

Abstract
Introduction:
Associations of physical exercise with Alzheimer disease (AD) biomarkers and cognitive functioning have been observed cross-sectionally. However, the effects of exercise on longitudinal change in AD biomarkers have not been thoroughly investigated. The current study examined whether individuals with higher baseline exercise exhibited less longitudinal change in AD biomarkers and cognitive functioning, and whether APOE and/or the BDNF genotypes moderated the effects of exercise on longitudinal changes.
Methods:
Clinically normal individuals completed a questionnaire on physical exercise over the prior 10-year period at baseline. Ninety-five individuals had serial CSF samples collected to examine Aβ42, ptau181 and total tau; 181 individuals underwent multiple assessments of amyloid PET imaging with Pittsburgh Compound B; 327 individuals underwent multiple cognitive assessments, including measures of episodic memory, executive functions, verbal fluency and processing speed.
Results:
Greater exercise was associated with less steep decline in processing speed. Baseline exercise did not robustly impact longitudinal change for any other outcomes. Neither APOE nor BDNF genotype robustly moderated the effect of exercise on trajectories of AD biomarkers or cognitive decline.
Interpretation:
Results suggest that self-reported physical exercise may be limited as a moderator of changes in AD biomarkers.
Keywords: aerobic exercise, memory, amyloid, neurofibrillary tangles
Introduction
Although Alzheimer disease (AD) is the most common form of dementia among older adults, there are currently no effective pharmacological treatments that can stop or significantly slow the disease. Research efforts have also targeted lifestyle factors that could potentially influence the course of the disease and may be particularly effective during the preclinical phase [1]. In particular, physical exercise has increasingly been associated with enhanced brain and cognitive health in older adults [2].
Physical activity, exercise and aerobic fitness are associated with less cognitive decline or decreased risk of dementia in observational longitudinal studies [3; 4]. These benefits have been observed across a variety of cognitive domains (e.g., executive functioning, processing speed, verbal fluency, attention, working memory). In addition, individuals with a genetic risk for developing AD (i.e., having an APOE ε4 allele) may especially benefit [e.g., 5].
While these findings are suggestive that physical activity or exercise may serve to slow cognitive decline and reduce risk of dementia, it is important to understand the degree to which they influence core AD pathology (i.e., amyloid deposition and neurofibrillary tangles). Several studies have reported beneficial effects of exercising on amyloid and tau deposition in transgenic AD mice [1]. However, there has been less research on the association between physical activity or exercise with AD pathology in humans.
Several biomarkers of AD pathology have been developed. Cerebrospinal fluid (CSF) biomarkers include Aβ42 as an estimation of levels of brain amyloid, ptau181 reflective of levels of phosphorylated tau (the main element of neurofibrillary tangles; [6]), and tau as indicative of neuronal injury. In addition, positron emission tomography (PET) can be used to measure fibrillar amyloid plaques using the Pittsburgh compound-B (PIB) radiotracer [7]. In some studies, more active individuals were found to have better biomarker profiles in terms of CSF Aβ42, ptau181, ptau181/Aβ42 and tau/Aβ42 ratios as well as PET-PIB [e.g., 8; 9], with some evidence of particular benefits for individuals with an APOE ε4 allele [10; 11]. However, other studies failed to show an association between self-reported physical activity and PET-PIB [e.g., 12; 13; 14]. One existing longitudinal study observed that higher baseline physical activity was associated with lower amyloid deposition at follow-up [15].
Another potential moderator of the effects of physical exercise relates to brain-derived neurotrophic factor (BDNF). BDNF increases after exercising and contributes to synaptic plasticity [e.g., 16]. Presence of the p.Met66 allele in the BDNF gene, compared to the Val allele, has been linked to reduced BDNF secretion [17]. In studies examining whether the p.Val66Met (rs6265) BDNF polymorphism moderates the relationship between physical activity and cognitive functioning, results have varied with regard to whether physical activity benefits are greater for p.Met66 carriers [e.g., 18] or for Val carriers [e.g., 19]. Whether the BDNF genotype moderates physical exercise associations with AD biomarkers in humans has been less examined.
The primary goals of this study were to examine a) whether higher baseline exercise is associated with less decline in AD biomarkers and cognitive functioning; b) whether APOE genotype moderates the association with a greater effect of exercise for APOE ε4-positive individuals; and c) whether BDNF genotype moderates the association with p.Met66 allele carriers benefitting less from exercise.
Method
Participants.
Older adults, 55 to 88 years-old, were recruited from the Knight Alzheimer’s Disease Research Center at Washington University. Participants age 65+ are followed annually with clinical and cognitive assessments; those 55–64 are followed with these assessments every 3 years. All participants have biomarker collection at baseline and every 3 years thereafter. The Clinical Dementia Rating (CDR) scale was used to determine absence or presence and severity of dementia when present (Morris 1993). All participants were clinically normal (CDR=0) at baseline assessments. All individuals who had baseline exercise data and either CSF (n=95), PET (n=181) or cognitive (n=327) data were included in order to maximize the sample size for each outcome (see Table 1 for sample characteristics). Nonetheless, there is substantial overlap in participants across the samples: 84 of 95 participants in the CSF sample are part of the PET-PIB sample, all of the 95 participants in the CSF sample are part of the cognitive sample, and 179 of 181 participants in the PET-PIB sample are part of the cognitive sample. Cross-sectional analyses of exercise associations with brain structure, CSF and PET-PIB have been published previously [20; 11; 9].
Table 1.
