

Abstract
Purpose:
Diffuse intrinsic pontine glioma (DIPG) bears a dismal prognosis. A genetically engineered brainstem glioma model harboring the recurrent DIPG mutation, ACVR1-G328V (mACVR1), was developed for testing an immune-stimulatory gene therapy.
Experimental Design:
We utilized the Sleeping Beauty transposase system to generate an endogenous mouse model of mACVR1 brainstem glioma. Histology was used to characterize and validate the model. We performed RNAseq analysis on neurospheres (NS) harboring mACVR1. mACVR1 NS were implanted into the pons of immune competent mice to test the therapeutic efficacy and toxicity of immune stimulatory gene therapy using adenoviruses expressing thymidine kinase (TK) and fms-like tyrosine kinase 3 ligand (Flt3L). mACVR1 NS expressing the surrogate tumor antigen ovalbumin were generated to investigate if TK/Flt3L treatment induces the recruitment of tumor-antigen specific T cells.
Results:
Histological analysis of mACVR1 tumors indicates that they are localized in the brainstem and have increased downstream signaling of bone morphogenetic pathway as demonstrated by increased phospho-smad1/5 and Id1 levels. Transcriptome analysis of mACVR1 NS identified an increase in the transforming growth factor beta (TGF-β) signaling pathway and the regulation of cell differentiation. Adenoviral delivery of TK/Flt3L in mice bearing brainstem gliomas resulted in anti-tumor immunity, recruitment of anti-tumor specific T cells and increased median survival.
Conclusions:
This study provides insights into the phenotype and function of the tumor immune microenvironment in a mouse model of brainstem glioma harboring mACVR1. Immune stimulatory gene therapy targeting the hosts’ anti-tumor immune response inhibits tumor progression and increases median survival of mice bearing mACVR1 tumors.
Keywords: Immune therapy, brainstem glioma, mouse model
Introduction
Diffuse intrinsic pontine glioma (DIPG) is an aggressive, diffusive brain tumor that originates in the pons and is diagnosed based on clinical and radiological criteria (1). DIPG occurs mostly in children of median age 6–7 years; it is an inoperable brain tumor and despite multiple trials testing various chemotherapeutic strategies, none have demonstrated a survival benefit. Thus, focal radiation remains the standard of care (1,2). The median overall survival for DIPG is 10.8 months, and the 2 year survival is ~5.2% (3). In 2014, four independent studies that comprised data from 195 DIPGs, identified Activin A receptor type I (ACVR1) as the most recurrently mutated gene following the histone H3 K27M mutation (4–7). Six recurrent, somatic mutations in ACVR1 were found in 24% of DIPG cases (8). The characteristics of DIPG patients presenting with the ACVR1 mutation include younger age of onset (~5 years of age) and longer overall survival time (4,9). Sequencing revealed that DIPG tumors have a lower mutation frequency compared to adult glioblastoma, and that classical tumor pathways such as the PI3K and p53 pathways are also altered in these tumors (10).
ACVR1 encodes a type 1 serine/threonine kinase that is part of the transforming growth factor beta (TGF-ß) family. It is noteworthy to mention that ACVR1 mutations are only found in DIPG tumors; ACVR1 is not mutated in any other cancers. Thus, uncovering the role of this gene mutation in DIPG tumorigenesis and disease progression could provide novel mechanistic insights useful to develop novel therapeutic targets.
Since DIPGs are not resectable and they are highly invasive, it would be advantageous to harness the power of the immune system to elicit effective anti-tumor immunity in DIPG patients. Immune mediated treatment modalities have yielded promising clinical benefits in melanoma, non-small-cell lung cancer, renal cell cancer, and prostate cancer (11–14). We have also previously shown the efficacy of an immune stimulatory gene therapy approach in several rat and mouse models of adult glioblastoma (15,16). This approach has recently completed its Phase I Clinical Trial accrual for the treatment of adult patients with newly diagnosed glioblastoma multiforme (World Health Organization (WHO) grade IV (NCT01811992). This immune-mediated gene therapy approach is based on adenoviral delivery of herpes simplex virus type 1-thymidine kinase (TK) and Fms-like tyrosine kinase 3 ligand (Flt3L). Upon administration of the prodrug, ganciclovir, proliferating tumor cells expressing TK undergo immunogenic cell death(15,16). Dying tumor cells, release damage-associated molecular patterns (DAMPs), such as high-mobility group B1 protein (HMGB1), calreticulin, and adenosine triphosphate (ATP) (15,17). Meanwhile, Flt3L elicits the recruitment of dendritic cells to the tumor microenvironment(15). HMGB1 released by dying tumor cells activates dendritic cells through Toll-like receptor 2 (TLR2) mediated signaling(15). Activated dendritic cells pick up the tumor antigens and traffic to the draining lymph nodes, where they generate a specific anti-tumor cytotoxic T-cell response (15,18). Herein, we aimed to test the efficacy of this immune stimulatory approach in an immunocompetent mouse model of mutant ACVR1 brainstem glioma.
To test the efficacy of this immune-mediated gene therapy approach in syngeneic mouse brainstem glioma models, we developed a genetically engineered mouse model encoding mutant ACVR1 using the Sleeping Beauty (SB) transposase system(19–22). SB is a transposase that is able to recognize inverted repeats/direct repeats (IR/DR) sites on DNA transposons and carry out a cut and paste reaction to integrate transposon DNA into a host genome(23). The SB system can be utilized to generate endogenous tumors that resemble gliomas through delivery of DNA transposons that encode oncogenes and tumor suppressors (19–22,24). Transcriptome analysis of ACVR1 mutant neurospheres identified an increase in the TGF-β signaling pathway and signaling pathways regulating the pluripotency of stem cells, and a decrease in the focal adhesion pathway. Treatment with TK/Flt3L immune stimulatory gene therapy significantly improved median survival compared to standard of care. The TK/Flt3L gene therapy induced a strong anti-tumor cytotoxic immune response demonstrated by an increase in the frequency of tumor antigen-specific CD8 T cells in mice treated with TK/Flt3L therapy when compared to saline controls. Our results suggest that immune-mediated gene therapy could be a promising therapeutic approach for DIPG.
Materials and Methods
Experimental model
All animal studies were conducted according to guidelines approved by the Institutional Animal Care and Use Committee at the University of Michigan. Animals were housed in an AAALAC accredited animal facility and had constant access to food and water; they were monitored daily for tumor burden. Males and females were used. The strain of mice utilized in the study was C57BL/6 (Jackson Laboratory, 000664).
A murine model of brainstem glioma was generated by employing the Sleeping Beauty (SB) transposon system to integrate plasmid DNA into the genome of postnatal day 1 (P1) mouse pups. The plasmids utilized were as follows: (i) SB Transposase and luciferase (pT2C-LucPGK-SB100X, henceforth referred to as SB/Luc) (ii) a short-hairpin against p53 (pT2-shp53-GFP4, henceforth referred to as shp53) or shp53-NO-GFP (iii) a constitutively active mutant of NRAS (pT2CAG-NRASV12, henceforth referred to as NRAS) with or without (4) mutant ACVR1 G328V (pkt-ACVR1-G328V-IRES-Katushka; henceforth referred to as mACVR1)(19,20,22). To create the mACVR1 plasmid we cloned pCMV5-ALK2-WT into pKT2-IRES-Katushka by blunt cloning. Then, we used the QuikChange II Site-Directed Mutagenesis Kit (Agilent, 200523) to introduce (c.983G>T, p.Gly328Val) mutation into pKT2-ALK2-WT-IRES-Katushka, to generate pKT2-ACVR1-G328V-IRES-Katushka (Addgene plasmid #77437). The resultant mACVR1 plasmid was confirmed by Sanger sequencing. SB/Luc, shp53 and NRAS plasmids were the generous gift of Dr. John Ohlfest (University of Minnesota, now deceased). The pCMV5-ALK2-WT plasmid was a generous gift from Jeff Wrana (Addgene plasmid #11741). All experiments were performed using post-natal day 1 (P1) or P2 wild-type C57BL/6 mice. The plasmid combinations injected were as follows: (1) (i) shp53 and NRAS (henceforth referred to as wt-ACVR1), (ii) shp53, NRAS, and ACVR1m. Mice were injected according to a previously described protocol (20) and described in detail in the Supplementary Methods.
Immunohistochemistry (IHC) of paraffin embedded brains
Immunhistochemistry staining was performed as previously described (20,22) and detailed in the Supplementary Methods. Antibody information is available in Supplementary Table S1.
Primary Neurospheres (NS)
Mouse neurospheres (NS) were generated from tumors that were developed using the SB system by injection of the following plasmid combinations (1) (i) shp53 and NRAS, or (ii) shp53, NRAS, and ACVR1m into the lateral ventricle (1.5 mm AP, 0.7 mm lateral, and 1.5 mm deep from the λ-suture) following previously described protocols and detailed in the Supplementary Methods (20–22,25). Western blot analysis and inhibitor treatment with LDN-214117 is also detailed in the Supplementary Methods and antibody information is available in Supplementary Table S1. SU-DIPG-VI and SU-DIPG-XX1 were obtained from Dr. Michelle Monje at Stanford University (Stanford, Ca) in accordance with an institutionally approved protocol at each institution. Experimental details of immunocytochemistry on DIPG cultures is available in the Supplementary Methods.
