

Abstract
Analyzing 12,361 all-cause cirrhosis cases and 790,095 controls from eight cohorts, we identify a common missense variant in the Mitochondrial Amidoxime Reducing Component 1 gene (MARC1 p.A165T) that associates with protection from all-cause cirrhosis (OR 0.91, p = 2.3*10−11). This same variant also associates with lower levels of hepatic fat on computed tomographic imaging and lower odds of physician-diagnosed fatty liver as well as lower blood levels of alanine transaminase (-0.025 SD, 3.7*10−43), alkaline phosphatase (-0.025 SD, 1.2*10−37), total cholesterol (-0.030 SD, p = 1.9*10−36) and LDL cholesterol (-0.027 SD, p = 5.1*10−30) levels. We identified a series of additional MARC1 alleles (low-frequency missense p.M187K and rare protein-truncating p.R200Ter) that also associated with lower cholesterol levels, liver enzyme levels and reduced risk of cirrhosis (0 cirrhosis cases for 238 R200Ter carriers versus 17,046 cases of cirrhosis among 759,027 non-carriers, p = 0.04) suggesting that deficiency of the MARC1 enzyme may lower blood cholesterol levels and protect against cirrhosis.
Author summary
Cirrhosis is a leading cause of death worldwide. However, the genetic underpinnings of cirrhosis remain poorly understood. In this study, we analyze twelve thousand individuals with cirrhosis and identify a common missense variant in a gene called MARC1 that protects against cirrhosis. Carriers of this missense variant also have lower blood cholesterol levels, lower liver enzyme levels and reduced liver fat. We identify an additional two low-frequency coding variants in MARC1 that are also associated with lower cholesterol levels, lower liver enzyme levels and protection from cirrhosis. Finally, we identify an individual homozygous for a predicted loss-of-function variant in MARC1 who exhibits very low blood LDL cholesterol levels. These genetic findings suggest that MARC1 deficiency may lower blood cholesterol levels and protect against cirrhosis, pointing to MARC1 as a potential therapeutic target for liver disease.
Introduction
Discovery of novel protective human genetic variation can identify new therapeutic targets for treatment of a given disease [1]. Targets with human genetic support are more than twice as likely to result in successful development of a therapeutic than targets without genetic support [2]. Indeed, identification of novel protective variants for coronary artery disease and type 2 diabetes, such as variation in ANGPTL3, ANGPTL4, and APOC3, has catalyzed the development of new therapeutics targeting these genes for treatment of metabolic disorders [3–6]. Although liver cirrhosis is a leading cause of death worldwide, no therapies currently exist to treat or delay the progression of cirrhosis [7]. Identification of novel protective variants for cirrhosis may therefore allow for the identification of new therapeutic targets with enhanced likelihood of successful clinical development for treatment of cirrhosis.
Cirrhosis is often considered to be the final stage of distinct pathogenic processes including excess alcohol consumption, viral infection and fatty liver secondary to obesity [8]. However, analyses of these separate processes have identified similar genetic determinants. For example, PNPLA3 p.I48M and TM6SF2 p.E40K, although initially identified as associated with hepatic steatosis [9,10], strongly predispose to the development of alcoholic cirrhosis [11], non-alcoholic cirrhosis [12,13] and hepatitis C-related cirrhosis [14,15]. The recently identified splice variant rs72613567 in HSD17B13 similarly protects against alcoholic cirrhosis, non-alcoholic cirrhosis and severe liver fibrosis among individuals with hepatitis C (S1 Fig) [16,17]. These individual findings suggest that analysis of an all-cause cirrhosis phenotype combining alcoholic and non-alcoholic causes may prove useful to find new protective genetic determinants.
In this study, we first examine whether known alcoholic and non-alcoholic cirrhosis variants associate with all-cause cirrhosis. After demonstrating that known alcoholic and non-alcoholic variants associate with all-cause cirrhosis, we leverage the increased power provided by analysis of all-cause cirrhosis to identify a novel common protective missense variant in MARC1. We further identify a low-frequency coding variant and a rare stop codon in MARC1 that form an allelic series associated with lower cholesterol levels, lower liver enzyme levels and protection from cirrhosis.
Results
Known alcoholic and non-alcoholic cirrhosis variants associate with all-cause cirrhosis
We first examined whether known alcoholic and non-alcoholic cirrhosis variants associate with all-cause cirrhosis. We created an all-cause liver cirrhosis phenotype in UK Biobank, combining the following ICD10 diagnostic codes: K70.2 (alcoholic fibrosis and sclerosis of the liver), K70.3 (alcoholic cirrhosis of the liver), K70.4 (alcoholic hepatic failure), K74.0 (hepatic fibrosis), K74.1 (hepatic sclerosis), K74.2 (hepatic fibrosis with hepatic sclerosis), K74.6 (other and unspecified cirrhosis of liver), K76.6 (portal hypertension), or I85 (esophageal varices). Using this definition, we identified 1,740 cases of all-cause cirrhosis in UK Biobank. We examined the association of all-cause cirrhosis with six genetic variants previously reported to be associated with alcoholic or non-alcoholic cirrhosis: PNPLA3 I48M, TM6SF2 E167K, MBOAT7 rs641738, HSD17B13 rs72613567, HFE C282Y and SERPINA1 E366K [11,16,18,19]. All six variants associated with all-cause cirrhosis in UK Biobank (S2 Fig). Each variant exhibited greater statistical significance with all-cause cirrhosis than with alcoholic or non-alcoholic subtypes in UK Biobank, with an average 30% gain in power by analyzing all-cause cirrhosis compared to non-alcoholic cirrhosis and an 87% gain in power compared to analyzing alcoholic cirrhosis.