Sample characteristics: AHA exercise groups.
| CSF | PET-PIB | Cognitive | ||||
|---|---|---|---|---|---|---|
| Low Exercise | High Exercise | Low Exercise | High Exercise | Low Exercise | High Exercise | |
| N | 62 | 33 | 122 | 59 | 211 | 116 |
| Age (M, SD) | 62±8 | 64±7 | 67±10 | 68±8 | 72±8 | 72±8 |
| Sex (F/M) | 43/19 | 18/15 | 85/37 | 33/26 | 136/75 | 64/52 |
| Education (M, SD) | 16±3 | 17±3 | 16±3 | 16±3 | 16±3 | 15±3 |
| Health composite (M, SD) | 1.2±1.0 | .8±.9* | 1±1 | 1±1 | 1±1 | 1±1 |
| APOE ε4 (−/+) | 37/25 | 24/9 | 85/37 | 40/19 | 134/77 | 83/33 |
| BDNF met (−/+) | 42/20 | 19/14 | 71/44 | 36/18 | 130/70 | 73/30 |
| Number follow-ups (M, SD) | 1.44±0.55 | 1.41±0.50 | 1.42±0.61 | 1.58±0.77 | 3.60±2.15 | 3.56±2.10 |
| Follow-up length, years (M, SD) | 3.28±1.45 | 3.66±1.12 | 3.74±2.40 | 4.23±2.47 | 6.09±2.28 | 6.01±2.19 |
Notes. BDNF genotype data was missing for 12 participants for the PET-PIB sample and 24 for the Cognitive sample. The BDNF subsamples were not significantly different from the APOE samples (all p’s > .59). Processing speed composite data were missing for 2 participants. Baseline exercise occurred within 5.33 years (M=1.36, SD=1.22) of baseline CSF, within 7.25 years (M=1.74, SD=1.81) years of PET-PIB, and within 7.87 years (M=1.22, SD=1.00) of Cognitive assessment. Continuous variables were compared using t-tests and categorical variables were compared using a chi-square test.
= p<.05.
Measurement of physical exercise.
Validity.
History of engaging in a walking, running, and jogging exercise program for the past 10 years was assessed with a validated questionnaire to estimate physical exercise [21]. The measure was significantly correlated with cardiorespiratory fitness measured via treadmill test in a sample of 5,063 individuals aged 18 to 80 years (r=.40-.61). Retrospective self-report of activity for a particular year and aerobic fitness for that year across 10 1-year assessment periods evidenced stable correlations, indicating that participants across the examined age range were capable of relatively accurate self-report over this extended time span [21].
Procedure.
The questionnaire was administered by telephone with participants reporting number of months per year, number of workouts per week, average number of miles per workout, and average time per mile for each year they engaged in a walking, running, or jogging program. Metabolic equivalent (MET) values were estimated using the compendium of physical activities to derive the physical exercise score [22; 21]. The average MET-hours/week over each of the past 10 1-year assessment periods was used as the index of physical exercise. Participants were divided into low and high exercise groups by American Heart Association criterion for moderate exercise levels (i.e., 7.5 MET-hrs, which corresponds to moderate intensity activity 5 days/week for 30 minutes).
Cerebrospinal fluid collection and processing.
Cerebrospinal fluid (20–30mL) was acquired by lumbar puncture at 8am after an overnight fasting period, as described previously [23]. Samples were gently inverted to avoid possible gradient effects, centrifuged at low speed, and aliquoted (0.5mL) into propylene tubes before freezing at −84°C. Levels of total tau, phosphorylated tau181 (ptau181), and Aβ42 were analyzed after a single thaw following initial freezing by enzyme-linked immunosorbent assay (Innotest; Fujirebio [formerly Innogenetics], Ghent, Belgium]. A single assay lot number was used, and longitudinal samples from a given individual were run on the same assay plate.
In vivo amyloid imaging with PET-PIB.
In vivo amyloid imaging with positron emission tomography (PET) with [11C]PiB was performed as described previously [24]. Simultaneously with the initiation of a 60-minute dynamic PET scan in 3-dimensional mode, approximately 12mCi of [11C]PiB was administered intravenously. Measured attenuation factors and a ramp filter were used to reconstruct dynamic PET images. Three-dimensional regions of interest were created based on individual’s structural MRI scans. To account for number of binding sites in expressing regional binding values, a binding potential for each region of interest was calculated. Mean Cortical Binding Potential (MCBP) value was obtained by averaging the binding potential values from the prefrontal cortex, gyrus rectus, lateral temporal, and precuneus regions of interest. Higher MCBP values represent greater presence of amyloid pathology.
Cognitive assessment.
The episodic memory domain consisted of the free recall score from the Free and Cued Selective Reminding Test [25], Logical Memory Delayed from the Wechsler Memory Scale (WMS)-R [26] or the WMS-III [27], and Associate Learning/Verbal Paired Associates from the WMS [28] or WMS-III [28]. The processing speed domain consisted of Trail Making A and Wechsler Adult Intelligence Scale-Revised Digit Symbol test [29]. The executive functions domain consisted of WMS Digit Span Forward and Backward [28], the difference between Trailmaking B and Trailmaking A scores, and WMS-III Letter-Number Sequencing [27]. The verbal fluency composite was created from Category Fluency (animals and vegetables) and Letter Word Fluency (F & L) tests.
In the case of multiple versions of a test (i.e., Logical Memory and Associate Learning/Verbal Paired Associates), raw scores from each subsample were standardized separately and then combined across subsamples. Cognitive composites were standardized to each visit independently and represent the average of available standardized scores from each of the tests comprising the composite.
APOE and BDNF genotyping.
Detailed procedures for genotyping have been described previously [30]. Briefly, DNA samples were genotyped using Illumina genotyping arrays. All samples and genotypes underwent quality control before the analysis. The BDNF p.Val66Met SNP (rs6265) genotype was extracted from the imputed PLINK file and coded as 0 (Val/Val homozygotes) or 1 (Met allele carriers). APOE genotyping for both rs429358 (ABI#C_3084793_20) and rs7412 (ABI#C_904973_10) was done with TaqMan assays as described previously [31]. APOE genotype was coded as 0 (ε4 non-carrier) or 1 (ε4 carrier).
Statistical analyses.
Covariates.
Baseline age, education, sex, delay between baseline exercise and the first measurement of the outcome variable, CDR change, and health composite were covariates. The CDR change variable coded for any increase in CDR over time from baseline CDR=0 (CSF sample: N=3; PET-PIB sample: N=23, Cognitive sample: N=86) versus no change in CDR. Health composite represented accumulated count of current or past instances of: stroke, diabetes, seizures, traumatic brain injury, hypertension, Huntington’s disease, Parkinson’s disease, cardiovascular disease, and depression.
