

Abstract
The maternal environment during pregnancy is critical for fetal development and perinatal perturbations can prime offspring disease risk. Here, we briefly review evidence linking two well-characterized maternal stressors – psychosocial stress and infection – to increased neuropsychiatric risk in offspring. In the current climate of increasing obesity and globalization of the Western-style diet, maternal overnutrition emerges as a pressing public health concern. We focus our attention on recent epidemiological and animal model evidence showing that, like psychosocial stress and infection, maternal overnutrition can also increase offspring neuropsychiatric risk. Using lessons learned from the psychosocial stress and infection literature, we discuss how altered maternal and placental physiology in the setting of overnutrition may contribute to abnormal fetal development and resulting neuropsychiatric outcomes. A better understanding of converging pathophysiological pathways shared between stressors may enable development of interventions against neuropsychiatric illnesses that may be beneficial across stressors.
Keywords: Developmental origins of health and disease, Maternal stress, Maternal diet, Maternal obesity, Maternal immune activation, Inflammation, Allostatic load, Neuropsychiatric risk, Autism spectrum disorders, Schizophrenia, Anxiety, Depression
1. Introduction
The focus of this review is on neuropsychiatric risk associated with in utero exposure to maternal environmental stressors. The field of stress research has largely focused on the health effects of psychosocial or physiological stressors on hypothalamic–pituitary–adrenal (HPA) axis function in adult humans and animal model systems. Yet it has become increasingly clear that the HPA axis is not the only regulatory system sensitive to the effects of stress, and that these effects not only act on adults, but also occur throughout the lifespan. The emergence of the concepts of “allostasis” and “allostatic overload” has broadened the definition of environmental stressors and study of pathophysiological mechanisms across a range of regulatory systems. In parallel, the developmental origins of health and disease (DOHaD) hypothesis has provided a developmental context.
The DOHaD, which emerged as a broadening of the “Barker hypothesis” and was named for epidemiologist David Barker, posits that early life environmental exposures can pattern risk for and severity of later life disease. Since Barker’s initial studies on the cardiovascular and metabolic effects of low birth weight (Barker, 2007; Barker and Osmond, 1986), epidemiological studies have associated exposure to under- and overnutrition (Armitage et al., 2008; Calkins and Devaskar, 2011), psychosocial stress and trauma (King et al., 2012; Van den Bergh et al., 2017; Vaiserman, 2015), infection (Bilbo and Schwarz, 2012; Izvolskaia et al., 2018), and toxins (Young et al., 2018; Drwal et al., 2019) during development with adverse effects on offspring across the lifespan and across a range of organ systems. To understand this mechanistically, we consider the concept of allostatic load. Pregnancy can be considered a state of allostasis, where the maternal-fetal dyad must balance dual goals of maternal and fetal stability across all organ systems, and across the developmental trajectory (Power and Schulkin, 2012). Additional environmental challenges during this time could result in allostatic overload that not only affects the health of the mother, but also impacts the health and resilience of the developing fetus.
It is clear that the developing brain, like other organ systems, is vulnerable to the effects of an adverse early environment (Giussani, 2011). Maternal adversity has been linked to a spectrum of neuropsychiatric problems – from neurodevelopmental diseases to psychiatric disorders to late life neurodegenerative disease – and some suggest that many brain diseases should be considered at least partially neurodevelopmental in origin (Levitt and Veenstra-VanderWeele, 2015; Heindel and Vandenberg, 2015). Here, we begin with an overview of the epidemiological and animal model evidence for two maternal stressors, psychosocial stress and infection, in priming offspring neuropsychiatric risk. In the current climate of increasing obesity and globalization of the Western-style diet model, maternal overnutrition is an emerging and pressing public health issue. We spend the majority of the review discussing the epidemiological and animal model evidence linking maternal overnutrition with increased neuropsychiatric risk in offspring. The overlapping neuropsychiatric consequences for offspring associated with these three maternal stressors suggests pathophysiological convergence, in line with our current understanding of allostatic overload. We discuss putative mechanisms linking maternal overnutrition and offspring neuropsychiatric risk, highlighting potential converging pathways and lessons learned from the infection and stress fields, and how existing model systems and current technologies may lead to better understanding of the multifactorial etiologies of neuropsychiatric illnesses. Finally, identification of common pathophysiological pathways shared between stressors opens the door to potential interventions that may be beneficial across stressors. We conclude with a brief discussion of potential future directions for the field and implications for intervention.
2. Maternal psychosocial stress & immune activation
The contributions of perinatal stress (Van den Bergh et al., 2017; Bale, 2005; Bale et al., 2010; Boersma et al., 2014; Entringer et al., 2015; Chan et al., 2018) and maternal immune activation (Bilbo and Schwarz, 2012; Meyer et al., 2009; Solek et al., 2018; Meyer, 2014; Estes and McAllister, 2016) to offspring neuropsychiatric risk have been extensively reviewed. As such, we will touch on these topics only briefly as a framework to approach the less well-studied role of maternal overnutrition in neuropsychiatric vulnerability.
While it was long established that exposure to stress in adulthood negatively affects mental health, few studies evaluated risks of stress during gestation. Studies of children exposed in utero to disasters, like the 1986 Chernobyl accident or the 1988 Quebec ice storm, provided some of the first associations between perinatal stress exposure and neuropsychiatric outcomes (Boersma et al., 2014; Kolominsky et al., 1999; Laplante et al., 2008). Numerous other epidemiological studies have since expanded this association to prenatal psychosocial stressors of various type, duration, gestational timing, and severity, as well as a range of outcomes. However, recurring limitations in human studies include the difficulty in dissociating prenatal from early postnatal stress and reliance on retrospective self-report of stress experience, which may not adequately account for additional moderating variables.
In recent decades, a large number of studies using animal model systems have allowed for more controlled evaluation of the association between prenatal stress (PNS), timing and severity of stress exposure (Weinstock, 2017; Boersma and Tamashiro, 2015), and neuropsychiatric impairment in offspring. Broadly, offspring exposed to PNS demonstrate behavioral changes relevant to neuropsychiatric disease including but not limited to depression-like behavior (Morley-Fletcher et al., 2003; Morley-Fletcher et al., 2004), anxiety-like behavior (Vallee et al., 1997), alterations in stress coping style (Boersma et al., 2014), cognitive impairment (Vallee et al., 1999), decreased social interaction (Lee et al., 2007), and increased aggression (de Souza et al., 2013). While behavioral changes are consistently reported, phenotype appears highly dependent on type, degree, and timing of maternal stress, as well as sex and age of offspring at evaluation (Boersma et al., 2014). This variance highlights the importance of considering type and severity of stressor when interpreting offspring outcomes from PNS (Lesage et al., 2001), while also pointing to potential critical windows of exposure and thresholds of risk. Among the first investigations of time- and sex-dependent effects of PNS used mild chronic variable stress (CVS) applied to pregnant mice during early, mid, or late gestation (Mueller and Bale, 2006; Mueller and Bale, 2007; Mueller and Bale, 2008). For male offspring, only those exposed to early gestational PNS demonstrated neurobehavioral impairments as adults (Mueller and Bale, 2007; Mueller and Bale, 2008), which were accompanied by increased HPA axis responsivity (Mueller and Bale, 2008). Similarly, only those female offspring exposed to early gestational stress showed changes in behavior, but they exhibited improved learning performance, the opposite outcome compared to their male littermates (Mueller and Bale, 2007; Mueller and Bale, 2008). A growing body of work supports these findings suggesting that females may be less sensitive to the effects of PNS (Bale and Epperson, 2015). That is not to say that female offspring are completely resistant to stress. Instead, gestational stress, particularly in early development, seems to promote a more insidious risk for affective disorders in females compared to risk of autism spectrum disorders (ASDs), behavioral, and intellectual disability in males (Bronson and Bale, 2016; Davis and Pfaff, 2014; Sandman et al., 2013). This dovetails with the sexual dimorphism in neuropsychiatric presentation: among males, the prevalence rates of neurodevelopmental disorders such as ASDs, attention deficit hyperactivity disorder (ADHD), and earlier-onset schizophrenia tend to be higher, whereas females have an increased prevalence of affective disorders (Davis and Pfaff, 2014; McLean et al., 2011; Hantsoo and Epperson, 2017; Fombonne, 2009; Bauermeister et al., 2007; Loomes et al., 2017; Zhang et al., 2012; Froehlich et al., 2007). Moving forward, understanding sexual dimorphism in the developing brain and further elucidating the effects of gestational timing and sex on offspring exposed to maternal stress remains an important direction for the field.
Several mechanisms have been proposed to underlie the relationship between maternal stress and offspring neuropsychiatric phenotypes. The most widely studied are alterations of the HPA axis, including potentially sex-specific changes in the circadian cycle of corticosterone (CORT), HPA axis responsivity, and impaired HPA axis negative feedback (Henry et al., 1994; Barbazanges et al., 1996; Maccari et al., 1995; Koehl et al., 1999). Within the brain, PNS is associated with decreased glucocorticoid and mineralocorticoid receptor binding capacities (Maccari et al., 1995; Koehl et al., 1999), which may alter hippocampal neurogenesis (Gould et al., 1992; Rodriguez et al., 1998; Montaron et al., 1999) and neuronal proliferation (Lemaire et al., 2000; Fujioka et al., 2006; Kawamura et al., 2006). Other mechanisms proposed to mediate the relationship between PNS and brain function include, but are not limited to, alterations in immune regulation and cytokines (Hantsoo et al., 2019), metabolic signaling (Tamashiro et al., 2009), placental function (Bronson and Bale, 2016), neurotrophic signaling (Ruf and Preissner, 2017), microbiome composition (Jasarevic et al., 2015), and epigenetic regulation (Boersma and Tamashiro, 2015), highlighting the diverse and complex actions of early life stress.
While many have focused on effects of PNS, others have studied another common early-life exposure, maternal immune activation (MIA), and there is now abundant evidence of overlapping neuropsychiatric risks between these two “stressors.” Though much of the early epidemiological work focused on links between maternal infection and schizophrenia (Mednick et al., 1988; Patterson, 2009; Meyer, 2014; Patterson, 2009) additional associations have been identified with ASDs (Patterson, 2011), developmental delay (Zerbo et al., 2013), cognitive impairment (Richetto and Riva, 2014; Knuesel et al., 2014), and affective disorders (Simanek and Meier, 2015; Murphy et al., 2017; Parboosing et al., 2013). Again similar to PNS, both clinical epidemiology and translational studies suggest that neuropsychiatric phenotypes associated with maternal infection appear to be influenced by gestational timing, duration, and severity of infectious stressor, as well as age and sex of the offspring (Brown and Meyer, 2018; Estes and McAllister, 2016; Schwartzer et al., 2013; Murray et al., 2019; de Souza et al., 2015; Giovanoli et al., 2015; Vernon et al., 2015). Overall, it appears that early immune stress leads to more extensive neuropsychological consequences, but that late gestation remains another critical and specific window for the effects of exposure (Meyer et al., 2007).
Mechanistically, recent evidence suggests that it is immune system activation, and not a specific pathogen, that is primarily responsible for negative effects on developing offspring (Estes and McAllister, 2016). This observation has been leveraged in pre-clinical studies that bypass specific pathogenic infection and instead directly activate the immune response with the viral mimetic polyinosinic:polycytidylic (poly[I:C]), the bacterial endotoxin lipopolysaccharide (LPS), or specific immune components such as cytokines. Doing so allows for study of generalizable and direct effects of MIA while tightly controlling the type, timing, duration, and intensity of the immune activation (Meyer et al., 2009; Brown and Meyer, 2018; Meyer et al., 2009). Offspring in these studies exhibit deficits in sensorimotor gating, attention, learning and memory, and social behavior, in addition to increased repetitive, anhedonic, and anxiety-like behaviors (Meyer, 2014; Estes and McAllister, 2016; Patterson, 2009; Knuesel et al., 2014). These changes have been linked to aberrations in brain structure and function including decreased cortical thickness, increased ventricular volume, and decreased regional volumes in the hippocampus, amygdala, and striatum (Estes and McAllister, 2016; Patterson, 2009; Piontkewitz et al., 2011). Emerging evidence suggests altered circuit connectivity and altered serotonergic, dopaminergic, and GABAergic signaling in some of these regions (Meyer et al., 2009; Estes and McAllister, 2016; Dickerson and Bilkey, 2013; Missault et al., 2019). At the synaptic level, reported changes include decreased dendritic spine density, shortened dendritic length, reductions in pre- and post-synaptic proteins, and deficits in synaptic transmission and long-term potentiation (Giovanoli et al., 2015; Choi et al., 2016; Coiro et al., 2015; Zhang and van Praag, 2015; Patrich et al., 2016). At the cellular level, there is evidence for altered neurogenesis and neuronal migration, as well as alterations in microglia and astroglia (Solek et al., 2018; de Souza et al., 2015; Zhang et al., 2018; Smolders et al., 2018; Prins et al., 2018; Liu et al., 2013; Oskvig et al., 2012).
While individual application of single immune factors has been shown to be sufficient to induce neuropsychiatric alterations (Smith et al., 2007; Ponzio et al., 2007), the immune system is in constant interplay with other biological systems including the placenta, HPA axis, gut microbiome, metabolism, oxidation, neurotrophic signaling, and epigenetic regulation, and changes within each of these systems are implicated in MIA (Estes and McAllister, 2016; Oskvig et al., 2012; Hsiao and Patterson, 2011; Udagawa and Hino, 2016; Gilmore et al., 2005; Babri et al., 2014; Reisinger et al., 2015; Simoes et al., 2018; Lammert et al., 2018). This reflects the complex nature of the etiology of neuropsychiatric risk, but it also points to commonalities between MIA and PNS in producing similar behavioral phenotypes – a concept we will return to at the end of this review.
3. Maternal overnutrition
3.1. Background and summarized clinical findings
While psychosocial stress and infection are among the most widely studied stressors experienced during pregnancy, another major stressor has emerged in the current climate of a global obesity epidemic: maternal overnutrition. Nearly two-thirds of women of reproductive age in the United States are overweight or obese, and rapidly climbing obesity rates in developing countries indicate that this is a worldwide public health problem (Flegal et al., 2010; Hillemeier et al., 2011; Chen et al., 2018). Obesity and overweight are associated with a range of metabolic and cardiovascular comorbidities which, in pregnant women, increase risk for additional complications including pre-eclampsia and gestational diabetes mellitus (GDM) (Sohlberg et al., 2012; Mission et al., 2015; Stang and Huffman, 2016). Exposure to maternal obesity, high-energy diet, and associated metabolic consequences are consistently associated with a multitude of adverse health outcomes in offspring (Nguyen et al., 2015; Rivera et al., 2015; Maftei et al., 2015; Olson et al., 2009; Brekke et al., 2007), including neuropsychiatric disease (Rivera et al., 2015; Olfson et al., 2014; Boyle et al., 2011). Here we review epidemiological and animal model evidence for the association between these maternal conditions and offspring neuropsychiatric risk.
Epidemiological studies suggest a relationship between maternal overnutrition and both risk for and severity of several neurodevelopmental and psychiatric illnesses, including but not limited to ASDs, ADHD, affective disorders, and schizophrenia (Rivera et al., 2015) (Table 1). Maternal overnutrition is additionally associated with cognitive impairments in children and adolescents, and has been suggested to predispose the development of later-life cognitive dementias (Contu and Hawkes, 2017). However, it is important to acknowledge that across these studies there exists no consensus on how to determine or quantify “maternal overnutrition.” The most common determinations are based on body mass index (BMI, both pre- or post-pregnancy), weight gain during pregnancy (gestational weight gain, GWG), prepregnancy type 2 diabetes mellitus (T2DM), and the presence of gestational metabolic disorders including GDM (Rivera et al., 2015; Edlow, 2017). Given the high comorbidity and significant interplay between these conditions – and the resulting overlap in animal model systems used to investigate associated neuropsychiatric outcomes in offspring – we have grouped this discussion under “maternal overnutrition” as an umbrella terminology.
Table 1.
