

Abstract
Background:
Longevity gene klotho (KL) is associated with age-related phenotypes including lifespan, cardiometabolic disorders, cognition, and brain morphology, in part, by conferring protection against inflammation. We hypothesized that the KL/inflammation association might be altered in the presence of psychiatric stress and operate via epigenetic pathways. We examined KL polymorphisms, and their interaction with posttraumatic stress disorder (PTSD) symptoms, in association with KL DNA methylation in blood. We further examined KL DNA methylation as a predictor of longitudinal changes in a peripheral biomarker of inflammation (C-reactive protein; CRP).
Methods:
The sample comprised 309 white non-Hispanic military veterans (93.5% male; mean age: 32 years, range: 19-65; 30% PTSD per structured diagnostic interview); 111 were reassessed approximately two years later.
Results:
Analyses revealed a methylation quantitative trait locus at rs9527025 (C370S, previously implicated in numerous studies of aging) in association with a Cytosine-phosphate-Guanine site (cg00129557; B = −.65, p = 1.29 X 10−20), located within a DNase hypersensitivity site in the body of KL. There was also a rs9527025 x PTSD severity interaction (B = .004, p = .035) on methylation at this locus such that the minor allele was associated with reduced cg00129557 methylation in individuals with few or no PTSD symptoms while this effect was attenuated in those with elevated levels of PTSD. Path models revealed that methylation at cg00129557 was inversely associated with CRP over time (B = −.14, p = .005), controlling for baseline CRP. There was also an indirect effect of rs9527025 X PTSD on subsequent CRP via cg00129557 methylation (indirect B = −.002, p = .033).
Conclusions:
Results contribute to our understanding of the epigenetic correlates of inflammation in PTSD and suggest that KL methylation may be a mechanism by which KL genotype confers risk vs. resilience to accelerated aging in those experiencing traumatic stress.
Keywords: PTSD, inflammation, methylation, accelerated aging, klotho
1. Introduction
Polymorphisms in the gene klotho (KL) have been associated with lifespan and age-related phenotypes in numerous preclinical and clinical studies (Kuro-o et al., 1997; Kurosu et al., 2005; Dubal et al., 2014; Abraham et al., 2016). The gene and the protein it encodes were named for the mythological Greek goddess, Clotho, who was said to have spun the thread of human life (Hamilton, 2011). The gene and its protein have been linked to a wide variety of age-related processes including cardiometabolic function (Arking et al., 2002), recovery following experimentally-induced cardiac damage (Ramez et al., 2019), kidney function (Drew et al., 2017), late-life cognition (Dubal et al., 2014; Yokoyama et al., 2015), white matter integrity (Chen, Sloane et al., 2013; King, Rosene & Abraham, 2012), motor function (Kuro-o et al., 1997), and lifespan (Kuro-o et al., 1997; Kurosu et al., 2005; Dubal et al., 2014). Greater KL expression in old age is thought to confer protection against age-related declines in neural integrity and cognition (Dubal et al., 2014).
KL exerts its neuro- and cardio-protective effects by reducing inflammation (Hui et al., 2017; Maekawa et al., 2009; Xia et al., 2016; Zhu et al., 2018; Zeldich et al., 2019) and oxidative stress (Yamamoto et al., 2005; Zeldich et al., 2014). Inflammation and oxidative stress are well known biomarkers of cellular aging, with increasing levels of each known to play a mechanistic role in age-related adverse outcomes and cellular senescence (Ridker et al., 2002; Frasca & Blomberg, 2016; Fülöp et al., 2016). KL is known to suppress nuclear factor κB (NF-κB) signaling via inhibition of IκB phosphorylation and expression of tumor necrosis factor alpha (TNFα; Guo et al., 2018; Maekawa et al., 2009), which in turn reduces oxidative stress and endothelial cell inflammation (Maekawa et al., 2009). In vivo, KL expression (i.e., mRNA; Martín-Núñez et al., 2017) and circulating KL proteins (Prystupa et al., 2016) tend to be inversely correlated with pro-inflammatory biomarkers, including C-reactive protein (CRP), while in vitro studies have shown KL to reduce inflammatory cytokines (Sedighi et al., 2019). In the choroid plexus, the anatomical barrier between the periphery and brain ventricles, KL has been found to play a pivotal role in preventing peripheral inflammatory molecules from reaching the central nervous system (Zhu et al., 2018). The KL protein also influences endocrine fibroblast growth factor pathways that impact phosphate and calcium metabolism (Kuro-o, 2019) and insulin regulation (Donate-Correa et al., 2016), and KL impacts Wnt signaling, which is critical for determination of cellular fate and progenitor cell senescence (Liu et al., 2007).
Environmental factors are also known to contribute to cellular aging. Traumatic stress and other forms of psychological stress are positively associated with age-related conditions, including inflammation and cardiometabolic pathology (for reviews, see Miller, Lin et al., 2018; Wolf & Morrison, 2017). Posttraumatic stress disorder (PTSD) has been associated with elevated CRP, interleukin (IL)-6, IL1β, and TNFα (Miller, Lin et al., 2018; Miller, Maniates et al., 2018; Passos et al., 2015; Michopoulos et al., 2015; Rosen et al., 2017). Its association with CRP, an acute-phase protein that responds quickly to inflammation and helps to activate innate immunity to bacterial infection (Black et al., 2004; Sproston & Ashworth, 2018), is not consistent across all studies (Miller, Lin et al., 2018). However, this association is of particular interest because chronically elevated CRP is a marker for cardiovascular disease risk (Ridker et al., 2002) and a signature of inflammation-related biological aging (Frasca & Blomberg, 2016). A better understanding of the mechanisms by which PTSD relates to CRP could shed light on novel treatment approaches to reduce inflammation and associated age-related comorbidities.
PTSD is also positively associated with epigenome-wide DNA methylation [DNAm] markers of advanced cellular age (e.g., advanced “epigenetic age” also known as “advanced DNAm age,” Wolf et al., 2016; Wolf, Logue et al., 2018; Wolf, Maniates et al., 2018; Wolf, Logue et al., 2019). In a recent study that forms part of the basis for this one, we found that the strength of the association between traumatic stress and both advanced epigenetic age and CRP was impacted by KL genotype (Wolf, Morrison et al., 2019) such that the association was accentuated among those with the minor frequency allele (A) of the KL SNP rs9315202. This SNP was also found to interact with PTSD to predict reduced integrity of two white matter tracts that connect prefrontal and limbic regions (Wolf, Morrison et al., 2019). The SNP is located just downstream of the gene. The minor allele (C) of a second KL polymorphism, rs398655, was associated with reduced epigenetic age among those with chronic pain. That SNP is upstream of the gene and was previously associated with better cognitive function (Mengel-From et al., 2016). The minor allele (C) of a third KL SNP, rs9527025 (also known as C370S, located in an exonic coding region), was directly associated with slowed epigenetic age. This latter finding was consistent with many prior studies suggesting that this SNP, which is part of a haplotype comprised of SNPs in 100% linkage disequilibrium (LD) referred to as KL-VS, is associated with improved health in humans across various markers of biological aging, including cognition (Dubal et al., 2014; Yokoyama et al., 2015), prefrontal cortex morphology, and longevity (Arking et al., 2002, 2005; Revelas et al., 2018). One study suggested that the minor allele at rs9527025 altered the protein product of the gene in such a way as to enhance its co-receptor functioning for fibroblast growth factor signaling (Zhou et al., 2013). The direct molecular consequences of rs9315202 and rs398655 are unknown.
To our knowledge, no studies have yet examined the possible role of KL methylation in mediating the relationship between KL genotype and inflammatory processes associated with aging. Research on KL methylation published to date has been limited in scope and focused primarily on cancer, kidney-related, and brain aging phenotypes. Those studies suggest that hypermethylation of the KL promotor region is associated with reduced KL expression (e.g., Lee et al., 2010; Rubinek et al., 2012; King et al., 2012) and is a signature of cancerous tumors (Lee et al., 2010; Rubinek et al., 2012), cancer-related death (Zhu et al., 2019), and kidney fibrosis (Azuma et al., 2012). One study expanded this work to cognitive aging and found that KL promoter hypermethylation was positively associated with mild cognitive impairment in a Chinese sample (Luo et al., 2015). However, this effect was not replicated in a second Chinese sample in the same study. Finally, a study in rhesus monkeys found increased kl methylation in the promoter region of prefrontal white matter in older monkeys and also found that this alteration was associated with decreased kl transcription, possibly contributing to brain aging phenotypes (e.g., impaired cognition, lack of myelin integrity; King et al., 2012). However, the role of KL methylation in human biological aging remains an open question.
