.
Conclusions:
These results demonstrate that highly engineered C’ dots are promising drug delivery vehicles capable of navigating the complex physiological barriers observed in a clinically-relevant brain tumor model.
Introduction
Despite breakthroughs in the treatment of solid tumor malignancies, treatment options for primary gliomas and metastatic central nervous system (CNS) tumors remain limited due in part to variable and poor transport of small molecule drugs across the blood brain barrier (BBB), a significant obstacle to the delivery, penetration, diffusion, and retention of drugs within CNS tumors (1,2). Significant off-target effects contribute to dose-limiting toxicities of targeted agents that could otherwise offer significant therapeutic potential (3,4). In an attempt to overcome these technical hurdles, a variety of nanodelivery vehicles have emerged that aim to abrogate acute toxicity while improving precision drug delivery, efficacy, and therapeutic index (5).
The introduction of targeted small molecule inhibitors (SMIs) has revolutionized cancer treatment for tumors with genomically-defined sensitivities. However, as these patients live longer with successful treatment of systemic disease, there has been an increased incidence of brain metastasis and cases where the CNS is the only site of uncontrolled progression (3,4). Newer SMIs offer the promise of higher selectivity and potency, but BBB penetration and dose-limiting toxicity remain major limitations to successful therapy (6-13). This suggests that improving BBB penetration and distribution may augment the therapeutic potential for drugs previously thought to lack efficacy in the CNS. Moreover, current standard of care treatment approaches for glioblastoma, the most common primary brain malignancy, have led to a stagnation in cancer survival rates, with a median survival of less than 15 months and a 5-year survival rate of only 5% (14). Small molecule pathway inhibitor drugs with promising preclinical data have failed to show efficacy against GBM in clinical trials of unselected population to date (15,16). For instance, dasatinib is a highly potent second-generation adensosine triphosphate (ATP)-competitive inhibitor shown to be effective against multiple protein tyrosine kinases, including platelet derived growth factor receptor (PDGFR) and the Src family of kinases, in vitro. However, clinical trial results with dasatinib have been disappointing, failing to demonstrate a survival benefit at clinically acceptable doses (17). Possible reasons include a poor understanding of Src signaling in GBM making enrichment of patient populations difficult(18), as well as the presence of drug efflux pumps (i.e. p-glycoprotein, breast cancer resistance protein) expressed in the CNS leading to low levels of dasatinib accumulation(19). For the long list of receptor tyrosine kinase inhibitors whose clinical use is limited by off-target or wild-type receptor toxicities, it is critical to explore mechanisms to improve the tumor delivery and therapeutic index of existing drugs (9-11).
The growing concern over limitations driving inefficient therapeutic delivery to CNS malignancies has led to an explosive growth in nanodelivery systems, with the hope that such agents may overcome impediments of limited drug transport across the BBB, high interstitial pressures, and diffusion of drugs beyond sites of initial deposition (20-23). Given their versatility and tunability, particle-based delivery vehicles can offer a viable solution - not only to potentially overcome these limitations, but to enhance the accumulation, distribution, and retention (ADR) of therapeutics at sites of disease. Specifically, the physicochemical characteristics, including size, shape, charge, and surface chemistries, of such materials during particle synthesis are known to modulate these and other biological properties. However, despite the enormous body of research focused on the development of a variety of CNS drug delivery vehicles including polymeric nanoparticles (24-26), iron oxide nanoparticles (27), gold nanoparticles (28-30), liposomes (31), micelles (32,33), quantum dots (34), and silica nanoparticles (35), consensus has yet to be achieved on clinically-promising design features (Supplemental Table 1). Nearly all nanodelivery vehicles are larger than 20 nm diameter, which can limit extravasation from pores of the tumor neovasculature and impede diffusion through the tumor interstitium (36). Furthermore, the imaging information derived from many of these systemically-administered probes is largely of a qualitative nature, thereby precluding accurate assessments of probe uptake at sites of disease and off-target sites (e.g., liver) in a time-dependent manner to inform particle designs and tailor treatment planning efforts.
While the introduction of targeting moieties on the surfaces of particle-based probes in the form of antibodies (or their fragments) (37), peptides (38), and aptamers (39) have been shown to enhance target tissue penetration and distribution in brain tumor models, sufficiently detailed and time-dependent biological investigations needed to improve understanding of structure-function relationships for different classes of therapeutic platforms at disease sites are surprisingly scarce. Studies demonstrating increased accumulation and distribution of therapeutic platforms are also often limited to in vitro assessments and/or lack quantitation with respect to in vivo particle distributions throughout the bulk tumor area (40-42). Additionally, it is not well understood how changes in particle surface chemistry affect therapeutic product accumulation, retention, tissue diffusion, and distribution over extended time intervals (>24 hours). It is therefore important to explore new high-resolution in vivo imaging strategies that can carefully assess the influence of surface modifications, such as the attachment of therapeutics or the conjugation of targeting ligands, on particle ADR and pharmacokinetics (PK) using clinically-relevant models of CNS disease.
Herein, we employed a genetically engineered mouse model of glioblastoma (mGBM), known to effectively recapitulate the heterogenous breakdown of the blood-brain-barrier observed in patients (43). We aimed to determine the physiochemical factors impacting the ADR by modifying the surface chemistry of a clinically-promising ultrasmall fluorescent core-shell silica nanoparticle, termed Cornell prime dots (or C’ dots) (44). We assessed the feasibility of this particle probe as a dual-modality (PET-optical) platform to determine whether its distinctly favorable physicochemical features, pharmacological properties, and modular nature could be exploited to overcome key biological limitations plaguing native small molecule drugs. We chose to functionalize its surface with varying densities of cyclized Arg-Gly-Asp (cRGD) peptides to create cRGD-PEG-Cy5-C’ dots (abbreviated cRGD-C’ dots) for targeting αv integrins, a well described target of tumor neovasculature and gliomas (45). Comparison of ADR across a range of targeting ligand densities attached to C’ dots, as well as to non-binding cyclic Arg-Ala-Asp scrambled peptide control particles (cRAD-C’ dots), demonstrated an enhanced penetration, diffusion, and ADR for high ligand density cRGD-C’ dots at extended time frames. Expanding on these findings, cRGD-C’ dots were subsequently engineered for drug delivery, using previously described methods for the conjugation of SMIs to the C’ dot surface (46). Dasatinib was utilized as a prototype SMI drug for the inhibition of PDGFR signaling and illustrated how the PK and other key biological properties of the free drug could be favorably altered by conjugating drug-linker constructs to the particle surface while preserving its activity. The work presented highlights the successive tailoring of particle surface chemistry to achieve a first-in-kind renally-clearable, targeted delivery system – one that exhibits controlled pharmacological properties, high intratumoral ADR, and in vivo target inhibition in a genetically-engineered glioma model for precision drug delivery and treatment.
Materials and Methods
Flow cytometry.
RCAS/tv-a derived glioma (DXFM) cells were plated at a density of 5×105 cells per well in a 6-well plate and allowed to attach overnight in DMEM-high glucose media supplemented with 10% FBS, 1.8 g/L sodium bicarbonate, and 1% penicillin/streptomycin. Cells were subsequently treated with 100 nM solutions of cRAD-C’ dot or cRGD-C’ dot containing varying ligand numbers (n=6, 15–20 and n=6,14,18, respectively) and allowed to incubate for 4 hours at 37°C. Following incubation, cells were washed twice in full media, transferred to flow cytometry tubes, and stained with 0.05 mg/ml DAPI for live/dead cell determination. Flow cytometry analysis was carried out in the MSKCC Flow Cytometry Core Facility (New York, NY) on a BD LSRFortessa flow cytometer (BD Biosciences, San Jose, CA). Results of triplicate samples were displayed as percent of Cy5+ (C’ dot+) cells or median fluorescent intensities using Prism 7 software (GraphPad, La Jolla, CA).