Summarized clinical studies associating maternal overnutrition with neuropsychiatric risk to offspring. Studies referenced are limited to the present review.
| Offspring outcome | Measure of maternal overnutrition | Ref # | Concluded association | Study design | Notes |
|---|---|---|---|---|---|
| ↑ Risk and Severity of ASDs | ↑ Pre-pregnancy BMI | (Reynolds et al, 2014) | Yes | Cohort | |
| (Moss and Chugani, 2014) | Yes | Cohort | |||
| (Krakowiak et al, 2012) | Yes | Case-Control | |||
| (Dodds et al., 2011) | Yes | Cohort | |||
| (Getz et al., 2016) | Yes | Case-Control | |||
| (Gardner et al., 2015) | Inconclusive | Cohort | Population-based association disappears in matched sibling analysis | ||
| (Li et al., 2016) | Inconclusive | Case-Control | Association in combination with maternal diabetes | ||
| (Bilder et al., 2013) | No | Cohort, Case-Control | |||
| ↑ Gestational weight gain (GWG) | (Dodds et al., 2011) | Yes | Cohort | ||
| (Gardner et al., 2015) | Yes | Cohort | |||
| (Bilder et al., 2013) | Yes | Cohort, Case-Control | |||
| (Reynolds et al., 2014) | No | Cohort | |||
| Metabolic Complications (Diabetes, | (Krakowiak et al., 2012) | Yes | Case-Control | ||
| Hypertension, Pre-Eclampsia) | (Dodds et al., 2011) | Yes | Cohort | ||
| (Wallace et al., 2008) | Yes | Cohort | |||
| (Li et al., 2016) | Yes | Case-Control | |||
| (Reynolds et al., 2014) | No | Cohort | |||
| ↑ Risk of Cognitive | ↑ Pre-pregnancy BMI | (Basatemur et al., 2013) | Yes | Cohort | |
| Impairment | (Bliddal et al., 2014) | Yes | Cohort | ||
| (Casas et al., 2013) | Yes | Cohort | |||
| (Craig et al., 2013) | Yes | Case-Control | |||
| (Eriksen et al., 2013) | Yes | Cohort | |||
| (Hinkle et al., 2012) | Yes | Cohort | |||
| (Huang et al., 2014) | Yes | Cohort | |||
| (Neggers et al., 2003) | Yes | Cohort | |||
| (Brion et al., 2011) | Inconclusive | Cohort | Inconsistent between cohorts; potential confounder effect | ||
| (Li et al., 2016) | Inconclusive | Case-Control | Association in combination with maternal diabetes | ||
| (Krzeczkowski et al., 2018) | Inconclusive | Cohort | Association disappears when accounting for maternal diet | ||
| Metabolic Complications (Diabetes, Hypertension, Pre-Eclampsia) | (Li et al., 2016) | Yes | Case-Control | ||
| (Krzeczkowski et al., 2018) | Inconclusive | Cohort | Association disappears when accounting for maternal diet | ||
| ↑ Risk of Anxiety or Depression | ↑ Pre-pregnancy BMI | (Van Lieshout et al., 2013) | Yes | Cohort | |
| (Mina et al., 2017) | Yes | Cohort | |||
| (Rodriguez, 2010) | Yes | Cohort | |||
| (Robinson et al., 2013) | Yes | Cohort | |||
| ↑ Risk of Schizophrenia | ↑ Pre-pregnancy BMI | (Jones et al., 1966) | Yes | Cohort | |
| (Schaefer et al., 2000) | Yes | Cohort | |||
| ↑ GWG | (Kawai et al., 2004) | Yes | Case-Control | ||
| Metabolic Complications (Diabetes, Hypertension, Pre-Eclampsia) | (Cannon et al., 2002) | Yes | Meta-analysis |
Despite a growing body of clinical evidence for a correlation between many of these markers of maternal metabolic state and adverse neuropsychiatric outcomes, not all studies support this. As an example, while several studies have linked high pre-pregnancy BMI with risk for ASD (Reynolds et al., 2014; Moss and Chugani, 2014; Krakowiak et al., 2012; Dodds et al., 2011; Getz et al., 2016), one study suggested that this association is due to residual confounds from familial risk factors (Gardner et al., 2015), and other studies found no association between BMI proximal to pregnancy onset and ASD (Bilder et al., 2013). GWG, while less studied, is consistently associated with ASD in all but one study (Reynolds et al., 2014), and numerous studies have identified metabolic complications prior to and during pregnancy, such as pre-existing diabetes, GDM, or hypertension, with ASD (Krakowiak et al., 2012; Dodds et al., 2011; Gardner et al., 2015; Bilder et al., 2013; Wallace et al., 2008). At least in part, these discrepancies likely arise from the fact that clinical studies are often difficult to control and confounders often difficult to identify. Animal models on the other hand, while less directly translatable, have allowed for more controlled studies aimed at disentangling the potential mechanistic players and pathways.
3.2. Animal models
The majority of animal studies in this field use dietary manipulations that result in metabolic disturbances in the dam prior to and/or during pregnancy that can affect the developing fetus. These diets vary in their macronutrient content, but in general they are palatable, energy-dense, high in fat, and, in some cases, high in sugar (Winther et al., 2018; Zambrano and Nathanielsz, 2017; Musial et al., 2017; Mucellini et al., 2014; Bolton et al., 2017; Sun et al., 2012; Niculescu and Lupu, 2009; Tozuka et al., 2010; Wright et al., 2011). Diet exposure is usually continued until the end of gestation or lactation, at which point offspring are most commonly weaned onto a control diet. There is strong evidence that maternal overnutrition changes both behavior and brain physiology in offspring (Table 2). In the next several sections, behavioral and brain changes will be discussed as they relate to features of human neurodevelopmental and psychiatric disease.
Table 2.
Findings from animal models of maternal overnutrition. Models of maternal overnutrition are presented with resulting changes to offspring behavior, brain, and peripheral outcomes; maternal physiology; and placental physiology. Studies referenced are limited to the present review.
| Ref | Species (Strain) | Model | Offspring Behavioral Phenotype | Offspring Physiology, Central | Offspring Physiology, Peripheral | Maternal Physiology | Placental Physiology | Notes |
|---|---|---|---|---|---|---|---|---|
| (Winther et al., 2018) | Rat [Sprague- Dawley (SD)] | 60% High-Fat Diet (HFD), 8 w pre-conception (PC) through gestation (G) and lactation (L) | Anxiety-like behaviour | Hippocampus
|
Pre- / at weaning
|
|
_ | |
| (Musial et al., 2017) | Mouse (C57) | 30% kcal fat, 36% kcal sugar (HFHS diet), through G Dam sacrifice G16 or G19 |
- | - | Fetal, G16
|
G16
|
|
|
| (Sun et al., 2012) | Rat(SD) | 60% HFD, through G and L; Offspring cross-fostered to same- or opposite-diet dam at PI
|
- | Hypothalamus, pre- /atweaning
|
Pre- /at weaning, L HFD
|
|
- | Differential effects by exposure window: ↑ impact on offspring metabolic phenotype with L HFD |
| (Niculescu and Lupu, 2009) | Mouse (C57) | 60% HFD, 10 w PC through G Dam sacrifice G17 |
- | Hippocampus, fetal
|
Fetal
|
|
- | |
| (Tozuka et al., 2010) | Mouse (C57) | 57.5% HFD, 6 w PC through G and L until P16 | Cognitive impairment (adolescent) | Hippocampus, adolescent
|
- |
|
- | Cognitive phenotype normalized by adulthood |
| (Wright et al., 2011) | Rat (Wistar) | Cafeteria diet, 11 w PC through G and L 8 experimental groups limiting HFD to PC, G, L, or a combination of exposure windows (e.g. PC & L only) | Decreased anxiety-like behavior (L exposure, ♂) | - |
|
|
- | Differential effects by exposure window |
| (Cordner et al., 2019) | Rat (SD) | 60% HFD, through G and L | Cognitive impairment | Hippocampus, weaning
|
|
- | - | |
| (White et al., 2009) | Rat (Long Evans) | 60% HFD, 4 w PC through G and L Offspring placed as adults onto HFD or Con | Cognitive impairment (HFD offspring exposed to adult HFD) | With maternal HFD
|
|
|
- | Maternal + offspring diet exposure |
| (Can et al., 2012) | Rat(SD) | 65% HFD, through G and L Offspring weaned onto respective dam diet | Anxiety-like behavior Depression-like behavior Cognitive impairment | Hippocampus
|
|
- | - | Maternal + offspring diet exposure |
| (Robb et al., 2017) | Rat (SD) | 45% HFD, 10 w PC through G and L | Cognitive impairment (adolescent ♂, trend in adult ♂) | - |
|
|
Cognitive phenotype normalized by adulthood; Sex differences: ↑ effect ♂ | |
| (Moser et al., 2017) | Rat (Long Evans) | 60% HFD, 6 w PC through G and L | Cognitive impairment | - | Pre- /at weaning
|
|
- | |
| (Page et al., 2014) | Rat (SD) | 45% HFD, 1 m PC through G and L Offspring weaned onto HFD or Con |
Cognitive impairment (maternal HFD exposure) | Hippocampus, maternal HFD exposure
|
Maternal HFD exposure
|
- | - | |
| (Bilbo and Tsang, 2010) | Rat (SD) | 60% high saturated fat HFD (SFD) or 60% high trans fat HFD (TFD), 4 w PC through G and L | Anxiety-like behavior (SFD or TFD ♂) Improved cognitive performance (SFD or TFD ♂) | Hippocampus, neonate
|
Pre- /at weaning
|
|
||
| (Graf et al., 2016) | Mouse (C57) | 60% HFD, 8 w PC through G and L | Cognitive impairment (♂) | Pre- /at weaing
|
Pre- /at weaning
|
|
- | Sex differences: ↑ effect ♂ |
| (Kang et al., 2014) | Mouse (C57) | 60% HFD,
|
Anxiety-like behavior (♀; HFD-HFD, HFD-Con) Social interaction deficits (♀; HFD-HFD only) Hyperactivity (♂; HFD-HFD, HFD-Con) |
Adult
|
|
|
- | Timing of exposure: ↑ impact PC/G v L HFD, partial mitigation with L switch |
| (Buffington et al., 2016) | Mouse (C57) | 60% HFD, 8 w PC through G and L | Social interaction deficits |
|
|
|
- | |
| (Sasaki et al., 2013) | Rat (Long Evans) | 60% HFD, 4 w PC through G and L | Anxiety-like behavior | Amygdala
|
Pre- /at weaning
|
|
- | Timing of offspring effect: altered phenotype in adolescent (Sasaki et al., 2014) v adult animals |
| (Sasaki et al., 2014) | Rat (Long Evans) | 60% HFD, 4 w PC through G and L | Decreased anxiety-like behavior (adolescent) | Amygdala, adolescent
|
Pre- /at weaning
|
|
- | Timing of offspring effect: altered phenotype in adolescent v adult (Sasaki et al., 2013) animals |
| (Giriko et al., 2013) | Rat (Wistar) | 52% HFD, through L | Delayed reflex development (early life) Depression-like behavior (adult) |
- | Pre- / at weaning
|
|
- | |
| (Lin et al., 2015) | Rat (SD) | 45% HFD, 10 d PC through G and L Adult offspring subjected to chronic unpredictable mild stress (CUMS) | Sensitization to stress-induced depression-like behavior |
|
|
Maternal HFD ↑ sensitization to stress | ||
| (Zieba et al., 2019) | Mouse (C57) | 43% HFD, 5 w PC through G and L | Facilitation of PPI | - |
|
|
- | |
| (Wolfrum and Peleg-Raibstein, 2018) | Mouse (C57) | 60% HFD, 3 w PC through G and L | Cognitive impairment (adult, ↑ severity in aged adult)Facilitation of PPI (adult, aged adult) | Ventral hippocampus, adult
|
- | - | - | |
| (Song et al., 2017) | Rat(SD) | 60% HFD, through G | - | - | - |
|
|
|
| (Sun et al., 2014) | Rat(SD) | 60% HFD, through G and L | - | Hypothalamus, pre- /at weaning
|
Pre- / at weaning
|
|
- | |
| (Schmitz et al., 2018) | Mouse (C57) | 60% HFD, 9–11 w PC through G and L | - | Hippocampus, adult
|
Pre- /at weaning
|
|
- | |
| (Ford et al., 2009) | Sheep | 150% of recommended intake sheep diet by body weight, 60 d PC through G Dam sacrifice mid-gestation (G75 of 150 d G course) | - | - | Fetal
|
|
||
| Mouse (C57) | 60% HFD, through G | - | - | Fetal |
|
|
||
| (Qiao et al., 2015) |
|
|
|
|||||
| (Sanders et al., 2014) | Mouse (C57) | 45% HFD, 8 w PC through G | - | Hypothalamus, fetal
|
Fetal
|
|
- | |
| (Edlow et al., 2018) | Mouse (C57) | 60% HFD, 12–14 w PC through G | - | Fetal
|
Fetal
|
|
|
|
| (Kim et al., 2016) | Mouse (C57) | 45% HFD, 8 w PC through G | - | Hypothalamus, fetal
|
- | - | - | |
| (D’Asti et al., 2010) | Rat(SD) | 30%* by weight high n-6 fat HFD OR 30% by weight high n-3 fat HFD, GD 14 through L;* By % weight; % kcal fat not reported | - | Hypothalamus, pre-weaning
|
Pre- / at weaning
|
|
- | |
| (Abuaish et al., 2018) | Rat (Long Evans) | 60% HFD, 3 w PC through G and L | Anxiety-like behavior (neonate) | PVN, pre- /at weaning
|
Pre- /at weaning
|
|
- | |
| (Walker et al, 2008) | Rat (SD) | 30%* by weight HFD, GD 14 through L; * By % weight; % kcal fat not reported | - | - | Pre- /at weaning
|
- | - | |
| (Niu et al, 2019) | Rat (SD) | 27.5%* by weight HFD, 10 d PC through G and L; *By % weight; % kcal fat not reported | - | Hypothalamus
|
|
- | - | |
| (Frias et al., 2011) | Non-Human Primate (Japanese macaque) | 32% HFD, ≤ 4y PC through G | - | - | Fetal, sensitive/obese
|
Sensitive/obese dams
|
Sensitive/obese dams
|
|
| (Thompson etal., 2017) | Non-Human Primate (Japanese macaque) | 32% HFD + calorically dense treats, ≥ 1 y PC through G and L; Offspring weaned to HFD or Con | Anxiety-like behavior (juvenile, with maternal HFD) Increased stereotypic behaviors (juvenile, postweaning HFD) | Dorsal raphe, juvenile
|
Pre- /at weaning
|
|
- | Differential effects of maternal and postweaning HFD |
| (Hayes et al., 2014) | Rat (SD) | 45% HFD, 16 w PC through G | - | - | - |
|
|
|
| (Mark et al., 2011) | Rat (Wistar) | 45% HFD, through G | - | - | Fetal
|
|
|
|
| (Sullivan et al., 2010) | Non-Human Primate (Japanese macaque) | 32% HFD + calorically dense treats, ≤ 4 y PC through G | Anxiety-like behavior (♀, infant) | Dorsal raphe, fetal
|
- | - | - | |
| (Peleg-Raibstein et al., 2012) | Mouse (C57) | 60% HFD, 3 w PC through G and L | Anxiety-like behavior |
Dorsal hippocampus
|
- | - | - | |
| (Mao et al., 2010) | Mouse (NIH Swiss) | 60% HFD, 30–35 w PC through G (sacrifice at mid-gestation) | - | - | - |
|
|
|
| (Thompson et al., 2018) | Non-Human Primate (Japanese macaque) | 32% HFD + calorically dense treats, ≥ 1 y PC through G and L | Anxiety-like behavior (juvenile) | - |
|
- | - |
3.2.1. Cognitive impairment
Cognitive impairment is a common feature of several neuropsychiatric and neurodevelopmental diseases, and in some cases is predictive of both severity and trajectory of disease course (Liu-Seifert et al., 2015; Pitteri et al., 2017; Treuer and Tohen, 2010; Stirling et al., 2003).
We have shown that as adults, male rat offspring from dams that were fed a 60% HFD during gestation and lactation are cognitively impaired. Male offspring exhibit slower learning acquisition and impaired memory retention in the Barnes maze as well as decreased novel object preference in the novel object recognition test in comparison to control offspring (Cordner et al., 2019). These behavioral changes were accompanied by decreased hippocampal brain-derived neurotrophic factor (BDNF), insulin receptor, and leptin receptor mRNA at weaning, and persistent downregulation of insulin receptor and leptin receptor in adulthood (Cordner et al., 2019). While BDNF is widely known for its neurotrophic properties, insulin and leptin signaling within the hippocampus are also key regulators of both neural development and function, and have been implicated in impaired cognition associated with adult consumption of HFDs (Van Doorn et al., 2017; Ferrario and Reagan, 2018; Cordner and Tamashiro, 2015). Evidence suggests that maternal overnutrition may additionally sensitize offspring to cognitive impairment associated with adult HFD exposure. Two studies have demonstrated impaired spatial cognition in maternal HFD rats that were also weaned onto HFD (White et al., 2009; Can et al., 2012). In one of these studies, White and colleagues found no significant deficit in maternal HFD rats who were weaned onto standard chow or in offspring of chow-fed dams weaned onto HFD, illustrating the potentially additive effects of both maternal and offspring exposure (White et al., 2009). Given the known comorbidity between obesity and cognitive impairment (Dye et al., 2017), it is critical for investigators to consider whether phenotypic outcomes are a direct consequence of developmental exposure to an insult (e.g. maternal overnutrition causes cognitive deficit) or indirectly related through a different, co-existing phenotype (e.g. maternal overnutrition causes offspring obesity which in turn results in cognitive deficits) (Tamashiro et al., 2009; Trandafir and Temneanu, 2016).
The lack of cognitive impairment seen from maternal HFD alone in White et al., in contrast to our findings, also highlights some inconsistency in the literature. Several studies have now examined aspects of learning and memory in maternal HFD offspring with somewhat conflicting results. Like in the PNS and MIA literature, these discrepancies suggest how differences in experimental design – type, timing, and duration of maternal diet, species and strain differences, and how and when offspring are evaluated – can alter phenotypic outcomes. To the latter point, two studies thus far have found impaired spatial cognitive performance in adolescent but not adult offspring of HFD-fed dams. In one, adolescent offspring also exhibit increased peroxidized lipid accumulation, decreased BDNF, and impaired dendritic arborization within the hippocampus that is normalized by adulthood, suggesting a transient effect of maternal HFD (Tozuka et al., 2010). In the second study, while adult offspring from HFD-fed dams did not meet statistical criteria for cognitive impairment, the authors note that male offspring trend towards impaired performance (Robb et al., 2017). Given the findings by White and others, the question remains whether adult offspring from either study would demonstrate cognitive impairment if challenged with HFD themselves.
Other discrepancies include the nature of cognitive deficit, revealing nuances to seemingly similar outcomes. Two studies demonstrating impaired Morris water maze performance in adult offspring exposed to maternal HFD illustrate subtle variations in outcomes both classified as cognitive impairment (Moser et al., 2017; Page et al., 2014). In Page et al., offspring exhibited impaired retention but intact acquisition of learning, whereas Moser and colleagues found impaired acquisition with no deficits in retention upon acquisition. Other discrepancies in the literature are more striking, with at least one study finding improved spatial learning in adult offspring from HFD-fed dams (Bilbo and Tsang, 2010). Fewer studies have evaluated recognition memory, but with similarly inconsistent results, again perhaps reflecting differences in model and design. Consistent with our findings, in one study of mice exposed to maternal HFD, adult males exhibited impaired recognition memory, though this effect was absent in females (Graf et al., 2016). However, other studies of maternal overnutrition have found no deficits in offspring recognition memory (Winther et al., 2018; Moser et al., 2017).
