

Distinct dimeric forms of PU.1 mutually antagonize to control the transcriptionally active dose via a disordered PEST domain.
Abstract
Transcription factors comprise a major reservoir of conformational disorder in the eukaryotic proteome. The hematopoietic master regulator PU.1 presents a well-defined model of the most common configuration of intrinsically disordered regions (IDRs) in transcription factors. We report that the structured DNA binding domain (DBD) of PU.1 regulates gene expression via antagonistic dimeric states that are reciprocally controlled by cognate DNA on the one hand and by its proximal anionic IDR on the other. The two conformers are mediated by distinct regions of the DBD without structured contributions from the tethered IDRs. Unlike DNA-bound complexes, the unbound dimer is markedly destabilized. Dimerization without DNA is promoted by progressive phosphomimetic substitutions of IDR residues that are phosphorylated in immune activation and stimulated by anionic crowding agents. These results suggest a previously unidentified, nonstructural role for charged IDRs in conformational control by mitigating electrostatic penalties that would mask the interactions of highly cationic DBDs.
INTRODUCTION
Eukaryotic transcription factors are highly enriched in intrinsically disordered regions (IDR), which are sequences that do not adopt a stably structured conformation but are nevertheless essential for activity. Compared with only ~5% in prokaryotes and archaea, more than 80% of eukaryotic transcription factors have extended IDRs (1). In the unicellular baker’s yeast (Saccharomyces), transcription factors comprise the most prodigious functional category of disorder-encoding proteins (2). In multicellular organisms, ~50% of all residues in eukaryotic factors from model animals (humans, Drosophila) and plants (Arabidopsis) map to disordered regions (3). Clearly, IDRs constitute a major component in eukaryotic gene regulation, and it is therefore important to define their contributions to the molecular properties of transcriptional factors.
While IDRs are generally diverse in sequence, charge characteristics confer specific properties to transcription factor IDRs. For example, positively charged tails mediate diffusion along DNA (4) and ubiquitination by E3 ligases of several transcription factors, notably p53 (5). More common, however, are negatively charged (“acidic”) IDRs such as transactivation domains, which recruit basal factors such as TFIIB and TATA binding protein to the promoter (6, 7), and signaling moieties such as PEST domains that are rich in Glu and Asp residues (8, 9). While IDRs exhibit sequence-dependent conformational preferences on their own, these preferences are also modified by folded domains to which they are tethered (10). In transcription factors, IDRs are highly enriched around DNA binding domains (DBDs) (11), which display electrostatically biased surfaces to their surroundings. Their DNA contact surfaces are typically rich in positively charged residues while exposing neutral or even negatively charged residues elsewhere. Because DBDs alone represent an incomplete context in functional regulation, our aim is to elaborate the mechanism by which charged, particularly acidic IDRs regulate the recognition of tethered DBDs with each other as well as with target DNA.
As a model system for understanding the impact of intrinsically disordered tethers in transcription factors, the ETS family protein PU.1 exemplifies the most common (known as type I) configuration (3), in which its eponymous DBD of ~90 residues comprises the only well-folded structure. The remaining ~170 and 12 residues that flank N- and C-terminally, respectively, are intrinsically disordered sequences. The extended N-terminal IDR consists of an acidic transactivation domain (human residues 1 to 80), a Q-rich domain (residues 81 to 116), and a highly negatively charged PEST domain (residues 117 to 165), all of which are characteristic disordered regions in eukaryotic factors (9). This domain architecture, conserved among PU.1 orthologs, commends PU.1 as an ideal model from which more complex transcription factor architectures may be approached.
In addition to the canonical attributes representative of eukaryotic transcription factors, PU.1 is also specifically required for life. During hematopoiesis, all circulating blood cells are ultimately derived from a small population of self-renewing stem cells. PU.1 is a master regulator that is required for the renewal of the hematopoietic stem cells (12) and, in collaboration with other factors, directs their differentiation to every major myeloid and lymphoid lineage. Aberrant PU.1 activity is associated with lymphomas (13), myeloma (14), leukemias (15), and Alzheimer’s disease (16). Most recently, PU.1 was also identified as the key trigger for tissue fibrosis (17). Genetic and pharmacologic interventions targeted at PU.1 have established its therapeutic potential in acute myeloid leukemia (18, 19) and fibrotic diseases (17). Mechanisms that govern the molecular interactions of PU.1 are therefore relevant to developmental genetics and multiple therapeutic areas including hematology/oncology, immunology, neurology, and rheumatology.
Despite its biological significance, detailed knowledge of the molecular properties of PU.1 has been limited to its structured ETS domain. PU.1 therefore exemplifies the “incomplete context problem” in structural biology, which we have now tackled by addressing the role of the N- and C-terminal IDRs in the behavior of the ETS domain. The data reveal that these tethered IDRs critically control the propensities of the ETS domain to form discrete dimers with and without cognate DNA. These dimeric states, which are conformationally distinct, establish a novel regulatory mechanism that enables negative feedback in PU.1 transactivation. In addition to implications on PU.1 autoregulation in vivo, these results address a general class of problems in which negatively charged IDRs, which are abundant in transcription factors as transactivation and other functional domains, exert direct functional control at the protein/DNA level.
RESULTS
The DNA binding (ETS) domain of PU.1 represents its only structured domain, whose 1:1 complex with cognate DNA (Fig. 1A) is structurally conserved in this family of transcription factors. However, in contrast with other ETS members, the ETS domain of PU.1 (ΔN165) forms 2:1 complexes at single DNA cognate sites in biophysical assays (20, 21). Measurements of self-diffusion by protein-observed diffusion ordered spectroscopy (DOSY) nuclear magnetic resonance (NMR) showed that inclusion of the N-terminal PEST domain (ΔN117) maintained the DNA binding modes accessible to the ETS domain (Fig. 1B). Specifically, DOSY titrations of both ΔN117 and ΔN165 with DNA oligomers harboring a single cognate binding site showed two distinct bound states, with the minima in diffusion coefficient occurring sharply at a DNA:protein ratio of 0.5, corresponding to a 2:1 complex. The single minima at DNA:protein = 0.5 excluded the formal possibility of a 2:2 complex or nonspecific binding. If PU.1 were binding DNA nonspecifically beyond the 1:1 complex at equilibrium (i.e., in an unsaturable manner proportional to the concentration of free protein), then the minima in diffusion coefficient would occur at the lowest DNA:protein ratios where protein would be at greatest excess relative to DNA. Independently, protein-into-DNA titrations showed that ΔN117 enhanced the affinity of the 1:1 complex (KD1) by more than twofold and reduced the affinity of the 2:1 complex (KD2) relative to ΔN165 by about fourfold (Fig. 1B). Taking the ratio KD2/KD1 as an index of cooperativity in DNA binding, 2:1 complex formation by ΔN117 was therefore more negatively cooperative than ΔN165. The PEST domain, therefore, preserved the intrinsic binding modes of the ETS domain of PU.1, namely, a 1:1 and 2:1 complex with cognate DNA, while modulating their affinities in solution.
Fig. 1. PU.1 transactivation is regulated by negative feedback.
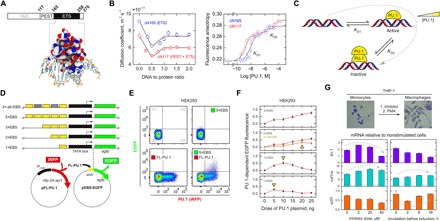
(A) The ETS domain (ΔN165) is the only structured and minimal DNA binding unit (PDB: 1PUE). (B) Left: DOSY NMR titrations of ΔN117 and ΔN165 with cognate DNA yielding equivalence points at DNA:protein = 0.5 and 1.0, corresponding to 2:1 and 1:1 complexes, respectively. The absence of single global minima at DNA:protein = 1:1 formally excludes the possibility of a 2:2 complex. Right: Fluorescence anisotropy titrations with labeled cognate DNA. Both ΔN117 and ΔN165 form a 2:1 complex at a single DNA site with different negative cooperativity as defined by the ratio of the two sequential dissociation constants KD1 and KD2 (dashed lines; see Materials and Methods). Parametric values are given in table S1. (C) Scheme of negative feedback in PU.1 trans-regulation. A mechanistic link between dimerization and negative feedback predicts a reduction in PU.1 activity under conditions permissive of an inactive 2:1 complex. (D) Synthetic PU.1-dependent EGFP reporters. A minimal TATA box was driven by enhancers composed only of tandem EBS (yellow blocks) spaced 20 bp apart. Hatched blocks represent mutated sites. (E) Representative flow cytometric data of untreated HEK293 cells and upon transfection with a constant dose of the 5×EBS reporter and/or up to 25 ng of an expression plasmid encoding full-length PU.1 (see Materials and Methods). Quadrant Q2 contained the EGFP-positive cells to be counted out of all PU.1-expressing cells (Q2 + Q3). (F) EGFP fluorescence in Q2 taken over the summed fluorescence in Q2 + Q3 at 24 hours after cotransfection of the EGFP reporter plasmid and the indicated dose of PU.1 expression plasmid. Each data point represents the means ± SE of triplicate or more samples. (G) RT-PCR measurements of pu.1, csf1ra, and e2f1 mRNA abundance (relative to gapdh) in THP-1 cells induced with PMA following exposure to either doses of a PU.1 inhibitor for 2 hours (left) or a fixed dose of 20 μM inhibitor for various periods (right). Cells were visualized at ×40 magnification after Giemsa staining.
PU.1 is self-regulated by negative feedback
In tissues that natively express PU.1, such as macrophages, PU.1 activity is highly inducible (22). The 1:1 complex formed by ETS domains represents the established trans-regulatory complex for ETS transcription factors. Little is understood about the functional nature of the 2:1 complex, for which no ETS analog is known, although its negative cooperative relationship with the 1:1 complex suggests an inactive species (Fig. 1C). To solve this puzzle, we measured PU.1 transactivation in cells using an enhanced green fluorescent protein (EGFP) reporter gene under the control of various synthetic enhancer elements consisting only of tandem copies of the λB motif (Fig. 1D), a PU.1-specific ETS binding site (EBS) derived from the lymphoid Igλ2-4 enhancer (GenBank X54550). Each consecutive site was spaced by 20 base pairs (bp), or two helical turns, such that bound proteins were arrayed on the same helical face to facilitate the recruitment of the transcriptional machinery. In addition, as the 2:1 complex was known to require an extended site size relative to the monomer (20), presenting the bound protein along one helical face would amplify site-site interactions and DNA perturbations, thus rendering most manifest the functional effects of the 2:1 complex.