Similar methods to those described above were used for temperature-dependent C’ dot uptake studies. cRGD-C’ dots with varying ligand densities were again incubated with DXFM cells in 6-well plates. However, individual plates were incubated at either 4°C, 25°C, or 37°C for 4 hours. Following incubation with the C’ dots, cells were prepared for analysis in a manner analogous to that mentioned previously.
Fluorescent imaging of frozen GBM brain sections.
Whole brains mounted in Tissue-Tek O.C.T were sectioned at a thickness of 10 μm on an Avantik Cryostatic Microtome (Avantik Biogroup, Springfield Township, NJ). Sections were evaluated for tumor based on altered cellular density and vascular permeability determined by visualization of the Hoechst and FITC-Dextran fluorescence signals. Tumor was confirmed present on sections via H&E staining. Images were captured on a BX60 fluorescence microscope (Olympus America Inc., Center Valley, PA) equipped with a motorized stage (Prior Scientific Instruments Ltd., Rockland, MA) and CC12 camera (Olympus) at 10X magnification. Whole brain images were obtained by acquiring numerous fields that were aligned using MicroSuite Biologic Suite (version 2.7, Olympus America Inc., Center Valley, PA). Brightfield and fluorescent images were acquired using the appropriate filter cube sets.
Tumor diffusion calculations were carried out using ImageJ software (Ver. 2.0-rc-43/1.51h). Briefly, both H&E and fluorescence images were first set to the proper scale. Regions of interest (ROIs) were drawn around the entire tumor (H&E) as well as any detectable Cy5 signal (optical). Dividing the measured Cy5 area by the total tumor area and multiplying by 100 provided the percent of tumor diffused with particle. These computed values were then plotted using Prism 7 (GraphPad, La Jolla, CA).
In vivo static PET imaging in RCAS/tv-a PDGFR-β mGBM mice.
For PET imaging, adult brain tumor-bearing mice (n=6) were i.v. injected with 200–300 μCi (7.4–11.1 MBq) 89Zr-cRGD-C’ dots. Approximately 5 min prior to the acquisition of PET images, mice were anesthetized via inhalation of 2% isoflurane (Baxter Healthcare, Deerfield, IL)/oxygen gas mixture and placed on the scanner bed; anesthesia was maintained using 1% isoflurane/oxygen gas mixture. PET imaging was performed in a small-animal PET scanner (Focus 120 microPET; Concorde Microsystems, Knoxville, TN) at 0.5 and 24 h post-injection. An energy window of 350−700 keV and a coincidence timing window of 6 ns were used. Data were sorted into 2D histograms by Fourier rebinning, and transverse images were reconstructed by filtered back-projection into a 128 × 128 × 63 (0.72 × 0.72 × 1.3 mm3) matrix. The PET imaging data was normalized to correct for non-uniformity of response, dead-time count losses, positron branching ratio, and physical decay to the time of injection; no attenuation, scatter, or partial-volume averaging corrections were applied. The counting rates in the reconstructed images were converted to activity concentrations (percentage injected dose per gram of tissue, %ID/g) by use of a system calibration factor derived from the imaging of a mouse-sized water-equivalent phantom containing 89Zr. ROI analyses of the PET data were performed using IRW software.
At 72-hours post-i.v. administration of 89Zr-cRGD-C’ dots, whole brain tissue samples were collected and frozen in Tissue-Tek O.C.T for sectioning. As described previously, the presence of brain tumors was first confirmed via H&E, followed by imaging of 89Zr-cRGD-C’ dot accumulation via fluorescence microscopy (Cy5) and autoradiography (89Zr). Autoradiography signal intensity and total tumor area was calculated by ROI analysis using ImageJ software (Ver. 2.0-rc-43/1.51h) across all subjects and plotted using Prism 7 software (GraphPad, La Jolla, CA).
p-S6RP Immunohistochemistry.
Frozen sections were air dried for 30 min at room temperature (RT), followed by fixation in 3% paraformaldehyde for 15 min at RT in a humidified chamber. Sections were then washed 2X with wash buffer (1X TBS/0.1% Tween-20) for 5 min. Endogenous peroxides were blocked using BLOXALL (SP-6000, Vector Labs, Burlingame, CA) for 10 min at RT. Sections were then washed 2X with wash buffer for 5 min, prior to the addition of the blocking solution (5% Normal Goat Serum/wash buffer). Blocking solution was incubated with sections for 1 hr at RT. Blocking solution was removed from sections and anti-pS6RP (#2211, Cell Signaling, Danvers, MA) primary antibody was added at a dilution of 1:200 in blocking solution. Sections were incubated overnight at 4°C in a humidified chamber. Sections were then washed 3X with wash buffer, followed by addition of anti-rabbit secondary antibody (BA-1000, Vector Labs, Burlingame, CA) at a dilution of 1:300 in blocking solution and incubated at RT for 30 min. Tissue sections were then washed 3X for 5 min. Staining detection was performed using Vectastain ABC HRP Kit (PK-4000, Vector Labs, Burlingame, CA) and ImmPACT DAB Peroxidase Substrate (SK-4105, Vector Labs, Burlingame, CA) according to the manufacturer’s instructions. Sections were then counterstained using VECTOR Hematoxylin QS (H-3404, Vector Labs, Burlingame, CA), rinsed 2X in dH2O, and coverslips mounted using VectaMount AQ Aqueous Mounting Medium (H-5501, Vector Labs, Burlingame, CA). Whole-section brightfield images were acquired as described previously in these methods. Signal intensity quantification was carried out on threshold-adjusted images using ImageJ analysis software, and adaptation of previously reported methods (47). Briefly, the sum of signal intensities across five high power fields within the tumor area, as delineated by H&E staining, were acquired. These values were then averaged and corrected for the area of the ROI. Lastly, the data for treated sections (45 uM cRGD-Das-NDC) was normalized to the untreated control group.
Results
Design and modular properties of ultrasmall silica nanoparticles
Ultrasmall silica core-shell nanoparticles, referred to as Cornell dots (C dots) (48,49), as well as the newer generation Cornell Prime dots (C’ dots) manufactured in water-based environments (48,50,51), enable exquisite control over particle architecture and composition. Ultrasmall C’ dots are core-shell hybrid silica nanoparticles composed of a rigid silica core encapsulating deep-red/near-infrared (NIR) fluorescent dyes. The particle surface is covalently modified by polyethylene glycol (PEG) chains to confer in vivo stability and facilitate renal clearance (Figure 1a) (51,52), Targeting moieties such as cyclic (arginine-glycine-aspartic acid-D-tyrosine-cysteine) (cRGDY) are conjugated to the surface of C’ dots for active cancer targeting (44,53) (Figure 1a). C’ dots exhibit homogeneous particle morphology (48) (Figure 1b), narrow size dispersion (54) (Figure 1c and d), and high product purity (48,55,56) (Figure 1c). The average hydrodynamic size of C’ dots is around 6–7 nm by fluorescence correlation spectroscopy (FCS, Figure 1d), endowing C’ dots with unique pharmacokinetic properties, as previously described (46,53). C’ dots can be surface-conjugated with radioisotopes, e.g. 124I or 89Zr, for serial, quantitative positron emission tomography (PET)-computed tomography (CT) imaging of time-varying particle distributions and ADR (56) (Figure 1a). Small molecular drugs have been attached to C’ dot surfaces via a peptide-based enzyme-cleavable linker group (46). Resulting C’ dot NDCs exhibit efficient drug release kinetics in the presence of model proteases, and are stable in various biological media, rendering C’ dots a promising drug-delivery platform for cancer therapeutics (Figure 1a). However, in-vivo results of such C’ dot based NDCs have not been reported to date.
Figure 1. Structure of cRGD-functionalized and Dasatinib (Das)-bound NDCs.
