

Abstract
Background:
Clinical and epidemiological studies have shown that obesity is associated with asthma and that these associations differ by asthma subtypes. Little is known about the shared genetic components between obesity and asthma.
Objective:
To identify shared genetic associations between obesity-related traits and asthma subtypes in adults.
Methods:
A cross-trait genome-wide association study (GWAS) was performed using 457,822 individuals of European ancestry from the UK Biobank. Experimental evidence to support the role of genes significantly associated with both obesity-related traits and asthma via GWAS was sought using results from obese vs. lean mouse RNA-seq and RT-PCR experiments.
Results:
We found a substantial positive genetic correlation between BMI and later-onset asthma defined by asthma age of onset at 16 years of age or older (Rg =0.25, P=9.56×10−22). Mendelian Randomization analysis provided strong evidence in support of BMI causally increasing the risk of asthma. Cross-trait meta-analysis identified 34 shared loci among 3 obesity-related traits and 2 asthma subtypes. GWAS functional analyses identified potential causal relationships between the shared loci and GTEx tissue eQTLs, shared immune- and cell differentiation-related pathways between obesity and asthma. Finally, RNA-seq data from lungs of obese versus control mice found that two genes (ACOXL and MYL6) from the cross-trait meta-analysis were differentially expressed, and these findings were validated by RT-PCR in an independent set of mice.
Conclusions:
Our work identified shared genetic components between obesity-related traits and specific asthma subtypes, reinforcing the hypothesis that obesity causally increases the risk of asthma, and identifying molecular pathways that may underlie both obesity and asthma.
Keywords: obesity, metabolic disorder, asthma subtypes, shared genetics, genome-wide association study, RNA-seq, RT-PCR
Graphical Abstract
Capsule Summary:
This large-scale genome-wide cross-trait study discovers shared genetic etiology of obesity- and glycemic-related traits with specific asthma subtypes in adults and reinforces the idea of genetic instruments of obesity causally increases the risk of asthma.
Introduction
Asthma is a common chronic disease that accounts for a substantial burden of disease worldwide1. Metabolic disorders, such as obesity and type 2 diabetes (T2D), have become highly prevalent globally and in the US2, and numerous studies have linked metabolic traits with increased asthma risk3, 4. Asthma is a highly heterogeneous disease; both age of onset and atopic status play important roles in currently recognized asthma subtypes5. Early-onset asthma is highly heritable and associated with atopy, while later-onset asthma is often non-atopic and resistant to treatment6. Recent studies indicate that different asthma subtypes are related disproportionally with metabolic traits, such as obesity7. For example, obesity appears to have a stronger association with non-atopic asthma than atopic asthma8.
Metabolic disorders and asthma are highly heritable traits, and the parallel rise in prevalence of metabolic disorders (especially obesity) and asthma worldwide suggests these conditions share genetic and environmental risk factors1. Understanding the shared genetic architecture across various complex traits has been a topic of recent interest. For example, shared genetic risk factors have been identified between sleep disturbance and metabolic traits9, and type 2 diabetes and coronary heart diseases10. Additionally, we previously performed a cross-trait genome-wide association study (GWAS) to identify shared genetic variants among asthma and allergic diseases11, Alzheimer’s disease and metabolic traits12, and chronic obstructive pulmonary diseases and cardiovascular traits13. Previous studies that have investigated shared genetic associations between obesity-related traits (i.e., obesity and high-density lipoprotein [HDL]) and asthma14-16 have found that genes such as GNPDA2, PTPRD and ROBO1 are associated with both body-mass index (BMI and asthma, these studies were limited by small sample size). Therefore, it remains to be determined to what extent the phenotypic association between obesity-related traits and asthma is due to genetic effects, and whether the shared genetic factors with obesity differ among main asthma subtypes: early-onset and later-onset asthma. In the current study, we conducted a large-scale GWAS cross-trait analysis between asthma (early-onset, later-onset asthma, atopic asthma and non-atopic asthma) and obesity traits (i.e., BMI, BMI adjusted waist-to-hip ratio [WHRadjBMI], and BMI adjusted waist circumferences [WCadjBMI]) using UK Biobank and other publicly available GWAS data for obesity-related metabolic traits, including T2D from the DIAGRAM Consortium17, fasting glucose (FG) and fasting insulin (FI) from the MAGIC Consortium18, and blood lipids (i.e. low-density lipoproteins [LDL], HDL, total cholesterol [TC], triglycerides [TG]) from the ENGAGE Consortium19.
Methods
Study Population and Design
The UK Biobank study has been previously described in detail11, 20. All participants provided informed consent to the UK Biobank. Our overall study design is shown in Figure 1. To identify genetic variants that contributed to self-reported obesity traits and doctor-diagnosed asthma, we performed GWAS using phenotype data provided for UK Biobank participants (N = 487,409). We restricted subjects to 457,822 individuals of European ancestry (457,822 with BMI measures, 457,690 with WHR and WC measures, 13,435 with early-onset asthma, 33,418 with later-onset asthma, 28,862 with non-atopic asthma, 23,982 with atopic asthma) with high-quality genotyping and complete phenotype/covariate data. We did not remove related samples because we used a linear mixed model (LMM) method for phenotype-genotype association analysis that appropriately accounted for relatedness21. Detailed demographic information is provided in Supplementary Table 1.
Figure 1.

Overall study design.
UKBB: UK Biobank; LDSC: LD score regression. GSMR: generalized summary data-based Mendelian Randomization; LCV: Latent Causal Variable model
Ascertainment of asthma in UK Biobank
Asthma subjects were subset in two ways: by age of onset and atopic status. Early-onset asthma was defined as an individual with doctor-diagnosed asthma that developed before 16 years of age and later-onset asthma as doctor-diagnosed asthma that developed at 16 years of age or older. Non-atopic asthma was defined as doctor-diagnosed asthma without hayfever/allergic rhinitis or eczema. Atopic asthma was defined as doctor-diagnosed asthma with hayfever/allergic rhinitis or eczema. UK Biobank data field 6154 was used to determine doctor-diagnosed asthma or allergic diseases; data field 3786 was used to determine age of asthma onset. There were 21,434 cases who had later-onset and non-atopic asthma; 15,172 cases who had later-onset and atopic asthma; 6,640 cases who had early-onset and non-atopic asthma; and 7,999 cases who had early-onset and atopic asthma.
GWAS analysis
Genome-wide genotyping was performed in UK Biobank participants using the UK Biobank Axiom array and UK BiLEVE Axiom™ Array from Affymetrix20. Genotype imputation was performed using the Haplotype Reference Consortium panel22. We selected variants that did not deviate from Hardy-Weinberg Equilibrium (P>1×10−6), have per-variant missing call rates <10%, have per-sample missing rate <10%, and did have minor allele frequency >1% and imputation quality score >0.8. Detailed quality control (QC) and imputation information is provided in the Supplementary Appendix. We used BOLT-LMM v2.321 to conduct association tests. For continuous outcomes, we carried out LMM association analyses while adjusting for age, age squared, sex, genotyping array and twenty ancestry principal components (PCs) to assess association between the inverse normally transformed phenotype residuals and imputed genotype dosages. For binary phenotypes, we performed LMM association analyses while adjusting for age, sex, genotyping array, twenty ancestry PCs. The effect estimate output of BOLT-LMM linear regression was transformed into log odds ratio.
After association analyses, we used the PLINK clumping function (parameters: --clump-p1 5×10−8 --clump-p2 1×10−5 --clump-r2 0.2 --clump-kb 500) to determine top loci that were independent of each other (i.e., variants with p-value less <1×10−5, r2 >0.2 and within 500kb of a given peak's clump).
