

Abstract
High fidelity human mitochondrial DNA polymerase (Pol γ) contains two active sites, a DNA polymerization site (pol) and a 3′−5′ exonuclease site (exo) for proofreading. Although separated by 35 Å, coordination between the pol and exo sites is crucial to high fidelity replication. The biophysical mechanisms for this coordination are not completely understood. To understand the communication between the two active sites, we used a statistical-mechanical model of the protein ensemble to calculate the energetic landscape and local stability. We compared a series of structures of Pol γ, complexed with primer/template DNA, and either a nucleotide substrate or a series of nucleotide analogues, which are differentially incorporated and excised by pol and exo activity. Despite the nucleotide or its analogues being bound in the pol, Pol γ residue stability varied across the protein, particularly in the exo domain. This suggests that substrate presence in the pol can be “sensed” in the exo domain. Consistent with this hypothesis, in silico mutations made in one active site mutually perturbed the energetics of the other. To identify specific regions of the polymerase that contributed to this communication, we constructed an allosteric network connectivity map that further demonstrates specific pol–exo cooperativity. Thus, a cooperative network underlies energetic connectivity. We propose that Pol γ and other dual-function polymerases exploit an energetic coupling network that facilitates domain–domain communication to enhance discrimination between correct and incorrect nucleotides.
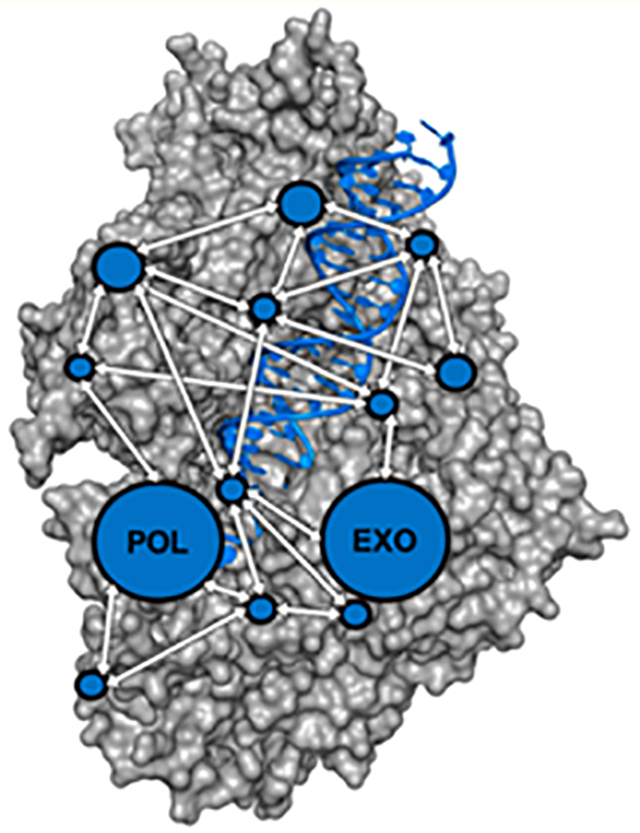
Graphical Abstract
INTRODUCTION
Human mitochondrial DNA polymerase (Pol γ) is responsible for replicating mitochondrial DNA and is vital for organelle function. Pol γ is a heterotrimeric holoenzyme that consists of a catalytic subunit, Pol γA, and a dimeric accessory subunit, Pol γB. Pol γ is a high fidelity polymerase, and all enzymatic activity is fulfilled by Pol γA including polymerization (pol), proofreading (3′−5′ exonuclease, exo), and 5′-deoxyribose phosphate lyase activities for base excision repair. Although having no enzymatic activity itself, Pol γB regulates all Pol γA activities.1–3
The pol domain adopts a canonical right-hand configuration similar to other Pol I Family members, with subdomains called palm, fingers, and thumb. The palm houses the pol catalytic active site, and the fingers are responsible for binding to DNA and incoming deoxynucleotide triphosphates (dNTPs). The thumb likely plays an important role in directing the primer/template to either pol or exo active sites that are located on two ends of the molecule.4 Coordination of pol and exo activity contributes substantially to replication fidelity of Pol γ.
Mutations in the POLG gene are one of the most common causes of inherited mitochondrial disease in several major phenotypes of neurodegenerative disease that include childhood myocerebrohepatopathy spectrum disorders, Alpers syndrome, ataxia neuropathy spectrum disorders, myoclonus epilepsy myopathy sensory ataxia, and progressive external ophthalmoplegia.5,6 To date, more than 170 Pol γ mutations have been associated with disease symptoms.7 This is believed to be due to impaired Pol γ function leading to deficits in mitochondrial replication and repair. Ultimately, deficits disrupt mitochondrial function, which is essential in neurons for energy production.8 Crystal structures of Pol γ and its ternary complex structures provide considerable information to rationalize many mutations; nonetheless, certain mutations are inexplicable. While structures may explain the local effects of mutations, they cannot explain how mutations in pol or exo active sites, separated by 35 Å, can mutually affect each other’s activity. For example, mutations in the finger subdomain can reduce Pol γ DNA synthesis efficiency and elevate exonuclease activity, and mutations in the exo simultaneously reduce exonuclease and polymerase activities.9,10 In addition, one of the most common mutations A467T, distal (~40 Å) to either active site, inhibits pol as well as exo activity.11 The phenotypes of these mutants indicate that the two active sites are functionally connected and may be allosterically regulated. However, the structural and molecular basis for such long-range connectivity is unknown.