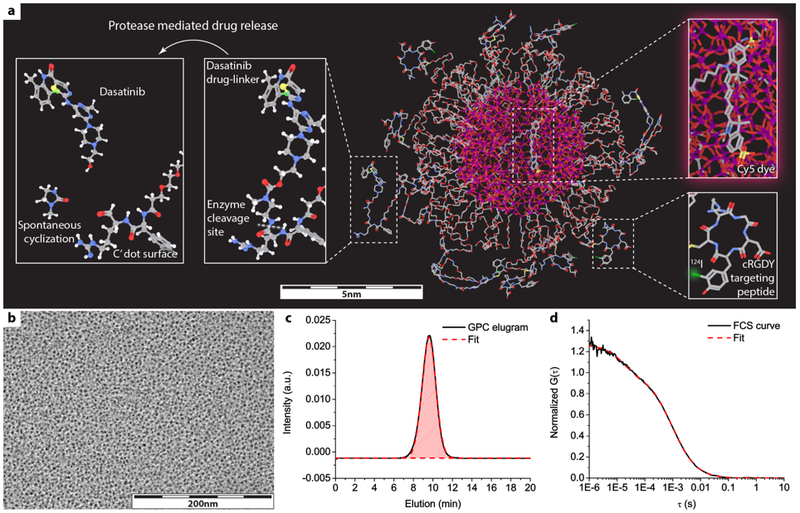
a) 3D molecular rendering of dasatinib (das)-NDC, in which Cy5 dyes are encapsulated within a silica core, cancer-targeting cRGDY peptides are attached on particle surface, and Dasatinib (Das) drug-linker molecules conjugated to the particle surface via enzymatically cleavable linkers. Radioisotope 124I can be further attached to cRGDY peptide, endowing C’ dots with PET imaging capabilities. Silicon, oxygen, carbon, nitrogen, sulfur, iodine, chloride, hydrogen atoms are colored by purple, red, gray, blue, yellow, green, light green, and white, respectively. Hydrogen atoms are only displayed in the zoomed-in images of drug-linker for the better visualization. b-d) representative TEM image (b), GPC elugram with fit (c), and FCS curve with fit (d) of C’ dots.
cRGD-C’ dots demonstrate increased cellular binding and uptake in αν integrin-expressing RCAS/tv-a primary glioblastoma cells.
Integrins are well-known transmembrane receptors recognized to be overexpressed in a number of tumor types, as well as neo-angiogenic vessels (49,57). To demonstrate integrin expression in the RCAS/tv-a driven model of glioblastoma, we conducted immunofluorescence staining on murine brain tumor tissue sections obtained from mice with confirmed GBM. The results depicted in Figure 2a demonstrate a high level of αv integrin (red) expression throughout the tumor area, as well as along the endothelium of the tumor neovasculature, defined by CD31 expression (green). Utilizing mGBM single cell suspensions derived from murine RCAS/tv-a GBM, we next sought to understand 1) whether an integrin-targeting peptide was necessary to increase cellular binding and internalization of the particles and 2) optimal ligand densities needed to maximize targeted uptake. To answer these questions, C’ dots were functionalized with varying densities of the αv integrin-targeting cyclic-arginine-glycine-aspartic acid peptide (cRGD; n=6–18) or scrambled peptide control, cyclic-arginine-alanine-aspartic acid (cRAD; n = 6–20). Results demonstrated that high cRGD ligand densities (n = 14, 18) led to an increase in the percentage of cells staining positively for cRGD-C’ dots (Cy5+) (Figure 2b) and an amplified cellular median fluorescence intensity (Supplemental Figure 1a) as determined by flow cytometry. Minor increases in positively staining cells (Figure 2b) and median fluorescence values (Supplemental Figure 1a) were observed for cRAD-C’ dots as a function of increasing ligand density, however the percentage and median intensity values were significantly less than those observed with cRGD-C’ dots. The enhanced cellular binding of cRGD-C’ dots demonstrated the importance of an active targeting moiety and thus became the focus of subsequent studies.
Figure 2. cRGD-C’ dots demonstrate a ligand-dependent increase in cellular binding and uptake in αν integrin-expressing primary cells from RCAS/tv-a glioblastoma.

a) Immunofluorescence staining of tissue harvested from an RCAS/tv-a tumor bearing mouse. Integrin αν expression (red), is demonstrated throughout the tumor and along endothelial cells lining its neovasculature. Tumor neovasculature was visualized by anti-CD31 (green) antibody. Cell nuclei are counterstained with DAPI (blue). b) Comparison of cRGD-C’ dot and cRAD-C’ dot cell binding by flow cytometry. RCAS/tv-a derived glioma cells were incubated with cRAD-C’ dots or cRGD-C’ dots with varying ligand densities (n=6-20 and n=6-18, respectively) and analyzed (Student’s T-test, *** p = 0.0008, **** p < 0.0001). c) Temperature-dependent uptake of cRGD-C’ dot nanoparticles in RCAS/tv-a derived glioma cells. cRGD-C’ dot particles displaying 6, 14, and 18 cRGD per particle were incubated with glioma cells at 4, 25, and 37°C for 4 hours. Cell uptake was measured using flow cytometry (Student’s T-Test, **** p < 0.0001). d & e) High-resolution confocal microscopy of cRGD- and cRAD-C’ dot (n=18) uptake in glioma cells, respectively. Cells were treated with 100 nM cRGD-C’ dots (d) or cRAD-C’ dots (e) (red) in the presence of 1 mg/ml FITC-labeled 70kDa dextran (FITC-Dextran, green) for 4 hours. Treated cells were then fixed, counterstained with DAPI (blue). (Scale bars = 20 μm)
We next sought to determine whether cRGD-C’ dots were internalized non-specifically in mGBM cells, or through an energy-dependent pathway, such as receptor-mediated endocytosis. To demonstrate that C’ dots were internalized via endocytosis, cells were incubated with 100 nM solutions of C’ dots with varying cRGD ligand densities. Incubation was performed for 4 hours at 4°C, 25°C, or 37°C, and subsequently analyzed via flow cytometry. Results indicated that reductions in incubation temperature led to a decrease in both the number of Cy5+ cells (Figure 2c), as well as cellular median fluorescence intensities (Supplemental Figure 1b), suggesting a receptor-mediated mechanism of cRGD-C’ dot uptake (45). Lastly, visualization of cRGD-C’ dot (n = 18) intracellular accumulation was performed via high resolution confocal microscopy. mGBM cells treated with 100 nM cRGD-C’ dots for 4 hours showed robust accumulation of Cy5 signal in cells, likely localized to endolysosomes as shown for prior cancer cell types (58), with a predominant perinuclear distribution (Figure 2d, Supplemental Figure 1c) when compared to cRAD-C’ dot (n=15–20) treated GBM cells (Figure 2e, Supplemental Figure 1d). Together, these results confirmed that cRGD-C’ dots underwent internalization, and that this occurs primarily through energy-dependent mechanisms of uptake.
Overall, these data indicated that the inclusion of an integrin targeting peptide (cRGD) on the C’ dot platform led to an increase in the level of cellular binding and internalization in mGBM cells. Observed increases were found to be dependent on targeting ligand density, where 18 cRGD peptides per particle were shown to maximize intracellular accumulations. This ligand density was therefore selected for in vivo studies.
C’ dot-exposed mGBM distributions closely mimic EPR-driven dispersal patterns at early times post-injection.