1.1. Study aims
The primary aim of this study was to address this question by examining the effects of the three previously implicated KL variants (rs9315202, rs398655, and rs9527025) and PTSD on KL methylation levels. The second aim was to examine if KL methylation, in turn, contributed to changes in a peripheral marker of inflammation (CRP) over time, such that the association between PTSD and CRP was indirect, via KL methylation. This study was based on the same sample of white non-Hispanic military veterans with a high prevalence of PTSD in which we previously reported KL genotype X PTSD effects on measures of biological aging (Wolf, Morrison et al., 2019). This study expanded on that prior cross-sectional investigation in that we examined potential molecular mechanisms (KL methylation) that may link KL genotype and PTSD to changes in a biomarker of aging (inflammation) over time.
2. Methods
2.1. Participants and Procedure
Participants in cross-sectional analyses in this study were 309 white non-Hispanic military veterans (93.5% male, mean age = 32 years, Table 1) who deployed as part of the Global War on Terror; 111 of these veterans were included in the longitudinal analyses based on a follow-up assessment that was completed on average approximately two years later (M = 1.96 years, SD = .65 years, range = 0.98 to 4.78 years).1 Analyses were restricted to this ancestral group (from a larger baseline sample of n = 458 veterans of mixed ancestry) due to genetic population stratification and insufficient sample size to proceed with these analyses in other ancestral groups. Veterans participated in The Translational Research Center for TBI and Stress Disorders (TRACTS) research protocol (McGlinchey et al., 2017), an on-going longitudinal study of deployment stress and psychological, cognitive, metabolic, and neurological health among veterans aged 18 – 65 years. Recruitment efforts and exclusion criteria were described in detail previously (McGlinchey et al., 2017). In short, veterans were excluded primarily due to serious active psychopathological processes that posed an immediate risk (e.g., suicidality) or prevented data collection, presence of neurological condition other than that related to mild traumatic brain injury, and due to standard neuroimaging exclusionary criteria. Participants completed psychological interviews and cognitive testing administered by doctoral-level staff; they also had peripheral blood drawn by a certified phlebotomist into 2 X 10ml EDTA tubes for DNA extraction and metabolic and inflammation panels and underwent magnetic resonance imaging of the brain. The most common trauma exposure type was combat (64.08%), with the sudden and unexpected loss of a friend or loved one next most common (41.42%). On average, participants had experienced 1.84 different types of traumatic experiences (SD: 1.73). The study was reviewed and approved annually by the hospital institutional review board.
Table 1.
Participant Characteristics
| Baseline (n = 309) | Follow-up (n = 111) | |
|---|---|---|
| Variable | M (SD) or n (%) | M (SD) or n (%) |
| Male | 289 (93.5) | 101 (91.0) |
| Age (years) | 32.04 (8.40) | 33.97 (8.86) |
| Current PTSD dx | 198 (64.1) | 56 (50.5) |
| Current PTSD sev | 52.45 (29.62) | 43.51 (29.93) |
| CRP (mg/dL) | .23 (.24) | .18 (.12) |
Note. Raw CRP values are reported here but log-transformed CRP values were analyzed. Dx = diagnosis; sev = severity; CRP = C-reactive protein.
2.2. Measures
2.2.1. PTSD.
PTSD was assessed with the Clinician Administered PTSD Scale (CAPS) for DSM-IV (Blake et al., 1995). The CAPS was administered by doctoral-level psychologists, and both symptom severity ratings and PTSD diagnosis were tabulated according to standard scoring rules (Weathers et al., 1999). Diagnoses were based on expert consensus team review of the CAPS. Descriptive statistics pertaining to PTSD are provided in Table 1. Analyses focused on PTSD symptom severity ratings in the past 30 days (current PTSD severity).
2.2.2. CRP.
As described elsewhere (Miller, Maniates et al., 2018), serum CRP levels were obtained from a commercial laboratory (Quest Diagnostics; Cambridge, MA) with a nephelometric assay using CRP monoclonal antibodies. Samples were shipped to the lab the same day they were collected and processed within 24 hours. Lab procedures were standardized by the International Federation of Clinical Chemistry and Laboratory Medicine, the Bureau Communautaire de Référence, and the College of American Pathologists. The analytic sensitivity of the test was 0.10 mg/dL. Regrettably, the clinical lab did not provide information regarding inter/intra-assay variation. Descriptive statistics for CRP are listed in Table 1. The reference range for normal values is < .80 mg/dL. The CRP values for one participant at Time 1 (T1) and for a different participant at Time 2 (T2) were set to missing as they were extreme outliers (12 SDs beyond the mean).
2.3. DNA-Related Methods and Statistical Processing
DNA was extracted from whole blood and genotyped on the Illumina HumanOmni2.5-8 array as described in Logue et al. (2013) and in the Supplementary Materials. We restricted our analysis to genetically confirmed white non-Hispanic subjects identified using SNP weights (Chen, Pollack et al., 2013) according to the PTSD-GWAS consortium criteria (Logue et al. 2015). Principal components (PCs) reflecting ancestry were computed within the white non-Hispanic subsample for inclusion as covariates in analyses (see Supplementary Materials). These PCs were included to prevent false positives due to unmeasured population substructure within the white non-Hispanic subjects. The three SNPs that were evaluated in this study were in Hardy-Weinberg equilibrium (all p > .35).
DNAm was interrogated using the Illumina Infinium MethylationEPIC array (approximately 850K sites) per manufacturer’s protocol (Supplementary Materials). For subjects assessed at a single time point (cross-sectional cohort), sample positions on the methylation arrays were balanced for PTSD diagnostic status and sex. For longitudinal DNAm data, sample position on the arrays was balanced for PTSD diagnostic status, sex, and time (i.e., the T1 and T2 DNA samples from the same subject were included on the same array). We applied the Psychiatric Genomics Consortium PTSD Epigenetics workgroup pipeline for methylation data screening, processing, cleaning, and quality control procedures (Ratanatharathorn et al., 2017), which we have used in the past (see e.g. Sadeh et al. 2016), and which we will only briefly describe here. Missingness filters were used on CpG sites (>10%) and subjects (>5%). Sites cross hybridizing to non-autosomal chromosomes were excluded. DNAm data were normalized with the beta mixture quantile dilation (BMIQ) method (Pidsley et al., 2013), and batch correction was performed using the ComBat method (Leek et al., 2017). The identity of the samples was confirmed by examining genomic variants assessed on the EPIC array and matching genotypes to the corresponding SNP array. The proportions of different white blood cell (WBC) types in each blood sample were estimated from the methylation data, inclusive of CD8-T, CD4-T, natural killer, B-cells, and monocytes (Aryee et al., 2014; Fortin et al., 2017). Additional details pertaining to DNA extraction for the cross-sectional and longitudinal cohorts and the statistical pipelines are provided in Wolf et al. (2016), Wolf, Logue et al. (2019), and in the Supplementary Materials.
2.4. Data Analyses
We first evaluated the three SNPs as candidate methylation quantitative trait loci (meQTLs; i.e. methylation associated SNPs), by testing their association (using hierarchical regression) with 10 KL DNAm loci which passed cleaning and variability filters in the cross-sectional dataset of n = 309 white non-Hispanic military veterans. Linear regression was used given the dimensional nature of DNAm data as the response variable; we focused on identification of meQTLs so as to identify SNPs that were regulatory with respect to KL DNAm. DNAm probes were selected for analysis if they met all quality control criteria (Wolf, Logue et al., 2019; Ratanatharathorn et al., 2017) and had sufficient variability (range > .10, per Logue et al., 2017). This model included the covariates age, sex, WBCs estimated from the DNAm data, the first three ancestry substructure PCs, as well as the main effect of current PTSD symptom severity in a first step of the model. The SNP was entered in a second step of the model to determine the unique contribution of each SNP. As we found significant correlations across the 10 KL DNAm loci, a Bonferroni correction for the evaluation of SNP to DNAm associations would be overly conservative. Therefore, we used a permutation procedure to derive multipletesting corrected p-values that accounted for the three SNPs and 10 DNAm loci in the model. This procedure permuted the genotypes randomly between subjects (with 10,000 replicates) while preserving the correlational structure within the SNPs and within the DNAm loci. Each replicate was analyzed, and the minimum p-value across SNPs and CpGs was noted. The percentile of the uncorrected p-value in this minimum-p distribution is the corrected p-value. These analyses were conducted in RStudio v.1.0.153 running R version 3.6.1. After determining which SNPs were associated with KL DNAm probes in step 2 of the regression, we built upon the model described above by adding a third step to the cross-sectional model that included the interaction between PTSD severity and SNPs identified as meQTLs in the first set of analyses to determine if the association between PTSD symptom severity and DNAm differed as a function of genotype.