Pol γ is a major off-target for nucleoside analogue reverse transcriptase inhibitors (NRTIs) designed to inhibit pathogenic human virus HIV, which contributes to their toxicity.12 NRTIs are prodrugs that must be enzymatically converted to a triphosphate form intracellularly and incorporated by a polymerase into the 3′-end of a growing DNA primer. The pol and exo active sites of Pol γ recognize NRTIs differently.13 The catalytic efficiency (kpol/Kd) of cytosine-based NRTIs Zalcitabine (ddC), Lamivudine (3TC), and Emtricitabine (FTC) (Figure 1) varies over 5 orders of magnitude relative to the natural substrate deoxycytidine triphosphate (dCTP), dCTP ≫ (+)-FTCTP > ddCTP > (+)-3TCTP > (–)-3TCTP ≫ (–)-FTCTP, but the excision rates from primer-DNA by the exo site are in a different order, dC > (+)-3TC ≫ (–)-FTC ≫ ddC.14,15
Figure 1.

Structures of natural substrate deoxycytidine and nucleoside reverse transcriptase inhibitors (NRTI).
The precise mechanism by which incorrect substrates are differentially recognized is not completely known. The pol and exo sites are separated by 35 Å, suggesting that communication must be mediated either by a “path” connecting the two active sites or by a large conformational change. Revealing such a connecting path is not only important for understanding the fidelity of the DNA polymerase but also important for developing low toxicity antiviral polymerase inhibitors. The development of analogues that are recognized by HIV reverse transcriptases but rejected by Pol γ, either by pol or by exo, would be a breakthrough that increases the efficacy and/or decreases the toxicity of NRTI therapy.
We have determined several crystal structures of Pol γ, but they have not yet provided a mechanism for how the two active sites communicate and how that may impact polymerase fidelity. We report here computational studies that detect energetic connectivity using a semiempirical statistical mechanical method encoded in the COREX algorithm. We compared a group of Pol γ ternary complex crystal structures with primer/template DNA, and either a substrate dNTP or a NRTI (Figure 1).4,16 Our study identifies potential two-way communication between the pol and exo domains. As seen in other allosteric proteins, site–site coupling does not necessarily involve a direct pathway, a series of discrete deformations in contiguous amino acids, but rather occurs through a more diffuse cooperative network that involves all of the subdomains known to be relevant to catalytic activity. Our computational approach detected long-range intramolecular connectivity that cannot be directly revealed by crystal structures alone.
METHODS
COREX Calculations
C source code of COREX was obtained from Professor Vincent Hilser (Johns Hopkins University). COREX calculations were performed on crystal structures of Pol γ holoenzyme ternary complexes with either a nucleotide or an HIV reverse transcriptase inhibitor and a 24/28 nt primer/template DNA 5′-d d CATACCGTGACCGGGAGCAAAAGC-3′ and 5′-GCTTTTGCTCCCGGTCACGGTATGGAGC-3′ (Table S1). Structures were prepared by removing nonprotein atoms as well as the accessory subunit, Pol γB. To prevent overestimating contributions of residues with high flexibility (B-factor > 100), we eliminated the side chains of these residues and only use their backbone atoms for calculations. These residues are E481, D482, F610, Y649, V742, S759, and C760. In silico mutants were constructed in COOT.17 The rotamer configuration for each substitution was selected such that there was no steric clash. Thermostable Bacillus DNA Polymerase I large fragment (Pol I BF) structures were prepared similarly.
Briefly, COREX generates a conformational ensemble from a given three-dimensional structure. This is accomplished by systematically unfolding small “windows” of residues in the fully folded structure. The unfolding of that set of residues exhibits a change in solvent accessible surface area (SASA), composed of the area gained from unfolding the residues plus any newly exposed interfacial surface.18 This interfacial surface is key to the algorithm’s ability to model distant cooperative unfolding events that might be involved in long-distance communication. The free energy (ΔGi) of each conformational permutation i, or microstate, is then calculated on the basis of previously determined empirical equations that link changes in SASA to thermodynamic parameters.18 Probabilities (Pi) of any given microstate are then determined using the partition function Q, which is the sum of all microstate energies (eq 1):
| (1) |
where R is the universal gas constant and T is temperature in kelvin. The primary output used is a residue stability or folding constant, Kf,j, defined as the ratio of the summed probability in which a particular residue j is folded (∑Pfj) over all microstates divided by the summed probability of all states in which that residue is unfolded (∑Pufj) as shown in eq 2:
| (2) |
This value is an estimated equilibrium constant for unfolding at every residue position, which can be converted to a free energy of stability by the relation ΔG = –RT ln Kf,j. As such, these calculations have been verified by direct comparison to hydrogen exchange protection factors.19
The Monte Carlo sampling version of COREX was used, and the source was further modified to allow for the large size of Pol γA by modifying the following variables: Max_Res to 6000, Max_Atoms to 50000, and Max_Nunits to 1000. Specific parameters used for the calculation include a window size of 50 residues, a minimum window size of 40 residues, and 5000 samples per partition. The partition refers to the “sliding window” approach used in COREX to generate microstates. The first partition is the first 50 residues from the N-terminus of Pol γA, and each following 50 residues is another window, for a total of 24 windows that can be either “folded” or “unfolded”. The windows then were incremented by one residue, and another 24 windows were generated. The sliding was iterated until the first window on the N-terminus was moved to the 50th residue, generating a total of 50 partitions. We sampled 5000 microstates in each partition for a total of 250 000 microstates in the simulated ensemble. To test the sufficiency of sampling, crystal structures were also run with the same parameters but a larger sampling size of 50 000. Residues 505–539 were under-sampled, and no values were calculated, due to their highly solvent accessible surface area. Pol I binary complexes were run similarly with 1000 samples per partition.
Residue Stability Variance
Differences between the different ternary complexes or in silico mutations were obtained by computing the standard deviation of residue stability (Kf,j) between the same residue across all structures, for all residues in Pol γA.
