

Abstract
Comparative algal genomics often relies on predicted genes from de novo assembled genomes. However, the artifacts introduced by different gene‐prediction approaches, and their impact on comparative genomic analysis remain poorly understood. Here, using available genome data from six dinoflagellate species in the Symbiodiniaceae, we identified methodological biases in the published genes that were predicted using different approaches and putative contaminant sequences in the published genome assemblies. We developed and applied a comprehensive customized workflow to predict genes from these genomes. The observed variation among predicted genes resulting from our workflow agreed with current understanding of phylogenetic relationships among these taxa, whereas the variation among the previously published genes was largely biased by the distinct approaches used in each instance. Importantly, these biases affect the inference of homologous gene families and synteny among genomes, thus impacting biological interpretation of these data. Our results demonstrate that a consistent gene‐prediction approach is critical for comparative analysis of dinoflagellate genomes.
We implemented a customized, comprehensive workflow (Appendix S1 and Fig. S1 in the Supporting Information) to predict protein‐coding genes in six published draft Symbiodiniaceae genomes: Breviolum minutum (Shoguchi et al. 2013), Symbiodinium tridacnidorum, Cladocopium C92 (Shoguchi et al. 2018), Symbiodinium microadriaticum (Aranda et al. 2016), Cladocopium goreaui and Fugacium kawagutii (Liu et al. 2018). These draft genome assemblies, generated largely using short‐read sequence data, remain fragmented (e.g., N50 lengths range from 98.0 Kb for C. goreaui to 573.5 Kb for S. microadriaticum); we treated these genome assemblies independently as is standard practice. The published genes from these four studies were predicted using three different approaches: (a) ab initio using AUGUSTUS (Stanke et al. 2006) guided by transcriptome data (Shoguchi et al. 2013, 2018); (b) ab initio using AUGUSTUS guided by a more‐stringent selection of genes (Aranda et al. 2016); and (c) a more‐thorough approach incorporating evidence from transcriptomes, machine learning tools, homology to known sequences, and ab initio methods (Liu et al. 2018). Because repetitive regions are commonly removed prior to gene prediction, multi‐copy genes are sometimes misidentified as repeats and excluded from the final predictions (see Appendix S1). To address this issue, we adapted the workflow from Liu et al. (2018) to ignore inferred repeats in the final step that integrates multiple evidence sources using EVidenceModeler (Haas et al. 2008). To minimize the potential contaminants in the published draft genomes and their impact on gene prediction, we adopted a robust, two‐phase strategy for identifying contaminant sequences among the assembled genome scaffolds. This was based on shared similarity to known genome sequences from bacterial, archaeal, and viral sources, and irregular G+C content among the assembled scaffolds (Fig. S2 in the Supporting Information); see Appendix S1 and Figure S1 for detail of this overall workflow. We then compared, for each genome, the published genes in the remaining scaffolds against the predicted genes in these same scaffolds using our approach. Specifically, we assessed metrics of predicted genes and the inference of homologous gene families and conserved synteny within a phylogenetic context.
For simplicity, hereinafter, we refer to the published genes as α genes and those predicted in this study as β genes. Compared to α genes, the structure of β genes (based on the distribution of intron lengths) resembles more closely the structure of dinoflagellate genes inferred using transcriptome data (Fig. S3 in the Supporting Information). These results suggest that β genes are likely more biologically realistic. Variation between α and β genes was assessed using 10 metrics: number of predicted genes per genome; average gene length; number of exons per genome; average exon length; number of introns per genome; average intron length; proportion of GT splice‐donor site, proportion of GC splice‐donor site, number of intergenic regions; and average length of intergenic regions.