A longitudinal path (i.e., regression) analysis was performed next to examine SNP and PTSD severity main effects and SNP X PTSD interactions on T1 DNAm, and to simultaneously examine the intermediate effect of T1 DNAm on T2 CRP, controlling for T1 levels of CRP. This mediation model examined the indirect effects of the SNP, PTSD, and their interactions on T2 inflammation via T1 DNAm (i.e., a direct test of our hypothesis that KL DNAm was a mechanism linking KL genotype X PTSD to later CRP). Because we controlled for baseline levels of CRP, the model predicted residualized change in CRP (e.g., levels of the T2 biomarker that were not simply due to the T1 level of the same biomarker, Allison, 1990; Shutz, 1989). The direct effects of the SNP, PTSD, and their interaction on T2 inflammation were also included in path models to account for potential effects that did not operate via KL DNAm, as were the direct effects of several covariates (age, sex, top three PCs). Although our primary mediation models focused on residualized change in CRP (as the only T2 variable included in the model), we also conducted additional analyses in which we included T2 PTSD and T2 methylation and allowed for the direct effects of the SNP, T1 CRP, T1 PTSD and T1 methylation on each of these variables in order to more comprehensively assess autoregressive and cross-variable longitudinal associations in the data. Path models were examined in Mplus 8.2 (Muthén & Muthén, 1998-2018) using an estimator that is robust to non-normality (MLR). Model fit was evaluated using established guidelines (Hu & Bentler, 1999). Unstandardized regression coefficients are presented in the text unless otherwise noted. The path models were conducted in the subset of 111 participants with follow-up data.
3. Results
3.1. SNP, PTSD, and SNP X PTSD Effects on KL DNAm: Cross-sectional Sample
Table 2 shows the descriptive statistics pertaining to KL variants and DNAm loci that were evaluated. Table S1 lists the full results for each model predicting KL DNAm. Results revealed two meQTLs: The KL-VS SNP (rs9527025) evidenced corrected significant main effect associations with methylation levels at cg027066582 (B = −.55, p = 1.3 X 10−19, corrected p < .0001) and cg00129557 (B = −.65, p = 1.29 X 10−20, corrected p < .0001; Table S1). These loci are 215 bp apart within a DNase hypersensitivity site in the body of KL (GRCh37 chr13:33607361-33607915 as identified in the ENCODE-project database wgEncodeRegDnaseClusteredV3 as accessed through UCSC Genome Browser). They are highly correlated (r = .69 in these data), hence they likely represent association between rs9527025 and methylation across a broad genomic region, and are not independent effects. Given the high correlation between the two associated KL methylation loci, we proceeded to conduct our primary analyses with just cg00129557, which showed the strongest association with rs9527025 (but replicated results with cg02706658 and also examined effects predicting mean methylation values across the two loci as a potential indicator of methylation in this region, see Supplementary Materials for details). Figure 1 shows the location of the three SNPs, cg00129557, and the DNase hypersensitivity site. Table 3 shows the zero-order correlations between the primary variables in the path model.3
Table 2.
Characteristics of Examined KL SNPs and Methylation Loci
| SNPs |
Methylation Loci |
|||||
|---|---|---|---|---|---|---|
| Variable | MAF | Minor/Major Allele | Location (bp) | Mean (M values) | SD (M values) | Range (raw beta values) |
| SNPs | ||||||
| rs398655 | 40.71 | C/A | 33587652 | |||
| rs9527025 | 17.15 | C/G | 33628193 | |||
| rs9315202 | 25.64 | A/G | 33642016 | |||
| Methylation | ||||||
| cg00129557 | −3.70 | .77 | .22 | |||
| cg02706658 | −3.33 | .63 | .23 | |||
| cg09886946 | −3.03 | .38 | .13 | |||
| cg17806623 | −2.64 | .27 | .12 | |||
| cg18056695 | −3.61 | .46 | .14 | |||
| cg19154940 | 3.53 | .49 | .14 | |||
| cg20672059 | 2.48 | .23 | .11 | |||
| cg23584087 | 3.91 | .64 | .15 | |||
| cg25223823 | 2.56 | .40 | .22 | |||
| cg26325430 | 4.06 | .58 | .12 | |||
Note. M values (log 2 logit transformed beta-values) were analyzed in this study. The raw beta range (proportion of methylated DNA) is presented in this table as this was used to exclude loci with insufficient variability. All SNPs examined were located on chromosome 13 and the base pair location is referenced to the GRCH37.p13 build. The MAF of each SNP was consistent with that reported for the CEU reference sample in 1000 Genomes. SNPs = single nucleotide polymorphism; bp = base pair; MAF = minor allele frequency; SD = standard deviation.
Figure 1.
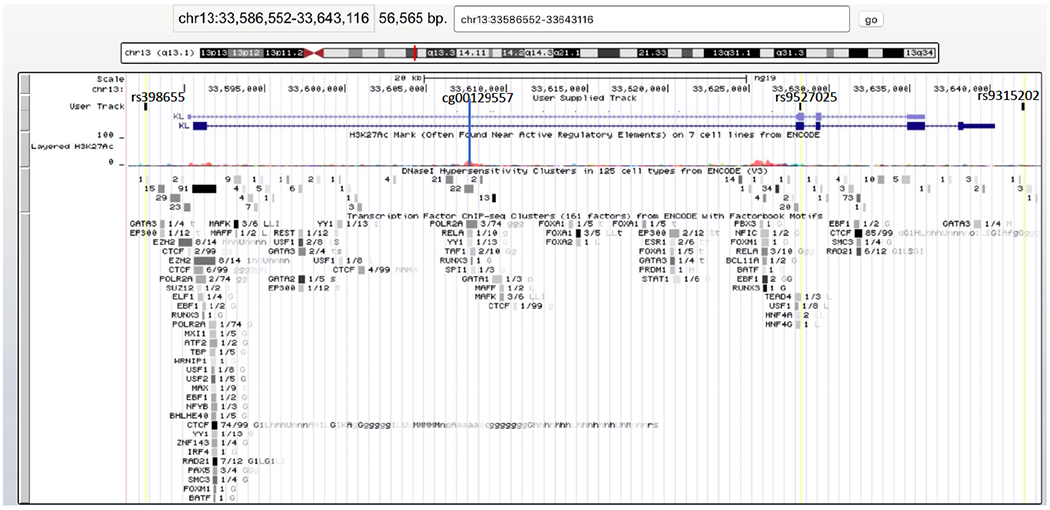
The figure shows the region of the KL gene, including the three SNPs that were evaluated (yellow lines labeled by rs #), cg00129557, and the DNase hypersensitivity site just below this DNAm locus (as indicated by the vertical blue line). The figure was generated using the University of California, Santa Cruz (UCSC) Genome Browser (Kent et al., 2002).
Table 3.
Bivariate Associations Among Key Study Variables
| Variable | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1. rs9527025 | ||||
| 2. cg00129557 at T1 | −.45*** | |||
| 3. PTSD severity at T1 | .08 | .06 | ||
| 4. CRP at T1 | −.01 | −.09 | .17**td | |
| 5. CRP at T2 | −.05 | −.22* | .13 | .43*** |
Note. N = 309 for cross-sectional associations and n = 111 for longitudinal associations. PTSD = posttraumatic stress disorder; CRP = C-reactive protein, T1 = time 1, T2 = time 2.
p < .05.
p < .01.
p < .001.
The same SNP (rs9527025) also evidenced a nominally significant main effect on cg19154940 (B = .12, p = .03, corrected p = .54; Table S1). We were not able to detect meQTL effects for the other two SNPs examined, although rs398655 was nominally associated with methylation values at cg00129557, cg02706658, cg17806623, and cg09886946, but not at a level that survived multiple testing correction (Table S1).