Allosteric Connectivity Map
To construct the allosteric network connectivity map using the single site thermodynamic mutation algorithm,22 the ensemble data generated as described above were used. A 1 kcal/mol stabilizing energy, Φ, then was applied to each microstate in which a given residue, k, was found folded. Then we estimated how perturbing residue k affected the residue stability of the jth residue in the protein, Kf,j, as encompassed in eq 3.
| (3) |
RESULTS
Crystal Structures
3-D structures of catalytic subunit Pol γA were taken from holoenzyme ternary complexes containing an identical 24/28 nucleotide primer/template DNA with a 3′-dideoxy-terminated primer to prevent elongation, and a dNTP or an analogue: dCTP, or a triphosphate form of cytosine-based NRTI, Zalcitabine (ddC), Emtricitabine (–)-FTC, or the chiral enantiomer (+)-FTC (Table S1). Pol γA in the crystal structures is an exonuclease-deficient double mutant (D198A/E200A).
COREX Analysis of Pol γ Complexes Containing dCTP or NRTIs
To investigate energetic fluctuations as a possible cause for the different kinetics and binding affinities, residue stability constants (ln Kf,j, eq 2) were calculated for the four Pol γ ternary complex structures. Each complex was sampled with 250 000 microstates. To ensure sufficient sampling, we computed ln Kf,j for the structure at a larger sampling size of 2 500 000 microstates. The results from the two calculations are consistent (Figure S1), suggesting that sampling of 250 000 microstates sufficiently captured features of the entire ensemble.
Population standard deviation (S.D.) of residue stability was determined to reveal the difference in stability between the four different ligand-bound Pol γ ternary complexes (Figure 2). Neglecting the termini, because they tend to be highly flexible and under-sampled, six regions named ranges A–F were shown to contain variability greater than the mean (Figure 2A,B).
Figure 2.

Different dNTP/NRTI perturb local and nonlocal regions of Pol γ. (A) The hypervariable regions are shown in the table along with their domain, subdomain, and motif associations.20,21 (B) Population standard deviation (S.D.) of residue stability for each complex is plotted as the regional average of 30 amino acids, and the horizontal line is the average SD. The N and C termini were not considered hypervariable and so are shown instead as dotted lines. Gray is the N-terminal domain (1–170), green the 3′−5′ exonuclease (170–440), orange the thumb subdomain (440–475 and 785–815), blue the spacer (475–815), red the palm (815–910 and 1095–1239), and purple is the fingers subdomain (910–1095) of pol. (C,D) Ranges A–F are mapped onto the ternary structure (PDB: 4ZTZ) and colored based upon domain assignment (DNA not shown), Pol γA is shown in gray, and Pol γB subunits are shown in light blue. The perturbed thumb domain interacts with Pol γB subunit, which may affect enzyme processivity. (D) Most perturbed residues lie between the two active sites. Active site residues are colored in blue and labeled according to their respective active site, while the dCTP substrate is shown as sticks.
While any changes in residue stability are meaningful, we focus on the regions that exhibit high variability. We mapped these regions onto the ternary complex containing dCTP (PDB: 4ZTZ).21 The biological significance of the perturbed regions is immediately obvious (Figure 2A–D). Ranges A and B are found within the exo domain in conserved motifs.20 Range B contains exo motif III and a portion of the thumb subdomain, and range C includes the remainder of the thumb. Ranges D–F comprise parts of both the pol catalytic palm and the fingers subdomain that binds to DNA and incoming nucleotide (Figure 2C,D). Interestingly, both local and distal energetic residue stability fluctuates with different ligands (dNTP/NRTIs). For example, on the backside of the catalytic subunit, behind where DNA binds, a portion of the thumb domain is found to be perturbed. This region also interacts with one of the accessory subunits, Pol γB (Figure 2C). Nonetheless, most of the perturbed residues lie between the pol and exo active sites where the elongating primer/template DNA resides (Figure 2D, DNA not shown). While perturbation of residues in the local pol active site would be expected, the largest variations were seen in the remote exo domain, in range B. This implies a compensatory coupling between exo and pol sites. The results suggest the existence of a cooperative network that transmits information from the active site to one or more regions of the protein.
The stability constant Kf,j provides an orthogonal measurement to the structural information in a crystal. The complexes with dCTP and ddCTP are highly similar to a RMSD of 0.019 Å (Table S2). However, due to subtle differences in side chain conformation that affect SASA (Figure S2), the residue stability differences can be revealed by Kf,j values (Figure S3).
Impact of in Silico Mutations on the Energetic Coupling between pol and exo Sites
To further explore pol–exo communication, we took advantage of naturally occurring POLG mutations associated with human diseases to probe this potential connectivity. We chose deleterious exo motif III and thumb mutations, G426S, L424G/G431 V, L428P/A467T, and A467T, as well as pol finger subdomain mutations, Y955C, A957S, and Y955C/A957S. In Pol γ, binding of incoming nucleotide dNTP is accomplished by two helices: the O-helix that interacts with the dNTP and the O1-helix that stabilizes the helix-turn-helix motif.
Y955 is located on the nucleotide binding O-helix and is proposed to be involved in dNTP selection. A957 is located on the connecting loop between the O- and O1-helix. Both helices undergo open-to-closed conformational changes when the correct dNTP is bound.4
Mutations A957S and A957P were analyzed by pre-steady-state kinetics on a single nucleotide incorporation assay and electrophoretic mobility shift assay.23 The results show that the substitutions of A957 with Serine or Proline reduced catalytic efficiency by 1.8- and 60-fold, respectively, largely due to reduced dNTP binding in the pol site. Mutant Y955C displayed a similar phenotype, with a 46-fold reduced KM to the incoming nucleotide without significantly altering catalytic rates.24 Thus, both Y955 and A957 contribute substantially to substrate dNTP binding affinity. As they do not directly interact with dNTP, their effects are likely mediated by alteration of the conformational ensemble.
