

Summary
In mammalian genomes, a subset of genes is regulated by genomic imprinting, resulting in silencing of one parental allele. Imprinting is essential for cerebral cortex development, but prevalence and functional impact in individual cells is unclear. Here, we determined allelic expression in cortical cell types and established a quantitative platform to interrogate imprinting in single cells. We created cells with uniparental chromosome disomy (UPD) containing two copies of either the maternal or the paternal chromosome; hence, imprinted genes will be 2-fold overexpressed or not expressed. By genetic labeling of UPD, we determined cellular phenotypes and transcriptional responses to deregulated imprinted gene expression at unprecedented single-cell resolution. We discovered an unexpected degree of cell-type specificity and a novel function of imprinting in the regulation of cortical astrocyte survival. More generally, our results suggest functional relevance of imprinted gene expression in glial astrocyte lineage and thus for generating cortical cell-type diversity.
Keywords: cerebral cortex, genomic imprinting, uniparental chromosome disomy, mosaic analysis with double markers, genetic mosaic, astrocyte, scRNA-seq, cell type diversity, epigenetics, apoptosis
Graphical Abstract
Highlights
-
•
Uniform allelic expression of imprinted genes in major forebrain cell types
-
•
Cortical cell-type-specific expression levels of imprinted genes
-
•
Cell-type-specific transcriptional responses in uniparental chromosome disomy (UPD)
-
•
Cortical cell-type-specific phenotype in cells with UPD
Laukoter et al. analyze prevalence and functional impact of genomic imprinting, an epigenetic phenomenon resulting in the silencing of one parental allele, in cerebral cortex development at the single-cell level. They find a high degree of cell-type specificity and a novel function of imprinting in cortical astrocyte development.
Introduction
The cerebral cortex is composed of an extraordinary number of neuronal and glial cell types assembling into cortical circuits that account for cognitive abilities. Remarkable heterogeneity in the cortical cell types has been described (Ecker et al., 2017; Lein et al., 2017; Zeng and Sanes, 2017), yet the identity of neuronal classes is largely hardwired genetically (Lodato and Arlotta, 2015). The mechanisms generating cortical cell-type diversity are not well understood. However, efforts employing RNA sequencing (RNA-seq) at the single-cell level indicate that developmentally regulated transcriptional programs play critical roles in establishing the full spectrum of cortical cell fates (Mayer et al., 2018; Mi et al., 2018; Nowakowski et al., 2017; Telley et al., 2016, 2019).
The control of precise transcriptional programs establishing cortical cell fates includes epigenetic mechanisms (Amberg et al., 2019). For instance, DNA methylation represents a critical epigenetic mark modifying DNA-protein interactions and thus controlling transcriptional states and cellular identity (Albert et al., 2017; Gray et al., 2017; Luo et al., 2017). Although many DNA methylation regulatory mechanisms involve large-scale and global chromatin modulation, some cues act at highly specific locations. In particular, differential DNA methylation at imprinting control regions serves as a fundamental mechanism of genomic imprinting. Imprinting is an epigenetic phenomenon and results in monoallelic parent-of-origin-specific gene expression (Barlow and Bartolomei, 2014; Ferguson-Smith, 2011). Thus, certain genes are only expressed from the paternally inherited allele and others are only expressed from the maternally inherited allele. The most characteristic feature of imprinted genes is reflected in their cardinal gene-dosage sensitivity. Whether and how allelic expression, and therefore imprinted gene dosage, is regulated at the single-cell level and whether imprinting contributes mechanistically to the generation of transcriptional and/or phenotypic cell-type diversity are unknown.
Although the overall number of imprinted genes is relatively small (<1%) (Tucci et al., 2019; Williamson et al., 2013), many imprinted genes are prominently expressed during neural development and in the adult brain (Andergassen et al., 2017; Babak et al., 2015; Perez et al., 2015). The preferential expression of the maternal or the paternal allele of certain genes suggests widespread implications for the development and function of the brain. Indeed, genetic deletion of individual imprinted genes results in various neuronal and behavioral deficits (Perez et al., 2016; Peters, 2014; Wilkinson et al., 2007). However, many phenotypes with loss of imprinted gene function have been analyzed at the whole-animal and/or global tissue level. Thus, the functional role of imprinting, and therefore the regulated expression of imprinted gene dosage, at the individual-cell level is poorly understood (Barlow and Bartolomei, 2014; Chess, 2016; Huang et al., 2018; Perez et al., 2016; Tucci et al., 2019).
The investigation of genomic imprinting at the organismal level has vastly benefited from the analysis of mice carrying uniparental chromosome disomy (UPD, somatic cells containing either two copies of the maternal or paternal chromosome) (Cattanach and Kirk, 1985; Ferguson-Smith et al., 1991; Schulz et al., 2006). Because one parental allele is duplicated and the other is not present in cells carrying UPD, imprinted genes are in principle either 2-fold overexpressed or not expressed. Several imprinting phenotypes in mice, as well as certain human disorders, are due to UPD and resulting imbalances of imprinted gene expression (Peters, 2014; Yamazawa et al., 2010). Two prominent examples affecting the brain include Prader-Willi and Angelman syndromes (Buiting et al., 2016; Horsthemke and Wagstaff, 2008; Mabb et al., 2011). Cell-type-specific and/or allelic expression strength of imprinted genes could contribute to overall phenotype and clinical manifestation in conditions with deregulated imprinted gene expression in UPD (Buiting et al., 2016; Cassidy and Driscoll, 2009; Horsthemke and Wagstaff, 2008; LaSalle et al., 2015; Mabb et al., 2011). However, the lack of experimental approaches allowing the interrogation and phenotypic analysis upon deregulated imprinted gene expression at the single-cell level has thus far precluded the investigation of cell-type specificity.
Here, we first used single-cell RNA sequencing (scRNA-seq) to map and quantitatively assess allelic expression strength in genetically defined major forebrain cell types at single-cell resolution. We then exploited the potential of UPD and established a quantitative assay to probe genomic imprinting at unprecedented single-cell resolution in the developing cortex using MADM (mosaic analysis with double markers) technology (Hippenmeyer et al., 2010, 2013; Zong et al., 2005). By capitalizing upon the MADM assay, we determined the prevalence and phenotypic cell-type specificity of imprinted gene dosage in the developing cerebral cortex at the single-cell level.
Results
Analysis of Allelic Expression in Genetically Defined Major Forebrain Cell Types
Previous studies have established genome-wide allelic expression maps (allelomes) in many organs and tissues (Andergassen et al., 2017; Babak et al., 2015; Bonthuis et al., 2015; Gregg et al., 2010; Perez et al., 2015) using well-established genetic differences (single-nucleotide polymorphisms, SNPs) in the F1 generation of crosses between distinct mouse strains (Figure 1A). These efforts proved extremely useful to identify tissue-specific imprinted gene expression but lacked the cellular resolution to determine cell-type-specific allelic expression. To this end, we set out to first analyze the allelomes of genetically defined cell types compared with whole tissue. We focused on cortical projection neurons, interneurons, and olfactory bulb (OB) granule cells and crossed Emx1- and Nkx2.1-Cre drivers to fluorescent Z/EG and Ai14 reporter lines, respectively, all in the C57BL/6J (B6) genetic background. These B6-Cre/reporter mice were then crossed to CAST/EiJ (CAST) mice with the father in B6 and the mother in CAST (initial cross), or vice versa (reverse cross). We used 2 biological replicates for both crosses (Table S1A; Figure 1A). Next, labeled cells from F1 of the preceding crosses were isolated by fluorescence-activated cell sorting (FACS) followed by RNA-seq and allelic expression analysis using Allelome.PRO (Andergassen et al., 2015) to determine genome-wide allelic gene expression (Figure 1B). For global imprinted gene identification, we used a false discovery rate (FDR) cutoff of 1% and an allelic expression ratio cutoff of 0.7, indicating a 70/30 ratio of expressed/silent allele (Andergassen et al., 2017). To refine this definition, we separated genes showing canonical (allelic ratio cutoff of 0.95) and biased (allelic ratio cutoff between 0.95 and 0.7) imprinted expression (Figure 1A). We confirmed cell-type identity in our samples using principal-component analysis (Figure S1A) and marker gene expression (Figure S1B). To identify cell-type-specific differences in imprinted gene expression, we focused our analysis on 25 genes with imprinted expression in embryonic and adult whole mouse brain (Andergassen et al., 2017; Perez et al., 2015; Figure 1C). Most (20/25, or 80%) showed uniform canonical allelic expression (i.e., no switching of parental allele-specific expression) in all informative cell types, as well as in whole tissue (Figure 1D). We next plotted the allelic maternal expression/paternal expression (mat/pat) ratios for several representative maternally (Rian and Meg3) and paternally (Sgce and Snrpn) expressed imprinted genes (Figure 1E). Only 5/25 genes appeared to show biased imprinted expression (Ago2, Cdkn1c, Grb10, Impact, and Inpp5f). Of these 5 genes, Grb10 is known to switch promoter usage and thus imprinted expression developmentally and cell type specifically (Plasschaert and Bartolomei, 2015; Yamasaki-Ishizaki et al., 2007), and Cdkn1c shows almost exclusive imprinted expression with only one cell-type exception (OB, mat/pat ratio of 0.940 and cutoff of 0.95). Next, we investigated Ago2, Impact, and Inpp5f and found marked cell-type-specific variation in the allelic mat/pat ratios, contrasting with canonical imprinted expression (Figure 1E). In summary, most (80%) expressed imprinted genes exhibit canonical imprinting in all major, genetically defined, cortical cell types, with a smaller fraction (20%) showing expression bias.
Figure 1.

Uniform Allelic Expression of Imprinted Genes in Major Forebrain Cell Types
(A) Strategy for cell-type-specific allelic expression analysis. Left: overview of parental Z/EG;Emx1Cre/+ and Ai14;Nkx2.1-Cre+/− reporter in a B6 genetic background and CAST mouse strains. Images depict neocortex (CX), hippocampus (HC), and OB (insets) in experimental B6/CAST mice with labeling of Emx1+ (Z/EG;Emx1Cre/+) and Nkx2.1+ (Ai14;Nkx2.1-Cre+/−) cell lineages in overview and at higher magnification (boxed areas in overview) at P0. GFP expression (green) and tdT expression (red) are indicated. Nuclei were stained using DAPI (blue). Cortical layers are indicated (roman numerals). Scale bar: overview, 500 μm; magnification, 60 μm; OB insets, 600 μm. Middle: breeding scheme for generating F1 B6xCAST hybrids with expected SNPs in F1. Right: expected relative allelic SNP expression upon FACS and RNA-seq analysis for biallelically expressed genes (BAE), canonical imprinted genes, and genes with expression bias.
(B) Experimental strategy for the analysis of allelic expression in bulk samples from whole tissue, Emx1+, and Nkx2.1+ lineages from CX, HC, and OB at P0 using the Allelome.PRO pipeline.
(C) Heatmap showing allelic expression of 25 known imprinted genes in whole tissue (whole), Emx1+, and Nkx2.1+ cell types in CX, HC, and OB. mat (red); pat (blue); BAE (green); NI/B6, not informative or expression bias toward B6 allele (white). The mat and pat labeling within individual boxes indicates genes with consistently higher expression in that direction below the allelic ratio cutoff (0.7).
(D) Number of genes in (C) with biased expression or canonical imprinting.
(E) Allelic ratio (mat/pat) of genes with canonical imprinting (Sgce, Snrpn, Rian, and Meg3) or biased expression (Impact, Ago2, and Inpp5f).
(F) Experimental strategy for allelic expression analysis in single cells of Emx1+ lineage at P0 and P42 using the Allelome.PRO pipeline. Numbers of cells analyzed from B6xCAST and reciprocal CASTxB6 crosses are indicated.
(G) Heatmap displays expression of a representative set of marker genes for classification of individual cell types in the Emx1+ lineage. Cell types (columns) and genes (rows) were ordered arbitrarily after hierarchical clustering. Colored bars above the heatmap indicate different cell types: aIPC (pink), astros (purple), oligos (orange), NI (gray), NII (black).
(H) Allelic expression of selected genes in single cells in defined cell types: Ncam1 and Fgfr2, biallelic expression; Meg3, canonical mat; Snrpn, canonical pat; Inpp5f and Impact, biased pat. Numbers indicate informative cells.
See also Figure S1.
Allelic Imprinted Gene Expression in Cortical Cell Types at the Single-Cell Level
Biased imprinted gene expression can arise either from uniformly skewed expression in all cells within a population or from a major population showing exclusive imprinted expression from one parental allele and a minority population switching parental alleles and showing exclusive expression of the opposite parental allele (Chess, 2016; Huang et al., 2018; Perez et al., 2016). To discriminate between these possibilities we isolated single cells from the cortical Emx1+ lineage of F1 progeny from B6 and CAST parents, as described earlier, at two developmental time points, postnatal day (P) 0 and P42 using FACS (initial and reverse cross) (Figures 1F and S1C). Next, we performed scRNA-seq using SMARTer technology. Upon quality control, we identified 404 cells from both crosses (223 B6xCAST and 181 CASTxB6; the maternal strain is written on the left). We classified all informative cells into 5 classes using hierarchical clustering of gene expression (Figure 1G; Table S1B; STAR Methods). Clustering did not result in major bias with respect to the direction of B6xCAST cross in any class (Figure S1C). We separated neurons into two groups, with neuron I (NI, nascent projection neurons) and neuron II (NII, mature projection neurons) originating mainly from P0 and P42, respectively (Figure S1C). Astrocyte intermediate progenitor cells (aIPCs) were mainly observed at P0, whereas mature astrocytes (astros) and oligodendrocytes (oligos) were mostly identified at P42 (Figure S1C). Using a modified version of Allelome.PRO, we calculated allelic mat/pat ratios of the 25 known imprinted genes as described earlier and two control (i.e., biallelically expressed) genes (Ncam1 and Fgfr2) (STAR Methods). Our analysis revealed that the parental bias of all investigated imprinted genes was present at the single-cell level (Table S2). Importantly, biased paternal expression of Inpp5f and Impact at the single-cell level was detected in all major cell types (Figure 1H), similar to our observation at the bulk level (Figure 1E). In contrast, almost exclusive expression from the maternal or the paternal allele was detected in each informative cell for selected genes with canonical imprinted expression (maternal, Meg3 and Rian; paternal, Snrpn) (Figure 1H). Highly expressed genes that are not subject to genomic imprinting, e.g., Fgfr2 and Ncam1, were found to be expressed either from both parental alleles (i.e., biallelic, green bar in Figure 1H) or from one of the parental alleles in equal amounts of single cells (red/blue bars in Figure 1H), consistent with the observation and concept of transcriptional bursts (Larsson et al., 2019). In summary, we found uniform canonical imprinted gene expression across distinct cell types, which is in contrast to the idea of cell-type-specific variation of biased expression. Both canonical expression and biased expression of the respective analyzed imprinted genes were observed in all different cortical cell types with no detectable allele switching.
Quantitative Assessment of Imprinted Gene Expression Levels in Major Forebrain Cell Types
In the above analysis, we noticed that although relative ratios of allelic expression were rather uniform across cell types, absolute imprinted gene expression levels were not. Extreme examples included Rasgrf1 and Magel2, which were not informative in allelic expression analysis because of low expression in several (i.e., 2–4) cell types (Figure 1C, white boxes). These findings prompted us to comparatively investigate the expression levels of all 25 well-characterized imprinted genes listed earlier in all distinct cortical cell types. We first re-analyzed the bulk RNA-seq data of the preceding B6xCAST crosses (Figure 2A). We plotted the relative expression levels in a heatmap to reveal similarities and differences in expression profiles across specific cortical cell types (Figure 2B). This analysis indicated marked differences in the expression of most analyzed imprinted genes across distinct cell types. We next plotted the normalized read counts of Impact, which shows similar expression levels in all cortical cell types. In contrast, the normalized read counts of Meg3 and Grb10 revealed substantial differences of expression in distinct cortical cell types (Figure 2C). To corroborate these findings, we calculated a cell-type specificity index based on differential gene expression (bulk) (see STAR Methods). This analysis identified progressively increasing but significant cell-type-specific imprinted expression levels for 84% of the investigated 25 imprinted genes (Figure 2D). Next, we analyzed cell-type-specific expression of imprinted genes at the single-cell level and re-analyzed the data from scRNA-seq of the B6xCAST crosses (Figure 2E). Normalized expression of 20 informative imprinted genes, visualized in a heatmap, indicated that even in individual cells, imprinted gene expression varies strongly across distinct cortical cell types (Figure 2F). Furthermore, normalized expression values for three genes—Impact, similar expression, and Meg3 and Grb10, cell-type-specific expression—supported the preceding observation (Figure 2G). Based on the highest fraction of cumulative expression, we calculated a specificity index for single cells (single cell) (see STAR Methods). Strikingly, 13/20 genes showed significant differential expression among the 5 cortical cell types (Figure 2H, indicated with asterisks next to the gene name, Monocle2, adjusted p value (padj) < 0.05). Altogether, we found that imprinted genes with uniform canonical or biased allelic expression exhibit significant variation in absolute expression levels across cortical cell types.