Linkage disequilibrium score regression (LDSC) analysis
LDSC estimates genetic correlation between the true causal effects of two traits (ranging from −1 to 1) from summary statistics using the fact that the GWAS effect size estimate for each SNP incorporates the effects of all SNPs in linkage disequilibrium with that SNP. LDSC applies a self-estimated intercept during the analysis to account for shared subjects between studies11, 23.
Analysis of sex-specific effects
Previous studies have shown that the association between asthma and metabolic traits differs by sex24 and that girls with early age at menarche have lower lung function and increased risk of asthma in adulthood25. Thus, we performed sex-stratified analyses to evaluate the genetic correlation between obesity traits and asthma subtypes according sex and without the effect of age at menarche in female obesity traits and asthma.
Partitioned genetic correlation analysis
To characterize the genetic correlation at the level of functional categories, we estimated genetic correlations between metabolic traits and asthma (later-onset and non-atopic) using partitioned LDSC in 11 annotation categories: transcribed region, super enhancer, intron, transcription factor binding sites (TFBS), DNaseI digital genomic footprinting (DGF) region, DNase I hypersensitivity sites (DHSs), fetal DHS, and histone marks H3K4me1, H3K4me3, H3K9ac, and H3K27ac26, 27. For each annotation category, we re-calculated LD scores for SNPs assigned to that particular category and then used the annotation-specific LD scores for estimating the metabolic trait-asthma genetic correlation.
Genetic causal inference analysis
We used generalized summary data-based Mendelian Randomization (GSMR)28 and Latent Causal Variable (LCV)29 to infer putative direction of causality between BMI and asthma phenotypes. While both GSMR and LCV perform causal inference based on GWAS summary statistics, GSMR estimates effect of exposure on outcome using SNPs significantly associated with exposure28, whereas LCV estimates genetic causality proportion (GCP) using all common SNPs and is robust to confounding by pleiotropy29. A significant effect of exposure on outcome and a GCP close to 1 are strong evidence of causal relationship. Because samples shared across GWAS induce bias in causal inference30, we avoided sample overlap by using summary statistics obtained from our UK Biobank analysis for asthma phenotypes and publicly available summary statistics from the GIANT consortium for BMI31. We did not use other obesity-related phenotypes (i.e., WHRadjBMI and WCadjBMI) for causal inference analyses, as these phenotypes are adjusted for heritable covariates and are thus prone to collider bias32, 33. We removed strand-ambiguous SNPs and SNPs in the MHC region (chr6:25-34M) only in MR analyses due to its strong pleotropic effect.
Cross-trait meta-analysis
Cross Phenotype Association (CPASSOC) combines effect estimates and standard errors of GWAS summary statistics to test the hypothesis of association between a SNP with two traits34. We used heterogonous version of CPASSOC (SHet) that is based on a fixed effect model and is more powerful when there is a heterogonous effect present between studies, which is common35. SHet uses the sample size for a trait as a weight and accounts for correlation due to overlapping or related subjects within and among different studies.
Fine mapping credible-set analysis
In order to identify the most credible genes within the clumping results, we identified a credible set of variants that were 99% likely to contain the causal variant at each of the shared loci obtained from the cross-trait meta-analysis using the Bayesian-likelihood fine-mapping algorithm36. This algorithm maps the primary signal using a flat prior with steepest descent approximation.
Overrepresentation enrichment analysis
To identify biological pathways that significant genes after cross-trait meta-analysis (Pmeta<5×10−8) were involved in, we used the WebGestalt tool37 to assess overrepresented enrichment of gene sets in Gene Ontology (GO) biological process categories.
Colocalization analysis
We used the R package coloc38 to determine whether cross-trait meta-analysis association signals of shared loci co-localized at the same variant. After extracting summary association data for variants within 500kb of the index SNP at each of the shared loci, we calculated the posterior probability that the two traits were associated with different causal variants (H3) or that the two traits were associated and shared one common causal variant (H4). Loci were considered to be co-localized if the posterior probability H4 was >0.7. In addition, since the GTEx eQTL signals by themselves are pervasive, we conducted the colocalization analysis between cross-trait meta-analysis results and 48 single GTEx tissue cis-eQTL (version 7) to determine if the same genetic variants were related to expression and the diseases.
Mouse lung gene expression changes with obesity
We obtained two independent measures of differential lung expression, one with RNA-Seq and one with RT-PCR, to identify gene expression changes in lung between (1) mice on a high fat diet (HFD) vs. a low fat diet (LFD) mice and (2) autosomal recessive diabetes mutation (db/db) vs. wild-type mice. Such differentially expressed genes were sought as providing evidence linking obesity to asthma vis-à-vis potential changes in lung tissue. A detailed description of gene expression analyses is provided in the Supplementary Appendix.
Results
Single Trait GWAS Results
We identified 1,636 genome-wide significant independent loci associated with BMI, 1,074 with WHRadjBMI, 1,260 with WCadjBMI, 77 with later-onset asthma, 219 with early-onset asthma, 101 with non-atopic asthma, and 198 with atopic asthma (Supplementary Table 2-8, Supplementary Figure 1-7). The genomic inflation factor intercepts from LDSC were 1.12 for BMI, 1.19 for WHRadjBMI, 1.17 for WCadjBMI, 1.03 for later-onset asthma, 1.06 for early-onset asthma, 1.04 for non-atopic asthma and 1.05 for atopic asthma (Supplementary Figure 8-14). Given the small genomic inflation factor intercept values, we attributed the lift-off of QQ plots (high lambda GC) to polygenicity and not population stratification.
Genetic Correlation between Metabolic Traits and Asthma
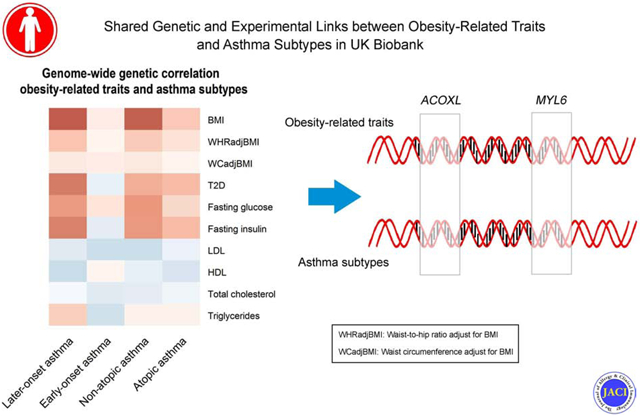
We estimated the genetic correlation of metabolic traits with four asthma subtypes. As shown in Figure 2 and Supplementary Table 9, obesity traits were positively genetically correlated with later-onset asthma (Rg=0.25, P=9.56×10−22 for BMI; and Rg=0.08, P=1.19×10−3 for WHRadjBMI), but not with early-onset asthma. Sex-specific analyses found no significant difference between the later-onset asthma/BMI genetic correlation in males and females, but did find a significant difference between later-onset asthma and WHRadjBMI genetic correlation (Supplementary Table 10-11). When we extended our analysis to atopic and non-atopic asthma, we observed a positive genetic correlation between obesity traits and non-atopic asthma (Rg=0.24 , P=5.75×10−17 for BMI; and Rg=0.07, P=0.02 for WHRadjBMI), but only a modest association between BMI and atopic asthma (Rg=0.08, P=1.70×10−3). In addition, T2D, FG, and FI had significant positive genetic correlations with later-onset asthma (Rg: 0.20, 0.15, and 0.19; P: 1.39×10−6, 0.02, and 9.21×10−3, respectively) and non-atopic asthma (Rg: 0.12, 0.16, and 0.15; P: 4.19×10−3, 5.72×10−3, and 0.04, respectively) but not with early-onset asthma. We did not find significant genetic correlations between lipids (i.e., HDL, LDL, TC, TG) and any asthma subtypes. To evaluate whether certain annotation categories contributed disproportionately to the observed whole-genome genetic correlation between later-onset/non-atopic asthma and obesity-related traits, we performed LDSC for 11 functional annotation categories (Supplementary Table 12-13). Generally, the partitioned LDSC by functional categories were consistent and had similar magnitude as genome-wide genetic correlations. For example, BMI was significantly correlated with later-onset asthma (Rg ranges from 0.21 to 0.25, P<0.05/110) and non-atopic asthma (Rg ranges from 0.18 to 0.26, P<0.05/110) for nine functional categories (i.e., transcribed region, TFBS, DGF, DHSs, fetal DHS, H3K4me1, H3K4me3, H3K9ac, and H3K27ac) after correcting for multiple comparisons. The Intron category had Rg=0.37 but was not statistically significant after multiple comparisons correction (P= 0.0043). No functional category had a disproportionate contribution to the genetic correlation between later-onset/non-atopic asthma and obesity-related traits. Thus, our results provided evidence that obesity-related traits and later-onset/non-atopic asthma are genetically correlated and motivated us to further investigate the genetic components shared between these traits.