Residue stability and its SD were calculated for both sets of exo (G426S, L424G/G431 V, L428P/A467T, and A467T) and pol mutations (Y955C, A957S, and Y955C/A957S) relative to the wild-type (wt) complex such that the variance in residue stability is a relative measurement of deviation from the wt complex. Exo mutants and pol mutants, relative to wt, are shown in green and red, respectively (Figure 3A). Exo mutants show local fluctuations in range B. However, large energetic variation is unexpectedly seen in the distal fingers, adjacent to the pol active site (Figure 3A–C). We also observed that a perturbation in the pol domain affects the exo, as shown by the red peaks in ranges A and B (Figure 3A). The finger residues ~900–1050, referred to as the new hypervariable region (NHR), were the most perturbed residues of the pol domain. This span of residues includes portions of ranges D and E that were identified as hypervariable across structures with different substrates bound. When previously comparing different crystal structures, the residues between ranges D and E comprised one of the least variable portions of the protein. However, here we show that both catalytic site pol or exo mutations affect this region. This suggests that this region is important intrinsically for pol–exo communication, although it is not differentially affected by cytosine-based NRTI binding. Also, exo mutant perturbations had a pattern similar to that of pol mutants, implying a similar pattern of cooperativity (Figure 3A).
Figure 3.

In silico pol and exo mutations suggest cooperativity between active sites. We find that either exo or pol mutations perturb local and nonlocal regions. This includes residues adjacent to active site residues. (A) Residue stability SD is plotted with finger and exo domain mutations in red and green, respectively. A regional average of 30 amino acids was used. (B,C) Hypervariable regions displayed are ranges A, B, and residues 900–1050 referred to as the new hypervariable region (NHR). These are mapped on the Pol γ complex according to the domain/subdomain coloring scheme.
These results provide additional supporting evidence that the two active sites are energetically coupled.
Allosteric Network Connectivity Map Using Single Site Thermodynamic Mutations
Thus far, we have demonstrated there is energetic coupling between Pol γ pol and exo by observing the mutual change of stability in one active site when the other is perturbed by mutations or different ligands. To investigate whether such mutual effects are specific to the active sites, we thermodynamically perturbed each residue in our COREX ensemble using a single site thermodynamic mutation algorithm (eq 3).22 If a region remote to a perturbed region alters its energetics in response to the perturbation, it is said to have cooperativity with the perturbed region. If the energetic changes of the remote and perturbed regions are in sync, the two sites are positively coupled; if the energetic changes are in anti-sync, the two sites are negatively coupled. In all other situations, the two sites are not related. The diagonal area of the map represents self-cooperativity of local regions of 10–20 residues.
The operation yielded an allosteric network connectivity map that reveals that not all regions or residues are cooperative with other residues, and that only limited regions show connectivity. Nearly all residues in the pol finger subdomain are positively coupled to regions within the exo and the N-terminal domain (Figure 4). This includes residues corresponding to,or near those, in which we found to be hypervariable either by in silico mutations or with different ligands such as range B/motif III, the exo active site (D198/200), and residues 300–375. Additionally, fingers subdomain mutations selected, Y955 and A957, are located in the region that is highly coupled with the exo domain (Figure 4). This region also contains mutation R964C, which was discovered in AIDS patients with elevated NRTI toxicity.25 This mutation displays only slightly reduced pol activity but nearly abolished exo activity,9 confirming the specificity of energetic coupling between the pol and exo sites. The biochemical results suggest drug toxicity may be caused by defective exo activity as the erroneously incorporated NRTI cannot be excised, due to disrupted pol–exo communication.
Figure 4.

Allosteric network connectivity map reveals a cooperative network. The X-axis represents perturbed residues and the Y-axis denotes the corresponding affected residues. A color scale is shown on the right where red is highly positively cooperative, blue is negatively cooperative, and green represents no cooperativity; units are kcal/mol. Shown above are mutational clusters identified by Nurminen et al.7
Other regions showing coupling are mutational clusters that have been identified in patients. Previously defined clusters are pol active site and its environs (1), upstream DNA-binding channel (2), altered exo:pol ratio (3), distal accessory subunit interface (4), and protein–protein interactions (5).7 Mutation cluster 3 is hypothesized to regulate pol and exo activities, and contains the “partitioning loop” believed to partition DNA in either active site.4 Portions of cluster 3 are found within the fingers subdomain, a region that is highly coupled with large regions of the exo and N-terminal domains (Figure 4). This analysis supports our results that either differential ligand binding or mutations can perturb both pol and exo domains. It also suggests active site communication is specific, as perturbations of many other regions do not change the energetics of either active site (Figure 4). Conversely, most cluster 1 mutations found in the pol domain, which are believed to only affect the pol, appear not to affect other domains, as expected. Similarly, cluster 4 substitution mutations do not affect other residues in the A subunit. As this cluster does not appear to be cooperative with other regions in Pol γA, perhaps it most likely affects the accessory subunit Pol γB, as predicted.
The spacer domain appears to be highly self-cooperative, as demonstrated by the large stretch of ~150 amino acids highly positively coupled (the red block in the center of Figure 4). This is particularly interesting as this region is believed to stabilize the interaction with DNA as well as the accessory subunits. Self-cooperativity of this large stretch may facilitate rapid and large conformational changes upon DNA binding.
Some mutational clusters fall into regions that appear highly coupled to the entire protein, including most of clusters 2 and 5. Cluster 2 mutations affect DNA binding, suggesting that mutations in the cluster might disrupt coordination of DNA binding with conformational changes necessary for correct nucleotide selection. Although separated by primary sequence, these regions are structurally adjacent and interact both with DNA and with the processivity subunit (Figure S4). This raises the possibility that allostery in the overall system may in part be mediated by DNA itself, a conclusion supported by the large conformational changes seen when comparing the holoenzyme structure, which contains no ligand or DNA (PDB ID: 3IKM), to the ternary complex (PDB ID: 4ZTZ).