Figure 2.

Imprinted Genes Show Distinct Expression Levels in Different Cortical Cell Types
(A) Overview of genetically defined cortical cell types that were analyzed in small bulk samples at P0.
(B) Heatmap depicting relative expression levels of imprinted genes in different cortical cell types.
(C) Normalized expression levels of three imprinted genes: Impact, Meg3, and Grb10. Note the uniform (Impact) but highly variable (Meg3 and Grb10) expression levels in distinct cell types.
(D) Cell-type specificity index (bulk), with a low specificity score indicating more uniformity and a high specificity score reflecting increasing variance of cell-type-specific expression levels.
(E) Overview of expression analysis at the single-cell level in aIPC (pink), astrocytes (purple), oligodendrocytes (orange), NI (gray), and NII (black).
(F) Heatmap depicting expression of 20 (informative) imprinted genes in distinct cell types.
(G) Expression levels of Impact, Meg3, and Grb10 in single cells.
(H) Cell-type specificity index (single cell) based on the highest fraction of cumulative expression.
MADM Can Generate UPD to Probe Genomic Imprinting at the Single-Cell Level
The preceding findings show that imprinted gene expression strength varies significantly across distinct cortical cell types. How relevant is the absolute expression of imprinted genes in a particular cell type? To address this question, it is imperative to modulate the expressed dose of imprinted genes in a cell-type-specific manner while maintaining endogenous transcriptional control of gene expression. Currently, the prime assay fulfilling the preceding criteria is UPD. We therefore set out to exploit the potential of UPD and established a quantitative assay to probe genomic imprinting at single-cell resolution in the developing cortex using MADM technology (Hippenmeyer et al., 2010, 2013; Zong et al., 2005; Figures 3A and S2). MADM can generate UPD via Cre/LoxP-dependent mitotic recombination at G2 phase in dividing stem cells, followed by X segregation of recombined chromosomes. G2-X events produce near-complete UPD of particular chromosomes carrying the MADM cassettes in genetically defined cell types (Hippenmeyer et al., 2013; Laukoter et al., 2020). As a consequence, imprinted genes located on such chromosomes are expected to be homozygosed and show imbalanced imprinted gene expression, i.e., either a 2-fold increase in expression or no expression (Schulz et al., 2006). MADM-induced UPD thus represents a unique functional assay with the possibility of both loss of function (LOF) and gain of function (GOF) of imprinted gene dose. Furthermore, cells with UPD can be visualized in vivo with distinct fluorescent colors (Figures 3A, S2, and S3A–S3L), e.g., maternal UPD (matUPD) in red (tdTomato [tdT+]) and paternal UPD (patUPD) in green (GFP+). Fluorescently labeled cells with UPD can be compared in situ to control cells (carrying one maternal and one paternal chromosome) labeled in yellow (tdT+/GFP+) in genetic mosaic animals (Figures 3A, S2, and S3A–S3L). In summary, our experimental paradigm provides a unique platform to systematically generate and isolate MADM-induced UPDs in genetically defined cell types and with single-cell resolution. Because MADM-induced UPDs exhibit predictable imbalances in imprinted gene expression, ensuing cell-type-specific phenotypes can be analyzed at unprecedented transcriptomic and cellular resolution.
Figure 3.

MADM-Induced UPD Results in Highly Cell-Type-Specific Transcriptional Responses
(A) MADM events generate UPD labeled in fluorescent green (patUPD) and red (matUPD) and yellow control cells. Predicted gene expression levels of mat (red) and pat (blue) canonical imprinted genes in MADM-labeled cells with UPD, compared with BAE (biallelically expressed gene; black). See also Figure S2.
(B) Isolation and analysis of distinct classes of genetically defined (Emx1+ and Nkx2.1+) MADM-labeled cells with UPD of chr7, chr11, or chr12 from developing CX, HC, and OB. Cells with MADM-induced UPDs were isolated by FACS, followed by RNA-seq and differential gene expression analysis.
(C) Relative expression, depicted as matUPD/patUPD fold change (log2), of imprinted genes in distinct cell types upon MADM-induced UPD. Ndn, paternally expressed on chr7; Grb10, maternally expressed on chr11; Meg3, maternally expressed on chr12. Significant differential expression in matUPD/patUPD is indicated (padj < 0.01, DESeq2, asterisks).
(D) Heatmap depicting the expression of select marker genes for excitatory projection neurons (Pax6 and Tbr1), inhibitory interneurons (Erbb4 and Dlx1), and olfactory granule cells (Dlx1).
(E) Hierarchical clustering based on global gene expression of all samples (n = 153), including matUPD, patUPD, and controls of chr7, chr11, and chr12. The tree structure indicates large differences between cell types but small differences between control and UPD samples within a cell type. Asterisks indicate selected 100% bootstrap probability of sample clustering. See also Figure S3.
(F) Principal-component analysis (PCA) of gene expression in Emx1+ and Nkx2.1+ cell types with matUPD or patUPD of chr7 and control in CX, HC, and OB. Data points reflect individual biological samples.
(G) Relative expression levels of imprinted genes (Cdkn1c, maternally expressed; Ndn and Snrpn, paternally expressed) in distinct cell types with matUPD or patUPD of chr7 and control. Expression values are shown relative to the mean expression in control cells for each cell type. Values above 3 were removed (see STAR Methods).
(H) Number of significantly DEGs (padj < 0.05, DESeq2) in matUPD/control (light gray) and patUPD/control (dark gray) in different cell types.
(I) Genomic location of DEGs on chr7. Dots (black) indicate individual genes; predicted imprinted domains are also indicated (gray).
(J) Fraction of DEGs located on chr7 (black) and all other chromosomes (white).
(K) Heatmap depicting scores for DEGs. Gene clusters are based on hierarchical clustering, and most significant terms for GO enrichment (p < 0.05, hypergeometric test) are indicated. See also Figure S4.
MADM-Induced UPD Reveals Cell-Type-Specific Transcriptional Responses to Imbalanced Imprinted Gene Expression
We focused our analysis of UPD on chromosome 7 (chr7), chromosome 11 (chr11), and chromosome 12 (chr12), which all harbor well-studied clusters of imprinted genes (Williamson et al., 2013). By using Emx1- and Nkx2.1-Cre drivers, we genetically targeted MADM-induced UPD to excitatory projection neurons and inhibitory interneurons in developing neocortex and hippocampus and to OB granule cells (Figures 3B and S3A–S3L). To validate our assay and assess the consequences of imbalanced imprinted gene expression in the previously listed cell types with UPD, we first analyzed their transcriptomes. Fluorescently labeled cells carrying distinct UPDs and control cells were isolated by FACS, followed by RNA extraction and library preparation for RNA-seq. We isolated between 1,000 and 10,000 cells, depending on MADM recombination efficiency (Figures S3A–S3L). For sequencing, we processed 2–4 biological replicates of forward crosses (matUPD in red and patUPD in green) and reverse crosses (matUPD in green and patUPD in red). A total of 153 samples were used for analysis (Table S1C; STAR Methods). We performed differential gene expression analysis and first analyzed the expression status of known imprinted genes. The expression of paternally expressed Ndn located on chr7, maternally expressed Grb10 located on chr11, and maternally expressed Meg3 located on chr12 displayed skewed expression in patUPD and matUPD (Figure 3C). These results validated our experimental approach, because both matUPD- and patUPD-mediated changes in imprinted gene expression occurred as predicted. Next, we determined whether the UPD-mediated change in imprinted gene dose influenced cell fate and identity. To this end, we determined the expression status of groups of select marker genes, which are characteristic of excitatory projection neurons (e.g., Pax6 and Tbr1) and inhibitory interneurons (e.g., Erbb4 and Dlx1) in neocortex and hippocampus and of granule cells in OB (e.g., Dlx1) (Figures 3D and S3M). We confirmed sample identity (i.e., cell type) but did not find bias in the expression state of the tested marker genes that correlated with UPD status. We also performed hierarchical clustering analysis of all sequenced samples and found that the tissue and genetic identity, but again not the state of UPD, defined significant clustering (asterisks in Figure 3E; STAR Methods). In summary, MADM-induced UPD of chr7, chr11, or chr12 did not affect cell-fate specification of cortical excitatory and inhibitory neurons based on marker gene expression. In contrast, UPD results in highly imbalanced expression of imprinted genes located on the respective chromosomes.
Even though the preceding experiments validated our experimental approach, we evaluated the effects of imbalanced imprinted gene expression in greater detail and greater depth. We thus sequenced a higher number of replicates and used SMARTer technology to remove potential bias from a varying number of cells present in each sample. We focused our analysis on chr7, because it carries a large number of imprinted genes, including some that have been shown to regulate cortical development (Amberg et al., 2019; Perez et al., 2016; Tucci et al., 2019; Williamson et al., 2013). Upon RNA-seq, we performed principal-component analysis of the 64 samples (Table S1D) that passed quality control (Figure 3F). We found that clustering predominantly resulted from cell-type identity and not UPD, reinforcing the preceding findings. Differential imprinted gene expression analysis (matUPD/patUPD) confirmed the allele-specific expression pattern (i.e., bias) of 15 imprinted genes on chr7 (Figure S4A). We also plotted the relative expression levels in cells with matUPD and patUPD and control cells for the following representative imprinted genes: Ndn, Cdkn1c, and Snrpn (Figure 3G).
To systematically assess the consequences of imbalanced imprinted gene expression, we monitored global transcriptional states as an initial proxy for cellular responses to UPD. We performed differential gene expression analysis between control and matUPD or patUPD samples. We found great variance among distinct cell types, with numbers of significantly differentially expressed genes (DEGs) ranging from 18 to 1,202 (Figure 3H, padj < 0.05, DESeq2). The transcriptional response to UPD included highly cell-type-specific components, because 23%–61% of DEGs were unique to a single cell type and/or UPD (Figure S4B). We found no significant enrichment of DEGs close to known imprinted regions on chr7 (Figure 3I, hypergeometric test), or on chr7, carrying the MADM cassettes (Figure 3J, hypergeometric test). Altogether, these findings indicate that MADM-induced UPD of chr7 results in genome-wide transcriptional responses, with their extent showing high (orders of magnitude) variability across cortical cell types.
Next, we plotted in a heatmap the differential expression score of 3,413 genes that were significantly differentially expressed in matUPD/control and/or patUPD/control comparisons (Figure 3K, padj < 0.05). We also performed hierarchical clustering of DEGs based on their differential expression score pattern (STAR Methods) and identified 10 clusters, of which 8 clusters (1–4 and 6–9) largely consisted of DEGs that were specific to only one particular cell type and 2 clusters (5 and 10) consisted mainly of DEGs that showed differential expression in multiple cortical cell types (Figure 3K). Perhaps in contrast to expectations, matUPD and patUPD of the same cell type appeared to show more similar transcriptional changes than UPDs in the same direction for different cell types (Figure S4C). To gain insight into how transcriptional changes in MADM-induced UPD could translate into cellular phenotypes, we performed Gene Ontology (GO) enrichment analysis of each gene cluster (numbered in Figure 3K). The GO analysis revealed several significant terms associated with various neural developmental and physiological processes, but the terms were highly specific for distinct cortical cell types (Figure 3K; Table S3).
Single-Cell Transcriptome Analysis of Neocortical Emx1+ Lineage with chr7 UPD
So far, we have mapped transcriptional changes in response to UPD in genetically defined cortical cell classes in bulk (still reflecting a mix of individual cell types) and at one time point (i.e., P0). To increase the resolution of our analysis, we isolated single-cell transcriptomes and at distinct developmental stages (embryo, birth, early postnatal, and adult). We carried out scRNA-seq as described earlier but in cells restricted to the neocortical Emx1+ lineage and carrying UPD of chr7. We collected individual cells with matUPD and patUPD and control cells from initial and reverse crosses at embryonic day (E) 15, P0, P7, P14, and P42 by FACS (Figure 4A). Upon scRNA-seq and quality control, we identified 1,153 cells for further analysis (530 cells with matUPD, 532 cells with patUPD, and 91 control cells) (Table S1E). We reduced the dimensionality of our data by uniform manifold approximation and projection (UMAP) and cell clustering (STAR Methods). We could identify all major cell types (radial glial progenitors [RGPs], neurons, olfactory bulb neuroblasts [OBNBs], oligodendrocytes [oligos], and astrocytes [astros] by marker gene expression (Figure 4B; Table S1E). The clusters defined by distinct cell types included comparable numbers of cells with matUPD and patUPD (Figure 4C). We classified single cells according to their developmental age for each cell type (Figure 4D).
Figure 4.

Developmental Time Course Analysis of chr7 UPD Single-Cell Transcriptomes
(A) Experimental outline for analysis of single-cell transcriptomes in the Emx1+ lineage with MADM-induced UPD of chr7 at E15, P0, P7, P14, and P42.
(B) Heatmap displays expression of a representative set of marker genes for the classification of individual cell types in the Emx1+ lineage. Colored bars above the heatmap indicate different cell types: RGPs (cyan), neurons (light and dark gray), OBNBs (light and dark blue), oligos (orange), astros (purple).
(C) Fractions of cells with matUPD and patUPD in distinct cell types.
(D) Age distribution (E15, white; P0, light gray; P7, gray; P14, dark gray; P42, black) of analyzed cells indicated as the relative fraction in distinct cell types.
(E–P) Re-clustering of RGPs, neuronal cells, oligos, and astros. (E, I, and M) UMAP dots indicate individual cells. Neuronal cells (n = 717) were separated into 7 developmental clusters; RGP, OBNB, and neuronal classes (E). oligos (n = 143) were separated into 4 developmental clusters; oligodendrocyte progenitor cells (OPCs) and newly formed and myelinating oligos (I). astros (n = 290) were separated into 4 developmental clusters; aIPCs, immature astros, and mature astros (M). See also Figure S5. (F, J, and N) Violin plots show distribution of imprinted gene expression (Ndn, Peg3, Snrpn: paternally expressed; Cdkn1c: maternally expressed) in single cells (black dots) with matUPD (red, left side) and patUPD (blue, right side) from neurons (F), oligodendrocytes (J), and astrocytes (N). (G, K, and O) Number of DEGs in matUPD and patUPD at defined developmental stages (padj < 0.2, likelihood-ratio test) in neurons (G), oligodendrocytes (K), and astrocytes (O). (H, L, and P) Number of significantly enriched GO terms (p < 0.01, hypergeometric test) in apoptosis, growth/cell cycle, and synapse groups in neurons (H), oligodendrocytes (L), and astrocytes (P).
Next, we performed refined analyses (STAR Methods) separately for 717 neurons, RGPs, and OBNBs (Figure 4E); 143 oligodendrocytes (Figure 4I); and 290 astrocytes (Figure 4M). We reconstructed expected developmental trajectories, with 3–4 distinct states for each lineage (Figures 4E, 4I, 4M, S5A, S5C, and S5E; Table S1E). Trajectories corresponded well to the developmental time of origin and marker gene expression (Figure S5). Next, we investigated the expression levels of imprinted genes in all cell clusters. Consistent with our earlier allelic expression data, we found higher expression of Ndn, Peg3, and Snrpn (paternally expressed) in cells with patUPD and higher expression of Cdkn1c (maternally expressed) in single cells with matUPD (Figures 4F, 4J, and 4N). Some genes, e.g., Snrpn, Ndn, and Cdkn1c, showed marked cell-type-specific expression changes during development (Figures 4F and 4N).
To analyze developmental transcriptional responses upon UPD, we performed differential gene expression analysis between matUPD and patUPD cells in each cell cluster. In neuronal cells and oligodendrocytes, we identified between 56 and 436 DEGs, with no consistent bias toward one UPD (Figures 4G and 4K, padj < 0.2, likelihood-ratio test; Table S4). In contrast, astrocytes showed the most dramatic changes, with up to 2,079 DEGs. We noticed a bias toward higher numbers of DEGs in patUPD at later stages (Figure 4O, padj < 0.2, likelihood-ratio test; Table S4). To gain information about putative phenotypes because of deregulated gene expression, we performed GO enrichment analysis (Table S4). We focused the analysis on a set of well-established imprinted gene functions (genes located on chr7) relevant for neural development: regulation of cell cycle and growth (Huang et al., 2013; Martín et al., 2015; Zhang et al., 1997), apoptosis (Broad et al., 2009; Huang et al., 2013; Kurita et al., 2006), and synaptic connectivity (Judson et al., 2016; Li et al., 1999, 2016; Rotaru et al., 2018; Wallace et al., 2012). The distribution of significantly enriched GO terms within these 3 categories was distinct for different cell types (Figures 4H, 4L, and 4P). Whereas no trend was obvious in neuronal cells, oligodendrocytes showed consistent enrichment for synapse-containing terms, and astrocytes showed consistent enrichment for all three categories: synapse-, cell cycle/growth-, and apoptosis-containing terms.