When transiently transfected into PU.1-negative human embryonic kidney (HEK) 293 cells, the reporters were negligibly activated by endogenous transcription factors, including other ETS family proteins (Fig. 1E). Cotransfection of an expression plasmid encoding full-length PU.1, which was independently tracked by a cotranslating infrared fluorescent protein (iRFP) marker, yielded EGFP fluorescence in a dose-dependent manner. We established a dosing range for the PU.1 expression plasmid that gave a linear variation in PU.1 abundance in HEK293 cells within the physiologically inducible range found in PU.1-expressing myeloid cells (fig. S1). In this configuration, PU.1-dependent transactivation was quantified as the fraction of iRFP-positive cells that were also EGFP positive (Fig. 1E). The functional outcome of an inactive, negatively cooperative 2:1 complex would be a bell-shaped reporter dose-response as the enhancer, which varied in density and spacing of EBS (i.e., cis-regulatory syntax), became saturated with nonproductively (2:1) bound PU.1. In the alternative, the reporter signal would dose-dependently settle to a saturable level, depending on the level at which the 2:1 complex retained activity relative to the 1:1 complex. The synthetic λB reporters were therefore well suited to interrogate cellular PU.1 activity, free from the requirement or interference from other promoter-specific cofactors, at the protein/DNA level.
At equivalent PU.1 doses, all enhancer configurations showed graded reporter expression in step with the density of EBS at each enhancer (Fig. 1F). This was consistent with an expected multivalent effect with respect to PU.1 binding sites. However, EGFP expression increased monotonically only with enhancers harboring tandem 3× and 5× EBS. Upon peaking at intermediate PU.1 doses, the 1× and 2× enhancers were repressed by further increases in PU.1. To determine whether the reversal in transactivation involved PU.1 interactions at the enhancer, we mutated the even-numbered sites in the 5×EBS reporter to generate a 3×EBS variant in which the cognate sites doubled in spacing (Fig. 1D). The resultant 3×-alt-EBS reporter exhibited lower transactivation than the more densely spaced 3×EBS, and its reporter signal also no longer increased monotonically (Fig. 1F). The spacing effect, therefore, demonstrated that the functional reversal could not be due solely to PU.1 interactions away from the DNA, which would be inert to syntax changes at the DNA. The observation of bell-shaped dose response for the 1× and 2× enhancers, but not the 3× or 5×EBS enhancers, suggested additive perturbations of the local DNA structure, which were amplified by the helical spacing of the sites. This interpretation was supported by previous DNA footprinting of the PU.1 ETS domain, which showed strong differences between the singly and doubly PU.1-bound DNA (20). Alternatively, binding at the higher-density sites might exhaust a required co-repressing factor for the 2:1 complex. However, this possibility was discounted by the different dose responses exhibited by the 3×EBS and 3×-alt-EBS, which had the same site density, and the occurrence in a cell line (HEK293) that does not natively use PU.1 in gene regulation. Because net transactivation activity was reduced under conditions corresponding to population of the 2:1 complex, the evidence suggested that the 2:1 complex lost activity relative to the 1:1 complex. Thus, manipulation of enhancer syntax (density and spacing) demonstrated negative feedback in PU.1 transactivation in a manner consistent with self-titration of the transcriptionally active 1:1 complex by an inactive dimer bound to DNA.
To extend our functional results to a more physiologic context, we evaluated the impact of graded PU.1 inhibition on three PU.1 target genes in THP-1 cells, a widely used human monocyte/macrophage model. Cells were treated with a PU.1 inhibitor (fig. S2), as a function of dose or incubation period, before stimulation with phorbol 12-myristate 13-acetate (PMA) to mimic PU.1 induction during myeloid differentiation. As PU.1 targets, we examined the pu.1 (Spi-1) gene itself, which is autoregulated (23); csf1ra, a PU.1 target that encodes the α subunit of the colony-stimulating factor receptor; and e2f1, which is negatively regulated by PU.1 (24). We first tested the effect of dose-dependent inhibition of PU.1 for a fixed period of 2 hours on the transcription of these genes by reverse transcription polymerase chain reaction (RT-PCR) (table S2). Expression of pu.1 and csf1ra, both positively regulated PU.1 targets, was increased by lower doses of inhibitor before marked reduction to ~50% at higher doses, yielding bell-shaped profiles (Fig. 1G). In the case of negatively regulated e2f1, expression was further inhibited across the dosage range of inhibitor tested. Trans-regulation of all three genes upon dose-dependent inhibition of PU.1 was consistent with an increase in PU.1 activity associated with the relief of negative feedback.
To assess the impact of PU.1 inhibition temporally, we tested a fixed dose of inhibitor (20 μM) over time, up to 16 hours before PMA induction. While PU.1 expression gave a bell-shaped dose response at 2 hours of inhibitor exposure, continued exposure at an intermediate (derepressing) dose became strictly inhibitory (Fig. 1G). In contrast, derepression in csf1ra expression continued for 8 hours. Expression of the negative-regulated e2f1 gene, which was dose-dependently reduced at 2 hours of PU.1 inhibition, began to increase by 8 hours of inhibitor exposure. These results thus demonstrated a dynamic nature to the negative feedback that corresponded to the specific effect of PU.1 on the target gene (peaks in activated genes or troughs in repressed genes). The opposing behavior of csf1ra and e2f1 expression, in accordance to their opposite dependence on PU.1, supported the physiologic relevance of PU.1 negative feedback. Last, the latency exhibited by the two target genes relative to the autoregulated pu.1 gene suggested a combined effect between changes in PU.1 availability at the expression level and competition for binding at the DNA level.
In summary, the expression profiles of pu.1, csf1ra, and e2f1 showed that graded PU.1 inhibition led to nonmonotonic changes in trans-regulatory activity in a manner consistent with derepression of negative feedback. Together with the dependence of the synthetic λB reporter on PU.1 dose and enhancer syntax (site density and spacing), the data support the biophysically observed 2:1 complex as a functionally relevant species in the cell and motivate specific interest in how the ETS domain dimerizes in its native structural context.
The PU.1 PEST domain is an IDR that modulates the stability of the 2:1 DNA complex
Comparison of DNA binding by ΔN117 and ΔN165 shows that the N-terminally tethered PEST domain enhanced the affinity of the 1:1 complex but reduced the affinity of the 2:1 complex (Fig. 1B). To better understand the influence of the PEST domain on DNA recognition by PU.1, we first established whether the PEST domain was disordered in the cognate complex by comparing the 1H-15N heteronuclear single quantum coherence spectroscopy (HSQC) fingerprint region of DNA-bound ΔN165 and ΔN117 (Fig. 2A). As with ΔN165 (20), the unbound and 1:1 complex gave well-dispersed spectra, while >80% of the cross peaks for 2:1-bound ΔN117 were broadened out (fig. S3). The similar behavior by the two constructs indicated that broadening was not due to the larger size of the 2:1 complex, in which case broadening would be exacerbated for ΔN117. In 1:1-bound ΔN165, whose resonances were well resolved, 88 of the 95 assigned residues overlapped with ΔN117, with all PEST residues clustered around 8.2 ± 0.2 parts per million (ppm) on the 1H dimension, a chemical shift characteristic of disordered structures. Because this region also represented the residues that were detected in HSQC of the 2:1 complex (fig. S3), the evidence suggested similar changes in the chemical environment for the structured ETS domain (represented by the dispersed resonances in intermediate exchange) between free and DNA-bound states of ΔN117 and ΔN165. Thus, the local structure of the ETS domain was not altered upon DNA binding by the flanking residues, and the PEST domain behaved as a disordered tether in the ETS/DNA complex.
Fig. 2. The intrinsically disordered PEST domain modifies DNA recognition by PU.1.
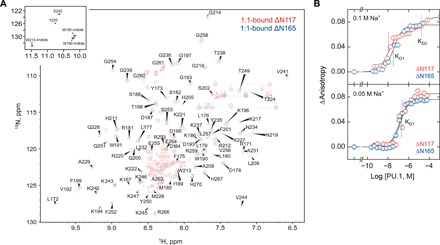
(A) 1H-15N HSQC of ΔN117 and ΔN165 in the 1:1 complex with cognate DNA. Assignment of the ΔN165 spectrum was 90% complete. (B) DNA binding by ΔN165 and ΔN117 in 0.1 M and 0.05 M NaCl, showing the impact of the PEST domain on the cooperativity of 2:1 complex formation. Parametric values of the equilibrium dissociation constants are given in table S1.
Ligand binding to DNA is generally sensitive to electrostatic interactions. To better understand the impact of the disordered PEST domain on 2:1 complex formation, we probed the electrostatic contribution to site-specific binding by ΔN165 and ΔN117 (Fig. 2B). Reducing Na+ concentration from 0.15 M (as shown in Fig. 1B) to 0.10 M did not affect 2:1 binding by ΔN117. However, the biphasic binding indicative of strongly negatively cooperative formation of the 2:1 complex for ΔN165 was abolished as the anisotropy values showed. A further reduction to 0.05 M salt resulted in monophasic transitions to the 2:1 complex by both constructs. Notably, binding weakened with decreasing Na+ concentration and therefore could not reflect simple electrostatic effects on DNA binding. These observations indicated that additional unbound species must regulate DNA recognition by PU.1 and that these species were salt sensitive and controlled by the disordered PEST domain.
The disordered PEST promotes PU.1 homodimerization
The isolated ETS domain, ΔN165, forms a feeble dimer without DNA, as judged by heteronuclear NMR (25) as well as static and dynamic light scattering (21). To determine the role of the disordered PEST domain in PU.1 dimerization without DNA, we examined several hydrodynamic parameters, which are highly sensitive to self-association, of ΔN117 as a function of concentration (Fig. 3A). DOSY NMR spectroscopy revealed a marked concentration dependence for the apparent diffusion coefficient. The profile was described by a two-state monomer-dimer equilibrium (detailed in Materials and Methods) with a dissociation constant below 10 μM (table S1). To assess concentrations below 50 μM, which was limiting for NMR, we performed intrinsic Trp fluorescence anisotropy measurements, which is sensitive to rotational diffusion. ΔN117 exhibited a substantial change in steady-state anisotropy that was also described by a two-state dimer with a dissociation constant at below 10 μM. In contrast, ΔN165 showed no change. The localization of the three Trp residues in the structured ETS domain of both constructs represented further evidence that the concentration-dependent changes in ΔN117 involved the ETS domain. Last, high-precision densimetry showed a concentration-dependent transition by ΔN117 that was again described by two-state dimerization. (Because density varies directly with concentration, density-detected transitions sit on sloped baselines as opposed to the flat baselines in spectrometric titrations.) Relative to the diffusion probes, the densimetric titration gave a higher dissociation constant, 35 ± 15 μM. As a control, ΔN165 gave a concentration-independent partial specific volume (from the slope, see Materials and Methods) of 0.77 ± 0.01 ml/g, a value characteristic of structured globular proteins. Multiple orthogonal probes therefore described a reversible ΔN117 dimer that was considerably more avid than ΔN165.
Fig. 3. The disordered PEST domain in PU.1 drives dimerization in the absence of DNA.