Utilizing an experimental design that employed concurrent intravital stains and timed injections to create a real-time map of the blood brain barrier, we were able to evaluate particle distributions relative to the BBB at various time points post-treatment. This allowed for a non-invasive, quantifiable, and time-dependent method for assessing distribution and brain tumor penetration beyond that attributable to the enhanced permeability and retention (EPR) effect, a property characterized by preferential delivery of drugs to tumors (59). Animal models of GBM were created using well-established RCAS/tv-a methodologies (43) (Supplemental Scheme 1a). Mice with confirmed mGBM were treated systemically with cRAD- or cRGD-C’ dots and sacrificed 3 hours post-treatment (Supplemental Scheme 1b). We utilized a 70 kDa FITC-labeled dextran molecule (FITC-Dextran) that is of comparable size to the C’ dot as a measure of BBB permeability (60,61) At 3 hours post-injection, tumor-specific Cy5 signal (red) distribution closely corresponded to that observed for the intravenously-administered permeability marker, FITC-Dextran (green), demonstrating strong co-localization in regions of disrupted BBB breakdown (Supplemental Figure 2a, b). These results were equivalent for both cRAD- (Supplemental Figure 2a) and cRGD-C’ dot treated animals (Supplemental Figure 2b), suggesting that the in vivo distribution at this early time-point is largely dependent on the EPR effect without additive effects from integrin targeting. High magnification imaging (60x) was performed for subcellular localization of cRGD-C’ dots (Supplemental Figure 2c). These high-resolution images demonstrated strong Cy5 signal (red) in close proximity to nuclei stained by Hoechst (blue), suggestive of intracellular localization of cRGD-C’ dots. Furthermore, co-localization of FITC-Dextran (green) with particles suggested cellular internalization was occurring through the endo-lysosomal pathway (62).
Enhanced tumor accumulation, retention, and distribution beyond BBB breakdown in cRGD-C’ dot treated mGBM.
A qualitative comparison of particle uptake between cRGD- and cRAD-C’ dot-treated mGBM showed an increase in the ADR of the targeted particle at 96-hours post-administration (Figure 3a & b, respectively). Fluorescence microscopy demonstrated enhanced particle distribution (Figure 3a2, a6) across the total tumor area, as well as increased distribution beyond areas of BBB breakdown (Figure 3a3, a7), for cRGD-C’ dots in comparison to cRAD-functionalized particles (Figure 3b). Furthermore, 124I-labeled cRGD-C’ dots (Figure 3a4) demonstrated increased particle accumulation relative to cRAD-C’ dots (Figure 3b4) in areas of disease, as shown by MR imaging (Figure 3a8 & 3b8, respectively). Together, these results provided qualitative support for the use of a cRGD targeting moiety on the C’ dot surface as a means of increasing particle accumulation, distribution, and retention.
Figure 3. Enhanced cRGD-C’ dot particle retention and increased diffusion that extends beyond regions of BBB breakdown at 96 hours post-treatment.

RCAS/tv-a tumor bearing mice were treated in vivo with 124I labeled cRGD- or cRAD-C’ dots 96h prior to sacrifice (p.t.s.) followed by FITC-Dextran 3h p.t.s. a) mGBM tumors were frozen, sectioned, and confirmed via H&E staining (a1, a5). Tumors were evaluated using fluorescence microscopy to determine the distribution of 124I-cRGD-C’ dots (a2, a6) and the permeability marker FITC-Dextran (a3, a7). 124I-cRGD-C’ dots retain high in vivo radiostability as demonstrated by colocalization of autoradiography signal (a4) with particle Cy5 fluorescence signal (a2). Figure a8 demonstrates a representative MRI image, confirming the presence of tumor (red circle) prior to treatment initiation. b) mGBM tumor areas (b1, b5) were evaluated for the distribution of 124I-cRAD-C’ dots (b2, b6) in comparison to FITC-Dextran (b3, b7). 124I-cRAD-C’ dots demonstrate strong colocalization of 124I signal (b4) with Cy5 particle signal (b2). MRI imaging confirmation of mGBM tumor (b8, red circle). (Scale bars = 1 mm)
A quantitative approach utilizing region of interest (ROI) analysis was employed to measure fluorescence signal in relation to the total tumor area. Clear enhancement of cRGD-C’ dot distribution and retention within the defined tumor area was observed when compared with that shown for cRAD-C’ dot-treated tumors at 96-hours post-injection (Figure 4a). Comparison of ROI areas for cRAD- and cRGD-C’ dot signal (Cy5), in relation to the total tumor area, as defined by H&E staining, demonstrated a significant enhancement of coverage offered by cRGD-C’ dots over that of cRAD-C’ dots (Figure 4b).
Figure 4. Quantitative analysis of particle distribution relative to total tumor area and BBB breakdown demonstrates increased particle diffusion with cRGD targeting at 96 hours post-treatment.
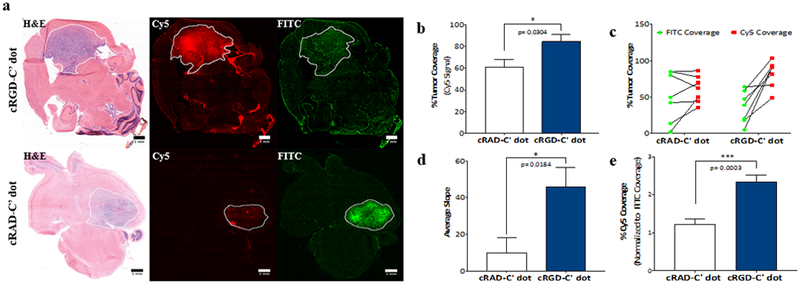
RCAS-tumor bearing mice were treated with cRGD- or cRAD-C’ dots and sacrificed 96 hours later. FITC-Dextran was administered 3 hours prior to sacrifice. Fluorescence analysis of FITC and Cy5 signal on frozen sections was performed and total tumor areas were measured using ROI analysis of H&E staining. a) Representative images of mGBM brain sections of mice treated with cRGD- (top row) versus cRAD-C’ dots (bottom row) 96 hours post-treatment. b) Comparison of cRAD-C’ dot and cRGD-C’ dot tumor coverage as measured by Cy5 fluorescent signal (Student’s T-test, * p = 0.0304). c) Tumor coverage of administered FITC-Dextran and cRAD- or cRGD-C’ dots across all experimental animals. d) Average slope comparison of cRAD- and cRGD-C’ dot treated animals depicted in Figure 4c (Student’s T-test, * p = 0.0184). e) Cy5 percent tumor coverage normalized to average FITC signal in both cRAD- and cRGD-C’ dot treated tumors (Student’s t test, *** p = 0.0003). (Scale bars = 1 mm)
We next examined the extent to which cRAD- and cRGD-C’ dots distributed beyond areas of BBB breakdown, as demonstrated by the diffusion pattern of FITC-dextran, within GBM tumors (Figure 4a, right column). Highlighting areas where fluorescent particles were free to diffuse as a result of an altered BBB made it possible to measure any enhanced distribution of C’ dots. Results of this analysis revealed a trend in the data, with cRGD-C’ dots distributed more uniformly throughout the tumor volume than that observed for FITC-Dextran alone, which showed a more locally distributed, punctate pattern of particle signal. In comparison, the distribution of cRAD-C’ dots closely mimicked that of the permeability marker across all mice (Figure 4c). The slope of each line depicted in Figure 4d was then calculated, allowing for a measure of enhanced coverage beyond that of BBB breakdown across groups. This analysis demonstrated a >4.5-fold increase in cRGD-C’ dot coverage (slope = 45.9 ± 10.4) relative to that seen for cRAD-C’ dots (slope = 9.8 ± 8.2). This indicates cRGD-C’ dots distribute to a much greater degree, and beyond areas which are freely diffusible, than that found for cRAD-C’ dots. Finally, the percentage of Cy5 coverage was evaluated based on the average FITC-Dextran coverage across each treatment group (Figure 4e). This result again demonstrated that the addition of cRGD to the C’ dot platform enhanced coverage of the total tumor area to a greater extent than that observed for the scrambled (cRAD-C’ dot) control.
These results provide quantitative support to the observed increase in cRGD-C’ dot coverage relative to that found with cRAD-C’ dots within mGBM. In addition, it was shown that αv integrin-targeting C’ dots were capable of distributing beyond areas of BBB breakdown, a property critically needed for augmenting particle-based therapeutic delivery and penetration of CNS malignancies.