RNA-seq
RNA-sequencing was performed in collaboration with the University of Michigan sequencing core. Total RNA was isolated from tumor NS using the RNeasy Plus Mini Kit (Qiagen, 74134) and 100 ng of purified RNA were sent for analysis. The libraries were prepared using RiboGone (Takara, 634836) and TruSeq Stranded Total RNA Human/Mouse/Rat (Illumina, 20020596) with 100 ng of input and 13 PCR cycles. Sequencing was performed by the UM DNA Sequencing Core, using the Illumina Hi-Seq platform. RNA-seq analysis is available in the Supplementary Methods.
Implantable syngeneic murine brainstem glioma models
Female C57BL/6 mice between 6–8 weeks were used for all implantation experiments. Intracranial tumors were generated by stereotaxic injection of 1000 ACVR1m tumor NS into the pons using a 5 μl Hamilton syringe with a removable 33-gauge needle with the following coordinates: (0.8mm posterior; 1.00mm lateral to the λ-suture and 5mm deep). Animals were anesthetized, then the skin over the incision site was cut and retracted, and a burr hole was drilled into one side of the skull using a 0.45 mm drill bit corresponding to the pons coordinates. Tumor NS were delivered in a 2 μl volume after holding the needle in place for 2 minutes. Each injection was performed over the course of 7 minutes; the needle was left in place for an additional minute before being slowly withdrawn from the brain. Intra-tumoral injection of adenoviral vectors and radiation treatment, flow cytometry, T cell proliferation analysis, and the cytotoxic T cell assay are detailed in the Supplementary Methods. Details of complete blood cell count and serum chemistry analysis, neuropathological analysis, and H&E staining of liver sections are elaborated in the Supplementary Methods.
Statistical Analysis
All data were analyzed using GraphPad Prism version 8, or R (version 3.1.3). All animal studies were carried out with at least 3 animals per group (specified in each experiment). The statistical test used is indicated in each figure. A p ≤ 0.05 was considered significant.
Results
Brainstem gliomas induced by transforming neural progenitor cells using the Sleeping Beauty (SB) transposase system
To assess the impact of activating ACVR1 mutations in brainstem glioma pathogenesis and response to therapeutics, we generated a genetically engineered mouse model of brainstem glioma using the Sleeping Beauty (SB) transposase system (19,20). We induced brainstem tumors by activation of the receptor tyrosine kinase (RTK)-RAS- phosphatidylinositol 3-kinase (PI3K) pathway, which is upregulated in a large percentage of DIPGs, and through inactivation of TP53, also commonly mutated in DIPG (3,26,27). This was achieved through the delivery of the following plasmids: NRASV12, a short hair pin targeting tumor protein p53 (TP53) (shP53), and SB transposase/firefly luciferase, with or without ACVR1G328V (Fig.1A, Supplementary Fig. 1S) into the fourth ventricle of neonatal mice. We used bioluminescence imaging to monitor transfection efficiency and tumor development (Fig.1B). The two experimental groups were: (1) Wild-type ACVR1 (wt-ACVR1) [NRASV12 and shp53) and (2) mutant ACVR1 (mACVR1) (NRASV12, shp53, ACVR1G328V). The median survival (MS) of mice in the mACVR1 group was 127 days post injection (dpi), while the median survival for the wt-ACVR1 group was MS=85 dpi; p = 0.0014, Mantel-Cox test (Fig. 1 C). All tumors, regardless of ACVR1 mutation status, displayed high cellularity, nuclear atypia, invasive features, and grew in the brainstem (Fig. 1D). All tumors expressed oligodendrocyte transcription factor 2 (Olig2) (Fig. 1E), glial fibrillary acidic protein (GFAP) (Fig. 1F), nestin (Fig. 1 G) and transcription factor Sox2 (Fig. 1 H). Both wt-ACVR1 and mACVR1 tumors were positive for the proliferative marker, Ki67 (Fig. 1 I), phosphorylated extracellular signal-regulated kinase (pERK) 1/2 (Fig. 1J) and phospho-MEK1/2 (Supplementary Fig. S2A). Expression of pERK 1/2 (Supplementary Fig. S2B) and pMEK1/2 (Supplementary Fig. S2C) was also confirmed on human DIPG cultures: SU-DIPG VI (wt-ACVR1) and SU-DIPG XX1 (mACVR1).
Figure 1. Generation of mutant (mAcvr1) mouse brainstem glioma model using the Sleeping Beauty (SB) transposase system.
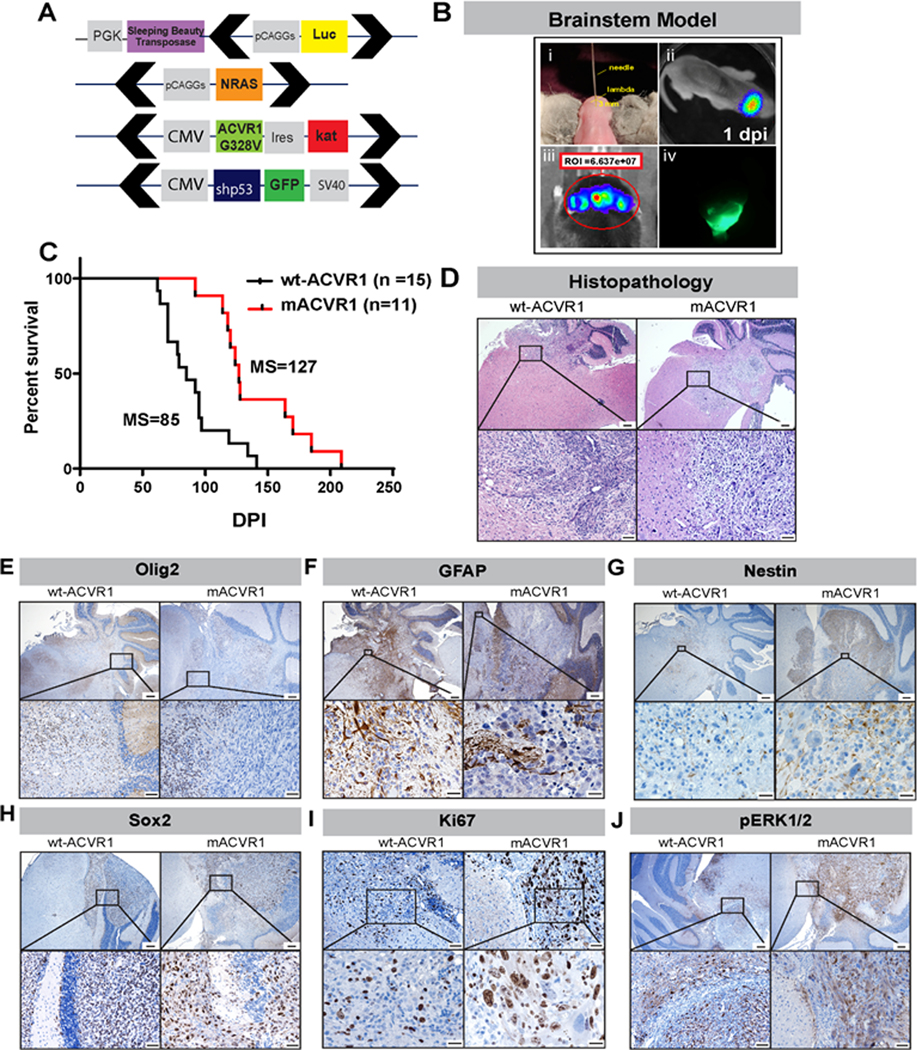
(A) Schematic representation of SB transposase and transposon plasmids used to develop gliomas in the brainstem. Black arrows indicate position of inverted repeat and direct repeat (IR/DR) sequences which flank specific DNA sequences (Luciferin, NRAS GV12, shP53-GFP, ACVR1 G328V-IRES-Katushka). (B) Schematic representation of brainstem glioma model. (i) One day old pups were injected in the fourth ventricle (3 mm posterior to the λ-suture and 3 mm deep) with plasmid cocktail. (ii) Bioluminescence imaging at 1-day post injection (dpi) confirming the efficiency of the in vivo transfection. (iii) Bioluminescence imaging obtained when mice displayed signs of neurological deficits confirms the presence of a large tumor. (iv) Fluorescence image of GFP+ brainstem tumor. (C) Kaplan-Meier survival curve for genetically engineered mice bearing wt-ACVR1 (n=15) mACVR1 (n= 11) tumor; MS: median survival. Data were analyzed using log-rank (Mantel-Cox) test; ** p < 0.01. (D) Hematoxylin and eosin stained paraffin embedded SB brainstem tumor sections (wt-ACVR1 and mACVR1). Scale bar in the upper panel images is 200 μm. Scale bar in the bottom panel images is 20 μm. (E-J) Immunohistochemistry (IHC) staining for: (E) Olig2, an oligodendrocyte marker (scale bar in the upper panel image is 200 μm, scale bar for the bottom panel images is 50 μm) (F) GFAP, an astrocyte marker (scale bar in the upper panel image is 200 μm, scale bar for the bottom panel images is 20 μm) (G) Nestin, a neural stem cell marker (scale bar in the upper panel image is 200 μm, scale bar for the bottom panel images is 20 μm) (H) Sox2, a transcription factor that plays an important role in the maintenance of neural stem cells (scale bar in the upper panel image is 200 μm, scale bar for the bottom panel images is 50 μm) (I) Ki67, a marker of proliferating cells (scale bar in the upper panel image is 200 μm, scale bar for the bottom panel images is 20 μm) and (J) phosphorylated ERK-1/2 protein (pERK1/2) (scale bar in the upper panel image is 200 μm, scale bar for the bottom panel images is 50 μm).