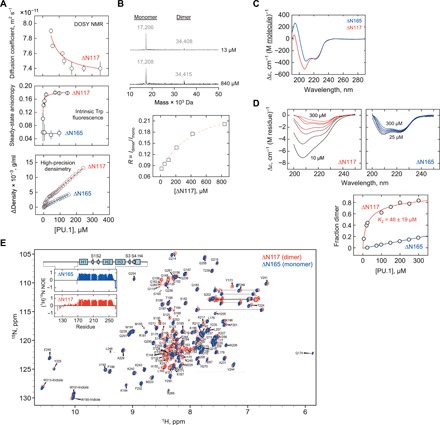
(A) Concentration-dependent changes in hydrodynamic and volumetric properties by DOSY NMR, intrinsic Trp fluorescence anisotropy, and high-precision densimetry of ΔN117 in 0.15 M Na+ at 25°C. Red curves represent fits of the data to a two-state monomer-dimer transition. (B) Representative zero-charge ESI mass spectra of ΔN117 at 13 and 840 μM total concentration, normalized to the height of the monomer (17 kDa) peak. The ratios of the integrated dimer-to-monomer intensities (molecular weight shown) were French-curved to guide the eye. (C) Far-UV CD spectra of ΔN117 and ΔN165 at 25 μM, plotted on a per-molecule basis to highlight the contribution of the N-terminal residues. (D) Concentration-dependent, per-residue spectra of ΔN117 and ΔN165 (left). Dimerization as revealed by singular value decomposition of the ΔN117 spectra and fitted to a two-state transition. (E) 1H-15N HSQC of 400 μM ΔN117 and ΔN165 at 0.15 NaCl. Under these conditions, ΔN117 was predominantly dimeric and ΔN165 was monomeric. The assignments shown are for ΔN117. Inset: {1H}15N-NOE for ΔN117 and ΔN165.
We pause to note that concentration dependence of the equilibrium constant (and melting temperatures) rules out monomolecular interactions, such as conformational changes without association. Local conformational changes can and do produce changes in diffusion and volumetric parameters, but this behavior without an intermolecular component cannot depend on total concentration at thermodynamic equilibrium. Artefacts such as aggregation during the experiments are unlikely based on the linear posttransition baselines for all three probes (DOSY NMR, fluorescence anisotropy, and density). Independent evaluation of purified ΔN117 by SDS–polyacrylamide gel electrophoresis (PAGE) and mass spectrometry (MS) (fig. S4) also confirmed the absence of detectable contamination and aggregation. At a deeper level of analysis, the two-state self-association model, given by Eq. 7 in Materials and Methods that fitted the titration data in Fig. 3A, is an nth-order polynomial, where n is the stoichiometry of the oligomer. The value of n (= 2 for dimer), which is fixed in the fitting, imposes a severe constraint on the shape of the titration to which the model may adequately fit. As detailed elsewhere (26), oligomers n ≥ 3 invariably show sigmoidal (S-shaped) transitions. Only a two-state dimer exhibits nonsigmoidal profiles on linear concentration scales, precisely as constructed in Fig. 3A and observed in the data. On this basis, the biophysical evidence is unambiguous in showing homodimerization of PU.1 without DNA, and the range of dissociation constants yielded by the different probes reflected the distinct molecular properties they sampled.
To further strengthen this evidence, we resolved ΔN117 by electrospray ionization (ESI)–MS up to a concentration of 840 μM. Using an established maximum entropy procedure (27), peaks corresponding to both monomeric and dimeric PU.1 were observed in deconvoluted zero-charge mass spectra (Fig. 3B). The integrated intensities of the two species were quantitative, but they did not correspond to solution conditions in the other experiments. This was due to the technique’s requirement for a volatile buffer (NH4HCO3), species-dependent ionization efficiency, and the potential for ionization-induced dissociation of the complex. Notwithstanding, the ratio of dimer-to-monomer intensities varied in favor of the dimeric species with increasing total protein concentration (Fig. 3B, bottom). The concentration dependence excluded the possibility that either species could represent a static contaminant but rather corresponded to a ΔN117 monomer and dimer at dynamic equilibrium.
To gain insight into the conformational structure of the free PU.1 dimer, we interrogated ΔN165 and ΔN117 by circular dichroism (CD) and NMR spectroscopy. At an identically low concentration (25 μM), a net contribution of coil content due to the PEST domain was apparent (Fig. 3C). With increasing concentration, ΔN165 showed a spectral shift but without an endpoint at 300 μM. In contrast, the corresponding spectra for ΔN117 (weighted by contributions from the disordered PEST domain) underwent a nonsigmoidal transition that, unlike ΔN165, was substantially completed at 300 μM (Fig. 3D). Model fitting of the far–ultraviolet (UV) CD spectra, which are sensitive to secondary structure content, to a two-state dimer yielded a dissociation constant of K2 = 46 ± 19 μM. As an analysis of full CD spectra by singular value decomposition rendered more structural information than the other titration probes in Fig. 3A, we will use the CD-fitted K2 for comparison with other PU.1 constructs and solution conditions.
To probe the local structure of the PU.1 dimer, we compared the 1H-15N HSQC fingerprint of 400 μM ΔN117 and ΔN165, concentrations at which the preceding experiments showed that ΔN117 was predominantly dimeric, while ΔN165 remained monomeric (Fig. 3E; compare to Fig. 3A). Dispersed cross-peaks for the two constructs mostly overlapped within experimental uncertainty (inset). PEST domain residues were clustered at 8.2 ± 0.2 ppm. {1H}15N-NOE (nuclear Overhauser effect) measurements confirmed that the ETS residues in ΔN117 remained well ordered throughout, similarly as ΔN165, while PEST residues exhibited much lower values as a group (Fig. 3E, inset). Thus, the ΔN117 dimer was a fuzzy complex in which the PEST domain did not deviate from a tethered IDR to the structured ETS domain.
Dimeric forms of PU.1 with and without DNA are nonequivalent
The 2:1 complex formed by PU.1 at a single cognate site suggested that the PU.1 dimer was asymmetric, as a symmetric dimer that exposes the DNA contact surfaces would logically yield a 2:2 complex. However, this stoichiometry was excluded by the DOSY titration data, which showed two inflections with the least diffusive species at a DNA:protein ratio of 1:2, corresponding to the 2:1 complex (Fig. 1B). Unbound PU.1 also formed a homodimer, which could logically arise only if the complex was symmetric. Experimentally, a symmetric dimer was strongly inferred by a single set of 1H-15N signals for unbound ΔN117 at high concentrations (Fig. 3E). Moreover, the CD-detected structure of PU.1 showed negligible changes upon titration by DNA (Fig. 4A), in contrast with the self-titration in the absence of DNA (Fig. 3D). These clues suggested that DNA-bound and free PU.1 dimerized into distinct conformers.
Fig. 4. Mutations demonstrate nonequivalent PU.1 dimers with and without DNA.
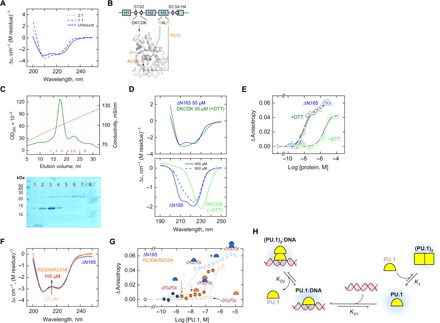
(A) Far-UV CD spectra of the DNA-bound ΔN165 upon subtracting the spectrum of the cognate DNA acquired under identical conditions (75 μM and 0.15 M Na+). (B) Residues involved in the DKCDK mutant and in the binding-deficient mutant (R230A/R233A). The structure is homology-modeled against the cocrystal 1PUE. (C) Purification of the DKCDK mutant by ion exchange chromatography under nonreducing conditions. Lysate was loaded at 0.5 M NaCl and extensively washed before elution over a linear gradient to 2 M NaCl. SDS-PAGE of purified fractions is shown. Fractions containing primarily monomer (e.g., 1 and 2) or dimers (e.g., 5 onwards) were concentrated and dialyzed separately into buffer containing 0.15 M NaCl with or without 5 mM DTT, respectively. (D) CD spectra of the DKCDK monomer (top) and dimer (bottom) under various conditions with wild-type ΔN165 as reference. The spectrum for the DKCDK monomer was less well resolved due to the presence of DTT, which contributed to the total absorption of the sample at 50 μM protein. See text for details. (E) Fluorescence anisotropy measurements of cognate DNA binding by monomeric and dimeric DKCDK with wild-type ΔN165 as reference. (F) CD spectrum of 25 to 100 μM of the R230A/R233A mutant, with ΔN165 at 25 μM as reference. (G) DNA loading by the R230A/R233A mutant in the presence of wild-type ΔN165 (solid symbols). Concentrations of the mutant and wild-type protein that individually failed to bind DNA collaborated to bind DNA as a heterocomplex. (H) Proposed model for the formation of two nonequivalent PU.1 dimers: an asymmetric one in the 2:1 DNA complex and a symmetric one without DNA.
To test these notions, we constructed a constitutive ETS dimer via insertion of a single Cys residue into ΔN165, which did not harbor this amino acid, between residues 194 and 195 (Fig. 4B). We targeted this position given its turn conformation in the known structures of the ETS monomer [Protein Data Bank (PDB): 5W3G] and the 1:1 complex (1PUE), and its reported involvement in 2:1 complex formation by heteronuclear NMR (20). Purification of this mutant, termed DKCDK, by ion exchange chromatography under nonreducing conditions eluted monomer and its cystine-linked dimer at >1 M NaCl (Fig. 4C). Fractions containing predominantly monomer or dimer were separately dialyzed into a buffer containing 0.15 M NaCl with or without 5 mM dithiothreitol (DTT), respectively. In the absence of DNA, the far-CD spectrum of the DKCDK monomer (maintained with 5 mM DTT) overlapped closely with the spectrum of ΔN165 (Fig. 4D) and formed the 1:1 complex with cognate DNA similarly as wild-type ΔN165, indicating that the Cys insertion was nonperturbative in the DKCDK monomer (Fig. 4E). In stark contrast, the cystine-linked DKCDK dimer exhibited a CD spectrum that was altogether unlike PU.1 at equivalent molar concentrations (400 μM). It bore some similarity to a spectrum for ΔN165 at the highest concentration available (800 μM), which contained a greater fraction of dimeric PU.1 (dashed spectrum in Fig. 4D). However, the dimeric DKCDK spectrum was further redshifted by ~7 nm and ~15% more intense. Moreover, the DKCDK dimer bound cognate DNA >100-fold more poorly than wild-type ΔN165 (Fig. 4E). Thus, the DKCDK mutant showed that a symmetric configuration was severely perturbed in conformation without DNA and unlike DNA-bound wild-type ΔN165 (compare to Fig. 4A). Together with a deficiency in DNA binding, the DKCDK mutant demonstrated that the wild-type DNA-bound dimer was not a symmetric species in contrast with the unbound PU.1 dimer.