Dosage-Sensitive Imprinted Gene Expression Regulates Cortical Astrocyte Development
The preceding results show that UPD leads to highly cell-type-specific, genome-wide transcriptional responses, which could translate into distinct cellular phenotypes. Strikingly, cortical astrocytes show by far the highest number of DEGs and significantly enriched GO terms compared with projection neurons and oligodendrocytes. We thus began phenotypic analysis with a focus on the astrocyte lineage. Because we observed a high number of GO terms related to cell cycle and apoptosis in single cells of astrocyte lineage, we first analyzed absolute numbers of aIPCs (Beattie et al., 2017). Around birth (P0), the number of BLBP+ aIPCs with patUPD was significantly increased when compared with aIPCs with matUPD (Figures 5A–5C; Table S7A). These data indicate that aIPC and subsequent cortical astrocyte development may be regulated differently in cells with distinct UPDs. Parenchymal astrocytes with patUPD were relatively increased compared with astrocytes with matUPD in brains of 3-week-old mice (P21) (Figures 5D–5F; Table S7A). The increased relative number of astrocytes with patUPD was apparent from P7 onward but did not further increase from P21 up to three months, and we could not detect signs of astrocytoma formation (Figure 5G). In contrast, the relative numbers of projection neurons with patUPD or matUPD at P0 and P21 were ∼1, indicating that RGP-mediated neurogenesis occurs equally, regardless of the UPD state.
Figure 5.

Neocortical Astrocytes with chr7 patUPD Are Relatively Increased to chr7 matUPD
(A and B) Analysis of brain lipid-binding protein (BLBP) (white) expression in developing cortical plate in MADM-7GT/TG;Emx1-Cre+/− at P0. matUPD cells are labeled in red (tdT+), patUPD cells are labeled in green (GFP+), and nuclei are labeled in blue (A) or unlabeled (B).
(C) Quantification of the fraction (%) of BLBP+/MADM+ double-positive cells of the total number of MADM-labeled cells with matUPD (orange) and patUPD (green).
(D–F) Analysis of the MADM-labeling pattern (cross section hemisphere (D) and boxed area (E)) and relative abundance (F) of matUPD (red, tdT+) and patUPD (green, GFP+) in CX in P21 MADM-7GT/TG;Emx1-Cre+/− mice.
(G) Quantification of the fraction (%) of MADM-labeled astrocytes of the total number of MADM-labeled cells with matUPD (orange) and patUPD (green) at P7, P14, P21, and 3 months.
(H) Quantification of the fraction (%) of Ki67+/MADM+ astrocytes of the total MADM-labeled astrocytes with matUPD (orange) and patUPD (green).
(I) Quantification of the fraction (%) of EdU+/MADM+ astrocytes of the total MADM-labeled astros with matUPD (orange) and patUPD (green).
Bars and error bars represent mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 t test. Cortical layers are indicated (roman numerals). Scale bar: 50 μm (A and B), 25 μm (b′–b″′, h′, h″, i′, and i″), 500 μm (D), and 60 μm (E). See also Figure S6.
Next, we analyzed marker expression and morphology of astrocytes with chr7 matUPD and patUPD. We found no differences in marker gene expression (Figures S6A–S6F), branching pattern (Figures S6G–S6K), or cellular volume (Figure S6L). To assess whether differences in proliferation rate could explain differential abundance of astros with matUPD versus patUPD, we (1) stained cryosections for Ki67 at P7 (Figure 5H; Table S7A) and (2) injected 5′-ethynyl-2′-deoxyuridine (EdU) at P4/P6, with analysis of incorporation at P21 (Figure 5I; Table S7A). These experiments revealed a slight increasing, albeit non-significant, trend in patUPD/Ki67+ and patUPD/EdU+ when compared with matUPD/Ki67+ and matUPD/EdU+ double-positive cells.
Imprinted Genes on chr7 Associate with Deregulated Gene Networks Implicated in Growth and Apoptosis in Cortical Astrocytes with chr7 UPD
The preceding results indicate that astrocytes with chr7 patUPD have a growth and/or survival advantage over astrocytes with matUPD. To refine and deepen the analysis, we isolated pure populations of cortical astrocytes with UPD at different time points for RNA-seq, affording higher sensitivity compared with scRNA-seq. To enrich for cortical astrocytes, we combined MADM with a LacZ transgene driven by the human GFAP promoter that marks the cortical astrocyte lineage (Brenner et al., 1994). We isolated LacZ+/tdT+ and LacZ+/GFP+ chr7 UPD astrocytes at P0 and P14 and LacZ−/tdT+ and LacZ−/GFP+ neurons at P0 as control, using FACS and followed by RNA-seq (Figure 6A). After quality control, we identified 2–4 replicates for each matUPD and patUPD astrocytes at P0 and P14, respectively (Table S1F; STAR Methods).
Figure 6.

Transcriptome Analysis in Cortical Astrocytes with chr7 UPD Reveals Deregulated Gene Networks Modulating Growth and Apoptosis
(A) Strategy to FACS-enrich cortical astrocytes with chr7 UPD using hGFAP promoter-driven LacZ transgene at P0 and P14 for RNA-seq and differential gene expression analysis.
(B) Normalized expression of marker genes for neurons (Camk2b and Mapt) and astrocytes (Serpine2a and Glast).
(C) Heatmap showing differential expression of 9 imprinted genes in matUPD/patUPD astrocytes at P0 and P14. Genes with higher expression in matUPD cells (log2 fold change > 0) are marked in red, and genes with higher expression in patUPD cells (log2 fold change < 0) are marked in blue. Asterisks mark significant differential expression (padj < 0.05, DESeq2).
(D) Number of DEGs (padj < 0.1, DESeq2) in astrocytes with matUPD (light gray) or patUPD (dark gray) at P0 and P14.
(E) Number of significantly enriched GO terms (p < 0.1, hypergeometric test) related to apoptosis, growth/cell cycle, and synapse.
(F and G) Merged top significant gene subnetworks identified by PhenomeExpress (p = 0.001) at P0/P14 that associate with deregulated imprinted genes in astrocytes with chr7 UPD. Each circle represents an individual gene. Imprinted genes are in red (mat) and blue (pat).
(F) Genes detected in P0, P14, or both subnetworks are in pink, purple, or gray, respectively. Large circles indicate significant deregulation (padj < 0.1, DESeq2, DEG) at P0 and/or P14. Small circles indicate expressed genes (EGs).
(G) Deregulated gene subnetworks highlighting genes involved in apoptosis (black), cell cycle/growth (green), or both (dark green).
The effectiveness of our approach was validated by high expression of neuronal markers (Camk2b and Mapt) in LacZ− compared with LacZ+ samples, and high expression of astrocytic markers (Serpine2a and Glast) in LacZ+ compared with LacZ− samples (Figure 6B). Next, we performed differential gene expression analysis in matUPD/patUPD LacZ+ astrocyte samples at P0 and P14. We found 9 known imprinted genes on chr7, with the expected expression bias toward patUPD or matUPD samples (Figure 6C). Overall, we identified 51 and 549 significantly DEGs at P0 and P14, respectively, with a bias toward higher numbers in patUPD (Figure 6D, padj < 0.1, DESeq2; Table S5). GO term enrichment analysis revealed many terms associated with growth and apoptosis (Figure 6E; Table S5), corroborating our findings from scRNA-seq.
To connect imprinted genes to the observed phenotype (increased relative numbers of astrocytes with chr7 patUPD), we used PhenomeExpress (Soul et al., 2015). PhenomeExpress combines information from differential gene expression with protein-protein interaction networks to identify statistically significant subnetworks. We first performed PhenomeExpress analysis separately for P0 and P14 astrocyte samples and merged networks with the largest number of genes from both time points (Figure 6F; STAR Methods). This analysis produced a single connected network with 275 genes, of which 75 genes showed significant differential expression at P0 and/or P14 (padj < 0.1, DESeq2), including 5 imprinted genes (Snrpn, Peg12, Cdkn1c, Ndn, and Peg3) (Figure 6F). Genes that were present at both P0/P14 time points in the preceding network included Nos3 and Sarm1, which are critically involved in apoptosis (de la Monte et al., 2000; Mukherjee et al., 2015; Osterloh et al., 2012). To gain a more holistic understanding of the gene groups present in the network, we performed GO enrichment analysis. Besides apoptosis-related terms, we identified terms related to cell cycle and growth in the top enriched GO terms (Figure 6G; Table S6). In summary, the preceding analysis revealed connections of 5 imprinted genes (located on chr7) to deregulated gene networks associated with growth and apoptosis in astrocytes with chr7 UPD.
Increased Relative Numbers of Astrocytes with chr7 patUPD Emerge Independent of Igf2
The preceding results imply a mechanism of growth and/or survival advantage of astrocytes with chr7 patUPD over cells with matUPD. To functionally test such a hypothesis, we carried out MADM-based candidate gene analysis. First, we focused on imprinted paternally expressed Igf2 (located on chr7), with the rationale that in liver, Igf2 is the major component driving massive growth dominance of hepatocytes with chr7 patUPD (Hippenmeyer et al., 2013). As such, increased Igf2 dosage in the astrocyte lineage with patUPD (2× Igf2 dose) could result in a growth advantage when compared with matUPD (no Igf2 expression because of imprinting). We tested such a possibility and introduced an Igf2 null allele into distinct MADM paradigms (Figures 7A and S7). We first introduced the Igf2 null allele from the father and compared this with control (Figures 7A–7G). Because of imprinting (no expression from the maternal allele), the offspring upon paternal deletion reflects full Igf2 knockout (KO) (DeChiara et al., 1991), which would predict an equalizing patUPD growth advantage. In contrast, introduction of the Igf2 mutation from the mother (Figures 7A and 7H–7J) would not change Igf2 expression levels (i.e., 2× paternal Igf2 and no mat); therefore, the patUPD advantage would remain. Lastly, introduction of the Igf2 deletion allele from both parents results in full Igf2 KO (Figures 7A and 7K–7M), again predicting equalization of the patUPD growth advantage. To our surprise, however, the relative numbers of astrocytes with chr7 patUPD compared with matUPD remained increased in all preceding MADM paradigms, regardless of the status of Igf2 (Figures 7D, 7G, 7J, and 7M; Table S7B). These findings indicate no major role for Igf2 in promoting a growth advantage and thus higher relative numbers of cortical astrocytes with patUPD.
Figure 7.

Genetic Dissection of Igf2 and Bax in Cortical Astrocyte Genesis and Survival
(A–M) Schematics of Igf2 expression in MADM-deletion paradigms, corresponding to predictions of growth/astrocyte genesis (?) and ratios (patUPD/matUPD [PP/MM]) in cells with matUPD (orange) and patUPD (green) (A); experimental MADM-labeling in CX at P21 in overview (B, E, H, and K) and at higher resolution (C, F, I, and L); and quantification of PP/MM ratios of cortical astrocytes (D, G, J, and M) in control, MADM-7GT/TG;Emx1-Cre+/− (B–D); paternal (E–G) or maternal (H–J) Igf2 deletion, MADM-7GT/TG,Igf2;Emx1-Cre+/−; and Igf2 full KO, MADM-7GT,Igf2/TG,Igf2;Emx1-Cre+/− (K–M). Loss of Igf2 in astrocytes has no effect on PP/MM ratios, because comparisons of values in all Igf2 deletion paradigms relative to control were non-significant.
(N–Z) Schematics of Bax ablation in MADM-deletion paradigms, corresponding predictions of astrocyte survival (?) and ratios (PP/MM) in cells with matUPD (orange) and patUPD (green) (N); experimental MADM-labeling in CX at P21 in overview (O, R, U, and X) and at higher resolution (P, S, V, and Y); and quantification of PP/MM ratios of cortical astrocytes (Q, T, W, and Z) in control, MADM-7GT/TG;Emx1-Cre+/− (O–Q); paternal (R–T) or maternal (U–W) Bax deletion, MADM-7GT/TG,Bax;Emx1-Cre+/−; and Bax cKO, MADM-7GT,Bax/TG,Bax;Emx1-Cre+/− (X–Z). In maternal Bax deletion (W) and Bax cKO animals, ratios of astrocytes (PP/MM) equalized to ∼1 and showed significant differences compared with control (Q) (mat deletion to control, ∗∗p = 0.0061; cKO to control, ∗p = 0.0207; t test). Bars and error bars represent mean ± SEM.
Cortical layers (C, F, I, L, P, S, V, and Y) are indicated (roman numerals). Scale bar: 50 μm (B, E, H, K, O, R, U, and X) and 60 μm (C, F, I, L, P, S, V, and Y). See also Figure S7.
Loss of Bax Equalizes Increased Numbers of Astrocytes with patUPD Relative to matUPD
In the preceding analysis, we could connect imprinted genes located on chr7 to gene networks functionally implicated in the regulation of apoptosis (Figure 6). Therefore, we tested whether increased numbers of astrocytes with chr7 patUPD could result from a survival advantage. To this end, we introduced a conditional allele for the pro-apoptotic gene Bax into distinct MADM paradigms (Figures 7N and S7). Because Bax is not subject to imprinting, we could generate MADM mosaics with, for example, homozygous Bax−/− mutant patUPD and wild-type matUPD cells, and vice versa (Figure 7N). Deletion of Bax in patUPD astrocytes did not change the relatively increased numbers of patUPD compared with matUPD, similar to in control (Figures 7O–7T; Table S7B). In contrast, ablation of Bax in astrocytes with chr7 matUPD equalized the increased numbers of patUPD astrocytes relative to matUPD (Figures 7U–7W; Table S7B). These findings indicate that astrocytes with chr7 matUPD had a survival disadvantage when compared with patUPD and that loss of Bax rescued matUPD cells. To corroborate these results, we analyzed Bax conditional knockout (cKO) in the Emx1+ lineage, in which both patUPD and matUPD astros lack Bax expression. We found that in this genetic paradigm, the increased ratio of patUPD/matUPD was equalized with a value of ∼1 (Figures 7X–7Z; Table S7B). In summary, the neocortical astrocyte lineage with patUPD has a survival advantage over cells with matUPD, and elimination of pro-apoptotic Bax from matUPD equalized increased patUPD/matUPD ratios.
Discussion
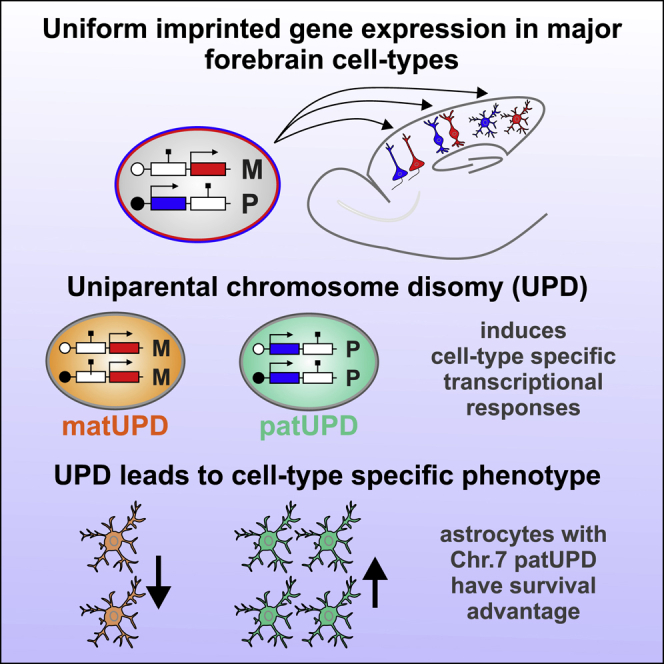
Genomic imprinting controls the allelic expression of a subset of dosage-sensitive genes in a parent-of-origin-dependent manner. Here we uncovered allelic expression of all imprinted genes in the major cortical cell types during development and in adult with unprecedented single-cell resolution. Our data show that the control of imprinting acts independently of cell type but that expression strength is highly cell type specific (Figures 8A and 8B). To assess whether the preceding findings are functionally relevant, we probed changes in imprinted gene dosage by UPD and with single-cell phenotypic analysis. We found highly cell-type-specific transcriptional responses precipitating in unique cellular phenotypes in response to UPD (Figures 8C and 8D). We discuss our findings in the general context of imprinting control and function in health and disease and the role and implications of imprinted gene expression in cortical astrocyte development.
Figure 8.