Energetic Coupling in Another Pol I Family Polymerase, Pol I BF
To investigate whether or not our findings in Pol γ are innate to the structural features of this family of polymerases, we performed the same analyses on another Pol I Family member, thermostable Bacillus DNA Polymerase I large fragment (Pol I BF). Pol I BF shares the same right-hand conformation with fingers (residues 656–815), thumb (residues 496–595), and palm (residues 617–655 and 830–869) subdomains that constitute the pol domain. It also contains a vestigial 3′−5′ exonuclease domain (302–468).26,27
To better understand pol–exo communication under physiological situations, we compared Pol I BF bound to either a correctly matched or a mismatched primer/template DNA. A mismatch in pol, resulting from incorporation of an incorrect nucleotide, should engage the exo site for mismatch removal. Comparison of the energetics of Pol I BF with a matched or mismatched primer/template DNA, using the binary complex crystal structures (Table S3),28 reveals that differences are concentrated in four regions: I (residues 350–388), II (residues 478–515), III (residues 581–658), and IV (689–780 and 802–817) (Figure 5A,B). Region I contains the conserved exo motif III, and region IV is in the fingers subdomain. The fingers subdomain undergoes conformational changes with each nucleotide addition. It adapts mostly an open conformation in the absence of dNTP, and changes to ajar and or closed conformations in the presence of an incorrect or a correct nucleotide, respectively.27,29,30 Our results in Figure 4 suggest that in most conformations, captured by our ensemble calculations, the fingers domain is strongly coupled with the exo motif III region. The results from Pol I BF are in good agreement with those from Pol γ, suggesting that this mode of communication is conserved within Pol I family members.
Figure 5.

Pol I BF mismatches show similarly perturbed domains as Pol γ. (A) Residue stability is plotted as a regional average of 15 residues, and the following domains and subdomains are displayed above: 3′−5′exonuclease domain (302–468), fingers (656–815), thumb (496–595), and palm (617–655 and 830–869). Mean residue stability is shown as a horizontal line in black. (B) Regions shown to be hypervariable are mapped on the crystal structure with the DNA not shown (PDB: 1L3U). Hypervariable regions (residue stability greater than the mean) are colored based upon domain association. Catalytic residues are in blue.
DISCUSSION
Allosteric Regulation of pol and exo Sites
Pol γ pol and exo sites have distinct enzymatic activities that are highly coordinated during DNA replication. This coordination is critical for correct dNTP discrimination. The pol site selects for the correct nucleotide and catalyzes phosphodiester bond formation to extend the primer DNA, while the exo hydrolyzes the primer 3′-end. If a correct nucleotide is incorporated, the enzyme retains the primer DNA in the pol for continued synthesis. If an incorrect nucleotide is incorporated, mismatched primer/template DNA is transferred to the exo for removal. Crosstalk between these sites in DNA polymerases is known to be an important contributor to the fidelity of DNA replication. The biophysical basis for signal transduction from misincorporation in the pol to the distal exo site is not fully understood. Pol and exo site coordination is also critical for discrimination of NRTIs from normal dNTPs. Otherwise, misincorporation of an NRTI into mitochondrial DNA can result in drug toxicity. Understanding active site coordination and its role in discriminating between these inhibitors is critical to both improving drug efficacy and decreasing toxicity.
In this study, we report positive cooperativity between the fingers subdomain of the pol and conserved motif III of the exo domain. Perturbation of the pol, by either mismatched primer, NRTI inhibitor, or a mutation, can stimulate a corresponding perturbation in the exo, indicating energetic coupling between the two regions. Similar findings in Pol I BF suggest that this communication may be intrinsic to their conserved structure and sequence, in two evolutionarily divergent but structurally homologous polymerases. The positive cooperativity between the pol finger subdomain and exo is particularly intriguing as the fingers directly interacts with the incoming dNTP and contributes to correct nucleotide selection through open-closed conformational changes.31
A multitude of factors contribute to correct nucleotide dNTP selection including hydrogen bonding with the templating nucleobase, steric interactions, nucleotide sugar conformation, and polymerase conformational changes.27,31,32 Nucleotides can also influence polymerase conformation in a series of distinct steps both before and during covalent bond formation.33 Even nucleotide interactions with DNA template and solvent can have profound impacts on nucleotide selection.34,35 Quantum mechanical/molecular modeling studies of human Pol β revealed that residues surrounding the pol active site, referred to as base-binding residues, contribute to the transition state of dNTP incorporation.36–38 Furthermore, the study found incorrect dNTP can perturb the pol active site and form a nonproductive transition state, indicating that both dNTP and pol residues contribute to stability and reactivity. Our conclusions are not only in agreement with those concerning Pol β, but they also suggest that ligand binding and perturbations in the pol can be propagated to the distal exo site. Pol β is a DNA repair polymerase without exo activity. Whether changes to its pol site are propagated to other regions is unknown.
For dual active site polymerases, DNA transfer during replication between pol and exo active sites is necessary to achieve high fidelity. Our work here suggests that inter- and intradomain cooperativity may facilitate two innate fidelity checkpoints: pol–exo sampling and finger subdomain conformational changes (open/closed/ajar), respectively.31,39 Studies of Escherichia coli DNA Pol I Klenow fragment (KF) showed that both DNA intramolecular and intermolecular transfer between active sites coexist, but intramolecular transfer is the faster of the two pathways.39 Importantly, mismatched DNA preferentially associates with the exo site, and even more so in the presence of dNTP. In agreement with our study, this suggests that pol–exo cooperativity may facilitate transitioning the polymerase to its proofreading mode when bound to mismatched DNA, and to its synthesis mode when bound to correctly matched DNA, and dNTP ligand can further influence this coordination.