The variation in the animal literature is reflected in the epidemiological literature. While several studies thus far have found an association between maternal overnutrition and cognitive problems in children, effect size and quality of cognitive deficit vary between studies (Basatemur et al., 2013; Bliddal et al., 2014; Brion et al., 2011; Casas et al., 2013; Craig et al., 2013; Eriksen et al., 2013; Hinkle et al., 2012; Huang et al., 2014; Li et al., 2016; Neggers et al., 2003; Krzeczkowski et al., 2018). Overnutrition in children is independently associated with reduced IQ, and while many of these studies adjusted for child BMI, BMI is only one index of metabolic dysregulation (Wraw et al., 2018). Given the known comorbidity between metabolic and cognitive impairment and the demonstrated influence of maternal metabolic dysregulation on the programming and regulation of offspring metabolism, dissociating direct and indirect maternal effects remains a priority.
3.2.2. Autism spectrum disorder (ASD)-like phenotypes
ASD in human patients has a considerably heterogeneous presentation largely characterized by impaired social interaction and patterns of restricted, repetitive behaviors (Ha et al., 2015). Female mice exposed to maternal HFD until weaning have decreased sociability compared to controls, though this difference was not seen in male siblings or in females whose mothers were switched to a control diet upon delivery (Kang et al., 2014). Decreased sociability in female offspring was associated with increased microglial activation and levels of pro-inflammatory cytokines IL-1β and TNF-α within the brain, reminiscent of ASD-like behavior and associated cytokine changes in models of maternal immune activation (Estes and McAllister, 2016). In a similar model of murine HFD-induced maternal obesity, male offspring (females were not tested) display increased repetitive behavior and a complement of socially deficient behavior including fewer reciprocal interactions, decreased sociability, and no preference for social novelty (Buffington et al., 2016). Mechanistically, a synergistic interaction between oxytocin and dopamine is implicated in the processing of social cues (Modi and Young, 2012). These mice also demonstrated reduced oxytocin immunoreactivity in the paraventricular nucleus (PVN) of the hypothalamus and an absence of social interaction-induced long-term potentiation in ventral tegmental dopamine neurons, known projection targets for oxytocin-expressing PVN neurons (Melis et al., 2007). Taken together, these findings support the growing body of epidemiological evidence associating maternal overnutrition to ASD risk (Reynolds et al., 2014; Moss and Chugani, 2014; Krakowiak et al., 2012; Dodds et al., 2011; Getz et al., 2016; Gardner et al., 2015; Bilder et al., 2013; Wallace et al., 2008) and begin to link this risk to inflammatory and neuroendocrine mediated changes
3.2.3. Anxiety- and depression-like phenotypes
Several studies have associated maternal overnutrition with increased anxiety-like behavior in offspring (Wright et al., 2011; Bilbo and Tsang, 2010; Kang et al., 2014; Sasaki et al., 2013; Sasaki et al., 2014). Bilbo and Tsang found that adult male rat offspring exposed to maternal HFD swim faster within the MWM, suggesting an anxiety-like motivation to escape (Bilbo and Tsang, 2010). These animals also spent less time in the open arms of the EPM, supporting an anxiety-like phenotype. Female rats in this model did not exhibit anxiety-like behavior, and in fact several studies have demonstrated sexual dimorphism in the anxiety phenotype. These differences suggest sex effects in brain programming and its behavioral consequences, an assertion that appears to be true in other behavioral phenotypes associated with maternal overnutrition such as locomotor hyperactivity, but may also be indicative of variations in study design (Rivera et al., 2015; Kang et al., 2014; Sasaki et al., 2013). Few studies have evaluated females, and in studies that have, the female estrous cycle, which is known to affect behavior, is often not accounted for (Frye et al., 2000; D’Souza and Sadananda, 2017). There is also the possibility that some behavioral measures used to assess anxiety have different sensitivities in male versus female offspring. For example, Sasaki et al. found increased anxiety-like behavior in both male and female offspring of HFD dams, but this was specific to the elevated plus maze in females and the open field in males (Sasaki et al., 2013). In both males and females, increased anxiety-like behavior was accompanied by decreased baseline corticosterone, slower returns to baseline levels following an acute stress (restraint) challenge, increased gene expression of the glucocorticoid receptor within the amygdala, and altered gene expression of cytokines within limbic areas of the brain, suggesting a similar underlying physiology to sexually dimorphic anxiety-like behavioral phenotypes. In a follow up study, the same group found that as adolescents, male and female offspring of maternal HFD dams have decreased anxiety-like behavior, suggesting that maternal overnutrition may have age-dependent influences on anxiety-like behavior (Sasaki et al., 2014). Fewer studies have evaluated depression-like behavior in offspring exposed to maternal overnutrition, though existing data suggest that maternal overnutrition may have a role in programming this set of behaviors as well. Two studies have found that as adults, rats exposed to maternal HFD exhibit depression-like behavior in the FST (Can et al., 2012; Giriko et al., 2013). Another study evaluating mild CVS in adult offspring exposed to maternal HFD found that maternal HFD sensitized offspring to stress-induced depression-like behavior, with stressed HFD offspring demonstrating reduced sucrose preference, decreased locomotor activity, and increased FST immobility in comparison to stressed offspring from dams fed a standard diet (Lin et al., 2015). In contrast, another group found that maternal HFD may actually mitigate stress-induced behavioral alterations in offspring exposed to early life stress, suggesting a potentially divergent interaction between discrete stressors across the course of development (Rincel et al., 2016).
Epidemiological studies suggest a link between maternal overnutrition and affective problems in children. To date multiple cohort studies have associated maternal overnutrition with increased internalizing pathology, including anxiety and depression symptomatology (Van Lieshout et al., 2013; Mina et al., 2017; Rodriguez, 2010; Robinson et al., 2013). Increasing maternal pre-pregnancy BMI attenuates the normal decline of internalizing symptoms across childhood and adolescence (Van Lieshout et al., 2013). While a positive association between maternal BMI and internalizing psychopathology emerged at age 8, this association increased in strength across childhood through age 17. These results suggest the continued potential for increased anxiety and depression in adults exposed to maternal HFD, but long-term prospective epidemiological studies on adult offspring remain to be conducted.
3.2.4. Schizophrenia-like phenotypes
Despite epidemiological evidence for increased schizophrenia risk in the offspring of obese mothers, few studies have examined models of maternal overnutrition in the context of schizophrenia-like behavior. Contrary to epidemiological data, one study in mouse dams fed HFD found facilitation of prepulse inhibition (PPI) in adult offspring, and no changes in cognition, social behavior, or locomotor behavior (Zieba et al., 2019). Another murine study also found improved PPI in adult offspring, however this was accompanied by cognitive impairment (Wolfrum and Peleg-Raibstein, 2018). It is possible that altered maternal care behavior during the lactation period may partially play into these results. In both studies, HFD exposure was continued until the end of lactation. We have previously shown in rats that dams fed HFD during gestation and lactation exhibit altered maternal care behavior, spending more time nursing their pups and doing so in a more optimal nursing posture (Purcell et al., 2011). In turn, while studies are limited, early-life maternal care appears to influence later-life sensorimotor gating. For example, one study in mice found that adult offspring of dams who spent less time engaged in pup-licking behavior during lactation exhibited impaired PPI compared to offspring of high pup-licking dams (Pedersen et al., 2011). While the limited evidence available creates a largely speculative discussion, it further highlights the importance of distinguishing between the prenatal and early postnatal environments.
In addition, the discrepancy between epidemiological work associating maternal overnutrition and schizophrenia and the limited animal literature suggests a gap that remains to be filled. Two cohort studies have associated elevated maternal BMI with multifold increased risk for schizophrenia in offspring (Jones et al., 1966; Schaefer et al., 2000), and one meta-analysis calculated that children of mothers with diabetes were over 7 times more likely to develop schizophrenia (Cannon et al., 2002). Kawai and colleagues report that schizophrenia risk increases by 24% for every maternal BMI unit increase in early pregnancy and 19% per unit in late pregnancy (Kawai et al., 2004). Considerable work remains to be done to evaluate schizophrenia-relevant phenotypes in animal models of overnutrition.
3.2.5. Summary and future directions
Maternal overnutrition has a significant long-term influence on brain function and behavior in offspring. Existing data suggest that maternal overnutrition is a risk factor for development of behavioral deficits consistent with a number of neuropsychiatric disorders. As with stress and infection, variation in outcomes could stem from differences in type and length of maternal exposure. As an example, obesity or HFD consumption during the pre-conception period alone has been shown to affect oocyte metabolism and morphology (Reynolds et al., 2015). In addition, the comorbidity between metabolic dysregulation and behavioral aberration calls to question whether behavioral phenotypes in offspring are due directly to maternal exposure or indirect, secondary to maternal programming of metabolic dysregulation in offspring. Future studies may be focused on sex differences, determining the degree of interdependence between cognitive and metabolic phenotypes, and determining critical periods of neuropsychiatric risk and resilience in response to maternal overnutrition. Despite these remaining questions, it is apparent that, like stress and infection, maternal overnutrition potentiates offspring neuropsychiatric risk. In the next sections, we examine putative mechanisms underlying this relationship.
4. Potential mechanisms linking maternal overnutrition and offspring neuropsychiatric outcomes
A range of mechanisms linking maternal nutrition to neuropsychiatric outcomes in offspring have been proposed. Like in both maternal psychosocial stress and maternal infection, these include changes to the HPA axis and neuroendocrine communication, metabolic hormones, immune regulation, placental structure and function, neurotrophic signaling, microbiome, and epigenetics. Here we will overview the rationale and evidence for some of the more well studied mechanisms and discuss how they may interact with one another (Table 2).
4.1. Metabolic regulation
Maternal overnutrition consistently results in metabolic impairment in the dam. We have shown that rat dams exposed to a HFD (60% kcal fat) during gestation only have increased plasma leptin by G10, lower plasma adiponectin by G14, impaired glucose tolerance and increased plasma insulin in a glucose tolerance test at G15, and increased adiposity at G21 (Song et al., 2017). Offspring in this model also exhibit metabolic impairment despite being weaned onto a standard chow diet (Tamashiro et al., 2009; Sun et al., 2012; Sun et al., 2014; Sun et al., 2013). While overnutrition induced metabolic impairment in dams is likely important in patterning metabolic programming in offspring, evidence from our lab and others suggests it also has a role in patterning brain development and function (Cordner et al., 2019; Sun et al., 2014; Treesukosol et al., 2014). In addition to their role in energy balance and metabolism, metabolic hormones including insulin, leptin, and ghrelin have been shown to cross the blood brain barrier and act as key regulators of neural development and plasticity (Van Doorn et al., 2017; Ferrario and Reagan, 2018; Fadel et al., 2013; Bouret, 2017). Here we will discuss insulin and leptin as potential candidates linking maternal overnutrition and alterations in offspring brain and behavior.
The brain is densely populated with insulin receptors (Insr) (Abbott et al., 1999; Werther et al., 1987; Kar et al., 1993). Importantly, these receptors are not only found in areas of the brain involved in peripheral metabolic control, such as the hypothalamus, but also in regions relevant to cognition, motivation, and mood regulation (Ferrario and Reagan, 2018; Werther et al., 1987; Kar et al., 1993). These receptors bind peripheral insulin, which crosses the blood brain barrier throughout life (Banks et al., 2012; Banks, 2004). Receptor density is highest during early development, when their activation is postulated to play critical roles in a variety of functions including neurogenesis, neuronal maturation, and synaptogenesis (Kar et al., 1993; Chiu and Cline, 2010). In adulthood, regional insulin signaling is important not only for regulation of energy homeostasis (Schwartz et al., 2000), but also for behavior. Within the hippocampus, insulin signaling regulates structural and functional plasticity via facilitation of glutamatergic and GABAergic activity to promote cognition (Ferrario and Reagan, 2018). Recent evidence suggests that insulin signaling within the hippocampus and amygdala may also coordinate anxiety-like behavior; virally-mediated knockdown of insulin and insulin-like growth factor 1 receptors within either the hippocampus or amygdala increase anxiety-like behavior in adult mice (Soto et al., 2019). Finally, emerging evidence suggests a role for insulin signaling in coordinating neuro-transmitter activity within the nucleus accumbens – ventral tegmental area reward network (Ferrario and Reagan, 2018; Stouffer et al., 2015).
Exposure to overnutrition in adulthood is widely associated with central insulin resistance and resulting cognitive impairment (Fadel and Reagan, 2016). Recent evidence suggests that maternal overnutrition may program similar changes to offspring (Cordner et al., 2019; Schmitz et al., 2018). Insulin does not appear to cross the placental barrier, but glucose does. When glucose is transported in high amounts such as in the context of maternal overnutrition, it has been shown to increase production of insulin by the fetus, thereby exposing the developing fetal brain to a hyperinsulinemic environment (Sullivan et al., 2014; Ford et al., 2009; Oken and Gillman, 2003). Increased insulin action during this period may alter the development of neural circuitry involved in higher order processing (Sullivan et al., 2014). Further, it may prime for future insulin resistance within the offspring brain. A recent study demonstrated that maternal obesity in mice results in hippocampal insulin resistance within adult offspring along with decreased markers of neurogenesis, functional plasticity, and synaptic plasticity (Schmitz et al., 2018). We have shown that cognitive impairment in adult rat offspring exposed to maternal HFD during pregnancy and lactation is preceded by decreased hippocampal expression of Insr and glucose transporter 1 (Slc2a1) at P21, with continued downregulation of Insr in adulthood, though we have not yet evaluated downstream markers of insulin resistance in the brain (Cordner et al., 2019).
Unlike insulin, maternal leptin can directly cross the placenta to act on the developing fetal brain (Luo et al., 2013; Djiane and Attig, 2008). Leptin acts through overlapping signaling cascades with insulin to promote neurodevelopment and cognitive function, and changes to leptin signaling during early life have been shown to alter the development of neural circuitry (Fadel and Reagan, 2016; Djiane and Attig, 2008; Bouret, 2010). We have shown that rat offspring exposed to maternal HFD also exhibit downregulation of hippocampal leptin receptor (Lepr) expression that persists into adulthood (Cordner et al., 2019). These animals additionally exhibit downregulated Lepr in the hypothalamus along with impaired signaling (Sun et al., 2012). It remains to be seen whether hippocampal expression changes in Insr and Lepr within the maternal HF diet model translate to signaling deficits, and the relative contributions of each to altered behavior.
4.2. Immune-related changes
Inflammatory mechanisms have a role in mediating maternal overnutrition-induced changes to brain and behavior. Obesity is considered a state of low-grade inflammation, and increasing adiposity is associated with greater circulating levels of both pro-inflammatory cytokines and markers of systemic inflammation (Hotamisligil, 2006; Howell and Powell, 2017; Segovia et al., 2014; Das, 2001). Overnutrition is additionally associated with inflammation at the level of the placenta, which may be stimulated not only through systemic inflammatory influences but also directly through activation of placental toll-like receptor (TLR)4 (Howell and Powell, 2017; Challier et al., 2008; Lackey and Olefsky, 2016; Basu et al., 2011; Roberts et al., 2011). TLR4, traditionally known to respond to bacterial endotoxin, also binds to and is activated by saturated fatty acids such as those found in HFDs, triggering an inflammatory cascade characterized by production and secretion of pro-inflammatory cytokines (Howell and Powell, 2017; Yang et al., 2016; Yang et al., 2015). In addition, the placenta expresses endogenous lipases, and in obesity possesses increased lipid storage capacity, suggesting that placental TLR4 activation in a state of maternal overnutrition may be induced by increases in both systemically circulating and local fatty acids (Pathmaperuma et al., 2010; Varastehpour et al., 2006; Qiao et al., 2015; Gauster et al., 2011). Obese women have a 3- to 9-fold increase in placental TLR4 expression, which positively correlates with pro-inflammatory interleukin(IL)-6 and IL-8 levels within both systemic circulation and the placenta (Yang et al., 2016). It is unclear to what degree systemic and placental inflammation contribute to fetal inflammation, though it is suggested that both may play a role. Evidence from MIA models suggests that both placental-derived and maternal circulating pro-inflammatory cytokines contribute to cytokine accumulation in the fetal brain (Ponzio et al., 2007; Hsiao and Patterson, 2011; Dahlgren et al., 2006).
While cytokines are well known for their roles in immune system regulation, strong evidence for their functions in neurodevelopment and function suggests that this may be a mechanism through which maternal overnutrition primes neuropsychiatric risk. First, cytokines have pleiotropic roles during normal neurodevelopment, including promotion of neurogenesis, neuronal migration, axon guidance, synapse formation, circuitry refinement, and synaptic plasticity (Deverman and Patterson, 2009). These functions are under tight temporal and spatial regulation over the course of development, and altering the trajectory and magnitude of availability, localization, and balance between different cytokines could disrupt the neurodevelopmental trajectory. Increased pro-inflammatory cytokine levels within fetuses exposed to maternal overnutrition are associated with impaired cognition, increased anxiety-like behavior, and decreased social behavior (Rivera et al., 2015; White et al., 2009; Bilbo and Tsang, 2010; Sullivan et al., 2014; Sanders et al., 2014).
Second, an inflammatory milieu in early life may alter the development and function of astrocytes and microglia, the brain’s resident immunocompetent cells that also perform essential neurodevelopmental functions including support of neuronal proliferation and differentiation, angiogenesis and vascular guidance, apoptosis and debris clearance, synaptogenesis, patterning, and pruning, myelination, and establishment of connectivity. Increased fetal brain inflammation associated with maternal overnutrition may prime microglia towards excessive production of pro-inflammatory cytokines and altered function, which may not only have consequences for the neurodevelopmental trajectory in utero but also persistent consequences, including increased sensitivity to future insult (Bilbo and Schwarz, 2009). For example, Edlow et al. found that maternal obesity in mice primes fetal microglia to overrespond to an immune challenge (Edlow et al., 2018). Bilbo and Tsang demonstrated further that offspring of maternal HFD-fed dams not only have increased microglial activation and Il-1β production within the hippocampus at birth, but also present as adults with increased hippocampal microglial activation at baseline and greater reactivity to an immune challenge, accompanied by increased anxiety-like behavior (Bilbo and Tsang, 2010). Maternal obesity increases proliferation of astrocytes within the hypothalamus, a relationship that is postulated to be mediated by IL-6 (Kim et al., 2016). Emerging evidence also implicates complement proteins and major histocompatibility complex class I molecules in mediating neurodevelopment and these may also be altered in maternal overnutrition (Estes and McAllister, 2015; Hsiao, 2013).
