

Abstract
As the most common form of arthritis, osteoarthritis (OA) has become a major cause of severe joint pain, physical disability, and quality of life impairment in the affected population. To date, precise pathogenesis of OA has not been fully clarified, which leads to significant obstacles in developing efficacious treatments such as failures in finding disease-modifying OA drugs (DMOADs) in the last decades. Given that diarthrodial joints primarily display the weight-bearing and movement-supporting function, it is not surprising that mechanical stress represents one of the major risk factors for OA. However, the inner connection between mechanical stress and OA onset/progression has yet to be explored. Mitochondrion, a widespread organelle involved in complex biological regulation processes such as adenosine triphosphate (ATP) synthesis and cellular metabolism, is believed to have a controlling role in the survival and function implement of chondrocytes, the singular cell type within cartilage. Mitochondrial dysfunction has also been observed in osteoarthritic chondrocytes. In this review, we systemically summarize mitochondrial alterations in chondrocytes during OA progression and discuss our recent progress in understanding the potential role of mitochondria in mediating mechanical stress-associated osteoarthritic alterations of chondrocytes. In particular, we propose the potential signaling pathways that may regulate this process, which provide new views and therapeutic targets for the prevention and treatment of mechanical stress-associated OA.
Keywords: Osteoarthritis, Chondrocyte, Mitochondria, Mechanical stimuli, Mechanotransduction, Mitonuclear communication
Graphical Abstract
Introduction
Osteoarthritis (OA) is a degenerative disease majorly characterized by the progressive breakdown of joint cartilage and abnormal bone remodeling. First recognized in the late eighteenth century [1], OA now has become the most common joint disorder. According to the data collected by United States (U.S.) Centers for Disease Control and Prevention in 2015, OA affected 54.4 million adult people in the U.S., and the number is expected to reach 78.4 million (25.9% of the projected total adult population) by 2040 [2, 3]. With the high predominance of overweight and obesity, both the prevalence and incidence of OA are expected to rise [4].
Clinical symptoms and radiographic changes of OA include chronic pain, stiffness, tenderness, synovial inflammation, degeneration of ligaments, hypertrophy of the joint capsule, joint space narrowing, crepitus formation, subchondral bone sclerosis, thinning of the cartilage, and changes in periarticular muscles, nerves, bursa, and local fat pads [5]. Therefore, OA has recently been indicated as the failure of the joint organ [6], meaning all the joint elements are affected and contribute to the disease pathogenesis. However, degeneration of articular cartilage still represents the key feature in OA pathology.
Pathological changes of OA are complicated with multifactorial etiologies. Aging, obesity, genetic predisposition, excessive loading, trauma, metabolic syndrome, gender, and hormone profile are all identified to be closely related to the progression of OA [7]. But the precise pathogenesis of OA is still beyond the scope of current knowledge. Consequently, pharmacological treatments are limited to symptom relief and function restoration rather than slowing or stopping the progression of the disease [8]. The only end-point treatment for late-stage OA is joint replacement surgery [9].
OA has the highest prevalence in weight-bearing joints such as knees, hips, and the spine [10]. In fact, it has been determined that mechanical loading is one of the major risk factors of OA. The novel structure of the synovial joints allows mechanical loadings to be dispersed and absorbed. Particularly, the articular cartilage is able to reduce shock and friction to hard tissues beneath. Depending on different intensity, frequency, and duration, mechanical loading plays different roles in the health of cartilage [11]. Moderate mechanical loading is a requirement for chondrocytes to maintain a homeostatic balance between catabolic and anabolic processes. Also, moderate mechanical loading can promote the growth of chondrocytes and cartilage formation by regulating proliferation related processes such as inducing the release of growth factors or transiting latent transforming growth factors (TGF) into its active form [12, 13]. Lacking physiological loading, like in spinal cord-injured patients, the knee cartilage experiences an accelerated degeneration [14]. In contrast, overloading and chronic overuse may shift this balance to catabolic processes, leading to cartilage degradation and onset of osteoarthritis [15]. For instance, the maximum pressure endured by the human knee during a normal gait ranges from 3 to 9 MPa [16]. If exceeded, mechanical loading can cause degenerative changes in the cartilage [16, 17]. Likewise, frequency is another crucial factor. Immutable or static loading, in the physiological range, fails to elicit a chondrogenic response [18]. On the contrary, moderate dynamic mechanical loading, with load frequencies exceeding 0.2 Hz, promotes cartilage growth and matrix synthesis [19]. In vitro studies further identified that cyclic compression at frequencies of 0.01 to 1 Hz and strain amplitudes of 1% to 5% stimulated aggrecan biosynthesis [15]. Low-level cyclic tibial compression after joint injury was also found to attenuate post-traumatic osteoarthritis (PTOA) progression in mice [20]. However, the mechanotransduction mechanism in the context of chondrocytes is still poorly understood. Understanding the interplay between mechanical loading and other biological factors within the joint is necessary to gain insight into the OA disease initiation and progression and in order to find targets for therapeutic intervention. In this review, we pay special attention to the mitochondria and discuss their roles in orchestrating mechanical stimuli and other physiological signals.