mACVR1 brain stem gliomas exhibit elevated levels of phosphorylated Smad1/5
We next investigated whether tumors encoding mutant ACVR1 exhibited activated Smad1/5/8 transcription factors, downstream mediators of ACVR1 signaling (Fig. 2A). We observed that, indeed, mACVR1 brainstem gliomas displayed elevated levels of phosphorylated (phospho)-smad1/5 (Fig. 2B). This correlated with increased levels of the downstream canonical target gene, inhibitor of DNA binding 1 (Id1) (Fig. 2C). Tumor neurospheres (NS) expressing mutant ACVR1 (mACVR1 NS) also exhibited elevated levels of phospho-smad1/5 (Fig. 2D) and elevated levels of Id1 (Fig. 2D). To test the effect of a specific inhibitor of ACVR1, LDN-214117 on phospho-Smad1/5 signaling (Fig. 2E), we treated mACVR1 NS with increasing concentrations of LDN-214117. We observed decreased levels of phospho-Smad1/5 and Id2, while the levels of total Smad1 remain unchanged. Expression of ID1 was also confirmed on human DIPG cultures: SU-DIPG VI (wt-ACVR1) and SU-DIPG XX1 (mACVR1) (Supplementary Fig. S2D). Over-expression of wt-ACVR1 did not result in activation of the phospho-Smad1/5 pathway, suggesting that phopho-Smad1/5 activation is only activated in the mACVR1 modal (Supplementary Fig. S3).
Figure 2. ACVR1G328V activates the TGF-ß /Smad signaling pathway in vivo and in vitro.
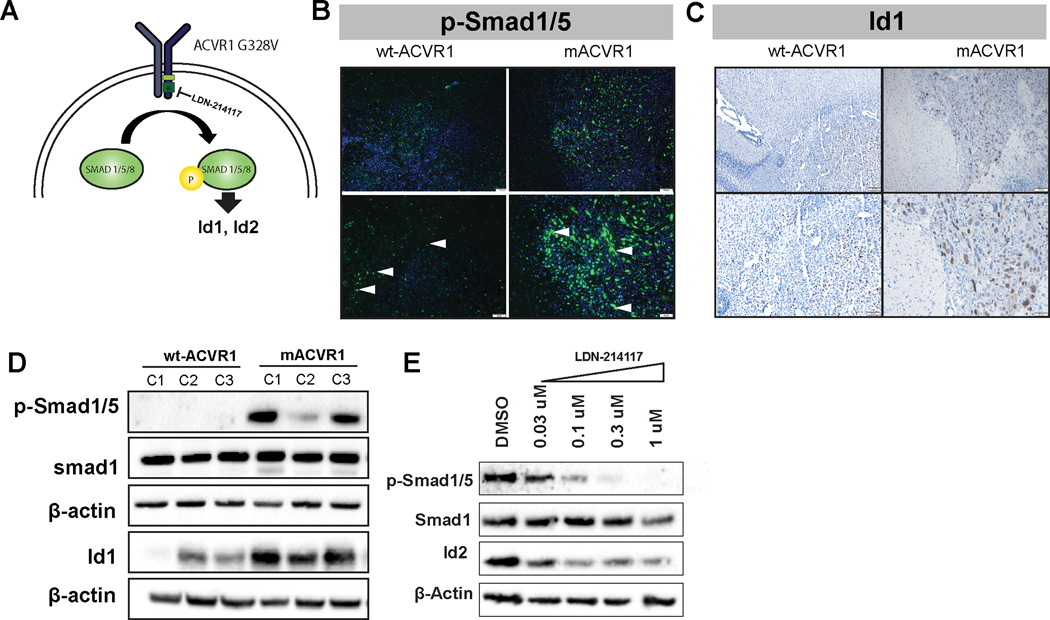
(A) Diagram representing the TGF-ß/Smad signaling pathway. When ACVR1 is mutated this pathway is constitutively active regardless of the presence or absence of BMP ligand. LDN-214117 inhibits ACVR1 kinase activity. (B) Immunofluorescence (IF) staining for phospho-Smad1/5 in paraffin embedded SB brainstem tumor sections (wt-ACVR1 and mACVR1). Scale bar in the upper panel image is 100 μm, scale bar for the bottom panel images is 50 μm. White arrows indicate positive expression. (C) Immunohistochemistry (IHC) staining for Id1 protein in paraffin embedded SB brainstem tumor sections (wt-ACVR1 and mACVR1). Scale bar in the upper panel image is 100 μm, scale bar for the bottom panel images is 50 μm. (D) WB assay was performed on three different clones of wt-ACVR1 and mACVR1 NS to assess the protein level of phopho-Smad1/5, Smad1, and Id1, a downstream Smad1/5 regulated gene. ß-actin: loading control. C1, C2, C3 represent the clones of NS used. (E) Western blot assay was performed on mACVR1 NS treated with LDN-214117 (0.03–1uM) and blotted for Smad1/5, Smad1, and Id2; ß-actin: loading control.
mACVR1 differentially regulates TGF-beta pathway and pathways related to stem cell maintenance and focal adhesion
RNAseq analysis of mACVR1 vs wt-ACVR1 NS identified genes that were differentially regulated (1.5-fold; FDR-corrected P < 0.05) (Fig. 3A). Gene ontology (GO) terms that were over represented in the set of differentially expressed genes (DE) included: developmental processes, cell migration, neuron differentiation, neurogenesis, nervous system development, cellular developmental process, cell development, cell proliferation, neuron development, cell surface receptor signaling pathway, neuron projection development, cell adhesion, positive regulation of developmental process, and positive regulation of cell differentiation (FDR correction; minimum 30 DE genes per term) (Fig. 3B). The top three pathways that were impacted by the mutation in ACVR1 were focal adhesion (FDR corrected, p=0.004), the TGF-beta signaling pathway (FDR corrected, p=0.004), and signaling pathways regulating pluripotency of stem cells (FDR corrected, p=0.009) (Fig. 3C). Over expressed genes within the TGF-Beta pathway included ACVR1, and inhibitor of DNA binding genes Id1, Id2, and Id3 (Fig. 3 C). Gene set enrichment analysis (GSEA) suggests an enrichment in the response to BMP and the regulation of cell differentiation (Fig. 3 D–F). Since one of the top signaling pathways was involved the regulation of pluripotency in stem cells, we evaluated whether mACVR1 tumors express CD133, CD44, and Aldh1, stem cell markers. Intracranial wt-ACVR1 or mACVR1 tumors were established in the pons of adult mice using SB derived NS. The results demonstrate that mACVR1 tumors have increased expression of the cancer stem cell markers CD133 (p=0.0002) and CD44 (p=0.0112) (Fig. 4 A). We did not observe differences in the expression of another cancer stem cell marker, i.e., Aldehyde dehydrogenase 1 family, member A1 (Aldh1) (Fig. 4 A). We next investigated the tumor initiating potential of wt-ACVR1 and mACVR1 NS in vivo. With wt-ACVR1 NS, the minimum number of cells required to generate brainstem gliomas with 100% penetrance was 1,000 cells, whereas, with mACVR1 NS it was possible to generate brainstem gliomas with 100% penetrance using 500 cells (Fig. 4 B, C). These results indicate that mACVR1 NS have a greater tumor initiating potential.
Figure 3. ACVR1G328V upregulates genes involved in the TGF-ß /Smad signaling and pathways related to stem cell maintenance and focal adhesion.