To assess the feasibility of an alternative, asymmetric configuration in forming the 2:1 complex, which would involve the DNA contact surface, we then examined an R230A/R233A mutant in the DNA-recognition helix H3 of PU.1 (Fig. 4B). The double R→A mutant retained an indistinguishable CD spectrum as wild-type ΔN165 (Fig. 4F). At a subsaturating concentration of wild-type ΔN165, the addition of the mutant at a concentration that showed no DNA binding on its own nevertheless produced strong DNA loading (Fig. 4G). Such a result would most simply arise if the R→A mutant associated with the wild-type 1:1 complex to drive the 2:1 heterocomplex. The data thus pointed to an asymmetric PU.1 dimer in the 2:1 complex, in which the secondary structure content of PU.1 did not change significantly. Both features contrast sharply with the symmetric conformation required by the DNA-free dimer.
A synthesis of the evidence leads us to propose a model for PU.1 dimerization in the presence and absence of DNA (Fig. 4H). In terms of affinity, the 1:1 active complex is strongly favored (>102-fold) over either the 2:1 complex or the unbound dimer. Excess PU.1 drives one or the other dimeric state depending on the presence of DNA. The key cornerstone of this model is the nonequivalence of the two dimeric states. Specifically, the incompatibility of the free dimer with DNA binding means that a preexisting dimer cannot serve as an intermediate for the 2:1 complex. Thermodynamic insulation of the two dimeric species leads to a mutually antagonistic relationship, in which the formation of one species is favored at the expense of the other. ΔN117 illustrates this antagonism, as relative to ΔN165, the N-terminal PEST domain promotes dimerization without DNA and reduces the affinity of 2:1 complex formation (Fig. 1B). Together with enhancing the apparent affinity for 1:1 binding, the result is a widened concentration window for the 1:1 complex for ΔN117.
The electrostatic basis of PU.1 dimerization
The ETS domain as embodied by ΔN165 is highly enriched in Lys and Arg residues, with an isoelectric point (pI) of 10.5. Dimerization should, therefore, be highly sensitive to salt concentration. Contrary to the expectation that the dimer would be stabilized at high salt, which would screen electrostatic repulsion, the opposite was observed. CD-detected self-titration of ΔN165 at 50 mM Na+ showed a nearly complete two-state transition (Fig. 5A) but not at 150 mM Na+ (compare to Fig. 3A). The low-salt spectra, extended in wavelength to 190 nm and protein concentration to 800 μM because of the reduced Cl− level, showed the same transition characteristics as acquired at 150 mM Na+ (fig. S5), indicating that the same transition was inspected at both salt concentrations. Although the transition at 50 mM Na+ corresponded to a dissociation constant of ~200 μM, it was still fivefold higher than that for ΔN117 in 150 mM Na+ (Fig. 5B). The data, therefore, reaffirmed the stimulatory role of the disordered PEST domain in dimerization of the ETS domain while revealing an electrostatic basis in the unbound PU.1 dimer.
Fig. 5. Dimerization of the DBD of PU.1 is electrostatically mediated and conformationally destabilizing.
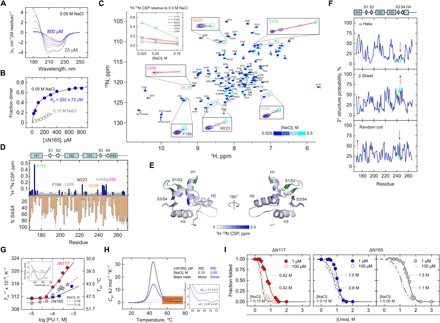
(A) CD-detected titration of ΔN167 at 50 mM NaCl from 25 to 800 μM. (B) Analysis of the titration by singular value decomposition yields a two-state transition with a dissociation constant K2 of 202 ± 72 μM. (C) 1H-15N HSQC as a function of salt from 25 to 500 mM NaCl. Residues with the strongest 1H-15N CSPs (Y173, M223, G239, V244, and L248) are boxed. Inset: Salt dependence of the CSPs of these residues. (D) Summary of the residue CSPs with the average %SASA from the unbound NMR monomer, 5W3G. Residues above a 0.5-ppm cutoff are colored in dark blue, and the subset of internal residues (<35% SASA, based on the termini) is marked with yellow circles. Residues implicated in the DNA-bound dimer are marked with green circles. (E) Mapping of the high-CSP residues to 5W3G. Green residues mark known residues involved in the DNA-bound dimer (20). (F) Chemical shift–derived secondary structure prediction via the CSI using 1H and 15N signals. The color scheme follows the HSQC in (C). Regions with significant changes in secondary structure are marked by arrows. (G) Near-UV CD-detected thermal melting of ΔN117 and ΔN165. Two salt concentrations were evaluated for ΔN165 (blue and gray). Inset: Representative near-UV CD spectra. (H) DSC thermograms (solid) for ΔN165 under conditions (salt and concentrations) in which the protein was primarily monomeric or dimeric. The ΔCp values are given in kJ (mol monomer)−1 K−1. Dashed curves represent the two-state transition for a monomer (black) and dimer (blue). (I) Trp fluorescence-detected denaturation by urea of ΔN117 and ΔN165 at two monomer concentrations. Curves represent fit to the linear extrapolation model for a two-state dimer. The marked concentrations represent urea concentration at 50% unfolding.
The sensitivity of the ETS dimer to salt allowed us to access the local structural changes in the DNA-free ETS dimer by NMR spectroscopy. 1H-15N HSQC spectra of ΔN165 with 0.5 to 0.025 M NaCl (Fig. 5C) revealed a panel of residues with significant chemical shift perturbations (CSPs). Taking the spectrum acquired in 0.5 M NaCl as the reference for monomeric PU.1, the CSPs exhibited a well-ordered salt dependence (Fig. 5C, inset). The salt-induced CSPs were plotted as a function of residues (Fig. 5D), and a cutoff of 0.05 was applied to identify the residues most affected by electrostatic interactions. These perturbed residues were spatially diffuse, as a formal mapping to the unbound PU.1 structure demonstrated (Fig. 5E) and did not overlap with the known residues involved in 2:1 complex formation (20). We also examined the transverse spin relaxation (T2) properties of the methyl proton peaks in the 1H spectra as a global representation of the tumbling of ΔN165 at different NaCl concentrations (fig. S6). The effective T2* relaxation values for the three characteristic methyl 1H peaks at 0.025 M NaCl were up to ~25% lower than at 0.5 M NaCl and well beyond experimental error. This result indicated that the salt-induced CSPs reflected the formation of a slower tumbling dimer.
To correlate the NMR data with the CD-detected changes, we used the heteronuclear chemical shifts to infer secondary structure via the chemical shift index (CSI) (28). The CSI results corroborated the CD-detected loss of α-helical and gain in β/coil content and furthermore localized these changes to helix 1 (H1) and the loop between β sheet 3 (S3) and β sheet 4 (S4) (Fig. 5F). Local H1 unwinding accounted for the CSPs observed near the N terminus of ΔN165, including the particularly strong CSP at Y173, while the loop between S3 and S4 gained β-sheet structure.
Dimeric PU.1 is conformationally destabilized relative to the constituent monomer
The N-terminal IDR promotes a structurally perturbative PU.1 dimer in the absence of DNA. To reveal the underlying conformational thermodynamics of the PU.1 dimer, we performed thermal melting experiments over a range of protein concentrations, using the near-UV CD spectrum from 250 to 300 nm as a probe. The thermal transition was analyzed from a singular value decomposition of the full spectra at each concentration and fitted to a two-state model. The apparent melting temperature (Tm) dropped with increasing concentration in step with the propensity for dimer formation (Fig. 5G). ΔN117 suffered a larger drop than ΔN165 over a ~10-fold increase in concentration. A reduction in salt concentration, which drove dimerization, similarly caused a larger drop in Tm for ΔN165 (0.15 versus 0.05 M Na+; Fig. 5G, dashed line).
The presentation of Fig. 5G as Tm−1 versus the logarithm of concentration implies that steeper slopes correspond to lower enthalpies (heats) of dissociation/unfolding, which relate to the quality of conformational interactions. To rigorously define the conformational thermodynamics of the PU.1 dimer, we performed differential scanning calorimetry (DSC) experiments on ΔN165 under conditions (salt and protein concentrations) where the quantitatively major population was either monomer or dimer (Fig. 5H, all values on a per-mole monomer basis). The thermograms showed a much greater calorimetric molar enthalpy (area under the curve) for the monomer (300 μM at 0.15 M Na+) than dimer (500 μM at 0.05 M Na+). In addition to enthalpy, DSC yields heat capacity changes (ΔCp, difference in the pre- and posttransition baselines) that inform on changes in water-accessible surface area. The ΔN165 monomer exhibited a ΔCp of 3.1 ± 0.3 kJ/(mol K), in good agreement with the structure-based value of 3.3 kJ/(mol K) from the NMR structure of the PU.1 monomer (29). In contrast, the ΔN165 dimer exhibited a significantly reduced ΔCp of 0.97 ± 0.27 kJ/(mol K). Assuming identical thermally unfolded states, the differences in heat capacity changes indicated that ΔN165 was less well folded than the monomer.
To probe the effect of the N-terminal IDR on the conformational stability of the PU.1 ETS domain, we performed chemical denaturation experiments with urea, which could be reported by intrinsic tryptophan fluorescence at much lower protein concentrations than DSC. At a strictly monomeric concentration (1 μM) at 0.15 M Na+, ΔN117 was only slightly more sensitive to urea, as judged by the urea concentration at 50% unfolding, than ΔN165 (Fig. 5I). At 100 μM concentration, at which ΔN117 is mostly dimeric but ΔN165 remains monomeric, ΔN117 became significantly more sensitive to urea, suggesting highly perturbative interactions between the PEST and ETS domains. A conformationally perturbed ΔN117 dimer was also implied by its volumetric properties. The posttransition density slope in Fig. 3A yields a partial specific volume of ΔN117 of 0.85 ± 0.01 ml/g, which is atypically high for structured globular proteins and suggests altered molecular packing and hydration properties. ΔN165 was more stable at 1 μM at 0.05 M Na+ than at 0.15 M Na+, an observation consistent with the ~2°C higher apparent Tm for 10 μM ΔN165 over the same Na+ concentrations (Fig. 5G). The ΔN165 monomer and dimer were therefore opposite in their conformational stabilities with respect to salt, underlining the structure perturbation by salt or the anionic PEST domain.
In summary, spectroscopic and calorimetric measurements showed that the PU.1 ETS dimer was destabilized with respect to unfolding relative to its monomeric constituents. Structural considerations aside, conformational destabilization contributes to the DNA binding deficiency of the apo ETS dimer. A destabilized dimeric state implied favorable concentration-dependent interactions within the unfolded ensemble over the folded state. The ability of the anionic PEST domain to promote formation of the unbound dimer in ΔN117 therefore further suggests a basis in mitigating the electrostatic repulsion among the cationic ETS domains.