Cell-Type Specificity of Genomic Imprinting in Cerebral Cortex
(A) Uniform allelic expression of imprinted genes in major forebrain cell types, including projection neurons, interneurons and astrocytes in CX and HC and olfactory granule cells.
(B) Imprinted genes show highly variable expression levels in distinct cortical cell types.
(C) UPD results in imbalanced imprinted gene expression, which in turn leads to highly cell-type-specific but global transcriptional changes.
(D) UPD results in highly cell-type-specific phenotypes, revealing a novel function for dosage-sensitive imprinted gene expression in cortical astrocyte development. The astrocytes with chr7 patUPD show a survival advantage compared with chr7 matUPD astrocytes.
Cell-Type Specificity of Allelic Expression
Imprinting controls animal development, and disturbed imprinted gene expression is associated with human disease (Monk et al., 2019; Peters, 2014; Tucci et al., 2019). Many previous studies aimed to identify all imprinted genes and to map their allelic expression pattern at the individual organ or tissue level in both mouse and human (Andergassen et al., 2017; Babak et al., 2015; Baran et al., 2015; DeVeale et al., 2012; Gregg et al., 2010; Perez et al., 2015). These pioneering works provided an invaluable tissue map of imprinted genes, which can be classified into three broader categories (Chess, 2016; Huang et al., 2018; Perez et al., 2016): (1) Imprinted genes with exclusive (all or none) allelic expression, whereby one parental allele is silenced; the genes that fall into this category are also called canonical imprinted genes. (2) Genes that show biased allelic expression with detectable expression of both parental alleles, also called non-canonical imprinted expression. (3) Genes that switch expression of their parental alleles in different tissues and/or during development. Genes belonging to the third category include Dlk1. Importantly, it has been shown that the loss of Dlk1 imprinting (i.e., change in overall imprinting and expression status) serves a critical function for postnatal neurogenesis (Ferrón et al., 2011). Along the same lines, changes in biased imprinted Magel2 gene expression result in distinct animal behavior (Stacher Horndli et al., 2019). Thus, dynamic changes in imprinting translate into biologically relevant functions. The breadth of such phenomena, among all imprinted genes, and whether they are common or restricted to distinct cell types are unknown. Our allelic expression data show that in all major forebrain cell types (including excitatory and inhibitory neurons, OB granules, and glial cells), imprinting is rather stable. We observed no major loss of imprinted expression and no allele switching, indicating no widespread role for these imprinting control mechanisms, at least during cortical development. However, it will be important to extend the analysis of allelic expression at the individual-cell level to more, ideally all, cellular classes in the entire developing brain.
The emergence of biased imprinted expression at the population level and its existence in single cells has been subject to speculation and debate (Chess, 2016; Huang et al., 2018; Perez et al., 2016). One scenario postulates the concurrency of two distinctly sized populations of cells with opposite allelic expression patterns (Huang et al., 2018; Perez et al., 2016). However, efforts to obtain conclusive insights suffered from technical challenges. For instance, fluorescence in situ hybridization (Ginart et al., 2016) or reporter fusion proteins (Judson et al., 2014; Stelzer et al., 2016) provided some evidence for individual genes but lack the throughput required to systematically analyze all imprinted genes. RNA-seq of genetically distinct alleles at the single-cell level promised a powerful approach (Deng et al., 2014) but thus far lacks the depth to conclusively determine canonical and biased imprinted gene expression at the single-cell level (Lin et al., 2016; Santoni et al., 2017). Here we used crosses of genetically defined inbred mouse strains, with genetic fluorescent reporters and in combination with scRNA-seq, and explored the cortical Emx1+ cell lineage in the developing and adult cerebral cortex. Our approach for the first time revealed allelic expression of all imprinted genes at the single-cell level in a series of well-defined cell types. Our data also unambiguously demonstrated that allelic expression in single cells follows the pattern observed at the population level for both canonical and biased imprinting. We therefore conclude that genomic imprinting modulates the probability of expression from one parental allele independent of the cell type. This is in line with our results from bulk RNA-seq, which revealed that the tissue-specific expression control acts in parallel with genomic imprinting. Future efforts with the goal of establishing a single-cell brain atlas (Ecker et al., 2017; Regev et al., 2017), in combination with systematic allelic mapping, will further the generality of our findings across all cell types.
Cell-Autonomous and Cell-Type-Specific Transcriptional Responses in UPD
Imprinted gene expression renders a cell functionally haploid for particular genes and therefore vulnerable to genetic defects that may cause diseases in human. Besides mutations and epigenetic alterations, UPDs are important underlying causes for human syndromes involving imprinted genes (Peters, 2014; Yamazawa et al., 2010). However, UPDs have been exploited successfully to identify imprinted genes (Bittel et al., 2005; Schulz et al., 2006) and to investigate the function of the parental genomes in embryonic and brain development (Allen et al., 1995; Cattanach and Kirk, 1985; Ferguson-Smith et al., 1991; Keverne et al., 1996). Despite this initial interest in UPDs, virtually nothing is known about the cellular responses to these structural chromosome aberrations. Technical limitations so far only allowed the investigation of UPD at the whole-animal level; they lacked the resolution to obtain insights at the cellular level. Another major drawback in the analysis of UPD in whole animals is reflected in the key importance of many imprinted genes in nutrient transfer during pregnancy (Barlow and Bartolomei, 2014). Thus, the phenotypic interpretation of UPD at the individual-cell level is confounded by putative whole-animal systemic effects. MADM technology provides a solution and is to date the sole technology that can produce UPD sparsely in genetic mosaic animals within genetically defined cell populations (Hippenmeyer et al., 2013; Laukoter et al., 2020). Here we used the MADM approach to systematically probe the consequences of UPD at the single-cell level in neocortical Emx1+ cell lineages. Our results revealed that UPD leads to global genome-wide transcriptional changes that were highly cell type specific. In other words, the same UPD induced in different cell types leads to distinct changes in gene expression.
In a broader context, our findings could be relevant for understanding of the etiology of syndromic imprinting diseases. In effect, many neurological imprinting disorders caused by UPD lead to multiple symptoms that likely emerge from functional deficits in several brain areas and therefore affect distinct cell types. In this context, UPD of mouse chr7, which includes syntenic stretches to human chromosome 15 (chr15) that are causal for Prader-Willi and Angelman syndromes (Bervini and Herzog, 2013; Mabb et al., 2011), leads to deregulation of many genes and enrichment of several GO terms associated with synaptic signaling. In summary, our results contribute to understanding of the intricate interplay between common and cell-type-specific responses because of UPD. It will be important in the future to assess putative differences in neuronal activity in distinct parental UPDs and to correlate putative phenotypical manifestations at the physiological level to the observed transcriptional changes. Such efforts hold the potential to obtain unprecedented and possibly general mechanistic insights into the etiology of neurological and psychiatric imprinting disorders associated with UPD.
Imprinted Gene Expression Regulates Cortical Astrocyte Production
The function of imprinted genes has been mainly studied using genetic full/global tissue KO (Amberg et al., 2019; Perez et al., 2016; Tucci et al., 2019). However, the functional requirement of imprinted genes in single cells is mostly unknown. Here we analyzed phenotypic attributes due to UPD of chr7 during the development of the neocortex. Although neurogenesis appeared normal, cortical astrocyte development was distinct in cells carrying patUPD compared with matUPD. About two times more astrocytes with patUPD were observed relative to matUPD. Several mechanistic models could explain these differences. Igf2-mediated selective growth stimulation of astrocytes with patUPD was not involved, unlike in liver hepatocytes (Hippenmeyer et al., 2013). However, astrocytes with patUPD appeared to have a survival advantage over astrocytes with matUPD, because loss of pro-apoptotic Bax in matUPD in the MADM paradigm equalized relative ratios of patUPD/matUPD astrocytes to ∼1. Bax is located on chr7 but is not an imprinted gene and is not dosage sensitive. Thus, differential expression of paternal versus maternal Bax alleles is not the primary cause for the observed patUPD astrocyte survival advantage—unlike in the case of Bcl-x, which shows biased imprinted expression (Perez et al., 2015). How does imbalanced imprinted gene expression translate to a survival advantage in astrocytes with chr7 patUPD? Imprinted genes may act within broader imprinted gene networks (Al Adhami et al., 2015; Varrault et al., 2006). Given the cell-type-specific expression strength and resulting genome-wide transcriptional changes in UPD, it is intriguing to speculate that imprinted genes could play key roles at critical hubs in highly cell-type-specific gene networks that in turn modulate or instruct the intracellular state and/or cellular phenotype. Indeed, in our gene network analysis, we found 5 imprinted genes on chr7 (and therefore deregulated in UPD) that could be connected to major gene hubs regulating apoptosis and/or cell survival. We also found a large number of deregulated genes, and with a bias toward patUPD at later stages (P14) of astrocyte development, when patUPD were significantly overrepresented relative to matUPD. Thus, the large amount of patUPD-expressed genes likely reflects a gene expression signature of a surviving astrocyte population that is missing from matUPD samples. These data may indicate an apoptosis protective function of imprinted genes during astrocyte development. Ndn and Cdkn1c, two imprinted genes that we identified in UPD-associated gene networks, have been reported to be involved in cortical neuron survival during development (Hasegawa and Yoshikawa, 2008; Imaizumi et al., 2020; Laukoter et al., 2020). It will thus be important in future studies to investigate precise roles of Cdkn1c and/or Ndn in astrocyte survival and/or apoptosis in normal and UPD cell states.
More generally, the analysis of UPD-associated cellular phenotypes can be extended in the future to any mouse organ or tissue, provided that appropriate tissue- and/or cell-type-specific Cre drivers are available. With the completion of a genome-wide library of MADM mice, in which MADM cassettes have been inserted on all mouse autosomes (Contreras et al., 2020), MADM technology holds the potential to systematically probe imprinting at the single-cell level using UPD in any cell type and across the entire mouse genome.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GFP - Chick | Aves Labs Inc. | Cat#GFP-1020; RRID:AB_10000240 |
| RFP - Rabbit | MBL | Cat#PM005; RRID:AB_591279 |
| tdTomato - Goat | Sicgen Antibodies | Cat#AB8181-200; RRID:AB_2722750 |
| BLBP - Rabbit | Millipore | Cat#AB9558; RRID:AB_2314014 |
| S100b – Mouse | Sigma-Aldrich | Cat#S2532; RRID:AB_477499 |
| GFAP – Rabbit | Dako | Cat#Z0334; RRID:AB_10013382 |
| Ki67 – Rabbit | Abcam | Cat#AB15580; RRID:AB_443209 |
| Alexa Fluor 488 Anti-Chicken IgG | Jackson ImmunoResearch Labs | Cat#703-545-155; RRID:AB_2340375 |
| Cy3 Anti-Rabbit IgG | Jackson ImmunoResearch Labs | Cat#711-165-152; RRID:AB_2307443 |
| Cy3 Anti-Goat IgG | Jackson ImmunoResearch Labs | Cat#705-165-147; RRID:AB_2307351 |
| Alexa Fluor 647 Anti-Rabbit IgG | Jackson ImmunoResearch Labs | Cat#711-605-152; RRID:AB_2340624 |
| Alexa Fluor 647 Anti-Rabbit IgG | Molecular Probes | Cat#A31573; RRID:AB_2536183 |
| Alexa Fluor 647 Anti-Mouse IgG | Jackson ImmunoResearch Labs | Cat#715-605-151; RRID:AB_2340863 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Papain Vial Source | Worthington | Cat#PAP2 |
| DNase Vial Source | Worthington | Cat#D2 |
| Inhibitor Vial Source | Worthington | Cat#OI-BSA |
| Critical Commercial Assays | ||
| QuantSeq Library Prep Kit FWD | Lexogen | Cat#015.96 |
| Illumina TruSeq Stranded Total RNA LT - (with Ribo-Zero TM Gold) | Illumina | Cat#RS-122-2301 |
| Nextera XT library preparation kit | Illumina | Cat#FC-131-1096 |
| SMARTer Stranded Total RNA Sample Prep Kit – Low Input Mammalian | Clonetech | Cat#634861 |
| Agencourt RNAclean XP | Beckman Coulter | Cat#A66514 |
| Click-iT Alexa Fluor 647 imaging kit | Thermo Fisher Scientific | Cat#C10340 |
| FACS Blue LacZ beta Galactosidase detection kit | Abcam | Cat#ab189815 |
| Deposited Data | ||
| RNA-Seq of B6/CAST crosses in major forebrain cell types (bulk and single cell) | This study | http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152716 |
| RNA-Seq of MADM induced UPDs of Chr. 7, 11, 12 (QuantSeq) | This study | http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152716 |
| RNA-Seq of MADM induced UPD of Chr. 7 (SMARTer) | This study | http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152716 |
| Single cell RNA-Seq of Emx1 positive cells in MADM induced UPD of Chr. 7 | This study | http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152716 |
| RNA-Seq of enriched astrocytes with MADM induced UPD of Chr. 7 (SMARTer) | This study | http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152716 |
| Experimental Models: Organisms/Strains | ||
| Mouse: MADM-7-GT | The Jackson Laboratory | RRID:IMSR_JAX:021457 |
| Mouse: MADM-7-TG | The Jackson Laboratory | RRID:IMSR_JAX:021458 |
| Mouse: MADM-11-GT | The Jackson Laboratory | RRID:IMSR_JAX:013749 |
| Mouse: MADM-11-TG | The Jackson Laboratory | RRID:IMSR_JAX:013751 |
| Mouse: MADM-12-GT | The Jackson Laboratory | RRID:IMSR_JAX:021460 |
| Mouse: MADM-12-TG | The Jackson Laboratory | RRID:IMSR_JAX:021461 |
| Mouse: Emx1-Cre | The Jackson Laboratory | RRID:IMSR_JAX:005628 |
| Mouse: Nkx2.1-Cre | The Jackson Laboratory | RRID:IMSR_JAX:008661 |
| Mouse: Z/EG | The Jackson Laboratory | RRID:IMSR_JAX:004178 |
| Mouse: Ai14 | The Jackson Laboratory | RRID:IMSR_JAX:007914 |
| Mouse: FVB/NJ | The Jackson Laboratory | RRID:IMSR_JAX:001800 |
| Mouse: CAST/EiJ | The Jackson Laboratory | RRID:IMSR_JAX:000928 |
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: Igf2+/− | (DeChiara et al., 1990) | N/A |
| Mouse: Baxflox | The Jackson Laboratory | RRID: IMSR_JAX: 006329 |
| Mouse: XGFAP-lacZ | The Jackson Laboratory | RRID: IMSR JAX: 003487 |
| Software and Algorithms | ||
| ZEN Digital Imaging for Light Microscopy | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html#introduction |
| FACS Diva | BD Biosciences | N/A |
| Graphpad Prism 7.0 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| IMARIS 9.2.4 | Bitplane | https://imaris.oxinst.com/products/imaris-for-neuroscientists |
| STAR v2.5.0c | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| picard toolkit v.2.16.0 | N/A | https://broadinstitute.github.io/picard/ |
| Bedtools v2.26.0 | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| Samtools v1.3.1 | (Li et al., 2009) | https://github.com/samtools/samtools |
| Allelome.PRO | (Andergassen et al., 2015) | https://sourceforge.net/projects/allelomepro/ |
| Allelome.PRO v0.2 | This study | https://sourceforge.net/projects/allelomepro/ |
| Cufflinks v2.2.1 | (Trapnell et al., 2010) | http://cole-trapnell-lab.github.io/cufflinks/ |
| R v3.4.4/3.6.1 | N/A | https://www.r-project.org/ |
| DESeq2 v1.16.1/1.26.0 | (Love et al., 2014) | http://www.bioconductor.org/ |
| Limma v3.32.2 | (Ritchie et al., 2015) | http://www.bioconductor.org/ |
| Monocle v2.4.0 | (Trapnell et al., 2014) | http://www.bioconductor.org/ |
| Dendextend v1.5.2 | (Galili, 2015) | https://cran.r-project.org/web/packages/dendextend/index.html |
| Pvclust v2.0 | N/A | https://cran.r-project.org/web/packages/pvclust/index.html |
| clusterProfiler v3.4.4/3.14.3 | (Yu et al., 2012) | http://www.bioconductor.org/ |
| Seurat v3.1.4 | (Stuart et al., 2019) | http://www.bioconductor.org/ |
| phenomeExpress | (Soul et al., 2015) | https://github.com/soulj/PhenomeExpress |
| igraph v1.2.4.2 | (Csardi and Nepusz, 2006) | https://igraph.org |
| Cytoscape 3.7.2 | (Shannon et al., 2003) | https://cytoscape.org |
| Other | ||
| FACS Aria III | BD Biosciences | N/A |
| LSM 800 Confocal | Zeiss | N/A |
| Cryostat Cryostar NX70 | Thermo Fisher Scientific | N/A |
| Bioanalyzer | Agilent | N/A |
| Qubit Fluorometer | Thermo Fisher Scientific | N/A |
| HiSeq 2500 | Illumina | N/A |
| HiSeq 3000/4000 | Illumina | N/A |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Simon Hippenmeyer (simon.hippenmeyer@ist.ac.at).