MARC1 p.A165T associates with protection from cirrhosis, fatty liver, elevated liver enzymes and elevated blood cholesterol levels
Having established that an analysis of all-cause cirrhosis would provide improved statistical power, we sought to identify novel genetic determinants of all-cause cirrhosis through a discovery genome-wide association analysis followed by replication (Fig 1). In the discovery analysis, we analyzed 3,754 all-cause cirrhosis cases and 444,791 controls from five cohorts (S1 Table). Baseline characteristics and distribution of ancestry in each cohort are provided (S2 Table, S3 Table). Mean ages across the studies ranged from 45 years to 57 years. Proportion of female gender was similar in hospital and population-based studies (53.8% to 54.6%), but lower in the alcoholic cirrhosis case-control studies (8.0% and 27.2%). The proportion of individuals with cirrhosis was higher in Partners Biobank (3.9%), a hospital-based cohort, than UK Biobank (0.4%) or ARIC (0.9%), population-based cohorts.
Fig 1. Study design.
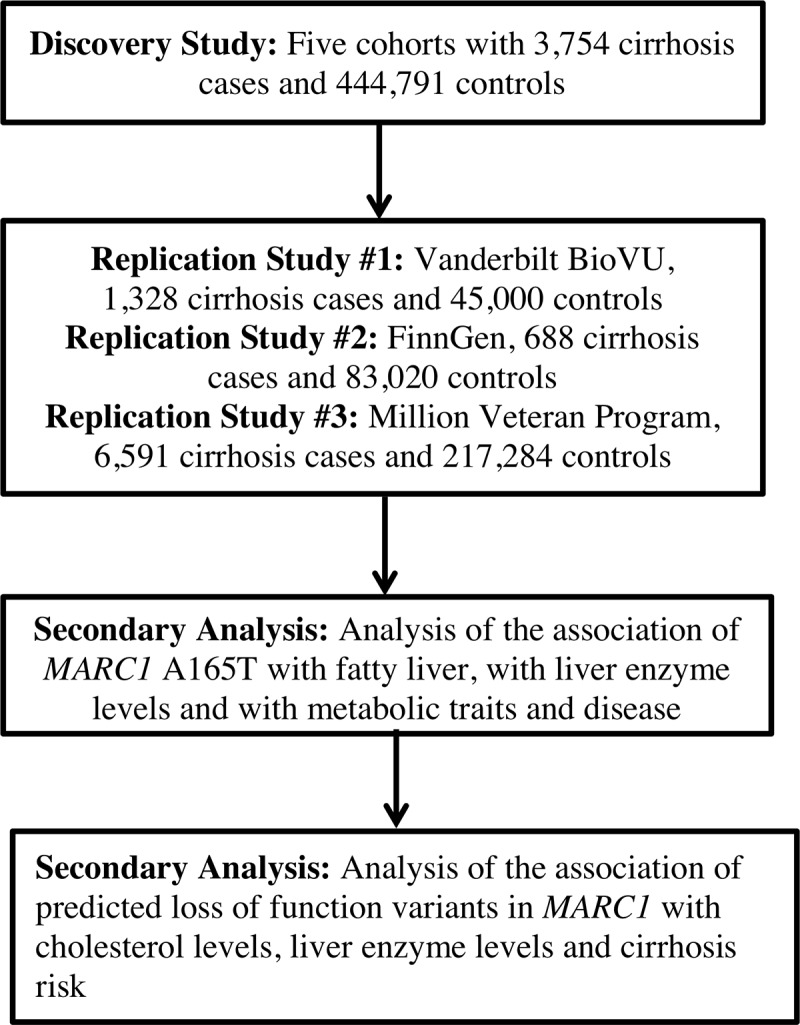
We tested the association of 14 million genetic variants with minor allele frequency > 0.1% with all-cause cirrhosis in both additive and recessive models. No evidence of genomic inflation was observed (lambda 1.02, S3 Fig). We replicated known associations of PNPLA3, TM6SF2, HFE and HDS17B13 variants with cirrhosis at genome-wide significance (Table 1). No other variants were associated with all-cause cirrhosis at genome-wide significance.
Table 1. DNA sequence variants associated with all-cause cirrhosis in the discovery analysis.
| Model | Variant | CHR | EA | EAF | Gene | Annotation | OR | p-value |
|---|---|---|---|---|---|---|---|---|
| Additive | rs738409 | 22 | G | 26% | PNPLA3 | Missense: p.I48M | 1.47 | 2.2*10−67 |
| Additive | rs58542926 | 19 | T | 7% | TM6SF2 | Missense: p.E167K | 1.42 | 9.7*10−24 |
| Recessive | rs1800562 | 6 | A | 3% | HFE | Missense: p.C282Y | 3.2 | 1.3*10−14 |
| Additive | rs72613567 | 4 | TA | 22% | HSD17B13 | Splice Variant | 0.82 | 4.5*10−8 |
| Additive | rs2642438 | 1 | A | 25% | MARC1 | Missense: p.A165T | 0.87 | 8.7*10−7 |
CHR: chromosome, EA: effect allele, EAF: effect allele frequency
The lead coding variant at sub-genome wide significance was a common missense variant in MARC1 (p.A165T) that was associated with lower risk of all-cause cirrhosis (OR 0.87, p = 8.7*10−7, minor allele frequency 25%). We sought replication of this observation in three independent studies: BioVU, FinnGen Consortium and Million Veteran Program. MARC1 p.A165T associated with protection from cirrhosis in BioVU (OR 0.92, p = 0.045), in FinnGen (0.89, p = 0.044) and in the Million Veteran Program (OR 0.92, p = 7.4*10−5). When the statistical evidence from the discovery and replication studies are combined, MARC1 p.A165T associated with protection from cirrhosis at a significance level exceeding genome wide significance (OR 0.91, p = 2.3*10−11, Fig 2). No evidence of heterogeneity in the association of MARC1 p.A165T with all-cause cirrhosis in the discovery analysis and replication analyses was observed (test for heterogeneity p = 0.64). In UK Biobank, MARC1 p.A165T associated with protection from both alcoholic (OR 0.86, p = 0.05) and non-alcoholic (OR 0.85, p = 0.0002) cirrhosis subtypes. MARC1 p.A165T also associated with protection in cohorts where cirrhosis was ascertained using ICD codes (OR 0.91, p = 1.9x10-10) as well as in cohorts where cirrhosis was ascertained using clinical evaluation (OR 0.88, p = 0.04, p interaction = 0.65, S4 Fig).