89Zr-cRGD-C’ dots accumulate specifically in mGBM while demonstrating rapid systemic clearance
Implementing newly described methods for radiolabeling C’ dot platforms with 89Zr (56), a radionuclide that residualizes stably within cells after internalization (63), we next evaluated the utility of 89Zr labeled DFO-cRGD-C’ dots (i.e., 89Zr-cRGD-C’ dots) as a particle-based imaging tracer for serial PET imaging using the RCAS/tv-a mGBM model (Figure 5). Representative maximum intensity projections (MIP) of a single mouse at an early (0.5 hour) and a late (24 hour) time point demonstrated rapid renal clearance and retained tumor uptake (Figure 5a, Supplemental Video 1a-b). High specific uptake (~7% ID/g) was noted in mGBM at 24 hours post-injection (Figure 5a, top row), with a dramatic decrease in whole body signal when compared to imaging performed at 0.5-hours (Figure 5a, bottom row). Whole brain ex vivo analysis of 89Zr-cRGD-C’ dot treated mice demonstrated specific particle accumulation in areas of tumor visualized by both Cy5 and 89Zr signal localization (Figure 5b-c). Finally, particle activity observed on autoradiography showed a positive correlation with overall tumor area (Figure 5d) and was dramatically higher than that found in normal brain (Figure 5e).
Figure 5. Accumulation of 89Zr-cRGD-C’ dots in mGBM and rapid systemic clearance.

a) Representative maximum projected intensity PET image of a mGBM mouse treated with 89Zr-cRGD-C’ dots. Images were acquired at 0.5 hours (top row) and 24 hours (bottom row) after intravenous particle injection. Accumulation of 89Zr-cRGD-C’ dot in the brain was measured to be ~7% ID/g. b) Histological analysis of whole brain extracted from mouse in Figure 5a, showing H&E confirmation of tumor and particle distribution demonstrated by optical (Cy5) and autoradographic (89Zr) imaging. c) Histological analysis of 5 mice treated with intravenous 89Zr-cRGD-C’ dots (72 hours post-injection) illustrating tumor (H&E) and particle localization (Cy5 and autoradiography; scale bars= 1 mm). d) Analysis of average autoradiography signal intensity as a function of overall tumor area demonstrates a positive correlation (R2 = 0.6474). e) Tumor to normal brain analysis of mice from Figure 5c demonstrating a >150-fold increase in 89Zr-cRGD-C’ dot accumulation in tumor as compared to surrounding normal brain (Tumor = 164.713, Normal = 1.1364; Student’s t test, ** p = 0.0029).
Together these data demonstrate that 89Zr-cRGD-C’ dots can be utilized for molecularly-targeted PET imaging of the RCAS/tv-a mGBM model. Radiolabeled particles demonstrated “target-or-clear” capabilities with rapid renal clearance (56), relative lack of reticuloendothelial system uptake (i.e., liver), and retained brain tumor uptake. Favorable pharmacokinetic and biodistribution profiles show increased uptake of 89Zr-cRGD-C’ dots in areas of tumor, thus resulting in enhanced tumor-to-normal brain uptake ratios. These results highlight the utility of 89Zr-cRGD-C’ dots as a viable PET tracer for the imaging of CNS malignancies.
Dasatinib-NDC inhibits PDGFR signaling in mouse and human-derived glioma cells in vitro
Implementing previously described synthetic methods to link small molecular inhibitors to the C’ dot surface (46), dasatinib was successfully conjugated to the integrin-targeting particle probe to create a theranostic, cRGD-C’ dot nanoparticle drug conjugate, or cRGD-Das-NDC. cRGD-Das-NDC was synthesized utilizing a methylethylenediamino-carbamate attached to a dipeptide (Phe-Arg) (Supplemental Figure 3, 4a), such that a protease can liberate the terminal primary amine of the methylethylenediamine (Supplemental Figure 4a2). Next, an intra-molecular cyclization occurs (Supplemental Figure 4a3), leading to the subsequent release of dasatinib and a methyl-urea by-product (Supplemental Figure 4a4). As expected, this linker design exhibits a two-step release mechanism: a very fast initial protease-dependent event, and a much slower pH-dependent intra-molecular cyclization event that results in the release of the native dasatinib drug conformation.
We sought to demonstrate that dasatinib released from the NDC retains its efficacy by performing Western blot analysis of glioma cells, derived from both RCAS/tv-a and human tumors, following treatment with cRGD-Das-NDCs (Supplemental Figure 4b-d). Cells were exposed to increasing concentrations (10 nM-10 uM) of cRGD-Das-NDC or the native dasatinib and subsequently stimulated with PDGF-BB. Investigating the ability of cRGD-Das-NDCs to inhibit PDGFR signaling in DXFM cells, we successfully demonstrated reductions in the levels of p-Akt and p-S6RP (Supplemental Figure 4b), as well as p-Src and p-PRAS40 (Supplemental Figure 4c). The reduction of phosphorylation levels occurred across all markers in a dose-dependent manner, and were similar to those observed when using the native drug. In addition to effective inihibition of PDGFR singaling in cells derived from a murine origin, we also chose to evaluate the effectiveness of cRGD-Das-NDCs in a human glioma model by the use of TS543 neurosphere cultures (Supplemental Figure 4d). Again, treatment of TS543 cells with cRGD-Das-NDCs demonstrated a dose-dependent reduction in the levels of p-PDGFRα that was equal to, or better than results observed following treatment with free dasatinib.
Together, these results confirm the effectiveness of cRGD-Das-NDCs in their ability to inhibit PGFR signaling by reducing the level of active effectors in both PI3K (i.e. Akt, PRAS40, S6RP) and Src pathways. In addition, these effects were confirmed in both murine and human-derived models of glioma, thus demonstrating an effective delivery platform that performs irrespective of species or genetic background.
Targeted NDCs retain increased distributive properties and inhibit downstream signaling in the RCAS/tv-a mGBM model.
Dasatinib-functionalized NDCs, both targeted (cRGD-) and non-targeted (cRAD-), were first evaluated for their ability to replicate the tumor distribution profiles that had been observed when utilizing drug-free cRGD- or cRAD-C’ dot platforms. Tumor distributions of cRGD- and cRAD-Das-NDC particles were again analyzed using fluorescence microscopy 96 hours after NDC administration. The data generated showed enhanced coverage of the tumor by cRGD-NDCs (Figure 6a1-a4), as against that observed with cRAD-Das-NDCs (Figure 6a5-a8). These results corroborated our previous findings of a targeting moieties’ ability to enhance tumor penetration, accumulation, distribution and retention of C’ dots in comparison to non-targeting particle controls. Subsequent studies evaluating the biological properties of NDCs in vivo were therefore performed using cRGD-Das-NDC particles.
Figure 6. Distribution of Dasatinib-NDC in GBM and effective inhibition of downstream PDGFR-β signaling.

a) RCAS/tv-a tumor bearing mice were treated with RGD-targeted Das-nanoparticle conjugate (cRGD-Das-NDC, red, top row) or non-targeted nanoparticle drug conjugate (cRAD-Das-NDC, red, bottom row) and sacrificed at 96 hours post-treatment. Injection of FITC-Dextran (green) and Hoechst (blue) occurred 3 hours and 10 minutes prior to sacrifice, respectively. Frozen sections were imaged using fluorescence microscopy. Representative samples demonstrate Cy5 distribution and retention consistent with previous studies of non-drug conjugated particle. Panels 1-4: cRGD-Das-NDC treated mGBM demonstrating H&E (1) and corresponding Hoechst (2), FITC (3) and Cy5 (5) distributions. Similarly, Panels 5-8 show a representative cRAD-Das-NDC treated mGBM with H&E (5), Hoechst (6), FITC (7) and Cy5 (8) distributions. b) mGBM animals were treated with a single dose of 45 μM cRGD-Das-NDC dose (top row), or left untreated (bottom row), and sacrificed 24 hours later. Frozen brain tumor sections were evaluated for cRGD-Das-NDC therapeutic efficacy by p-S6RP immunohistochemistry. Clear reduction of p-S6RP intensity was observed following administration of cRGD-Das-C’ Dot. C) Quantification of p-S6RP tumor staining intensity across all control (n=5) and cRGD-Das-NDC treated tumors (n=6). Displayed are the results of the evaluation of 5 high power fields per tumor (Student’s T-test, * p = 0.0212).