Pol and exo active sites are spatially separated by 35 Å. This begs the question: How does their long-range coupling occur? Our COREX calculations are based on an ensemble model that proposes allostery is the result of changes in the conformational distribution, rather than a distinct linear pathway of contiguous amino acids or really any clearly defined “path”.40 Allosteric regulation of remote sites can be attributed to ligand-induced entropic changes and thermodynamic fluctuation, and/or to transitions in structural ensembles. Other studies also suggest that “paths” for allostery are diffuse, nonlinear, and nonlocal; they have also been described as an elastic network where soft collective modes of motion enable allosteric regulation of protein systems.41–44
Equilibrium of Conformational Populations
COREX allows estimation of a protein’s local stability at single residue resolution.18 An advantage is the generation of a thermodynamic ensemble of conformational microstates that reasonably models the additional degrees of freedom not obvious in crystal structures. For example, DNA polymerases undergo conformational fluctuations, mutations, DNA, and or dNTP binding influences the proportions of predominant conformations.30,45 We simulate these many possible conformations, constructed an appropriate partition function, and weighted them by their respective probabilities. COREX has also been used in describing cryptic long-range energetic connectivity in other systems, revealing the energetic consequences of subtle structural differences.46–48 Our analyses of Pol γ suggest that ensemble-based energetic coupling facilitates coordination between different functional domains.
New Evaluation Tool for Pol γ Mutations
Pol γ mutations have been implicated in many human diseases. Deficient polymerase activity reduces mitochondrial DNA replication, decreases mitochondrial DNA copy number, and hampers cellular energy supply. Our studies suggest that COREX is particularly useful in analyzing mutational effects. Starting with the ground state of the enzyme in its native structure, the algorithm, although unable to provide dynamics, readily reveals that perturbations of one region can affect the stability of other regions.
Pol γ mutations associated with disease are located in and around the active sites of pol and exo, as well as at noncatalytic sites. While crystal structures are invaluable in analyzing local effects of amino acid substitutions, they do not explain long-range effects of mutations and either intra- or interdomain communication. Our approach here suggests mutations or ligands that affect residue stability in distal regions do so through population changes in the conformational ensemble. As a result, COREX affords a new approach for mutational analyses. Perturbations can be revealed computationally to propagate over long distances across the molecule via newly exposed interfaces without the initiating and terminal sites being in direct contact. For example, the connectivity map analyses presented here show that the stability of some localized clusters of disease-associated mutants, distal to either active site, is associated with the stability of the entire enzyme as well as pol–exo communication. COREX can therefore provide an energetic explanation for the dysfunction of these Pol γ mutants. As a major adverse target for NRTIs, Pol γ mutations can alter the drug toxicity in different patients.49,50 Our results here should be an effective tool to screen potential NRTI response in the more than 400 single nucleotide polymorphisms (SNPs) in POLG (http://exac.broadinstitute.org/gene/ENSG00000140521). The network of local and nonlocal perturbations induced by POLG variants and NRTIs, as revealed by COREX, may facilitate personalized treatments for patients based on their POLG genotype.
CONCLUSION
This study provides the first examination of energetic coupling, where perturbations in the pol site are sensed by the exo, in the high-fidelity mitochondrial DNA polymerase, Pol γ. We demonstrated that substrate nucleotide or nucleotide analogue (NRTIs), as well as Pol γ amino acid substitutions, perturb a complex network, resulting in local and nonlocal perturbations. Our results have provided a biophysical basis for communication between pol and exo sites of Pol γ, suggesting that COREX is a powerful tool to analyze mutational effects of both local and remote regions of a protein.
Supplementary Material
ACKNOWLEDGMENTS
We thank V. J. Hilser, J. C. Lee, and L. C. Sowers for helpful comments. The work was supported in part by grants from the NIH (GM110591 and AI134611) to Y.W.Y. and from the Sealy and Smith Foundation to Sealy Center for Structural Biology at UTMB.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b04655.
COREX Monte Carlo calculation reproducibility; differences in SASA between structures; differences in residue stability; diffusely cooperative residues; Pol γ structures used; differences in RMSD; and Pol I BF structures used (PDF)
REFERENCES
- (1).Gray H; Wong TW Purification and Identification of Subunit Structure of the Human Mitochondrial DNA Polymerase. J. Biol. Chem 1992, 267, 5835–5841. [PubMed] [Google Scholar]
- (2).Johnson AA; Johnson KA Exonuclease Proofreading by Human Mitochondrial DNA Polymerase. J. Biol. Chem 2001, 276, 38097–38107. [DOI] [PubMed] [Google Scholar]
- (3).Johnson AA; Johnson KA Fidelity of Nucleotide Incorporation by Human Mitochondrial DNA Polymerase. J. Biol. Chem 2001, 276, 38090–38096. [DOI] [PubMed] [Google Scholar]