Materials Availability
All published reagents and mousselines will be shared upon request within the limits of the respective material transfer agreements.
Data and Code Availability
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE152716 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152716). The analysis software modified for this paper (Allelome.PRO v0.2) is available at http://sourceforge.net/projects/allelomepro/.
Experimental Model and Subject Details
Mouse Lines
All mouse colonies were maintained in accordance with protocols approved by institutional animal care and use committee, institutional ethics committee and the preclinical core facility (PCF) at IST Austria. Experiments were performed under a license approved by the Austrian Federal Ministry of Science and Research in accordance with the Austrian and EU animal laws (license numbers: BMWF-66.018/0007-II/3b/2012 and BMWFW-66.018/0006-WF/V/3b/2017).
Mice with specific pathogen free status according to FELASA recommendations (Mähler Convenor et al., 2014) were bred and maintained in experimental rodent facilities (room temperature 21 ± 1°C [mean ± SEM]; relative humidity 40%–55%; photoperiod 12L:12D). Food (V1126, Ssniff Spezialitäten GmbH, Soest, Germany) and tap water were available ad libitum.
Mouse lines with MADM cassettes inserted on Chr. 7, Chr. 11, and Chr. 12 (Hippenmeyer et al., 2010, 2013) (MADM-7-GT JAX stock # 021457, MADM-7-TG JAX stock # 021458, MADM-11-GT JAX stock # 013749, MADM-11-TG JAX stock # 013751, MADM-12-GT JAX stock # 021460, MADM-12-TG JAX stock # 021461); Emx1-Cre (Gorski et al., 2002) (JAX stock # 005628); Nkx2.1-Cre (Xu et al., 2008) (JAX stock # 008661); Z/EG (Novak et al., 2000) (JAX stock # 004178); Ai14 (Madisen et al., 2010) (JAX stock # 007914); Igf2+/− (DeChiara et al., 1990) and Baxflox (Takeuchi et al., 2005) (JAX stock #006329); XGFAP-LacZ (Brenner et al., 1994) (JAX stock #003487) have been described previously. FVB (JAX stock # 001800), CAST/EiJ (JAX stock # 000928) and C57BL/6J (JAX stock # 000664) were purchased from commercial vendors. We have not observed any influence of sex on the results in our study, and all experiments and analyses were thus carried out using animals of both sexes.
Analysis of F1 animals from intercrosses of mice in FVB (for whole tissue) or CAST/EiJ (for genetically defined Emx1+ and Nkx2.1+ cells-types) genetic background with C57BL/6J mice (in combination with Emx1-Cre;Z/EG or Nkx2.1-Cre;Ai14, respectively) for quantitative allelic expression experiments, in bulk and at single cell level, were carried out at P0 and P42.
All MADM-induced UPD analyses in animals as described below were carried out in a mixed CD1-C57BL/6J genetic background. Animals from forward (matUPD in red; patUPD in green) and/or reverse (matUPD in green and patUPD in red) crossing schemes were used for analysis and data acquisition. For initial sequencing experiments, animals with MADM-induced UPD of Chr. 7, Chr. 11 and Chr. 12 (in combination with Emx1-Cre and Nkx2.1-Cre) were analyzed at P0. For sequencing of small amounts (Chr. 7 UPD in combination with Emx1-Cre and Nkx2.1-Cre) using SMARTer technology, MADM animals were analyzed at P0. For single-cell sequencing experiments, MADM animals with Chr. 7 UPD in combination with Emx1-Cre were analyzed at E15, P0, P7, P14 and P42. Phenotypic analysis in vivo was performed at P0 (MADM-7, 11, and 12, each in combination with Emx1-Cre and Nkx2.1-Cre). Detailed phenotypic time course analysis of MADM-7 in combination with Emx1-Cre was performed at P0, P7, P14, P21 and 3 months. Isolation and analysis of astrocytes from animals with MADM-induced UPD of Chr. 7, LacZ-labeled using XGFAP-LacZ, was performed at P0 and P14. Genetic epistasis experiments of MADM-induced Chr. 7 UPD in combination with Emx1-Cre and with Igf2 or Bax were all performed at P21.
Method Details
Isolation of Tissue and Immunohistochemistry
Mice were deeply anesthetized through injection of a ketamine/xylazine/acepromazine solution (65 mg, 13 mg and 2 mg/kg body weight, respectively) and unresponsiveness was confirmed through pinching in the paw. The diaphragm of the mouse was opened from the abdominal side to expose the heart. Cardiac perfusion was performed with ice-cold PBS followed immediately by 4% PFA prepared in PB buffer (Sigma-Aldrich). Brains were removed and further fixed in 4% PFA o/n to ensure complete fixation. Brains were cryopreserved with 30% sucrose (Sigma-Aldrich) solution in PBS for approximately 48 hours. Brains were then embedded in Tissue-Tek O.C.T. (Sakura). For adult time points, 45μm coronal sections were collected in 24 multi-well dishes (Greiner Bio-one) and stored at −20°C in antifreeze solution (30% v/v ethyleneglycol, 30% v/v glycerol, 10% v/v 0.244M PO4 buffer) until used. Adult brain sections were mounted onto Superfrost glass-slides (Thermo Fisher Scientific), followed by 3 wash steps (5min) with PBS. Tissue sections were blocked for 30 minutes in a buffer solution containing 5% normal donkey serum (Thermo Fisher Scientific), 0.3% Trition X-100 in PBS. Primary antibodies were in blocking buffer and incubated o/n at 4°C. Sections were washed 3 times for 5 minutes each with PBT (0.3% Triton X-100 in PBS) and incubated with corresponding secondary antibody diluted in PBT for 1 hour. Sections were washed 2 times with PBT and once with PBS. Nuclear staining was done using 10min incubation with PBS containing 2.5% DAPI (Thermo Fisher Scientific). Sections were embedded in mounting medium containing 1,4-diazabicyclooctane (DABCO; Roth) and Mowiol 4-88 (Roth) and stored at 4°C. Tissue from postnatal day zero (P0) was directly transferred into ice-cold 4% PFA and kept o/n at 4°C. Cryopreservation and embedding was done as described for adult brains. Early postnatal brains were sectioned with 30μm and directly mounted onto Superfrost glass-slides (Thermo Fisher Scientific) and immunohistochemistry was performed as described above for adult brains.
EdU Labeling Experiments
Proliferation of astrocytes was assessed by EdU incorporation. Experiments were based on the use of the Click-iT Alexa Fluor 647 imaging kit (Thermo Fisher). Reagents were reconstituted according to the user manual. Intraperitoneal EdU injections were performed at P4 and P6 (1mg/ml EdU stock solution; 30-40μl per mouse). Tissue collection was done at P21, followed by imunohistochemistry as described above with one exception. The Click-iT imaging kit was used (according to the instruction manual) to visualize the EdU signal before the DAPI staining was performed.
Preparation of Single Cell Suspension and FACS
Experimental animals were sacrificed and brain areas of interest (neocortex, hippocampus and olfactory bulb) were dissected. Single cell suspensions were prepared by using Papain containing L-cysteine and EDTA (vial 2, Worthington), DNase I (vial 3, Worthington), Ovomucoid protease inhibitor (vial 4, Worthington), EBSS (Thermo Fisher Scientific), DMEM/F12 (Thermo Fisher Scientific), FBS (Thermo Fisher Scientific) and HS (Thermo Fisher Scientific). All vials from Worthington kit were reconstituted according to the manufacturer’s instructions using EBSS. The dissected brain areas were directly placed into Papain-DNase solution (20units/ml papain and 1000 units DNase). Enzymatic digestion was performed for 30min at 37°C in a shaking water bath. Next, solution 2 (EBSS containing 0.67mg Ovomucoid protease inhibitor and 166.7 U/ml DNase I) was added, the whole suspension was thoroughly mixed and centrifuged for 5min at 1000rpm at RT. Supernatant was removed and cell pellet was resuspended in solution 2. Trituration with p1000 pipette was performed to mechanically dissolve any remaining tissue parts. DMEM/F12 was added to the cell suspension as a washing solution, followed by a centrifugation step of 5min with 1500rpm at RT. Cells were resuspended in media (DMEM/F12 containing 10% FBS and 10% HS) and kept on ice until sorted. Right before sorting, cell suspension was filtered using a 40μm cell strainer. FACS was performed on a BD FACS Aria III using 100 nozzle and keeping sample and collection devices (1.5ml tubes or 96-well plate) at 4°C. Duplet exclusion was performed to ensure sorting of true single cells. For bulk cell analysis, cells were sorted directly into either Isolation Buffer (IB; Lexogen) if more than 2000 sorted cells/per color were expected or if samples yielded fewer labeled cells into custom made lysis buffer (30nM TRIS pH 8, 10nM EDTA pH 8, 1% SDS and 200 μg/μL Proteinase K). For bulk MADM samples, GFP+, tdT+, and GFP+/tdT+ cells were collected. For quantitative allelic expression experiments Emx1-Cre+/GFP+ and Nkx2.1-Cre+/tdT+ cells were isolated. For single cell analysis individual GFP+, tdT+ or GFP+/tdT+ cells were used.
RNA Extraction and cDNA Library Preparation of MADM Samples for RNA Sequencing
Data for Figures 3C–3E. Samples containing more than 2000 sorted cells were processed using the SPLIT kit (Lexogen) following the manufacturer’s instructions. Samples containing fewer cells were sorted into custom made lysis buffer (described above). Directly after sorting, samples were incubated for 30min at 37°C. Total volume was filled to 250μl using RNase-free H2O (Thermo Fisher Scientific) followed by addition of 750μl Trizol LS (Thermo Fisher Scientific). Samples were mixed by 5 times inverting. After a 5min incubation step at RT, the entire solution was transferred into a MaXtract tube (QIAGEN). 200μl chloroform (Sigma-Aldrich) was added, followed by 3 times 5sec vortexing and 2min incubation at RT. Samples were centrifuged for 2min with 12000rpm at 18°C. Supernatant was transferred to a new tube and isopropanol (Sigma-Aldrich) was added in a 1:1 ratio. For better visibility of the RNA pellet 1μl GlycoBlue (Thermo Fisher Scientific) was added and entire solution was mixed by vortexing (3x 5sec). Samples were left for precipitation o/n at −20°C. After precipitation samples were centrifuged for 20min with 14000rpm at 4°C. Supernatant was removed and RNA pellet was washed with 70% ethanol, followed by a 5min centrifugation step (14000rpm at 4°C). RNA pellet was resuspended in 12,5μl RNase-free H2O. RNA quality was analyzed using Bioanalyzer RNA 6000 Nano (Agilent) and RNA 6000 Pico kit (Agilent) following the manufacturer’s instructions. RNA samples were stored at −80°C until further use. cDNA libraries were prepared using QuantSeq 3′mRNA library preparation kit (Lexogen) following the manufacturer’s instructions. To amplify libraries correctly RT-PCR was performed as indicated in the protocol using PCR Add-on kit (Lexogen). cDNA library size distribution was analyzed using Bioanalyzer DNA High Sensitivity kit (Agilent) according to the manufacturer’s instructions. cDNA library concentration was measured using Qubit. Libraries were pooled according to library length and concentration. RNA sequencing was performed by VBCF GmbH on Illumina platforms.
Preparation of Single Cell Suspension and FACS of Astrocytes
Single cell suspension was prepared using the same reagents and the same concentrations as described above. However volumes of reagents were decreased since cortices of single P0 and P14 animals were processed as individual replicates and centrifugation steps were increased to 8min. Except for the last step (resuspension of cells in media) the protocol for preparing the single cell suspension was performed as outlined above. The LacZ staining was performed using FACS Blue LacZ betaGalactosidase Detection Kit (Abcam). LacZ substrate stock solution was prepared according to the instruction manual. To generate the staining solution LacZ substrate was diluted 1:50 with DMEM/F12 (Thermo Fisher Scientific). Cell pellet was resuspended with 100μl of LacZ staining solution and incubated for 25min at 37°C on a thermoshaker. Staining reaction was stopped by adding 100μl of media (DMEM/F12 containing 10% FBS and 10% HS) followed by a centrifugation step (1500rpm for 8min). Supernatant was removed and cells were resuspended in media. Samples were kept in the dark at room temperature until sorting was started. Before sorting, cell suspension was filtered using a 40μm cell strainer. As outlined above FACS was performed on a BD FACS Aria III using 100 nozzle. To ensure specificity of LacZ staining, a negative sample was always processed and sorting gates were adjusted accordingly.
RNA Extraction and cDNA Library Preparation of Allelome Samples for RNA Sequencing
For ‘whole tissue’ analysis cortex, hippocampus and olfactory bulb were dissected from newborn P0 mice from FVB/CAST crosses (see Table S1A for details). RNA extraction was performed with Trizol LS (Thermo Fisher Scientific) as described above. After each purification step, RNA quality and quantity was measured using Agilent Bioanalyzer. 500ng of total RNA were treated with DNase1 (Applied Biosystems) and purified using Ampure RNAClean XP beads. 200ng of the purified RNA was used for library preparation using the Illumina TruSeq Stranded Total RNA LT - (with Ribo-Zero TM Gold) - according to the manufacturer’s protocol. For cell type specific allelome analysis, B6/CAST crosses were used (see Table S1A for details). 7-40ng of total RNA from FACS sorted cells were used for library preparation with the Clonetech SMARTer Stranded Total RNA Sample Prep Kit – Low Input Mammalian (with Ribogone), according to the manufacturer’s protocol with 19 cycles of library amplification. If the concentration of the total RNA sample was too low, RNA was immobilized on Agencourt RNAclean XP beads according to the manufacturer’s protocol. After washing and drying of the beads the RNA was eluted in 5 μL water and the whole volume (including beads) was used in the RiboGone reaction. All samples were sequenced by VBCF GmbH on Illumina platforms. FVB/CAST samples were sequenced as 125bp paired end (NOTE: only the reverse read was used for analysis). All B6/CAST samples were sequenced 50bp single end.
cDNA Library Preparation and Sequencing of Low Cell Numbers or Single Cells
RNA sequencing was performed as described (Picelli et al., 2014). In brief, either purified RNA (data in Figures 3F–3K), up to 400 cells (data in Figure 6) or single cells (Figures 1G and 1H, 2F–2H, and 4) were used as input. Cells were processed as single cell suspension (described above) and sorted directly into 96 well plates (Bio-Rad) at 4°C (single cell) or up to 400 cells were collected at 4°C and transferred into 96 well plates (Bio-Rad). Cells were sorted in 4μl lysis buffer (0.2% Triton X-100, 2U/μl RNase Inhibitor [Clonetech]). Plates for single cell sequencing were sealed with AlumaSeal, (SIGMA) and quick-frozen on dry ice. All plates were kept at −80°C until further processing. Double stranded full-length cDNA was prepared using Smart-seq2. For P0 cells with MADM-induced UPD of Chr. 7 (sample_id starting with ‘S’ in Table S1E), efficiency of cDNA production was tested by qPCR for fluorescent protein expression (not shown) and 96 cells from each color were chosen to be converted to RNA-seq libraries using the Nextera XT DNA Library Prep Kit (Illumina). All other libraries were prepared using custom reagents (VBCF GmbH) and libraries from a 96 well plate were pooled, diluted and sequenced on Illumina platforms at the Biomedical Sequencing Facility (BSF) at CeMM (sample_id starting with ‘S’ in Table S1E) or at the VBCF NGS Unit (https://www.viennabiocenter.org/facilities/).
Quantification and Statistical Analysis
Analysis of MADM-Labeled Brains
Sections were imaged using an inverted LSM800 confocal microscope (Zeiss) and processed using Zeiss Zen Blue software. Confocal images were imported into Photoshop software (Adobe) and MADM-labeled cells were manually counted based on respective marker expression as described previously (Beattie et al., 2017). Statistical analysis was performed in Graphpad Prism 7.0. All data used for quantification of MADM-labeled tissue is compiled in Table S7.
Astrocyte Morphology Analysis
Detailed analysis of astrocyte morphology was done as described previously (Beattie et al., 2017) with some modifications. Lower layer astrocytes that expressed tdT+ or GFP+ were imaged with a 63x oil objective. Sholl analysis was performed to measure astrocyte branching complexity. 3D reconstruction and analysis was done using the Filament tracer algorithm of the IMARIS software. Total cell volume of astrocytes was assessed from the 3D structure.