Figure 2.

Genome-wide genetic correlation between 4 types of asthma and metabolic traits
The color of each box scales with the magnitude of the genetic correlation. Pairs of traits with nominal significant genetic correlation (p < 0.05) are marked by 1 star, and pairs of traits with significant genetic correlation after correcting for multiple testing (p < 0.05/40) are marked by 2 stars.
Causal Inference
We performed causal inference between BMI (GIANT consortium) and UK Biobank asthma phenotypes using GSMR and LCV. A strong positive causal effect of BMI on asthma phenotypes (later-onset asthma, non-atopic asthma, and atopic asthma) but not vice versa was observed with GSMR (Table 1). For example, the estimated effect of BMI on later-onset asthma was significant with an OR=1.21 (P=6.3×10−7), but the estimated effect of later-onset asthma on BMI was not (OR=1.00; P=0.70), where the odds ratio (OR) in the former case represents the increase in the odds of having asthma, per standard deviation increase in BMI. Our estimated causal effect of BMI on asthma was comparable to that obtained by Skaaby et al. (OR=1.07, 95% CI: [1.03, 1.10] based on a GWAS of sample size N=162,124 and 26 instruments)39, and that obtained by Granell et al. (OR=1.55, 95% CI: [1.16, 2.07] based on a GWAS of sample size N=4,835 and 32 instruments)40.
Table 1.
Estimates of causal effect size and genetic causality proportion (GCP) for BMI and asthma phenotypes, obtained by 2-sample bi-directional GSMR and LCV, respectively
| Trait 1 | Trait 2 | Causal Odds Ratio (S.E.) | Genetic Causality Proportion (S.E.) |
||
|---|---|---|---|---|---|
| BMI | Later-onset Asthma | forward | 1.21 (0.04) | P=6.3×10−7 | 0.90 (0.09) |
| reverse | 1.00 (0.01) | P=0.70 | |||
| BMI | Non-atopic Asthma | forward | 1.10 (0.04) | P=0.04 | 0.86 (0.11) |
| reverse | 1.00 (0.006) | P=0.75 | |||
| BMI | Atopic Asthma | forward | 1.20 (0.03) | P=8.4×10−6 | 0.68 (0.21) |
| reverse | 1.00 (0.01) | P=0.76 | |||
BMI GWAS is from GIANT consortium. The “forward” denotes trait 1 is the exposure and trait 2 the outcome; and the “reverse” denotes trait 2 as the exposure and trait 1 the outcome. Here, we used the default GSMR outlier p-value threshold of 0.01. Causal effect size estimates obtained using a more exclusive threshold (p<0.1) can be found in Supplementary Table 34.
Because BMI and asthma are polygenic traits11, 31, inferred causal relationships between them are prone to confounding due to pleiotropy. Therefore, we also applied LCV, a method robust to pleiotropy confounding, to infer the genetic causality proportion (GCP) between BMI and asthma phenotypes. GCP is a unitless measure of causality and equal to 1 under full causality. Reassuringly, we found that the LCV estimates of GCP for all inferred causal relationships were close to 1 (Table 1). For example, the estimated GCP for the causal direction BMI → later-onset asthma was 0.90 (S.E. 0.09), not significantly different from 1. Taken together, both GSMR and LCV provided strong evidence in support of the putative model that BMI causally increases the risk of asthma. Detailed causal inference results are provided in Supplementary Table 14.
Cross-Trait Associations Between Metabolic Traits and Asthma
In total, we identified 8 independent loci shared between BMI and later-onset asthma and 10 shared between BMI and non-atopic asthma (Psingle trait<1×10−5 and Pmeta<5×10−8) (Table 2). The credible set of SNPs for each of these shared loci was also identified (Supplementary Tables 15 and 16). Our results found both similarities and differences in the shared genetic components of obesity-related traits with later-onset and non-atopic asthma. Three out of the 8 significant shared loci between BMI and later-onset asthma were also significantly shared between BMI and non-atopic asthma at the same top variant (including 12q13.2 [ERBB3, index SNP: rs4759229], 1p35.2 [COL16A1, index SNP: rs6681149], and 17q25.1 [UNC13D, index SNP: rs111365807]). Interestingly, the strongest shared signal of BMI with later-onset and non-atopic asthma was at chromosome 12q13.2 (ERBB3, index SNP: rs4759229) (Pmeta=1.67×l0−23 for later-onset asthma and Pmeta=1.75×10−21 for non-atopic asthma). Other shared genetic components between BMI and later-onset asthma included 1q24.1 (POU2F1 or Oct-1), 2q13 (ACOXL), 3q26.32 (TBL1XR1), 4q12 (near USP46), and 6q24.2 (PLAGL1). POU2F1 (Oct-1) and TBL1XR1 are both involved in glucocorticoid receptor regulation, while loss of TBL1XR1 drives glucocorticoid resistance41, and thus, may play a functional role in the glucocorticoid resistance observed in obese asthmatics42. Shared loci between BMI and non-atopic asthma were 1p36.23 (RERE), 6p21.32 (near HLA-DQB1), 7p21.1 (near ABCB5), 11q13.2 (ACTN3), 17q21.1 (PSMD3), 17q25.3 (near TIMP2 and USP36), and 18q12.3 (near PIK3C3), genes involved in cell cycle processes including cellular differentiation, apoptosis, or proliferation43-45.
Table 2.