We next sought to evaluate the in vivo efficacy of cRGD-Das-NDCs to inhibit downstream pathway signaling in our murine model of GBM. Previously, we demonstrated that our drug-linker construct is capable of being cleaved from C’ dots by enzymes such as cathepsin B (46). Interestingly, cathepsin B expression has been shown to increase in higher grade gliomas (64), is found in lysosomes, on cell surface membranes, involves cancer cells and endothelial cells comprising brain tumors, and may be secreted into the extracellular milieu (65,66).We therefore administered a single dose of 45 uM cRGD-Das-NDC to mice harboring RCAS/tv-a mGBM. Twenty-four hours after administration, whole brain tissues were extracted and subjected to immunohistochemical staining for the detection of p-S6RP, a downstream regulator of the PI3K pathway (67). Visual observation showed an appreciable reduction in the p-S6RP staining intensity between untreated control mice (Figure 6b, top row, Supplemental Figure 5a) and those treated with 45 uM cRGD-Das-NDC (Figure 6b, bottom row, Supplemental Figure 5b). Region of interest analysis across all treated and control mice confirmed there was a significant reduction in staining intensity as a result of cRDG-Das-NDC treatment (Figure 6c).
These results confirm that cRGD-Das-NDCs retain an increased ADR over the cRAD-functionalized NDCs (cRAD-Das-NDC). In vivo administration and successful delivery of dasatinib was further confirmed by p-S6RP immunohistochemistry, where an appreciable inhibition of phosphorylation was observed across all treated mGBM in comparison with untreated controls. It should be noted that we do recognize the time discrepancy between the analysis of this in vivo study and our previous in vitro work to demonstrate release of effective drug. We attribute the more rapid appearance of functional dastinib in brain tumors to be a result of tumor microenvironment components that may aid in cleavage and cyclization of the dasatinib precursor (68). Together, these results demonstrate cRGD functionalization increases the accumulation, diffusion, and retention of C’ dots in a model of mGBM that displays heterogenous BBB breakdown.
Discussion
In this study we have focussed on the delivery of a drug carrier across a partially disrupted BBB rather than on the distinct challenges of crossing an intact barrier. A partially disrupted BBB reflects the majority of clinical cases of CNS tumor progression where contrast enhancing tumors are detectable and growing. We used a tumor model that (1) is known to generate a heterogeneous BBB and (2) is known to respond to the small molecule inhibitor dasatinib, providing a biomarker of target inhibition as an end effect of the NDC. We have aimed to characterize how functional surface modifications alter the behavior of the C’ dot in the challenging microenvironment of a high grade brain tumor. The glioma model provides a uniquely high contrast between tumor and normal brain tissue when assessing NDC delivery via fluorescence, autoradiography or PET. While our model is a CNS primary tumor, we expect the results to be relevant to other tumor types which disrupt the BBB. Brain metastases may be a better target for NDC development in the specific case where the primary tumor is sensitive to small molecule drugs. At the moment, no effective small molecule inhibitor has emerged for gliomas. Although our mouse glioma model is sensitive to dasatinib, we do not suggest that improvements in delivery of this particular inhibitor will benefit patients with gliomas. Within the larger efforts to develop “magic bullet” therapeutics for cancer, our current study is focused on the magic rather than the bullet.
With obstacles such as the BBB limiting the delivery of therapeutics to malignant brain tumors, the enhanced tumor distribution and drug carrying capabilities of our platform may provide a means by which drug accumulation can be preferentially enhanced at sites of disease without significant off-target accumulations, thereby improving treatment efficacy and therapeutic index. An initial Phase 1 imaging study is now underway to investigate cRGD-C’ dot accumulation in patients diagnosed with primary gliomas or CNS metastasis (Supplemental Figure 6).
This study aimed to elucidate the delivery of C’ dots to a mGBM model, along with their intratumoral and intracellular distributions, and to further investigate C’ dots as a tunable platform for precision drug delivery. Our findings demonstrated that targeting of αv integrins with cRGD increased the accumulation, retention, and diffusion of C’ dots throughout the tumor interstitium, leading to a more uniform distribution of this platform within malignant brain tumor tissues, in particular, beyond regions of BBB breakdown. The modular design of C’ dots allowed for multimodal imaging of these particles as both optical probes and radiotracers through encapsulation of fluorescent dyes and surface attachment of various radiolabels such as 89Zr and 124I.
The ultimate goal of this study was to assess the potential of C’ dots as a cancer-targeted ultrasmall vehicle for precision drug delivery, penetration, and ADR at the target site while minimizing off-target effects. We have shown that SMIs can be attached to C’ dots via a cleavable linker, with retention of in vivo pharmacologic activity against a specific target. Diffusion and intratumoral retention of NDCs suggests that C’ dots can be tailored to efficiently deliver small molecule therapies with enhanced PK. Given the current challenges of systemic therapies in their ability to penetrate heterogenous tumors, these results suggest a mechanism whereby precision drug delivery can overcome technical hurdles associated with existing small molecule therapies and provide new opportunities for drugs with otherwise unacceptable systemic toxicity, PK, and/or target tissue uptake. These findings, while not optimized for dose or drug loading, lay the foundation for further investigation of C’ dots as potent drug delivery vehicles to treat both primary and metastatic disease to the CNS.
Supplementary Material
Statement of Translational Relevance.
Despite breakthroughs in the treatment of solid tumor malignancies, successful treatment of malignant brain tumors remains elusive, in part due to a heterogeneously-permeable blood-brain barrier that hinders drug delivery and penetration, as well as accumulation, diffusion and retention. Herein, we demonstrate the favorable in vivo biological properties of integrin-targeting, ultrasmall, core-shell multimodal (PET-optical) silica nanoparticles, Cornell prime dots (C’ dots), in a genetically engineered mouse model of glioblastoma. We further underscore their potential utility as a precision drug delivery vehicle for effective treatment of brain tumors following attachment of a small molecule drug. Taken together, these data support the use of targeted nanoparticle-drug conjugates as clinically-promising platforms for precision drug delivery, laying the framework for investigating its utility as a targeted theranostic for treating malignant brain tumors. These results have since led to the initiation of a Phase 1 clinical trial focusing on targeted C’ dot accumulation in high-grade gliomas.
Aknowledgements:
We would like to acknowledge that this study was funded by grants from the National Institutes of Health (1U54 CA199081-01 to M. Bradbury and U. Wiesner) and Sloan Kettering Institute (Core Grant P30 CA008748CCSG to M. Bradbury).
Footnotes
Conflict of Interest: M.S.B. and U.W. hold interest in Elucida Technologies, Inc., which has licensed IP from Cornell and MSKCC on C’ dots and their application in oncology.