The C-terminal IDR is required for PU.1 dimerization without DNA
In addition to the N-terminal IDR, the structured ETS domain of PU.1 is also tethered at the C terminus to a shorter, 12-residue disordered segment (residues 259 to 270), as apparent in the unbound PU.1 monomer structure (fig. S7A) (29). Far-UV CD spectra at 0.15 M Na+ showed that hPU.1(117-258) and hPU.1(165-258), termed sΔN117 and sΔN165, respectively (fig. S7B), lacked the secondary structure changes characteristic of ΔN117 and Δ165 across comparable concentrations (fig. S7C; compare to Fig. 3D). sΔN117 was also much less sensitive to urea over the same protein concentration range as ΔN117, and sΔN165 showed no change relative to ΔN165. In contrast to their dimeric deficiency without DNA, sΔN117 and sΔN165 were both intact with respect to dimerization with cognate DNA (fig. S7D). While sΔN117 formed 1:1 and 2:1 DNA complexes with the same affinities as ΔN117 in 0.15 M NaCl within experimental error, sΔN165 was a significantly poorer DNA binder than ΔN165 (table S1). In particular, 2:1 complex formation was less negatively cooperative for sΔN165, with the concentration window (KD2/KD1) for the 1:1 complex only ~65% that for ΔN165 (fig. S7D). Last, unlike ΔN165 at 0.05 M Na+, sΔN165 showed a negligible propensity to dimerize and exhibited biphasic binding with cognate DNA (fig. S7E; compare to Fig. 2B). The divergent impact of removing the C-terminal IDR on dimerization with and without DNA stood in clear agreement with our concept of nonequivalent dimeric states for PU.1 and the structural distinctiveness of the two states.
Phosphomimetic substitutions of the N-terminal IDR reinforce the dimeric propensity of the DNA-free PU.1 dimer
Characteristic of many IDRs flanking DBDs, the N-terminal PEST domain in PU.1 is highly enriched in Glu and Asp residues (pI 3.5), in sharp contrast with the positively charged DBD (pI 10.5) to which it is tethered. The foregoing structural and thermodynamic evidence strongly suggests that the acidic IDR interacts with the ETS domain and shifts it toward dimer formation. Functional studies have established a panel of Ser residues in the PEST domain, including residues 130, 131, 140, and 146 (human numbering), which are multiply phosphorylated in cells (30, 31). Phosphoserines at these positions would enhance the anionic charge density by a substantial amount from −11 (3 Asp + 8 Glu) to −17 (~−1.5 per phosphoserine). Because these residues are disordered, we made phosphomimetic substitutions of these residues to Asp, generating a di-substituted (140 and 146, termed D2ΔN117) and tetra-substituted mutant (termed D4N117), to probe their general charge-dependent effects (Fig. 6A). Far-UV CD spectra showed that the phosphomimetic substitutions progressively drove the affinity of the DNA-free dimer, and the resultant dimers appeared to harbor greater random coil content than their wild-type counterpart (Fig. 6B; compare to Fig. 3D). In DNA binding experiments, the di-substituted mutant D2ΔN117 behaved approximately as wild-type ΔN117, while the affinity of the 2:1 complex for the tetra-substituted mutant D4ΔN117 was ~15-fold lower than that for wild-type ΔN117 (table S1). Stimulation of the unbound dimer was therefore associated with a marked reduction in the affinity of the 2:1 complex by D4ΔN117 (Fig. 6C). As a result, the selective effect on the 2:1 complex in D4ΔN117 resulted in greater negative cooperativity (i.e., increasing KD2/KD1) in the dimerization of DNA-bound PU.1. In turn, the concentration window for the 1:1 complex widened more than fourfold for D4ΔN117 relative to wild-type ΔN117.
Fig. 6. Phosphomimetic substitutions at the PEST domain and charged crowding demonstrate a general electrostatic basis of IDR/ETS interactions.
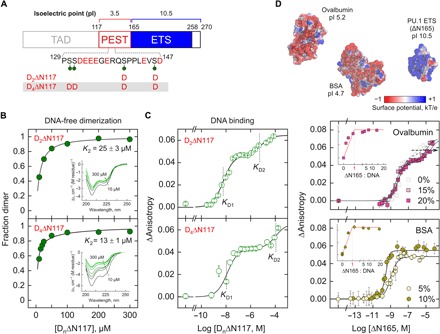
(A) Schematic of the phosphorylated Ser residues in human PU.1, marked by green pins. The Ser→Asp substituted positions in D2ΔN117 and D4ΔN117 are shown. (B) D2ΔN117 and D4ΔN117 exhibit enhanced dimeric propensities without DNA relative to ΔN117 (compare to Fig. 3D). (C) D2ΔN117 and D4ΔN117 are progressively impaired in 2:1 DNA complex formation. (D) The anionic crowders ovalbumin and BSA modulate DNA binding by ΔN117 in a similar way as the phosphomimetic substitutions on ΔN117 to an extent that correlates with their sizes and low pIs. Inset: Stoichiometric determination using 1 μM DNA (first binding transition in the case of ovalbumin). The spacing of the ordinates is identical to main plots. The surface potentials of the structures were computed using the Adaptive Poisson-Boltzmann Solver (APBS) at 0.15 NaCl.
The reinforcing effects of multiple phosphomimetic substitutions in the disordered PEST domain strongly suggest that it influences the behavior of the ordered ETS domain via a generally electrostatic, nonstructurally specific mechanism. To further establish this notion, specifically the absence of dependence on structurally specific interactions, we tested the effect of crowding concentrations (in the range of 102 g/liter) of ovalbumin or bovine serum albumin (BSA) on DNA binding by ΔN165 (Fig. 6D). These two anionic proteins share pIs (pI = 5.2 and 4.7 for albumin and BSA, respectively) that are close to the PEST domain (pI = 3.5) but present well-formed globular structures. If PEST/ETS interactions involved structurally specific interactions between the two domains, the anionic crowders should differ significantly from the PEST domain in their effects on DNA recognition by the ETS domain. DNA binding in the presence of up to 20% (w/v) ovalbumin showed little effect on the 1:1 complex (Fig. 6D, inset) while progressively decreasing the affinity of 2:1 binding. This behavior mirrored closely the phosphomimetic mutants, and similarly, the more pronounced biphasic appearance in the presence of ovalbumin was a result of the increased negative cooperativity and widening concentration window for the 1:1 complex. With BSA, an even more anionic crowder, the effect was correspondingly more pronounced. A concentration of 5% suppressed 2:1 binding at 10−5 M, an almost 105-fold molar excess of ΔN165 over DNA. Only the 1:1 complex was formed (inset). In contrast, the neutral crowder PEG 8K preserved biphasic DNA binding (fig. S8A), showing that the effects of BSA and ovalbumin were not due to volume exclusion from crowding alone and highlighting the importance of charge. To test our model’s prediction that BSA would, therefore, promote the PU.1 dimer, we evaluated ΔN165 labeled with 5-fluoroTrp by 19F NMR in the presence of BSA. The three tryptophan residues in ΔN165 underwent distinct CSPs with 5% BSA under conditions that gave monomers in dilute solution (fig. S8B). These changes reflected conformational perturbations consistent with unbound dimer formation. Thus, phosphomimetic substitutions and acidic crowding supported nonmicrostructural electrostatic field interactions on the ETS domain as the basis of the PEST-stimulated dimerization in the absence of DNA.
DISCUSSION
PU.1 is a markedly inducible transcription factor during hematopoiesis and immune stimulation (22). Open-source repositories such as the Human Protein Atlas show that the expression of PU.1 varies among a panel of resting cell lines by ~25-fold. Independently, single-cell cytometry shows that the abundance of PU.1 transcript in unstimulated murine bone marrow cells ranges from less to 5% to ~50% that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (32), a housekeeping glycolytic enzyme that is present at ~70 μM in the cell (33). Induction by ligands such as retinoic acid (34) or bacterial endotoxins (35) increases PU.1 expression another 10-fold or more. Depending on the combination of cell line, physiology, and the presence of stimulatory ligands, cellular PU.1 abundance varies by a multiplier comparable to the ratio of the two dissociation constants KD2/KD1 (102- to 103-fold) of the 1:1 and 2:1 PU.1/DNA complexes.
In patients and animal models, PU.1 dosage is well established as critical to hematopoietic physiology and dysfunction in vivo (15, 36). Dosage effects have been extensively defined in terms of expression, but relatively little is understood about direct dosage effects on transactivation at the protein/DNA level. In this study, manipulation of enhancer syntax in HEK293 cells, which do not use PU.1, demonstrated negative feedback in ectopic PU.1 trans-regulation, independent of modifying interactions with tissue-specific coactivators. The recapitulation of negative feedback, manifested by dose-dependent derepression of endogenous PU.1 in myeloid THP-1 cells, strongly supports functional relevance in native PU.1-dependent gene regulation. Characterization of the attributable species, a 2:1 DNA complex, revealed two nonequivalent dimeric states that are reciprocally controlled by DNA and the IDRs tethered to the structured DBD. Under physiologic salt conditions, structural alterations that bias unbound PU.1 toward dimerization (e.g., full phosphomimetic substitutions of the N-terminal IDR) oppose dimerization of DNA-bound PU.1. Conversely, alterations that abrogate PU.1 dimerization (e.g., truncation of the C-terminal IDR) promote the formation of the 2:1 DNA complex. Only at low salt conditions (e.g., 50 mM Na+) are the DNA-free and DNA-bound dimers both favored, and the 1:1 complex is not populated. The tethered IDRs do not appear to become part of the structured dimer but determine preference between the two dimeric states such that the DNA-free dimer remains essentially cryptic without both terminal IDRs.
If the 2:1 complex represents the structural basis of negative feedback, what functional role does the unbound PU.1 dimer play? The thermodynamic relationships among the various states accessible to PU.1 (Fig. 4H), critically the nonequivalent free and DNA-bound dimer, suggest a novel “push-pull” mechanism of PU.1 autoregulation by distinct pools of dimeric protein. By antagonizing the 2:1 complex, we postulate that the unbound dimer suppresses negative feedback and dynamically increases the circulating dose of transcriptionally active PU.1. This model affords for the first time a unifying basis for the PU.1-activating effects of PEST phosphorylation by casein kinase II and protein kinase C (31, 37, 38), as well as the PU.1-inactivating effects of phosphorylation inhibition by oncogenic transcription factors (39), by biasing PU.1 conformations toward or away from the unbound dimer.
Earlier in vitro studies reporting on dimers of the ETS domain (20, 21, 25), including our own, did not appreciate their functional significance. The solution NMR structure of the unbound monomer (5W3G) reflects the incomplete context afforded by the ETS domain alone (i.e., ΔN165) at physiologic ionic strength (0.15 M Na+/K+). The dissociation constant for the PU.1 dimer in dilute solution (10−5 M) should not be misinterpreted as denoting a physiologically irrelevant interaction. The complex formed by PU.1 and its partner GATA-1 is functionally critical in cell lineage specification during myeloid differentiation in vivo (40), but its equilibrium dissociation constant in dilute solution was 10−4 M as determined by NMR spectroscopy (41). This and other examples show how volume exclusion and other crowding effects favor interactions in vivo relative to dilute solution. Facilitated diffusion along genomic DNA is also expected to promote cognate occupancy beyond the affinity for oligomeric targets from free solution.