Cross-trait meta-analysis results between 2 types of asthma and 3 obesity-related metabolic traits (Pmeta<5×10−8, single trait P<1×10−5)
| Model | SNP | Genome position | Effect Allele |
Ref Allele |
P1 | P2 | PMeta | Genes within clumping region |
|---|---|---|---|---|---|---|---|---|
| Later-onset asthma and BMI | rs6427082 | chr1:167179419-167393635 | T | A | 1.00×10−07 | 1.80×10−06 | 2.66×10−13 | POU2F1 |
| rs6681149 | chr1:32152518-32228004 | G | A | 6.70×10−06 | 3.30×10−10 | 2.91×10−13 | BAI2,COL16A1,MIR4254 | |
| rs72836344 | chr2:111861838-112023842 | G | T | 2.10×10−06 | 3.80×10−06 | 3.62×10−09 | ACOXL,BCL2L11 | |
| rs12493005 | chr3:176869498-176928657 | C | T | 7.00×10−06 | 3.00×10−06 | 2.44×10−11 | TBL1XR1 | |
| rs7670374 | chr4:53272753-53392903 | G | A | 4.10×10−06 | 5.70×10−07 | 4.89×10−10 | Intergenic region | |
| rs6900923 | chr6:144277568-144318433 | C | G | 3.70×10−06 | 1.30×10−06 | 4.74×10−10 | PLAGL1 | |
| rs4759229 | chr12:56379060-56609885 | A | G | 1.60×10−09 | 1.10×10−13 | 1.67×10−23 | ERBB3,ESYT1,IKZF4,MYL6, MYL6B,PA2G4,RAB5B,RNF4 1,RPL41,RPS26,SMARCC2,S UOX,ZC3H10 | |
| rs111365807 | chr17:73757836-73953404 | G | C | 3.90×10−07 | 1.40×10−07 | 7.07×10−12 | ACOX1,FBF1,GALK1,H3F3B,MIR4738,MRPL38,TRIM47,TRIM65,UNC13D,UNK,WBP2 | |
| Later-onset asthma and WHRadjBMI | rs72836349 | chr2:111861838-112023842 | T | C | 2.20×10−06 | 1.50×10−13 | 1.02×10−18 | ACOXL,BCL2L11 |
| rs1787663 | chr11:65211979-65342981 | C | A | 5.90×10−06 | 2.30×10−06 | 6.35×10−10 | FAM89B,LTBP3,MALAT1,MIR548AR,MIR548BA,MIR612,SCYL1,SSSCA1,SSSCA1-AS1 | |
| rs4616071 | chr11:111366844-111443192 | C | G | 1.40×10−06 | 1.40×10−06 | 5.85×10−12 | BTG4,C11orf88,LAYN,MIR34B,MIR34C | |
| rs10152595 | chr15:67468525-67661784 | C | G | 1.10×10−11 | 1.80×10−11 | 2.56×10−20 | AAGAB,IQCH,SMAD3 | |
| rs4842921 | chr15:84497207-84637651 | G | A | 2.70×10−06 | 3.30×10−06 | 7.60×10−11 | ADAMTSL3 | |
| rs8103278 | chr19:46219145-46374916 | G | A | 1.70×10−06 | 9.90×10−07 | 1.30×10−11 | DMPK,DMWD,FBXO46,FOXA3,LOC388553,RSPH6A,SIX5,SYMPK | |
| Later-onset asthma and FG | rs174583 | chr11:61537529-61650747 | C | T | 4.60×10−16 | 1.69×10−08 | 4.45×10−17 | FADS1,FADS2,FADS3,FEN1,MIR611,MIR1908,MIR6746,MYRF,TMEM258 |
| Non-atopic asthma and BMI | rs1381928 | chr1:8378599-8895866 | G | A | 2.30×10−07 | 2.00×10−06 | 3.90×10−10 | RERE,SLC45A1 |
| rs6681149 | chr1:32152518-32228004 | G | A | 2.80×10−06 | 3.30×10−10 | 3.17×10−13 | BAI2,COL16A1,MIR4254 | |
| rs9275570 | chr6:32428062-32681631 | G | A | 2.90×10−18 | 2.60×10−06 | 1.81×10−19 | HLA-DQA1,HLA-DQB1,HLA-DRB1,HLA-DRB5,HLA-DRB6 | |
| rs28423374 | chr7:20501204-20638155 | T | C | 9.90×10−06 | 3.70×10−07 | 2.42×10−11 | Intergenic region | |
| rs540874 | chr11:66151649-66483265 | A | G | 1.60×10−06 | 5.20×10−06 | 4.09×10−09 | ACTN3,BBS1,CCDC87,CCS,CTSF,DPP3,MRPL11,NPAS4,PELI3,RBM4,RBM4B,RBM14,RBM14-RBM4,SPTBN2,ZDHHC24 | |
| rs4759229 | chr12:56379060-56609885 | A | G | 6.90×10−09 | 1.10×10−13 | 1.75×10−21 | ERBB3,ESYT1,IKZF4,MYL6,MYL6B,PA2G4,RAB5B,RNF41,RPL41,RPS26,SMARCC2,SUOX,ZC3H10 | |
| rs111365807 | chr17:73757836-73953404 | G | C | 1.00×10−07 | 1.40×10−07 | 9.81×10−12 | ACOX1,FBF1,GALK1,H3F3B,MIR4738,MRPL38,TRIM47,TRIM65,UNC13D,UNK,WBP2 | |
| rs3826331 | chr17:38128765-38218804 | T | C | 1.40×10−08 | 2.90×10−06 | 1.13×10−12 | CSF3,GSDMA,MED24,MIR6884,PSMD3,SNORD124,THRA | |
| rs9895419 | chr17:76667271-76858539 | A | G | 7.40×10−06 | 3.60×10−06 | 1.32×10−10 | CYTH1,TIMP2,USP36 | |
| rs8089834 | chr18:39468982-39697096 | T | C | 7.60×10−06 | 4.20×10−09 | 1.46×10−13 | PIK3C3 | |
| Non-atopic asthma and WHRadjBMI | rs10152595 | chr15:67468525-67661784 | C | G | 2.80×10−12 | 1.80×10−11 | 1.40×10−20 | AAGAB,IQCH,SMAD3 |
| rs112104755 | chr18:20580534-20580534 | A | G | 3.50×10−06 | 9.00×10−06 | 6.50×10−10 | RBBP8 | |
| rs11213940 | chr11:111366844-111776891 | C | A | 4.30×10−09 | 1.60×10−06 | 1.73×10−13 | ALG9,BTG4,C11orf1,C11orf88,FDXACB1,LAYN,MIR34B,MIR34C,PPP2R1B,SIK2 | |
| rs12491785 | chr3:168679771-168905214 | C | T | 2.50×10−07 | 7.10×10−08 | 1.10×10−12 | MECOM | |
| rs73084574 | chr7:20344126-20344126 | G | A | 9.10×10−06 | 3.20×10−08 | 4.39×10−12 | LOC101927769 | |
| rs73086541 | chr7:20371232-20478448 | C | A | 1.40×10−08 | 3.10×10−11 | 6.90×10−18 | ITGB8,LOC101927811 | |
| rs73504817 | chr19:17152232-17229439 | T | C | 1.50×10−06 | 4.90×10−11 | 1.43×10−15 | HAUS8,MYO9B | |
| rs8103278 | chr19:46219145-46374916 | G | A | 4.40×10−06 | 9.90×10−07 | 5.12×10−11 | DMPK,DMWD,FBXO46,FOXA3,LOC388553,RSPH6A,SIX5,SYMPK | |
| Non-atopic asthma and FG | rs174583 | chr11:61543499-61617676 | C | T | 1.10×10−08 | 1.69×10−08 | 3.54×10−10 | FADS1,FADS2,FEN1,MIR611,MIR1908,MYRF,TMEM258 |
P1 is asthma (later-onset or non-atopic) single trait P-value, P2 is metabolic trait (BMI, WHRadjBMI or FG) single trait P-value. Pmeta is cross-trait meta-analysis P-value. FG is fasting glucose.