4.3. HPA axis and neuroendocrine hormones
The impact of early life insult on the development and function of the HPA axis has been demonstrated across several maternal stressors, including prenatal stress and immune activation. Neuroendocrine modulation may be a mechanism by which maternal overnutrition promotes neuropsychiatric risk in offspring (Sullivan et al., 2015). In response to stress, the PVN produces corticotropin releasing hormone (CRH) and arginine vasopressin, which stimulate the pituitary to release adrenocorticotropic hormone (ACTH) into systemic circulation. ACTH triggers the release of glucocorticoids from the adrenal cortex, which in turn act on brain regions including the pituitary, hypothalamus, hippocampus, and amygdala to regulate HPA axis activity and stress behavior. Extrahypothalamic populations of CRH in the amygdala and lateral bed nucleus of the stria terminalis are additionally thought to regulate fear and anxiety behavior (Schulkin et al., 1998). In addition to acute stress reactivity, aspects of the HPA axis demonstrate circadian biorhythmicity at baseline (Sage et al., 2001). It is suggested that maternal overnutrition affects both baseline HPA axis activity and stress responsivity in offspring, and that these changes may contribute to altered brain signaling and behavior (Sullivan et al., 2015).
In interpreting the mechanistic role of the HPA axis in behavioral outcomes within offspring exposed to maternal overnutrition, it is important to consider the developmental stage of the offspring at evaluation. In one study by Sasaki et al., adult offspring of rat dams fed a HFD during pregnancy and lactation exhibited decreased baseline corticosterone, increased glucocorticoid receptor expression in the amygdala, and increased anxiety-like behavior (Sasaki et al., 2013). In response to a restraint stress challenge, these offspring demonstrated a heightened corticosterone response followed by a slower return to baseline (Sasaki et al., 2013). Yet in a follow up study, this same group found that adolescent offspring exposed to the same maternal paradigm demonstrate decreased anxiety-like behavior and increased glucocorticoid receptor expression in the hippocampus but not the amygdala (Sasaki et al., 2014). Thus, these changes appear to be age-dependent, likely following the developmental trajectory of the HPA axis and maturation of feedback systems mediating corticosteroid sensitivity (McCormick et al., 2010; Romeo, 2013). Increased glucocorticoid receptor expression in the hippocampus is suggested to inhibit the HPA axis, whereas increased expression in the amygdala potentiates the stress response, demonstrating within this context the developmental and spatial relationship between HPA axis changes and behavior (Groeneweg et al., 2011). Additionally, it appears there may be a relationship between increased exploratory and risk-taking behaviors in adolescence and increased anxiety-like behaviors in adulthood, potentially implicating early stressor-induced alterations in the developmental trajectory of HPA regulation in different abnormal behaviors across development (Jacobson-Pick and Richter-Levin, 2010; Jacobson-Pick et al., 2011; McCormick and Green, 2013). Studies in neonate offspring are limited, but overall it appears that maternal overnutrition increases stress-induced corticosterone release and anxiety-like behavior in pre-weanlings, again reflecting the complexity of the developmental course (D’Asti et al., 2010; Abuaish et al., 2018). In conjunction with age, offspring sex is also of critical consideration as males and females have differential HPA axis responsivity at baseline as well as dimorphic development and maturation of stress circuitry beginning in gestation but continuing throughout life (Bale et al., 2010; Bale and Epperson, 2015).
The complex relationship between maternal overnutrition and neuroendocrine function not only exhibits spatial and temporal sensitivity in terms of developmental trajectory, but may also be highly dependent on type of maternal dietary intervention, diurnal rhythm for baseline measurements, and method of stress challenge. For example, Sasaki et al.’s finding of decreased basal corticosterone in adult offspring was conducted in the middle of the light cycle, whereas Walker and colleagues found increased basal corticosterone during the dark but not light cycle (Sasaki et al., 2013; Walker et al., 2008). Niu et al. sampled basal plasma corticosterone over a 24-h period and found an overall increase secondary to increases in both pulse frequency and amplitude of corticosterone secretion (Niu et al., 2019). Consistent with Sasaki’s group, however, Niu and colleagues went on to demonstrate that offspring have heighted stress responsivity, exhibiting increased corticosterone levels and slower negative feedback following acute restraint stress, and additionally that they exhibit attenuated habituation to repeated restraint stress (Niu et al., 2019). Niu also showed that maternal HFD-induced stress sensitization was dependent on the amygdala (Niu et al., 2019). Overall, while methodological considerations appear to greatly influence findings, maternal HFD appears to change the developmental trajectory and function of the HPA axis in offspring, and these changes may underlie offspring changes to brain and behavior.
5. The placenta as an integrator and propagator of allostatic load
It is important to note that metabolic, immune, and neuroendocrine pathways exhibit significant co-regulation and crosstalk. Pregnancy is a state of normally occurring allostasis, where the body must maintain maternal health while changing physiological set points towards protection and resource allocation for the developing fetus (Power and Schulkin, 2012). These changes occur in temporal sequence across the metabolic, neuroendocrine, and immune systems, and changes within each system may influence one another. Disruption to any of these axes by maternal overnutrition may potentiate disruptions within the other systems towards a multisystem allostatic overload that can only be conveyed to the developing fetus through one organ: the placenta. The placenta serves as both messenger and sentinel between the mother and developing fetus, but its function in that role is adversely impacted by systemic dysregulation. For example, the placental inflammation associated with maternal overnutrition is not only the result of increased cytokine production from adipose tissue and direct effects of fatty acids at the placenta, but also likely impacted by dysregulation of metabolic hormones that can act as adipokine immune mediators. Leptin, high in obese pregnancy, has structural similarity to IL-6 and pro-inflammatory action (Iikuni et al., 2008; Lappas et al., 2005); adiponectin, low in obese pregnancy, has anti-inflammatory action (Ouchi and Walsh, 2007). Leptin and insulin also act through converging pathways with cytokine signaling (Iikuni et al., 2008; Shoelson et al., 2006). Thus, in gestational diabetes for example, inflammation at the placenta can promote both placental insulin resistance and maternal hyperglycemia, potentially increasing the metabolic dysregulation experienced by the fetus (Feng et al., 2016). In another example of placental pathway convergence, placental 11β-hydroxysteroid dehydrogenase type 2, an enzyme that inactivates glucocorticoids and protects the fetus from exposure to excess maternal glucocorticoids, is inhibited by pro-inflammatory cytokines, representing a mechanism by which maternal overnutrition-associated inflammation may increase fetal exposure to glucocorticoids (Kossintseva et al., 2006). Thus, the placenta is itself vulnerable to the effects of overload via alterations in its structure and function thereby amplifying an environment of dysregulation in the fetal compartment. In this section, we will use lessons learned from the stress and infection literature to comment on how maternal overnutrition may increase risk for offspring psychopathology by altering placental structural integrity, nutrient transfer, and neurotrophic communication (Table 2).
5.1. Structural integrity and nutrient transfer
Placental structure and sufficiency are critical for proper growth and development of the fetus. Placental insufficiency as seen in excessive maternal inflammation is associated with spontaneous miscarriage. Yet even mild disruptions of placental architecture can alter the capacity of the placenta to provide oxygen and nutrients to the fetus, changing the trajectory of fetal growth and development and potentially promoting neuropsychiatric risk. This mechanism has been studied more extensively in the context of MIA, where there is loss of placental integrity, decreased blood perfusion to the fetus, and perinatal brain damage. Evidence suggests that maternal overnutrition may also alter placental cytoarchitecture and blood flow. In non-human primates (NHP), exposure to maternal HFD increases placental inflammation and decreases uterine blood flow volume even in lean, metabolically unimpaired mothers. In obese, hyperinsulinemic mothers, HFD exposure additionally reduces fetal compartment blood flow and induces placental infarctions, increasing frequency of stillbirth (Frias et al., 2011). Behaviorally, the offspring of HFD fed NHP mothers display greater anxiety despite weaning onto a low-fat control diet (Thompson et al., 2017). Studies in rodent models are consistent for placental structure changes, but mixed for blood flow capacity. Hayes et al. found that maternal HFD-induced obesity in rats increases placental inflammation, impairs vascular maturation, induces placental hypoxia, and compromises fetal growth and viability (Hayes et al., 2014), whereas Mark et al. showed decreased placental and fetal size and increased inflammation without changes to markers of vascular development (Mark et al., 2011). We have found that in rats provided maternal HFD the thickness of the placental labyrinth layer is reduced, suggesting reduced vascular capacity, but we have not examined hemodynamics in this model (Song et al., 2017). Even without frank changes to blood flow, reductions in placental thickness and integrity may alter the protective capacity of the placenta against systemic dysregulation.
The impact of maternal overnutrition on placental nutrient transfer has been more widely studied. The three major nutrient substrates for the growing fetus are glucose, lipids, and amino acids. Epidemiological and animal model studies suggest that maternal overnutrition may alter fetal exposure to these substrates by altering placental nutrient sensing, metabolism, and levels of nutrient transporters themselves (Song et al., 2017; Gallo et al., 2017). The variable interplay between reduced placental thickness and increased nutrient transporters may account for the range of aberrant birth weights associated with maternal overnutrition, from small for gestational age to macrosomia (Howell and Powell, 2017). This interplay may also reflect on the range of neuropsychiatric phenotypes associated with maternal overnutrition, as these nutrient substrates are also the building blocks for the developing brain.
5.2. Neurotrophic communication
In addition to its role as an arbiter of maternal signaling, the placenta is a significant source of neurotrophic signals to the developing fetal brain. Serotonin (5-HT) is produced by the placenta and is critical for normal brain development (Bonnin et al., 2011). In addition to its action in mood regulation, 5-HT signaling during fetal life has a significant role in promoting neurogenesis, neuronal migration, axon guidance, and synaptogenesis (Bonnin et al., 2011; Velasquez et al., 2013). Since the early fetal brain does not have the capacity to produce 5-HT, it is synthesized in the placenta from maternal l-tryptophan (TRP). Placental 5-HT is necessary for fetal brain development until the arrival of serotonergic projections from the dorsal raphe in mid-gestation (Bonnin et al., 2011). Too much or too little placental 5-HT availability during this early period leads to developmental alterations, including aberrant serotonergic outgrowth and abnormal development of the major thalamocortical axon tract (Goeden et al., 2016). A seminal study by Goeden et al. demonstrated that poly(I:C)-induced inflammation (a moderate immune challenge) in mouse dams increases placental synthesis of 5-HT via a transient increase in substrate availability of maternal TRP to the placenta and subsequent increase in the expression and biosynthetic activity of placental tryptophan hydro-xylase 1 (TPH1). Increased placental output of 5-HT accumulates in the fetal forebrain and blunts serotonergic axon outgrowth from the hind-brain (Goeden et al., 2016). Proinflammatory cytokine treatment has additionally been shown to reduce survival of 5-HT neurons within the dorsal raphe and substantia nigra (Hochstrasser et al., 2011). Impairment of central serotonergic signaling is implicated across several neuropsychiatric illnesses associated with maternal overnutrition including ASD, ADHD, and mood disorders.
Indeed, animal models suggest that maternal overnutrition may impair development of the brain serotonergic system, resulting in lower 5-HT production and signaling. Non-human primates exposed to maternal HFD have reduced gene expression in the 5-HT synthesis pathway in the dorsal raphe and decreased serotonergic immunoreactivity in the prefrontal cortex, with concomitant anxiety and hyperactivity (Sullivan et al., 2015; Sullivan et al., 2010). Rats exposed to maternal HFD have increased expression of the 5HT1A receptor within the ventral hippocampus, maybe a compensatory response to reduced 5-HT availability, leading to increased anxiety-like behavior (Peleg-Raibstein et al., 2012).
In addition to potential consequences from overnutrition-induced placental inflammation, insulin appears to be a regulator of placentally sourced 5-HT by modulating cell surface localization of serotonin transporter (SERT) within the placenta, suggesting a potential convergence of mechanisms by which maternal overnutrition may alter the availability of 5-HT to the fetal brain and subsequent serotonergic system development (Murthi and Vaillancourt, 2019).
Importantly, 5-HT synthesis accounts for less than 5% of normal TRP metabolism, most of which occurs through the kynurenine (KYN) pathway (Gal and Sherman, 1980). Placental KYN pathway components are also upregulated in inflammation, and there is evidence for KYN system alterations within the brains of offspring exposed to maternal HFD, suggesting another possible pathway through which maternal overnutrition may promote neuropsychiatric risk (Winther et al., 2018; Schwarcz et al., 2012; Williams et al., 2017; Campbell et al., 2015; Manuelpillai et al., 2005). In the study by Goeden et al., in addition to augmenting TRP availability and upregulating TPH1 gene expression, maternal poly(I:C) injection also upregulated placental gene expression of indoleamine 2,3-dioxygenase (IDO1), responsible for converting maternal TRP to KYN, and subsequently increased KYN concentrations within the fetal brain (Goeden et al., 2016). In another, more severe model of intrauterine infection and inflammation, Williams et al. report upregulation of the placental KYN pathway and increased fetal brain accumulation of KYN (Williams et al., 2017). Here, the authors found decreased placental TRP and an increased KYN:TRP ratio, suggesting that TRP was shunted away from 5-HT synthesis pathway, potentially (placental 5-HT was not measured) resulting in a decrease in placental 5-HT availability for the developing fetal brain. There has been limited examination of the placental KYN pathway in maternal overnutrition, but one clinical study found a positive correlation between BMI and plasma KYN in pregnant women (Groer et al., 2018). High BMI was also associated with decreased plasma TRP, suggesting a potential reduction in substrate availability as well as possible shunting towards KYN production at the level of the placenta, though exact placental changes remain to be determined (Groer et al., 2018). These examples illustrate that inflammation may have varying effects on placental 5-HT and that the severity of immune challenge or inflammatory response may dictate whether 5-HT production and signaling is decreased or increased. Nevertheless, such immune stressors ultimately lead to similar neuropsychiatric outcomes via an altered 5-HT system.
In addition to 5-HT, the placenta also produces BDNF which facilitates placental development and metabolism. Both maternal and placental BDNF cross the placental barrier into fetal circulation to reach the fetal brain (Kodomari et al., 2009; Antonakopoulos et al., 2018) where it plays a critical role in supporting neuronal survival, synaptic transmission, plasticity, growth, and repair (Reichardt, 2006). Maternal overnutrition decreases BDNF expression within the cortex and hippocampus of offspring, and the decrease in BDNF associates with cognitive impairment (Tozuka et al., 2010; Cordner and Tamashiro, 2015; Page et al., 2014; Arnold et al., 2014; Yamada-Goto et al., 2012). Maternal obesity has been shown to impair placental BDNF signaling (Prince et al., 2017). Additionally, elevated glucocorticoids associated with maternal stress has been shown to alter BDNF within the placenta and offspring brain (Suri and Vaidya, 2013; Kertes et al., 2017) and may thus contribute to maternal overnutrition and maternal stress associated behavioral deficits.
5.3. Sex differences and the placenta
The number of studies in which male and female offspring are both evaluated remain limited, but existing studies suggest sex differences in offspring behavior and physiology across stressors. Recent work has suggested that these differences may emerge in part at the level of the placenta, consistent with its role as an integrator and propagator of maternal stress. The placenta is sexually dimorphic, with differences emerging as early as the trophectoderm stage of embryogenesis (Bale, 2016; O’Connell et al., 2013; Sood et al., 2006). Studies across a variety of maternal insults have revealed sexually dimorphic placental responses to stressors (Bronson and Bale, 2016; Davis and Pfaff, 2014; Sandman et al., 2013; Mao et al., 2010; Howerton et al., 2013; Bronson and Bale, 2014; Dunn et al., 2011; Gabory et al., 2013; Clifton, 2010). In general, the XY placenta appears to be more sensitive to environmental change, whereas the XX placenta is relatively resistant (Bale, 2016), suggesting that such differential responses may, in part, underlie sexually divergent brain development and behavioral outcomes. As an example, Bale and colleagues have found that O-linked N-acetylglucosamine transferase (OGT), an X-linked gene expressed in the placenta, is decreased in response to maternal CVS during early gestation in both male and female offspring. Under normal conditions, OGT expression is lower in the XY placenta compared to XX placenta because OGT escapes X-inactivation and is biallelically expressed in XX placentas. Thus, maternal stress resulted in even lower OGT in stressed XY placentas compared to unstressed placentas from both sexes as well as stressed XX placentas (Howerton et al., 2013; Howerton and Bale, 2014). OGT plays a critical role in chromatin remodeling and reduced OGT was associated with significant transcriptional changes in the placenta and brain that was consistent with behavioral and physiological abnormalities in male, but not female, offspring exposed to maternal stress, as well as in unstressed mice with targeted placental deletion of OGT (Howerton et al., 2013; Howerton and Bale, 2014). The OGT story thus illustrates one way in which the placenta may mediate sex-specific effects of gestational stress on offspring.
6. Final perspectives
It is now clear that diverse maternal stressors including psychosocial stress, infection, and overnutrition result in overlapping neuropsychiatric outcomes for offspring. Considering the shared outcomes across diverse stressors, some have started to investigate common etiologies, and evidence supporting this idea is growing. As has long been recognized in the adult stress literature, it is increasing clear that the concepts of allostatic load and allostatic overload apply to maternal stressors, with aberrant neurodevelopment and neuropsychiatric risks to offspring emerging from stress effects on interacting maternal regulatory systems, the placenta, and the developing fetal brain (Fig. 1).