Linear mixed effects models.
A series of linear mixed-effects models were performed using the R nlme package [32] in the R statistical software [33]. The outcome measures were CSF Aβ42, CSF tau, CSF ptau181, MCBP, and cognitive composites. A full model included time (number of years between baseline and most recent follow-up), exercise group, and APOE (or BDNF) genotype, as well as all possible interaction terms. Time and intercept were random effects. In the next step, the highest order interaction term (i.e., three-way interaction between time, exercise group, and APOE (or BDNF) genotype) was excluded from the model. A likelihood ratio test determined whether models with and without the three-way interaction were significantly different. If models were not significantly different based on Chi-square (p<0.05), the term was dropped, otherwise, it was retained in the model. This was done for all three two-way interactions as well. Non-significant terms were excluded in a stepwise manner until a final model included significant higher-order terms and/or lowest-order terms.
Outliers.
Case-level outliers for outcomes were defined as residuals of the full model that were three standard deviations above or below the mean, and examined in terms of influence on model results. For outliers with Cook’s D value greater than 4/n, separate models were conducted with and without outliers. Results were similar unless otherwise noted in the Results section.
Results
Primary analyses: AHA-defined exercise groups.
Cross-sectional.
APOE ε4-positive individuals had lower baseline CSF Aβ42 (p=.002), as well as higher baseline CSF tau (p=.004), CSF ptau181 (p=.033) and MCBP (p<.001; Table 2; Figure 1A–D). Furthermore, there was a significant BDNF × exercise interaction for MCBP (p=.022; Supplementary Figure 1). Val homozygotes in the high exercise group had lower MCBP than those in the low exercise group (p=.046), but there was no effect of exercise for p.Met66-carriers (p=.081). Lastly, the high exercise group had better verbal fluency performance (p=.008; Table 3; Figure 2A–D).
Table 2.
Parameter estimates for AD biomarker outcomes: AHA exercise groups.
| Effect | APOE: Aβ42 | APOE: tau | APOE: Ptau181 | APOE: PET-PIB | BDNF: Aβ42 | BDNF: tau | BDNF: Ptau181 | BDNF: PET-PIB |
|---|---|---|---|---|---|---|---|---|
| Time | −35.4 (4.7)* | 5.4 (1.8)* | .9 (.3)* | .004 (.002)* | −35.3 (4.7)* | 8.3 (1.5)* | 1.4 (.2)* | .009 (.002)* |
| Exercise group | −48.1 (80.1) | −35.6 (30.1) | −2.0 (5.1) | −.02 (.03) | −3.0 (83.6) | −45.2 (30.3) | −3.1 (4.9) | −.07 (.03) |
| APOE or BDNF Genotype | −241.5 (75.4)* | 88.0 (29.8)* | 10.7 (5.0)* | .10 (.03)* | −62.9 (78.5) | 20.9 (28.5) | 5.6 (4.7) | .03 (.03) |
| CDR change | 254.6 (216.4) | 132.5 (80.0) | 16.6 (13.6) | .11 (.04)* | 252.9 (227.9) | 121.2 (81.1) | 14.2 (13.4) | .14 (.04)* |
| Sex | 39.4 (84.5) | 28.1 (31.5) | 7.0 (5.3) | −.04 (.03) | 87.8 (88.4) | 17.6 (31.6) | 5.9 (5.2) | −.05 (.03) |
| Age | −6.6 (5.0) | 6.1 (1.9)* | .7 (.3)* | .002 (.001) | −6.4 (5.2) | 6.1 (1.9)* | .8 (.3)* | .002 (.001) |
| Education | 14.0 (15.2) | −8.0 (5.7) | −1.2 (.9) | .002 (.004) | 20.3 (15.9) | −9.5 (5.7) | −1.4 (.9) | .002 (.005) |
| Health composite | −53.1 (38.3) | 10.0 (14.5) | 2.4 (2.4) | .0004 (.01) | −35.2 (39.9) | 4.6 (14.7) | 1.9 (2.4) | −.001 (.01) |
| Exercise - 1st Outcome Delay | 7.0 (30.4) | 2.3 (12.4) | −1.2 (2.0) | −.006 (.008) | 9.1 (32.2) | 1.3 (12.9) | −1.6 (2.1) | −.005 (.009) |
| Time × Exercise | --- | --- | --- | --- | --- | --- | --- | --- |
| Exercise × Genotype | --- | --- | --- | --- | --- | --- | --- | .14 (.06)* |
| Time × APOE Genotype | --- | 7.9 (2.9)* | 1.1 (.5)* | .02 (.004)* | --- | --- | --- | --- |
| Time × Exercise × Genotype | --- | --- | --- | --- | --- | --- | --- | --- |
Notes: Data represent parameter estimates (standard errors) for the final model;
= p<.05,
= p<.06; --- = not included in final model.
Figure 1.