Processing and Analysis of Bulk RNA-seq Data
For all analyses mouse genome sequence (GRCm38.p5), gene annotation conversion tables and Gencode M16 annotation in gtf format were downloaded from https://www.gencodegenes.org on 21 Feb 2018. For optimal representation of the long non-coding RNA Meg3 the spliced ENSMUST00000143836.7 transcript was modified to represent a single exon transcript. STAR (Dobin et al., 2013) (version 2.5.0c) index was prepared with “–genomeSAsparseD 2” and “–sjdbOverhang 37” parameters and the modified Gencode M16 gtf file (using the “–sjdbGTFfile” parameter). Only chromosomes 1-19, X, Y and M were used for the index building (all other sequences were removed). Raw reads were delivered as BAM files and converted to fastq format using “bamToFastq” from the bedtools suite (Quinlan and Hall, 2010) for all alignment steps. All downstream analyses were performed in R v3.4.4 (Figures 1, 2, and 3) and R v3.6.1 (Figures 4 and 6). Markers for projection neurons were selected from Greig et al. (2013) and Molyneaux et al. (2007), and markers for interneurons were selected from Taniguchi (2014). DESeq2 (Love et al., 2014) package was v1.16.1 (Figure 3) or v1.26.0 (Figure 6), limma (Ritchie et al., 2015) package was v3.32.2.
Bulk RNA-seq analyses in Figures 1 and 2 are based on sorted neuronal populations from B6/CAST crosses, unless indicated otherwise. Details on direction of crosses and replicates are listed in Table S1A. Figures 1C, S1A, and S1B: Twenty million reads (counting from the start of each BAM file) were aligned with STAR using the following parameters:–clip5pNbases 3,–outFilterMultimapNmax 1,–runThreadN 2,–outSAMtype BAM, SortedByCoordinate,–limitBAMsortRAM 3000000000,–quantMode GeneCounts. For Figures S1A and S1B, as well as differential expression analysis, count tables obtained by STAR were used for the analysis (3rd column, ∗.ReadsPerGene.out.tab files). Principal component analysis was performed on variance stabilized counts (DESeq2) using the “prcomp” function (package stats v3.4.3) on the top 500 most variable genes. For marker heatmap the median expression of biological replicates was determined from normalized counts as determined by the function “counts” (DESeq2). The heatmap was prepared using “pheatmap” with cluster_rows = T, cluster_cols = F, scale = ”row” parameters. To produce the allelic expression heatmap in Figure 1C BAM files were separated by strand using the “separate_BAM_strand.pl” from the Allelome.PRO package (Andergassen et al., 2015) using the ‘++,–‘ strand rule. The resulting BAM files were analyzed using Allelome.PRO with standard parameters (allelic ratio cutoff: 0.7, FDR: 1%). SNP data was obtained from ftp://ftp-mouse.sanger.ac.uk (mgp.v3.snps.rsIDdbSNPv137.vcf.bz2). As an annotation two bed6 files (for the genes on “+” and “-” strand) were prepared using the “gene” annotations from the Gencode M16 gtf file. We identified 25 genes that were found to show imprinted expression in the brain (Andergassen et al., 2017) and plotted their allelic expression bias derived from Allelome.PRO’s ∗locus_full.txt files. Heatmaps were produced using “geom_tile” from ggplot2 package and colors reflect allelic expression (red: maternal, blue: paternal, biallelic: green, gray: any other tag). In cases where a gene showed an “imprinting score” of either > 1 or < −1 in all replicates of one sample (indicating biased expression toward the maternal or paternal allele) but the allelic ratio was below the cutoff of 0.7, we added a MAT/PAT tag to indicate this consistent bias. For comparison we also included bulk tissue from FVB/CAST crosses in this analysis (“whole_tissue,” see Table S1A for details). For Figures 1D and 1E we extracted the “I_ratio” values from Allelome.PRO’s ∗locus_full.txt files for all single cell type datasets and classified genes as ‘biased imprinted expression’ when the respective I_ratio was < 0.95 (for a maternally expressed gene) or > 0.05 (for a paternally expressed gene) in one or more tissues. Conversely, a gene was classified as ‘canonical imprinted expression’ when all tissues showed an I_ratio of > = 0.95 (for a maternally expressed gene) or ≤ 0.05 (for a paternally expressed gene). Figure 1E shows individual I_ratio values for 3 genes with biased imprinted expression (Ago2, Impact, Inpp5f) and 4 genes with canonical imprinted expression (Sgce, Snrpn, Rian, Meg3).
For Figure 2B normalized counts (DESeq2) were used to prepare the median expression value for each gene in every cell type. The heatmap was plotted using ggplot2 package using relative expression values, normalized to the highest expressing cell type. In Figure 2C normalized counts obtained by “counts” (DESeq2), are plotted for selected genes. For Figure 2D differential expression was calculated with DESeq2 for all possible pairwise comparisons of the 5 cell types used in this analysis using contrasts (see Table S1A for details on replicates and genotype). The number of pairwise comparisons where individual imprinted genes showed significant differential expression (adjusted p value < 0.01) was used as the “specificity score.”
Figures 3C–3E: To limit batch effects that could be introduced by optimization of the QuantSeq KIT by the Lexogen company as well as by optimizing sequencing parameters for technical replicates, reads aligning to genes were counted in a 2 step process. In the first step reads were aligned to transcriptome space using STAR with ‘–clip5pNbases 12–outFilterMultimapNmax 1–outSAMmode BAM–quantMode TranscriptomeSAM GeneCounts’ parameters. The resulting BAM file was sorted by read name, using samtools (Li et al., 2009) v1.3.1 and the number of unique reads was counted using a custom script. This number was used to calculate the number of raw reads that is necessary to obtain 10 million reads aligning to the transcriptome (#total.reads.used reads in Table S1C). In the second step the number of reads was extracted randomly from the pool of raw reads using DownsampleSam from the picard toolkit (v.2.16.0) and ‘S = Chained R = 2401’ parameters. Note that this random read retrieval was necessary due to varying alignment efficiencies for technical sequencing replicates. Reads aligning to Gencode M16 genes were counted using STAR with “–outFilterMultimapNmax 1–outSAMtype None–outSAMmode None–quantMode GeneCounts–clip5pNbases 12” parameters. Two sets of samples (MADM-12; Nkx2.1-Cre HC and MADM-12; Emx1-Cre OB) consistently gave low % uniquely aligned reads and were removed from the subsequent analysis (samples marked: “used_in_analysis = FALSE” in Table S1C). To further reduce noise, genes with a mean expression of < 20 reads over all used samples (“used in analysis = TRUE” in Table S1C), were removed. For Figure 3C differential gene expression was analyzed using DESeq2. Count tables obtained by STAR were used for the analysis (3rd column, ∗.ReadsPerGene.out.tab files). For each MADM/Cell/Tissue set (maternal UPD, paternal UPD, control), a DESeqDataSet was created using the “DESeqDataSetFromMatrix” function and ’design = ∼group_id + UPD‘ (Table S1C). “DESeq” function was called with fitType = ”local” and parallel = T parameters. Results for matUPD / patUPD comparisons were obtained using contrasts and ’altHypothesis = ”greaterAbs,” “alpha =0.1” and ” lfcThreshold = 0”’ parameters. Fold changes and p values (∗ for adjusted p values < 0.01) for selected genes were plotted. Figure 3D: Batch correction was performed on variance stabilized count data (“varianceStabilizingTransformation,” DESeq2, parameters: “blind=T”) using “removeBatchEffect” (limma package) with ‘batch = “group_id,” model.matrix(∼tissue ∗ cre ∗ UPD)’ parameters. Note that all sample details are given in Table S1C. For marker heatmaps (Figures 3D and S3M) the mean expression of variance stabilized, batch corrected counts from all biological replicates of matUPD, patUPD and control in one cell type (MADM/tissue/cell) was calculated for each gene and normalized to the highest expressing cell type. The heatmap was prepared using the “pheatmap” package (v1.0.9) with ‘scale = ”none,” cluster_rows = T, cluster_cols = F’ parameters. Figure 3E: For hierarchical clustering all pairwise correlations of batch corrected variance stabilized counts were calculated (R function “cor,” parameter: method = ”spearman”) and converted to a distance matrix by subtracting the values from 1. Clustering was performed using R function “hclust” using ‘method = ”ward.D2”’ parameter. The branches of the resulting tree were ordered using the “rotate” function (package dendextend v1.5.2) (Galili, 2015) to group samples with similar origin and the dendrogram was plotted using the package dendextend. Bootstrap probabilities of hierarchical clustering were calculated with a modified version of the R package “pvclust” to allow the usage of function “cor” with parameter: ‘method = ”spearman”’ (pvclust v2.0).
For Figures 3F–3K, we sequenced 88 samples consisting of 5 different cell types with different UPD states for Chr. 7 (matUPD/patUPD/control, details on samples see Table S1D). We removed samples based on the following criteria: less than 900000 sequenced reads, correlation to the other biological replicates < 0.9 (using “cor” function with standard parameters), distance and position to other biological replicates in PCA plot. This resulted in 64 samples where each cell type/UPD state had 3-5 biological replicates. Count tables obtained by STAR were used for the analysis (2rd column, ∗.ReadsPerGene.out.tab files, note that the SMARTer v2 protocol, used here, does not preserve the strandedness of the mRNA). PCA analysis in Figure 3F was done as described above (Figure 1C) using batch corrected variance stabilized counts. All genes with a mean expression > 0 across all samples were used for analysis. Batch correction was performed as described above (Figure 3D). For Figure 3G normalized counts as determined by the function “counts” (from DESeq2 package) were centered to the median of the control samples for each cell type to correct for cell type specific expression differences. Note that for display, values above 3 were removed: Cdkn1c: 21.1, 7.3 (Emx1 OB matUPD), 5.0 (Emx1 OB patUPD).
Figures 3H–3K and S4A: Genes with mean expression > 10 across all samples were used in subsequent analyses. Differential expression between all possible pairwise comparisons of UPD states in all cell types was calculated using DESeq2 using contrasts, similar to the analysis described above for Figure 3 but without using “altHypothesis/alpha/lfcThreshold” parameters. Figure S4A: Differential expression results of matUPD/patUPD samples in each cell type are shown for selected known imprinted genes on Chr. 7. Heatmap was prepared using ggplot2 with adjusted p value < 0.1 as intense color and marked with an asterisk and intense color only marking adjusted p values > 0.1 (not significant). Colors were based on log2 fold changes where value < 0 are indicated as blue (higher in patUPD samples) and values > 0 are indicated as red (higher in matUPD samples). Figure 3H: Number of DEGs with an adjusted p value < 0.05 in the matUPD/wt and patUPD/wt comparisons for each cell type. Figure 3J: Using the same dataset of DEGs as in Figure 4D, the proportion of chromosomes in the respective dataset was plotted. The relative number of genes on Chr. 7 is indicated in black. To test an over-representation of any chromosome in any of these datasets we performed a hypergeometric test using clusterProfiler’s ‘enricher’ function (Yu et al., 2012) with a custom gene set (pvalueCutoff = 0.01, qvalueCutoff = 0.1). Figure 3I: DEGs (adjusted p value < 0.05) from pairwise comparisons matUPD/control and patUPD/control states were used in this analysis. The position of DEGs on Chr. 7 was plotted as their midpoint (start + (end-start)/2). To indicate hypothetical imprinted domains we used differentially methylated regions identified in the brain (Xie et al., 2012) and extended them by 7Mbp in each direction, because this represents the largest distance of a known imprinted gene to its imprint control element (Andergassen et al., 2017). These regions were plotted as gray shades. To identify whether DEGs were enriched within these hypothetical imprinted domains we performed a hypergeometric test using clusterProfiler’s ‘enricher’ function (Yu et al., 2012) with a custom gene set (pvalueCutoff = 0.01, qvalueCutoff = 0.1). Figure 3K: All genes with an adjusted p value of < 0.05 in at least one cell type in the matUPD/wt and patUPD/wt comparisons were used in this analysis. Note that only genes with a unique gene symbol annotation were used here. For each gene in each comparison a significance score was calculated as the log10 of the uncorrected p value. This score was corrected to be positive for genes with a log2 fold change > 0 (higher in the respective UPD sample) or negative for genes with a log2 fold change < 0 (higher in the wt sample). Scores were cut at a level of 5/-5 for better visibility in the resulting heatmap. This score matrix was used to prepare a hierarchical clustering of genes using the pheatmap package with ‘clustering_method = “ward.D2”’. The resulting tree was used to define 10 gene clusters using “cutree” with ‘k = 10’. These clusters were manually ordered (preserving the clustered order of genes within each cluster) and plotted using “pheatmap.” Genes in each cluster are given in tab “genes in clusters” in Table S3.
To identify gene ontology term enrichment in each of the 10 gene clusters identified above we used the “clusterProfiler” package (v3.4.4). Gene Symbol annotation were converted to Entrez Gene IDs using “bitr” and the GO term enrichment was calculated using ‘enrichGO’ with parameters: ‘OrgDb = org.Mm.eg.db (v3.4.1), keytype = “ENTREZID,” ont = ”ALL,” pool = T, readable = T, pvalueCutoff = 0.05, qvalueCutoff = 0.2, maxGSSize = 500’. As a gene “universe” we used all genes that were used in differential expression analysis. Top enriched GO terms (by adjusted p value) are indicated. Note that a full list of GO terms found to be significantly enriched in this analysis is given in Table S3. Figure S4B: We first identified genes that are significantly deregulated in matUPD/control or patUPD/control in each cell type with adjusted p value < 0.05. Next, we identified genes that are deregulated in only one UPD of a single cell type (red: matUPD, blue: patUPD), that are shared between the matUPD/control and patUPD/control comparisons of a single cell type (black) or that are shared in any other combination (white). Figure S4C: The score matrix described above (Figure 3K) was used to calculate the significance of clustering of the effect of the different UPDs in the different cell types (i.e., the matUPD/control and patUPD/control comparisons in each cell type) using “pvclust” with ‘method.dist = ”euclidean,” method.hclust = ”ward.D2,” nboot = 10000, parallel = T’. The resulting dendrogram is shown with AU (Approximately Unbiased, red) p value and BP (Bootstrap Probability, green) values.
Processing and Analysis of scRNA-seq Data
Reads from scRNA-seq were aligned using STAR with the same index as used for bulk sequencing but with the following parameters to create aligned BAM files:–outFilterMultimapNmax 1–outSAMstrandField intronMotif–outFilterIntronMotifs RemoveNoncanonical–outSAMtype BAM SortedByCoordinate–quantMode GeneCounts. Figure 1: Samples with less than 1M total reads were removed from further analysis. These BAM files were used to prepare gene expression matrices (for the same annotation used for bulk RNA-seq analysis) using cuffquant and cuffnorm from the cufflinks suite (v2.2.1) (Trapnell et al., 2010). A final expression matrix was prepared from “genes.fpkm_table” files in R. Small RNAs (gene symbols “Mir” and “Snord”) as well as transcripts of unknown annotation quality (gene symbols “Gm) were removed to reduce noise. Single cell analyses for Figures 1 and 2 were done using the Monocle package (v2.4.0) (Trapnell et al., 2014) for cell type identification or a modified version of Allelome.PRO (Andergassen et al., 2015) for allelic expression (see below). 744 single cells from crosses between C57/Bl6 and CAST were sequenced for this analysis (see Table S1B for details). The “CellDataSet” was prepared using the “newCellDataSet” function with parameters: lowerDetectionLimit = 0.1, expressionFamily = tobit(Lower = 0.1). Absolute transcript counts were determined using the “relative2abs” function with parameter: method = ”num_genes.” The final “CellDataSet” from absolute transcript counts was prepared with parameters: lowerDetectionLimit = 0.5, expressionFamily = negbinomial.size(). We removed cells with < 10000 or > 30000 total mRNAs reported, which left 404 cells with a median of 1735775 uniquely aligned reads and a median of 2597.5 expressed genes (min_expr = 0.1) that were used for all downstream analyses. We identified 1000 genes by t-SNE based clustering and differential expression analysis (Monocle2, genes ranked by adjusted p-value). We performed hierarchical clustering based on these 1000 genes using the “pheatmap” function with standard parameters and log2 size normalized expression values (Note: we added a pseudo-count of 1 before log2 transforming the expression data). The resulting dendrogram was used to identify 7 groups using the “cutree” function. Based on marker gene expression we combined these 7 groups into 5 cell types. These groups and the expression pattern (plotted using “pheatmap” as above) for a set of marker genes is shown in Figure 1G. Figure 1H: Allelic expression analysis in single cells. BAM files were analyzed using a modified version of the Allelome.PRO pipeline (Andergassen et al., 2015), (http://sourceforge.net/projects/allelomepro/). In brief the Allelome.PRO pipeline was modified to allow faster processing of large datasets by allowing the use of pre-prepared annotation files (initial version of Allelome.PRO prepares these files each run of the pipeline and cause the majority of the run-time with many datasets). Additional modifications included the removal of hard coded cutoffs and the termination of the analysis at the reporting stage (i.e., before the calculation of FDR and report of gene lists, which are restricted to the use of 4 samples). For gene specific allelic expression analysis we inspected the “sample_locus_full.txt” files and focused our analysis on the same 25 known imprinted genes as in Figure 1C. For each cell we performed the following analysis: Only genes that were covered by more than 10 reads were kept (“total_reads” columns). Gene expression in each cell was classified into maternal (MAT)/paternal (PAT)/biallelic (BAE) according to the following criteria: MAT: I_score > = 1.3 AND I_ratio > = 0.7, PAT: I_score > = 1.3 AND I_ratio ≤ 0.3, BAE: either I_score < 1.3 or (I_score > = 1.3 AND I_ratio < 0.7 AND I_ratio > 0.3). The relative amount of cells in each allelic expression group was then plotted for each cell type (as determined in Figure 1G). Figure 1H shows 6 selected genes and results for all genes are shown in Table S2.