Fig. 1.
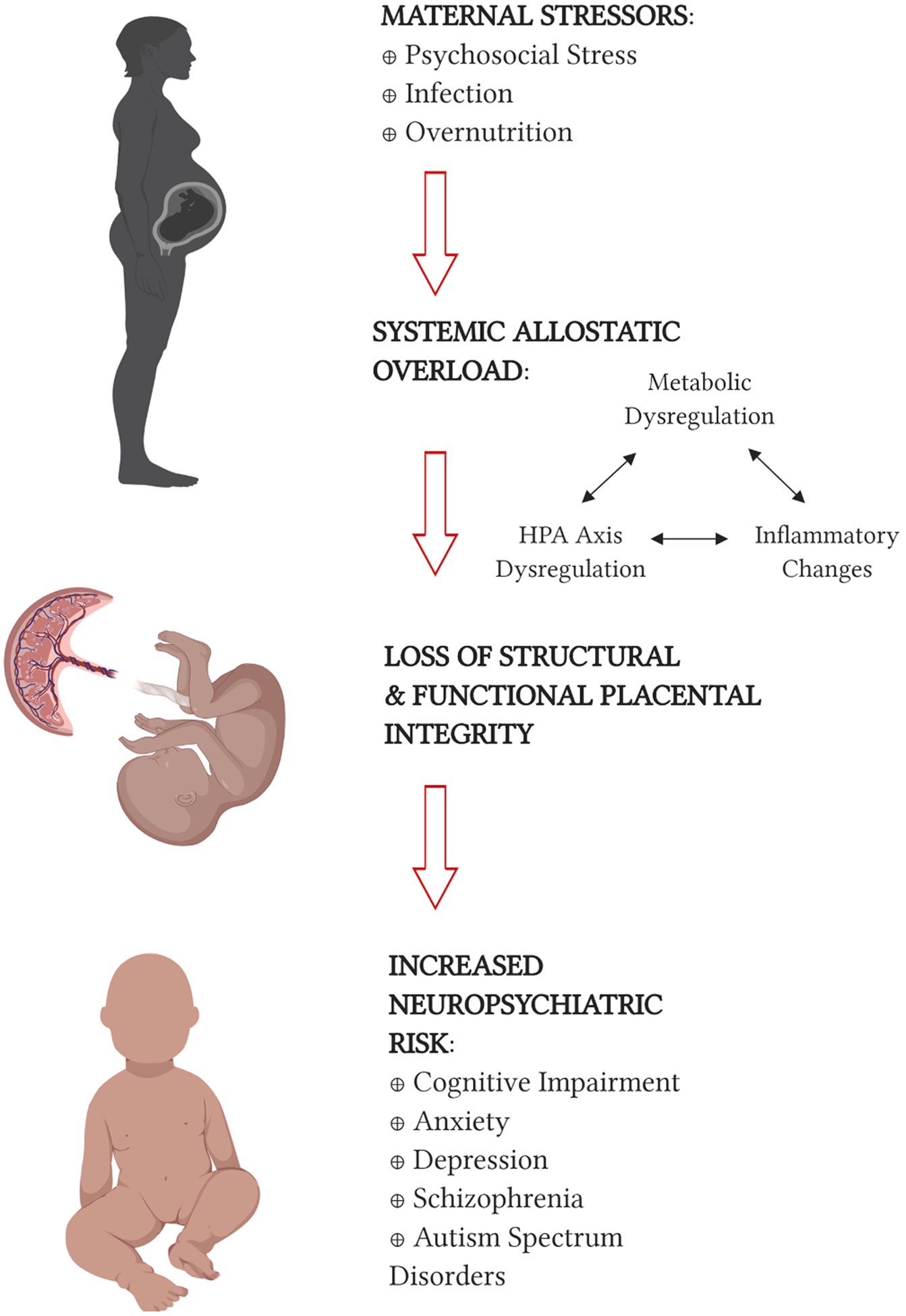
Different maternal environmental stressors lead to common neuropsychiatric risk profiles in offspring. Epidemiological and animal model studies associate maternal stress, infection, and altered nutrition with increased risk for an overlapping range of neuropsychiatric outcomes in offspring, including cognitive impairment, anxiety, depression, schizophrenia, and autism spectrum disorders. Evidence suggests that these stressors act via converging pathophysiological pathways characterized by systemic allostatic overload across regulatory systems including the hypothalamic–pituitary–adrenal (HPA) axis, metabolic signaling, and inflammatory pathways. The placenta acts as an integrator and propagator of allostatic overload, not only communicating the maternal environment to the fetus but also altering its own structural and functional capacity in response. The developing fetus, patterned in an environment of dysregulation, is programmed towards increased neuropsychiatric risk. Created with BioRender.com.
Common risks and mechanisms across various stressors have attracted greater attention and the information gained will be critical in identifying potential interventions that could apply to a wide variety of developmental “stressors”. Collaboration across disciplines and innovations in design and analysis that can better account for the ways in which different stressors overlap and interact with each other will facilitate advancement in the field. Importantly, individuals exposed a particular stressor are often the same individuals at highest risk for facing other stressors, suggesting that successful interventions should also be broad in scale. As such, behavioral interventions may not only be more accessible to those at risk, but may also allow for more widespread mechanistic action, functioning across multiple systems to broadly promote neuropsychiatric resilience.
In the interest of translating findings to humans, pre-clinical studies that incorporate individual differences and experiences rather than simply single-variable group effects are needed. Thompson and colleagues recently utilized multivariate analysis in a non-human primate model of maternal overnutrition, concluding that maternal pre-pregnancy adiposity and gestational HFD consumption exert unique effects on offspring behavior (Thompson et al., 2018). Thompson’s work demonstrates the ability to utilize more sophisticated statistical modeling to go beyond single variable associations towards a more complex, nuanced understanding of underlying etiology. Although resources limit extensive collection of multisystem variables across all studies, more robust collaboration across disciplines and open-source reporting of raw data from individual studies may offer alternatives. Expanding such databases would significantly increase our ability to compare between studies and stressors, identify promising etiological pathways, and design and test interventions.
Acknowledgements
The authors thank Dr. Timothy H. Moran for enlightening conversations and his continual mentoring and guidance. This work was supported by the National Institutes of Health (NIH) HD093338 (S. G. K.), the Greif Family Scholar Fund (Z. A. C.), NIH MH108944 (K. L. T.), and the Dalio Philanthropies.
References
- Abbott MA, Wells DG, Fallon JR, 1999. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J. Neurosci 19, 7300–7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abuaish S, Spinieli RL, McGowan PO, 2018. Perinatal high fat diet induces early activation of endocrine stress responsivity and anxiety-like behavior in neonates. Psychoneuroendocrinology 98, 11–21. [DOI] [PubMed] [Google Scholar]
- Antonakopoulos N, Iliodromiti Z, Mastorakos G, Iavazzo C, Valsamakis G, Salakos N, Papageorghiou A, Margeli A, Kalantaridou S, Creatsas G, Deligeoroglou E, Vrachnis N, 2018. Association between brain-derived neurotrophic factor (BDNF) levels in 2(nd) trimester amniotic fluid and fetal development. Mediators Inflamm. 2018, 8476217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage JA, Poston L, Taylor PD, 2008. Developmental origins of obesity and the metabolic syndrome: the role of maternal obesity. Front. Horm. Res 36, 73–84. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Lucki I, Brookshire BR, Carlson GC, Browne CA, Kazi H, Bang S, Choi BR, Chen Y, McMullen MF, Kim SF, 2014. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis 67, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babri S, Doosti MH, Salari AA, 2014. Strain-dependent effects of prenatal maternal immune activation on anxiety- and depression-like behaviors in offspring. Brain Behav. Immun 37, 164–176. [DOI] [PubMed] [Google Scholar]
- Bale TL, 2005. Is mom too sensitive? Impact of maternal stress during gestation. Front. Neuroendocrinol 26, 41–49. [DOI] [PubMed] [Google Scholar]
- Bale TL, 2016. The placenta and neurodevelopment: sex differences in prenatal vulnerability. Dialogues Clin. Neurosci 18, 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, Nemeroff CB, Reyes TM, Simerly RB, Susser ES, Nestler EJ, 2010. Early life programming and neurodevelopmental disorders. Biol. Psychiatry 68, 314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Epperson CN, 2015. Sex differences and stress across the lifespan. Nat. Neurosci 18, 1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, 2004. The source of cerebral insulin. Eur. J. Pharmacol 490, 5–12. [DOI] [PubMed] [Google Scholar]
- Banks WA, Owen JB, Erickson MA, 2012. Insulin in the brain: there and back again. Pharmacol. Ther 136, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbazanges A, Piazza PV, Le Moal M, Maccari S, 1996. Maternal glucocorticoid secretion mediates long-term effects of prenatal stress. J. Neurosci 16, 3943–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, 2007. The origins of the developmental origins theory. J. Int. Med 261, 412–417. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, 1986. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1, 1077–1081. [DOI] [PubMed] [Google Scholar]
- Basatemur E, Gardiner J, Williams C, Melhuish E, Barnes J, Sutcliffe A, 2013. Maternal prepregnancy BMI and child cognition: a longitudinal cohort study. Pediatrics 131, 56–63. [DOI] [PubMed] [Google Scholar]
- Basu S, Haghiac M, Surace P, Challier JC, Guerre-Millo M, Singh K, Waters T, Minium J, Presley L, Catalano PM, Hauguel-de Mouzon S, 2011 Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity (Silver Spring) 19 (2011) 476–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauermeister JJ, Shrout PE, Chavez L, Rubio-Stipec M, Ramirez R, Padilla L, Anderson A, Garcia P, Canino G, 2007. ADHD and gender: are risks and sequela of ADHD the same for boys and girls? J. Child. Psychol. Psychiatry 48, 831–839. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM, 2009. Early-life programming of later-life brain and behavior: a critical role for the immune system. Front. Behav. Neurosci 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM, 2012. The immune system and developmental programming of brain and behavior. Front. Neuroendocrinol 33, 267–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Tsang V, 2010. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 24, 2104–2115. [DOI] [PubMed] [Google Scholar]
- Bilder DA, Bakian AV, Viskochil J, Clark EA, Botts EL, Smith KR, Pimentel R, McMahon WM, Coon H, 2013. Maternal prenatal weight gain and autism spectrum disorders. Pediatrics 132, e1276–e1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliddal M, Olsen J, Stovring H, Eriksen HL, Kesmodel US, Sorensen TI, Nohr EA, 2014. Maternal pre-pregnancy BMI and intelligence quotient (IQ) in 5-year-old children: a cohort based study. PLoS One 9, e94498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma GJ, Moghadam AA, Cordner ZA, Tamashiro KL, 2014. Prenatal stress and stress coping style interact to predict metabolic risk in male rats. Endocrinology 155, 1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma GJ, Tamashiro KL, 2015. Individual differences in the effects of prenatal stress exposure in rodents. Neurobiol. Stress 1, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma GJ, Bale TL, Casanello P, Lara HE, Lucion AB, Suchecki D, Tamashiro KL, 2014. Long-term impact of early life events on physiology and behaviour. J. Neuroendocrinol 26, 587–602. [DOI] [PubMed] [Google Scholar]
- Bolton JL, Wiley MG, Ryan B, Truong S, Strait M, Baker DC, Yang NY, Ilkayeva O, O’Connell TM, Wroth SW, Sanchez CL, Swamy G, Newgard C, Kuhn C, Bilbo SD, Simmons LA, 2017. Perinatal western-type diet and associated gestational weight gain alter postpartum maternal mood. Brain Behav 7, e00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC, Blakely RD, Deneris ES, Levitt P, 2011. A transient placental source of serotonin for the fetal forebrain. Nature 472, 347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, 2010. Neurodevelopmental actions of leptin. Brain Res. 1350, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, 2017. Development of hypothalamic circuits that control food intake and energy balance. In: nd, and Harris RBS, (Eds.), Appetite and Food Intake: Central Control, Boca Raton (FL), pp. 135–154. [PubMed] [Google Scholar]
- Boyle CA, Boulet S, Schieve LA, Cohen RA, Blumberg SJ, Yeargin-Allsopp M, Visser S, Kogan MD, 2011. Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics 127, 1034–1042. [DOI] [PubMed] [Google Scholar]
- Brekke HK, van Odijk J, Ludvigsson J, 2007. Predictors and dietary consequences of frequent intake of high-sugar, low-nutrient foods in 1-year-old children participating in the ABIS study. Br. J. Nutr 97, 176–181. [DOI] [PubMed] [Google Scholar]
- Brion MJ, Zeegers M, Jaddoe V, Verhulst F, Tiemeier H, Lawlor DA, Smith GD, 2011. Intrauterine effects of maternal prepregnancy overweight on child cognition and behavior in 2 cohorts. Pediatrics 127, e202–e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson SL, Bale TL, 2014. Prenatal stress-induced increases in placental inflammation and offspring hyperactivity are male-specific and ameliorated by maternal antiinflammatory treatment. Endocrinology 155, 2635–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson SL, Bale TL, 2016. The placenta as a mediator of stress effects on neurodevelopmental reprogramming. Neuropsychopharmacology 41, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Meyer U, 2018. Maternal immune activation and neuropsychiatric illness: a translational research perspective. Am. J. Psychiatry 175, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, Costa-Mattioli M, 2016. Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell 165, 1762–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins K, Devaskar SU, 2011. Fetal origins of adult disease. Curr. Probl. Pediatr. Adolesc. Health Care 41, 158–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell BM, Pocivavsek A, Notarangelo FM, Parachikova AI, 2015. The Role of Kynurenine Pathway Metabolites in Neuropsychiatric Disorders In: Mittal S (Ed.), Targetting the Broadly Pathogenic Kynurenine Pathway. Springer, Cham, pp. 241–254. [Google Scholar]
- Can OD, Ulupinar E, Ozkay UD, Yegin B, Ozturk Y, 2012. The effect of simvastatin treatment on behavioral parameters, cognitive performance, and hippocampal morphology in rats fed a standard or a high-fat diet. Behav. Pharmacol 23, 582–592. [DOI] [PubMed] [Google Scholar]
- Cannon M, Jones PB, Murray RM, 2002. Obstetric complications and schizophrenia: historical and meta-analytic review. Am. J. Psychiatry 159, 1080–1092. [DOI] [PubMed] [Google Scholar]
- Casas M, Chatzi L, Carsin AE, Amiano P, Guxens M, Kogevinas M, Koutra K, Lertxundi N, Murcia M, Rebagliato M, Riano I, Rodriguez-Bernal CL, Roumeliotaki T, Sunyer J, Mendez M, Vrijheid M, 2013. Maternal pre-pregnancy overweight and obesity, and child neuropsychological development: two Southern European birth cohort studies. Int. J. Epidemiol 42, 506–517. [DOI] [PubMed] [Google Scholar]
- Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel-de Mouzon S, 2008 Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 29 (2008) 274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JC, Nugent BM, Bale TL, 2018. Parental advisory: maternal and paternal stress can impact offspring neurodevelopment. Biol. Psychiatry 83, 886–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Xu X, Yan Y, 2018. Estimated global overweight and obesity burden in pregnant women based on panel data model. PLoS One 13, e0202183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SL, Cline HT, 2010. Insulin receptor signaling in the development of neuronal structure and function. Neural. Dev 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR, 2016. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton VL, 2010. Review: Sex and the human placenta: mediating differential strategies of fetal growth and survival. Placenta 31 (Suppl), S33–S39. [DOI] [PubMed] [Google Scholar]
- Coiro P, Padmashri R, Suresh A, Spartz E, Pendyala G, Chou S, Jung Y, Meays B, Roy S, Gautam N, Alnouti Y, Li M, Dunaevsky A, 2015. Impaired synaptic development in a maternal immune activation mouse model of neurodevelopmental disorders. Brain Behav. Immun 50, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contu L, Hawkes CA, 2017. A review of the impact of maternal obesity on the cognitive function and mental health of the offspring. Int. J. Mol. Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordner ZA, Tamashiro KL, 2015. Effects of high-fat diet exposure on learning & memory. Physiol. Behav 152, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordner ZA, Khambadkone SG, Boersma GJ, Song L, Summers TN, Moran TH, Tamashiro KLK, 2019. Maternal high-fat diet results in cognitive impairment and hippocampal gene expression changes in rat offspring. Exp. Neurol 318, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig WY, Palomaki GE, Neveux LM, Haddow JE, 2013. Maternal body mass index during pregnancy and offspring neurocognitive development. Obstet. Med 6, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren J, Samuelsson AM, Jansson T, Holmang A, 2006. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr. Res 60, 147–151. [DOI] [PubMed] [Google Scholar]
- Das UN, 2001. Is obesity an inflammatory condition? Nutrition 17, 953–966. [DOI] [PubMed] [Google Scholar]
- D’Asti E, Long H, Tremblay-Mercier J, Grajzer M, Cunnane SC, Di Marzo V, Walker CD, 2010. Maternal dietary fat determines metabolic profile and the magnitude of endocannabinoid inhibition of the stress response in neonatal rat offspring. Endocrinology 151, 1685–1694. [DOI] [PubMed] [Google Scholar]
- Davis EP, Pfaff D, 2014. Sexually dimorphic responses to early adversity: implications for affective problems and autism spectrum disorder. Psychoneuroendocrinology 49, 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza MA, Centenaro LA, Menegotto PR, Henriques TP, Bonini J, Achaval M, Lucion AB, 2013. Prenatal stress produces social behavior deficits and alters the number of oxytocin and vasopressin neurons in adult rats. Neurochem. Res 38, 1479–1489. [DOI] [PubMed] [Google Scholar]
- de Souza DF, Wartchow KM, Lunardi PS, Brolese G, Tortorelli LS, Batassini C, Biasibetti R, Goncalves CA, 2015. Changes in astroglial markers in a maternal immune activation model of schizophrenia in wistar rats are dependent on sex. Front Cell Neurosci. 9, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman BE, Patterson PH, 2009. Cytokines and CNS development. Neuron 64, 61–78. [DOI] [PubMed] [Google Scholar]
- Dickerson DD, Bilkey DK, 2013. Aberrant neural synchrony in the maternal immune activation model: using translatable measures to explore targeted interventions. Front Behav. Neurosci 7, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djiane J, Attig L, 2008. Role of leptin during perinatal metabolic programming and obesity. J. Physiol. Pharmacol 59 (Suppl 1), 55–63. [PubMed] [Google Scholar]
- Dodds L, Fell DB, Shea S, Armson BA, Allen AC, Bryson S, 2011. The role of prenatal, obstetric and neonatal factors in the development of autism. J. Autism Dev. Disord 41, 891–902. [DOI] [PubMed] [Google Scholar]
- Drwal E, Rak A, Gregoraszczuk EL, 2019. Review: polycyclic aromatic hydrocarbons (PAHs)-Action on placental function and health risks in future life of newborns. Toxicology 411, 133–142. [DOI] [PubMed] [Google Scholar]
- D’Souza D, Sadananda M, 2017. Estrous cycle phase-dependent changes in anxiety- and depression-like profiles in the late adolescent wistar-kyoto rat. Ann. Neurosci 24, 136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GA, Morgan CP, Bale TL, 2011. Sex-specificity in transgenerational epigenetic programming. Horm Behav. 59, 290–295. [DOI] [PubMed] [Google Scholar]