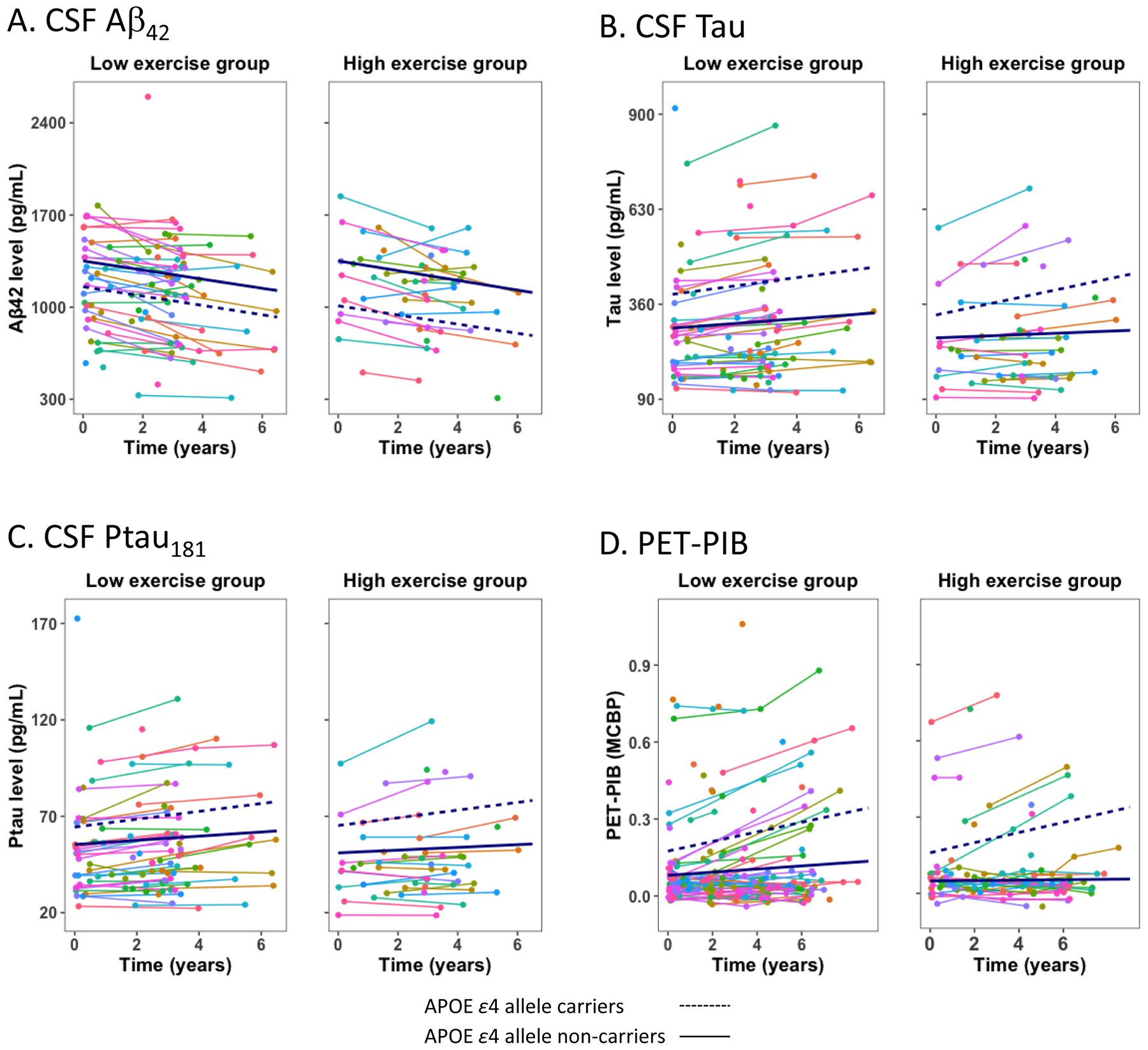
Exercise engagement and AD biomarkers. Spaghetti plots of individual trajectories. Plots depict regression lines for low and high exercise groups defined by AHA criteria for moderate exercise levels.
Table 3.
Parameter estimates for cognitive outcomes: AHA exercise groups.
| Effect | APOE: Verbal Fluency | APOE: Episodic Memory | APOE: Processing Speed | APOE: Executive Functions | BDNF: Verbal Fluency | BDNF: Episodic Memory | BDNF: Processing Speed | BDNF: Executive Functions |
|---|---|---|---|---|---|---|---|---|
| Time | −.04 (.007)* | −.02 (.008)* | −.08 (.01)* | −.02 (.006)* | −.04 (.007)* | −.03 (.007)* | −.08 (.01)* | −.02 (.006)* |
| Exercise group | .23 (.09)* | .05 (.08) | .06 (.10) | .05 (.08) | .21 (.09)* | .05 (.08) | .05 (.10) | .03 (.08) |
| APOE or BDNF Genotype | −.02 (.09) | −.02 (.10) | −.04 (.10) | .001 (.08) | −.02 (.09) | −.002 (.08) | −.03 (.10) | .02 (.08) |
| CDR change | −.84 (.10)* | −1.25 (.09)* | −.54 (.11)* | −.56 (.09)* | −.87 (.10)* | −1.28 (.09)* | −.45 (.11)* | −.57 (.09)* |
| Sex | .27 (.09)* | .35 (.08)* | .23 (.10)* | .15 (.09) | .26 (.09)* | .37 (.08)* | .21 (.11)* | .10 (.09) |
| Age | −.009 (.005) | −.001 (.005) | .006 (.006) | −.007 (.005) | −.007 (.005) | .001 (.005) | .01 (.006) | −.005 (.005) |
| Education | .08 (.02)* | .08 (.01)* | .05 (.02)* | .06 (.01)* | .08 (.02)* | .08 (.01)* | .05 (.02)* | .06 (.01)* |
| Health composite | −.02 (.04) | .001 (.03) | −.09 (.04)* | −.01 (.04) | −.02 (.04) | .02 (.03) | −.09 (.04)* | −.02 (.04) |
| Exercise - 1st Outcome Delay | .03 (.04) | −.11 (.04)* | −.02 (.06) | .06 (.04) | .03 (.05) | −.09 (.04)* | −.02 (.06) | .04 (.04) |
| Time × Exercise | --- | --- | .04 (.02)* | --- | --- | --- | .04 (.02)* | --- |
| Exercise × Genotype | --- | --- | --- | --- | --- | --- | --- | --- |
| Time × Genotype | --- | −.03 (.01)* | --- | --- | --- | --- | --- | --- |
| Time × Exercise × Genotype | --- | --- | --- | --- | --- | --- | --- | --- |
Notes: Data represent parameter estimates (standard errors) for the final model;
= p<.05,
= p<.06; --- = not included in final model.
Figure 2.

Exercise engagement and cognitive decline. Spaghetti plots of individual trajectories. Plots depict regression lines for low and high exercise groups defined by AHA criteria for moderate exercise levels.
Longitudinal.