Fig 2. Association of MARC1 p.A165T with cirrhosis and fatty liver in discovery and replication datasets.

OR, odds ratio; SD, standard deviation; CI, 95% confidence interval; MVP, Million Veterans Program; FHS, Framingham Heart Study; MESA, Multi-ethnic Study of Atherosclerosis.
Next, we examined whether MARC1 p.A165T associated with fatty liver (definitions provided in S4 Table). MARC1 p.A165T was associated with reduced hepatic fat on computed tomographic imaging in both the Framingham Heart Study (FHS) and Multi-ethnic Study of Atherosclerosis (MESA) cohorts (-0.08 SD, p = 8.2*10−6). MARC1 p.A165T also associated with reduced risk of physician-diagnosed fatty liver in three biobank studies (OR 0.83, p = 1.90*10−8).
Having established that MARC1 p.A165T associates with protection from all-cause cirrhosis as well as fatty liver, we tested association of this variant with plasma biomarkers–ALT (alanine transaminase), AST (aspartate transaminase), ALP (alkaline phosphatase), total cholesterol, LDL (low-density lipoprotein) cholesterol, HDL (high-density lipoprotein) cholesterol, and triglycerides. MARC1 p.A165T associated with lower ALT levels (-0.025 SD, p = 3.7*10−43), AST levels (-0.013 SD, p = 1.8*10−11) and ALP levels (-0.025 SD, p = 1.2*10−37). MARC1 p.A165T also associated with lower total cholesterol (-0.030, p = 1.9*10−36) and LDL cholesterol (-0.027, p = 5.1*10−30). MARC1 p.A165T associated with higher triglyceride levels (0.013 SD, p = 3.0*10−9) and lower HDL cholesterol levels (-0.028 SD, p = 1.3*10−30) but did not associate with blood pressure, BMI or waist-to-hip ratio (S5 Table).
MARC1 p.A165T is not associated with coronary artery disease
Variant alleles in PNPLA3 and TM6SF2 that decrease risk of cirrhosis have been reported to increase risk for coronary artery disease (CAD) [20]. This raises the possibility that treatment of cirrhosis may have adverse cardiovascular effects. We therefore examined whether MARC1 p.A165T associates with increased CAD risk. In contrast to PNPLA3 and TM6SF2, neither MARC1 p.A165T nor HSD17B13 rs72613567 associated with risk of CAD (S5 Fig) [21]. In a phenome wide association study in UK Biobank, MARC1 p.A165T associated with a lower risk of gallstones (OR 0.96, p = 0.0006) and an elevated risk of gout (OR 1.06, p = 0.001, S6 Fig).
Identification of a series of protective alleles in MARC1
We investigated whether loss or gain of MARC1 function might be responsible for the protection from cirrhosis and the reduced levels of liver enzymes and cholesterol observed. We leveraged a rare nonsense mutation observed early in the MARC1 gene (p.R200Ter). In a combined analysis of UK Biobank, Partners Biobank, MESA, Framingham, and Million Veteran Program, no cases of cirrhosis were observed among 238 carriers of MARC1 R200Ter compared to 17,046 cases of cirrhosis among 759,027 non-carriers (odds ratio 0, p = 0.04).
We assembled sequence data for MARC1 in 45,493 individuals and identified 94 carriers of MARC1 p.R200Ter and 21 carriers of other predicted loss of function variants (S6 Table, S7 Table). Carriers of R200Ter and other loss of function variants in MARC1 had lower total cholesterol levels (-0.16 SD, p = 0.04) and ALT levels (-0.24 SD, p = 0.02, Table 2).
Table 2. An allelic series of variants in MARC1 which associate with lower total cholesterol levels, alanine transaminase levels and reduced risk of cirrhosis.
| MARC1 Variant | MAF | Total Cholesterol, Effect in SD (CI) | Alanine Transaminase, Effect in SD (CI) | Cirrhosis, Odds Ratio (CI) |
|---|---|---|---|---|
| A165T | 29% |
-0.030 (-0.035, -0.025), p = 1.9x10-36 |
-0.025 (-0.028, -0.021) p = 3.7x10-43 |
0.91 (0.88, 0.93), p = 2.3x10-11 |
| M187K | 1.1% |
-0.053 (-0.074, -0.032), p = 7x10-7 |
-0.032 (-0.053, -0.012), p = 0.001 |
0.75 (0.60, 0.95), p = 0.01 |
| R200Ter | 0.009% |
-0.16 (-0.32, -0.01), p = 0.04 |
-0.24 (-0.43, -0.05), p = 0.02 |
0 (0, 0.90), p = 0.04 |
Minor allele frequency refers to minor allele frequency in UK Biobank. MAF, minor allele frequency; Dir., direction; SD, standard deviation; OR, odds ratio
R200Ter is enriched among individuals of Ashkenazi Jewish ancestry (0.4% frequency) compared to European ancestry (0.006% frequency). Therefore, in a sensitivity analysis, we matched each R200Ter carrier to the three individuals in UK Biobank who were most similar in ancestry. Carriers of R200Ter had lower ALT levels than non-carriers matched in ancestry (-0.26 SD, p = 0.03). R200Ter similarly associated with lower ALT levels (-0.24 SD, p = 0.04) and total cholesterol levels (-0.23 SD, p = 0.05) when individuals of European and Ashkenazi Jewish ancestry were analyzed separately. In Million Veteran Program, no cases of cirrhosis were observed among 15 R200Ter carriers of European ancestry and no cases of cirrhosis were observed among 6 R200Ter carriers of Ashkenazi Jewish ancestry.