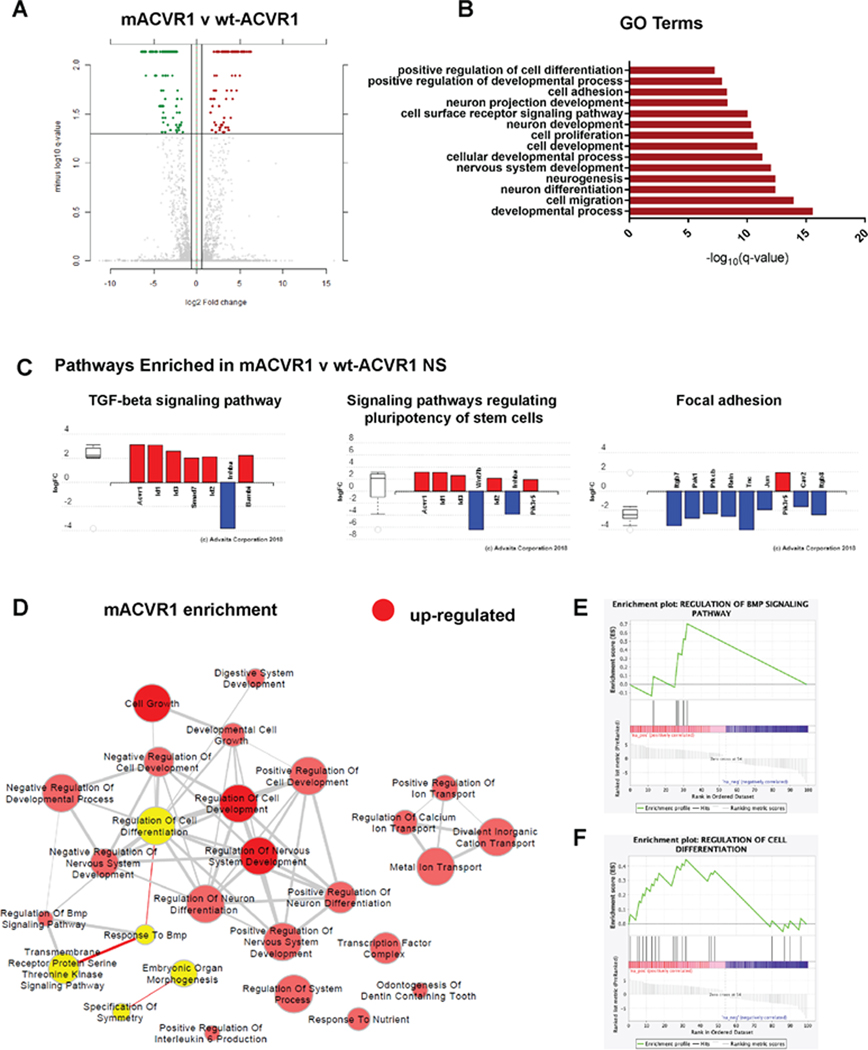
(A) Differential gene expression in mACVR1 tumors analyzed by RNA-seq. Volcano plot comparing differentially expressed genes in mACVR1 versus wt-ACVR1 mouse NS. The log 10 (FDR corrected p-values); q-values were plotted against the log 2 (Fold Change: FC) in gene expression. Genes upregulated by ≥ 1.3 fold and with a FDR corrected p-value < 0.05 are depicted as red dots; genes that were downregulated by ≥1.3 fold and with a FDR corrected p-value < 0.05 are depicted as green dots. (B) Gene Ontology (GO) enrichment analysis performed using iPathwayGuide for up- and down-regulated genes. The bar graph shows the GO biological process, selected for relevance to phenotype, that are differentially expressed between mACVR1 versus wt-ACVR1 NS. The –log10 q values of GO terms were plotted; FDR corrected P value = q value. (C) Differentially expressed pathway genes associated with mACVR1 in mACVR1 versus wt-ACVR1 mouse NS. Plot of the log (FC) of genes with an FDR corrected p-value < 0.05. (D) Pathway enrichment maps of DE genes in mACVR1 versus wt-ACVR1 NS. Clusters of nodes depicted in red illustrate differentially upregulated pathways resulting from GSEA (P < 0.05; FDR < 0.5). The yellow highlighted nodes indicate up-regulated GO terms containing ACVR1. (E) GSEA enrichment plot of regulation of BMP signaling pathway genes identified by RNA-Seq analysis in mACVR1 versus wt-ACVR1 mouse NS. (F) GSEA enrichment plot of regulation of cell differentiation genes identified by RNA-Seq analysis in mACVR1 versus wt-ACVR1 mouse NS.
Figure 4. Increased expression of stem cell markers and greater tumor initiation potential in mACVR1 NS compared to WT ACVR1 NS.
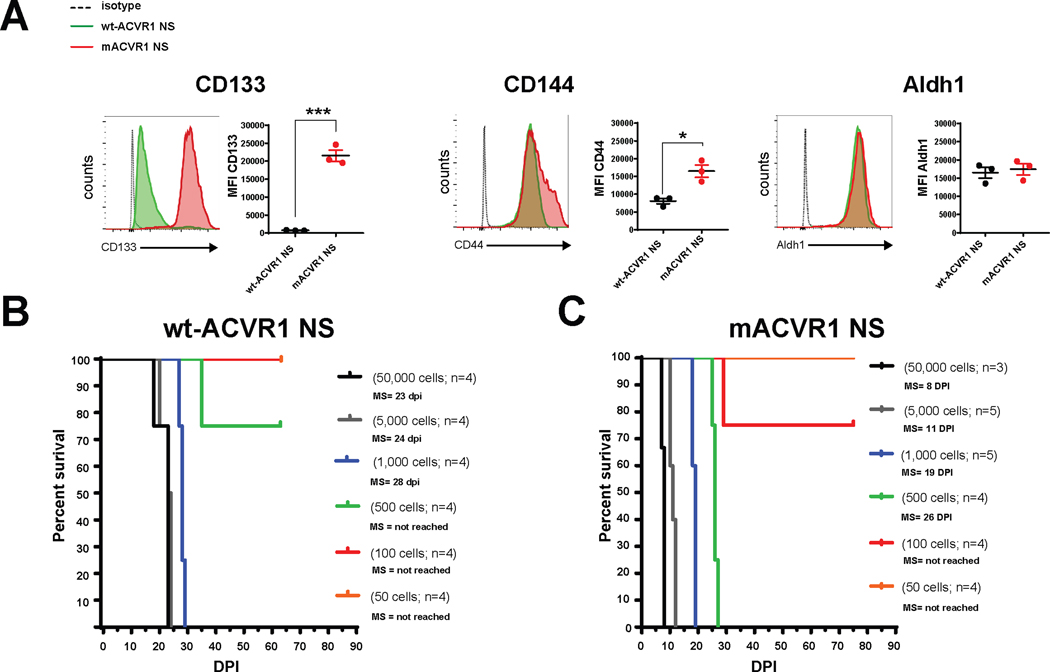
(A) Levels of CD133, CD44, and Aldh1 in the (tumor microenvironment) TME of mice bearing wt-ACVR1 or mACVR1 was assessed when animals displayed signs of tumor burden. Representative histograms display each marker’s expression (green = wt-ACVR1, red = mACVR1). MFI = mean fluorescence intensity; * p < 0.05; *** p < 0.001; unpaired t test. Bars represent mean ± standard error of the mean (SEM) (n = 3 biological replicates). (B-C) In vivo tumor initiation capacity. Kaplan-Meier survival curves for mice intracranially implanted with different numbers of wt-ACVR1 (B) or mACVR1 NS (C) as indicated in the plot. MS: median survival.
Pre-clinical Testing of Immune Stimulatory Gene Therapy using SB derived NS
Due to the invasive nature of brainstem gliomas and their refractive response to current therapies we wanted to assess the efficacy of immune stimulatory TK/Flt3L gene therapy using an immune competent, intracranial mouse model of brainstem glioma. To generate this model, we utilized NS expressing mACVR1. The release of DAMPs is crucial for the success of TK/Flt3L mediated therapy, therefore, we first assessed whether mACVR1 NS released DAMPs in vitro after treatment with GCV alone, TK alone, or GCV+TK, with, or without 3 Gray (Gy) of irradiation (IR). We found that mACVR1 NS treated with GCV+TK released increased levels of calreticulin (p<0.0001; Fig. 5A), high mobility group box 1 protein (HMGB1) (p<0.0001; Fig. 5A), adenosine triphosphate (ATP) (p = 0.0071; Fig. 5A). We also observed that the combination of GCV+TK with IR further increased the release of calreticulin (p<0.0001; Fig. 5A), HMGB1 (p=0.0375; Fig. 5A), and ATP (p = 0.0158; Fig 5A) release by the mACVR1 NS.
Figure 5. Efficacy of Immune-Stimulatory Gene Therapy.
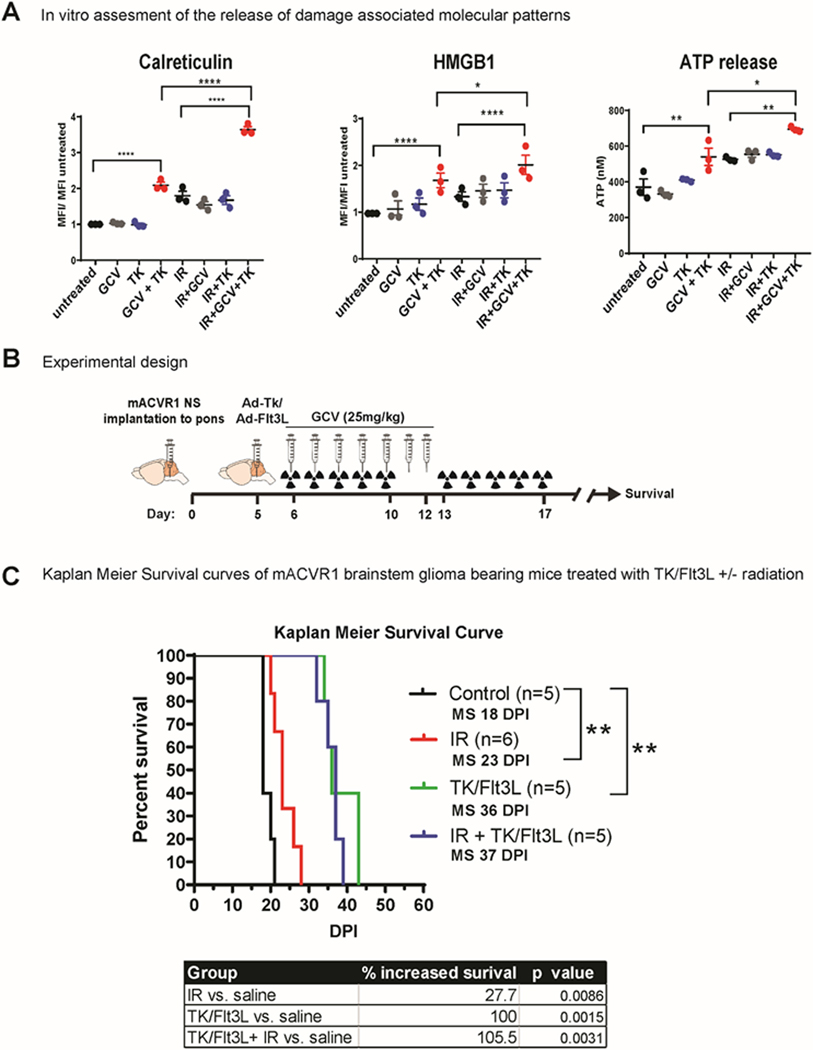
(A) wt-ACVR1 or mACVR1 NS were treated with TK (500 MOI) and/ or 3 Gray (Gy) ionizing radiation (IR), followed by GCV (25 μM). Levels of calreticulin or HMGB1 on tumor NS were assessed via flow cytometry analysis 24hrs post treatment. MFI = mean fluorescence intensity; * p < 0.05; **** p < 0.0001; two-way ANOVA, followed by Turkey’s multiple comparisons. Levels of ATP released into the media were assessed using a colorimetric assay. (B) Experimental design showing mice bearing mACVR1 brainstem gliomas treated with saline or TK/Flt3L gene therapy on day 5 post tumor implantation, followed by intraperitoneal administration of GCV (25 mg/kg) on day 6–12. Radiation was administered at a dose of 2Gy/Day for 10 days (total = 20 Gy) 5 days’ post tumor implantation; mice were monitored for survival. (C) Kaplan Meier survival analysis for mice bearing mACVR1 tumors treated with saline (n = 5), IR (n = 6), TK/Flt3L+ GCV (n=5), or IR+TK/Flt3L+GCV (n=5). Data were analyzed using log-rank (Mantel-Cox) test; ** p < 0.005.