We next expanded the model examining rs9527025 in association with cg00129557 by adding the PTSD severity X SNP term in a third step. This analysis revealed a significant interaction between PTSD severity and rs9527025 in association with DNAm at cg00129557 (B = .004, p = .035; Table 4; Figure 2). The minor allele of rs9527025 (C) was associated with less methylation at cg00129557 when PTSD severity was low, but this effect was attenuated at higher levels of PTSD severity. The residuals from the model were examined and found to be normally distributed. Results held when this model was rerun using dominant coding for the SNP, which compared those with 0 copies vs. 1 or 2 copies of the minor allele at this locus (B for SNP = −.69, p < .001; B for SNP X PTSD = .006, p = .015). In analyses described in the Supplementary Materials, we added potential confounds to our cross-sectional model (the main and interactive effects of current cigarette use, major depressive disorder, alcohol-use disorder, and body mass index) and found that none accounted for the significant main effect of rs9527025 or the interaction between this SNP and PTSD severity.
Table 4.
Effects of rs9527025 and PTSD on KL DNAm at cg00129557
| cg00129557 |
|||
|---|---|---|---|
| Variable | B | SE | p |
| Step 1: Covariates | |||
| Age | −.01 | .01 | .054 |
| Sex | −.21 | .16 | .198 |
| CD8-T | 3.24 | 1.20 | .007 |
| CD4-T | 4.47 | .91 | 1.63 X 10−06 |
| NK | 6.28 | 1.40 | 1.00 X 10−05 |
| B cells | 3.44 | 1.91 | .072 |
| Mono | −1.44 | 1.72 | .404 |
| PC1 | −.90 | .79 | .257 |
| PC2 | .69 | .79 | .382 |
| PC3 | −.94 | .77 | .221 |
| PTSD sev | −.001 | .001 | .641 |
| Step 2: SNP | |||
| rs9527025 | −.65 | .07 | 1.29 X 10−20 |
| Step 3: IX w/PTSD sev | |||
| rs9527025 | .004 | .002 | .035 |
Note. Beta values (i.e., regression coefficients) are unstandardized. Main effect results for other SNPs and CPGs that were either not associated with each other or were only nominally significant are shown in Table S1. NK = natural killer; mono = monocytes; PC = principal component; PTSD sev = posttraumatic stress disorder severity; SNP = single nucleotide polymorphism, w = with; IX = interaction.
Figure 2.
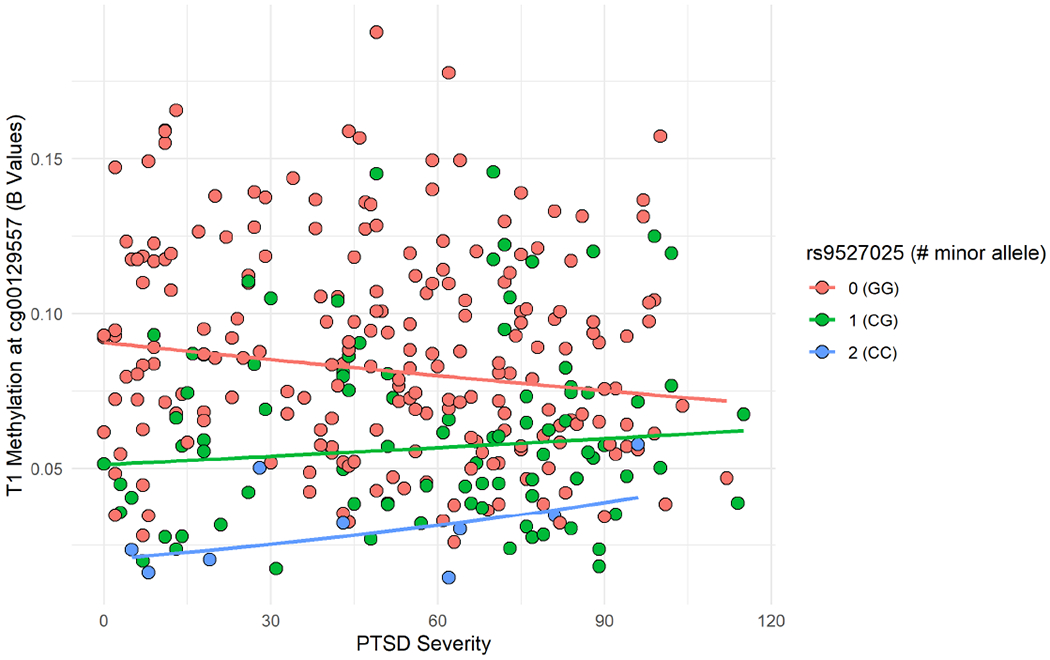
The figure shows the nature of the interaction between KL SNP rs9527025 and PTSD symptom severity on raw (beta) methylation levels at the KL locus cg00129557 for n = 308 participants in this cross-sectional analyses. The protective effects of the minor allele (C) were attenuated at high levels of PTSD symptom severity. This SNP is located on chromosome 13 at 33,628,193 bp; cg00129557 is located on chromosome 13 at 33,607,618 bp. There were 9 participants homozygous for the minor allele (CC), 88 heterozygous (CG), and 211 (GG) homozygous for the major allele.
3.2. Path Models Predicting Residualized Change in Peripheral Inflammation: Longitudinal Subsample
The first path analysis evaluated the main and interactive effects of the KL-VS SNP (rs9527025) and PTSD severity on residualized change in CRP values at T2 via T1 methylation levels at cg00129557 (i.e., indirect effects), controlling for age, sex, and the first three ancestry substructure PCs. Because the number of participants with two copies of the minor allele at this locus was small in the longitudinal subsample (n = 2), these analyses proceeded with dominant allele coding (n = 78 with 0 copies of minor allele vs. n = 33 with 1-2 copies). The direct effects of the SNP and PTSD (and their interaction) on residualized change in CRP values was also included in the model. Results revealed that T1 methylation at this locus was associated with residualized change in CRP values over time (B = −.14, p = .005). Methylation at this locus was predicted by the SNP (B = −1.20, p < .001) and by the interaction between the SNP and PTSD severity in this subset of the data (B = .01, p = .004; Figure 3 for standardized results). The indirect effect of the interaction term on residualized change in CRP values via methylation at this locus was also significant (B = −.002, standardized β = −.13, p = .033). In total, the model explained 30% of the variance in CRP values at T2 and 24% of the variance in methylation at cg00129557. We added to this model by including the estimated WBCs as covariates of methylation at this locus. Again, the pattern of results was unchanged with a significant indirect effect of the interaction term on residualized change in CRP values over time (B = −.002, standardized β = −.11, p = .04). The fit of the path models is shown in Table S2. Fit statistics suggested that all models were consistent with good model fit.
Figure 3.
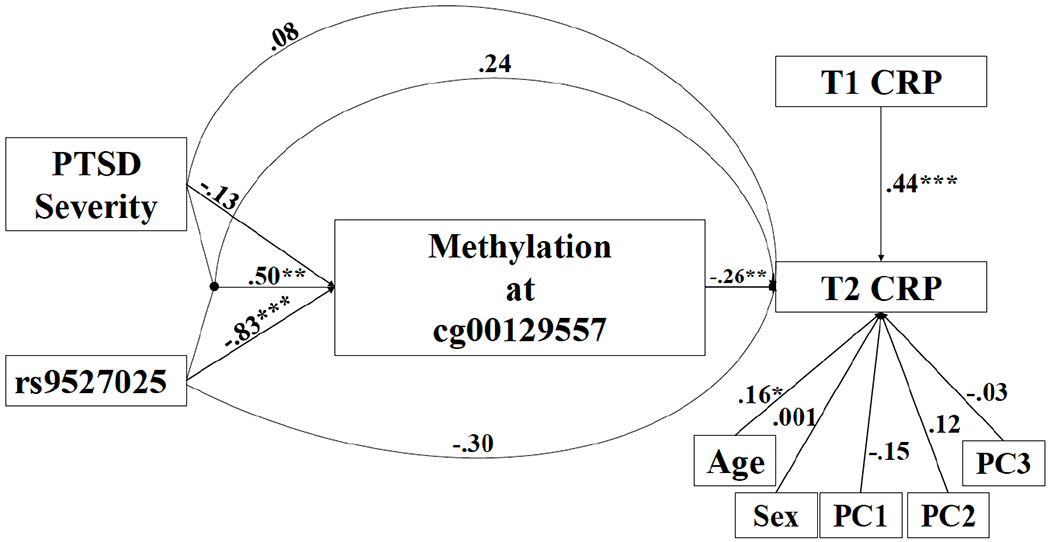
The figure shows the results of the path model examining the direct and indirect effects of the KL-VS SNP (rs9527025), PTSD severity, and their interaction on residualized changes in CRP levels over the course of approximately two years via methylation at the KL locus cg00129557 (n = 111). Standardized coefficients are presented.