As the tethered IDRs remain disordered in fuzzy PU.1 dimers with and without DNA, their formation is therefore unrelated to paradigms such as induced fit, conformational selection, or fly-casting mechanisms that involve order-disorder transitions by the IDRs (42, 43). Instead, the charged intrinsic disorder in PU.1 is involved in through-space electrostatic interactions. In the absence of the N-terminal IDR, dimerization of the highly cationic ETS domain (ΔN165) is favored by low salt. Conformational destabilization of the resultant dimer under these conditions suggests an electrostatic penalty arising from charge-charge repulsion of the DBDs. The stimulatory effect of the negatively charged IDR on the DNA-free dimer, therefore, arises from attenuation of this repulsive penalty of association. A nonstructurally specific basis is borne out by the similarly favorable effects of reinforcing negative charges via phosphomimetic substitutions in ΔN117 as anionic crowders on ΔN165. As phosphoserines confer higher charge density (~−1.5) than carboxylates, phosphorylation is expected to exert an even greater effect than the phosphomimetic substitutions. Notably, despite its nominal designation as a proteasome-recruiting signal, the PEST domain does not target PU.1 for metabolic turnover, but it is associated with a local role in dimerization and protein-protein partnerships such as with the lymphoid-specific factor IRF4 (44).
The properties of the flanking IDRs on DNA binding as revealed in this study highlight the divergent roles played by intrinsic disorder within the ETS transcription factor family, which is united by eponymous, their structurally homologous DBDs. Many ETS members are controlled by autoinhibition, a mechanism that specifically involves short flanking helices in the unbound state that unfolds and disrupts DNA binding allosterically or at the DNA contact interface (45). High-affinity binding to native promoters requires coactivators or homodimerization at tandem sites to displace autoinhibitory helices, forming with positive cooperativity 2:2 complexes (46). The regulatory strategy is activation through recruitment by other coactivators. In Ets-1, the paradigm autoinhibited ETS member, disordered elements in the serine-rich region (SRR) domain upstream of the autoinhibitory helices further modulate the regulatory potency of the autoinhibitory helices. Progressive phosphorylation of the SRR domain reinforces autoinhibition (47). PU.1, and likely its proximal ETS relatives, upends this paradigm. Lacking autoinhibitory domains, high-affinity DNA binding is the default behavior. The disordered elements flanking its structured ETS domain regulate DNA binding by modifying negative feedback. PU.1 is the recruiter in protein-protein partnerships such as IRF4 (44). Phosphorylation of the disordered PEST domain promotes the persistence of the active 1:1 complex and has been established as broadly stimulatory (30, 31, 37, 39). These contrasting features help frame in molecular and functional terms the evolutionary divergence in the ETS family, one of the most ancient families of transcription regulators in metazoan evolution.
As a final remark, both dimeric forms of PU.1 represent highly novel structures. Asymmetric DNA-bound dimers are known in the case of the zinc finger protein HAP1 (48). Zinc fingers are obligate dimers with a 2:2 subunit-to-DNA subsite configuration. Asymmetry in the HAP1 dimer is directed by the polarity of the DNA subsites bound to the subunits. The asymmetric 2:1 complex with PU.1 involves only a single DNA site without significant change in conformation. Functional deficiency of the 2:1 complex as evidenced by the cellular experiments in Fig. 1, therefore, suggests perturbation of DNA structure relative to the singly bound state or denial of specific surfaces of the 1:1 complex to form the transcriptional machinery. In contrast with the localized surface implicated in the 2:1 complex, NMR evidence shows that the residues involved are diffusely distributed with many buried in the PU.1 monomer, leading to a conformationally destabilized dimer. The CSPs observed in the DNA-free dimer, namely, H1 and the wing (S3/S4), were also recently observed for the interaction of PU.1 with a disordered peptide from the SRR domain of Ets-1 (29). These regions may, therefore, represent interaction hotspots for protein/protein partnerships for PU.1 in the absence of DNA. Beyond the minimal ETS domain, the short C-terminal IDR acts in concert with the PEST domain to reinforce the dimerizing properties of the ETS domain. Structurally, this suggests that the two IDRs likely interact physically, either antagonistically in the monomeric state or cooperatively in the dimeric state. In the cytoplasm, the ETS domain mediates nuclear import of PU.1 (49), so dimerization may also help regulate subcellular trafficking. Further studies to solve the structures of dimeric PU.1 and map their distributions in subcellular compartments will define their dynamics in vivo and contributions to target gene expression.
MATERIALS AND METHODS
Molecular cloning
DNA encoding fragments of human PU.1 encompassing the ETS domain with and without various segments of its N- and C-terminal IDRs were synthesized by Integrated DNA Technologies (IDT) (Midland, IA) and subcloned into the Nco I/Hind III sites of pET28b (Novagen). For truncated constructs harboring the PEST domain (i.e., ΔN117, sΔN117, D2ΔN117, and D4ΔN117), Cys118 was mutated to Ser to facilitate purification and biophysical experiments. Full-length PU.1 used in cell-based experiments was fully wild type. Various PU.1-sensitive enhancer sequences as described in the text were also purchased from IDT and inserted between the Age I/Bgl II sites of pD2EGFP (Clontech, CA). All constructs were verified by Sanger sequencing.
Cell culture
THP-1 and HEK293 cells were purchased from the American Type Culture Collection and were routinely cultured in RPMI 1640 and Dulbecco’s modified Eagle’s medium, respectively, supplemented with 10% heat-inactivated fetal bovine serum. Where indicated, cells were induced with a single dose of PMA at 16 nM for 72 hours (final dimethyl sulfoxide concentration: 0.1%, v/v). All cell lines were maintained at 37°C under 5% CO2.
Cellular reporter assays
Cellular PU.1 transactivation was measured using a PU.1-dependent EGFP reporter construct under the control of a minimal enhancer harboring only cognate binding sites for PU.1. In PU.1-negative HEK293 cells, the reporter was transactivated in the presence of an expression plasmid encoding wild-type full-length PU.1 and a cotranslating iRFP marker (18). Cells (7 × 104) were seeded in 24-well plates and cotransfected with a cocktail consisting of the EGFP reporter plasmid (250 ng) and up to 25 ng of expression plasmids for full-length PU.1, using jetPRIME reagent (Polyplus, Illkirch, France) according to the manufacturer’s instructions. The total amount of plasmid was made up to 500 ng with empty pcDNA3.1(+) vector. Twenty-four hours after transfection, cells were trypsinized and analyzed by flow cytometry using an FCS Fortessa instrument (BD Biosciences). Live cells were gated for iRFP and EGFP fluorescence using reporter and full-length PU.1 only controls, respectively, in FlowJo (BD Biosciences) before computing the total fluorescence of the dually fluorescent population.
RT-PCR experiments
Following extraction of total RNA using a spin column kit (Omega) and RT (Thermo Fisher Scientific), RT-PCR reactions were performed on a QuantStudio 3 instrument (Applied Biosystems) with SYBR Green PCR Master Mix (Thermo Fisher Scientific). Expression levels of genes were normalized to gapdh. The primer sequences used for pu.1, csf1ra, e2f1, and gapdh are given in table S2.
Protein expression and purification
Heterologous overexpression in BL21(DE3)pLysS Escherichia coli was performed as previously described (20). In brief, expression cultures in LB or M9 media (the latter containing 15NH4HCl or U-13C6-glucose as required) were induced at an optical density (OD600) of 0.6 with 0.5 mM isopropyl β-d-1-thiogalactopyranoside for 4 hours at 25°C. Uniformly 15N- and 15N/13C-labeled constructs were expressed in appropriate M9-based media. Harvested cells were lysed in 10 mM NaH2PO4/Na2HPO4 (pH 7.4) containing 0.5 M NaCl by sonication. After centrifugation, cleared lysate was loaded directly onto a HiTrap Sepharose SP column (GE) in 10 mM NaH2PO4/Na2HPO4 (pH 7.4) containing 0.5 M NaCl. After extensive washing in this buffer, the protein was eluted in a gradient at ~1 M NaCl in phosphate buffer. Purified protein was dialyzed extensively into various buffers, as described in the text, and diluted as needed with dialysate. Protein concentrations were determined by UV absorption at 280 nm.
DNA binding experiments
DNA binding by protein was measured by steady-state fluorescence polarization of a Cy3-labeled DNA probe encoding the optimal PU.1 binding sequence 5′-AGCGGAAGTG-3′. In brief, 0.5 nM of DNA probe was titrated with protein in a 10 mM tris-HCl buffer (pH 7.4) containing 0.1% (w/v) BSA and NaCl at concentrations as stated in the text. Steady-state anisotropies 〈r〉 were measured at 595 nm in 384-well black plates (Corning) in a Molecular Dynamics Paradigm plate reader with 530-nm excitation. The signal represented the fractional bound DNA probe (Fb), scaled by the limiting anisotropies of the ith bound 〈ri〉 and unbound states 〈r0〉, as follows
| (1) |
where Fb as a function of total protein concentration was fitted to various models as follows. In all cases, the independent variable was the total titrant concentration as taken.
For DNA binding, the two stepwise dissociation constants describing the formation of the 1:1 and 2:1 PU.1/DNA complexes are
| (2) |
where P and D denote PU.1 and DNA. In this analysis, the binding affinities were not further constrained by interactions of the unbound states. The ratio of KD2/KD1 = ω defines the nature of the cooperativity of the 2:1 complex in the paradigm of McGhee and von Hippel. Values of ω below, equal to, or above unity denote positively cooperative, noncooperative, and negatively cooperative formation of the 2:1 complex with respect to the 1:1 complex, respectively.
In direct titrations of the DNA probe by PU.1, the observed anisotropy change represented the summed contributions of the two complexes as expressed by Eq. 1. The most efficient approach is to determine binding in terms of the unbound protein, P. The solution, which is cubic in [P], is
| (3) |
where the subscript “t” represents the total concentration of the referred species. [P] was solved numerically from Eq. 3, rather than analytically via the cubic formula, to avoid failure due to loss of significance. With [P] in hand, [D], [PD], and [P2D] were computed from Eq. 2 and the corresponding equations of state. In the limit of no formation of the 2:1 complex (i.e., KD2 → ∞), Eq. 3 simplifies to a quadratic, corresponding to formation of only the 1:1 complex
| (4) |
Competition ESI-MS
Analyses were performed on a Waters Q-TOF (quadrupole orthogonal acceleration–time-of-flight) micro mass spectrometer equipped with an ESI source in positive ion mode. Samples were dialyzed extensively against 0.01 M NH4HCO3 (pH 8) and introduced into the ion source by direct infusion at a flow rate of 5 μl/min. The instrument operation parameters were optimized as follows: capillary voltage of 2800 V, sample cone voltage of 25 V, extraction cone voltage of 2.0 V, desolvation temperature of 90°C, source temperature of 120°C, and collision energy of 3.0 V. Nitrogen was used as nebulizing and drying gas on a pressure of 50 and 600 psi, respectively. MassLynx 4.1 software was used for data acquisition and deconvolution. A multiply charged spectra were acquired through a full scan analysis at mass range from 300 to 3000 Da and then deconvoluted by a maximum entropy procedure (27) to the zero-charge spectra presented. Samples were diluted with dialysate to different concentrations for acquisition and data processing under the same conditions.