- Dye L, Boyle NB, Champ C, Lawton C, 2017. The relationship between obesity and cognitive health and decline. Proc. Nutr. Soc 76, 443–454. [DOI] [PubMed] [Google Scholar]
- Edlow AG, 2017. Maternal obesity and neurodevelopmental and psychiatric disorders in offspring. Prenat Diagn 37, 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlow AG, Glass RM, Smith CJ, Tran PK, James K, Bilbo S, 2018. Placental macrophages: a window into fetal microglial function in maternal obesity. Int. J. Dev. Neurosci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entringer S, Buss C, Wadhwa PD, 2015. Prenatal stress, development, health and disease risk: a psychobiological perspective-2015 Curt Richter Award Paper. Psychoneuroendocrinology 62, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen HL, Kesmodel US, Underbjerg M, Kilburn TR, Bertrand J, Mortensen EL, 2013. Predictors of intelligence at the age of 5: family, pregnancy and birth characteristics, postnatal influences, and postnatal growth. PLoS One 8, e79200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes ML, McAllister AK, 2015. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci 16, 469–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes ML, McAllister AK, 2016. Maternal immune activation: implications for neuropsychiatric disorders. Science 353, 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadel JR, Reagan LP, 2016. Stop signs in hippocampal insulin signaling: the role of insulin resistance in structural, functional and behavioral deficits. Curr. Opin. Behav. Sci 9, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadel JR, Jolivalt CG, Reagan LP, 2013. Food for thought: the role of appetitive peptides in age-related cognitive decline. Ageing Res. Rev 12, 764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Su R, Song Y, Wang C, Lin L, Ma J, Yang H, 2016. Positive correlation between enhanced expression of TLR4/MyD88/NF-kappaB with insulin resistance in placentae of gestational diabetes mellitus. PLoS One 11, e0157185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CR, Reagan LP, 2018. Insulin-mediated synaptic plasticity in the CNS: anatomical, functional and temporal contexts. Neuropharmacology 136, 182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Ogden CL, Curtin LR, 2010. Prevalence and trends in obesity among US adults, 1999–2008. JAMA 303, 235–241. [DOI] [PubMed] [Google Scholar]
- Fombonne E, 2009. Epidemiology of pervasive developmental disorders. Pediatr. Res 65, 591–598. [DOI] [PubMed] [Google Scholar]
- Ford SP, Zhang L, Zhu M, Miller MM, Smith DT, Hess BW, Moss GE, Nathanielsz PW, Nijland MJ, 2009. Maternal obesity accelerates fetal pancreatic beta-cell but not alpha-cell development in sheep: prenatal consequences. Am. J. Physiol. Regul. Integr. Comp. Physiol 297, R835–R843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frias AE, Morgan TK, Evans AE, Rasanen J, Oh KY, Thornburg KL, Grove KL, 2011. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 152, 2456–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froehlich TE, Lanphear BP, Epstein JN, Barbaresi WJ, Katusic SK, Kahn RS, 2007. Prevalence, recognition, and treatment of attention-deficit/hyperactivity disorder in a national sample of US children. Arch. Pediatr. Adolesc. Med 161, 857–864. [DOI] [PubMed] [Google Scholar]
- Frye CA, Petralia SM, Rhodes ME, 2000. Estrous cycle and sex differences in performance on anxiety tasks coincide with increases in hippocampal progesterone and 3alpha,5alpha-THP. Pharmacol. Biochem. Behav 67, 587–596. [DOI] [PubMed] [Google Scholar]
- Fujioka A, Fujioka T, Ishida Y, Maekawa T, Nakamura S, 2006. Differential effects of prenatal stress on the morphological maturation of hippocampal neurons. Neuroscience 141, 907–915. [DOI] [PubMed] [Google Scholar]
- Gabory A, Roseboom TJ, Moore T, Moore LG, Junien C, 2013. Placental contribution to the origins of sexual dimorphism in health and diseases: sex chromosomes and epigenetics. Biol. Sex Differ 4, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal EM, Sherman AD, 1980. L-kynurenine: its synthesis and possible regulatory function in brain. Neurochem. Res 5, 223–239. [DOI] [PubMed] [Google Scholar]
- Gallo LA, Barrett HL, Dekker Nitert M, 2017. Review: placental transport and metabolism of energy substrates in maternal obesity and diabetes. Placenta 54, 59–67. [DOI] [PubMed] [Google Scholar]
- Gardner RM, Lee BK, Magnusson C, Rai D, Frisell T, Karlsson H, Idring S, Dalman C, 2015. Maternal body mass index during early pregnancy, gestational weight gain, and risk of autism spectrum disorders: results from a Swedish total population and discordant sibling study. Int. J. Epidemiol 44, 870–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauster M, Hiden U, van Poppel M, Frank S, Wadsack C, Hauguel-de Mouzon S, Desoye G, 2011. Dysregulation of placental endothelial lipase in obese women with gestational diabetes mellitus. Diabetes 60, 2457–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz KD, Anderka MT, Werler MM, Jick SS, 2016. Maternal pre-pregnancy body mass index and autism spectrum disorder among offspring: a population-based case-control study. Paediatr. Perinat. Epidemiol 30, 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore JH, Jarskog LF, Vadlamudi S, 2005. Maternal poly I: C exposure during pregnancy regulates TNF alpha, BDNF, and NGF expression in neonatal brain and the maternal-fetal unit of the rat. J. Neuroimmunol 159, 106–112. [DOI] [PubMed] [Google Scholar]
- Giovanoli S, Notter T, Richetto J, Labouesse MA, Vuillermot S, Riva MA, Meyer U, 2015. Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J Neuroinflammation 12, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giriko CA, Andreoli CA, Mennitti LV, Hosoume LF, Souto Tdos S, Silva AV, Mendes-da-Silva C, 2013. Delayed physical and neurobehavioral development and increased aggressive and depression-like behaviors in the rat offspring of dams fed a high-fat diet. Int. J. Dev. Neurosci 31, 731–739. [DOI] [PubMed] [Google Scholar]
- Giussani DA, 2011. The vulnerable developing brain. Proc. Natl. Acad. Sci. USA 108, 2641–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeden N, Velasquez J, Arnold KA, Chan Y, Lund BT, Anderson GM, Bonnin A, 2016. Maternal inflammation disrupts fetal neurodevelopment via increased placental output of serotonin to the fetal brain. J. Neurosci 36, 6041–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Cameron HA, Daniels DC, Woolley CS, McEwen BS, 1992. Adrenal hormones suppress cell division in the adult rat dentate gyrus. J. Neurosci 12, 3642–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf AE, Lallier SW, Waidyaratne G, Thompson MD, Tipple TE, Hester ME, Trask AJ, Rogers LK, 2016. Maternal high fat diet exposure is associated with increased hepcidin levels, decreased myelination, and neurobehavioral changes in male offspring. Brain Behav. Immun 58, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeneweg FL, Karst H, de Kloet ER, Joels M, 2011. Rapid non-genomic effects of corticosteroids and their role in the central stress response. J. Endocrinol 209, 153–167. [DOI] [PubMed] [Google Scholar]
- Groer M, Fuchs D, Duffy A, Louis-Jacques A, D’Agata A, Postolache TT, 2018. Associations among obesity, inflammation, and tryptophan catabolism in pregnancy. Biol. Res. Nurs 20, 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S, Sohn IJ, Kim N, Sim HJ, Cheon KA, 2015. Characteristics of brains in autism spectrum disorder: structure, function and connectivity across the lifespan. Exp. Neurobiol 24, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantsoo L, Epperson CN, 2017. Anxiety disorders among women: a female lifespan approach. Focus (Am Psychiatr Publ) 15, 162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantsoo L, Kornfield S, Anguera MC, Epperson CN, 2019. Inflammation: a proposed intermediary between maternal stress and offspring neuropsychiatric risk. Biol. Psychiatry 85, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes EK, Tessier DR, Percival ME, Holloway AC, Petrik JJ, Gruslin A, Raha S, 2014. Trophoblast invasion and blood vessel remodeling are altered in a rat model of lifelong maternal obesity. Reprod. Sci 21, 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel JJ, Vandenberg LN, 2015. Developmental origins of health and disease: a paradigm for understanding disease cause and prevention. Curr. Opin. Pediatr 27, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry C, Kabbaj M, Simon H, Le Moal M, Maccari S, 1994. Prenatal stress increases the hypothalamo-pituitary-adrenal axis response in young and adult rats. J. Neuroendocrinol 6, 341–345. [DOI] [PubMed] [Google Scholar]
- Hillemeier MM, Weisman CS, Chuang C, Downs DS, McCall-Hosenfeld J, Camacho F, 2011. Transition to overweight or obesity among women of reproductive age. J. Womens Health (Larchmt) 20, 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle SN, Schieve LA, Stein AD, Swan DW, Ramakrishnan U, Sharma AJ, 2012. Associations between maternal prepregnancy body mass index and child neurodevelopment at 2 years of age. Int. J. Obes. (Lond) 36, 1312–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser T, Ullrich C, Sperner-Unterweger B, Humpel C, 2011. Inflammatory stimuli reduce survival of serotonergic neurons and induce neuronal expression of indoleamine 2,3-dioxygenase in rat dorsal raphe nucleus organotypic brain slices. Neuroscience 184, 128–138. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, 2006. Inflammation and metabolic disorders. Nat. 444, 860–867. [DOI] [PubMed] [Google Scholar]
- Howell KR, Powell TL, 2017. Effects of maternal obesity on placental function and fetal development. Reproduction 153, R97–R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howerton CL, Bale TL, 2014. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 111, 9639–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howerton CL, Morgan CP, Fischer DB, Bale TL, 2013. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc. Natl. Acad. Sci. USA 110, 5169–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, 2013. Immune dysregulation in autism spectrum disorder. Int. Rev. Neurobiol 113, 269–302. [DOI] [PubMed] [Google Scholar]
- Hsiao EY, Patterson PH, 2011. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun 25, 604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Yu X, Keim S, Li L, Zhang L, Zhang J, 2014. Maternal prepregnancy obesity and child neurodevelopment in the Collaborative Perinatal Project. Int. J. Epidemiol 43, 783–792. [DOI] [PubMed] [Google Scholar]
- Iikuni N, Lam QL, Lu L, Matarese G, La Cava A, 2008. Leptin and inflammation. Curr. Immunol. Rev 4, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izvolskaia M, Sharova V, Zakharova L, 2018. Prenatal programming of neuroendocrine system development by lipopolysaccharide: long-term effects. Int. J. Mol. Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson-Pick S, Audet MC, Nathoo N, Anisman H, 2011. Stressor experiences during the juvenile period increase stressor responsivity in adulthood: transmission of stressor experiences. Behav. Brain Res 216, 365–374. [DOI] [PubMed] [Google Scholar]
- Jacobson-Pick S, Richter-Levin G, 2010. Differential impact of juvenile stress and corticosterone in juvenility and in adulthood, in male and female rats. Behav. Brain Res 214, 268–276. [DOI] [PubMed] [Google Scholar]
- Jasarevic E, Rodgers AB, Bale TL, 2015. A novel role for maternal stress and microbial transmission in early life programming and neurodevelopment. Neurobiol. Stress 1, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PB, Rantakallio P, Hartikainen AL, Isohanni M, Sipila P, 1998. Schizophrenia as a long-term outcome of pregnancy, delivery, and perinatal complications: a 28-year follow-up of the 1966 north Finland general population birth cohort. Am. J. Psychiat 155 (1998) 355–64. [DOI] [PubMed] [Google Scholar]
- Kang SS, Kurti A, Fair DA, Fryer JD, 2014. Dietary intervention rescues maternal obesity induced behavior deficits and neuroinflammation in offspring. J. Neuroinflammation 11, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S, Chabot JG, Quirion R, 1993. Quantitative autoradiographic localization of [125I]insulin-like growth factor I, [125I]insulin-like growth factor II, and [125I]insulin receptor binding sites in developing and adult rat brain. J. Comp. Neurol 333, 375–397. [DOI] [PubMed] [Google Scholar]
- Kawai M, Minabe Y, Takagai S, Ogai M, Matsumoto H, Mori N, Takei N, 2004. Poor maternal care and high maternal body mass index in pregnancy as a risk factor for schizophrenia in offspring. Acta Psychiatr. Scand 110, 257–263. [DOI] [PubMed] [Google Scholar]
- Kawamura T, Chen J, Takahashi T, Ichitani Y, Nakahara D, 2006. Prenatal stress suppresses cell proliferation in the early developing brain. Neuroreport 17, 1515–1518. [DOI] [PubMed] [Google Scholar]
- Kertes DA, Bhatt SS, Kamin HS, Hughes DA, Rodney NC, Mulligan CJ, 2017. BNDF methylation in mothers and newborns is associated with maternal exposure to war trauma. Clin. Epigenetics 9, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Glendining KA, Grattan DR, Jasoni CL, 2016. Maternal obesity leads to increased proliferation and numbers of astrocytes in the developing fetal and neonatal mouse hypothalamus. Int. J. Dev. Neurosci 53, 18–25. [DOI] [PubMed] [Google Scholar]
- King S, Dancause K, Turcotte-Tremblay AM, Veru F, Laplante DP, 2012. Using natural disasters to study the effects of prenatal maternal stress on child health and development. Birth Defects Res. C Embryo. Today 96, 273–288. [DOI] [PubMed] [Google Scholar]
- Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, Toovey S, Prinssen EP, 2014. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol 10, 643–660. [DOI] [PubMed] [Google Scholar]
- Kodomari I, Wada E, Nakamura S, Wada K, 2009. Maternal supply of BDNF to mouse fetal brain through the placenta. Neurochem. Int 54, 95–98. [DOI] [PubMed] [Google Scholar]
- Koehl M, Darnaudery M, Dulluc J, Van Reeth O, Le Moal M, Maccari S, 1999. Prenatal stress alters circadian activity of hypothalamo-pituitary-adrenal axis and hippocampal corticosteroid receptors in adult rats of both gender. J. Neurobiol 40, 302–315. [PubMed] [Google Scholar]
- Kolominsky Y, Igumnov S, Drozdovitch V, 1999. The psychological development of children from Belarus exposed in the prenatal period to radiation from the Chernobyl atomic power plant. J. Child Psychol. Psychiatry 40, 299–305. [PubMed] [Google Scholar]
- Kossintseva I, Wong S, Johnstone E, Guilbert L, Olson DM, Mitchell BF, 2006. Proinflammatory cytokines inhibit human placental 11beta-hydroxysteroid dehydrogenase type 2 activity through Ca2+ and cAMP pathways. Am. J. Physiol. Endocrinol. Metab 290, E282–E288. [DOI] [PubMed] [Google Scholar]
- Krakowiak P, Walker CK, Bremer AA, Baker AS, Ozonoff S, Hansen RL, Hertz-Picciotto I, 2012. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 129, e1121–e1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzeczkowski JE, Boylan K, Arbuckle TE, Dodds L, Muckle G, Fraser W, Favotto LA, Van Lieshout RJ, Group MS, 2018. Neurodevelopment in 3–4year old children exposed to maternal hyperglycemia or adiposity in utero. Early Hum. Dev 125, 8–16. [DOI] [PubMed] [Google Scholar]
- Lackey DE, Olefsky JM, 2016. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol 12, 15–28. [DOI] [PubMed] [Google Scholar]
- Lammert CR, Frost EL, Bolte AC, Paysour MJ, Shaw ME, Bellinger CE, Weigel TK, Zunder ER, Lukens JR, 2018. Cutting edge: critical roles for microbiota-mediated regulation of the immune system in a prenatal immune activation model of autism. J. Immunol 201, 845–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante DP, Brunet A, Schmitz N, Ciampi A, King S, 2008. Project Ice Storm: prenatal maternal stress affects cognitive and linguistic functioning in 5 1/2-year-old children. J. Am. Acad. Child Adolesc. Psychiatry 47, 1063–1072. [DOI] [PubMed] [Google Scholar]
- Lappas M, Permezel M, Rice GE, 2005. Leptin and adiponectin stimulate the release of proinflammatory cytokines and prostaglandins from human placenta and maternal adipose tissue via nuclear factor-kappaB, peroxisomal proliferator-activated receptor-gamma and extracellularly regulated kinase 1/2. Endocrinology 146, 3334–3342. [DOI] [PubMed] [Google Scholar]
- Lee PR, Brady DL, Shapiro RA, Dorsa DM, Koenig JI, 2007. Prenatal stress generates deficits in rat social behavior: Reversal by oxytocin. Brain Res. 1156, 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire V, Koehl M, Le Moal M, Abrous DN, 2000. Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc. Natl. Acad. Sci. USA 97, 11032–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage J, Blondeau B, Grino M, Breant B, Dupouy JP, 2001. Maternal under-nutrition during late gestation induces fetal overexposure to glucocorticoids and intrauterine growth retardation, and disturbs the hypothalamo-pituitary adrenal axis in the newborn rat. Endocrinology 142, 1692–1702. [DOI] [PubMed] [Google Scholar]