As shown in Table S1 in the Supporting Information, the metrics for α and β genes differed substantially. The number of α genes per genome was much higher in Breviolum minutum, Cladocopium C92, Symbiodinium microadriaticum, and S. tridacnidorum, and showed greater variation (mean = 46,698; minimum = 25,109; maximum = 69,018) than that of β genes (mean = 32,048; minimum = 25,808; maximum = 39,006). This is likely due to the more‐stringent criteria used by our workflow to delineate protein‐coding genes. The larger variation in the number of α genes is likely due to the biases arising from the distinct prediction methods and not from assembly artifacts, because the same genome assembly for each species was used to independently derive α and β genes. Most genes in dinoflagellates are constitutively expressed irrespective of growth conditions (Moustafa et al. 2010, Liew et al. 2017), thus transcriptome support for the predicted genes provides a reasonable overview of true positives (i.e., that the predicted genes were transcribed). As shown in Table S1, most predicted genes (>60% of genes in each genome) analyzed in this study were supported by transcriptome evidence (BLASTn, E ≤ 10−10). In general, β genes are better supported than are the α genes.
Variation in the ten observed metrics among α and β genes was also assessed using PCA (Fig. 1a). The α genes spread greater than the β genes along principal component 1 (PC1, between −0.54 and 0.46), with those based on AUGUSTUS‐predominant workflows distinctly separated (PC1 < −0.10; Fig. 1a). The β genes are distributed more narrowly on PC1 (between 0 and 0.28) and more widely along principal component 2 (PC2; between −0.56 and 0.20). Interestingly, the distribution of genes along PC2 exhibits a pattern that is consistent with our current understanding of the phylogeny of these six species (Fig. 1b). Specifically, the Symbiodinium species are clearly separated from the others along PC2 (Fig. 1a) and the two Cladocopium species are clustered more closely based on β, rather than α genes. Therefore, PC1 (explaining 51.46% of the variance) largely reflects the variation introduced by distinct gene‐prediction methods, whereas the distribution along PC2 (explaining 25.91% of the variance) is likely attributable to the phylogeny of these species. This result suggests that variation among α genes is predominantly due to methodological biases, and that these biases are larger compared to those of β genes. Variation in the latter appears to be more biologically relevant and consistent with Symbiodiniaceae evolution. This observation suggests that using a consistent gene‐prediction approach allows the associated metrics of the predicted genes to be used to assess the biological relatedness of these genomes.
Figure 1.
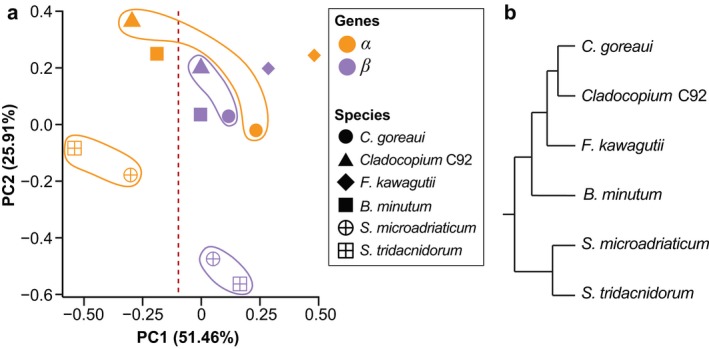
Variation among α and β genes from six Symbiodiniaceae genomes. (a) PCA plot based on ten metrics of the predicted genes, shown for the α genes in orange, and the β genes in purple, for each of the six genomes (noted in different symbols) as indicated in the legend. The two Cladocopium and the two Symbiodinium species were highlighted for clarity. (b) Tree topology depicting the phylogenetic relationship among the six taxa, based on LaJeunesse et al. (2018). [Color figure can be viewed at http://wileyonlinelibrary.com]
Genomes that are phylogenetically closely related are expected to share greater synteny than those that are more distantly related. We followed Liu et al. (2018) to define a collinear syntenic gene block as a region common to two genomes in which five or more genes are coded in the same order and orientation. These gene blocks were identified using SynChro (Drillon et al. 2014) at Delta = 4. Overall, 298 collinear syntenic blocks (implicating 1721 genes) between any genome‐pairs were identified among α genes, compared to 429 blocks (implicating 2509 genes) among β genes (Fig. 2, a and b). Based on the α genes comparison (Fig. 2a), Symbiodinium microadriaticum and S. tridacnidorum shared the largest number of syntenic blocks (98 and 582 genes, respectively), whereas S. microadriaticum and Fugacium kawagutii shared the fewest (1 and 6 genes, respectively). Surprisingly, S. tridacnidorum and Cladocopium C92 shared 16 blocks (98 genes). This close relationship is not evident between any other pair of genomes from these two genera (e.g., only 3 blocks implicating 15 genes between S. microadriaticum and Cladocopium goreaui). This observation may be explained by the fact that α genes from these two genomes were predicted using the same method (Shoguchi et al. 2018). In contrast, based on the β genes comparison (Fig. 2b), the number of syntenic blocks shared between any Symbiodinium and Cladocopium species did not vary to the same extent (e.g., 7 blocks [38 genes] between S. tridacnidorum and Cladocopium C92, and 10 blocks [55 genes] between S. microadriaticum and C. goreaui). The higher‐than‐expected number of collinear syntenic blocks of α genes between S. tridacnidorum and Cladocopium C92 can be explained by the distinct gene structure (i.e., exon configurations) of these genes relative to that of β genes (Fig. S4 in the Supporting Information).