*p < .05. **p < .01. ***p < .001
We also reran this model using additive genotype coding at this locus and found that the results were nearly identical, with the exception of an additional direct effect of rs9527025 on residualized changes in CRP (B = −.26, p = .049). The indirect effect of the SNP X PTSD on residualized changes in CRP via methylation at cg00129557 remained unchanged (B = −.002, p = .044; standardized β = −.13, p = .037).
Next, we added T2 methylation and T2 PTSD severity to the model and examined the direct effects of rs9527025, T1 PTSD severity, T1 CRP and T1 cg00129557 levels on these T2 variables. We found that T2 PTSD severity was predicted by T1 PTSD severity (B = .80, p < .001), but not by T1 CRP, methylation, or the SNP (Figure S1 for standardized path results). T2 methylation at cg00129557 was associated with T1 methylation at this same locus (B = .30, p = .002) and evidenced a residual association with rs9527025 (B = −.48, p = .002) that was not accounted for by the effect of this SNP on T1 methylation at this locus. There were no effects of T1 PTSD or CRP on methylation at this locus at T2. Other than these autoregressive effects and the additional effect of the SNP on T2 methylation, the results of the analysis were otherwise unchanged from that without these T2 variables included in the model. Specifically, the indirect effect of PTSD severity X rs9527025 on residualized change in CRP at T2 via T1 methylation at cg00129557 remained significant (B = −.002, p = .042, standardized β = −.13, p = .035). Results for the correlated DNAm locus (cg02706658) are shown in Figure S2.
4. Discussion
The primary finding of this study was that effects of the KL genetic variant rs9527025 and PTSD on residualized changes in inflammation over a two-year interval are mediated, at least in part, by KL methylation. Many of the age-related effects of KL are thought to operate through inflammatory pathways (Hui et al., 2017; Maekawa et al., 2009), and the results of this study implicate methylation at the cg00129557 locus as a causal factor. Though the minor allele of rs9527025 is known to confer protection against a host of age-related phenotypes (Arking et al., 2002) and was similarly associated with decreased CRP levels over time (and with reduced KL methylation at cg00129557) in this study, we found that this SNP also interacted with PTSD severity such that its protective effect was diminished in those with greater PTSD symptom severity. Specifically, those without substantial PTSD symptoms exhibited reduced methylation at this locus, consistent with the main effect of the genotype. In contrast, those with greater PTSD severity evidenced higher levels of methylation at this locus, despite having the protective variant, and this was associated with increased inflammation. These changes likely operate via reduced KL expression, as supported by data from the Genotype-Tissue Expression (GTEx) Project’s online database (version 8 release).4 Specifically, although gene expression was not examined in this study, the GTEx reference data suggested that rs9527025 is an expression quantitative trait loci (eQTL), with the minor allele predictive of lower KL expression in whole blood (normalized effect size [NES] = −.13, p = 8.1 X 10−5) in 670 subjects. Per GTEx, the SNP also showed significant associations with KL expression in at least two brain regions, including nucleus accumbens (NES = .18, p = .01, n = 202) and hippocampus (NES = −.11, p = .03, n = 165), further supporting the relevance of this SNP for alterations in KL expression in the periphery and brain.
Epigenetic modifications are thought to reflect both genetic and environmental influences and here we found that PTSD interacted with genotype to predict KL methylation (and indirectly, residualized CRP values over time). One way to conceptualize this association is to consider symptoms of PTSD as part of the environment that interacts with at risk genotypes (i.e., G X E) to promote cellular aging. Evaluation of genotype in an at-risk PTSD population could provide useful clinical information as to who is at greatest risk for increasing inflammation. This could encourage targeting of lifestyle and pharmacological interventions to those with the greatest need. As well, the role of cg00129557 in linking PTSD and genotype to inflammation may prove useful in developing new pharmacological therapies to reduce inflammation in those at risk (e.g., personalized medicine).
To our knowledge, this is the first study to link DNAm at cg00129557 to inflammation. This locus is located mid-way through the gene in a region identified as a “DNase hypersensitive site” per the Encode project (GRCh37 chr13:33607361-33607915). Hypersensitive sites are regions that are easily cleaved by the nuclease DNAse I. They occur at open ends of chromatin allowing for binding of transcriptional factors and as such, they are thought to be regulatory (Thurman et al., 2012). Increased methylation at these sites is associated with reduced chromatin binding availability, and therefore, reduced expression (Thurman et al., 2012). Thus, methylation at cg00129557 may be a mechanism linking the KL-VS SNPs to various age-related phenotypes identified in the literature previously, but replication in other samples and cell types is clearly needed.
The identification of a KL DNase hypersensitivity site that links genotype to inflammation further highlights the importance of pursuing KL-related interventions. Previous studies suggest that circulating KL is increased as a function of exercise (Ramez et al., 2019; Amaro-Gahete et al., 2019), and that KL expression is increased by cholesterol-lowering medications (Janić et al., 2019). Targeting KL promoter region using CRISPR technology has also been associated with alterations in KL expression in neural and kidney cell lines (Chen et al., 2018), suggesting a possible therapeutic approach for increasing KL levels and protecting against neurobiological health decline. Thus, there may be multiple avenues to pursue in the development of interventions to alter the modifiable aspects of KL expression and potentially protect against a range of age-related adverse cardiac, renal, and cognitive health outcomes.
The association between KL and CRP is important as CRP has been shown to interact with the NF-κB signaling pathway to promote greater inflammation and reactive oxygen species (Zhong et al., 2015). CRP can be used as a marker for “inflammaging” (Frasca & Blomberg, 2016), in which low, but chronic, levels of inflammation increase risk for age-related disease, health decline, and mortality (Franceschi et al., 2000). For example, in a large study of adults aged 65 or older, CRP was found to be negatively associated with functional ability (e.g., activities of daily living), and cognition, and it predicted mortality in survival analyses (Puzianowska-Kuźnicka et al., 2016). Similarly, CRP was one of the strongest correlates of having multiple physical health diagnoses in an epidemiological sample of over 50,000 adults (Stepanova et al., 2015). CRP has also been shown to predict worse psychiatric outcomes in depression (Eswarappa et al., 2019), although this finding is not consistent (Chu et al., 2019; Lamers et al., 2019), suggesting that its predictive value may be more meaningful for physical rather than psychological health outcomes. The association between PTSD and CRP has also been inconsistent in the literature, with some studies finding evidence of this and others reporting null effects (see Miller, Lin et al., 2018 for a review). The results of this study raise the possibility that one reason for variability in these findings is that the association between PTSD and CRP is dependent on genotype. Collectively, results contribute to our understanding of the importance of CRP as a risk-factor for age-related morbidity and mortality, particularly in populations at risk by virtue of their psychiatric status and genetic makeup.
4.1. Study limitations
There are several limitations to this study that should be considered in evaluating these results. First, although the EPIC array is currently the most comprehensive commercially available BeadChip, assaying more than 850K sites, its coverage of the genome is not complete, and it may well be that there are other sites near cg00129557 (and cg02706658) in the hypersensitivity site that alone, or in aggregate, are causally linked to CRP. Similarly, we could not determine whether the causal SNP in this case was actually rs9527025 or some other genetic variant in LD with this SNP. This may be resolved in the future by examining PTSD cohorts with other ancestries where patterns of LD may differ or via sequencing of the entire KL gene. DNAm data were obtained from whole blood, thus we could not examine cell type specific methylation patterns and instead (and as is standard) covaried for estimated cell type. It is also unknown if results would generalize to other tissue (e.g., neural tissue). We did not obtain other inflammatory markers, which would allow for a more comprehensive examination of the effects of PTSD and KL on inflammation and inflammaging. Results from our cohort of white non-Hispanic mostly male veterans may not generalize to other demographic groups or other PTSD samples. The longitudinal analyses were conducted over a relatively short period of time (approximately two years), which may be too short an interval to observe substantial changes in biomarkers and the follow-up sample was small, limiting our statistical power. As well, there are numerous unmeasured potential confounds (such as new disease onset, unmeasured daily life stressors occurring between T1 and T2, and participation in mental health treatment) that could affect our longitudinal results. We also did not obtain mRNA or KL protein levels on these participants, so could not directly test the impact of KL methylation on expression. We did not have access to a suitable replication dataset and it is critical to test if these effects replicate in independent cohorts. Given these concerns, additional research is necessary to examine the replicability of these results and to test their robustness in the broader PTSD population.