The mitochondrion is generally seen as an energy factory that converts nutritional molecules into adenosine triphosphate (ATP) via oxidative phosphorylation [21]. It is a double-membrane-enclosed organelle composed of the outer membrane, intermembrane space, and inner membrane (Figure 1) [22]. As a semiautonomous organelle, mitochondrion has its own independent genome called mitogenome, which encodes 13 proteins, 22 transfer RNAs, and 2 mitochondrial ribosome-coding RNAs [23]. The mitochondrial outer membrane (MOM) encloses the entire organelle and contains porins and enzymes that are involved in diverse activities, such as the elongation of fatty acids, oxidation of epinephrine, and the degradation of tryptophan [24]. Increased permeabilization of MOM results in the leakage of proteins from the intermembrane space to the cytosol, eventually leading to apoptosis [25]. The inner membrane is extensively folded and compartmentalized, forming numerous invaginations, known as cristae, to increase total inner membrane surface area. Unlike the outer membrane, the inner membrane does not contain porins, and is highly impermeable. Only several types of molecules such as oxygen, carbon dioxide, and water can freely travel through [26]. Enzymes involved in ATP production are embedded on the inner membrane, including NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), cytochrome c reductase (complex III), cytochrome c oxidase (complex IV), and ATP synthase (sometimes referred to as complex V) [27].
Figure 1. Overview of mitochondrial structure and components.
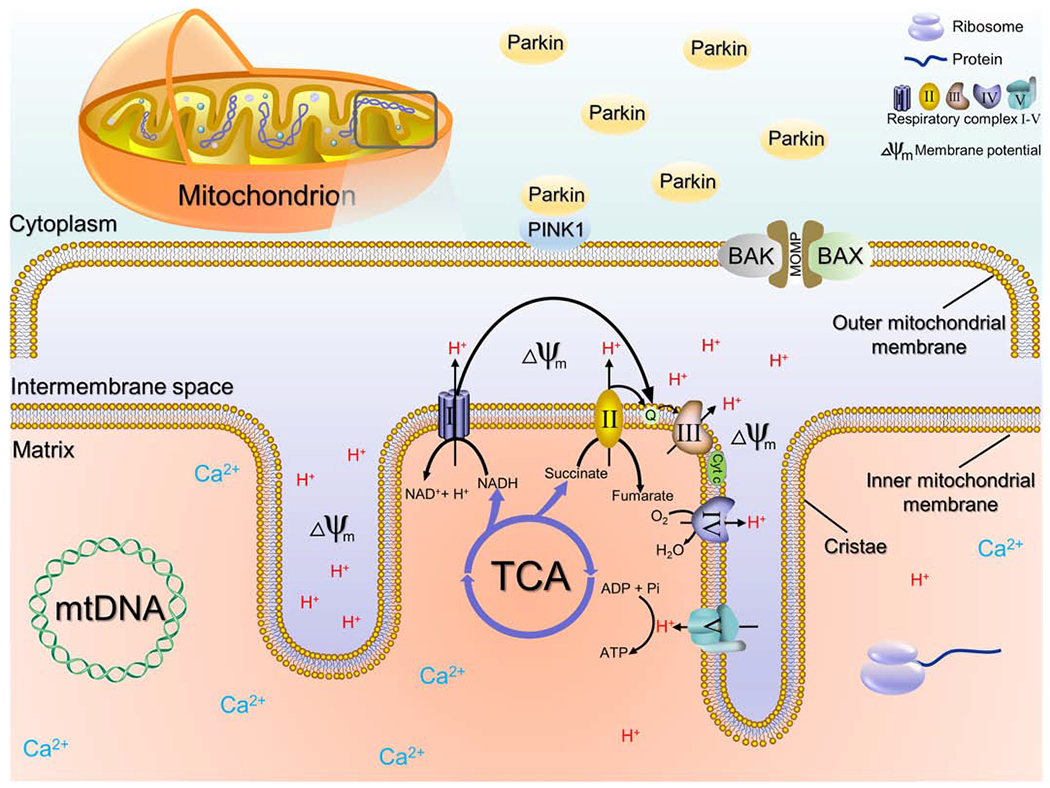
The mitochondrion is composed of outer membrane, intermembrane space, and inner membrane. Enzymes involved in ATP production are embedded on the inner membrane, including NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), cytochrome c reductase (complex III), cytochrome c oxidase (complex IV), and ATP synthase (sometimes referred to as complex V). Irons, mitochondrial DNA (mtDNA) and protein synthesis related structures like ribosomes exist in the mitochondrial matrix. PTEN-induced putative kinase protein 1 (PINK1) exists on the outer membrane of all mitochondria. Mitochondrial damage and membrane potential loss lead to the accumulation of PINK1 on the impaired mitochondria, which then recruits the E3 ubiquitin ligase parkin from the cytosol. PINK1 phosphorylates and activates Parkin, which in turn, ubiquitylates mitochondrial proteins. Finally, disrupted mitochondria are engulfed by isolation membranes and fuse with lysosomes to form autophagosomes. Mitochondrial damage also activates BCL-2-associated X protein (BAX) and BCL-2 antagonist or killer (BAK), which in turn initiate the permeabilization of MOM. Increased permeabilization permits the leakage of proteins from the intermembrane space to the cytosol, eventually leading to apoptosis.