- (4).Szymanski MR; Kuznetsov VB; Shumate C; Meng Q; Lee Y-SY-S; Patel G; Patel S; Yin YW Structural Basis for Processivity and Antiviral Drug Toxicity in Human Mitochondrial DNA Replicase. EMBO J. 2015, 34 (14), 1959–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Rahman S; Copeland WC POLG-Related Disorders and Their Neurological Manifestations. Nat. Rev. Neurol 2019, 15 (1), 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wong LC; Naviaux RK; Brunetti-pierri N; Zhang Q; Schmitt ES; Truong C; Milone M; Cohen BH; Wical B; Ganesh J; Basinger AA; Burton BK; Swoboda K; Gilbert DL; Vanderver A; Saneto RP; Maranda B; Arnold G; Abdenur JE; Waters PJ; Copeland WC Molecular and Clinical Genetics of Mitochondrial Diseases Due to POLG Mutations. Hum. Mutat 2008, 29 (9), E150–E172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Nurminen A; Farnum GA; Kaguni LS Pathogenicity in POLG Syndromes: DNA Polymerase Gamma Pathogenicity Prediction Server and Database. BBA Clin. 2017, 7 (February), 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zheng X; Boyer L; Jin M; Mertens J; Kim Y; Ma L; Ma L; Hamm M; Gage FH; Hunter T Metabolic Reprogramming during Neuronal Differentiation from Aerobic Glycolysis to Neuronal Oxidative Phosphorylation. Elife 2016, 5, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kasahara T; Ishiwata M; Kakiuchi C; Fuke S; Iwata N; Ozaki N; Kunugi H; Minabe Y; Nakamura K; Iwata Y; Fujii K; Kanba S; Ujike H; Kusumi I; Kataoka M; Matoba N; Takata A; Iwamoto K; Yoshikawa T; Kato T Enrichment of Deleterious Variants of Mitochondrial DNA Polymerase Gene (POLG1) in Bipolar Disorder. Psychiatry Clin. Neurosci 2017, 71 (8), 518–529. [DOI] [PubMed] [Google Scholar]
- (10).Bratic A; Kauppila TES; Macao B; Grönke S; Siibak T; Stewart JB; Baggio F; Dols J; Partridge L; Falkenberg M; Wredenberg A; Larsson NG Complementation between Polymerase- and Exonuclease-Deficient Mitochondrial DNA Polymerase Mutants in Genomically Engineered Flies. Nat. Commun 2015, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chan SSL; Longley MJ; Copeland WC; Carolina N Common A467T Mutation in the Human Mitochondrial DNA Polymerase (POLG) Compromises Catalytic Efficiency and Interaction with the Accessory Subunit * 2005, 280, 31341–31346. [DOI] [PubMed] [Google Scholar]
- (12).Lewis W; Day BJ; Copeland WC Mitochondrial Toxicity of Nrti Antiviral Drugs: An Integrated Cellular Perspective. Nat. Rev. Drug Discovery 2003, 2 (10), 812–822. [DOI] [PubMed] [Google Scholar]
- (13).Glesby MJ Overview of Mitochondrial Toxicity of Nucleoside Reverse Transcriptase Inhibitors. Int. AIDS Soc 2002, 42–46. [Google Scholar]
- (14).Anderson KS A Transient Kinetic Approach to Investigate Nucleoside Inhibitors of Mitochondrial DNA Polymerase γ. Methods 2010, 51 (4), 392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hanes JW; Johnson KA Exonuclease Removal of Dideoxycytidine (Zalcitabine) by the Human Mitochondrial DNA Polymerase. Antimicrob. Agents Chemother 2008, 52 (1), 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sohl CD; Szymanski MR; Mislak AC; Shumate CK; Amiralaei S; Schinazi RF; Anderson KS; Yin YW; Sohl CD; Szymanski MR; Mislak AC; Shumate CK; Amiralaei S Probing the Structural and Molecular Basis of Nucleotide Selectivity by Human Mitochondrial DNA Polymerase γ. Proc. Natl. Acad. Sci. U. S. A 2015, 113 (5), E662–E662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hilser VJ; Freire E Structure-Based Calculation of the Equilibrium Folding Pathway of Proteins. Correlation with Hydrogen Exchange Protection Factors. J. Mol. Biol 1996, 262 (5), 756–772. [DOI] [PubMed] [Google Scholar]
- (19).Hilser VJ; García-Moreno E B; Oas TG; Kapp G; Whitten ST A Statistical Thermodynamic Model of the Protein Ensemble. Chem. Rev 2006, 106 (5), 1545–1558. [DOI] [PubMed] [Google Scholar]
- (20).Shevelev IV; Hübscher, U. The 3′−5′ Exonucleases. Nat. Rev. Mol. Cell Biol 2002, 3 (5), 364–375. [DOI] [PubMed] [Google Scholar]
- (21).Lee YS; Kennedy WD; Yin YW Structural Insight into Processive Human Mitochondrial DNA Synthesis and Disease-Related Polymerase Mutations. Cell 2009, 139 (2), 312–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hilser VJ; Dowdy D; Oas TG; Freire E The Structural Distribution of Cooperative Interactions in Proteins: Analysis of the Native State Ensemble. Proc. Natl. Acad. Sci. U. S. A 1998, 95 (17), 9903–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sohl CD; Kasiviswanathan R; Copeland WC; Anderson KS Mutations in Human DNA Polymerase γ Confer Unique Mechanisms of Catalytic Deficiency That Mirror the Diease Severity in Mitochondrial Disorder Patients. Hum. Mol. Genet 2013, 22 (6), 1074–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ponamarev MV; Longley MJ; Nguyen D; Kunkel TA; Copeland WC Active Site Mutation in DNA Polymerase γ Associated with Progressive External Ophthalmoplegia Causes Error-Prone DNA Synthesis. J. Biol. Chem 2002, 277 (18), 15225–15228. [DOI] [PubMed] [Google Scholar]
- (25).Yamanaka H; Gatanaga H; Kosalaraksa P; Matsuoka-Aizawa S; Takahashi T; Kimura S; Oka S Novel Mutation of Human DNA Polymerase γ Associated with Mitochondrial Toxicity Induced by Anti-HIV Treatment. J. Infect. Dis 2007, 195 (10), 1419–1425. [DOI] [PubMed] [Google Scholar]
- (26).Kiefer JR; Mao C; Hansen CJ; Basehore SL; Hogrefe HH; Braman JC; Beese LS Crystal Structure of a Thermostable Bacillus DNA Polymerase I Large Fragment at 2.1 A Resolution. Structure 1997, 5 (1), 95–108. [DOI] [PubMed] [Google Scholar]