To test whether other coding variants in MARC1 may influence liver disease risk, we conditioned on A165T and tested whether other variants in MARC1 associated with ALT levels and cholesterol. We identified a low-frequency missense variant in MARC1 p.M187K (1.0% frequency in Europeans) that also associated with lower total cholesterol levels (-0.053 SD, 7*10−7) and lower ALT levels (-0.032 SD, p = 0.001). This variant also associated with protection from cirrhosis (OR 0.75, p = 0.01). Similar to MARC1 p.A165T, p.M187K also associated with protection from physician-diagnosed fatty liver (OR 0.76, p = 0.02) and with protection from any liver disease (either cirrhosis or fatty liver, OR 0.75, p = 0.001). MARC1 p.A165T, p.M187K and p.R200Ter form an allelic series which protect against elevated cholesterol levels, elevated ALT levels and risk of cirrhosis (Table 2).
Identification of an individual homozygous for a predicted loss-of-function variant in MARC1
We wondered if homozygous MARC1 deficiency might be tolerated in humans. From exome sequencing data in the T2D-GENES consortium, we identified an individual who was homozygous for the loss-of-function variant R200Ter. Although we did not have liver enzyme measurements for this individual, a blood lipid profile was available.
This individual had low LDL cholesterol levels (46 mg/dl, Table 3) despite not receiving any lipid lowering therapy. However, the participant had elevated triglyceride levels (375 mg/dl, Table 3). These results suggest that homozygous MARC1 deficiency is both tolerated and results in a lipid phenotype (lower LDL cholesterol levels but higher triglyceride levels) similar to the common A165T variant.
Table 3. Blood lipids in an individual homozygous for a predicted loss-of-function variant in MARC1.
Blood lipids were measured in fasting state.
| Genotype | Homozygous for R200Ter |
|---|---|
| Age | 50 |
| Total Cholesterol, mg/dl | 159 |
| LDL Cholesterol, mg/dl | 46 |
| HDL Cholesterol, mg/dl | 38 |
| Triglycerides, mg/dl | 375 |
Discussion
Here, we identify MARC1 A165T as a novel genetic determinant of fatty liver and all-cause cirrhosis. We further identify an allelic series of coding variants in MARC1, including M187K and R200Ter, which associate with lower liver enzymes levels, lower blood cholesterol levels and protection from liver disease. These findings suggest that therapeutic MARC1 antagonism may be useful for prevention and treatment of liver disease.
MARC1 encodes Mitochondrial Amidoxime-Reducing Component 1, a molybdenum-containing enzyme [22]. It contains an N-terminal transmembrane helix that anchors the protein to the outer mitochondrial membrane, with the enzymatic domain of MARC1 located in the cytosol [23]. The crystal structure of MARC1 was recently described [24]. The molybdenum cofactor is coordinated in a solvent exposed center by predominantly positively charged amino acids. The A165 residue lies within an alpha helix in the N-terminal domain of MARC1. The function of MARC1 is unknown, however, it has been reported to activate N-hydroxylated prodrugs [25], reduce nitrite to produce nitric oxide [26] and detoxify trimethylamine N-oxide [27].
The mechanism by which MARC1 may contribute to liver damage and cirrhosis is unclear. The lack of association of MARC1 p.A165T and HSD17B13 rs72613567 with CAD (in contrast to PNPLA3 and TM6SF2) suggests that pharmacologic treatment of cirrhosis and hepatic steatosis may not universally cause excess cardiovascular risk.
A limitation of the current study is that the cohorts analyzed were overwhelmingly of European ancestry (83% in UK Biobank). Analysis of trans-ethnic cohorts may clarify whether variation in MARC1 contributes to cirrhosis protection among individuals of non-European ancestry. Analysis of biopsy-diagnosed samples may also clarify whether, like HSD17B13 [16,17], MARC1 has differing associations with fatty liver, non-alcoholic steatohepatitis and cirrhosis. Additionally, in this report, we do not provide any biochemical or functional evidence linking MARC1 to liver disease; such studies are warranted and underway.
Despite the substantial burden of disease posed by cirrhosis worldwide [28], identification of genetic risk factors has been limited relative to other common diseases such as type 2 diabetes, CAD or inflammatory bowel disease. Here we show that joint analysis of alcoholic and non-alcoholic cirrhosis cases from multiple cohorts increases statistical power to identify genetic variants that influence cirrhosis, to identify novel therapeutic targets and to further our understanding of this disease.
Methods
Association of known alcoholic and non-alcoholic cirrhosis variants with all-cause cirrhosis in UK Biobank
To examine whether known alcoholic and non-alcoholic cirrhosis variants associate with all-cause cirrhosis, we tested the association of six known cirrhosis variants (PNPLA3 I48M, TM6SF2 E167K, MBOAT7 rs641738, HSD17B13 rs72613567, HFE C282Y and SERPINA1 E366K[11,16,18,19]) with all-cause cirrhosis in UK Biobank (hospitalization or death due to ICD codes K70.2, K70.3, K70.4, K74.0, K74.1, K74.2, K74.6, K76.6, or I85). To examine whether this approach increased power relative to examining subtypes of alcoholic and non-alcoholic cirrhosis, we compared the significance of the association of these variants with all-cause cirrhosis (their Z-scores) to the significance of the association of these variants separately with alcoholic cirrhosis and with non-alcoholic cirrhosis. Alcoholic cirrhosis was defined as physician-diagnosed alcoholic cirrhosis or alcoholic liver failure (ICD codes K70.2, K70.3 or K70.4). Non-alcoholic cirrhosis was defined as non-alcoholic cirrhosis (ICD codes K74.0, K74.1, K74.2, K74.6, K76.6 or I85) that occurred among individuals who drank less than fourteen alcoholic drinks per week. We excluded former drinkers (individuals who previously consumed alcohol but stopped) from analysis of non-alcoholic cirrhosis, as these individuals may have previously consumed alcohol but quit due to adverse effects [29]. We tested the association of each of the six variants with all-cause cirrhosis, alcoholic cirrhosis and non-alcoholic cirrhosis in UK Biobank using logistic regression adjusted for age, sex, ten principal components of ancestry and a dummy variable for array type.