The respiratory chain involved in generating energy also produces reactive oxygen species (ROS), making mitochondria the primary source of ROS in mammalian cells [28]. Excessive ROS production and resultant mitochondrial damage have been observed in a range of diseases. Interestingly, mitochondria also possess a ROS neutralizing mechanism. In particular, superoxide dismutase (SOD) is a critical antioxidant defense that alternately catalyzes the dismutation of the superoxide (O2−) radical into ordinary molecular oxygen (O2) and hydrogen peroxide (H2O2), protecting cells from oxidative damage [29]. In humans, three forms of superoxide dismutase are present, including SOD1, SOD2 and SOD3 [30].
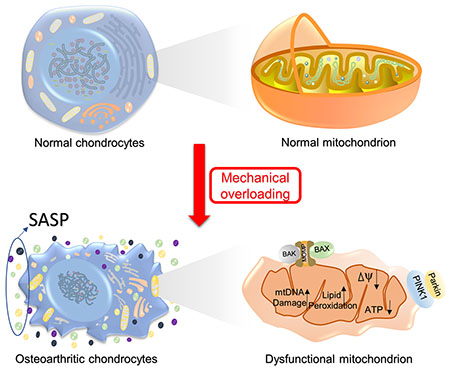
In healthy cartilage, chondrocytes maintain low mitochondrial content and slow rates of respiration in order to minimize oxidative damage [31]. During OA progression, increased proneness to aerobic respiration and mitochondrial DNA (mtDNA) damage, impaired mtDNA repair capacity, and decreased mitochondrial biosynthesis ability together induce mitochondrial dysfunction in OA pathogenesis [32]. Apart from energy generation, mitochondria also produce ‘non-energetic’ intracellular signals that are able to influence the gene expression in nucleus. The dysfunctional mitochondria in osteoarthritic chondrocytes may cause a detrimental impact on a number of cellular processes and metabolic pathways [33]. Interestingly, increasing research on the response of chondrocytes to mechanical loading has also revealed the important role of mitochondria in this process [34, 35]. Taken together, we propose that mitochondria play a critical role in mediating the mechanical stress-associated osteoarthritic changes in chondrocytes.
1. Mitochondrial dysfunction and OA pathogenesis
In addition to energy production, mitochondrial function is also associated with inflammation regulation, apoptosis induction, calcium metabolism, and the generation of ROS, and reactive nitrogen species (RNS) [36, 37]. Recent investigations have yielded new evidence on the relation between alterations of mitochondrial function and cartilage degeneration/OA pathogenesis, such as increased chondrocyte apoptosis, decreased type II collagen secretion, calcification of the cartilage matrix, and inflammation-mediated matrix breakdown [38].