Figure 2.
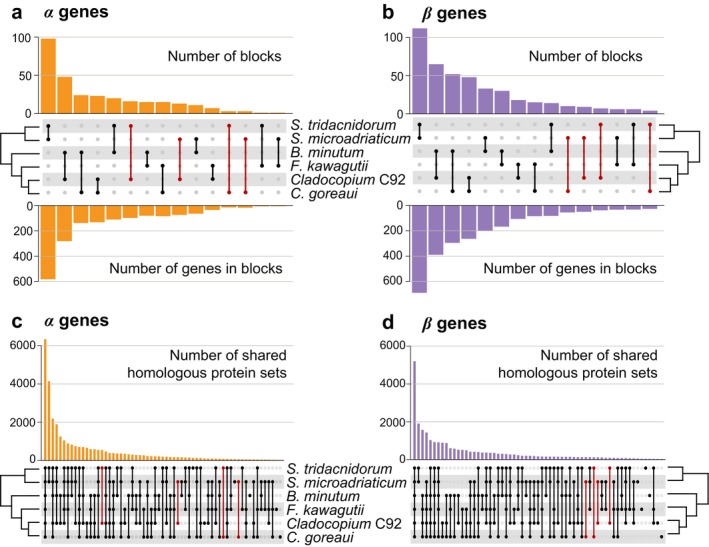
Conserved synteny and homologous sets among six Symbiodiniaceae genomes. The number of collinear syntenic gene blocks between each genome‐pair is shown for those inferred based on (a) α and (b) β genes; the upper bar chart shows the number of blocks, the lower bar chart shows the number of implicated genes in these blocks, and the middle panel shows the genome‐pairs corresponding to each bar with a line joining the dots that represent the implicated taxa. The number of homologous sets inferred from (c) α and (d) β genes is shown, in which the taxa represented in the set corresponding to each bar are indicated in the bottom panel. The most remarkable differences between (a) and (b), and (c) and (d), focusing on Symbiodinium and Cladocopium species, are highlighted in red. [Color figure can be viewed at http://wileyonlinelibrary.com]
To assess the impact of methodological biases on the delineation of homologous gene families, Orthofinder v2.3.1 (Emms and Kelly 2018) was used to infer “orthogroups” from protein sequences (i.e., homologous protein sets) encoded by the α and β genes (Fig. 2, c and d). More homologous sets were inferred among the α genes (31,426) than among the β genes (25,933), likely due to the higher number of total α genes. Genomes from closely related taxa are expected to share more homologous sequences (and therefore more sets) than those that are phylogenetically distant. Most of the identified homologous sets (6,337 from α genes; 5,201 from β genes) contained sequences from all analyzed taxa; these represent core gene families of Symbiodiniaceae. Similar to the results of the synteny analysis described above, the pattern of homologous sets shared between members from Symbiodinium and Cladocopium varies among the α genes (Fig. 2c). For instance, 549 homologous sets are shared only between S. tridacnidorum and Cladocopium C92, compared to 62 between C. goreaui and S. microadriaticum. In contrast, the corresponding number of homologous sets inferred based on β genes is closer to each other (Fig. 2d) that is, 93 between S. tridacnidorum and Cladocopium C92, and 119 between C. goreaui and S. microadriaticum.