4.2. Conclusions
These limitations notwithstanding, our results provide initial evidence of longitudinal effects of KL genotype and PTSD symptom severity on residualized changes in CRP over time via KL methylation. Results help to address questions regarding individual differences in inflammatory outcomes in PTSD samples by suggesting that associations between PTSD and CRP levels, which are well documented in the literature (Eswarappa et al., 2019; Rosen et al., 2017; Michopoulos et al., 2015), are at least partially dependent on KL genotype and methylation status. This highlights the importance of identifying the subpopulations of individuals who are at greatest risk for adverse PTSD-related health outcomes, including premature aging. Efforts to alter the transcriptional effects of circulating levels of KL (that has been shed from transmembrane KL; Chen et al., 2018) may ultimately prove beneficial in mitigating the health burden associated with psychiatric stress, and in promoting health and wellbeing into old age.
Supplementary Material
Funding and Acknowledgments
This work was supported by Merit Review Award Number I01 CX-001276-01 to EJW from the United States (U.S.) Department of Veterans Affairs Clinical Science R&D (CSRD) Service, by the National Institute On Aging of the National Institutes of Health under Award Number R03AG051877 to EJW, and by a Presidential Early Career Award for Scientists and Engineers (PECASE 2013A), as administered by U.S. Department of Veterans Affairs Office of Research and Development, to EJW. Funding for this work was also supported by NIMH grant R21MH102834 to MWM and the Translational Research Center for TBI and Stress Disorders (TRACTS), a VA Rehabilitation Research and Development National Center for TBI Research (B3001-C). FGM’s contribution to this work was supported by National Institute of Mental Health award # 5T32MH019836-16. This research is the result of work supported with resources and the use of facilities at the Pharmacogenomics Analysis Laboratory, Research and Development Service, Central Arkansas Veterans Healthcare System, Little Rock, Arkansas. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 02/18/2020. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs, the National Institutes of Health, or the United States Government.
Footnotes
Data collection for this multi-wave protocol is on-going and is released for data analyses at various points in time. There were no statistical differences in PTSD severity or age among those who did versus did not yet return for follow-up, however, a greater percentage of women (65% vs. 40% of the men) returned for the follow-up by the time of the data release that was used in this study (p = .028). Sex was included as a covariate in all analyses.
cg refers to cytosine guanine and is the standard nomenclature for identifying DNAm loci.
We did not have access to a suitable complete dataset to test for replication of the meQTL involving rs9527025 and cg00129557. This locus was also not included in publicly available datasets that we identified. However, we had a subset of genotype and DNAm data available from an on-going study of veterans who screened positive for PTSD (n = 133; see Supplementary Materials) and we used this genetic data to examine replication of the meQTL. We found that this SNP and DNAm locus correlated with each other at r = −.46, p = 1.91 X 10−8, which is within .01 of the coefficient reported in Table 3.
Accessible at: https://www.gtexportal.org/home/
Disclosures
All authors report no financial or other conflicts of interest in relationship to the contents of this article. Filomene G. Morrison’s contribution to this work was completed as a post-doctoral fellow at Boston University. Dr. Morrison is currently an employee of BlackThorn Therapeutics.
References
- Abraham CR, Mullen PC, Tucker-Zhou T, Chen CD, & Zeldich E 2016. Klotho is a neuroprotective and cognition-enhancing protein In Vitam Horm. 101, 215–238. UK: Academic Press; 10.1016/bs.vh.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Allison PD 1990. Change scores as dependent variables in regression analysis. Sociol Methodol. 20, 93–114. 10.2307/271083. [DOI] [Google Scholar]
- Amaro-Gahete FJ, De-la-O A, Jurado-Fasoli L, Espuch-Oliver A, de Haro T, Gutiérrez A, &Castillo MJ. 2019. Exercise training increases the S-Klotho plasma levels in sedentary middle-aged adults: A randomised controlled trial. The FIT-AGEING study. J Sports Sci. 37, 2175–2183. 10.1080/02640414.2019.1626048. [DOI] [PubMed] [Google Scholar]
- Arking DE, Atzmon G, Arking A, Barzilai N, & Dietz HC 2005. Association between a functional variant of the Klotho gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circulation 96, 412–418. 10.1161/01.RES.0000157171.04054.30. [DOI] [PubMed] [Google Scholar]
- Arking DE, Krebsova A, Macek M, Arking A, Mian IS, Fried L, & Dietz HC 2002Association of human aging with a functional variant of Klotho. Proc Natl Acad Sci U S A. 99, 856–861. 10.1073/pnas.022484299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, &Irizarry RA 2014. Minfi: A flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma M, Koyama D, Kikuchi J, Yoshizawa H, Thasinas D, Shiizaki K, & Kusano E 2012. Promoter methylation confers kidney-specific expression of the Klotho gene. FASEB J. 26, 4264–4274. 10.1096/fj.12-211631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black S, Kushner I, & Samols D 2004. C-reactive protein. J Biol Chem. 279, 48487–48490. 10.1074/jbc.R400025200. [DOI] [PubMed] [Google Scholar]
- Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS, & Keane TM 1995. The development of a clinician-administered PTSD scale. J Trauma Stress 8, 75–90. 10.1002/jts.2490080106. [DOI] [PubMed] [Google Scholar]
- Chen C-Y, Pollack S, Hunter DJ, Hirschhorn JN, Kraft P, & Price AL 2013. Improved ancestry inference using weights from external reference panels. Bioinformatics 29, 1399–1406. 10.1093/bioinformatics/btt144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Sloane JA, Li H, Aytan N, Giannaris EL, Zeldich E, & Luebke JI 2013. The antiaging protein Klotho enhances oligodendrocyte maturation and myelination of the CNS. J Neurosci. 33, 1927–1939. 10.1523/JNEUROSCI.2080-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Zeldich E, Li Y, Yuste A, & Abraham CR 2018. Activation of the anti-aging and cognition-enhancing gene Klotho by CRISPR-dCas9 transcriptional effector complex. J Mol Neurosci. 64, 175–184. 10.1007/s12031-017-1011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu AL, Stochl J, Lewis G, Zammit S, Jones PB, & Khandaker GM 2019. Longitudinal association between inflammatory markers and specific symptoms of depression in a prospective birth cohort. Brain Behav Immun. 76, 74–81. 10.1016/j.bbi.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donate-Correa J, Martín-Núñez E, Delgado NP, de Fuentes MM, Arduan AO, Mora-Fernández C, & González JFN 2016. Implications of Fibroblast growth factor/Klotho system in glucose metabolism and diabetes. Cytokine Growth Factor Rev. 28, 71–77. https://doi.org/10.1016/j.cytogfr.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Drew DA, Katz R, Kritchevsky S, Ix J, Shlipak M, Gutiérrez OM, & Sarnak M 2017Association between soluble Klotho and change in kidney function: The health aging and body composition study. J Am Soc Nephrol. 28, 1859–1866. 10.1681/ASN.2016080828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Yokoyama JS, Zhu L, Broestl L, Worden K, Wang D, & Ho K 2014. Life extension factor Klotho enhances cognition. Cell Rep. 7, 1065–1076. 10.1016/j.celrep.2014.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarappa M, Neylan TC, Whooley MA, Metzler TJ, & Cohen BE 2019. Inflammation as a predictor of disease course in posttraumatic stress disorder and depression: A prospective analysis from the mind your heart study. Brain Behav Immun. 75, 220–227. 10.1016/j.bbi.2018.10.012. [DOI] [PubMed] [Google Scholar]