- Levitt P, Veenstra-VanderWeele J, 2015. Neurodevelopment and the origins of brain disorders. Neuropsychopharmacology 40, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Fallin MD, Riley A, Landa R, Walker SO, Silverstein M, Caruso D, Pearson C, Kiang S, Dahm JL, Hong X, Wang G, Wang MC, Zuckerman B, Wang X, 2016. The association of maternal obesity and diabetes with autism and other developmental disabilities. Pediatrics 137, e20152206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Shao B, Huang H, Zhou Y, Lin Y, 2015. Maternal high fat diet programs stress-induced behavioral disorder in adult offspring. Physiol. Behav 152, 119–127. [DOI] [PubMed] [Google Scholar]
- Liu YH, Lai WS, Tsay HJ, Wang TW, Yu JY, 2013. Effects of maternal immune activation on adult neurogenesis in the subventricular zone-olfactory bulb pathway and olfactory discrimination. Schizophr. Res 151, 1–11. [DOI] [PubMed] [Google Scholar]
- Liu-Seifert H, Siemers E, Price K, Han B, Selzler KJ, Henley D, Sundell K, Aisen P, Cummings J, Raskin J, Mohs R., 2015. Alzheimer’s disease neuroimaging, cognitive impairment precedes and predicts functional impairment in mild Alzheimer’s disease. J. Alzheimers Dis 47, 205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomes R, Hull L, Mandy WPL, 2017. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J. Am. Acad. Child. Adolesc. Psychiatry 56, 466–474. [DOI] [PubMed] [Google Scholar]
- Luo ZC, Nuyt AM, Delvin E, Fraser WD, Julien P, Audibert F, Girard I, Shatenstein B, Deal C, Grenier E, Garofalo C, Levy E, 2013. Maternal and fetal leptin, adiponectin levels and associations with fetal insulin sensitivity. Obesity (Silver Spring) 21, 210–216. [DOI] [PubMed] [Google Scholar]
- Maccari S, Piazza PV, Kabbaj M, Barbazanges A, Simon H, Le Moal M, 1995. Adoption reverses the long-term impairment in glucocorticoid feedback induced by prenatal stress. J. Neurosci 15, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maftei O, Whitrow MJ, Davies MJ, Giles LC, Owens JA, Moore VM, 2015. Maternal body size prior to pregnancy, gestational diabetes and weight gain: associations with insulin resistance in children at 9–10 years. Diabet. Med 32, 174–180. [DOI] [PubMed] [Google Scholar]
- Manuelpillai U, Ligam P, Smythe G, Wallace EM, Hirst J, Walker DW, 2005. Identification of kynurenine pathway enzyme mRNAs and metabolites in human placenta: up-regulation by inflammatory stimuli and with clinical infection. Am. J. Obstet. Gynecol 192, 280–288. [DOI] [PubMed] [Google Scholar]
- Mao J, Zhang X, Sieli PT, Falduto MT, Torres KE, Rosenfeld CS, 2010. Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proc. Natl. Acad. Sci. USA 107, 5557–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark PJ, Sisala C, Connor K, Patel R, Lewis JL, Vickers MH, Waddell BJ, Sloboda DM, 2011. A maternal high-fat diet in rat pregnancy reduces growth of the fetus and the placental junctional zone, but not placental labyrinth zone growth. J Dev Origins Health Disease 2, 63–70. [Google Scholar]
- McCormick CM, Green MR, 2013. From the stressed adolescent to the anxious and depressed adult: investigations in rodent models. Neuroscience 249, 242–257. [DOI] [PubMed] [Google Scholar]
- McCormick CM, Mathews IZ, Thomas C, Waters P, 2010. Investigations of HPA function and the enduring consequences of stressors in adolescence in animal models. Brain Cogn 72, 73–85. [DOI] [PubMed] [Google Scholar]
- McLean CP, Asnaani A, Litz BT, Hofmann SG, 2011. Gender differences in anxiety disorders: prevalence, course of illness, comorbidity and burden of illness. J. Psychiatr. Res 45, 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mednick SA, Machon RA, Huttunen MO, Bonett D, 1988. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch. Gen. Psychiatry 45, 189–192. [DOI] [PubMed] [Google Scholar]
- Melis MR, Melis T, Cocco C, Succu S, Sanna F, Pillolla G, Boi A, Ferri GL, Argiolas A, 2007. Oxytocin injected into the ventral tegmental area induces penile erection and increases extracellular dopamine in the nucleus accumbens and paraventricular nucleus of the hypothalamus of male rats. Eur. J. Neurosci 26, 1026–1035. [DOI] [PubMed] [Google Scholar]
- Meyer U, 2014. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol. Psychiatry 75, 307–315. [DOI] [PubMed] [Google Scholar]
- Meyer U, Yee BK, Feldon J, 2007. The neurodevelopmental impact of prenatal infections at different times of pregnancy: the earlier the worse? Neuroscientist 13, 241–256. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Fatemi SH, 2009. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci. Biobehav. Rev 33, 1061–1079. [DOI] [PubMed] [Google Scholar]
- Mina TH, Lahti M, Drake AJ, Raikkonen K, Minnis H, Denison FC, Norman JE, Reynolds RM, 2017. Prenatal exposure to very severe maternal obesity is associated with adverse neuropsychiatric outcomes in children. Psychol. Med 47, 353–362. [DOI] [PubMed] [Google Scholar]
- Missault S, Anckaerts C, Ahmadoun S, Blockx I, Barbier M, Bielen K, Shah D, Kumar-Singh S, De Vos WH, Van der Linden A, Dedeurwaerdere S, Verhoye M, 2019. Hypersynchronicity in the default mode-like network in a neurodevelopmental animal model with relevance for schizophrenia. Behav. Brain Res 364, 303–316. [DOI] [PubMed] [Google Scholar]
- Mission JF, Marshall NE, Caughey AB, 2015. Pregnancy risks associated with obesity. Obstet. Gynecol. Clin. North Am 42, 335–353. [DOI] [PubMed] [Google Scholar]
- Modi ME, Young LJ, 2012. The oxytocin system in drug discovery for autism: animal models and novel therapeutic strategies. Horm. Behav 61, 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaron MF, Petry KG, Rodriguez JJ, Marinelli M, Aurousseau C, Rougon G, Le Moal M, Abrous DN, 1999. Adrenalectomy increases neurogenesis but not PSANCAM expression in aged dentate gyrus. Eur. J. Neurosci 11, 1479–1485. [DOI] [PubMed] [Google Scholar]
- Morley-Fletcher S, Darnaudery M, Koehl M, Casolini P, Van Reeth O, Maccari S, 2003. Prenatal stress in rats predicts immobility behavior in the forced swim test. Effects of a chronic treatment with tianeptine. Brain Res 989, 246–251. [DOI] [PubMed] [Google Scholar]
- Morley-Fletcher S, Darnaudery M, Mocaer E, Froger N, Lanfumey L, Laviola G, Casolini P, Zuena AR, Marzano L, Hamon M, Maccari S, 2004. Chronic treatment with imipramine reverses immobility behaviour, hippocampal corticosteroid receptors and cortical 5-HT(1A) receptor mRNA in prenatally stressed rats. Neuropharmacology 47, 841–847. [DOI] [PubMed] [Google Scholar]
- Moser VC, McDaniel KL, Woolard EA, Phillips PM, Franklin JN, Gordon CJ, 2017. Impacts of maternal diet and exercise on offspring behavior and body weights. Neurotoxicol. Teratol 63, 46–50. [DOI] [PubMed] [Google Scholar]
- Moss BG, Chugani DC, 2014. Increased risk of very low birth weight, rapid postnatal growth, and autism in underweight and obese mothers. Am. J. Health Promot 28, 181–188. [DOI] [PubMed] [Google Scholar]
- Mucellini AB, Goularte JF, de Araujo da Cunha AC, Caceres RC, Noschang C, da Silva Benetti C, Silveira PP, Sanvitto GL, 2014. Effects of exposure to a cafeteria diet during gestation and after weaning on the metabolism and body weight of adult male offspring in rats. Br. J. Nutr 111, 1499–506. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL, 2006. Impact of prenatal stress on long term body weight is dependent on timing and maternal sensitivity. Physiol. Behav 88, 605–614. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL, 2007. Early prenatal stress impact on coping strategies and learning performance is sex dependent. Physiol. Behav 91, 55–65. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL, 2008. Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci 28, 9055–9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SK, Fineberg AM, Maxwell SD, Alloy LB, Zimmermann L, Krigbaum NY, Cohn BA, Drabick DAG, Ellman LM, 2017. Maternal infection and stress during pregnancy and depressive symptoms in adolescent offspring. Psychiatry Res. 257, 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KN, Edye ME, Manca M, Vernon AC, Oladipo JM, Fasolino V, Harte MK, Mason V, Grayson B, McHugh PC, Knuesel I, Prinssen EP, Hager R, Neill JC, 2019. Evolution of a maternal immune activation (mIA) model in rats: early developmental effects. Brain Behav. Immun 75, 48–59. [DOI] [PubMed] [Google Scholar]
- Murthi P, Vaillancourt C, 2019. Placental serotonin systems in pregnancy metabolic complications associated with maternal obesity and gestational diabetes mellitus. Biochim Biophys. Acta Mol. Basis Dis [DOI] [PubMed] [Google Scholar]
- Musial B, Vaughan OR, Fernandez-Twinn DS, Voshol P, Ozanne SE, Fowden AL, Sferruzzi-Perri AN, 2017. A Western-style obesogenic diet alters maternal metabolic physiology with consequences for fetal nutrient acquisition in mice. J. Physiol 595, 4875–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neggers YH, Goldenberg RL, Ramey SL, Cliver SP, 2003. Maternal prepregnancy body mass index and psychomotor development in children. Acta Obstet. Gynecol. Scand 82, 235–240. [DOI] [PubMed] [Google Scholar]
- Nguyen MU, Wallace MJ, Pepe S, Menheniott TR, Moss TJ, Burgner D, 2015. Perinatal inflammation: a common factor in the early origins of cardiovascular disease? Clin. Sci. (Lond) 129, 769–784. [DOI] [PubMed] [Google Scholar]
- Niculescu MD, Lupu DS, 2009. High fat diet-induced maternal obesity alters fetal hippocampal development. Int. J. Dev. Neurosci 27, 627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X, Wu X, Ying A, Shao B, Li X, Zhang W, Lin C, Lin Y, 2019. Maternal high fat diet programs hypothalamic-pituitary-adrenal function in adult rat offspring. Psychoneuroendocrinology 102, 128–138. [DOI] [PubMed] [Google Scholar]
- O’Connell BA, Moritz KM, Walker DW, Dickinson H, 2013. Sexually dimorphic placental development throughout gestation in the spiny mouse (Acomys cahirinus). Placenta 34, 119–126. [DOI] [PubMed] [Google Scholar]
- Oken E, Gillman MW, 2003. Fetal origins of obesity. Obes. Res 11, 496–506. [DOI] [PubMed] [Google Scholar]
- Olfson M, Blanco C, Wang S, Laje G, Correll CU, 2014. National trends in the mental health care of children, adolescents, and adults by office-based physicians. JAMA Psychiatry 71, 81–90. [DOI] [PubMed] [Google Scholar]
- Olson CM, Strawderman MS, Dennison BA, 2009. Maternal weight gain during pregnancy and child weight at age 3 years. Mat. Child Health J 13, 839–846. [DOI] [PubMed] [Google Scholar]
- Oskvig DB, Elkahloun AG, Johnson KR, Phillips TM, Herkenham M, 2012. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain. Behav. Immun 26, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi N, Walsh K, 2007. Adiponectin as an anti-inflammatory factor. Clin. Chim. Acta 380, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page KC, Jones EK, Anday EK, 2014. Maternal and postweaning high-fat diets disturb hippocampal gene expression, learning, and memory function. Am. J. Physiol. Regul. Integr. Comp. Physiol 306, R527–R537. [DOI] [PubMed] [Google Scholar]
- Parboosing R, Bao Y, Shen L, Schaefer CA, Brown AS, 2013. Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiatry 70, 677–685. [DOI] [PubMed] [Google Scholar]
- Pathmaperuma AN, Mana P, Cheung SN, Kugathas K, Josiah A, Koina ME, Broomfield A, Delghingaro-Augusto V, Ellwood DA, Dahlstrom JE, Nolan CJ, 2010. Fatty acids alter glycerolipid metabolism and induce lipid droplet formation, syncytialisation and cytokine production in human trophoblasts with minimal glucose effect or interaction. Placenta 31, 230–239. [DOI] [PubMed] [Google Scholar]
- Patrich E, Piontkewitz Y, Peretz A, Weiner I, Attali B, 2016. Maternal immune activation produces neonatal excitability defects in offspring hippocampal neurons from pregnant rats treated with poly I:C. Sci. Rep 6, 19106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH, 2009. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav. Brain Res 204, 313–321. [DOI] [PubMed] [Google Scholar]
- Patterson PH, 2011. Maternal infection and immune involvement in autism. Trends Mol. Med 17, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen CA, Vadlamudi S, Boccia ML, Moy SS, 2011. Variations in maternal behavior in C57BL/6J mice: behavioral comparisons between adult offspring of high and low pup-licking mothers. Front. Psychiatry 2, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg-Raibstein D, Luca E, Wolfrum C, 2012. Maternal high-fat diet in mice programs emotional behavior in adulthood. Behav. Brain Res 233, 398–404. [DOI] [PubMed] [Google Scholar]
- Piontkewitz Y, Arad M, Weiner I, 2011. Risperidone administered during asymptomatic period of adolescence prevents the emergence of brain structural pathology and behavioral abnormalities in an animal model of schizophrenia. Schizophr. Bull 37, 1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteri M, Romualdi C, Magliozzi R, Monaco S, Calabrese M, 2017. Cognitive impairment predicts disability progression and cortical thinning in MS: An 8-year study. Mult. Scler 23, 848–854. [DOI] [PubMed] [Google Scholar]
- Ponzio NM, Servatius R, Beck K, Marzouk A, Kreider T, 2007. Cytokine levels during pregnancy influence immunological profiles and neurobehavioral patterns of the offspring. Ann N Y Acad. Sci 1107, 118–128. [DOI] [PubMed] [Google Scholar]
- Power ML, Schulkin J, 2012. Maternal obesity, metabolic disease, and allostatic load. Physiol. Behav 106, 22–28. [DOI] [PubMed] [Google Scholar]
- Prince CS, Maloyan A, Myatt L, 2017. Maternal obesity alters brain derived neurotrophic factor (BDNF) signaling in the placenta in a sexually dimorphic manner. Placenta 49, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins JR, Eskandar S, Eggen BJL, Scherjon SA, 2018. Microglia, the missing link in maternal immune activation and fetal neurodevelopment; and a possible link in preeclampsia and disturbed neurodevelopment? J. Reprod. Immunol 126, 18–22. [DOI] [PubMed] [Google Scholar]
- Purcell RH, Sun B, Pass LL, Power ML, Moran TH, Tamashiro KL, 2011. Maternal stress and high-fat diet effect on maternal behavior, milk composition, and pup ingestive behavior. Physiol. Behav 104, 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L, Guo Z, Bosco C, Guidotti S, Wang Y, Wang M, Parast M, Schaack J, Hay WW Jr., Moore TR, Shao J, 2015. Maternal high-fat feeding increases placental lipoprotein lipase activity by reducing SIRT1 expression in mice. Diabetes 64, 3111–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt LF, 2006. Neurotrophin-regulated signalling pathways. Philos. Trans. R Soc. Lond. B Biol. Sci 361, 1545–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisinger S, Khan D, Kong E, Berger A, Pollak A, Pollak DD, 2015. The poly(I:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol. Ther 149, 213–226. [DOI] [PubMed] [Google Scholar]
- Reynolds KA, Boudoures AL, Chi MM, Wang Q, Moley KH, 2015. Adverse effects of obesity and/or high-fat diet on oocyte quality and metabolism are not reversible with resumption of regular diet in mice. Reprod. Fertil. Dev 27, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds LC, Inder TE, Neil JJ, Pineda RG, Rogers CE, 2014. Maternal obesity and increased risk for autism and developmental delay among very preterm infants. J. Perinatol 34, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richetto J, Riva MA, 2014. Prenatal maternal factors in the development of cognitive impairments in the offspring. J Reprod Immunol 104–105, 20–25. [DOI] [PubMed] [Google Scholar]
- Rincel M, Lepinay AL, Delage P, Fioramonti J, Theodorou VS, Laye S, Darnaudery M, 2016. Maternal high-fat diet prevents developmental programming by early-life stress. Transl. Psychiatry 6, e966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera HM, Christiansen KJ, Sullivan EL, 2015. The role of maternal obesity in the risk of neuropsychiatric disorders. Front. Neurosci 9, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb JL, Messa I, Lui E, Yeung D, Thacker J, Satvat E, Mielke JG, 2017. A maternal diet high in saturated fat impairs offspring hippocampal function in a sexspecific manner. Behav. Brain. Res 326, 187–199. [DOI] [PubMed] [Google Scholar]
- Roberts KA, Riley SC, Reynolds RM, Barr S, Evans M, Statham A, Hor K, Jabbour HN, Norman JE, Denison FC, 2011. Placental structure and inflammation in pregnancies associated with obesity. Placenta 32, 247–254. [DOI] [PubMed] [Google Scholar]