1.1. Cellular nutrient sensors and mitochondrial function
Cell metabolic state is constantly changing in response to various inner and outer physiological signals. In order to maintain homeostasis and appropriate cellular function, cells develop several nutrient-responsive regulatory systems which orchestrate an integrated physiological response exclusively to cellular energy fluctuations. These regulations are not restricted to one or two organelles but involve all of the components in the cell, together modulating cell metabolism, growth, differentiation, and aging [39]. Several molecules have been identified as the universal energy sensors in mammalian cells, which include: (1) the AMP-activated protein kinase (AMPK) [40, 41], (2) the target of rapamycin (mTOR) [42, 43], (3) the nicotinamide adenine dinucleotide (NAD)+-dependent deacetylase sirtuin 1 (SIRT1) and SIRT3 [44, 45], (4) the peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) [46, 47], (5) the PAS domain-regulated serine/threonine kinase (PASK) [48, 49], (6) the hexosamine biosynthetic pathway (HBP) [50–52], (7) peroxisome proliferator-activated receptor-a (PPARa) [53, 54], and (8) farnesoid X receptor (FXR) [55–57]. The unfolded protein response (UPR) also participates in the energy sensing [58, 59]. Functions and regulatory mechanisms of these molecular sensors are summarized in Table 1. The interplay among these pathways are also demonstrated in Figure 2. The complex regulatory network jointly regulates the mitochondrial metabolic adaptation, biogenesis, and mitochondrial-nuclear communication, maintaining the homeostasis [60–62].
Table 1:
Summary of energy sensors in mammalian cells.
| Sensor | Activator | Target genes | Effect | Reference |
|---|---|---|---|---|
| AMPK | ADP: ATP Ca2+ flux | Phosphorylates multiple metabolic genes | Inhibits anabolism, promotes catabolism, autophagy and mitophagy | [40, 41] |
| mTOR | Amino acids Growth factors | Phosphorylates S6 kinases (S6Ks), 4EBP1/2 and Akt | Inhibits autophagy, promotes cell growth and anabolism. | [42, 43] |
| SIRTs | NAD+: NADH | Deacetylates multiple transcription factors and co-factors | Inhibits anabolism and inflammation, promotes insulin secretion, influences hypothalamus/pituitary axis | [44, 45] |
| PGC-1α | Exercise, fasting, cold exposure, Ca2+ | PPARs, ERRα, FoxO1, NRF1 | Promotes gluconeogenesis, fatty acid oxidation and angiogenesis; Regulates circadian clock | [46, 47] |
| PASK | Glucose | Phosphorylates Pdx-1, eEF1A1, S6 and glycogen synthase (Gsy) | Promotes insulin secretion, glycogen synthesis, protein translation and lipid accumulation | [48, 49] |
| HBP | Glucose | O-GIcNAc modification of Sp1, PGC-1β, and NRF1 | Decreases mitochondrial function and oxidative phosphorylation; Increases leptin production | [50–52] |
| PPARs | Fatty acids and fatty acid derivatives | Fatty acid oxidation and autophagy related genes | Induces fatty acid oxidation, autophagic lipid degradation and lipophagy. | [53, 54] |
| FXR | Bile acids | Hal, Prodh, Cps1, Ass1, Glul | Suppresses gluconeogenesis and autophagy; Promotes cholesterol metabolism | [55–57] |
| UPR | Unfolded proteins | IRE1, eIF2, PERK, ATF6 | Regulates protein folding, ER biogenesis, autophagy, innate immunity and glucose metabolism | [58, 59] |
Figure 2. The regulatory network that mediates the association between mitochondrial dysfunction and other physiological activities.