- Fortin JP, Triche TJ Jr., & Hansen KD 2017. Preprocessing, normalization and integration of the Illumina HumanMethylationEpic array with Minfi. Bioinformatics 33, 558–560. 10.1093/bioinformatics/btw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Valensin S, Bonafe M, Paolisso G, Yashin AI, Monti D, & De Benedictis G 2000. The network and the remodeling theories of aging: Historical background and new perspectives. Exp Gerontol. 35, 879–896. 10.1016/S0531-55650000172-8. [DOI] [PubMed] [Google Scholar]
- Frasca D, & Blomberg BB 2016. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 17, 7–19. 10.1007/s10522-015-9578-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fülöp T, Dupuis G, Witkowski JM, & Larbi A 2016. The role of immunosenescence in the development of age-related diseases. Rev Invest Clin. 68, 84–91. [PubMed] [Google Scholar]
- González-Reimers E, Romero-Acevedo L, Espelosín-Ortega E, Martín-González MC, Quintero-Platt G, Abreu-González P, & Santolaria-Fernández F 2018. Soluble Klotho and brain atrophy in alcoholism. Alcohol. 53, 503–510. 10.1093/alcalc/agy037. [DOI] [PubMed] [Google Scholar]
- Guo Y, Zhuang X, Huang Z, Zou J, Yang D, Hu X, & Liao X 2018. Klotho Protects the Heart from Hyperglycemia-induced Injury by Inactivating ROS and NF-κB-mediated inflammation both in vitro and in vivo. Biochim Biophys Acta Mol Basis Dis. 1864, 238–251. 10.1016/j.bbadis.2017.09.029. [DOI] [PubMed] [Google Scholar]
- Hamilton E, 2011. Mythology: Timeless Tales of Gods and Heroes. London, UK: Hachette UK. [Google Scholar]
- Hu LT, & Bentler PM 1999. Cutoff criteria for fit indexes in covariance structure analysis:Conventional criteria versus new alternatives. Structural Equation Modeling: A Multidisciplinary Journal 6, 1–55. 10.1080/10705519909540118. [DOI] [Google Scholar]
- Hui H, Zhai Y, Ao L, Cleveland JC Jr, Liu H, Fullerton DA, & Meng X 2017. Klotho suppresses the inflammatory responses and ameliorates cardiac dysfunction in aging endotoxemic mice. Oncotarget 8, 15663–15676. 10.18632/oncotarget.14933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janić M, Lunder M, Novaković S, Škerl P, & Šabovič M 2019. Expression of longevity genes induced by a low-dose Fluvastatin and Valsartan combination with the potential to prevent/treat “aging-related disorders.” Int J Mol Sci. 20, 1844 10.3390/ijms20081844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, & Haussler D 2002. The human genome browser at UCSC. Genome Res. 12, 996–1006. 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GD, Rosene DL, & Abraham CR 2012. Promoter methylation and age-related downregulation of Klotho in rhesus monkey. Age(Dordr) 34, 1405–1419. 10.1007/s11357-011-9315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M 2019. The Klotho proteins in health and disease. Nat Rev Nephrol. 15, 27–44. 10.1038/s41581-018-0078-3. [DOI] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, & Iwasaki H 1997. Mutation of the mouse Klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51. 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, & Shimomura I 2005. Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833. 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers F, Milaneschi Y, Smit JH, Schoevers RA, Wittenberg G, & Penninx BW 2019. Longitudinal association between depression and inflammatory markers: results from the Netherlands study of depression and anxiety. Biol Psychiatry. 85, 829–837. 10.1016/j.biopsych.2018.12.020. [DOI] [PubMed] [Google Scholar]
- Lee DH, Blomhoff R, & Jacobs DR 2004. Review is serum gamma glutamyltransferase a marker of oxidative stress? Free Radic Res. 38, 535–539. 10.1080/10715760410001694026. [DOI] [PubMed] [Google Scholar]
- Lee J, Jeong DJ, Kim J, Lee S, Park JH, Chang B, & Kim KI 2010. The anti-aging gene KLOTHO is a novel target for epigenetic silencing in human cervical carcinoma. Mol Cancer 9, 109 10.1186/1476-4598-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Fertig EJ, Jaffe AE, Storey JD, … & Torres LC 2017. SVA: Surrogate Variable Analysis R package version 3.10. 10–18129. [Google Scholar]
- Leon J, Moreno AJ, Garay BI, Chalkley RJ, Burlingame AL, Wang D, & Dubal DB 2017. Peripheral elevation of a Klotho fragment enhances brain function and resilience in young, aging, and α-synuclein transgenic mice. Cell Rep. 20, 1360–1371. 10.1016/j.celrep.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, … & Gutkind JS 2007. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 317, 803–806. 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- Logue MW, Amstadter AB, Baker DG, Duncan L, Koenen KC, Liberzon I, Ressler KJ 2015. The psychiatric genomics consortium posttraumatic stress disorder workgroup: Posttraumatic stress disorder enters the age of large-scale genomic collaboration. Neuropsychopharmacology 40, 2287–2297. 10.1038/npp.2015.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue MW, Baldwin C, Guffanti G, Melista E, Wolf EJ, Reardon AF, … & Miller MW 2013. A genome-wide association study of post-traumatic stress disorder identifies the retinoid-related orphan receptor alpha RORA gene as a significant risk locus. Mol Psychiatry 18, 937–942. 10.1038/mp.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue MW, Smith AK, Wolf EJ, Maniates H, Stone A, Schichman SA, & Miller MW 2017. The correlation of methylation levels measured using Illumina 450k and EPIC BeadChips in blood samples. Epigenomics 9, 1363–1371. 10.2217/epi-2017-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Zhou X, Ji H, Ma W, Liu G, Dai D, & Duan S 2015. Population difference in the associations of KLOTH promoter methylation with mild cognitive impairment in Xinjiang Uygur and Han populations. PLoS One 10, e0132156 10.1371/journal.pone.0132156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa Y, Ishikawa K, Yasuda O, Oguro R, Hanasaki H, Kida I, & Rakugi H 2009. Klotho suppresses TNF-α-induced expression of adhesion molecules in the endothelium and attenuates NF-κB activation. Endocrine 35, 341–346. 10.1007/s12020-009-9181-3. [DOI] [PubMed] [Google Scholar]
- Martín-Núñez E, Donate-Correa J, López-Castillo Á, Delgado-Molinos A, Ferri C, Rodríguez-Ramos S, & Mora-Fernández C 2017. Soluble levels and endogenous vascular gene expression of KLOTHO are related to inflammation in human atherosclerotic disease. Clin Sci(Lond).131, 2601–2609. 10.1042/CS20171242. [DOI] [PubMed] [Google Scholar]
- McGlinchey RE, Milberg WP, Fonda JR, & Fortier CB 2017. A methodology for assessing deployment trauma and its consequences in OEF/OIF/OND veterans: The TRACTS longitudinal prospective cohort study. Int J Methods Psychiatr Res. 26 10.1002/mpr.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengel-From J, Soerensen M, Nygaard M, McGue M, Christensen K, & Christiansen L 2016Genetic variants in KLOTHO associate with cognitive function in the oldest old group. J Gerontol A Biol Sci Med Sci. 71, 1151–1159. 10.1093/gerona/glv163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michopoulos V, Rothbaum AO, Jovanovic T, Almli LM, Bradley B, Rothbaum BO, & Ressler KJ 2015. Association of CRP genetic variation and CRP level with elevated PTSD symptoms and physiological responses in a civilian population with high levels of trauma. Am J Psychiatry 172, 353–362. 10.1176/appi.ajp.2014.14020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW, Lin AP, Wolf EJ, & Miller DR 2018. Oxidative stress, inflammation, and neuroprogression in chronic PTSD. Harv Rev Psychiatry. 26, 57–69. https://dx.doi.org/10.1097%2FHRP.0000000000000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW, Maniates H, Wolf EJ, Logue MW, Schichman SA, Stone A, & McGlinchey R 2018. CRP polymorphisms and DNA methylation of the AIM2 gene influence associations between trauma exposure, PTSD, and C-reactive protein. Brain Behav Immun. 67, 194–202. 10.1016/j.bbi.2017.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthén LK, Muthén BO 1998-2018. Mplus User’s Guide 8th ed. Los Angeles, CA: Muthén & Muthén. [Google Scholar]
- Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, & Kauer-Sant’Anna M 2015. Inflammatory markers in post-traumatic stress disorder: A systematic review, meta-analysis, and meta-regression. Lancet Psychiatry 2, 1002–1012. 10.1016/S2215-03661500309-0. [DOI] [PubMed] [Google Scholar]