Genome wide association study for all-cause cirrhosis
We conducted a genome wide association for all-cause cirrhosis using five cohorts: UK Biobank, Partners Biobank, Atherosclerosis Risk in Communities study (ARIC) and summary statistics from two cohorts from a prior genome wide association study of alcoholic cirrhosis [11]. Definitions of cirrhosis used in each of the five cohorts are provided (S1 Table). We excluded cases of cirrhosis secondary to primary biliary cholangitis and primary sclerosis cholangitis as these autoimmune disorders are directed against the biliary (and not hepatic) parenchyma [30,31].
For UK Biobank, genotyping was performed using either the UK BiLEVE Axiom array or the UK Biobank Axiom array. Phasing and imputation were performed centrally, by UK Biobank, using the Haplotype Reference Consortium and a reference panel of UK 10K merged with the 1000 Genomes phase 3 panel. One related individual of each related pair of individuals, individuals whose genetic sex did not match self-reported sex and individuals with an excess of missing genotype calls or more heterozygosity than expected were excluded from analysis. For Partners Biobank, genotyping was performed using Illumina MEGA array. Variants were imputed to the HapRef consortium using the Michigan Imputation Server [32]. For ARIC, genotyping was performed using the Affymetrix 6.0 array. Variants were imputed to the HapRef consortium using the Michigan Imputation Server. We excluded any variants with an imputation quality < 0.3.
Genome wide association study in each cohort was performed using logistic regression with adjustment for age, sex and principal components of ancestry. In UK Biobank, Partners Biobank, ARIC, BioVU and FinnGen, ancestry was controlled for through inclusion of ten principal component of ancestry covariates. In MVP, ancestry was controlled for through inclusion of five principal component of ancestry covariates. In the AlcCir consortium case-control studies, ancestry was adjusted for by restricting analysis to White British and German populations. We tested the association of fourteen million variants with minor allele frequency of greater than 0.1% with cirrhosis in each cohort. PLINK was used for all analyses [33]. To combine estimates across cohorts, inverse variance fixed effects meta-analysis, as implemented by METAL, was used [34]. Quantile-quantile analysis was used to examine for the presence of population stratification. No evidence of inflation was observed (lambda 1.02; S2 Fig). Both additive and recessive analyses were performed.
Replication of the association of MARC1 p.A165T with cirrhosis and fatty liver
We replicated the association of MARC1 p.A165T with all-cause cirrhosis in three cohorts. First, we examined whether MARC1 p.A165T associates with physician-diagnosed all-cause cirrhosis in the Vanderbilt BioVU, a DNA databank linked to de-identified electronic health records. We identified 46,328 individuals of European ancestry with genome-wide genotyping and who were either cases or controls for all-cause cirrhosis. Using hospitalization or death due to ICD-9 (571.2, 571.5, 572.3, 456.0, 456.1, 456.2) or ICD-10 codes (K70.2, K70.3, K70.4, K74.0, K74.1, K74.2, K74.6, K76.6, I85) to define all-cause cirrhosis, 1,328 cases were identified. We identified 45,000 controls using the EHR-based PheWAS approach, which excludes related diseases based on ICD codes [35]. Logistic regression, with adjustment for age, sex and principal components of ancestry, was used to estimate the association of MARC1 p.A165T with cirrhosis in this dataset. Second, in FinnGen, we identified 688 cases of all-cause cirrhosis (ICD-10 K70.2, K70.3, K70.4, K74.0, K74.1, K74.2, K74.6, K76.6, I85) and 83020 controls. Logistic regression, as implemented in SAIGE [36], was used to test the association of MARC1 p.A165T with cirrhosis in this dataset while controlling for age, sex and relatedness within the sample. Third, in the Million Veteran Program, we identified 6,591 cases of all-cause cirrhosis (as defined above) and 217,284 controls among individuals of European ancestry. Logistic regression was also used to estimate the association of MARC1 p.A165T with cirrhosis in this dataset, with adjustment for age, sex and five principal components of ancestry.
To examine whether MARC1 p.A165T associates with fatty liver, we tested the association of this variant with fatty liver in five cohorts. In the Framingham cohort (Offspring Cohort and Third Generation Cohort), we examined whether MARC1 p.A165T associates with hepatic steatosis on CT imaging. 3284 individuals in Framingham with genotype data available underwent multidetector abdominal CT [37]. We measured hepatic steatosis by computing the liver-to-phantom ratio of the average Hounsfield units of three liver measurements to average Hounsfield units of three phantom measurements (to correct for inter-individual differences in penetration), as previously described [37]. We tested the association of the p.A165T variant with liver-to-phantom ratio with adjustment for age, sex and ten principal components of ancestry using a linear mixed model to control for relatedness among individuals. The liver-to-phantom ratio was subject to inverse normal transformation prior to analysis. In the Multi-Ethnic Study of Atherosclerosis cohort (MESA), 4195 individuals underwent multidetector CT. Hepatic steatosis was measured as the mean of three attenuation measurements (two in the right lobe of the liver and one in the left lobe). No phantom measurement was available for standardization. We therefore tested the association of the p.A165T variant with mean liver attenuation with adjustment for age, sex and ten principal components of ancestry. Mean liver attenuation was subject to inverse normal transformation prior to analysis. Individuals with higher liver fat have lower liver-to-phantom ratios and liver attenuation measurements. For interpretability, we therefore report all estimates in units of standard deviation increases in liver fat, with a one standard deviation increase in liver fat corresponding to a one standard deviation decrease in the liver-to-phantom ratio or mean liver attenuation.