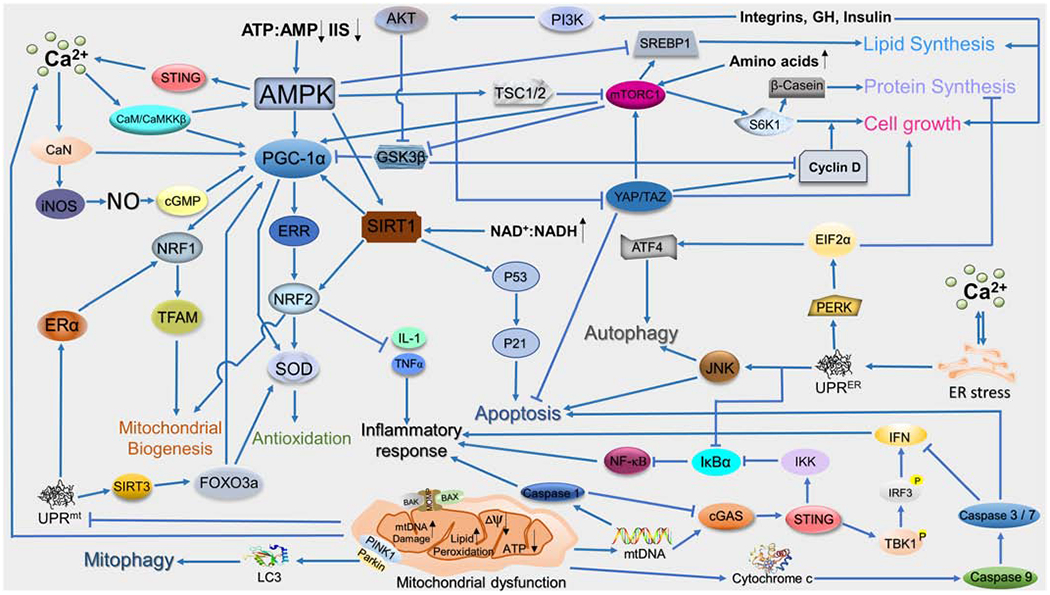
ADAMTS-5: A Disintegrin And Metalloproteinase with Thrombospondin Motifs 5; AKT: RAC-alpha serine/threonine-protein kinase; AMPK: 5′ AMP-activated protein kinase; CaMKKβ: Calcium/calmodulin-dependent protein kinase kinase 2; CaN: Calmodulin; Cdc42: Cell division control protein 42 homolog; COX: cyclooxygenase; eIFs: Eukaryotic initiation factors; ERR: Estrogen-related receptor; FAR: Focal adhesion kinase; MAPK: Mitogen-activated protein kinase; MCU: Mitochondrial calcium uniporter; NAD: Nicotinamide adenine dinucleotide; NFAT: Nuclear factor of activated T-cells; NRF1/2: Nuclear respiratory factor1/2; PI3K: Phosphatidylinositol 3-kinase ; RNS: Reactive nitrogen species; SIRT1: Silent information regulator 1; STING: Stimulator of interferon genes; SOD: Superoxide dismutase; TNF-α: Tumor necrosis factor α; UPR: Unfolded protein response; VDAC1: Voltage Dependent Anion Channel 1.
OA chondrocytes are sensitive to increased endoplasmic reticulum (ER) and mitochondrial stress, which may cause altered activity of energy biosensors and even cell apoptosis [63]. For example, biosensor deficiency in chondrocytes, such as the lack of AMPK, PGC-1α or the repression of SIRT1/AMPK pathway, accelerates the progression of OA in mice [64, 65]. Activation of AMPK, PGC-1α, SIRT1 or SIRT3 prevents the progression of osteoarthritis by preserving mitochondrial DNA integrity and function [66–69]. Autophagy is a protective cell survival mechanism, preventing cells from undergoing apoptosis induced by ER stress. mTOR is a major repressor of autophagy. Overexpression and phosphorylation of mTOR have been observed in human OA cartilage as well as animal OA models [70], which correlates with increased chondrocyte apoptosis and reduced expression of essential autophagy genes during OA. Inhibition of the PI3K/AKT/mTOR signaling pathway or persistently increasing UPR expression were shown to promote the autophagy of articular chondrocytes and attenuate inflammation response in OA chondrocytes [71–73].
1.2. Mitochondrial metabolism change and OA
Articular cartilage is an avascular dense tissue with limited permeability. Under normal physiological conditions, nutrition is supplied to chondrocytes mainly through diffusion, predominately from the synovial fluid (SF) and sparingly from subchondral bone marrow (BM) [74]. Asymmetric, decreasing oxygen and nutrient gradients therefore exist from the superficial to the deep zones and are affected by many factors such as cell density, cartilage thickness, mechanical loading, cell consumption rate, and nutrition concentration in synovial fluid [75]. The oxygen tension of synovial fluid in humans is 6.5–9.0% [76]. In knee joint cartilage, oxygen tension decreases from 5–7% in the superficial zone to less than 1% in the deep zone, a very low level compared to 13% in arterial blood [75]. Under such extremely hypoxic conditions, chondrocytes restrict metabolism to relatively low rates[27, 77]. Due to the limited availability of oxygen supplies, anaerobic glycolysis is thus the major energy source of chondrocytes, while mitochondrial oxidative phosphorylation account for no more than 25% of the ATP production [78, 79]. The relatively low mitochondrial content and slow rates of respiration are also helpful for chondrocytes to minimize oxidative damage [31].