Our results indicate that comparative genomics using the α genes (i.e., based on published genes) could lead to the inference that Symbiodinium tridacnidorum and Cladocopium C92 are more closely related to each other than is each of them with other isolates in their corresponding genus. In addition to the quality of genome assembly, the biases introduced by different gene‐prediction approaches can significantly impact the downstream comparative genomic analyses and affect subsequent biological interpretations. The impact of these biases intensified when comparing de novo assembled genomes of dinoflagellates, because genome sequences among closely related taxa (e.g., the symbiodiniacean taxa studied here) are known to be highly dissimilar (Lin et al. 2015, Liu et al. 2018, Stephens et al. 2019), and little reference data are available. In this situation, we urge the research community to consider a consistent gene‐prediction workflow when pursuing comparative genomics.
Competing interests
The authors declare no competing interests.
Author contribution
YC, RAGP, and CXC conceived the study and designed the experiments. YC conducted all computational analyses. All authors analyzed and interpreted the results. YC and RAGP prepared all figures, tables, and the first draft of this manuscript. YC, TGS, and RAGP provided analytical tools and scripts. All authors wrote, reviewed, commented on and approved the final manuscript.
Supporting information
Figure S1. Overview of our workflow for gene prediction in dinoflagellate genomes, including the two‐phase strategy for removing putative contaminant sequences from assembled genomes.
Figure S2. Distribution of G+C percentage of assembled genome scaffolds relative to scaffold length, shown for each of the six Symbiodiniaceae genomes analyzed in this study. Data points representing scaffolds that were identified as putative contaminants are highlighted in red.
Figure S3. Distribution of intron lengths in predicted genes from the six Symbiodiniaceae genomes. In each graph, the distribution of intron lengths among α genes (orange line), among β genes (purple line), and among transcript‐based genes (predicted using PASA and TransDecoder; red dashed line) are shown. The transcript‐based genes (see Appendix S1) were considered as a proxy for true gene structure.
Figure S4. Structural differences between α and β genes. (a) Four scenarios into which the structural differences are broadly categorized. (b) Among collinear syntenic blocks shared by Symbiodinium tridacnidorum and Cladocopium C92, the number of implicated α genes that fall into each of the four scenarios (relative to the corresponding β genes) is shown.
Table S1. Metrics of predicted genes in genomes of Symbiodiniaceae analyzed in this study.
Appendix S1. Overall customized approach used in this study for identifying putative contaminant sequences and predicting genes from dinoflagellate genomes.
Acknowledgements
RAGP is supported by an International Postgraduate Research Scholarship and a University of Queensland Centenary Scholarship. TGS was supported by an Australian Government Research Training Program Scholarship. This work was supported by two Australian Research Council grants (DP150101875 awarded to Mark Ragan, CXC and DB, and DP190102474 awarded to CXC and DB), and the computational resources of the Australian National Computational Infrastructure (NCI) Facility through the NCI Merit Allocation Scheme (project d85) awarded to CXC.
Data accessibility
Our customized gene‐prediction workflow for dinoflagellate genomes is available at https://github.com/TimothyStephens/Dinoflagellate_Annotation_Workflow. All genome data (after removal of microbial contaminants), the revised annotations, and all predicted gene and protein sequences from this study are available at https://doi.org/10.14264/uql.2019.745.