In three cohorts (Partners Biobank, UK Biobank, BioVU), we lacked CT imaging data to measure hepatic steatosis. We therefore tested the association of the MARC1 p.A165T variant with physician-diagnosed fatty liver in these cohorts (ICD codes K76.0 fatty change of liver, K75.81 non-alcoholic steatohepatitis) using logistic regression, adjusted for age, sex and ten principal components of ancestry. We pooled estimates of the association of MARC1 p.A165T with fatty liver across all five cohorts using fixed effects meta-analysis [34].
Association of MARC1 p.A165T with liver enzyme levels, metabolic traits and disease
We tested the association of p.A165T with metabolic traits using four different datasets. For serum levels of liver enzymes, we used data from UK Biobank (n = 386273), Partners Biobank (n = 26,471), Framingham (n = 3,288), LOLIPOP (n = 54,857) [38], BioBank Japan (n = 134,182) [39] where measures of serum alanine transaminase (ALT), aspartate transaminase (AST) and alkaline phosphatase (ALP) were available. We log transformed ALT, AST and ALP. We then conducted a linear regression analysis with adjustment for age, sex and ten principal components of ancestry. We pooled estimates across cohorts using inverse variance weighted fixed effects meta-analysis. For lipids [low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, triglycerides and total cholesterol], we used data from UK Biobank (n = 386,435) and the Global Lipids Genetics Consortium, a meta-analysis of 188 587 individuals of European descent [40]. This GWAS included 37 studies genotyped using the Illumina Metabochip array as well as an additional 23 studies genotyped using a variety of arrays. For BMI and WHRadjBMI we used data from the Genetic Investigation of ANthropometric Traits (GIANT) consortium [41,42]. For WHRadjBMI, data from 210,088 individuals of European ancestry were included. For BMI, data for 322,154 individuals of European ancestry were included. Individuals were genotyped using various arrays and imputed with the HapMap reference panel to 2.5 million SNPs. For blood pressure, we used data from UK Biobank. We tested the association of p.A165T with systolic blood pressure and diastolic blood pressure using linear regression with adjustment for age, sex and ten principal components of ancestry.
A phenome-wide association study of MARC1 p.A165T in UK Biobank was performed [43,44]. We tested the association of MARC1 p.A165T with diseases with more than one thousand cases in UK Biobank. Definitions for 31 different diseases analyzed in the phenome-wide association study are provided (S8 Table). The association of p.A165T with each disease was estimated using logistic regression with adjustment for age, sex, ten principal components of ancestry and a dummy variable for array type. A Bonferroni adjusted significance level of p < 0.0016 (0.05/31) was used.
Association of a rare stop codon in MARC1 with cholesterol levels, liver enzyme levels and cirrhosis
MARC1 p.A165T associated with lower total cholesterol levels, lower alanine transaminase levels and reduced risk of cirrhosis. To examine whether MARC1 deficiency may therefore protect against these three phenotypes, we leveraged a rare (frequency 0.009%) stop codon in MARC1 (Arg200Ter, rs139321832). This stop codon truncates the enzymatic domain of MARC1 before the catalytic site [24]. We tested the association of this rare stop variant with three phenotypes.
First, we examined the association of this stop codon in MARC1 with total cholesterol using data from the Global Lipids Genetics Consortium exome chip analysis (n = 283474) and UK Biobank (n = 386435) [20]. We also included additional rare predicted loss of function variants from two exome sequence datasets: the Myocardial Infarction Genetics consortium (n = 27034) and the T2D Genes consortium (n = 18456). Sequence data for MARC1 were extracted from exome sequencing performed in the MIGen Consortium and T2D Genes consortiums as previously described [45,46]. Estimates were adjusted for age, sex and five principle components of ancestry. We pooled estimates from all three data sources using inverse variance weighted fixed effects meta-analysis.
Second, we examined the association of the rare stop codon with ALT levels using data from UK Biobank and the Partners Biobank cohort. We tested for the association of this variant with inverse normal transformed ALT levels using linear regression with adjustment for age, sex and ten principal components of ancestry.
Third, we examined the association of the rare stop codon with cirrhosis risk using data from UK Biobank, Partners Biobank and Million Veteran Program. Odds ratios for cirrhosis were pooled across cohorts using meta-analysis. As this variant is more frequently observed in Ashkenazi Jewish ancestry than individuals of European ancestry, we examined whether this variant also associates with liver traits with individuals of Ashkenazi Jewish ancestry excluded. Individuals with Ashkenazi Jewish ancestry were identified using principal component analysis, as previously performed [47]. Self-reported Ashkenazi ancestry was plotted against all ten principal components in UK Biobank. As previously reported, self-reported Ashkenazi Jewish ancestry was found to segregate with principal component four. All individuals in this Ashkenazi Jewish cluster were excluded from analysis in a sensitivity analysis. We also examined whether this rare stop variant associated with protection from liver disease when each individual who carried it was matched to three individuals with the most similar principal components of ancestry.
Fourth, we examined whether homozygosity for predicted loss-of-function variants, as defined above, was tolerated. In UK Biobank, the Myocardial Infarction Genetics consortium and the T2D Genes Consortium, we examined the genotype of all loss-of-function variant carriers to determine if they were homozygous for a loss-of-function variant. For any homozygous individual, we examined if blood lipids, available in all three cohorts, differed from the corresponding general population.
Ethics statement
This research has been conducted using the UK Biobank resource and was approved by the UK Biobank application committee, application 7089.
Supporting information
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Acknowledgments
This research has been conducted using the UK Biobank resource, application 7089.
Data Availability
All genetic associations are reported in the manuscript. The raw data can be accessed upon application to UK Biobank (https://www.ukbiobank.ac.uk/using-the-resource/).