We identified 6 and 7 independent shared loci of WHRadjBMI with later-onset and non-atopic asthma, respectively (Table 2). The credible set of SNPs for these shared loci is provided in Supplementary Tables 17 and 18. Similar to the results of BMI, we observed 3 common loci shared among WHRadjBMI, later-onset asthma, and non-atopic asthma (two shared at the same index SNP, and one at different index SNP): 15q22.33 (SMAD3, index SNP: rs10152595, Pmeta=2.56×10−20 for later-onset asthma and Pmeta=1.40×10−20 for non-atopic asthma), 19q13.32 (FOXA3, index SNP: rs8103278, Pmeta=1.30×10−11 for later-onset asthma and Pmeta=5.12×10−11 for non-atopic asthma), and 11q23.1 (near LAYN, index SNP for later-onset asthma: rs4616071, index SNP for non-atopic asthma: rs11213940, Pmeta=5.85×10−12 for later-onset asthma and Pmeta=1.73×10−13 for non-atopic asthma).
We investigated shared loci for three glycemic metabolic traits (i.e., T2D, FG, and FI) that were genetically correlated with asthma (Table 2) and identified the credible set of SNPs for these shared loci (Supplementary Table 19-20). A locus at chromosome 11q12.2 was common for FG, later-onset asthma, and non-atopic asthma (FADS2, index SNP: rs174583 for FG with both asthma subtypes). Interestingly, FADS2 (index SNP: rs174584 for T2D with both asthma subtypes) was also a suggestive shared loci betweenT2D and both asthma subtypes when we set the single trait GWAS threshold at P<1×10−3 and Pmeta<5×10−8 (Supplementary Table 21-22). No significant shared loci were identified for FI with later-onset or non-atopic asthma.
Colocalization Analysis
To determine whether obesity traits colocalized at the shared loci with asthma, we performed colocalization38 analysis (Supplementary Table 23). Seven out of the eight independent loci that were in common between BMI and later-onset asthma colocalized at the same candidate causal variant (PPH4>0.7) and one (3q26.32) colocalized with different candidate causal variants (PPH3>0.7). Among the ten loci shared between BMI and non-atopic asthma, 4 colocalized at the same candidate causal variant within each loci (PPH4>0.7), three colocalized with different candidate causal variants (PPH3>0.7), and three loci did not colocalize. Most shared loci between WHRadjBMI and both asthma subtypes colocalized at the same candidate causal variants or colocalized with different candidate causal variants. Further, FG was colocalized at rs174583 with both later-onset and non-atopic asthma (PPH4=0.99).
We next conducted colocalization analysis to identify shared genetic components from the cross-trait meta-analysis between the two asthma subtypes and BMI (Supplementary Tables 24-25 and Supplementary Figures 15-16), WHRadjBMI (Supplementary Tables 26-27 and Supplementary Figures 17-18), and FG (Supplementary Tables 28-29 and Supplementary Figures 19-20), while including GTEx eQTL results across 48 tissues. We observed a significant amount of colocalized signals between metabolic traits and asthma in specific tissues, including skeletal muscle, esophagus mucosa and transformed fibroblast cells. The shared variant between BMI and asthma at 12q13.2 (rs4759229) was also a potential causal eQTL variant for ERBB3, MYL6B, or SUOX across 28 GTEx tissues.
Pathway Analysis
To investigate the biological pathways represented by shared genes, we assessed enrichment of independent loci for each trait and shared genes between obesity traits and asthma subtypes in GO biological process categories and observed several significant shared pathways (FDR: q <0.05) (Supplementary Table 30). Common pathways for genes shared between asthma and BMI or WHRadjBMI, included cell differentiation, cell proliferation, cell migration, and inflammatory response.
Lung Tissue Gene Expression in Obese Mice
We evaluated whether shared associated genes (Table 2) were differentially expressed in lung tissue using results from RNA-Seq studies of two obese mouse models: HFD mice and db/db genetically obese mice. Compared to their age-matched controls, both HFD and db/db mice are obese and exhibit innate airway hyperresponsiveness, a hallmark feature of asthma46. We found 3 genes from 34 loci to be differentially expressed in lung tissues for these mouse models compared to their corresponding controls. In the set of db/db vs. WT mice, we found that ACOXL was significantly differentially expressed (P=2.93×10−6) (Figure 3A and Supplementary Table 31). In HFD vs. LFD mice, genes near the top GWAS loci, MYL6 (sentinel SNP: rs4759229) (Figure 3B) and DPP3 (sentinel SNP: rs540874) were differentially expressed (P=7.99×10−5 and 1.28×10−5) (Supplementary Table 32).
Figure 3:

ACOXL and MYL6 expression in lean and obese mice
(A) RNA-seq detected ACOXL differential expression in lung tissue from wild-type (WT)(C57BL/6J) and db/db mice (n = 4 mice/group) (B) RNA-seq detected MYL6 differential expression in lung tissue from low fat diet (LFD) and high fat diet (HFD) mice (n = 4 mice/group). (C) RT-PCR detected ACOXL differential expression in lung tissue from WT and db/db mice (n = 6 mice/group). (D) RT-PCR detected MYL6 differential expression in lung tissue from LFD and HFD mice (n = 4 mice/group). (E) RT-PCR detected MYL6 expression in lung tissue from HFD mice at different time points (n = 4 mice/time point).
To confirm RNA-Seq findings for these 3 genes, we performed a RT-PCR in independent sets of mice. ACOXL gene expression decreased in lungs of db/db mice compared to WT mice (Figure 3C). Transcript levels of MYL6 in lung tissue from HFD mice which were fed a HFD for 4 months were increased significantly over lean control (1.38 ± 0.02-fold, P < 0.05, Figure 3D). We also found that mRNA levels of MYL6 in lung tissue reached peak values at 4 months after feeding WT mice a HFD diet, and this effect was attenuated at 5 months (Figure 3E). No significant difference of Dpp3 expression in the lung was found between HFD-fed and lean mice via RT-PCR.
Discussion
In the present study, we found positive genetic correlation between obesity traits and glycemic traits with later-onset and non-atopic asthma, but not with early-onset and atopic asthma. Sensitivity analysis showed a significantly higher genetic correlation between later-onset asthma and WHRadjBMI in male than female, but not for BMI and WCadjBMI, suggesting the differences in genetic background of obesity traits in relation to asthma. Cross-trait meta-analysis identified independent shared loci between obesity traits and asthma subtypes, highlighting the role of airway remodeling via changes of cell proliferation, differentiation, migration, apoptosis, and accumulation of extracellular matrices (ECM) proteins.
Our study found more evident genetic correlation of obesity traits with later-onset asthma and non-atopic asthma than with early-onset asthma and atopic asthma. In epidemiological studies, results for early-onset asthma were inconsistent. For example, Guibas et al. found that BMI was not associated with asthma in preschool children47, while a recent meta-analysis observed a significant association between high body weight and asthma in children48. Unlike early-onset asthma, later-onset asthma was usually non-atopic and occurred in parallel with obesity6. The most important difference between patients with atopic and non-atopic asthma is the different airway inflammation patterns49. Atopic asthma is characterized by high eosinophils, mast cells, and T lymphocytes, while non-atopic asthma displays high neutrophils and mast cells49. Muc et al. reported that the main change in obese asthmatics was the shifts of immune response from T helper (Th) 2 (a typical atopic immunological profile) to Th150. Thus, the type of airway inflammation driven mainly by neutrophil51 in obese subjects might partially explain the stronger relationship of obesity with later-onset and non-atopic asthma.
Obesity was closely related with metabolic disorders and age at menarche genetically and phenotypically. Although BMI was inversely correlated with age at menarche in the current study (Supplementary Table 26), sensitivity analysis after adjusting for age at menarche showed consistent genetic correlation between BMI and asthma (Supplementary Table 10). Further, even though both BMI and WHRadjBMI were consistently correlated with T2D, FG, FI, HDL, and TG (Supplementary Table 33), we only observed significant genetic correlations of later-onset and non-atopic asthma with glycemic metabolic traits (T2D, FG and FI) other than lipid traits, which indicated different shared genetic background with asthma for glucose metabolism and lipid metabolism.