References
- 1.Bohn JP, Pall G, Stockhammer G, Steurer M. Targeted Therapies for the Treatment of Brain Metastases in Solid Tumors. Targeted oncology 2016;11(3):263–75 doi 10.1007/s11523-015-0414-5. [DOI] [PubMed] [Google Scholar]
- 2.Jiang P, Mukthavaram R, Chao Y, Bharati IS, Fogal V, Pastorino S, et al. Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. Journal of translational medicine 2014;12:13 doi 10.1186/1479-5876-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nayak L, Lee EQ, Wen PY. Epidemiology of brain metastases. Curr Oncol Rep 2012;14(1):48–54 doi 10.1007/s11912-011-0203-y. [DOI] [PubMed] [Google Scholar]
- 4.Zimmermann S, Dziadziuszko R, Peters S. Indications and limitations of chemotherapy and targeted agents in non-small cell lung cancer brain metastases. Cancer treatment reviews 2014;40(6):716–22 doi 10.1016/j.ctrv.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Hassanzadeh P, Atyabi F, Dinarvand R. Application of modelling and nanotechnology-based approaches: The emergence of breakthroughs in theranostics of central nervous system disorders. Life Sci 2017;182:93–103 doi 10.1016/j.lfs.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer discovery 2014;4(9):1046–61 doi 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters S, Zimmermann S, Adjei AA. Oral epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer: comparative pharmacokinetics and drug-drug interactions. Cancer treatment reviews 2014;40(8):917–26 doi 10.1016/j.ctrv.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 8.Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer discovery 2013;3(12):1404–15 doi 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ou SH, Bazhenova L, Camidge DR, Solomon BJ, Herman J, Kain T, et al. Rapid and dramatic radiographic and clinical response to an ALK inhibitor (crizotinib, PF02341066) in an ALK translocation-positive patient with non-small cell lung cancer. J Thorac Oncol 2010;5(12):2044–6 doi 10.1097/JTO.0b013e318200f9ff. [DOI] [PubMed] [Google Scholar]
- 10.Forde PM, Rudin CM. Crizotinib in the treatment of non-small-cell lung cancer. Expert Opin Pharmacother 2012;13(8):1195–201 doi 10.1517/14656566.2012.688029. [DOI] [PubMed] [Google Scholar]
- 11.National Comprehensive Cancer Network. The NCCN Clinical Practice Guidelines in Oncology for Non-Small Cell Lung Cancer 2014.
- 12.Costa DB, Kobayashi S, Pandya SS, Yeo WL, Shen Z, Tan W, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29(15):e443–5 doi 10.1200/JCO.2010.34.1313. [DOI] [PubMed] [Google Scholar]
- 13.Kodama T, Hasegawa M, Takanashi K, Sakurai Y, Kondoh O, Sakamoto H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother Pharmacol 2014;74(5):1023–8 doi 10.1007/s00280-014-2578-6. [DOI] [PubMed] [Google Scholar]
- 14.Weller M, Stupp R, Hegi M, Wick W. Individualized targeted therapy for glioblastoma: fact or fiction? Cancer J 2012;18(1):40–4 doi 10.1097/PPO.0b013e318243f6c9. [DOI] [PubMed] [Google Scholar]
- 15.De Witt Hamer PC. Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro-oncology 2010;12(3):304–16 doi 10.1093/neuonc/nop068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mellinghoff IK, Schultz N, Mischel PS, Cloughesy TF. Will kinase inhibitors make it as glioblastoma drugs? Current topics in microbiology and immunology 2012;355:135–69 doi 10.1007/82_2011_178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lassman AB, Pugh SL, Gilbert MR, Aldape KD, Geinoz S, Beumer JH, et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro-oncology 2015;17(7):992–8 doi 10.1093/neuonc/nov011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schiff D, Sarkaria J. Dasatinib in recurrent glioblastoma: failure as a teacher. Neuro-oncology 2015;17(7):910–1 doi 10.1093/neuonc/nov086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. The Journal of pharmacology and experimental therapeutics 2009;330(3):956–63 doi 10.1124/jpet.109.154781. [DOI] [PubMed] [Google Scholar]
- 20.Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. Cancer research 1988;48(24 Pt 1):7022–32. [PubMed] [Google Scholar]
- 21.Bhowmik A, Khan R, Ghosh MK. Blood brain barrier: a challenge for effectual therapy of brain tumors. BioMed research international 2015;2015:320941 doi 10.1155/2015/320941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proceedings of the National Academy of Sciences of the United States of America 1994;91(6):2076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nance E, Zhang C, Shih TY, Xu Q, Schuster BS, Hanes J. Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local administration. ACS nano 2014;8(10):10655–64 doi 10.1021/nn504210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lam FC, Morton SW, Wyckoff J, Vu Han TL, Hwang MK, Maffa A, et al. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nature communications 2018;9(1):1991 doi 10.1038/s41467-018-04315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saucier-Sawyer JK, Seo YE, Gaudin A, Quijano E, Song E, Sawyer AJ, et al. Distribution of polymer nanoparticles by convection-enhanced delivery to brain tumors. Journal of controlled release : official journal of the Controlled Release Society 2016;232:103–12 doi 10.1016/j.jconrel.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He C, Cai P, Li J, Zhang T, Lin L, Abbasi AZ, et al. Blood-brain barrier-penetrating amphiphilic polymer nanoparticles deliver docetaxel for the treatment of brain metastases of triple negative breast cancer. Journal of controlled release : official journal of the Controlled Release Society 2017;246:98–109 doi 10.1016/j.jconrel.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 27.Stephen ZR, Kievit FM, Veiseh O, Chiarelli PA, Fang C, Wang K, et al. Redox-responsive magnetic nanoparticle for targeted convection-enhanced delivery of O6-benzylguanine to brain tumors. ACS nano 2014;8(10):10383–95 doi 10.1021/nn503735w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruan S, Xiao W, Hu C, Zhang H, Rao J, Wang S, et al. Ligand-Mediated and Enzyme-Directed Precise Targeting and Retention for the Enhanced Treatment of Glioblastoma. ACS applied materials & interfaces 2017;9(24):20348–60 doi 10.1021/acsami.7b02303. [DOI] [PubMed] [Google Scholar]
- 29.Huang R, Harmsen S, Samii JM, Karabeber H, Pitter KL, Holland EC, et al. High Precision Imaging of Microscopic Spread of Glioblastoma with a Targeted Ultrasensitive SERRS Molecular Imaging Probe. Theranostics 2016;6(8):1075–84 doi 10.7150/thno.13842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng C, Gao X, Xu J, Du B, Ning X, Tang S, et al. Targeting orthotopic gliomas with renal-clearable luminescent gold nanoparticles. Nano research 2017;10(4):1366–76 doi 10.1007/s12274-017-1472-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen CW, Yeh MK, Shiau CY, Chiang CH, Lu DW. Efficient downregulation of VEGF in retinal pigment epithelial cells by integrin ligand-labeled liposome-mediated siRNA delivery. International journal of nanomedicine 2013;8:2613–27 doi 10.2147/ijn.S39622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung EJ, Cheng Y, Morshed R, Nord K, Han Y, Wegscheid ML, et al. Fibrin-binding, peptide amphiphile micelles for targeting glioblastoma. Biomaterials 2014;35(4):1249–56 doi 10.1016/j.biomaterials.2013.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Johnson J, Peck A, Xie Q. Near infrared fluorescent imaging of brain tumor with IR780 dye incorporated phospholipid nanoparticles. Journal of translational medicine 2017;15(1):18 doi 10.1186/s12967-016-1115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X, Braun GB, Zhong H, Hall DJ, Han W, Qin M, et al. Tumor-Targeted Multimodal Optical Imaging with Versatile Cadmium-Free Quantum Dots. Advanced functional materials 2016;26(2):267–76 doi 10.1002/adfm.201503453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu J, Zhang X, Wen Z, Tan Y, Huang N, Cheng S, et al. Asn-Gly-Arg-modified polydopamine-coated nanoparticles for dual-targeting therapy of brain glioma in rats. Oncotarget 2016;7(45):73681–96 doi 10.18632/oncotarget.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nature reviews Clinical oncology 2010;7(11):653–64 doi 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wadajkar AS, Dancy JG, Roberts NB, Connolly NP, Strickland DK, Winkles JA, et al. Decreased non-specific adhesivity, receptor targeted (DART) nanoparticles exhibit improved dispersion, cellular uptake, and tumor retention in invasive gliomas. Journal of controlled release : official journal of the Controlled Release Society 2017;267:144–53 doi 10.1016/j.jconrel.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang T, Jiang M, Jiang D, Feng X, Yao J, Song Q, et al. Enhancing Glioblastoma-Specific Penetration by Functionalization of Nanoparticles with an Iron-Mimic Peptide Targeting Transferrin/Transferrin Receptor Complex. Molecular pharmaceutics 2015;12(8):2947–61 doi 10.1021/acs.molpharmaceut.5b00222. [DOI] [PubMed] [Google Scholar]