- Robinson M, Zubrick SR, Pennell CE, Van Lieshout RJ, Jacoby P, Beilin LJ, Mori TA, Stanley FJ, Newnham JP, Oddy WH, 2013. Pre-pregnancy maternal overweight and obesity increase the risk for affective disorders in offspring. J. Dev. Orig Health Dis 4, 42–48. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, 2010. Maternal pre-pregnancy obesity and risk for inattention and negative emotionality in children. J. Child. Psychol. Psychiatry 51, 134–143. [DOI] [PubMed] [Google Scholar]
- Rodriguez JJ, Montaron MF, Petry KG, Aurousseau C, Marinelli M, Premier S, Rougon G, Le Moal M, Abrous DN, 1998. Complex regulation of the expression of the polysialylated form of the neuronal cell adhesion molecule by glucocorticoids in the rat hippocampus. Eur. J. Neurosci 10, 2994–3006. [DOI] [PubMed] [Google Scholar]
- Romeo RD, 2013. The teenage brain: the stress response and the adolescent brain. Curr. Dir. Psychol. Sci 22, 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf W, Preissner KT, 2017. State of the Art 2017. J. Thromb. Haemost 15, 1239. [DOI] [PubMed] [Google Scholar]
- Sage D, Maurel D, Bosler O, 2001. Involvement of the suprachiasmatic nucleus in diurnal ACTH and corticosterone responsiveness to stress. Am. J. Physiol. Endocrinol. Metab 280, E260–E269. [DOI] [PubMed] [Google Scholar]
- Sanders TR, Kim DW, Glendining KA, Jasoni CL, 2014. Maternal obesity and IL-6 lead to aberrant developmental gene expression and deregulated neurite growth in the fetal arcuate nucleus. Endocrinology 155, 2566–2577. [DOI] [PubMed] [Google Scholar]
- Sandman CA, Glynn LM, Davis EP, 2013. Is there a viability-vulnerability tradeoff? Sex differences in fetal programming. J. Psychosom. Res 75, 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, de Vega WC, St-Cyr S, Pan P, McGowan PO, 2013. Perinatal high fat diet alters glucocorticoid signaling and anxiety behavior in adulthood. Neuroscience 240, 1–12. [DOI] [PubMed] [Google Scholar]
- Sasaki A, de Vega W, Sivanathan S, St-Cyr S, McGowan PO, 2014. Maternal high-fat diet alters anxiety behavior and glucocorticoid signaling in adolescent offspring. Neuroscience 272, 92–101. [DOI] [PubMed] [Google Scholar]
- Schaefer CA, Brown AS, Wyatt RJ, Kline J, Begg MD, Bresnahan MA, Susser ES, 2000. Maternal prepregnant body mass and risk of schizophrenia in adult offspring. Schizophr. Bull 26, 275–286. [DOI] [PubMed] [Google Scholar]
- Schmitz L, Kuglin R, Bae-Gartz I, Janoschek R, Appel S, Mesaros A, Jakovcevski I, Vohlen C, Handwerk M, Ensenauer R, Dotsch J, Hucklenbruch-Rother E, 2018. Hippocampal insulin resistance links maternal obesity with impaired neuronal plasticity in adult offspring. Psychoneuroendocrinol. 89, 46–52. [DOI] [PubMed] [Google Scholar]
- Schulkin J, Gold PW, McEwen BS, 1998. Induction of corticotropin-releasing hormone gene expression by glucocorticoids: implication for understanding the states of fear and anxiety and allostatic load. Psychoneuroendocrinology 23, 219–243. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ, 2012. Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci 13, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D Jr., Seeley RJ, Baskin DG, 2000. Central nervous system control of food intake. Nature 404, 661–671. [DOI] [PubMed] [Google Scholar]
- Schwartzer JJ, Careaga M, Onore CE, Rushakoff JA, Berman RF, Ashwood P, 2013. Maternal immune activation and strain specific interactions in the development of autism-like behaviors in mice. Transl. Psychiat 3, e240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia SA, Vickers MH, Gray C, Reynolds CM, 2014. Maternal obesity, inflammation, and developmental programming. Biomed. Res. Int 2014, 418975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB, 2006. Inflammation and insulin resistance. J. Clin. Invest 116, 1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanek AM, Meier HC, 2015. Association between prenatal exposure to maternal infection and offspring mood disorders: a review of the literature. Curr. Probl. Pediatr. Adolesc. Health Care 45, 325–364. [DOI] [PubMed] [Google Scholar]
- Simoes LR, Sangiogo G, Tashiro MH, Generoso JS, Faller CJ, Dominguini D, Mastella GA, Scaini G, Giridharan VV, Michels M, Florentino D, Petronilho F, Reus GZ, Dal-Pizzol F, Zugno AI, Barichello T, 2018. Maternal immune activation induced by lipopolysaccharide triggers immune response in pregnant mother and fetus, and induces behavioral impairment in adult rats. J. Psychiatr. Res 100, 71–83. [DOI] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH, 2007. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci 27, 10695–10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolders S, Notter T, Smolders SMT, Rigo JM, Brone B, 2018. Controversies and prospects about microglia in maternal immune activation models for neurodevelop-mental disorders. Brain Behav. Immun 73, 51–65. [DOI] [PubMed] [Google Scholar]
- Sohlberg S, Stephansson O, Cnattingius S, Wikstrom AK, 2012. Maternal body mass index, height, and risks of preeclampsia. Am. J. Hypertens 25, 120–125. [DOI] [PubMed] [Google Scholar]
- Solek CM, Farooqi N, Verly M, Lim TK, Ruthazer ES, 2018. Maternal immune activation in neurodevelopmental disorders. Dev. Dyn 247, 588–619. [DOI] [PubMed] [Google Scholar]
- Song L, Sun B, Boersma GJ, Cordner ZA, Yan J, Moran TH, Tamashiro KLK, 2017. Prenatal high-fat diet alters placental morphology, nutrient transporter expression, and mtorc1 signaling in rat. Obesity (Silver Spring) 25, 909–919. [DOI] [PubMed] [Google Scholar]
- Sood R, Zehnder JL, Druzin ML, Brown PO, 2006. Gene expression patterns in human placenta. Proc. Natl. Acad. Sci. USA 103, 5478–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto M, Cai W, Konishi M, Kahn CR, 2019. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. USA 116, 6379–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stang J, Huffman LG, 2016. Position of the academy of nutrition and dietetics: obesity, reproduction, and pregnancy outcomes. J. Acad. Nutr. Diet 116, 677–691. [DOI] [PubMed] [Google Scholar]
- Stirling J, White C, Lewis S, Hopkins R, Tantam D, Huddy A, Montague L, 2003. Neurocognitive function and outcome in first-episode schizophrenia: a 10-year follow-up of an epidemiological cohort. Schizophr Res 65, 75–86. [DOI] [PubMed] [Google Scholar]
- Stouffer MA, Woods CA, Patel JC, Lee CR, Witkovsky P, Bao L, Machold RP, Jones KT, de Vaca SC, Reith ME, Carr KD, Rice ME, 2015. Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat. Commun 6, 8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EL, Grayson B, Takahashi D, Robertson N, Maier A, Bethea CL, Smith MS, Coleman K, Grove KL, 2010. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J. Neurosci 30, 3826–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EL, Nousen EK, Chamlou KA, 2014. Maternal high fat diet consumption during the perinatal period programs offspring behavior. Physiol. Behav 123, 236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EL, Riper KM, Lockard R, Valleau JC, 2015. Maternal high-fat diet programming of the neuroendocrine system and behavior. Horm. Behav 76, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Purcell RH, Terrillion CE, Yan J, Moran TH, Tamashiro KL, 2012. Maternal high-fat diet during gestation or suckling differentially affects offspring leptin sensitivity and obesity. Diabetes 61, 2833–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Liang NC, Ewald ER, Purcell RH, Boersma GJ, Yan J, Moran TH, Tamashiro KL, 2013. Early postweaning exercise improves central leptin sensitivity in offspring of rat dams fed high-fat diet during pregnancy and lactation. Am. J. Physiol. Regul. Integr. Comp. Physiol 305, R1076–R1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Song L, Tamashiro KL, Moran TH, Yan J, 2014. Large litter rearing improves leptin sensitivity and hypothalamic appetite markers in offspring of rat dams fed high-fat diet during pregnancy and lactation. Endocrinology 155, 3421–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri D, Vaidya VA, 2013. Glucocorticoid regulation of brain-derived neurotrophic factor: relevance to hippocampal structural and functional plasticity. Neuroscience 239, 196–213. [DOI] [PubMed] [Google Scholar]
- Tamashiro KL, Terrillion CE, Hyun J, Koenig JI, Moran TH, 2009. Prenatal stress or high-fat diet increases susceptibility to diet-induced obesity in rat offspring. Diabetes 58, 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JR, Valleau JC, Barling AN, Franco JG, DeCapo M, Bagley JL, Sullivan EL, 2017. Exposure to a high-fat diet during early development programs behavior and impairs the central serotonergic system in juvenile non-human primates. Front. Endocrinol. (Lausanne) 8, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JR, Gustafsson HC, DeCapo M, Takahashi DL, Bagley JL, Dean TA, Kievit P, Fair DA, Sullivan EL, 2018. Maternal diet, metabolic state, and inflammatory response exert unique and long-lasting influences on offspring behavior in non-human primates. Front. Endocrinol. (Lausanne) 9, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozuka Y, Kumon M, Wada E, Onodera M, Mochizuki H, Wada K, 2010. Maternal obesity impairs hippocampal BDNF production and spatial learning performance in young mouse offspring. Neurochem. Int 57, 235–247. [DOI] [PubMed] [Google Scholar]
- Trandafir LM, Temneanu OR, 2016. Pre and post-natal risk and determination of factors for child obesity. J. Med. Life 9, 386–391. [PMC free article] [PubMed] [Google Scholar]
- Treesukosol Y, Sun B, Moghadam AA, Liang NC, Tamashiro KL, Moran TH, 2014. Maternal high-fat diet during pregnancy and lactation reduces the appetitive behavioral component in female offspring tested in a brief-access taste procedure. Am. J. Physiol. Regul. Integr. Comp. Physiol 306, R499–R509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuer T, Tohen M, 2010. Predicting the course and outcome of bipolar disorder: a review. Eur. Psychiat 25, 328–333. [DOI] [PubMed] [Google Scholar]
- Udagawa J, Hino K, 2016. Impact of maternal stress in pregnancy on brain function of the offspring. Nihon Eiseigaku Zasshi 71, 188–194. [DOI] [PubMed] [Google Scholar]
- Vaiserman AM, 2015. Epigenetic programming by early-life stress: evidence from human populations. Dev. Dyn 244, 254–265. [DOI] [PubMed] [Google Scholar]
- Vallee M, Mayo W, Dellu F, Le Moal M, Simon H, Maccari S, 1997. Prenatal stress induces high anxiety and postnatal handling induces low anxiety in adult offspring: correlation with stress-induced corticosterone secretion. J. Neurosci 17, 2626–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee M, MacCari S, Dellu F, Simon H, Le Moal M, Mayo W, 1999. Long-term effects of prenatal stress and postnatal handling on age-related glucocorticoid secretion and cognitive performance: a longitudinal study in the rat. Eur. J. Neurosci 11, 2906–2916. [DOI] [PubMed] [Google Scholar]
- Van den Bergh BRH, van den Heuvel MI, Lahti M, Braeken M, de Rooij SR, Entringer S, Hoyer D, Roseboom T, Raikkonen K, King S, Schwab M, 2017. Prenatal developmental origins of behavior and mental health: The influence of maternal stress in pregnancy. Neurosci. Biobehav. Rev [DOI] [PubMed] [Google Scholar]
- Van Doorn C, Macht VA, Grillo CA, Reagan LP, 2017. Leptin resistance and hippocampal behavioral deficits. Physiol. Behav 176, 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lieshout RJ, Robinson M, Boyle MH, 2013. Maternal pre-pregnancy body mass index and internalizing and externalizing problems in offspring. Can. J. Psychiatry 58, 151–159. [DOI] [PubMed] [Google Scholar]
- Varastehpour A, Radaelli T, Minium J, Ortega H, Herrera E, Catalano P, Hauguel-de Mouzon S, 2006. Activation of phospholipase A2 is associated with generation of placental lipid signals and fetal obesity. J. Clin. Endocrinol. Metab 91, 248–255. [DOI] [PubMed] [Google Scholar]
- Velasquez JC, Goeden N, Bonnin A, 2013. Placental serotonin: implications for the developmental effects of SSRIs and maternal depression. Front. Cell Neurosci 7, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon AC, So PW, Lythgoe DJ, Chege W, Cooper JD, Williams SC, Kapur S, 2015. Longitudinal in vivo maturational changes of metabolites in the prefrontal cortex of rats exposed to polyinosinic-polycytidylic acid in utero. Eur. Neuropsychopharmacol 25, 2210–2220. [DOI] [PubMed] [Google Scholar]
- Walker CD, Naef L, d’Asti E, Long H, Xu Z, Moreau A, Azeddine B, 2008. Perinatal maternal fat intake affects metabolism and hippocampal function in the offspring: a potential role for leptin. Ann. N Y Acad. Sci 1144, 189–202. [DOI] [PubMed] [Google Scholar]
- Wallace AE, Anderson GM, Dubrow R, 2008. Obstetric and parental psychiatric variables as potential predictors of autism severity. J. Autism. Dev. Disord 38, 1542–1554. [DOI] [PubMed] [Google Scholar]
- Weinstock M, 2017. Prenatal stressors in rodents: effects on behavior. Neurobiol. Stress 6, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Allen AM, Mendelsohn FA, 1987. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 121, 1562–1570. [DOI] [PubMed] [Google Scholar]
- White CL, Pistell PJ, Purpera MN, Gupta S, Fernandez-Kim SO, Hise TL, Keller JN, Ingram DK, Morrison CD, Bruce-Keller AJ, 2009. Effects of high fat diet on Morris maze performance, oxidative stress, and inflammation in rats: contributions of maternal diet. Neurobiol. Dis 35, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M, Zhang Z, Nance E, Drewes JL, Lesniak WG, Singh S, Chugani DC, Rangaramanujam K, Graham DR, Kannan S, 2017. Maternal inflammation results in altered tryptophan metabolism in rabbit placenta and fetal brain. Dev. Neurosci 39, 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winther G, Elfving B, Muller HK, Lund S, Wegener G, 2018. Maternal high-fat diet programs offspring emotional behavior in adulthood. Neuroscience 388, 87–101. [DOI] [PubMed] [Google Scholar]
- Wolfrum C, Peleg-Raibstein D, 2018. Maternal overnutrition leads to cognitive and neurochemical abnormalities in C57BL/6 mice. Nutr. Neurosci 1–12. [DOI] [PubMed] [Google Scholar]
- Wraw C, Deary IJ, Der G, Gale CR, 2018. Maternal and offspring intelligence in relation to BMI across childhood and adolescence. Int. J. Obes. (Lond) 42, 1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright T, Langley-Evans SC, Voigt JP, 2011. The impact of maternal cafeteria diet on anxiety-related behaviour and exploration in the offspring. Physiol. Behav 103, 164–172. [DOI] [PubMed] [Google Scholar]
- Yamada-Goto N, Katsuura G, Ochi Y, Ebihara K, Kusakabe T, Hosoda K, Nakao K, 2012. Impairment of fear-conditioning responses and changes of brain neurotrophic factors in diet-induced obese mice. J. Neuroendocrinol 24, 1120–1125. [DOI] [PubMed] [Google Scholar]
- Yang X, Haghiac M, Glazebrook P, Minium J, Catalano PM, Hauguel-de Mouzon S, 2015. Saturated fatty acids enhance TLR4 immune pathways in human trophoblasts. Hum. Reprod 30, 2152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Li M, Haghiac M, Catalano PM, O’Tierney-Ginn P, Hauguel-de Mouzon S, 2016. Causal relationship between obesity-related traits and TLR4-driven responses at the maternal-fetal interface. Diabetologia 59, 2459–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JL, Cai L, States JC, 2018. Impact of prenatal arsenic exposure on chronic adult diseases. Syst. Biol. Reprod. Med 64, 469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano E, Nathanielsz PW, 2017. Relative contributions of maternal Western-type high fat, high sugar diets and maternal obesity to altered metabolic function in pregnancy. J. Physiol 595, 4573–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbo O, Iosif AM, Walker C, Ozonoff S, Hansen RL, Hertz-Picciotto I, 2013. Is maternal influenza or fever during pregnancy associated with autism or developmental delays? Results from the CHARGE (CHildhood Autism Risks from Genetics and Environment) study. J. Autism. Dev. Disord 43, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XY, Chen DC, Xiu MH, Yang FD, Haile CN, Kosten TA, Kosten TR, 2012. Gender differences in never-medicated first-episode schizophrenia and medicated chronic schizophrenia patients. J. Clin. Psychiatry 73, 1025–1033. [DOI] [PubMed] [Google Scholar]
- Zhang J, Jing Y, Zhang H, Bilkey DK, Liu P, 2018. Maternal immune activation altered microglial immunoreactivity in the brain of postnatal day 2 rat offspring. Synapse e22072. [DOI] [PubMed] [Google Scholar]
- Zhang Z, van Praag H, 2015. Maternal immune activation differentially impacts mature and adult-born hippocampal neurons in male mice. Brain Behav. Immun 45, 60–70. [DOI] [PubMed] [Google Scholar]
- Zieba J, Uddin GM, Youngson NA, Karl T, Morris MJ, 2019. Long-term behavioural effects of maternal obesity in C57BL/6J mice. Physiol. Behav 199, 306–313. [DOI] [PubMed] [Google Scholar]