CD spectroscopy
Spectra were acquired in 10 mM NaH2PO4/Na2HPO4 (pH 7.4) plus NaCl as a function of concentration or temperature as indicated in the text in a Jasco J-810 instrument. Thermal denaturation experiments were performed at 45°C/hour with a response time of 32 s. The path length for near-UV scans was typically 1 or 0.1 mm for far-UV scans. Spectral analysis following blank subtraction and normalization with respect to path length and concentration was performed by singular value decomposition as follows.
For each experiment, a matrix A with column vectors represented by CD intensities at each protein concentration was factorized into the standard decomposition
| (5) |
where the left-singular unitary matrix U contained the orthonormal basis spectra ui scaled by the singular values σi from the diagonal matrix Σ. The row vectors in the right singular unitary matrix VT gave the concentration- or temperature-dependent contribution of each basis spectrum to the observed data and is termed as transition vectors vT in the text. For ease and clarity of presentation, the scaling due to Σ is captured into the transition vector, i.e., a = u(σvT) (matrix multiplication is associative), which has no effect on the fitted parameters. The transition vectors were fitted to titration models describing a two-state transition with dissociation constant K as follows
| (6) |
where X = σvT and the subscripts “1” and “n” refer to monomer and oligomer (n = 2 for dimerization), respectively. F1n is given by
| (7) |
where F1n is the fractional two-state 1-to-n oligomer at equilibrium and pt is the total protein concentration. As detailed elsewhere (26), a fundamental feature of Eq. 7 is that dimerization is uniquely nonsigmoidal on linear scales, which is diagnostic for two-state dimers. Any higher-order oligomer processes are invariably sigmoidal on linear scales.
Nuclear magnetic spectroscopy
NMR experiments were conducted at 25°C using Bruker BioSpin 500, 600, or 800 MHz spectrometers. For DOSY experiments, unlabeled protein and DNA were co-dialyzed in separate compartments against the required buffer, lyophilized, and reconstituted to 250 μM in 100% D2O before data acquisition at 500 MHz with a 5-mm total body irradiation probe. For two-dimensional (2D)/3D experiments, uniformly labeled ΔN165 and ΔN117 (± unlabeled DNA) were dialyzed against the required buffer at 11/10 excess concentration and adjusted 10% D2O at 400 to 700 μM protein. The dependence of the DOSY-derived self-diffusion coefficients on total protein concentration was fitted using Eq. 6, with X corresponding to the diffusion coefficients of the oligomer and monomer.
1H-15N correlated measurements were made using a phase-sensitive, double inept transfer with a garp decoupling sequence and solvent suppression (hsqcf3gpph19). Spectra were acquired with 1k × 144 data points and zero-filled to 4k × 4k. Steady-state heteronuclear {1H}15N-NOE was acquired at 600 MHz from the difference between spectra acquired with and without 1H saturation and a total recycle delay of 3 s. The data were processed with TopSpin 3.2 to extract peak intensities and fitted as single exponential decays.
Spectra were assigned with purified 13C/15N-labeled constructs in a standard suite of 3D experiments: HNCA, HNCACB, HN(CO)CACB, HNCO, and HN(CA)CO at 800 MHz using a 5-mm TCI cryoprobe for bound protein to DNA and at 600 MHz using a 5-mm QXI resonance probe for unbound protein. Spectra were processed using NMNRFx software, referenced to 4,4-dimethyl-4-silapentane-1-sulfonate (DSS), and peak picked/analyzed with NMRFAM-Sparky. Automated Assignments were made using the NMRFAM Pine server and verified manually.
Fluorescence-detected self-association and protein denaturation
The intrinsic fluorescence from three tryptophan residues in the PU.1 ETS domain was excited at 280 nm and detected at 340 nm with a slit of 15 nm for excitation and 20 nm for emission. Intensity data recorded in the vertical and horizontal polarizer positions were corrected for the grating factor and by blank subtraction. Concentration-dependent anisotropies were fitted to Eqs. 6 and 7, where X = 〈r〉. For denaturation studies, PU.1 at 100 μM and 1 μM in 10 mM tris-HCl buffer (pH 7.4) containing either 0.15 or 0.05 M NaCl was titrated with urea. Blank-subtracted intensity data were directly fitted with the linear extrapolation method.
Differential scanning calorimetry
Protein samples were exhaustively dialyzed against 10 mM NaH2PO4/Na2HPO4 (pH 7.4) and 0.05 M NaCl over 48 hours with at least three buffer changes. The final dialysate was reserved and used to rinse and fill the reference cell as well as diluent for the samples. Thermal scans were carried out at 45°C/hour from 10° to 80°C using a MicroCal VP-DSC instrument (Malvern). All scans were carried out only when the baseline was reproducibly flat. Thermograms were fitted to two-state transition models. Nonpolar and polar solvent-accessible surface area (SASA) for monomeric ΔN165 was estimated from the solution NMR structure 5W3G (29) based on a 1.4-Å probe. SASA for the unfolded state ensemble was provided by the ProtSA algorithm. The change in SASA in angstrom was converted to heat capacity change in kilojoule mol−1 Kelvin−1 using coefficients as follows
| (8) |
High-precision densimetry
Solution densities ρ were measured in 10 mM tris-HCl (pH 7.4) at 25°C, containing 150 mM NaCl using an Anton Paar model DMA-5000 vibrating tube densimeter with a precision of 1.5 × 10−6 g/ml. The partial molar volume of the solute V° was determined from the following relationship
| (9) |
where ρ0 is the density of the buffer, c is the molar solute concentration, and M is the molecular weight of the solute. For a two-state dimeric species, the observed density was analyzed as follows
| (10) |
where F1n is as defined by Eq. 7, with n = 2. Because the observed density varies with the concentration of any species, ρ1 and ρ2 are each treated as linear functions as described by Eq. 9.
Supplementary Material
Acknowledgments
We thank D. Beckett and W. D. Wilson for insightful discussions and L. McIntosh for providing a structure of the unbound PU.1 ETS monomer (5W3G) before publication. We also acknowledge with appreciation the many excellent suggestions from the reviewers. NMR data presented here were collected, in part, at the City University of New York Advanced Science Research Center (CUNY ASRC) Biomolecular NMR Facility. Funding: This investigation was supported by NSF grant MCB 15451600 and NIH grant R21 HL129063 to G.M.K.P. S.X., H.M.K., and S.E. were partially supported by GSU Molecular Basis of Diseases Fellowships. V.T.L.H. was supported by the GSU University Assistantship Program. Author contributions: S.L. and H.M.K. carried out cell-based studies. G.M.K.P. cloned the molecular constructs. S.X. and S.E. expressed and purified the recombinant protein constructs. S.X., S.E., J.M.A., and M.W.G. performed and analyzed the NMR experiments. S.W. performed and analyzed the data from the ESI-MS studies. V.L.T.H. performed the densimetric experiments and analyzed the volumetric data. S.X., M.K., G.L.F., and A.V.A. performed the binding, thermodynamic, and other spectroscopic experiments. S.X., M.W.G., and G.M.K.P. jointly designed the studies, analyzed data, composed the figures, and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaay3178/DC1
Table S1. DNA binding and self-association equilibrium constants of PU.1 constructs.
Table S2. Primers in RT-PCR experiments.
Fig. S1. Calibration of transgenic PU.1 dosage.
Fig. S2. Characterization of a peptide-based PU.1 inhibitor.
Fig. S3. 1H-15N HSQC-detected titration of ΔN117 and ΔN165 by cognate DNA.
Fig. S4. Purity of recombinant PU.1 constructs.
Fig. S5. Spectral analysis of far-UV CD of ΔN165 in 0.15 and 0.05 M NaCl.
Fig. S6. Salt-dependent line broadening of methyl protons in ΔN165.
Fig. S7 The short C-terminal IDR is required for DNA-free PU.1 dimerization.
Fig. S8. Effect of macromolecular crowding on dimerization of ΔN165 in the unbound and DNA-bound states.