We obtained strong and unequivocal evidence supporting the model that BMI causally increased the risk of asthma, providing insights into the pathological mechanisms of asthma. However, we emphasize that our inferred causal relationships are based on GWAS summary statistics from cross-sectional studies. Unadjusted confounders (e.g. population stratification) and genetic pleiotropy may bias the causal inference. In addition, longitudinal studies are necessary to confirm the inferred causal relationships.
According to previous studies, the hallmark pathological features of asthma includes airway inflammation and structure changes (or airway remodeling)52. While it is well known that obesity is not only mass loading but also a state of chronic low-grade inflammation53, it is reasonable to speculate that inflammation plays a role in it. Shared loci identified from cross-trait meta-analysis also supported the involvement of inflammation in the shared etiologies between obesity and asthma. For example, SMAD3 protein, encoded by SMAD3, is one of the intracellular mediators and important transducer of TGF-β signaling54 and TGF-β/SMAD3 signaling regulates inflammatory response and T cells activation and differentiation55, 56 ERBB3 encodes ERBB3 protein which is the receptor for neuregulin (NRG). Neuregulin-1 (NRG-1)/ERBB signaling plays a role in the control of proinflammatory activation of monocytes57. Additionally, we found a shared locus (6p21.32, HLA-DRB1-DQA1-DQB1) at human leukocyte antigen (HLA) region which was well-established in playing key roles in the immune system58. However, what interests us most was that the shared loci were implemented in cell proliferation, metaplasia, and ECM remodeling, all of which were reported to be important pathology of airway remodeling59. Firstly, except the involvement in inflammatory response, ERBB3 and SMAD3 are also important regulators of cell proliferation and differentiation. According to Polosa et al., ERBB3 and its family members occurred in bronchial epithelial cells to regulate epithelial repair and remodeling after mechanical damage to human bronchial epithelial cells in vitro60. DiRenzo et al. reported that elevated TGF-β/Smad3 stimulated the secretion of canonical Wnts which in turn enhanced SMC proliferation through β-catenin stabilization61. The blockade of TGF-β/SMAD3 was suggested to be beneficial for many diseases including obesity and asthma62, 63. Secondly, collagen and hyaluronan are components of ECM, and ECM deposition has been identified as an important pathogenic factor for severe asthma64. COL16A1 encodes type XVI collagen which is unregulated by TGF-β during tissue repair and causes the abnormal accumulation, a character for tissue fibrosis65. LAYN encodes a hyaluronan receptor. There was a significant correlation between the severity of asthma as measured by airway hyperresponsiveness and the hyaluronan (HA) levels in BAL fluid, serum, and lung tissue66. Thirdly, epithelial metaplasia is another feature of airway remodeling. FOXA3 was directly related with goblet cell metaplasia67 and body mass regulation68. Airway remodeling characterized by increased smooth muscle cell mass, goblet cell metaplasia, and ECM deposition was implicated in persistent airway hyperresponsiveness, excessive airflow narrowing, and resistance to high-intensity treatment and poor prognosis69, 70. Thus, airway remodeling might also be an explanation for treatment resistance and potential future therapeutic target for obese asthma patients who are characterized by more severe symptoms and poorer prognosis.
Surprisingly, we found some shared variants have opposite sign of effect estimates between obesity and asthma even though the overall genetic correlation is positive, which suggests the pathways shared by them are heterogeneous.
Finally, we found multiple top genes from cross-trait GWAS also showed differential gene expression in two independent mouse experiments, the RNA-seq and RT-PCR gene expression in two sets of mice. In the set of db/db vs WT, we identified ACOXL (Acyl-Coenzyme A oxidase-like) gene, which is proposed to participate in fatty acid β-oxidation, fatty acid metabolic process and oxidation reduction71. Fatty acid metabolism, particularly mitochondrial fatty acid oxidation (FAO) emerges as an important regulator of innate and adaptive immunity72. Obesity causes a decrease in FAO73, and increases inflammation72. The pro-inflammatory state found in obesity might be a critical player in the role of lipid mediators in asthmatic patients74.
In the set of HFD-fed vs lean controls, MYL6 (myosin light chain 6) is a protein-coding gene that encodes a myosin light chain and has important roles in airway smooth muscle (ASM) contractile function75. An increase in contractility of ASM is a contributing factor in the excessive airway narrowing in asthma76. The contractile response of ASM was enhanced in obese donors due to elevated intracellular calcium responses to contractile agonists77, resulting an increased risk of developing asthma.
We investigated the genome-wide genetic relationships of obesity-related traits with four asthma subtypes for the first time. The study has several important strengths. Firstly, the current study has large samples size and the ability to classify different types of asthma. Although sample overlapping was a concern for cross trait analysis, we used methods that are robust to such issue (including LDSC and SHet)11, 27, 34. Moreover, we used the strategy of restricting P<1×10−5 in single trait association as a secondary significance criterion in meta-analysis, which ensures the meta-analysis signals are at least suggestive signals with all of single traits. Finally, our computational analysis provides insights to the molecular mechanisms underlying co-morbid obesity and asthma. However, we also acknowledge potential limitations. First, we restricted the analysis to individuals of European ancestry in order to minimize the population stratification, thus, some findings in this study may not be generalizable to other racial/ethnical populations. Second, although cases of asthma and atopic diseases were identified using self-reported doctor diagnosed data, there was still possibility of misclassification of atopic asthma and non-atopic asthma and future studies using biomarkers (such as immunoglobulin E) in diagnosing atopic status are warranted to validate our findings. We also noted there is no clear-cut age threshold that defines early- and later-onset asthma, thus the definition based on age of 16 cut-point may contains heterogamous effect of asthma for young adult age. Third, we observed little evidence of significant genetic correlation between adult obesity and early-onset asthma; we encourage further studies to investigate the genetic correlation between childhood obesity and early-onset asthma.
Conclusion
In conclusion, the current study showed substantial evidence of positive genetic correlations of obesity traits and glycemic traits with later-onset and non-atopic asthma, which can further our understanding of the connection between obesity and different asthma subtypes. Our work reinforces common biological pathways related to immune and inflammatory systems between obesity and asthma and suggests new treatment avenues for subgroups of individuals with asthma.
Supplementary Material
Key Messages:
This large-scale cross-trait GWAS provides strong evidence for shared genetic components between obesity-related metabolic traits and asthma subtypes
The strongest positive genetic correlation was observed between obesity and later-onset asthma
Mendelian Randomization analysis provided strong evidence in support of BMI causally increasing the risk of asthma
Acknowledgments
This research was conducted using the UK Biobank Resource under application number 16549. Informed consent was received by all participants. We would like to thank the participants and researchers from the UK Biobank who significantly contributed or collected data.