Funding Statement
This research has been conducted using the UK Biobank resource, application 7089. This work was funded by the National Institutes of Health (R01 HL127564 to S.K.). Samples for the Leicester cohort were collected as part of projects funded by the British Heart Foundation (British Heart Foundation Family Heart Study, RG2000010; UK Aneurysm Growth Study, CS/14/2/30841) and the National Institute for Health Research (NIHR Leicester Cardiovascular Biomedical Research Unit Biomedical Research Informatics Centre for Cardiovascular Science, IS_BRU_0211_20033). NJS is supported by the British Heart Foundation and is a NIHR Senior Investigator. The Munich MI Study is supported by the German Federal Ministry of Education and Research (BMBF) in the context of the e:Med program (e:AtheroSysMed) and the FP7 European Union project CVgenes@target (261123). Additional grants were received from the Fondation Leducq (CADgenomics: Understanding Coronary Artery Disease Genes, 12CVD02).This study was also supported through the Deutsche Forschungsgemeinschaft cluster of excellence “Inflammation at Interfaces” and SFB 1123. The Italian Atherosclerosis, Thrombosis, and Vascular Biology (ATVB) Study was supported by a grant from RFPS-2007-3-644382 and Programma di ricerca Regione-Universita? 2010-2012 Area 1–Strategic Programmes–Regione Emilia-Romagna. Funding for the exome-sequencing project (ESP) was provided by RC2 HL103010 (HeartGO), RC2 HL102923 (LungGO), and RC2 HL102924 (WHISP). Exome sequencing was performed through RC2 HL102925 (BroadGO) and RC2 HL102926 (SeattleGO). The JHS is supported by contracts HHSN268201300046C, HHSN268201300047C, HHSN268201300048C, HHSN268201300049C, HHSN268201300050C from the National Heart, Lung, and Blood Institute and the National Institute on Minority Health and Health Disparities. Dr. Wilson is supported by U54GM115428 from the National Institute of General Medical Sciences. Exome sequencing in ATVB, PROCARDIS, Ottawa, PROMIS, Southern German Myocardial Infarction Study, and the Jackson Heart Study was supported by 5U54HG003067 (to Dr. Gabriel). Dr. Khera is supported by an institutional grant from the Broad Institute of MIT and Harvard (BroadIgnite), award numbers 1K08HG010155 and 5UM1HG008895 from the National Human Genome Research Institute, a Hassenfeld Scholar Award from Massachusetts General Hospital. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12: 581–594. 10.1038/nrd4051 [DOI] [PubMed] [Google Scholar]
- 2.King EA, Davis JW, Degner JF. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. Marchini J, editor. Genet PLoS. 2019;15: e1008489 10.1371/journal.pgen.1008489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dewey FE, Gusarova V, Dunbar RL, O'Dushlaine C, Schurmann C, Gottesman O, et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N Engl J Med. Massachusetts Medical Society; 2017;377: 211–221. 10.1056/NEJMoa1612790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gusarova V, O'Dushlaine C, Teslovich TM, Benotti PN, Mirshahi T, Gottesman O, et al. Genetic inactivation of ANGPTL4 improves glucose homeostasis and is associated with reduced risk of diabetes. Nat Commun. Nature Publishing Group; 2018;9: 2252–11. 10.1038/s41467-018-04611-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khetarpal SA, Zeng X, Millar JS, Vitali C, Somasundara AVH, Zanoni P, et al. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med. Nature Publishing Group; 2017;23: 1086–1094. 10.1038/nm.4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J Am Coll Cardiol. 2017;69: 2054–2063. 10.1016/j.jacc.2017.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu, European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. Journal of hepatology. 2018. pp. 406–460. 10.1016/j.jhep.2018.03.024 [DOI] [PubMed] [Google Scholar]
- 8.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371: 838–851. 10.1016/S0140-6736(08)60383-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40: 1461–1465. 10.1038/ng.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. Nature Publishing Group; 2014;46: 352–356. 10.1038/ng.2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buch S, Stickel F, Trépo E, Way M, Herrmann A, Nischalke HD, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet. 2015;47: 1443–1448. 10.1038/ng.3417 [DOI] [PubMed] [Google Scholar]
- 12.Speliotes EK, Butler JL, Palmer CD, Voight BF, GIANT consortium, MIGen Consortium, et al. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology. Wiley-Blackwell; 2010;52: 904–912. 10.1002/hep.23768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y-L, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JBS, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. Nature Publishing Group; 2014;5: 4309 10.1038/ncomms5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valenti L, Rumi M, Galmozzi E, Aghemo A, Del Menico B, De Nicola S, et al. Patatin-like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology. Wiley-Blackwell; 2011;53: 791–799. 10.1002/hep.24123 [DOI] [PubMed] [Google Scholar]
- 15.Liu Z, Que S, Zhou L, Zheng S, Romeo S, Mardinoglu A, et al. The effect of the TM6SF2 E167K variant on liver steatosis and fibrosis in patients with chronic hepatitis C: a meta-analysis. Sci Rep. Nature Publishing Group; 2017;7: 9273 10.1038/s41598-017-09548-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med. 2018;378: 1096–1106. 10.1056/NEJMoa1712191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.About F, Abel L, Cobat A. HCV-Associated Liver Fibrosis and HSD17B13. N Engl J Med. Massachusetts Medical Society; 2018;379: 1875–1876. 10.1056/NEJMc1804638 [DOI] [PubMed] [Google Scholar]
- 18.Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS , American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases Hepatology (Baltimore, Md: ). Wiley-Blackwell; 2011. pp. 328–343. 10.1002/hep.24330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dürr R, Caselmann WH. Carcinogenesis of primary liver malignancies. Langenbecks Arch Surg. 2000;385: 154–161. 10.1007/s004230050259 [DOI] [PubMed] [Google Scholar]