Having established that mACVR1 NS release calreticulin, HMGB1, and ATP in vitro following treatment with either IR or TK+GCV, we next implanted mACVR1 NS into the pons of immune competent adult C57BL/6 mice to generate a transplantable brainstem model amenable to pre-clinical therapeutic implementation. Brainstem tumors derived using the transplantable model are positive for Ki67 (proliferating cells), GFAP (astrocyte marker), Vimentin (astrocytes and ependymal cells), Iba1 (microglia), but negative for MBP (a marker of mature oligodendrocytes) (Supplementary Fig. S4). The tumors are also positive for pERK1/2 and pMEK1/2 demonstrating activation of the RTK-RAS-PI3K signaling pathway (Supplementary Fig. S4). We also observed expression of Id1 a downstream target gene of BMP-Smad1/5 signaling (Supplementary Fig. S4). Five days’ post intracranial tumor implantation into the pons (Fig. 5B), mice were assigned to four treatment groups as indicated in Figure 5C. Our results demonstrate that Ad-TK/Ad-Flt3L therapy is more effective in prolonging the median survival (MS) of mACVR1 brainstem glioma compared to standard of care alone (MS=36 days post implantation (dpi) for TK/Flt3L group vs. 23 dpi for IR group; p = 0.0014, Mantel-Cox test) or vs. saline control (MS=18dpi; p = 0.0015, Mantel-Cox test) (Fig. 5C). Our data also indicate that radiation does not have a detrimental effect on the efficacy of TK/Flt3L therapy (Fig. 5C). This experiment was validated in Supplementary Fig. S5.
We next aimed to investigate whether Ad-TK/Ad-Flt3L treatment recruits anti-brainstem glioma specific T cell infiltration into the tumor immune microenvironment (TME). To do this, we used the surrogate tumor antigen ovoalbumin (OVA)(18,28). Using OVA-expressing mACVR1 cells (mACVR1-OVA NS), we were able to quantify tumor specific CD8 T cells in the TME through the use of the SIINFEKL-H2Kb tetramer (Fig. 6A, B). The full gating strategy is presented in Supplementary Fig. S6. We observed a 3.8-fold increase in the frequency of tumor specific CD8 T cells in the TME after treatment with Ad-TK/Ad-Flt3L gene therapy (p = 0.0007; ***, Fig. 6B). To test the impact of Ad-TK/Ad-Flt3L gene therapy on the activation status of CD8 T cells in the TME, we stained effector T cells for the expression of IFNγ after re-stimulating CD8 T cells isolated from the TME with mACVR1-OVA NS-lysate for 24 hrs. Our data show that IFNγ is increased 3.77 fold (p < 0.0038; **) in CD8 T cells from Ad-TK/Ad-Flt3L gene therapy treated mice compared to saline controls (Fig. 6 C). To test if Ad-TK/Ad-Flt3L gene therapy affected antigen-specific T cell proliferation we labeled splenocytes with 5-(and 6)-carboxyfluorescein diacetate succinimidyl ester (CFSE), and stimulated them with the OVA cognate SIINFEKL peptide. Our results show that the percentage of T cells that proliferated in response to the SIINKEKL peptide was greater (3.2 fold, p < 0.0001; ****) in mice treated with Ad-TK/Ad-Flt3L gene therapy compared to the saline treated control group (Fig. 6D). We also measured IFNγ levels in supernatants of samples from the T cell proliferation assay and our results show IFNγ levels were 2 fold (p < 0.0001; ****) higher in the supernatant of splenocytes isolated from mice treated with Ad-Tk/Ad-Flt3L gene therapy compared to the saline treated control group, after stimulation with SIINFEKL peptide (Fig. 6D). Additionally, the cytolytic of T cells isolated from the spleen of animals treated with Ad-TK/Ad-Flt3L gene therapy was observed to be significantly higher (1.95 fold, p < 0.0001 at 20:1 ratio) (Fig. 6E) when compared with the saline treated group indicating that gene therapy significantly enhanced the cytolytic activity of splenic T cells. To determine if the cytolytic activity is mACVR1-OVA specific, we used B16F10 murine melanoma cells as a negative control. When splenic T cells from mACVR1-OVA bearing mice were co-cultured with mACVR1-OVA cells, we observed an increase (4.25 fold, p < 0.0001 at 20:1 ratio) in tumor cell death in comparison to when they were co-cultured with B16-F12 cells.
Figure 6. Immune stimulatory gene therapy induces tumor specific T-cell infiltration.
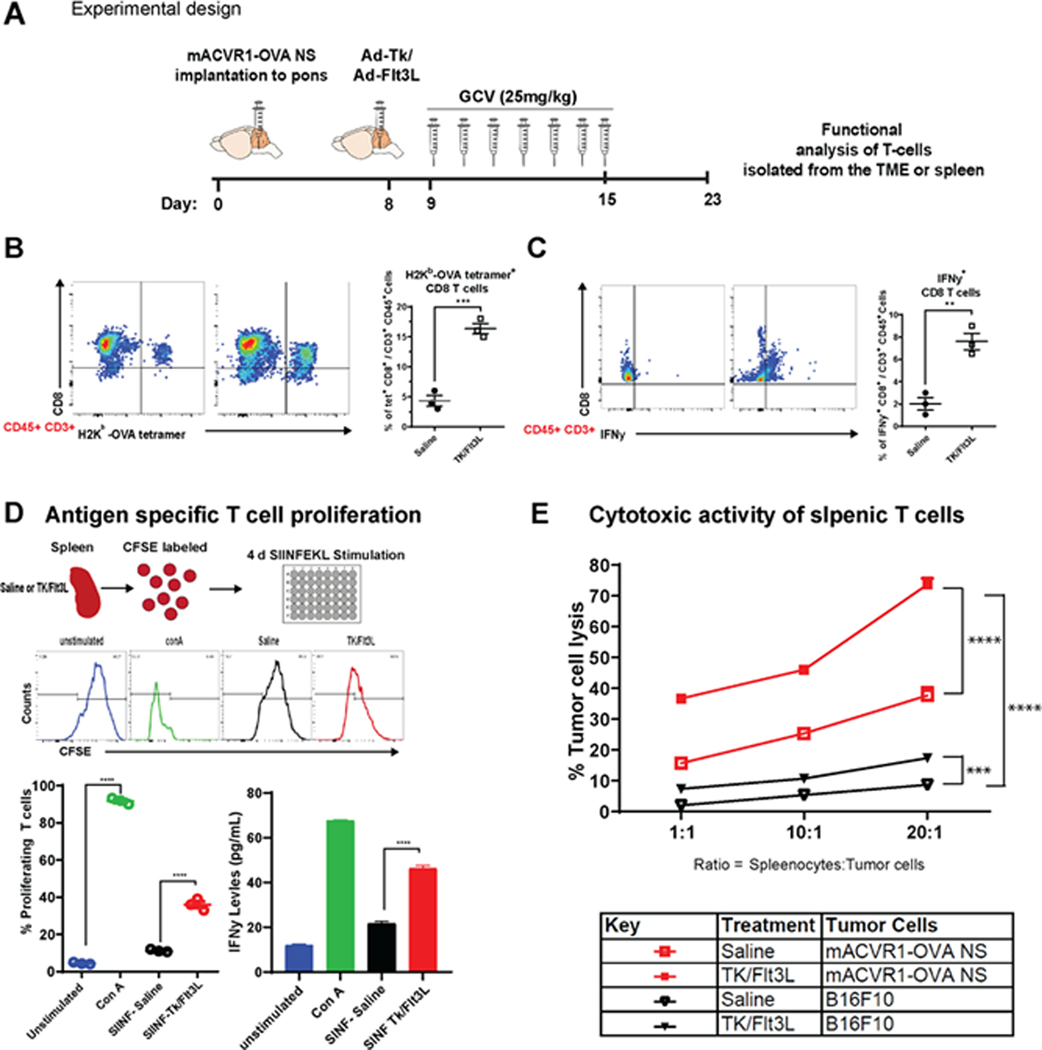
(A) Experimental design showing mice bearing mACVR1-OVA brainstem gliomas treated with saline or TK/FlT3L gene therapy on day 8 post implantation, followed by intraperitoneal administration of GCV (25 mg/kg) on day 9–15. (B) Tumor specific CD8 T cells within the TME of mACVR1-OVA tumors were analyzed by staining for SIINFEKL-Kb (SINF) tetramer. (C) Activation status of CD8 T cells within the TME was analyzed by staining for IFNγ after stimulation with tumor lysate. Representative flow plots for each group are displayed; ** p < 0.01, *** p < 0.001; unpaired t test. Bars represent mean ± SEM (n = 3 biological replicates, where resected tumors from 3 animals where pooled for the control group and 5 animals were pooled for the gene therapy group). (D) Experimental design showing splenocytes from untreated non tumor bearing mice, saline or TK/FlT3L treated mACVR1-OVA tumor bearing mice labeled with CFSE. Untreated non tumor bearing splenocytes were either stimulated with Concanavalin A (ConA) (positive control) or RPMI (negative control) cell culture media for four days. Splenocytes from mACVR1-OVA tumor bearing mice treated with saline or TK/Flt3L were stimulated with 100 nM of SIINFEEKL peptide for four days in culture to assess surrogate tumor antigen (OVA) specific T cell proliferation. IFNγ expression in supernatants from T cell proliferation assay was assessed by ELISA. Histograms show representative CFSE staining from unstimulated, ConA stimulated, or mACVR1-OVA stimulated splenocytes from TK/Flt3L treated mACVR1-OVA tumor bearing mice. Quantification of splenocytes undergoing T cell proliferation and ELISA; **** p < 0.0001; one-way ANOVA followed by followed by Turkey’s multiple comparisons. Bars represent mean ± SEM (n = 3 biological replicates). (E) Splenic T cells from saline or TK/Flt3L treated mACVR1-OVA tumor bearing mice were incubated with mACVR1-OVA NS or B16F10 melanoma cells for 24 hours at the indicated ratios. Lysis of tumor cells was assessed by using Annexin-V. Data were compared using two-way ANOVA test; *** p < 0.001, **** p < 0.0001. Bars represent mean ± SEM (n = 3 biological replicates).