References
- Aranda, M. , Li, Y. , Liew, Y. J. , Baumgarten, S. , Simakov, O. , Wilson, M. C. , Piel, J. et al. 2016. Genomes of coral dinoflagellate symbionts highlight evolutionary adaptations conducive to a symbiotic lifestyle. Sci. Rep. 6:39734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drillon, G. , Carbone, A. & Fischer, G. 2014. SynChro: a fast and easy tool to reconstruct and visualize synteny blocks along eukaryotic chromosomes. PLoS ONE 9:e92621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D. M. & Kelly, S. 2018. OrthoFinder2: fast and accurate phylogenomic orthology analysis from gene sequences. bioRxiv: 466201. [Google Scholar]
- Haas, B. J. , Salzberg, S. L. , Zhu, W. , Pertea, M. , Allen, J. E. , Orvis, J. , White, O. , Buell, C. R. & Wortman, J. R. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaJeunesse, T. C. , Parkinson, J. E. , Gabrielson, P. W. , Jeong, H. J. , Reimer, J. D. , Voolstra, C. R. & Santos, S. R. 2018. Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr. Biol. 28:2570–80. [DOI] [PubMed] [Google Scholar]
- Liew, Y. J. , Li, Y. , Baumgarten, S. , Voolstra, C. R. & Aranda, M. 2017. Condition‐specific RNA editing in the coral symbiont Symbiodinium microadriaticum . PLoS Genet. 13:e1006619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S. , Cheng, S. , Song, B. , Zhong, X. , Lin, X. , Li, W. , Li, L. et al. 2015. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 350:691–4. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Stephens, T. G. , González‐Pech, R. A. , Beltran, V. H. , Lapeyre, B. , Bongaerts, P. , Cooke, I. et al. 2018. Symbiodinium genomes reveal adaptive evolution of functions related to coral‐dinoflagellate symbiosis. Commun. Biol. 1:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustafa, A. , Evans, A. N. , Kulis, D. M. , Hackett, J. D. , Erdner, D. L. , Anderson, D. M. & Bhattacharya, D. 2010. Transcriptome profiling of a toxic dinoflagellate reveals a gene‐rich protist and a potential impact on gene expression due to bacterial presence. PLoS ONE 5:e9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoguchi, E. , Beedessee, G. , Tada, I. , Hisata, K. , Kawashima, T. , Takeuchi, T. , Arakaki, N. et al. 2018. Two divergent Symbiodinium genomes reveal conservation of a gene cluster for sunscreen biosynthesis and recently lost genes. BMC Genomics 19:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoguchi, E. , Shinzato, C. , Kawashima, T. , Gyoja, F. , Mungpakdee, S. , Koyanagi, R. , Takeuchi, T. et al. 2013. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 23:1399–408. [DOI] [PubMed] [Google Scholar]
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. & Morgenstern, B. 2006. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34:W435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, T. G. , González‐Pech, R. A. , Cheng, Y. , Mohamed, A. R. , Bhattacharya, D. , Ragan, M. A. & Chan, C. X. 2019. Polarella glacialis genomes encode tandem repeats of single‐exon genes with functions critical to adaptation of dinoflagellates. bioRxiv:704437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Overview of our workflow for gene prediction in dinoflagellate genomes, including the two‐phase strategy for removing putative contaminant sequences from assembled genomes.
Figure S2. Distribution of G+C percentage of assembled genome scaffolds relative to scaffold length, shown for each of the six Symbiodiniaceae genomes analyzed in this study. Data points representing scaffolds that were identified as putative contaminants are highlighted in red.
Figure S3. Distribution of intron lengths in predicted genes from the six Symbiodiniaceae genomes. In each graph, the distribution of intron lengths among α genes (orange line), among β genes (purple line), and among transcript‐based genes (predicted using PASA and TransDecoder; red dashed line) are shown. The transcript‐based genes (see Appendix S1) were considered as a proxy for true gene structure.
Figure S4. Structural differences between α and β genes. (a) Four scenarios into which the structural differences are broadly categorized. (b) Among collinear syntenic blocks shared by Symbiodinium tridacnidorum and Cladocopium C92, the number of implicated α genes that fall into each of the four scenarios (relative to the corresponding β genes) is shown.
Table S1. Metrics of predicted genes in genomes of Symbiodiniaceae analyzed in this study.
Appendix S1. Overall customized approach used in this study for identifying putative contaminant sequences and predicting genes from dinoflagellate genomes.
Data Availability Statement
Our customized gene‐prediction workflow for dinoflagellate genomes is available at https://github.com/TimothyStephens/Dinoflagellate_Annotation_Workflow. All genome data (after removal of microbial contaminants), the revised annotations, and all predicted gene and protein sequences from this study are available at https://doi.org/10.14264/uql.2019.745.