- 20.Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet. Nature Publishing Group; 2017;49: 1758–1766. 10.1038/ng.3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med. Massachusetts Medical Society; 2016;374: 1134–1144. 10.1056/NEJMoa1507652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Havemeyer A, Bittner F, Wollers S, Mendel R, Kunze T, Clement B. Identification of the missing component in the mitochondrial benzamidoxime prodrug-converting system as a novel molybdenum enzyme. J Biol Chem. American Society for Biochemistry and Molecular Biology; 2006;281: 34796–34802. 10.1074/jbc.M607697200 [DOI] [PubMed] [Google Scholar]
- 23.Klein JM, Busch JD, Potting C, Baker MJ, Langer T, Schwarz G. The mitochondrial amidoxime-reducing component (mARC1) is a novel signal-anchored protein of the outer mitochondrial membrane. J Biol Chem. American Society for Biochemistry and Molecular Biology; 2012;287: 42795–42803. 10.1074/jbc.M112.419424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kubitza C, Bittner F, Ginsel C, Havemeyer A, Clement B, Scheidig AJ. Crystal structure of human mARC1 reveals its exceptional position among eukaryotic molybdenum enzymes. Proc Natl Acad Sci USA. National Academy of Sciences; 2018;34: 201808576 10.1073/pnas.1808576115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gruenewald S, Wahl B, Bittner F, Hungeling H, Kanzow S, Kotthaus J, et al. The fourth molybdenum containing enzyme mARC: cloning and involvement in the activation of N-hydroxylated prodrugs. J Med Chem. American Chemical Society; 2008;51: 8173–8177. 10.1021/jm8010417 [DOI] [PubMed] [Google Scholar]
- 26.Sparacino-Watkins CE, Tejero J, Sun B, Gauthier MC, Thomas J, Ragireddy V, et al. Nitrite reductase and nitric-oxide synthase activity of the mitochondrial molybdopterin enzymes mARC1 and mARC2. J Biol Chem. American Society for Biochemistry and Molecular Biology; 2014;289: 10345–10358. 10.1074/jbc.M114.555177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider J, Girreser U, Havemeyer A, Bittner F, Clement B. Detoxification of Trimethylamine N-Oxide by the Mitochondrial Amidoxime Reducing Component mARC. Chem Res Toxicol. American Chemical Society; 2018;31: 447–453. 10.1021/acs.chemrestox.7b00329 [DOI] [PubMed] [Google Scholar]
- 28.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Elsevier; 2012;380: 2095–2128. 10.1016/S0140-6736(12)61728-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roerecke M, Rehm J. Ischemic heart disease mortality and morbidity rates in former drinkers: a meta-analysis. American journal of epidemiology. 2011;173: 245–258. 10.1093/aje/kwq364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu F, Tang R, Zuo X, Shi X, Wei Y, Zheng X, et al. A genome-wide association study identifies six novel risk loci for primary biliary cholangitis. Nat Commun. Nature Publishing Group; 2017;8: 14828 10.1038/ncomms14828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji S-G, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet. Nature Publishing Group; 2017;49: 269–273. 10.1038/ng.3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. Nature Publishing Group; 2016;48: 1279–1283. 10.1038/ng.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4: 7 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26: 2190–2191. 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26: 1205–1210. 10.1093/bioinformatics/btq126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. Nature Publishing Group; 2018;50: 1335–1341. 10.1038/s41588-018-0184-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Speliotes EK, Massaro JM, Hoffmann U, Foster MC, Sahani DV, Hirschhorn JN, et al. Liver fat is reproducibly measured using computed tomography in the Framingham Heart Study. J Gastroenterol Hepatol. Wiley/Blackwell (10.1111); 2008;23: 894–899. 10.1111/j.1440-1746.2008.05420.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, van der Harst P, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. Nature Publishing Group; 2011;43: 1131–1138. 10.1038/ng.970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanai M, Akiyama M, Takahashi A, Matoba N, Momozawa Y, Ikeda M, et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet. Nature Publishing Group; 2018;50: 390–400. 10.1038/s41588-018-0047-6 [DOI] [PubMed] [Google Scholar]
- 40.Global Lipids Genetics Consortium, Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45: 1274–1283. 10.1038/ng.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Mägi R, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. Nature Publishing Group; 2015;518: 187–196. 10.1038/nature14132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. Nature Publishing Group; 2015;518: 197–206. 10.1038/nature14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emdin C, Khera AV, Klarin D, Natarajan P, Zekavat SM, Nomura A, et al. Phenotypic Consequences of a Genetic Predisposition to Enhanced Nitric Oxide Signaling. Circulation. American Heart Association, Inc; 2018;137: 222–232. 10.1161/CIRCULATIONAHA.117.028021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emdin C, Khera AV, Chaffin M, Klarin D, Natarajan P, Aragam K, et al. Analysis of predicted loss-of-function variants in UK Biobank identifies variants protective for disease. Nat Commun. Nature Publishing Group; 2018;9: 1613 10.1038/s41467-018-03911-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Do R, Stitziel NO, Won H-H, Jørgensen AB, Duga S, Angelica Merlini P, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. Nature Publishing Group; 2015;518: 102–106. 10.1038/nature13917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khera AV, Won H-H, Peloso GM, Lawson KS, Bartz TM, Deng X, et al. Diagnostic Yield of Sequencing Familial Hypercholesterolemia Genes in Patients with Severe Hypercholesterolemia. J Am Coll Cardiol. 2016. 10.1016/j.jacc.2016.03.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Need AC, Kasperaviciute D, Cirulli ET, Goldstein DB. A genome-wide genetic signature of Jewish ancestry perfectly separates individuals with and without full Jewish ancestry in a large random sample of European Americans. Genome Biol. BioMed Central; 2009;10: R7–7. 10.1186/gb-2009-10-1-r7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Data Availability Statement
All genetic associations are reported in the manuscript. The raw data can be accessed upon application to UK Biobank (https://www.ukbiobank.ac.uk/using-the-resource/).