To evaluate any potential adverse effects of delivering gene therapy into the brainstem we performed a detailed histopathological analysis of brains from non-tumor bearing animals. After the animals were treated intracranially with Ad-TK/Ad-Flt3L, GCV was administered intraperitoneally one day after adenoviral injection for seven days. Brains were harvested for neuropathology analysis 24 hours after the last dose of GCV was administered. Architectural integrity was assessed by H&E staining and by immunohistochemistry using GFAP (astrocytes), MBP (myelin sheaths, oligodendrocytes) and Iba1 (microglia). No gross tissue abnormalities were observed in response to TK/Flt3L therapy compared to the saline controls (Supplementary Fig. S7). There was also no change in GFAP or MBP expression in response to TK/Flt3L therapy indicating that the brain architecture was unaffected (Supplementary Fig. S7). There was no increase in Iba1 expression in the animals treated with TK/Flt3L therapy indicating that gene therapy does not induce inflammation in normal brain tissue 8 days post treatment (Supplementary Fig. S7).
To assess potential systemic toxicity due to TK/Flt3L or IR treatment, we performed H&E staining on liver sections and complete hematological and serum biochemical analysis in tumor bearing animals from the saline, TK/Flt3L, and TK/Flt3L + IR treatment groups. The liver sections from all treatment groups had normal hepatocyte architecture and did not show signs of inflammation or necrosis (Supplementary Fig. S8A). The white blood cell counts were within normal range for the saline, TK/Flt3L, and TK/Flt3L + IR groups, but significantly decreased in the IR group in comparison to the saline treated group (p < 0.0001, Supplementary Fig. S8A). This is consistent with a study that found that whole brain radiation significantly decreased total white blood cell counts (29). Retrospective studies have found that total lymphocyte counts are associated with decreased response to immune checkpoint inhibitors in multiple cancers (30,31). Nevertheless, radiation has been shown to trigger immunogenic cell death which can sometimes have a favorable effect (17). We saw that radiation when combined with TK/Flt3L therapy did not have a detrimental effect on total white blood cell counts. Red blood cell, hemoglobin, hematocrit, platelet, lymphocyte, neutrophil, and monocyte counts were not significantly affected by TK/Flt3L or IR therapy (Supplementary Fig. S8B–I). We did not find any significant changes in important enzymes involved in liver (ALT, AST) and kidney (BUN) function as a result of TK /Flt3L or IR therapies (Supplementary Fig. S8J–L).
Discussion
DIPG remains an incurable tumor with a poor prognosis (32). The tumors originate in the pons and infiltrate into sensitive regions of the brainstem precluding surgical resection (33). DIPGs are also resistant to radiation and chemotherapy (34). DIPG studies involving the development and implementation of novel therapies have utilized patient derived xenograft models, where human DIPG cells are implanted into the brain of immune deficient mice or rats to establish intracranial tumors (35,36). One limitation with those models is that immune suppressed animals cannot be used to test immunotherapies or perform immune-related mechanistic studies. In this study, we utilized the SB transposase system to generate an endogenous mouse model of mutant ACVR1 brainstem glioma. ACVR1 is frequently mutated to ACVR1 G328V in DIPG (4–7). The SB system efficiently and reproducibly integrates plasmid DNA into the neural progenitor cells’ host chromosomal DNA of neonatal mice, allowing for the functional assessment of the role of candidate DIPG genes in promoting tumor progression (19–22). Mutations in ACVR1 confer ligand independent activation of ACVR1, which is a mediator of the BMP signaling pathway (4,5,37,38). We show that SB tumors are localized in the brainstem and have increased downstream signaling of BMP as demonstrated by increased phospho-smad1/5 levels and Id1 (Fig. 2B, C). Transcriptional profiling indicated an enrichment in the BMP signaling pathway in mACVR1 NS compared to wt-WT ACVR1 tumor NS (Fig. 3D). In addition, our tumors express Olig2, a biomarker for DIPG tumors that is highly expressed in 70%−80% of human DIPGs(39).
Our data also suggests that mACVR1 regulates stemness and tumor initiation potential. Interestingly, clonal evolution analysis of a human DIPG tumor found that the ACVR1 mutation was present along with the H3.1 K27M mutation in all tumor clones, in contrast to secondary mutations found only in some subclones, implicating mACVR1in DIPG tumor initiation (40). In addition, Hoeman C.M. and colleagues observed that ACVR1 R206H contributed to increased tumor incidence using the RCAS model driven by p53 loss and PDDGF-A, supporting our results that ACVR1 plays a role in tumor initiation (41). It was also recently reported that only amino acid substitutions of ACVR1 at the G328 residue confer a significant increase in survival in DIPG patients (9). Therefore, our data suggests independent roles for ACVR1 on survival and tumor progression.
Neurospheres (NS) harboring mACVR1 were implanted into the pons of adult immune competent mice, enabling the generation of transplantable DIPG models and amenable for preclinical therapeutic studies. This implantable model is more stringent for assessment of therapeutic efficacy than genetically engineered models since tumor growth rates and time of death vary less between animals. Currently, the treatment option for children with DIPG is limited to radiation to provide palliative care (1). Although radiation can temporarily provide symptomatic relief and extend survival by a few months, it can cause detrimental effects on the developing brain (42,43). Therefore, there is a dire need for new therapeutic interventions. This research focuses on the response of mACVR1 brainstem glioma tumors to immunotherapies.
There has been significant progress in the field of cancer immunotherapy leading to improvements in overall survival in many types of solid tumors (11–13,44,45). Advances in immunotherapies include the development of immune therapies targeting immune checkpoints, vaccine approaches against tumor antigens or dendritic cell vaccines designed to stimulate the adaptive immune response, adoptive cell therapy, oncolytic viral therapy, and immune stimulatory gene therapy (45–47). This has led to a growing number of clinical trials testing immunotherapies in DIPG (47).
In recent years, it has been established that activated immune cells are able to migrate and enter into the brain parenchyma, and that the brain has a functional lymphatic system that allows for transport of CNS-antigens to the draining lymph nodes (48,49). Thus, the brain is capable of mounting T cell-mediated adaptive immune responses, but, since there are very low numbers of local professional antigen presenting cells within the normal brain, it is not possible to prime a potent immune response against antigens localized in the brain parenchyma (50,51).
In adult glioblastoma (GBM) there is evidence of immune cell infiltration, but an immunosuppressive environment precludes effective anti-tumor immunity (46,52,53). GBM tumors establish an immunosuppressive environment by the release of immunosuppressive cytokines, such as TGF-β and IL-10, by the recruitment or induction of immunosuppressive cells, such as regulatory T cells, myeloid-derived suppressor cells, or tumor associated macrophages, and by the expression of immune checkpoint receptor ligands (52–58). In comparison to adult GBM, initial studies of the tumor microenvironment in DIPG have found that there is a low number of immune infiltrates in human DIPG tumors and that they do not express inflammatory cytokines and chemokines (59,60). These data provide support for the use of an immune modulatory therapeutic strategy to enhance the recruitment of immune cells into the tumor, with the aim of mounting an effective anti-DIPG immune response.