- 39.Monaco I, Camorani S, Colecchia D, Locatelli E, Calandro P, Oudin A, et al. Aptamer Functionalization of Nanosystems for Glioblastoma Targeting through the Blood-Brain Barrier. Journal of medicinal chemistry 2017;60(10):4510–6 doi 10.1021/acs.jmedchem.7b00527. [DOI] [PubMed] [Google Scholar]
- 40.Clark AJ, Davis ME. Increased brain uptake of targeted nanoparticles by adding an acid-cleavable linkage between transferrin and the nanoparticle core. Proceedings of the National Academy of Sciences of the United States of America 2015;112(40):12486–91 doi 10.1073/pnas.1517048112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miao D, Jiang M, Liu Z, Gu G, Hu Q, Kang T, et al. Co-administration of dual-targeting nanoparticles with penetration enhancement peptide for antiglioblastoma therapy. Molecular pharmaceutics 2014;11(1):90–101 doi 10.1021/mp400189j. [DOI] [PubMed] [Google Scholar]
- 42.van Rooy I, Mastrobattista E, Storm G, Hennink WE, Schiffelers RM. Comparison of five different targeting ligands to enhance accumulation of liposomes into the brain. Journal of controlled release : official journal of the Controlled Release Society 2011;150(1):30–6 doi 10.1016/j.jconrel.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 43.Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling Adult Gliomas Using RCAS/t-va Technology. Translational oncology 2009;2(2):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y, et al. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Science translational medicine 2014;6(260):260ra149 doi 10.1126/scitranslmed.3009524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benezra M, Phillips E, Overholtzer M, Zanzonico PB, Tuominen E, Wiesner U, et al. Ultrasmall integrin-targeted silica nanoparticles modulate signaling events and cellular processes in a concentration-dependent manner. Small (Weinheim an der Bergstrasse, Germany) 2015;11(14):1721–32 doi 10.1002/smll.201402331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoo B, Ma K, Zhang L, Burns A, Sequeira S, Mellinghoff I, et al. Ultrasmall dual-modality silica nanoparticle drug conjugates: Design, synthesis, and characterization. Bioorganic & medicinal chemistry 2015;23(22):7119–30 doi 10.1016/j.bmc.2015.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Zhao X, Zhang Z, Chen W, Zhang X. An immunohistochemistry study of Sox9, Runx2, and Osterix expression in the mandibular cartilages of newborn mouse. BioMed research international 2013;2013:265380 doi 10.1155/2013/265380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma K, Mendoza C, Hanson M, Werner-Zwanziger U, Zwanziger J, Wiesner U. Control of Ultrasmall Sub-10 nm Ligand-Functionalized Fluorescent Core–Shell Silica Nanoparticle Growth in Water. Chemistry of Materials 2015;27(11):4119–33 doi 10.1021/acs.chemmater.5b01222. [DOI] [Google Scholar]
- 49.Weis SM, Cheresh DA. alphaV integrins in angiogenesis and cancer. Cold Spring Harbor perspectives in medicine 2011;1(1):a006478 doi 10.1101/cshperspect.a006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma K, Wiesner U. Modular and Orthogonal Post-PEGylation Surface Modifications by Insertion Enabling Penta-Functional Ultrasmall Organic-Silica Hybrid Nanoparticles. Chemistry of Materials 2017;29(16):6840–55 doi 10.1021/acs.chemmater.7b02009. [DOI] [Google Scholar]
- 51.Ma K, Zhang D, Cong Y, Wiesner U. Elucidating the Mechanism of Silica Nanoparticle PEGylation Processes Using Fluorescence Correlation Spectroscopies. Chemistry of Materials 2016;28(5):1537–45 doi 10.1021/acs.chemmater.6b00030. [DOI] [Google Scholar]
- 52.Burns AA, Vider J, Ow H, Herz E, Penate-Medina O, Baumgart M, et al. Fluorescent silica nanoparticles with efficient urinary excretion for nanomedicine. Nano letters 2009;9(1):442–8 doi 10.1021/nl803405h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benezra M, Penate-Medina O, Zanzonico PB, Schaer D, Ow H, Burns A, et al. Multimodal silica nanoparticles are effective cancer-targeted probes in a model of human melanoma. The Journal of clinical investigation 2011;121(7):2768–80 doi 10.1172/jci45600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barteau KP, Ma K, Kohle FFE, Gardinier TC, Beaucage PA, Gillilan RE, et al. Quantitative Measure of the Size Dispersity in Ultrasmall Fluorescent Organic–Inorganic Hybrid Core–Shell Silica Nanoparticles by Small-Angle X-ray Scattering. Chemistry of Materials 2019;31(3):643–57 doi 10.1021/acs.chemmater.8b04369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen F, Ma K, Benezra M, Zhang L, Cheal SM, Phillips E, et al. Cancer-Targeting Ultrasmall Silica Nanoparticles for Clinical Translation: Physicochemical Structure and Biological Property Correlations. Chemistry of materials : a publication of the American Chemical Society 2017;29(20):8766–79 doi 10.1021/acs.chemmater.7b03033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen F, Ma K, Zhang L, Madajewski B, Zanzonico P, Sequeira S, et al. Target-or-Clear Zirconium-89 Labeled Silica Nanoparticles for Enhanced Cancer-Directed Uptake in Melanoma: A Comparison of Radiolabeling Strategies. Chemistry of materials : a publication of the American Chemical Society 2017;29(19):8269–81 doi 10.1021/acs.chemmater.7b02567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brooks PC, Stromblad S, Klemke R, Visscher D, Sarkar FH, Cheresh DA. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. The Journal of clinical investigation 1995;96(4):1815–22 doi 10.1172/jci118227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bradbury MS, Phillips E, Montero PH, Cheal SM, Stambuk H, Durack JC, et al. Clinically-translated silica nanoparticles as dual-modality cancer-targeted probes for image-guided surgery and interventions. Integrative biology : quantitative biosciences from nano to macro 2013;5(1):74–86 doi 10.1039/c2ib20174g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer research 1986;46(12 Pt 1):6387–92. [PubMed] [Google Scholar]
- 60.Natarajan R, Northrop N, Yamamoto B. Fluorescein Isothiocyanate (FITC)-Dextran Extravasation as a Measure of Blood-Brain Barrier Permeability. Current protocols in neuroscience 2017;79:9.58.1–9..15 doi 10.1002/cpns.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mayhan WG, Heistad DD. Permeability of blood-brain barrier to various sized molecules. The American journal of physiology 1985;248(5 Pt 2):H712–8. [DOI] [PubMed] [Google Scholar]
- 62.Humphries WHt, Szymanski CJ, Payne CK. Endo-lysosomal vesicles positive for Rab7 and LAMP1 are terminal vesicles for the transport of dextran. PloS one 2011;6(10):e26626 doi 10.1371/journal.pone.0026626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee FT, Scott AM. Immuno-PET for tumor targeting. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 2003;44(8):1282–3. [PubMed] [Google Scholar]
- 64.Rempel SA, Rosenblum ML, Mikkelsen T, Yan PS, Ellis KD, Golembieski WA, et al. Cathepsin B expression and localization in glioma progression and invasion. Cancer research 1994;54(23):6027–31. [PubMed] [Google Scholar]
- 65.Aggarwal N, Sloane BF. Cathepsin B: multiple roles in cancer. Proteomics Clinical applications 2014;8(5–6):427–37 doi 10.1002/prca.201300105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joyce JA, Hanahan D. Multiple roles for cysteine cathepsins in cancer. Cell cycle (Georgetown, Tex) 2004;3(12):1516–619 doi 10.4161/cc.3.12.1289. [DOI] [PubMed] [Google Scholar]
- 67.Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes & development 2008;22(4):436–48 doi 10.1101/gad.1627008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? British journal of cancer 2017;117(12):1736–42 doi 10.1038/bjc.2017.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.