- (27).Miller BR; Beese LS; Parish CA; Wu EY The Closing Mechanism of DNA Polymerase I at Atomic Resolution. Structure 2015, 23 (9), 1609–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Johnson SJ; Beese LS Structures of Mismatch Replication Errors Observed in a DNA Polymerase. Cell 2004, 116 (6), 803–816. [DOI] [PubMed] [Google Scholar]
- (29).Wu EY; Beese LS The Structure of a High Fidelity DNA Polymerase Bound to a Mismatched Nucleotide Reveals an “Ajar” Intermediate Conformation in the Nucleotide Selection Mechanism. J. Biol. Chem 2011, 286 (22), 19758–19767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hohlbein J; Aigrain L; Craggs TD; Bermek O; Potapova O; Shoolizadeh P; Grindley NDF; Joyce CM; Kapanidis AN Conformational Landscapes of DNA Polymerase I and Mutator Derivatives Establish Fidelity Checkpoints for Nucleotide Insertion. Nat. Commun 2013, 4, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Berezhna SY; Gill JP; Lamichhane R; Millar DP Single-Molecule Förster Resonance Energy Transfer Reveals an Innate Fidelity Checkpoint in DNA Polymerase I. J. Am. Chem. Soc 2012, 134 (27), 11261–11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Williams AA; Darwanto A; Theruvathu JA; Burdzy A; Neidigh JW; Sowers LC Impact of Sugar Pucker on Base Pair and Mispair Stability. Biochemistry 2009, 48 (50), 11994–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bermek O; Grindley NDF; Joyce CM Prechemistry Nucleotide Selection Checkpoints in the Reaction Pathway of DNA Polymerase I and Roles of Glu710 and Tyr766. Biochemistry 2013, 52 (36), 6258–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Petruska J; Sowers LC; Goodman MF Comparison of Nucleotide Interactions in Water, Proteins, and Vacuum: Model for DNA Polymerase Fidelity. Proc. Natl. Acad. Sci. U. S. A 1986, 83 (6), 1559–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Petruska J; Goodman MF; Boosalis MS; Sowers LC; Cheong C; Tinoco I Comparison between DNA Melting Thermodynamics and DNA Polymerase Fidelity. Proc. Natl. Acad. Sci. U. S. A 1988, 85 (17), 6252–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Xiang Y; Oelschlaeger P; Floria J; Goodman MF; Warshel A Simulating the Effect of DNA Polymerase Mutations on Transition-State Energetics and Fidelity: Evaluating Amino Acid Group Contribution and Allosteric Coupling for Ionized Residues in Human Pol †. Biochemistry 2006, 45, 7036–7048. [DOI] [PubMed] [Google Scholar]
- (37).Kamerlin SCLL; Warshel A; Kolmodin K The Empirical Valence Bond Model: Theory and Applications. Wiley Interdiscip. Rev. Comput. Mol. Sci 2011, 1 (1), 30–45. [Google Scholar]
- (38).Warshel A; Weiss RM An Empirical Valence Bond Approach for Comparing Reactions in Solutions and in Enzymes. J. Am. Chem. Soc 1980, 102 (20), 6218–6226. [Google Scholar]
- (39).Lamichhane R; Berezhna SY; Gill JP; Van Der Schans E; Millar DP Dynamics of Site Switching in DNA Polymerase. J. Am. Chem. Soc 2013, 135 (12), 4735–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cooper A; Dryden DTF Allostery without Conformational Change - A Plausible Model. Eur. Biophys. J 1984, 11 (2), 103–109. [DOI] [PubMed] [Google Scholar]
- (41).Wodak SJ; Paci E; Dokholyan NV; Berezovsky IN; Horovitz A; Li J; Hilser VJ; Bahar I; Karanicolas J; Stock G; Hamm P; Stote RH; Eberhardt J Allostery in Its Many Disguises: From Theory to Applications. Struct. Rev 2019, 27, 566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Buchenberg S; Sittel F; Stock G Time-Resolved Observation of Protein Allosteric Communication. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (33), E6804–E6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Motlagh HN; Wrabl JO; Li J; Hilser VJ The Ensemble Nature of Allostery. Nature 2014, 508 (7496), 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Urbanc B Protein Actions: Principles and Modeling by I. Bahar, R.L. Jernigan, and K.A. Dill. J. Biol. Phys 2017, 43 (4), 585–589. [Google Scholar]
- (45).Santoso Y; Joyce CM; Potapova O; Le Reste L; Hohlbein J; Torella JP; Grindley NDF; Kapanidis AN Conformational Transitions in DNA Polymerase I Revealed by Single-Molecule FRET. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Maillard RA; Liu T; Beasley DWC; Barrett ADT; Hilser VJ; Lee JC Thermodynamic Mechanism for the Evasion of Antibody Neutralization in Flaviviruses. J. Am. Chem. Soc 2014, 136 (29), 10315–10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Pan H; Lee JC; Hilser VJ Binding Sites in Escherichia Coli Dihydrofolate Reductase Communicate by Modulating the Conformational Ensemble. Proc. Natl. Acad. Sci. U. S. A 2000, 97 (22), 12020–12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Li J; White JT; Saavedra H; Wrabl JO; Motlagh HN; Liu K; Sowers J; Schroer TA; Thompson EB; Hilser VJ; Brad Thompson E; Hilser VJ Genetically Tunable Frustration Controls Allostery in an Intrinsically Disordered Transcription Factor. Elife 2017, 6, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Baruffini E; Ferrari J; Dallabona C; Donnini C; Lodi T Polymorphisms in DNA Polymerase γ Affect the MtDNA Stability and the NRTI-Induced Mitochondrial Toxicity in Saccharomyces Cerevisiae. Mitochondrion 2015, 20, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Chan SSL; Copeland WC DNA Polymerase Gamma and Mitochondrial Disease: Understanding the Consequence of POLG Mutations. Biochim. Biophys. Acta – Bioenerg 2009, 1787 (5), 312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.