We have previously demonstrated that combined immune stimulatory gene therapy mediated through the delivery of adenoviruses encoding herpes simplex virus type 1 thymidine kinase (TK) and fms-like tyrosine kinase 3 ligand (Flt3L) leads to tumor regression and long term survival in several rodent models of glioblastoma (15,16,61–63). This therapy is based on inducing tumor cell death though expression of suicide gene TK (64,65). Tumor antigens and damage-associated molecular pattern molecules (DAMPs), such as calreticulin, high mobility group box1 (HMGB1), and ATP, are released by dying tumor cells (17,66). The effectiveness of this combination therapy also relies on Flt3L to recruit dendritic cells into the tumor microenvironment, while the release of HMGB1 stimulates TLR2-dependent activation of dendritic cells (15,65,67). Activated dendritic cells can then transport antigens to the draining lymph nodes and induce tumor specific T cell responses (15,65). Initial results from the first in human Phase 1 clinical trial of combined adenoviral delivery of TK and Flt3L for the treatment of adult GBM are promising and report that the therapy was well tolerated (68). However, it has been established that the biology of DIPG and GBM is different (69). Therefore, it is necessary to assess the safety and efficacy of TK/Flt3L therapy in pre-clinical models of brainstem glioma. Herein, we demonstrate that treatment with TK/Flt3L gene therapy in mice bearing mACVR1 brainstem gliomas stimulates a strong anti-tumor cytotoxic immune response leading to a significant increase in survival (Fig. 5C). Treatment with TK/Flt3L increased the frequency of tumor-specific CD8 T cells in the tumor microenvironment and increased toxicity as demonstrated by enhanced IFNγ production (Fig. 6 B,C).
Additionally, delivery of TK/Flt3L into the normal brainstem did not induce any local or systemic cytotoxicity. Hematoxylin and eosin staining and immunostaining for GFAP (astoryctes), MBP (myelin sheaths, oligodendrocytes), and Iba1 (activated macrophages and microglia) was used to assess local toxicity, and we observed no architectural abnormalities or overt inflammation as a result of TK/Flt3L therapy. Histological examination of hematoxylin and eosin stained liver tissue did not show signs of inflammation, necrosis, or alterations in normal hepatocyte structure. Hematological toxicity, assessed by complete blood count and serum chemistry analysis, indicated that TK/Flt3L therapy did not induce any toxicity as values from a TK/Flt3L treated group were not significantly altered when compared to saline treated animals. Our results are consistent with other pre-clinical studies that report the brainstem can tolerate adenoviral mediated immunotherapy (70,71). Results from a clinical trial (NCT03178032) utilizing adenoviral vector delivery of an oncolytic virus into the pons of DIPG patients will also shed light on the feasibility and toxicity of intratumoral adenoviral delivery into the brainstem (72).
In conclusion, we demonstrate the amenability of the SB transposase to be used to develop genetically and histologically accurate models of DIPG expressing mutant ACVR1 and provide compelling evidence that warrants further development of conditionally cytotoxic immune stimulatory gene therapy for the treatment of DIPG. We anticipate that in the clinic, this approach could also be used in combination with immune checkpoint blockade to further enhance the therapeutic efficacy of the TK/Flt3L-mediated anti-brain stem glioma immune response.
Supplementary Material
Translational Relevance:
The therapeutic efficacy of anti-DIPG immune responses is limited due to a low number of immune cells in the tumor microenvironment. We have uncovered a novel treatment strategy that utilizes adenoviral delivery of therapeutic genes, thymidine kinase (TK) and fms tyrosine kinase 3 ligand (Flt3L) into the tumor, eliciting a reprograming of the host’s own immune system to recognize and kill tumor cells. We demonstrate that TK/Flt3L therapy generates an effective anti-tumor response and can be safely delivered into the brainstem. This treatment approach could provide a novel translational approach towards potentiating an anti-DIPG immune response to overcome the current limitations in the treatment of patients with DIPG.
Acknowledgments
We thank J. Ohlfest (University of Minnesota, deceased) for providing the SB model plasmids and Jeff Wrana for providing the wt-ACVR1 plasmid. We thank the support and academic leadership of Dr. Karin Muraszko, and the administrative support of Angela Collada. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Financial Support: Supported by National Institutes of Health/National Institute of Neurological Disorders & Stroke (NIH/NINDS) Grants R37-NS094804, R01-NS105556, R21-NS107894 to M.G.C.; NIH/NINDS Grants R01-NS076991, R01-NS082311, and R01-NS096756 to P.R.L.; NIH/NIBIB R01-EB022563 and NCI/UO1-CA-224160 to M.G.C. and P.R.L.; the Department of Neurosurgery; Leah’s Happy Hearts Foundation, ChadThough Foundation, Pediatric Brain Tumor Foundation, and Smiles for Sophie Forever Foundation to M.G.C. and P.R.L. RNA Biomedicine Grant F046166 to M.G.C. NIH/NINDS-F31NS103500 and Rackham Pre-doctoral Fellowship to F.M.M; S.C. is supported by NIH/NCI-T32-CA009676. MBGF is supported by the American Brain Tumor Association Basic Research Fellowship “in Memory of Bruce and Brian Jackson”.
Footnotes
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this article.
References
- 1.Cohen KJ, Jabado N, Grill J. Diffuse intrinsic pontine gliomas-current management and new biologic insights. Is there a glimmer of hope? Neuro Oncol 2017;19(8):1025–34 doi 10.1093/neuonc/nox021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frazier JL, Lee J, Thomale UW, Noggle JC, Cohen KJ, Jallo GI. Treatment of diffuse intrinsic brainstem gliomas: failed approaches and future strategies. J Neurosurg Pediatr 2009;3(4):259–69 doi 10.3171/2008.11.PEDS08281. [DOI] [PubMed] [Google Scholar]
- 3.Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017;32(4):520–37 e5 doi 10.1016/j.ccell.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, Philippe C, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 2014;46(5):457–61 doi 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 2014;46(5):462–6 doi 10.1038/ng.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 2014;46(5):444–50 doi 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 2014;46(5):451–6 doi 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor KR, Vinci M, Bullock AN, Jones C. ACVR1 mutations in DIPG: lessons learned from FOP. Cancer Res 2014;74(17):4565–70 doi 10.1158/0008-5472.CAN-14-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carvalho D, Taylor KR, Olaciregui NG, Molinari V, Clarke M, Mackay A, et al. ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Commun Biol 2019;2:156 doi 10.1038/s42003-019-0420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones C, Baker SJ. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer 2014;14(10) doi 10.1038/nrc3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363(8):711–23 doi 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 2016;375(19):1823–33 doi 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 13.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015;373(19):1803–13 doi 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363(5):411–22 doi 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 15.Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med 2009;6(1):e10 doi 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Candolfi M, Yagiz K, Foulad D, Alzadeh GE, Tesarfreund M, Muhammad AK, et al. Release of HMGB1 in response to proapoptotic glioma killing strategies: efficacy and neurotoxicity. Clin Cancer Res 2009;15(13):4401–14 doi 10.1158/1078-0432.CCR-09-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012;12(12):860–75 doi 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 18.Kamran N, Kadiyala P, Saxena M, Candolfi M, Li Y, Moreno-Ayala MA, et al. Immunosuppressive Myeloid Cells’ Blockade in the Glioma Microenvironment Enhances the Efficacy of Immune-Stimulatory Gene Therapy. Mol Ther 2017;25(1):232–48 doi 10.1016/j.ymthe.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiesner SM, Decker SA, Larson JD, Ericson K, Forster C, Gallardo JL, et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res 2009;69(2):431–9 doi 10.1158/0008-5472.CAN-08-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calinescu AA, Nunez FJ, Koschmann C, Kolb BL, Lowenstein PR, Castro MG. Transposon mediated integration of plasmid DNA into the subventricular zone of neonatal mice to generate novel models of glioblastoma. J Vis Exp 2015(96) doi 10.3791/52443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koschmann C, Calinescu AA, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med 2016;8(328):328ra28 doi 10.1126/scitranslmed.aac8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nunez FJ, Mendez FM, Kadiyala P, Alghamri MS, Savelieff MG, Garcia-Fabiani MB, et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci Transl Med 2019;11(479) doi 10.1126/scitranslmed.aaq1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izsvak Z, Ivics Z. Sleeping beauty transposition: biology and applications for molecular therapy. Mol Ther 2004;9(2):147–56 doi 10.1016/j.ymthe.