Figure 2F: Heatmap showing expression levels of known imprinted genes. The same list of genes as presented in Figure 2B was investigated, however Peg10, Zim1, Magel2, Kcnq1ot1, Mirg, Airn were removed because they were detectable in less than 10 cells. Expression was calculated as for the heatmap in Figure 1G and plotted using ggplot2. Coloring indicates the parental direction of expression as determined in Figure 1. Figure 2G: Expression values (log2 of size normalized expression values +1) of selected genes are shown for all cells of a given cell type. Figure 2H: The specificity of a gene to be expressed in a given cell type was calculated by first averaging the expression of each gene for all cells within each cell type (cell type expression). A relative expression value was calculated for each gene by dividing each cell type expression value by the sum of all cell type expression values of the respective gene. The maximum relative expression was plotted in the figure. Note that the closer the value gets to 1 the more exclusive a gene is expressed in a single cell type. Differential expression was calculated with differentialGeneTest function using fullModelFormulaStr = ‘∼label’ (see Table S1B on details on cell labels).
Figure 4: We sequenced 1604 cells with different UPD states for Chr. 7 (matUPD/patUPD/control). Read alignment and gene set used was similar as for Figure 1. Raw counts from STAR alignment were used to create a Seurat (v3.1.4) (Stuart et al., 2019) object with parameters: min.cells = 3, min.features = 200. Percentage of mitochondrial reads was determined with PercentageFeatureSet function with pattern = “ˆmt-” parameter. Cells were filtered by nFeature (> 2500 & < 7500, percent-mt (< 10) and & nCount_RNA (> 1e05, < 5e06). This filtering left 1153 cells with a median of 2030461 uniquely aligned reads and a median of 5191 expressed genes (nFeature_RNA in Table S1E) that were used for all downstream analyses.
Figures 4B–4D and S5: We followed the procedure as suggested by the Seurat vignettes (v3.1). In brief, normalization was performed with NormalizeData using normalization.method = “LogNormalize” and scale.factor = 10000 parameters. Dimensionality reduction, cluster identification and UMAP: FindVariableFeatures with selection.method = “vst” and nfeatures = 2000 parameters; ScaleData with features = all.genes parameter; RunPCA with features = VariableFeatures(object = [seurat object]) parameter; FindNeighbors dims = 1:10 parameter; FindClusters with resolution = 0.5 parameter; RunUMAP with dims = 1:10 parameter. Heatmaps were prepared with DoHeatmap function on selected lists of cluster marker genes determined with FindAllMarkers using only.pos = TRUE, min.pct = 0.25 and logfc.threshold = 0.25 parameters.
Figures 4E–4P: Cells from relevant clusters were extracted and re-analyzed with varying parameters for optimal separation of expected cell types and developmental states. Note that we removed 2/1 E15 cells from oligodendrocytes/astrocytes as they are unlikely to originate from the Emx1 lineage. Cluster numbers for initial clustering are given as ‘orig_seurat_clusters’ in Table S1E. Astrocytes (orig_seurat_clusters: 2, 3, 8): FindNeighbors with dims = 1:9 parameter; FindClusters with resolution = 0.5 parameter; RunUMAP with dims = 1:9 parameter. Oligodendrocytes (orig_seurat_clusters: 5, 9, 10): FindNeighbors with dims = 1:6 parameter; FindClusters with resolution = 0.5 parameter; RunUMAP with dims = 1:6 parameter. Neuronal cells (orig_seurat_clusters: 0, 1, 4, 6, 7): FindNeighbors with dims = 1:10 parameter; FindClusters with resolution = 0.5 parameter; RunUMAP with dims = 1:10 parameter. To plot expression of imprinted genes we used VlnPlot with parameters: slot = “data,” split.by = “UPD,” log = FALSE. Final cluster association is given as ‘cluster’ in Table S1E and abbreviations are OBNB: olfactory bulb neuroblast, RGP: radial glia progenitor, aIP: astrocyte intermediate progenitor, OPC: oligodendrocyte precursor cell. Colors of cells in Table S1E correspond to colors of clusters in Figure 4. Differential expression between matUPD and patUPD cells was performed using FindMarkers with parameters: test.use = ‘bimod’, min.pct = 0, logfc.threshold = 0.1. Results from differential gene expression for each cluster are given in Table S4. GO term enrichment was performed on genes with padj < 0.2 using enrichGO (clusterProfiler package v3.14.3) with parameters: minGSSize = 100, maxGSSize = 300, OrgDb = org.Mm.eg.db (v3.10.0), ont = “BP,” readable = TRUE. All GO terms with p value < 0.01 were used for further analyses and are listed in Table S4. For Figures 4H, 4L, and 4P, GO terms in indicated groups were counted based on the following keywords present in their description. Apoptosis: apop, death; growth/cell cycle: growth, cell cycle, mitosis; synapse: synapse, synaptic.
RNA-seq Analysis of FACS Enriched Astrocytes
Figure 6: We sequenced 28 samples including 20 samples enriched for astrocyte and 8 samples enriched for neuronal cells with matUPD or patUPD (details see Table S1F). Reads were aligned as described for single cells above and raw read counts reported by the STAR aligner were used. Samples were filtered based on clustering of biological replicates in PCA plot (mainly reflecting read coverage, #uniquely.aligned in Table S1F) which left 12 astrocyte samples and 7 neuronal samples (used_for_analysis = ‘Y’ in Table S1F). Figure 6B: Read counts are size factor normalized counts determined by function ‘counts’ with normalize = T parameter. For differential gene expression analysis we used DESeq2 (v1.26.0) and genes with a mean read count > 15 over all samples under investigation. Samples from P0 and P14 were analyzed separately using DESeqDataSetFromMatrix with design = ∼UPD parameter and DESeq with fitType = ”local,” quiet = T, betaPrior = F and parallel = T parameters. Differential expression statistics were obtained with ‘results’ and contrast = c(‘UPD’, ‘mat’, ‘pat’) parameter. Heatmap in Figure 6C was prepared by extracting log2 fold-changes as well as adjusted p value information from differential expression analysis. Genes with a fold-change > 0 (higher in matUPD) were colored red and < 0 (higher in patUPD) blue. Asterisks indicate adjusted p value < 0.05. GO enrichment analysis for genes with an adjusted p value < 0.1 was performed using enrichGO (clusterProfiler package v3.14.3) with OrgDb = org.Mm.eg.db (v3.10.0), ont = ‘BP’, minGSSize = 100, maxGSSize = 1000, pool = F and readable = T parameters. Counting significantly enriched GO terms in indicated groups was performed as described for Figure 4 but with a cutoff of p value < 0.1 for GO terms. Results for differential expression and GO enrichment are given in Table S5. For Figures 6F and 6G we obtained all relevant datasets and code examples from https://github.com/soulj/PhenomeExpress. To identify relevant phenotype IDs we downloaded the uberpheno annotation from http://compbio.charite.de/tl_files/HPO/uberpheno/ and selected all IDs where the description contained the term ’apoptosis’. Networks for the PhenomeExpress analysis were prepared following the PhenomeExpress examples using the differential expression data from DESeq2 and uncorrected p values. Significant subnetworks were identified using runPhenomeExpress with parameters: max_number = 30, sampleSize = 1000. Analysis was done separately for P0 and P14 and the top ranked PhenomeExpress networks (including largest number of genes) for each analysis (p value = 0.001) were extracted and merged using the ‘union’ function from igraph package (Csardi and Nepusz, 2006) with byname = T parameter. GO term enrichment for all genes present in the merged network was determined using enrichGO (clusterProfiler package v3.14.3) with parameters: pvalueCutoff = 0.2, qvalueCutoff = 0.9, minGSSize = 10, maxGSSize = 1000, OrgDb = org.Mm.eg.db (v3.10.0), ont = “BP,” readable = TRUE). Top 15 enriched GO terms were used for network annotation. All GO terms with padj < 0.01 are given in Table S6. Node/Vertex annotation was added in R as follows. Figure 6F: Color code indicates whether a gene was present in the PhenomeExpress network originating from P0 or P14 data. Node size indicates whether the respective gene showed differential expression (DEG) with padj < 0.1 at P0 and/or P14 as indicated in the legend. Figure 6G: Coloring was performed based on the association of the respective gene with the following significantly enriched GO terms. Apoptosis: positive regulation of cell death, positive regulation of apoptotic process, positive regulation of programmed cell death, neuron death; cell cycle/growth: regulation of cell cycle, mitotic cell cycle process (Table S6). Size was determined as for Figure 6F. Final network figure was prepared using Cytoscape 3.7.2 (Shannon et al., 2003).
Acknowledgments
We thank A. Heger (IST Austria Preclinical Facility), A. Sommer and C. Czepe (VBCF GmbH, NGS Unit), and A. Seitz and P. Moll (Lexogen GmbH) for technical support; G. Arque, S. Resch, C. Igler, C. Dotter, C. Yahya, Q. Hudson, and D. Andergassen for initial experiments and/or assistance; D. Barlow, O. Bell, and all members of the Hippenmeyer lab for discussion; and N. Barton, B. Vicoso, M. Sixt, and L. Luo for comments on earlier versions of the manuscript. This research was supported by the Scientific Service Units (SSU) of IST Austria through resources provided by the Bioimaging Facilities (BIF), Life Science Facilities (LSF), and Preclinical Facilities (PCF). A.H.H. is a recipient of a DOC fellowship (24812) of the Austrian Academy of Sciences. N.A. received support from the FWF Firnberg-Programm (T 1031). R.B. received support from the FWF Meitner-Programm (M 2416). This work was also supported by IST Austria institutional funds; a NÖ Forschung und Bildung n[f+b] life science call grant (C13-002) to S.H.; a program grant from the Human Frontiers Science Program (RGP0053/2014) to S.H.; the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007-2013) under REA grant agreement 618444 to S.H.; and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement 725780 LinPro) to S.H.
Author Contributions
S.H., S.L., and F.M.P. conceived the research. S.H., S.L., and F.M.P. designed all experiments and interpreted the data. S.L., F.M.P., R.B., N.A., A.H.H., T.P., and C.S. performed all experiments. F.M.P. performed all computational and bioinformatics analysis, with input from S.L. and S.H. S.H. wrote the manuscript with input from S.L. and F.M.P. All authors edited and proofread the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: July 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2020.06.031.
Supplemental Information
References
- Al Adhami H., Evano B., Le Digarcher A., Gueydan C., Dubois E., Parrinello H., Dantec C., Bouschet T., Varrault A., Journot L. A systems-level approach to parental genomic imprinting: the imprinted gene network includes extracellular matrix genes and regulates cell cycle exit and differentiation. Genome Res. 2015;25:353–367. doi: 10.1101/gr.175919.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert M., Kalebic N., Florio M., Lakshmanaperumal N., Haffner C., Brandl H., Henry I., Huttner W.B. Epigenome profiling and editing of neocortical progenitor cells during development. EMBO J. 2017;36:2642–2658. doi: 10.15252/embj.201796764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen N.D., Logan K., Lally G., Drage D.J., Norris M.L., Keverne E.B. Distribution of parthenogenetic cells in the mouse brain and their influence on brain development and behavior. Proc. Natl. Acad. Sci. USA. 1995;92:10782–10786. doi: 10.1073/pnas.92.23.10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg N., Laukoter S., Hippenmeyer S. Epigenetic cues modulating the generation of cell-type diversity in the cerebral cortex. J. Neurochem. 2019;149:12–26. doi: 10.1111/jnc.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andergassen D., Dotter C.P., Kulinski T.M., Guenzl P.M., Bammer P.C., Barlow D.P., Pauler F.M., Hudson Q.J. Allelome.PRO, a pipeline to define allele-specific genomic features from high-throughput sequencing data. Nucleic Acids Res. 2015;43:e146. doi: 10.1093/nar/gkv727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andergassen D., Dotter C.P., Wenzel D., Sigl V., Bammer P.C., Muckenhuber M., Mayer D., Kulinski T.M., Theussl H.C., Penninger J.M. Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. eLife. 2017;6:e25125. doi: 10.7554/eLife.25125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babak T., DeVeale B., Tsang E.K., Zhou Y., Li X., Smith K.S., Kukurba K.R., Zhang R., Li J.B., van der Kooy D. Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat. Genet. 2015;47:544–549. doi: 10.1038/ng.3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran Y., Subramaniam M., Biton A., Tukiainen T., Tsang E.K., Rivas M.A., Pirinen M., Gutierrez-Arcelus M., Smith K.S., Kukurba K.R., GTEx Consortium The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 2015;25:927–936. doi: 10.1101/gr.192278.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow D.P., Bartolomei M.S. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 2014;6:a018382. doi: 10.1101/cshperspect.a018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie R., Postiglione M.P., Burnett L.E., Laukoter S., Streicher C., Pauler F.M., Xiao G., Klezovitch O., Vasioukhin V., Ghashghaei T.H., Hippenmeyer S. Mosaic Analysis with Double Markers Reveals Distinct Sequential Functions of Lgl1 in Neural Stem Cells. Neuron. 2017;94:517–533. doi: 10.1016/j.neuron.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Bervini S., Herzog H. Mouse models of Prader-Willi Syndrome: a systematic review. Front. Neuroendocrinol. 2013;34:107–119. doi: 10.1016/j.yfrne.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Bittel D.C., Kibiryeva N., Talebizadeh Z., Driscoll D.J., Butler M.G. Microarray analysis of gene/transcript expression in Angelman syndrome: deletion versus UPD. Genomics. 2005;85:85–91. doi: 10.1016/j.ygeno.2004.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonthuis P.J., Huang W.C., Stacher Hörndli C.N., Ferris E., Cheng T., Gregg C. Noncanonical Genomic Imprinting Effects in Offspring. Cell Rep. 2015;12:979–991. doi: 10.1016/j.celrep.2015.07.017. [DOI] [PubMed] [Google Scholar]
- Brenner M., Kisseberth W.C., Su Y., Besnard F., Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J. Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad K.D., Curley J.P., Keverne E.B. Increased apoptosis during neonatal brain development underlies the adult behavioral deficits seen in mice lacking a functional paternally expressed gene 3 (Peg3) Dev. Neurobiol. 2009;69:314–325. doi: 10.1002/dneu.20702. [DOI] [PubMed] [Google Scholar]
- Buiting K., Williams C., Horsthemke B. Angelman syndrome—insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016;12:584–593. doi: 10.1038/nrneurol.2016.133. [DOI] [PubMed] [Google Scholar]
- Cassidy S.B., Driscoll D.J. Prader-Willi syndrome. Eur. J. Hum. Genet. 2009;17:3–13. doi: 10.1038/ejhg.2008.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattanach B.M., Kirk M. Differential activity of maternally and paternally derived chromosome regions in mice. Nature. 1985;315:496–498. doi: 10.1038/315496a0. [DOI] [PubMed] [Google Scholar]
- Chess A. Monoallelic Gene Expression in Mammals. Annu. Rev. Genet. 2016;50:317–327. doi: 10.1146/annurev-genet-120215-035120. [DOI] [PubMed] [Google Scholar]
- Contreras X., Davaatseren A., Amberg N., Hansen A.H., Sonntag J., Andersen L., Bernthaler T., Heger A., Johnson R., Schwarz L.A. A Genome-wide Library of MADM Mice for Single-Cell Genetic Mosaic Analysis. bioRxiv. 2020 doi: 10.1016/j.celrep.2021.109274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csardi G., Nepusz T. The Igraph Software Package for Complex Network Research. InterJournal Complex Syst. 2006;1695:1–9. [Google Scholar]
- de la Monte S.M., Sohn Y.K., Etienne D., Kraft J., Wands J.R. Role of aberrant nitric oxide synthase-3 expression in cerebrovascular degeneration and vascular-mediated injury in Alzheimer’s disease. Ann. N Y Acad. Sci. 2000;903:61–71. doi: 10.1111/j.1749-6632.2000.tb06351.x. [DOI] [PubMed] [Google Scholar]
- DeChiara T.M., Efstratiadis A., Robertson E.J. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature. 1990;345:78–80. doi: 10.1038/345078a0. [DOI] [PubMed] [Google Scholar]
- DeChiara T.M., Robertson E.J., Efstratiadis A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell. 1991;64:849–859. doi: 10.1016/0092-8674(91)90513-x. [DOI] [PubMed] [Google Scholar]