REFERENCES AND NOTES
- 1.Liu J., Perumal N. B., Oldfield C. J., Su E. W., Uversky V. N., Dunker A. K., Intrinsic disorder in transcription factors. Biochemistry 45, 6873–6888 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh G. P., Dash D., Intrinsic disorder in yeast transcriptional regulatory network. Proteins 68, 602–605 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Minezaki Y., Homma K., Kinjo A. R., Nishikawa K., Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J. Mol. Biol. 359, 1137–1149 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Vuzman D., Azia A., Levy Y., Searching DNA via a “Monkey Bar” mechanism: The significance of disordered tails. J. Mol. Biol. 396, 674–684 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Lohrum M. A. E., Woods D. B., Ludwig R. L., Bálint É., Vousden K. H., C-terminal ubiquitination of p53 contributes to nuclear export. Mol. Cell. Biol. 21, 8521–8532 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts S. G. E., Ha I., Maldonado E., Reinberg D., Green M. R., Interaction between an acidic activator and transcription factor TFIIB is required for transcriptional activation. Nature 363, 741–744 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Friedman J. S., Khanna H., Swain P. K., DeNicola R., Cheng H., Mitton K. P., Weber C. H., Hicks D., Swaroop A., The minimal transactivation domain of the basic motif-leucine zipper transcription factor NRL interacts with TATA-binding protein. J. Biol. Chem. 279, 47233–47241 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Rogers S., Wells R., Rechsteiner M., Amino acid sequences common to rapidly degraded proteins: The PEST hypothesis. Science 234, 364–368 (1986). [DOI] [PubMed] [Google Scholar]
- 9.Chevaillier P., Pest sequences in nuclear proteins. Int. J. Biochem. 25, 479–482 (1993). [DOI] [PubMed] [Google Scholar]
- 10.Mittal A., Holehouse A. S., Cohan M. C., Pappu R. V., Sequence-to-conformation relationships of disordered regions tethered to folded domains of proteins. J. Mol. Biol. 430, 2403–2421 (2018). [DOI] [PubMed] [Google Scholar]
- 11.X. Guo, M. L. Bulyk, A. J. Hartemink, Intrinsic disorder within and flanking the DNA-binding domains of human transcription factors, in Biocomputing 2012 (World Scientific, 2011), pp. 104–115. [PMC free article] [PubMed] [Google Scholar]
- 12.Staber P. B., Zhang P., Ye M., Welner R. S., Nombela-Arrieta C., Bach C., Kerenyi M., Bartholdy B. A., Zhang H., Alberich-Jordà M., Lee S., Yang H., Ng F., Zhang J., Leddin M., Silberstein L. E., Hoefler G., Orkin S. H., Göttgens B., Rosenbauer F., Huang G., Tenen D. G., Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Mol. Cell 49, 934–946 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuki H., Ueno S., Tatetsu H., Niiro H., Iino T., Endo S., Kawano Y., Komohara Y., Takeya M., Hata H., Okada S., Watanabe T., Akashi K., Mitsuya H., Okuno Y., PU.1 is a potent tumor suppressor in classical Hodgkin lymphoma cells. Blood 121, 962–970 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Tatetsu H., Ueno S., Hata H., Yamada Y., Takeya M., Mitsuya H., Tenen D. G., Okuno Y., Down-regulation of PU.1 by methylation of distal regulatory elements and the promoter is required for myeloma cell growth. Cancer Res. 67, 5328–5336 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Rosenbauer F., Wagner K., Kutok J. L., Iwasaki H., le Beau M. M., Okuno Y., Akashi K., Fiering S., Tenen D. G., Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat. Genet. 36, 624–630 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Gjoneska E., Pfenning A. R., Mathys H., Quon G., Kundaje A., Tsai L. H., Kellis M., Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature 518, 365–369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wohlfahrt T., Rauber S., Uebe S., Luber M., Soare A., Ekici A., Weber S., Matei A. E., Chen C. W., Maier C., Karouzakis E., Kiener H. P., Pachera E., Dees C., Beyer C., Daniel C., Gelse K., Kremer A. E., Naschberger E., Stürzl M., Butter F., Sticherling M., Finotto S., Kreuter A., Kaplan M. H., Jüngel A., Gay S., Nutt S. L., Boykin D. W., Poon G. M. K., Distler O., Schett G., Distler J. H. W., Ramming A., PU.1 controls fibroblast polarization and tissue fibrosis. Nature 566, 344–349 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antony-Debre I., Paul A., Leite J., Mitchell K., Kim H. M., Carvajal L. A., Todorova T. I., Huang K., Kumar A., Farahat A. A., Bartholdy B., Narayanagari S. R., Chen J., Ambesi-Impiombato A., Ferrando A. A., Mantzaris I., Gavathiotis E., Verma A., Will B., Boykin D. W., Wilson W. D., Poon G. M., Steidl U., Pharmacological inhibition of the transcription factor PU.1 in leukemia.1 in leukemia. J. Clin. Invest. 127, 4297–4313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durual S., Rideau A., Ruault-Jungblut S., Cossali D., Beris P., Piguet V., Matthes T., Lentiviral PU.1 overexpression restores differentiation in myeloid leukemic blasts. Leukemia 21, 1050–1059 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Esaki S., Evich M. G., Erlitzki N., Germann M. W., Poon G. M. K., Multiple DNA-binding modes for the ETS family transcription factor PU.1. J. Biol. Chem. 292, 16044–16054 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poon G. M. K., DNA binding regulates the self-association of the ETS domain of PU.1 in a sequence-dependent manner. Biochemistry 51, 4096–4107 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Kueh H. Y., Champhekar A., Nutt S. L., Elowitz M. B., Rothenberg E. V., Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science 341, 670–673 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H., Ray-Gallet D., Zhang P., Hetherington C. J., Gonzalez D. A., Zhang D. E., Moreau-Gachelin F., Tenen D. G., PU.1 (Spi-1) autoregulates its expression in myeloid cells. Oncogene 11, 1549–1560 (1995). [PubMed] [Google Scholar]
- 24.Solomon L. A., Podder S., He J., Jackson-Chornenki N. L., Gibson K., Ziliotto R. G., Rhee J., De Koter R. P., Coordination of myeloid differentiation with reduced cell cycle progression by PU.1 induction of MicroRNAs targeting cell cycle regulators and lipid anabolism. Mol. Cell. Biol. 37, e00013-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia X., Lee L. K., Light J., Palmer A. G. III, Assa-Munt N., Backbone dynamics of a short PU.1 ETS domain. J. Mol. Biol. 292, 1083–1093 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Poon G. M. K., Enhancement of oligomeric stability by covalent linkage and its application to the human p53tet domain: Thermodynamics and biological implications. Biochem. Soc. Trans. 35, 1574–1578 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Ferrige A. G., Seddon M. J., Green B. N., Jarvis S. A., Skilling J., Staunton J., Disentangling electrospray spectra with maximum-entropy. Rapid Commun. Mass Spectrom. 6, 707–711 (1992). [Google Scholar]
- 28.Berjanskii M. V., Wishart D. S., Application of the random coil index to studying protein flexibility. J. Biomol. NMR 40, 31–48 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Perez-Borrajero C., Lin C. S.-H., Okon M., Scheu K., Graves B. J., Murphy M. E. P., McIntosh L. P., The biophysical basis for phosphorylation-enhanced DNA-binding autoinhibition of the ETS1 transcription factor. J. Mol. Biol. 431, 593–614 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Huang W., Horvath E., Eklund E. A., PU.1, interferon regulatory factor (IRF) 2, and the nterferon consensus sequence-binding protein (ICSBP/IRF8) cooperate to activate NF1 transcription in differentiating myeloid cells. J. Biol. Chem. 282, 6629–6643 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Pongubala J. M., van Beveren C., Nagulapalli S., Klemsz M. J., McKercher S., Maki R. A., Atchison M. L., Effect of PU.1 phosphorylation on interaction with NF-EM5 and transcriptional activation. Science 259, 1622–1625 (1993). [DOI] [PubMed] [Google Scholar]
- 32.Warren L., Bryder D., Weissman I. L., Quake S. R., Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proc. Natl. Acad. Sci. U.S.A. 103, 17807–17812 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furfine C. S., Velick S. F., The acyl-enzyme intermediate and the kinetic mechanism of the glyceraldehyde 3-phosphate dehydrogenase reaction. J. Biol. Chem. 240, 844–855 (1965). [PubMed] [Google Scholar]
- 34.Mueller B. U., Pabst T., Fos J., Petkovic V., Fey M. F., Asou N., Buergi U., Tenen D. G., ATRA resolves the differentiation block in t(15;17) acute myeloid leukemia by restoring PU.1 expression. Blood 107, 3330–3338 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pedchenko T. V., Park G. Y., Joo M., Blackwell T. S., Christman J. W., Inducible binding of PU.1 and interacting proteins to the Toll-like receptor 4 promoter during endotoxemia. Am. J. Phys. Lung Cell. Mol. Phys. 289, L429–L437 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Will B., Vogler T. O., Narayanagari S., Bartholdy B., Todorova T. I., da Silva Ferreira M., Chen J., Yu Y., Mayer J., Barreyro L., Carvajal L., Neriah D. B., Roth M., van Oers J., Schaetzlein S., McMahon C., Edelmann W., Verma A., Steidl U., Minimal PU.1 reduction induces a preleukemic state and promotes development of acute myeloid leukemia. Nat. Med. 21, 1172–1181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lodie T. A., Savedra R. Jr., Golenbock D. T., Van Beveren C. P., Maki R. A., Fenton M. J., Stimulation of macrophages by lipopolysaccharide alters the phosphorylation state, conformation, and function of PU.1 via activation of casein kinase II. J. Immunol. 158, 1848–1856 (1997). [PubMed] [Google Scholar]
- 38.Hamdorf M., Berger A., Schule S., Reinhardt J., Flory E., PKC δ induced PU.1 phosphorylation promotes hematopoietic stem cell differentiation to dendritic cells. Stem Cells 29, 297–306 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Seshire A., Rößiger T., Frech M., Beez S., Hagemeyer H., Puccetti E., Direct interaction of PU.1 with oncogenic transcription factors reduces its serine phosphorylation and promoter binding. Leukemia 26, 1338–1347 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Arinobu Y., Mizuno S.-i., Chong Y., Shigematsu H., Iino T., Iwasaki H., Graf T., Mayfield R., Chan S., Kastner P., Akashi K., Reciprocal activation of GATA-1 and PU.1 marks initial specification of hematopoietic stem cells into myeloerythroid and myelolymphoid lineages. Cell Stem Cell 1, 416–427 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Liew C. W., Rand K. D., Simpson R. J. Y., Yung W. W., Mansfield R. E., Crossley M., Proetorius-Ibba M., Nerlov C., Poulsen F. M., Mackay J. P., Molecular analysis of the interaction between the hematopoietic master transcription factors GATA-1 and PU.1. J. Biol. Chem. 281, 28296–28306 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Fuxreiter M., Fuzziness in protein interactions—A historical perspective. J. Mol. Biol. 430, 2278–2287 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Shoemaker B. A., Portman J. J., Wolynes P. G., Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. U.S.A. 97, 8868–8873 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perkel J. M., Atchison M. L., A two-step mechanism for recruitment of Pip by PU.1. J. Immunol. 160, 241–252 (1998). [PubMed] [Google Scholar]
- 45.Currie S. L., Lau D. K. W., Doane J. J., Whitby F. G., Okon M., McIntosh L. P., Graves B. J., Structured and disordered regions cooperatively mediate DNA-binding autoinhibition of ETS factors ETV1, ETV4 and ETV5. Nucleic Acids Res. 45, 2223–2241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lamber E. P., Vanhille L., Textor L. C., Kachalova G. S., Sieweke M. H., Wilmanns M., Regulation of the transcription factor Ets-1 by DNA-mediated homo-dimerization. EMBO J. 27, 2006–2017 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pufall M. A., Lee G. M., Nelson M. L., Kang H.-S., Velyvis A., Kay L. E., Mc Intosh L. P., Graves B. J., Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science 309, 142–145 (2005). [DOI] [PubMed] [Google Scholar]
- 48.King D. A., Zhang L., Guarente L., Marmorstein R., Structure of a HAP1-DNA complex reveals dramatically asymmetric DNA binding by a homodimeric protein. Nat. Struct. Biol. 6, 64–71 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Kwok J. C., Perdomo J., Chong B. H., Identification of a monopartite sequence in PU.1 essential for nuclear import, DNA-binding and transcription of myeloid-specific genes. J. Cell. Biochem. 101, 1456–1474 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaay3178/DC1
Table S1. DNA binding and self-association equilibrium constants of PU.1 constructs.
Table S2. Primers in RT-PCR experiments.
Fig. S1. Calibration of transgenic PU.1 dosage.
Fig. S2. Characterization of a peptide-based PU.1 inhibitor.
Fig. S3. 1H-15N HSQC-detected titration of ΔN117 and ΔN165 by cognate DNA.
Fig. S4. Purity of recombinant PU.1 constructs.
Fig. S5. Spectral analysis of far-UV CD of ΔN165 in 0.15 and 0.05 M NaCl.
Fig. S6. Salt-dependent line broadening of methyl protons in ΔN165.
Fig. S7 The short C-terminal IDR is required for DNA-free PU.1 dimerization.
Fig. S8. Effect of macromolecular crowding on dimerization of ΔN165 in the unbound and DNA-bound states.