- Pidsley R, Wong CC, Volta M, Lunnon K, Mill J, & Schalkwyk LC 2013. A data-driven approach to preprocessing Illumina 450k methylation array data. BMC Genomics 14, 293 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prystupa A, browska A, Sak JJ, Tarach J., Toruń-Jurkowska A, Lachowska-Kotowska P., & Dzida G 2016. Concentrations of fetuin-A, osteoprotegerin and α-Klotho in patients with alcoholic liver cirrhosis. Exp Ther Med. 12, 3464–3470. 10.3892/etm.2016.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzianowska-Kuźnicka M, Owczarz M, Wieczorowska-Tobis K, Nadrowski P, Chudek J, Slusarczyk P, & Mossakowska M 2016. Interleukin-6 and C-reactive protein, successful aging, and mortality: The PolSenior Study. Immun Ageing 13, 21 10.1186/s12979-016-0076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramez M, Rajabi H, Ramezani F, Naderi N, Darbandi-Azar A, & Nasirinezhad F 2019. The greater effect of high-intensity interval training versus moderate-intensity continuous training on cardioprotection against ischemia-reperfusion injury through Klotho levels and attenuate of myocardial TRPC6 expression. BMC Cardiovasc Disord. 19, 118 10.1186/s12872-019-1090-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratanatharathorn A, Boks MP, Maihofer AX, Aiello AE, Amstadter AB, Ashley-Koch AE, & Garrett ME 2017. Epigenome-wide association of PTSD from heterogeneous cohorts with a common multi-site analysis pipeline. Am J Med Genet B Neuropsychiatr Genet. 174, 619–630. 10.1002/ajmg.b.32568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revelas M, Thalamuthu A, Oldmeadow C, Evans TJ, Armstrong NJ, Kwok JB,& Attia JR 2018. Review and meta-analysis of genetic polymorphisms associated with exceptional human longevity. Mech Ageing Dev. 175, 24–34. 10.1016/j.mad.2018.06.002. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Rifai N, Rose L, Buring JE, & Cook NR 2002. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 347, 1557–1565. 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- Rosen RL, Levy-Carrick N, Reibman J, Xu N, Shao Y, Liu M, & Marmor M 2017. Elevated C-reactive protein and posttraumatic stress pathology among survivors of the 9/11 World Trade Center attacks. J Psychiatr Res. 89, 14–21. 10.1016/j.jpsychires.2017.01.007. [DOI] [PubMed] [Google Scholar]
- Rubinek T, Shulman M, Israeli S, Bose S, Avraham A, Zundelevich A, & Wolf I 2012. Epigenetic silencing of the tumor suppressor Klotho in human breast cancer. Breast Cancer Res Treat. 133, 649–657. 10.1007/s10549-011-1824-4. [DOI] [PubMed] [Google Scholar]
- Sadeh N, Spielberg JM, Logue MW, Wolf EJ, Smith AK, Lusk J, & Salat DH 2016. SKA2 methylation is associated with decreased prefrontal cortical thickness and greater PTSD severity among trauma-exposed veterans. Mol Psychiatry 21, 357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedighi M, Baluchnejadmojarad T, Fallah S, Moradi N, Afshin-Majdd S, & Roghani M 2019. Klotho ameliorates cellular inflammation via suppression of cytokine release and upregulation of miR-29a in the PBMCs of diagnosed Alzheimer’s disease patients. J Mol Neurosci. 69, 157–165. 10.1007/s12031-019-01345-5. [DOI] [PubMed] [Google Scholar]
- Shutz RW 1989. Analyzing change In: Safrit MK, Wood TM. Eds., Measurement Concepts in Physical Education and Exercise Science pp. 207–28. Champaign, IL: Human Kinetics Books. [Google Scholar]
- Sproston NR, & Ashworth JJ 2018. Role of C-reactive Protein at sites of inflammation and infection. Front Immunol. 9, 754 10.3389/fimmu.2018.00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova M, Rodriguez E, Birerdinc A, & Baranova A 2015. Age-independent rise of inflammatory scores may contribute to accelerated aging in multi-morbidity. Oncotarget 6, 1414–1421. 10.18632/oncotarget.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, & Garg K 2012. The accessible chromatin landscape of the human genome. Nature 489, 75–82. 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weathers FW, Ruscio AM, & Keane TM 1999. Psychometric properties of nine scoring rules for the Clinician-Administered Posttraumatic Stress Disorder Scale. Psychol. Assess 11, 124 10.1037/1040-3590.11.2.124. [DOI] [Google Scholar]
- Wolf EJ, Logue MW, Hayes JP, Sadeh N, Schichman SA, Stone A, … Miller MW 2016. Accelerated DNA methylation age: Associations with PTSD and neural integrity. Psychoneuroendocrinology. 63, 155–162. 10.1016/j.psyneuen.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf EJ, Logue MW, Morrison FG, Wilcox ES, Stone A, Schichman SA, & Miller MW 2019. Posttraumatic psychopathology and the pace of the epigenetic clock: A longitudinal investigation. Psychol Med. 49, 791–800. 10.1017/S0033291718001411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf EJ, Logue MW, Stoop TB, Schichman SA, Stone A, Sadeh N, … Miller MW 2018. Accelerated DNA Methylation Age. Psychosom. Med 80, 42–48. 10.1097/psy.0000000000000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf EJ, Maniates H, Nugent N, Maihofer A, Armstrong D, Ratanatharathorn A, … Logue MW. 2018. Traumatic stress and accelerated DNA methylation age: A meta-analysis. Psychoneuroendocrinology. 92, 123–134. 10.1016/j.psyneuen.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf EJ, & Morrison FG 2017. Traumatic Stress and Accelerated Cellular Aging: From Epigenetics to Cardiometabolic Disease. Curr Psychiatry Rep. 19, 75 10.1007/s11920-017-0823-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf EJ, Morrison FG, Sullivan DR, Logue MW, Guetta RE, Stone A, & Miller MW 2019. The goddess who spins the thread of life: Klotho, psychiatric stress, and accelerated aging. Brain Behav Immun. 80, 193–203. 10.1016/j.bbi.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Zhang A, Jia Z, Gu J, & Chen H 2016. Klotho contributes to pravastatin effect on suppressing IL-6 production in endothelial cells. Mediators Inflamm. 2016 10.1155/2016/2193210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandi A, Kurosu H, & Kuro-o M 2005. Regulation of oxidative stress by the anti-aging hormone Klotho. J Biol Chem. 280, 38029–38034. 10.1074/jbc.M509039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama JS, Sturm VE, Bonham LW, Klein E, Arfanakis K, Yu L, Dubal DB 2015. Variation in longevity gene KLOTHO is associated with greater cortical volumes. Ann Clin Transl Neurol. 2, 215–230. 10.1002/acn3.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldich E, Chen CD, Boden E, Howat B, Nasse JS, Zeldich D & Ma Q 2019. Klotho is neuroprotective in the Superoxide Dismutase (SOD1 G93A) mouse model of ALS. J Mol Neurosci. 69, 1–22. 10.1007/s12031-019-01356-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldich E, Chen CD, Colvin TA, Bove-Fenderson EA, Liang J, Zhou TBT, & Abraham CR 2014. The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J Biol Chem. 289, 24700–24715. 10.1074/jbc.M114.567321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Cheng CF, Luo YZ, Tian CW, Yang H, Liu BR, & Liu SM 2015. C-reactive Protein stimulates RAGE expression in human coronary artery endothelial cells in vitro via ROS generation and ERK/NF-κB activation. Acta Pharmacol Sin. 36, 440–447. 10.1038/aps.2014.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou TBT, King GD, Chen C, & Abraham CR 2013. Biochemical and functional characterization of the klotho-VS polymorphism implicated in aging and disease risk. J Biol Chem. 288, 36302–36311. 10.1074/jbc.M113.490052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Cao X, Zhang X, Chen Q, Wen L, & Wang P 2019. DNA methylation-mediated Klotho silencing is an independent prognostic biomarker of head and neck squamous carcinoma. Cancer Manag Res. 11, 1383–1390. 10.2147/cmar.s188415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Stein LR, Kim D, Ho K, Yu GQ, Zhan L, & Mucke L 2018. Klotho controls the brain-immune system interface in the choroid plexus. Proc Natl Acad Sci U S A. 115, E11388–E11396. 10.1073/pnas.1808609115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.