- Deng Q., Ramsköld D., Reinius B., Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343:193–196. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- DeVeale B., van der Kooy D., Babak T. Critical evaluation of imprinted gene expression by RNA-Seq: a new perspective. PLoS Genet. 2012;8:e1002600. doi: 10.1371/journal.pgen.1002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker J.R., Geschwind D.H., Kriegstein A.R., Ngai J., Osten P., Polioudakis D., Regev A., Sestan N., Wickersham I.R., Zeng H. The BRAIN Initiative Cell Census Consortium: Lessons Learned toward Generating a Comprehensive Brain Cell Atlas. Neuron. 2017;96:542–557. doi: 10.1016/j.neuron.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson-Smith A.C. Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011;12:565–575. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith A.C., Cattanach B.M., Barton S.C., Beechey C.V., Surani M.A. Embryological and molecular investigations of parental imprinting on mouse chromosome 7. Nature. 1991;351:667–670. doi: 10.1038/351667a0. [DOI] [PubMed] [Google Scholar]
- Ferrón S.R., Charalambous M., Radford E., McEwen K., Wildner H., Hind E., Morante-Redolat J.M., Laborda J., Guillemot F., Bauer S.R. Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature. 2011;475:381–385. doi: 10.1038/nature10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili T. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics. 2015;31:3718–3720. doi: 10.1093/bioinformatics/btv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginart P., Kalish J.M., Jiang C.L., Yu A.C., Bartolomei M.S., Raj A. Visualizing allele-specific expression in single cells reveals epigenetic mosaicism in an H19 loss-of-imprinting mutant. Genes Dev. 2016;30:567–578. doi: 10.1101/gad.275958.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski J.A., Talley T., Qiu M., Puelles L., Rubenstein J.L., Jones K.R. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray L.T., Yao Z., Nguyen T.N., Kim T.K., Zeng H., Tasic B. Layer-specific chromatin accessibility landscapes reveal regulatory networks in adult mouse visual cortex. eLife. 2017;6:e21883. doi: 10.7554/eLife.21883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg C., Zhang J., Weissbourd B., Luo S., Schroth G.P., Haig D., Dulac C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science. 2010;329:643–648. doi: 10.1126/science.1190830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig L.C., Woodworth M.B., Galazo M.J., Padmanabhan H., Macklis J.D. Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 2013;14:755–769. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K., Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J. Neurosci. 2008;28:8772–8784. doi: 10.1523/JNEUROSCI.3052-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S., Youn Y.H., Moon H.M., Miyamichi K., Zong H., Wynshaw-Boris A., Luo L. Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron. 2010;68:695–709. doi: 10.1016/j.neuron.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S., Johnson R.L., Luo L. Mosaic analysis with double markers reveals cell-type-specific paternal growth dominance. Cell Rep. 2013;3:960–967. doi: 10.1016/j.celrep.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsthemke B., Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. A. 2008;146A:2041–2052. doi: 10.1002/ajmg.a.32364. [DOI] [PubMed] [Google Scholar]
- Huang Z., Fujiwara K., Minamide R., Hasegawa K., Yoshikawa K. Necdin controls proliferation and apoptosis of embryonic neural stem cells in an oxygen tension-dependent manner. J. Neurosci. 2013;33:10362–10373. doi: 10.1523/JNEUROSCI.5682-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.C., Bennett K., Gregg C. Epigenetic and Cellular Diversity in the Brain through Allele-Specific Effects. Trends Neurosci. 2018;41:925–937. doi: 10.1016/j.tins.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi Y., Furutachi S., Watanabe T., Miya H., Kawaguchi D., Gotoh Y. Role of the imprinted allele of the Cdkn1c gene in mouse neocortical development. Sci. Rep. 2020;10:1884. doi: 10.1038/s41598-020-58629-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson M.C., Sosa-Pagan J.O., Del Cid W.A., Han J.E., Philpot B.D. Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J. Comp. Neurol. 2014;522:1874–1896. doi: 10.1002/cne.23507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson M.C., Wallace M.L., Sidorov M.S., Burette A.C., Gu B., van Woerden G.M., King I.F., Han J.E., Zylka M.J., Elgersma Y. GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility. Neuron. 2016;90:56–69. doi: 10.1016/j.neuron.2016.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keverne E.B., Fundele R., Narasimha M., Barton S.C., Surani M.A. Genomic imprinting and the differential roles of parental genomes in brain development. Brain Res. Dev. Brain Res. 1996;92:91–100. doi: 10.1016/0165-3806(95)00209-x. [DOI] [PubMed] [Google Scholar]
- Kurita M., Kuwajima T., Nishimura I., Yoshikawa K. Necdin downregulates CDC2 expression to attenuate neuronal apoptosis. J. Neurosci. 2006;26:12003–12013. doi: 10.1523/JNEUROSCI.3002-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson A.J.M., Johnsson P., Hagemann-Jensen M., Hartmanis L., Faridani O.R., Reinius B., Segerstolpe Å., Rivera C.M., Ren B., Sandberg R. Genomic encoding of transcriptional burst kinetics. Nature. 2019;565:251–254. doi: 10.1038/s41586-018-0836-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaSalle J.M., Reiter L.T., Chamberlain S.J. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics. 2015;7:1213–1228. doi: 10.2217/epi.15.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukoter S., Beattie R., Pauler F.M., Amberg N., Nakayama K.I., Hippenmeyer S. Imprinted Cdkn1c genomic locus cell-autonomously promotes cell survival in cerebral cortex development. Nat. Commun. 2020;11:195. doi: 10.1038/s41467-019-14077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein E., Borm L.E., Linnarsson S. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science. 2017;358:64–69. doi: 10.1126/science.aan6827. [DOI] [PubMed] [Google Scholar]
- Li L., Keverne E.B., Aparicio S.A., Ishino F., Barton S.C., Surani M.A. Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science. 1999;284:330–333. doi: 10.1126/science.284.5412.330. [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Zhao P., Xu Q., Shan S., Hu C., Qiu Z., Xu X. The autism-related gene SNRPN regulates cortical and spine development via controlling nuclear receptor Nr4a1. Sci. Rep. 2016;6:29878. doi: 10.1038/srep29878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.Y., Huang S.C., Tung C.C., Chou C.H., Gau S.S., Huang H.S. Analysis of Genome-Wide Monoallelic Expression Patterns in Three Major Cell Types of Mouse Visual Cortex Using Laser Capture Microdissection. PLoS ONE. 2016;11:e0163663. doi: 10.1371/journal.pone.0163663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato S., Arlotta P. Generating neuronal diversity in the mammalian cerebral cortex. Annu. Rev. Cell Dev. Biol. 2015;31:699–720. doi: 10.1146/annurev-cellbio-100814-125353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C., Keown C.L., Kurihara L., Zhou J., He Y., Li J., Castanon R., Lucero J., Nery J.R., Sandoval J.P. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science. 2017;357:600–604. doi: 10.1126/science.aan3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb A.M., Judson M.C., Zylka M.J., Philpot B.D. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011;34:293–303. doi: 10.1016/j.tins.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mähler Convenor M., Berard M., Feinstein R., Gallagher A., Illgen-Wilcke B., Pritchett-Corning K., Raspa M., FELASA working group on revision of guidelines for health monitoring of rodents and rabbits FELASA recommendations for the health monitoring of mouse, rat, hamster, guinea pig and rabbit colonies in breeding and experimental units. Lab. Anim. 2014;48:178–192. doi: 10.1177/0023677213516312. [DOI] [PubMed] [Google Scholar]
- Martín Y., Cabrera E., Amoedo H., Hernández-Pérez S., Domínguez-Kelly R., Freire R. USP29 controls the stability of checkpoint adaptor Claspin by deubiquitination. Oncogene. 2015;34:1058–1063. doi: 10.1038/onc.2014.38. [DOI] [PubMed] [Google Scholar]
- Mayer C., Hafemeister C., Bandler R.C., Machold R., Batista Brito R., Jaglin X., Allaway K., Butler A., Fishell G., Satija R. Developmental diversification of cortical inhibitory interneurons. Nature. 2018;555:457–462. doi: 10.1038/nature25999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi D., Li Z., Lim L., Li M., Moissidis M., Yang Y., Gao T., Hu T.X., Pratt T., Price D.J. Early emergence of cortical interneuron diversity in the mouse embryo. Science. 2018;360:81–85. doi: 10.1126/science.aar6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux B.J., Arlotta P., Menezes J.R., Macklis J.D. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Monk D., Mackay D.J.G., Eggermann T., Maher E.R., Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019;20:235–248. doi: 10.1038/s41576-018-0092-0. [DOI] [PubMed] [Google Scholar]
- Mukherjee P., Winkler C.W., Taylor K.G., Woods T.A., Nair V., Khan B.A., Peterson K.E. SARM1, Not MyD88, Mediates TLR7/TLR9-Induced Apoptosis in Neurons. J. Immunol. 2015;195:4913–4921. doi: 10.4049/jimmunol.1500953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A., Guo C., Yang W., Nagy A., Lobe C.G. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- Nowakowski T.J., Bhaduri A., Pollen A.A., Alvarado B., Mostajo-Radji M.A., Di Lullo E., Haeussler M., Sandoval-Espinosa C., Liu S.J., Velmeshev D. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science. 2017;358:1318–1323. doi: 10.1126/science.aap8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh J.M., Yang J., Rooney T.M., Fox A.N., Adalbert R., Powell E.H., Sheehan A.E., Avery M.A., Hackett R., Logan M.A. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez J.D., Rubinstein N.D., Fernandez D.E., Santoro S.W., Needleman L.A., Ho-Shing O., Choi J.J., Zirlinger M., Chen S.K., Liu J.S., Dulac C. Quantitative and functional interrogation of parent-of-origin allelic expression biases in the brain. eLife. 2015;4:e07860. doi: 10.7554/eLife.07860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez J.D., Rubinstein N.D., Dulac C. New Perspectives on Genomic Imprinting, an Essential and Multifaceted Mode of Epigenetic Control in the Developing and Adult Brain. Annu. Rev. Neurosci. 2016;39:347–384. doi: 10.1146/annurev-neuro-061010-113708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. The role of genomic imprinting in biology and disease: an expanding view. Nat. Rev. Genet. 2014;15:517–530. doi: 10.1038/nrg3766. [DOI] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Plasschaert R.N., Bartolomei M.S. Tissue-specific regulation and function of Grb10 during growth and neuronal commitment. Proc. Natl. Acad. Sci. USA. 2015;112:6841–6847. doi: 10.1073/pnas.1411254111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M., Human Cell Atlas Meeting Participants The Human Cell Atlas. eLife. 2017;6:e27041. doi: 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotaru D.C., van Woerden G.M., Wallaard I., Elgersma Y. Adult Ube3a Gene Reinstatement Restores the Electrophysiological Deficits of Prefrontal Cortex Layer 5 Neurons in a Mouse Model of Angelman Syndrome. J. Neurosci. 2018;38:8011–8030. doi: 10.1523/JNEUROSCI.0083-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoni F.A., Stamoulis G., Garieri M., Falconnet E., Ribaux P., Borel C., Antonarakis S.E. Detection of Imprinted Genes by Single-Cell Allele-Specific Gene Expression. Am. J. Hum. Genet. 2017;100:444–453. doi: 10.1016/j.ajhg.2017.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz R., Menheniott T.R., Woodfine K., Wood A.J., Choi J.D., Oakey R.J. Chromosome-wide identification of novel imprinted genes using microarrays and uniparental disomies. Nucleic Acids Res. 2006;34:e88. doi: 10.1093/nar/gkl461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soul J., Hardingham T.E., Boot-Handford R.P., Schwartz J.M. PhenomeExpress: a refined network analysis of expression datasets by inclusion of known disease phenotypes. Sci. Rep. 2015;5:8117. doi: 10.1038/srep08117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacher Horndli C.N., Wong E., Ferris E., Bennett K., Steinwand S., Rhodes A.N., Fletcher P.T., Gregg C. Complex Economic Behavior Patterns Are Constructed from Finite, Genetically Controlled Modules of Behavior. Cell Rep. 2019;28:1814–1829. doi: 10.1016/j.celrep.2019.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer Y., Wu H., Song Y., Shivalila C.S., Markoulaki S., Jaenisch R. Parent-of-Origin DNA Methylation Dynamics during Mouse Development. Cell Rep. 2016;16:3167–3180. doi: 10.1016/j.celrep.2016.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O., Fisher J., Suh H., Harada H., Malynn B.A., Korsmeyer S.J. Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc. Natl. Acad. Sci. USA. 2005;102:11272–11277. doi: 10.1073/pnas.0504783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H. Genetic dissection of GABAergic neural circuits in mouse neocortex. Front. Cell. Neurosci. 2014;8:8. doi: 10.3389/fncel.2014.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telley L., Govindan S., Prados J., Stevant I., Nef S., Dermitzakis E., Dayer A., Jabaudon D. Sequential transcriptional waves direct the differentiation of newborn neurons in the mouse neocortex. Science. 2016;351:1443–1446. doi: 10.1126/science.aad8361. [DOI] [PubMed] [Google Scholar]
- Telley L., Agirman G., Prados J., Amberg N., Fièvre S., Oberst P., Bartolini G., Vitali I., Cadilhac C., Hippenmeyer S. Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science. 2019;364:eaav2522. doi: 10.1126/science.aav2522. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucci V., Isles A.R., Kelsey G., Ferguson-Smith A.C., Erice Imprinting Group Genomic Imprinting and Physiological Processes in Mammals. Cell. 2019;176:952–965. doi: 10.1016/j.cell.2019.01.043. [DOI] [PubMed] [Google Scholar]
- Varrault A., Gueydan C., Delalbre A., Bellmann A., Houssami S., Aknin C., Severac D., Chotard L., Kahli M., Le Digarcher A. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell. 2006;11:711–722. doi: 10.1016/j.devcel.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Wallace M.L., Burette A.C., Weinberg R.J., Philpot B.D. Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron. 2012;74:793–800. doi: 10.1016/j.neuron.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson L.S., Davies W., Isles A.R. Genomic imprinting effects on brain development and function. Nat. Rev. Neurosci. 2007;8:832–843. doi: 10.1038/nrn2235. [DOI] [PubMed] [Google Scholar]
- Williamson C.M., Blake A., Thomas S., Beechey C.V., Hancock J., Cattanach B.M., Peters J. MRC Harwell; 2013. Mouse Imprinting Data and References.http://www.mousebook.org/imprinting-gene-list [Google Scholar]
- Xie W., Barr C.L., Kim A., Yue F., Lee A.Y., Eubanks J., Dempster E.L., Ren B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–831. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Tam M., Anderson S.A. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J. Comp. Neurol. 2008;506:16–29. doi: 10.1002/cne.21529. [DOI] [PubMed] [Google Scholar]
- Yamasaki-Ishizaki Y., Kayashima T., Mapendano C.K., Soejima H., Ohta T., Masuzaki H., Kinoshita A., Urano T., Yoshiura K., Matsumoto N. Role of DNA methylation and histone H3 lysine 27 methylation in tissue-specific imprinting of mouse Grb10. Mol. Cell. Biol. 2007;27:732–742. doi: 10.1128/MCB.01329-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazawa K., Ogata T., Ferguson-Smith A.C. Uniparental disomy and human disease: an overview. Am. J. Med. Genet. C. Semin. Med. Genet. 2010;154C:329–334. doi: 10.1002/ajmg.c.30270. [DOI] [PubMed] [Google Scholar]
- Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H., Sanes J.R. Neuronal cell-type classification: challenges, opportunities and the path forward. Nat. Rev. Neurosci. 2017;18:530–546. doi: 10.1038/nrn.2017.85. [DOI] [PubMed] [Google Scholar]
- Zhang P., Liégeois N.J., Wong C., Finegold M., Hou H., Thompson J.C., Silverman A., Harper J.W., DePinho R.A., Elledge S.J. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387:151–158. doi: 10.1038/387151a0. [DOI] [PubMed] [Google Scholar]
- Zong H., Espinosa J.S., Su H.H., Muzumdar M.D., Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–492. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE152716 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152716). The analysis software modified for this paper (Allelome.PRO v0.2) is available at http://sourceforge.net/projects/allelomepro/.