Levels of CSF Aβ42 significantly decreased over time (p<.001), while levels of MCBP (p=.041), CSF tau (p=.004), and ptau181 (p=.002) significantly increased, consistent with disease progression. Significant decline in performance was also observed for all cognitive outcomes (verbal fluency: p<.001; episodic memory: p<.020; processing speed: p<.001; executive functions: p<.001). In addition, APOE ε4-positive individuals exhibited greater change over time compared to APOE ε4-negative individuals in terms of CSF tau (p=.009), CSF ptau181 (p=.021), MCBP (p<.001) and episodic memory (p=.026). Lastly, the only interaction involving time and exercise was for processing speed (p=.032). The high exercise group evidenced less decline (p=.009) compared to the low exercise group (p<.001).
Secondary analyses.
Age moderation.
Age did not significantly moderate effects of AHA-defined exercise group on longitudinal trajectories of cognitive performance or AD biomarkers (ps>.241 for the age × time × exercise interactions).
4-year delay subsample analyses.
Considering the long delay between baseline exercise and baseline measures of the outcome variables in the full samples (see Table 1), analyses were conducted on subsamples with a maximum of 4-years and with a maximum of 2-years between the baseline measures. Results were similar to the full sample AHA analyses in the 4-year delay subsample analyses (Supplementary Table 1 for sample characteristics) with one exception (Supplementary Tables 2, 3 and 10). The BDNF × exercise interaction for processing speed was significant with outliers removed (p=.048). There was a trend for p.Met66-carriers in the high exercise group to have higher processing speed performance compared to p.Met66-carriers in the low exercise group (p=.082), but there was no effect of exercise for the Val homozygotes (p=.470).
2-year delay subsample analyses.
In the 2-year delay subsample (Supplementary Table 4 for sample characteristics), results were similar to the full sample AHA analyses with three exceptions (Supplementary Tables 5, 6 and 10; Supplementary Figures 2–3). The main effect of exercise on verbal fluency (p=.107) and the APOE × time interaction for episodic memory (p=.063) were no longer significant. Finally, the time × exercise interaction for processing speed was significant only with outliers removed (p=.032).
Median split analyses.
In order to ensure roughly equal number of participants in exercise groups and thus enhance power, analyses were conducted with groups determined by a median split in the full sample (Supplementary Table 7 for sample characteristics). The median split values roughly correspond to engaging in moderate intensity activity 3 days/week for 31–39 minutes on average. Results were similar to primary analyses with AHA-defined groups with two exceptions (Supplementary Tables 8, 9 and 10; Supplementary Figures 4–5). The high exercise group had lower MCBP than the low exercise group (p=.043), which became non-significant with outliers removed (p=.113). In addition, there was an APOE × time × exercise group interaction for processing speed (p=.004). In APOE ε4-negative individuals, the high exercise group showed a less steep decline compared to the low exercise group (p=.004), whereas trajectories between exercise groups were not different in APOE ε4-positive individuals (p=.154).
Summary.
First, the secondary analyses revealed some additional, but inconsistent, cross-sectional effects of exercise group (i.e., BDNF × exercise for processing speed in 2-year subsample analyses; main effect of exercise for MCBP in the median split analyses). Second, the BDNF × exercise interaction for MCBP was observed in all analyses (Supplementary Figure 1, but the nature of the interaction varied (i.e., Val homozygotes benefiting from exercise in primary and median split analyses; p.Met66-carriers evidencing higher MCBP in the high exercise condition in 4-year and 2-year subsample analyses). Third, the time × exercise interaction for processing speed observed in the main analyses generally remained consistently present. Lastly, there was one indication that APOE genotype moderated the effects of exercise on the longitudinal trajectory of processing speed in the median split analyses.
Discussion
Longitudinal change was observed for all outcomes consistent with accumulation of AD pathology and cognitive decline. However, in the primary analyses, exercise only modulated change over time for processing speed. A series of secondary analyses were conducted to potentially provide insight into the minimal exercise effects in primary analyses and determine the robustness of observed effects. Overall, trajectories were not consistently affected by baseline exercise, with the exception of processing speed again. Furthermore, APOE and BDNF genotypes did not consistently modulate the relationship between exercise and rate of change.
The most consistent observation was that exercise modulated the rate of decline in processing speed, but with only one robust indication that APOE genotype influenced the effect of exercise. The limited current findings are in contrast with meta-analyses of longitudinal studies indicating higher levels of aerobic fitness or physical activity at baseline are associated with reduced risk of cognitive decline [3; 4], with effect sizes in the low-to-moderate range. The discrepant results might stem from differences in study design. Existing literature included in meta-analyses has generally examined categorical outcomes (i.e., decline vs no decline) at one follow-up and primarily used the MMSE. In contrast, the current study incorporated multiple time points as well as broad and robust estimation of specific cognitive domains. In fact, results have been mixed in the few studies that have actually examined cognitive trajectories in specific domains, with sample sizes ranging from 91 to over 10,000, follow-up duration ranging from 6–15 years and small effects in the studies with significant findings [5; 34; 35]. Notably, even a meta-analysis of intervention trials indicated modest beneficial effects on cognitive performance [36]. Overall, effects of physical activity on cognitive trajectories may be small and variable, and a meta-analysis on trajectories may be helpful in providing insight into important moderators of any protective effects of physical activity. Multimodal interventions incorporating exercise may be most effective [37].
The cross-sectional effect of exercise for the PET-based measure of amyloid deposition was only present in median split analyses, and there were no effects for the CSF-based variables. Furthermore, APOE status did not moderate associations. These findings are inconsistent with past work with a subsample of current participants [9; 11]. In fact, recent narrative reviews reveal mixed results in terms of exercise associations with either PET or CSF biomarkers cross-sectionally, or moderation by APOE status [1; 38]. It is conceivable that more objective measures of physical activity or aerobic fitness would be more sensitive than the self-report questionnaires used in most work. One study that incorporated accelerometry-measured physical activity observed associations of moderate intensity activity with CSF Aβ42, tau/Aβ42 and ptau181/Aβ42 [8]. However, one study that incorporated aerobic fitness measured by a graded maximum exercise test did not observe associations with PET or CSF measures [39]. Again, meta-analysis is needed to determine overall effect size as well as critical moderators of cross-sectional associations of physical activity or exercise with AD biomarkers.