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Calinescu AA, Yadav VN, Carballo E, Kadiyala P, Tran D, Zamler DB, et al. Survival and Proliferation of Neural Progenitor-Derived Glioblastomas Under Hypoxic Stress is Controlled by a CXCL12/CXCR4 Autocrine-Positive Feedback Mechanism. Clin Cancer Res 2017;23(5):1250–62 doi 10.1158/1078-0432.CCR-15-2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nunez FJ, Mendez FM, Garcia-Fabiani MB, Pardo J, Edwards M, Lowenstein PR, et al. Evaluation of Biomarkers in Glioma by Immunohistochemistry on Paraffin-Embedded 3D Glioma Neurosphere Cultures. J Vis Exp 2019(143) doi 10.3791/58931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 2010;28(18):3061–8 doi 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panditharatna E, Kilburn LB, Aboian MS, Kambhampati M, Gordish-Dressman H, Magge SN, et al. Clinically Relevant and Minimally Invasive Tumor Surveillance of Pediatric Diffuse Midline Gliomas Using Patient-Derived Liquid Biopsy. Clin Cancer Res 2018;24(23):5850–9 doi 10.1158/1078-0432.CCR-18-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Sanderson NS, Wawrowsky K, Puntel M, Castro MG, Lowenstein PR. Kupfer-type immunological synapse characteristics do not predict anti-brain tumor cytolytic T-cell function in vivo. Proc Natl Acad Sci U S A 2010;107(10):4716–21 doi 10.1073/pnas.0911587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexander TC, Butcher H, Krager K, Kiffer F, Groves T, Wang J, et al. Behavioral Effects of Focal Irradiation in a Juvenile Murine Model. Radiat Res 2018;189(6):605–17 doi 10.1667/RR14847.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho WJ, Yarchoan M, Hopkins A, Mehra R, Grossman S, Kang H. Association between pretreatment lymphocyte count and response to PD1 inhibitors in head and neck squamous cell carcinomas. J Immunother Cancer 2018;6(1):84 doi 10.1186/s40425-018-0395-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pike LRG, Bang A, Mahal BA, Taylor A, Krishnan M, Spektor A, et al. The Impact of Radiation Therapy on Lymphocyte Count and Survival in Metastatic Cancer Patients Receiving PD-1 Immune Checkpoint Inhibitors. Int J Radiat Oncol Biol Phys 2019;103(1):142–51 doi 10.1016/j.ijrobp.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Veldhuijzen van Zanten SEM, Baugh J, Chaney B, De Jongh D, Sanchez Aliaga E, Barkhof F, et al. Development of the SIOPE DIPG network, registry and imaging repository: a collaborative effort to optimize research into a rare and lethal disease. J Neurooncol 2017;132(2):255–66 doi 10.1007/s11060-016-2363-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016;131(6):803–20 doi 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 34.Hargrave D, Bartels U, Bouffet E. Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol 2006;7(3):241–8 doi 10.1016/S1470-2045(06)70615-5. [DOI] [PubMed] [Google Scholar]
- 35.Hashizume R, Smirnov I, Liu S, Phillips JJ, Hyer J, McKnight TR, et al. Characterization of a diffuse intrinsic pontine glioma cell line: implications for future investigations and treatment. J Neurooncol 2012;110(3):305–13 doi 10.1007/s11060-012-0973-6. [DOI] [PubMed] [Google Scholar]
- 36.Caretti V, Sewing AC, Lagerweij T, Schellen P, Bugiani M, Jansen MH, et al. Human pontine glioma cells can induce murine tumors. Acta Neuropathol 2014;127(6):897–909 doi 10.1007/s00401-014-1272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem 2010;147(1):35–51 doi 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- 38.Chaikuad A, Alfano I, Kerr G, Sanvitale CE, Boergermann JH, Triffitt JT, et al. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem 2012;287(44):36990–8 doi 10.1074/jbc.M112.365932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson JL, Muraleedharan R, Oatman N, Klotter A, Sengupta S, Waclaw RR, et al. The transcription factor Olig2 is important for the biology of diffuse intrinsic pontine gliomas. Neuro Oncol 2017;19(8):1068–78 doi 10.1093/neuonc/now299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M, et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nature Communications 2016;7:11185 doi 10.1038/ncomms11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoeman CM, Cordero FJ, Hu G, Misuraca K, Romero MM, Cardona HJ, et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nature Communications 2019;10(1) doi 10.1038/s41467-019-08823-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bledsoe JC. Effects of Cranial Radiation on Structural and Functional Brain Development in Pediatric Brain Tumors. Journal of Pediatric Neuropsychology 2015;2(1–2):3–13 doi 10.1007/s40817-015-0008-2. [DOI] [Google Scholar]
- 43.Jackson S, Patay Z, Howarth R, Pai Panandiker AS, Onar-Thomas A, Gajjar A, et al. Clinico-radiologic characteristics of long-term survivors of diffuse intrinsic pontine glioma. J Neurooncol 2013;114(3):339–44 doi 10.1007/s11060-013-1189-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2017;377(14):1345–56 doi 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang SS, Bandopadhayay P, Jenkins MR. Towards Immunotherapy for Pediatric Brain Tumors. Trends Immunol 2019;40(8):748–61 doi 10.1016/j.it.2019.05.009. [DOI] [PubMed] [Google Scholar]
- 46.Kamran N, Alghamri MS, Nunez FJ, Shah D, Asad AS, Candolfi M, et al. Current state and future prospects of immunotherapy for glioma. Immunotherapy 2018;10(4):317–39 doi 10.2217/imt-2017-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foster JB, Madsen PJ, Hegde M, Ahmed N, Cole KA, Maris JM, et al. Immunotherapy for pediatric brain tumors: past and present. Neuro Oncol 2019;21(10):1226–38 doi 10.1093/neuonc/noz077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest 2010;120(5):1368–79 doi 10.1172/JCI41911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015;523(7560):337–41 doi 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perry VH. A revised view of the central nervous system microenvironment and major histocompatibility complex class II antigen presentation. J Neuroimmunol 1998;90(2):113–21 doi 10.1016/s0165-5728(98)00145-3. [DOI] [PubMed] [Google Scholar]
- 51.Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience 1996;74(2):599–608 doi 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- 52.Rutledge WC, Kong J, Gao J, Gutman DA, Cooper LA, Appin C, et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin Cancer Res 2013;19(18):4951–60 doi 10.1158/1078-0432.CCR-13-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res 2011;17(13):4296–308 doi 10.1158/1078-0432.CCR-10-2557. [DOI] [PubMed] [Google Scholar]
- 54.Dutoit V, Migliorini D, Dietrich PY, Walker PR. Immunotherapy of Malignant Tumors in the Brain: How Different from Other Sites? Front Oncol 2016;6:256 doi 10.3389/fonc.2016.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res 2013;19(12):3165–75 doi 10.1158/1078-0432.CCR-12-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raychaudhuri B, Rayman P, Huang P, Grabowski M, Hambardzumyan D, Finke JH, et al. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J Neurooncol 2015;122(2):293–301 doi 10.1007/s11060-015-1720-6. [DOI] [PubMed] [Google Scholar]
- 57.Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol 2010;12(11):1113–25 doi 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol 2015;17(8):1064–75 doi 10.1093/neuonc/nou307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin GL, Nagaraja S, Filbin MG, Suva ML, Vogel H, Monje M. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol Commun 2018;6(1):51 doi 10.1186/s40478-018-0553-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lieberman NAP, DeGolier K, Kovar HM, Davis A, Hoglund V, Stevens J, et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro Oncol 2019;21(1):83–94 doi 10.1093/neuonc/noy145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ali S, King GD, Curtin JF, Candolfi M, Xiong W, Liu C, et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res 2005;65(16):7194–204 doi 10.1158/0008-5472.CAN-04-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.King GD, Muhammad AK, Curtin JF, Barcia C, Puntel M, Liu C, et al. Flt3L and TK gene therapy eradicate multifocal glioma in a syngeneic glioblastoma model. Neuro Oncol 2008;10(1):19–31 doi 10.1215/15228517-2007-045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ghulam Muhammad AK, Candolfi M, King GD, Yagiz K, Foulad D, Mineharu Y, et al. Antiglioma immunological memory in response to conditional cytotoxic/immune-stimulatory gene therapy: humoral and cellular immunity lead to tumor regression. Clin Cancer Res 2009;15(19):6113–27 doi 10.1158/1078-0432.CCR-09-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res 1986;46(10):5276–81. [PubMed] [Google Scholar]
- 65.Castro MG, Candolfi M, Wilson TJ, Calinescu A, Paran C, Kamran N, et al. Adenoviral vector-mediated gene therapy for gliomas: coming of age. Expert Opin Biol Ther 2014;14(9):1241–57 doi 10.1517/14712598.2014.915307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev 2012;249(1):158–75 doi 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Curtin JF, King GD, Barcia C, Liu C, Hubert FX, Guillonneau C, et al. Fms-like tyrosine kinase 3 ligand recruits plasmacytoid dendritic cells to the brain. J Immunol 2006;176(6):3566–77 doi 10.4049/jimmunol.176.6.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lowenstein PR, Orringer DA, Sagher O, Heth J, Hervey-Jumper SL, Mammoser AG, et al. First-in-human phase I trial of the combination of two adenoviral vectors expressing HSV1-TK and FLT3L for the treatment of newly diagnosed resectable malignant glioma: Initial results from the therapeutic reprogramming of the brain immune system. 2019;37(15_suppl):2019- doi 10.1200/JCO.2019.37.15_suppl.2019. [DOI] [Google Scholar]
- 69.Schroeder KM, Hoeman CM, Becher OJ. Children are not just little adults: recent advances in understanding of diffuse intrinsic pontine glioma biology. Pediatr Res 2014;75(1–2):205–9 doi 10.1038/pr.2013.194. [DOI] [PubMed] [Google Scholar]
- 70.Martinez-Velez N, Garcia-Moure M, Marigil M, Gonzalez-Huarriz M, Puigdelloses M, Gallego Perez-Larraya J, et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat Commun 2019;10(1):2235 doi 10.1038/s41467-019-10043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schuelke MR, Wongthida P, Thompson J, Kottke T, Driscoll CB, Huff AL, et al. Diverse immunotherapies can effectively treat syngeneic brainstem tumors in the absence of overt toxicity. J Immunother Cancer 2019;7(1):188 doi 10.1186/s40425-019-0673-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tejada S, Diez-Valle R, Dominguez PD, Patino-Garcia A, Gonzalez-Huarriz M, Fueyo J, et al. DNX-2401, an Oncolytic Virus, for the Treatment of Newly Diagnosed Diffuse Intrinsic Pontine Gliomas: A Case Report. Front Oncol 2018;8:61 doi 10.3389/fonc.2018.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.