Abbreviations Used:
- ASM
Airway smooth muscle
- BMI
Body mass index
- CPASSOC
Cross phenotype association
- db/db
Autosomal recessive diabetes mutation
- DGF
DNaseI digital genomic footprinting
- DHS
DNase I hypersensitivity site
- ECM
Extracellular matrices
- eQTL
Expression quantitative trait loci
- FAO
Fatty acid oxidation
- FG
Fasting glucose
- FI
Fasting insulin
- GCP
Genetic causality proportion
- GO
Gene Ontology
- GSMR
Generalized summary data-based Mendelian Randomization
- GWAS
Genome-wide association study
- HDL
High-density lipoprotein
- HFD
High fat diet
- HLA
Human leukocyte antigen
- LCV
Latent Causal Variable
- LDL
Low-density lipoproteins
- LDSC
LD score regression
- LFD
Low fat diet
- QC
Quality control
- T2D
Type 2 diabetes
- TC
Total cholesterol
- TFBS
Transcription factor binding sites
- TG
Triglycerides
- WC
Waist circumference
- WHR
Waist-to-hip ratio
- WT
Wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: The authors declare no competing interests
URLs. UK Biobank, http://biobank.ctsu.ox.ac.uk/; 1000 Genomes Project, http://www.1000genomes.org/; Haplotype Reference Consortium, http://www.haplotype-reference-consortium.org/; BOLT-LMM, https://data.broadinstitute.org/alkesgroup/BOLT-LMM/; PLINK, https://www.cog-genomics.org/plink2; LDSC, https://github.com/bulik/ldsc; CPASSOC, http://hal.case.edu/zhu-web/; GTEx, http://www.gtexportal.org; WebGestalt, http://www.webgestalt.org/option.php; Latent Causal Variable Model,https://github.com/lukejoconnor/LCV/; R, https://www.r-project.org/; Roadmap Epigenomics,http://www.roadmapepigenomics.org/; Coloc, https://github.com/chr1swallace/coloc
DATA AVAILABILITY
UK Biobank summary GWAS statistics will be available at the UK Biobank website (http://biobank.ctsu.ox.ac.uk).
References
- 1.Martinez FD, Vercelli D. Asthma. The Lancet 2013; 382:1360–72. [DOI] [PubMed] [Google Scholar]
- 2.Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world--a growing challenge. N Engl J Med 2007; 356:213–5. [DOI] [PubMed] [Google Scholar]
- 3.Holguin F, Bleecker ER, Busse WW, Calhoun WJ, Castro M, Erzurum SC, et al. Obesity and asthma: an association modified by age of asthma onset. J Allergy Clin Immunol 2011; 127:1486–93 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ford ES. The epidemiology of obesity and asthma. J Allergy Clin Immunol 2005; 115:897–909; quiz 10. [DOI] [PubMed] [Google Scholar]
- 5.Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med 2010; 181:315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Nijs SB, Venekamp LN, Bel EH. Adult-onset asthma: is it really different? Eur Respir Rev 2013; 22:44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeong A, Imboden M, Hansen S, Zemp E, Bridevaux PO, Lovison G, et al. Heterogeneity of obesity-asthma association disentangled by latent class analysis, the SAPALDIA cohort. Respir Med 2017; 125:25–32. [DOI] [PubMed] [Google Scholar]
- 8.Appleton SL, Adams RJ, Wilson DH, Taylor AW, Ruffin RE, North West Adelaide Health Study T. Central obesity is associated with nonatopic but not atopic asthma in a representative population sample. J Allergy Clin Immunol 2006; 118:1284–91. [DOI] [PubMed] [Google Scholar]
- 9.Lane JM, Liang J, Vlasac I, Anderson SG, Bechtold DA, Bowden J, et al. Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nat Genet 2017; 49:274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao W, Rasheed A, Tikkanen E, Lee JJ, Butterworth AS, Howson JMM, et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat Genet 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu Z, Lee PH, Chaffin MD, Chung W, Loh PR, Lu Q, et al. A genome-wide cross-trait analysis from UK Biobank highlights the shared genetic architecture of asthma and allergic diseases. Nat Genet 2018; 50:857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Z, Lin Y, Li X, Driver JA, Liang L. Shared genetic architecture between metabolic traits and Alzheimer's disease: a large-scale genome-wide cross-trait analysis. Hum Genet 2019; 138:271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu Z, Wang X, Li X, Lin Y, Shen S, Liu CL, et al. Genetic overlap of chronic obstructive pulmonary disease and cardiovascular disease-related traits: a large-scale genome-wide cross-trait analysis. Respir Res 2019; 20:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yiallouros PK, Kouis P, Kolokotroni O, Youhanna S, Savva SC, Dima K, et al. Shared genetic variants between serum levels of high-density lipoprotein cholesterol and wheezing in a cohort of children from Cyprus. Ital J Pediatr 2016; 42:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melen E, Himes BE, Brehm JM, Boutaoui N, Klanderman BJ, Sylvia JS, et al. Analyses of shared genetic factors between asthma and obesity in children. J Allergy Clin Immunol 2010; 126:631–7 e1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hallstrand TS, Fischer ME, Wurfel MM, Afari N, Buchwald D, Goldberg J. Genetic pleiotropy between asthma and obesity in a community-based sample of twins. J Allergy Clin Immunol 2005; 116:1235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott RA, Scott LJ, Magi R, Marullo L, Gaulton KJ, Kaakinen M, et al. An Expanded Genome-Wide Association Study of Type 2 Diabetes in Europeans. Diabetes 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010; 42:105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Surakka I, Horikoshi M, Magi R, Sarin AP, Mahajan A, Lagou V, et al. The impact of low-frequency and rare variants on lipid levels. Nat Genet 2015; 47:589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015; 12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loh P-R, Kichaev G, Gazal S, Schoech AP, Price AL. Mixed model association for biobank-scale data sets. bioRxiv 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016; 48:1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics C, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015; 47:291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sood A Sex differences: implications for the obesity-asthma association. Exerc Sport Sci Rev 2011; 39:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macsali F, Real FG, Plana E, Sunyer J, Anto J, Dratva J, et al. Early age at menarche, lung function, and adult asthma. Am J Respir Crit Care Med 2011; 183:8–14. [DOI] [PubMed] [Google Scholar]
- 26.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 2015; 47:1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015; 47:1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Z, Zheng Z, Zhang F, Wu Y, Trzaskowski M, Maier R, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun 2018; 9:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Connor LJ, Price AL. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat Genet 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol 2016; 40:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015; 518:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity Analyses for Robust Causal Inference from Mendelian Randomization Analyses with Multiple Genetic Variants. Epidemiology 2017; 28:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aschard H, Vilhjalmsson BJ, Joshi AD, Price AL, Kraft P. Adjusting for heritable covariates can bias effect estimates in genome-wide association studies. Am J Hum Genet 2015; 96:329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu X, Feng T, Tayo BO, Liang J, Young JH, Franceschini N, et al. Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am J Hum Genet 2015; 96:21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Z, Anttila V, Smoller JW, Lee PH. Statistical power and utility of meta-analysis methods for cross-phenotype genome-wide association studies. PLoS One 2018; 13:e0193256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wellcome Trust Case Control C, Maller JB, McVean G, Byrnes J, Vukcevic D, Palin K, et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet 2012; 44:1294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallace C Statistical testing of shared genetic control for potentially related traits. Genet Epidemiol 2013; 37:802–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skaaby T, Taylor AE, Thuesen BH, Jacobsen RK, Friedrich N, Mollehave LT, et al. Estimating the causal effect of body mass index on hay fever, asthma and lung function using Mendelian randomization. Allergy 2018; 73:153–64. [DOI] [PubMed] [Google Scholar]