There were no indications of exercise modulating longitudinal decline in AD biomarkers. One prior longitudinal examination observed that higher physical activity at baseline had beneficial associations with plasma Aβ42 at both 9-year and 13-year follow-ups [15]. The studies differ in several potentially important ways. The Stillman [15] study had a longer follow-up period than current work, but did not directly examine rate of change. Furthermore, plasma Aβ42 was examined, rather than CSF or PET. Lastly, leisure time physical activity over a 1-week period was the predictor variable. Given these differences in the limited work thus far, it is difficult to draw conclusions regarding any protective effects of physical activity or exercise on AD biomarkers over the long-term. Notably, one intervention trial failed to find an effect of 16 weeks of exercise on CSF Aβ42, tau or ptau181 in individuals with mild AD [40]. Overall, although animal studies provide support for an exercise effect on core AD pathology, results from human studies are inconsistent and inconclusive with limited examination of factors that may moderate any protective effects [1].
The inconsistency of the cross-sectional interactive BDNF genotype and exercise effects can be considered in the context of variability in the nature of the interactions observed in literature. That is, some studies observed a differential benefit of physical activity for Val carriers [e.g., 19], whereas others noted a particular benefit for p.Met66 carriers [e.g., 18]. This lack of consistency could in part reflect unreliability of the underlying phenomenon. However, the literature is relatively sparse in this domain. This highlights the need for further examination with objective measures of exercise, consideration of potential moderators such as age, and sufficiently large samples that permit cross-validation to systematically assess the robustness of potentially small interactive effects.
Strengths of this study include use of a validated measure of long-term exercise, incorporation of both CSF and PET measures of AD pathology, broad and robust measures of cognitive functioning, and multiple time points for all outcome measures to estimate rates of change. There are also several limitations that should be considered. Firstly, exercise was assessed via a self-report measure that is significantly but not perfectly correlated with cardiorespiratory fitness. In addition, exercise was only assessed at one time point prior to the outcome measures, hence, it is possible that participants changed their exercise behavior in the years following baseline measurements. Lastly, the long-term beneficial effect of exercising on rates of change might not be large enough to be detected with our sample size. Due to these limitations and the large number of examined models, even significant results reported should be interpreted with caution.
In summary, our results suggest that self-reported physical exercise may not be a robust moderator of longitudinal trajectories of AD biomarkers and cognitive functioning, except perhaps for processing speed. Neither APOE nor BDNF genotypes had consistent influence on this main finding. Future observational studies should repeatedly measure exercise objectively while following people longitudinally to obtain information about exercise behavior, AD pathology and cognitive functioning over time. Additionally, examining the effect of exercise in conjunction with other moderators could provide knowledge about individual and additive contribution of different factors to protection against development of AD pathology. Ultimately, large clinical trials with multiple time points over long follow-up periods, preferably in the preclinical AD phase, are needed to demonstrate whether exercise may actually reduce cognitive decline by directly modulating core AD pathology.
Supplementary Material
Supplementary Figure 1. Cross-sectional interaction between BDNF genotype and exercise group for PET-PIB. Error bars represents standard error. A) There was a greater benefit of exercise on MCBP for Val homozygotes (p=.046) than for Met carriers (p=.081; BDNF × exercise interaction, p=.022). B) There was a greater benefit of exercise on MCBP for Met carriers (p=.053) than for Val homozygotes (p=.122; BDNF × exercise interaction, p=.019). C) There was a greater benefit of exercise on MCBP for Met carriers (p=.009) than for Val homozygotes (p=.255; BDNF × exercise interaction, p=.017). D) There was a greater benefit of exercise on MCBP for Val homozygotes (p=.005) than for Met carriers (p=.542; BDNF × exercise interaction, p=.028).
Supplementary Figure 2. Exercise engagement and AD biomarkers: 2-year baseline-outcome delay subsamples. Plots depict regression lines for low and high exercise groups defined by AHA criteria for moderate exercise levels. Data for PET-PIB represent results with outliers removed.
Supplementary Figure 3. Exercise engagement and cognitive decline: 2-year baseline-outcome delay subsamples. Plots depict regression lines for low and high exercise groups defined by AHA criteria for moderate exercise levels. Data for processing speed represent results with outliers removed.
Supplementary Figure 4. Exercise engagement and AD biomarkers: median split groups. Plots depict regression lines for low and high exercise groups defined by median split.
Supplementary Figure 5. Exercise engagement and cognitive decline: median split groups. Plots depict regression lines for low and high exercise groups defined by median split. The APOE × time × exercise interaction was significant for processing speed (p=.004), but not for verbal fluency (p=.081), episodic memory (p=.200) or executive functions (p=.333).
Acknowledgements
Funding for this study was provided by the National Institute on Aging R01AG043434, P50AG005681, P01AG003991, P01AG026276; UL1TR000448; P30NS098577; R01EB009352; the Barnes-Jewish Hospital Foundation, and the Charles and Joanne Knight Alzheimer Research Initiative of the Washington University Knight Alzheimer Disease Research Center (ADRC). The authors thank the participants, investigators, and staff of the Knight ADRC, the Clinical Core for participant assessments, the Genetics Core for genotyping, the Biomarker Core for CSF analysis and the Imaging Core for PET analysis.
Anne M Fagan is supported by NIH grants including P50AG005681, P01AG003991, P01AG026276 and UF01AG03243807. She is on the Scientific Advisory Boards for Roche Diagnostics, AbbVie and Genentech and consults for Araclon Biotech/Griffols, Biogen, and DiamiR.
John C Morris is funded by NIH grants # P50AG005681; P01AG003991; P01AG026276 and UF1AG032438. Neither Dr. Morris nor his family owns stock or has equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company.
Tammie L Benzinger is funded by NIH grants P01AG003991, P01AG003991, P01AG026276, P50AG005681, UF1AG032438, U01AG042791, RF1AG053550, R01NS095773, R01AG056466, R01AG053548, R01AG053503, R01AG057536, R01AG052550, R01AG046179, P01AG052350, R01AG053267, U01AG059798, and R56AG061900. She is an investigator in clinical trials sponsored by Avid Radiopharmaceuticals, Eli Lilly, Roche, and Biogen.
Jason Hassenstab is funded by NIH grants including R01AG057840; P50AG005681; P01AG003991; P01AG026276 and UF1AG032438. He serves on scientific advisory boards for Lundbeck, Biogen, and Takeda and serves on a Data Safety and Monitoring Board for Eisai.
Carlos Cruchaga is funded by NIH# R01AG044546, P01AG003991, RF1AG053303, RF1AG058501, and U01AG058922. CC receives research support from Biogen, EISAI, Alector and Parabon, and is a member of the advisory board of ADx Healthcare, Halia Terapeutics and Vivid Genomics
References
- [1].Brown BM, Peiffer J, & Rainey-Smith SR (2019). Exploring the relationship between physical activity, beta-amyloid and tau: A narrative review. Ageing research reviews. [DOI] [PubMed] [Google Scholar]
- [2].Hillman CH, Erickson KI, & Kramer AF (2008). Be smart, exercise your heart: exercise effects on brain and cognition. Nature Reviews Neuroscience, 9(1), 58–65. [DOI] [PubMed] [Google Scholar]
- [3].Blondell SJ, Hammersley-Mather R, & Veerman JL (2014). Does physical activity prevent cognitive decline and dementia?: A systematic review and meta-analysis of longitudinal studies. BMC public health, 14(1), 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sofi F, Valecchi D, Bacci D, Abbate R, Gensini GF, Casini A, & Macchi C (2011). Physical activity and risk of cognitive decline: a meta‐analysis of prospective studies. Journal of internal medicine, 269(1), 107–117. [DOI] [PubMed] [Google Scholar]
- [5].Pizzie R, Hindman H, Roe C, Head D, Grant E, Morris JC, & Hassenstab JJ (2014). Physical activity and cognitive trajectories in cognitively normal adults: the adult children study. Alzheimer disease and associated disorders, 28(1), 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sunderland T, Linker G, Mirza N, Putnam KT, Friedman DL, Kimmel LH, … & Bartko JJ (2003). Decreased β-amyloid1–42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. Jama, 289(16), 2094–2103. [DOI] [PubMed] [Google Scholar]
- [7].Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, … & Avsen B (2004). Imaging brain amyloid in Alzheimer’s disease using the novel PET tracer, PIB. Annals of Neuroogyl, 55, 781–789. [DOI] [PubMed] [Google Scholar]
- [8].Law LL, Rol RN, Schultz SA, Dougherty RJ, Edwards DF, Koscik RL, … Okonkwo OC (2018). Moderate intensity physical activity associates with CSF biomarkers in a cohort at risk for Alzheimer’s disease. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring, 10, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liang KY, Mintun MA, Fagan AM, Goate AM, Bugg JM, Holtzman DM, … Head D (2010). Exercise and Alzheimer’s disease biomarkers in cognitively normal older adults: Exercise and AD Biomarkers. Annals of Neurology, 68(3), 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brown BM, Peiffer JJ, Taddei K, Lui JK, Laws SM, Gupta VB, … & Rainey-Smith SR (2013). Physical activity and amyloid-β plasma and brain levels: results from the Australian Imaging, Biomarkers and Lifestyle Study of Ageing. Molecular psychiatry, 18(8), 875–882. [DOI] [PubMed] [Google Scholar]
- [11].Head D, Bugg JM, Goate AM, Fagan AM, Mintun MA, Benzinger T, … Morris JC (2012). Exercise Engagement as a Moderator of the Effects of APOE Genotype on Amyloid Deposition. Archives of Neurology, 69(5), 636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Landau SM, Marks SM, Mormino EC, Rabinovici GD, Oh H, O’Neil JP, … Jagust WJ (2012). Association of lifetime cognitive engagement and low β-Amyloid Deposition. Archives of Neurology, 69(5), 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Okonkwo OC, Schultz SA, Oh JM, Larson J, Edwards D, Cook D, … Sager MA (2014). Physical activity attenuates age-related biomarker alterations in preclinical AD. Neurology, 83(19), 1753–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Roberts RO, Lowe VJ, … Jack CR (2012). Effect of lifestyle activities on Alzheimer disease biomarkers and cognition. Annals of Neurology, 72(5), 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stillman CM, Lopez OL, Becker JT, Kuller LH, Mehta PD, Tracy RP, & Erickson KI (2017). Physical activity predicts reduced plasma β amyloid in the Cardiovascular Health Study. Annals of clinical and translational neurology, 4(5), 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gómez-Pinilla F, Ying Z, Roy RR, Molteni R, & Edgerton VR (2002). Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. Journal of neurophysiology, 88(5), 2187–2195. [DOI] [PubMed] [Google Scholar]
- [17].Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, … Weinberger DR (2003). The BDNF val66met Polymorphism Affects Activity-Dependent Secretion of BDNF and Human Memory and Hippocampal Function. Cell, 112(2), 257–269. [DOI] [PubMed] [Google Scholar]
- [18].Brown BM, Castalanelli N, Rainey-Smith SR, Doecke J, Weinborn M, Sohrabi HR, … & Peiffer JJ (2019). Influence of BDNF Val66Met on the relationship between cardiorespiratory fitness and memory in cognitively normal older adults. Behavioural Brain Research, 362, 103–108. [DOI] [PubMed] [Google Scholar]
- [19].Thibeau S, McFall GP, Wiebe SA, Anstey KJ, & Dixon RA (2016). Genetic factors moderate everyday physical activity effects on executive functions in aging: Evidence from the Victoria Longitudinal Study. Neuropsychology, 30(1), 6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bugg JM, & Head D (2011). Exercise moderates age-related atrophy of the medial temporal lobe. Neurobiology of Aging, 32(3), 506–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bowles HR, FitzGerald SJ, Morrow JR Jr, Jackson AW, & Blair SN (2004). Construct validity of self-reported historical physical activity. American Journal of Epidemiology, 160(3), 279–286. [DOI] [PubMed] [Google Scholar]
- [22].Ainsworth BE, Haskell WL, Whitt MC, Irwin ML, Swartz AM, Strath SJ, … Emplaincourt PO (2000). Compendium of physical activities: an update of activity codes and MET intensities. Medicine and Science in Sports and Exercise, 32(9; SUPP/1), S498–S504. [DOI] [PubMed] [Google Scholar]