- 40.Granell R, Henderson AJ, Evans DM, Smith GD, Ness AR, Lewis S, et al. Effects of BMI, fat mass, and lean mass on asthma in childhood: a Mendelian randomization study. PLoS Med 2014; 11:e1001669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones CL, Bhatla T, Blum R, Wang J, Paugh SW, Wen X, et al. Loss of TBL1XR1 disrupts glucocorticoid receptor recruitment to chromatin and results in glucocorticoid resistance in a B-lymphoblastic leukemia model. J Biol Chem 2014; 289:20502–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ali Z, Ulrik CS. Obesity and asthma: a coincidence or a causal relationship? A systematic review. Respir Med 2013; 107:1287–300. [DOI] [PubMed] [Google Scholar]
- 43.Hayakawa T, Yamashita K, Ohuchi E, Shinagawa A. Cell growth-promoting activity of tissue inhibitor of metalloproteinases-2 (TIMP-2). J Cell Sci 1994; 107 ( Pt 9):2373–9. [DOI] [PubMed] [Google Scholar]
- 44.Waerner T, Gardellin P, Pfizenmaier K, Weith A, Kraut N. Human RERE is localized to nuclear promyelocytic leukemia oncogenic domains and enhances apoptosis. Cell Growth Differ 2001; 12:201–10. [PubMed] [Google Scholar]
- 45.Wilson BJ, Saab KR, Ma J, Schatton T, Putz P, Zhan Q, et al. ABCB5 maintains melanoma-initiating cells through a proinflammatory cytokine signaling circuit. Cancer Res 2014; 74:4196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnston RA, Theman TA, Lu FL, Terry RD, Williams ES, Shore SA. Diet-induced obesity causes innate airway hyperresponsiveness to methacholine and enhances ozone-induced pulmonary inflammation. J Appl Physiol (1985) 2008; 104:1727–35. [DOI] [PubMed] [Google Scholar]
- 47.Guibas GV, Manios Y, Xepapadaki P, Moschonis G, Douladiris N, Mavrogianni C, et al. The obesity-asthma link in different ages and the role of body mass index in its investigation: findings from the Genesis and Healthy Growth Studies. Allergy 2013; 68:1298–305. [DOI] [PubMed] [Google Scholar]
- 48.Papoutsakis C, Priftis KN, Drakouli M, Prifti S, Konstantaki E, Chondronikola M, et al. Childhood overweight/obesity and asthma: is there a link? A systematic review of recent epidemiologic evidence. J Acad Nutr Diet 2013; 113:77–105. [DOI] [PubMed] [Google Scholar]
- 49.Amin K, Ludviksdottir D, Janson C, Nettelbladt O, Bjornsson E, Roomans GM, et al. Inflammation and structural changes in the airways of patients with atopic and nonatopic asthma. BHR Group. Am J Respir Crit Care Med 2000; 162:2295–301. [DOI] [PubMed] [Google Scholar]
- 50.Muc M, Mota-Pinto A, Padez C. Association between obesity and asthma - epidemiology, pathophysiology and clinical profile. Nutr Res Rev 2016; 29:194–201. [DOI] [PubMed] [Google Scholar]
- 51.Scott HA, Gibson PG, Garg ML, Wood LG. Airway inflammation is augmented by obesity and fatty acids in asthma. Eur Respir J 2011; 38:594–602. [DOI] [PubMed] [Google Scholar]
- 52.Saglani S, Lloyd CM. Novel concepts in airway inflammation and remodelling in asthma. Eur Respir J 2015; 46:1796–804. [DOI] [PubMed] [Google Scholar]
- 53.Peters U, Suratt BT, Bates JHT, Dixon AE. Beyond BMI: Obesity and Lung Disease. Chest 2018; 153:702–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J 1999; 18:1280–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, et al. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol 2010; 185:842–55. [DOI] [PubMed] [Google Scholar]
- 56.Anthoni M, Fyhrquist-Vanni N, Wolff H, Alenius H, Lauerma A. Transforming growth factor-beta/Smad3 signalling regulates inflammatory responses in a murine model of contact hypersensitivity. Br J Dermatol 2008; 159:546–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ryzhov S, Matafonov A, Galindo CL, Zhang Q, Tran TL, Lenihan DJ, et al. ERBB signaling attenuates proinflammatory activation of nonclassical monocytes. Am J Physiol Heart Circ Physiol 2017; 312:H907–H18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gough SC, Simmonds MJ. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr Genomics 2007; 8:453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bergeron C, Tulic MK, Hamid Q. Airway remodelling in asthma: from benchside to clinical practice. Can Respir J 2010; 17:e85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Polosa R, Puddicombe SM, Krishna MT, Tuck AB, Howarth PH, Holgate ST, et al. Expression of c-erbB receptors and ligands in the bronchial epithelium of asthmatic subjects. J Allergy Clin Immunol 2002; 109:75–81. [DOI] [PubMed] [Google Scholar]
- 61.DiRenzo DM, Chaudhary MA, Shi X, Franco SR, Zent J, Wang K, et al. A crosstalk between TGF-beta/Smad3 and Wnt/beta-catenin pathways promotes vascular smooth muscle cell proliferation. Cell Signal 2016; 28:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab 2011; 14:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov 2012; 11:790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Royce SG, Tan L, Koek AA, Tang ML. Effect of extracellular matrix composition on airway epithelial cell and fibroblast structure: implications for airway remodeling in asthma. Ann Allergy Asthma Immunol 2009; 102:238–46. [DOI] [PubMed] [Google Scholar]
- 65.Grassel S, Tan EM, Timpl R, Chu ML. Collagen type XVI expression is modulated by basic fibroblast growth factor and transforming growth factor-beta. FEBS Lett 1998; 436:197–201. [DOI] [PubMed] [Google Scholar]
- 66.Ghosh S, Samarasinghe AE, Hoselton SA, Dorsam GP, Schuh JM. Hyaluronan deposition and colocalization with inflammatory cells and collagen in a murine model of fungal allergic asthma. Inflamm Res 2014; 63:475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen G, Korfhagen TR, Karp CL, Impey S, Xu Y, Randell SH, et al. Foxa3 induces goblet cell metaplasia and inhibits innate antiviral immunity. Am J Respir Crit Care Med 2014; 189:301–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adler-Wailes DC, Alberobello AT, Ma X, Hugendubler L, Stern EA, Mou Z, et al. Analysis of variants and mutations in the human winged helix FOXA3 gene and associations with metabolic traits. Int J Obes (Lond) 2015; 39:888–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Durrani SR, Viswanathan RK, Busse WW. What effect does asthma treatment have on airway remodeling? Current perspectives. J Allergy Clin Immunol 2011; 128:439–48; quiz 49-50. [DOI] [PubMed] [Google Scholar]
- 70.Girodet PO, Dournes G, Thumerel M, Begueret H, Dos Santos P, Ozier A, et al. Calcium channel blocker reduces airway remodeling in severe asthma. A proof-of-concept study. Am J Respir Crit Care Med 2015; 191:876–83. [DOI] [PubMed] [Google Scholar]
- 71.Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol 2006; 7 Suppl 1:S12 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Namgaladze D, Brune B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim Biophys Acta 2016; 1861:1796–807. [DOI] [PubMed] [Google Scholar]
- 73.Fucho R, Casals N, Serra D, Herrero L. Ceramides and mitochondrial fatty acid oxidation in obesity. FASEB J 2017; 31:1263–72. [DOI] [PubMed] [Google Scholar]
- 74.Wendell SG, Baffi C, Holguin F. Fatty acids, inflammation, and asthma. J Allergy Clin Immunol 2014; 133:1255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao N, Huang J, He W, Zhu M, Kamm KE, Stull JT. Signaling through Myosin Light Chain Kinase in Smooth Muscles. 2013; 288:7596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.An SS, Bai TR, Bates JHT, Black JL, Brown RH, Brusasco V, et al. Airway smooth muscle dynamics: a common pathway of airway obstruction in asthma. 2007; 29:834–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Orfanos S, Jude J, Deeney BT, Cao G, Rastogi D, Zee Mv, et al. Obesity increases airway smooth muscle responses to contractile agonists. 2018; 315:L673–L81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.