- [23].Fagan AM, Younkin LH, Morris JC, Fryer JD, Cole TG, Younkin SG, & Holtzman DM (2000). Differences in the Aβ40/Aβ42 ratio associated with cerebrospinal fluid lipoproteins as a function of apolipoprotein E genotype. Annals of Neurology, 48(2), 201–210. [PubMed] [Google Scholar]
- [24].Mintun MA, LaRossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, … Morris JC (2006). [11C]PIB in a nondemented population: Potential antecedent marker of Alzheimer disease. Neurology, 67(3), 446–452. [DOI] [PubMed] [Google Scholar]
- [25].Grober E, Buschke H, Crystal H, Bang S, & Dresner R (1988). Screening for dementia by memory testing. Neurology, 3, 900–903. [DOI] [PubMed] [Google Scholar]
- [26].Wechsler D (1987). Manual: Wechsler Memory Scale-Revised. San Antonio, Texas: Psychological Corporation. [Google Scholar]
- [27].Wechsler D (1997). Wechsler Memory Scale (3rd ed.): Administration and scoring manual. San Antonio, TX: Psychological Corporation. [Google Scholar]
- [28].Wechsler D, & Stone CP (1973). Manual: Wechsler Memory Scale. New York: Psychological Corporation. [Google Scholar]
- [29].Wechsler D (1981). WAIS-R manual: Wechsler adult intelligence scale-revised. San Antonio, Texas: Psychological Corporation. [Google Scholar]
- [30].Cruchaga C, Chakraverty S, Mayo K, Vallania FLM, Mitra RD, Faber K, … for the NIA-LOAD/NCRAD Family Study Consortium. (2012). Rare Variants in APP, PSEN1 and PSEN2 Increase Risk for AD in Late-Onset Alzheimer’s Disease Families. PLoS ONE, 7(2), e31039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cruchaga C, Kauwe JSK, Mayo K, Spiegel N, Bertelsen S, Nowotny P, … Goate AM (2010). SNPs Associated with Cerebrospinal Fluid Phospho-Tau Levels Influence Rate of Decline in Alzheimer’s Disease. PLoS Genetics, 6(9), e1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pinheiro J, Bates D, DebRoy S, Sarkar D and R Core Team (2018). nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1–137, https://CRAN.R-project.org/package=nlme. [Google Scholar]
- [33].RStudio Team (2015). RStudio: Integrated Development for R. RStudio, Inc., Boston, MA: URL http://www.rstudio.com/. [Google Scholar]
- [34].Hamer M, Terrera GM, & Demakakos P (2018). Physical activity and trajectories in cognitive function: English Longitudinal Study of Ageing. J Epidemiol Community Health, 72(6), 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sabia S, Dugravot A, Dartigues JF, Abell J, Elbaz A, Kivimäki M, & Singh-Manoux A (2017). Physical activity, cognitive decline, and risk of dementia: 28-year follow-up of Whitehall II cohort study. Bmj, 357, j2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Smith PJ, Blumenthal JA, Hoffman BM, Cooper H, Strauman TA, Welsh-Bohmer K, … & Sherwood A (2010). Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosomatic medicine, 72(3), 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, … & Lindström J (2015). A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. The Lancet, 385(9984), 2255–2263. [DOI] [PubMed] [Google Scholar]
- [38].Frederiksen KS, Gjerum L, Waldemar G, & Hasselbalch SG (2019). Physical Activity as a Moderator of Alzheimer Pathology: A Systematic Review of Observational Studies. Current Alzheimer Research, 16(4), 362–378. [DOI] [PubMed] [Google Scholar]
- [39].Schultz SA, Boots EA, Almeida RP, Oh JM, Einerson J, Korcarz CE, … & Bendlin BB (2015). Cardiorespiratory fitness attenuates the influence of amyloid on cognition. Journal of the International Neuropsychological Society, 21(10), 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jensen CS, Portelius E, Siersma V, Høgh P, Wermuth L, Blennow K, … & Simonsen AH (2016). Cerebrospinal fluid amyloid beta and tau concentrations are not modulated by 16 weeks of moderate-to high-intensity physical exercise in patients with Alzheimer disease. Dementia and geriatric cognitive disorders, 42(3–4), 146–158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Cross-sectional interaction between BDNF genotype and exercise group for PET-PIB. Error bars represents standard error. A) There was a greater benefit of exercise on MCBP for Val homozygotes (p=.046) than for Met carriers (p=.081; BDNF × exercise interaction, p=.022). B) There was a greater benefit of exercise on MCBP for Met carriers (p=.053) than for Val homozygotes (p=.122; BDNF × exercise interaction, p=.019). C) There was a greater benefit of exercise on MCBP for Met carriers (p=.009) than for Val homozygotes (p=.255; BDNF × exercise interaction, p=.017). D) There was a greater benefit of exercise on MCBP for Val homozygotes (p=.005) than for Met carriers (p=.542; BDNF × exercise interaction, p=.028).
Supplementary Figure 2. Exercise engagement and AD biomarkers: 2-year baseline-outcome delay subsamples. Plots depict regression lines for low and high exercise groups defined by AHA criteria for moderate exercise levels. Data for PET-PIB represent results with outliers removed.
Supplementary Figure 3. Exercise engagement and cognitive decline: 2-year baseline-outcome delay subsamples. Plots depict regression lines for low and high exercise groups defined by AHA criteria for moderate exercise levels. Data for processing speed represent results with outliers removed.
Supplementary Figure 4. Exercise engagement and AD biomarkers: median split groups. Plots depict regression lines for low and high exercise groups defined by median split.
Supplementary Figure 5. Exercise engagement and cognitive decline: median split groups. Plots depict regression lines for low and high exercise groups defined by median split. The APOE × time × exercise interaction was significant for processing speed (p=.004), but not for verbal fluency (p=.081), episodic memory (p=.200) or executive functions (p=.333).