However, in osteoarthritic cartilage, a different situation is observed. Due to cartilage thinning, microcracks, and even disappearance in OA, chondrocytes in the deep zone are directly exposed to the synovial fluid, while cells in the superficial zone are easily exposed to the biochemical factors generated by subchondral bone. Additionally, the increased number of microfractures and vascular channels within the subchondral bone may lead to the leakage of components in subchondral bone marrow in to the cartilage [80, 81]. Therefore, the originally established oxygen tension and molecular gradients in the healthy cartilage are completely altered [75, 82]. Under a relatively high oxygen tension environment, chondrocytes will change their metabolic state to a higher ratio of aerobic metabolism, producing more ROS as byproducts [83, 84]. Moreover, the expression of SOD2significantly decreases in OA chondrocytes at both gene and protein levels, further increasing the accumulation of ROS [85]. Massively accumulated ROS results in DNA damage, lipid peroxidation, oxidation of amino acids in proteins, which eventually triggers cell apoptosis [83].
1.3. mtDNA haplogroups and OA
Exposed to different stresses such as excessive ROS, mtDNA has very high rates of mutation and sequence evolution [86]. In fact, mtDNA with very different sequences can coexist alongside their normal counterparts without causing pathological changes. This situation is also referred to as “heteroplasmy” [87]. The mutations in mtDNA are maternally inherited and have accumulated throughout human history, primarily shaped by climate selection as humans migrated into colder living environments [88]. After a long period of evolution, the presence of a particular set of single nucleotide polymorphisms (SNPs) accumulated sequentially along radiating maternal lineages, forming so-called haplogroups [89]. As affected by climate and many other environmental factors, mtDNA haplogroups are continent-specific[90]. Briefly, haplogroups HV, IWX, JT, and KU account for >90% of European mitochondrial mtDNA variation, haplogroups A, C, D, G, Y and Z represent >75% of the mtDNA variability in Siberia, haplogroups A, B, C, D, and X encompass 100% of the mtDNA variation in native Americans, macrohaplogroups N and M contributed equally to mtDNA radiation in southeastern, Central Asia, and Africa. Furthermore, the three most ancient mtDNA haplogroups are L0, L1 and L2 [88, 91]. These accumulated mutations, including synonymous, non-synonymous, and non-coding mutations, affect gene transcription, protein synthesis, and functional changes of complex I to V, resulting in different susceptibilities to insults in multiple human diseases [86, 92].
mtDNA haplogroups have been demonstrated to influence many aspects of OA, including OA incidence, prevalence, and progression [93] (Table 2). The 8-year longitudinal osteoarthritis initiative (OAI) and cohort hip and cohort knee (CHECK) cohorts show that mtDNA haplogroup J is associated with a lower incident rate of knee OA [94]. Investigations on the relationship between mtDNA haplogroups and the prevalence of OA reveals that the mtDNA haplogroup J and T have a decreased risk of knee OA in Spain and UK populations respectively [95, 96]. Furthermore, in a population study from China, mtDNA haplogroup B seems to be a protective factor against knee OA [97]. mtDNA haplogroup T and mtDNA cluster JT show a lower risk of radiographic knee OA progression [98, 99]. The protective role of these haplogroups may be attributed to low mitochondrial respiration and glycolysis, and reduced production of free radical and pro-inflammatory cytokines such as IL-33, IL-6 and TGFB2 [100], low grade of apoptosis, as well as decreased expression of the mitochondrially related pro-apoptotic gene BCL2 binding component 3 (BBC3) [94]. As not all patients carrying these haplogroups respond in the same way or are protected from OA, the precise identification of molecular mechanism of each haplogroup is needed to finally use haplogroups as a means to predict OA.
Table 2:
Influence of mitochondrial haplogroups.
| mtDNA Haplogroup | Mutation Type | Protein affected | Effect | Related to OA | Ref. |
|---|---|---|---|---|---|
| Haplogroup H | Synonymous | Complex I Complex IV |
Increased ROS production and expression of IL33 and NF-κB; Increased risk of retinopathy and neuropathy. | - | [86, 92] |
| Haplogroup I | Synonymous | Complex I | Increased risk of Amyotrophic Lateral Sclerosis. | - | [86, 92] |
| Haplogroup W | Synonymous | Complex I | Increased longevity. Protection against Parkinson’s disease. | - | [86, 92] |
| Haplogroup X | Synonymous | Complex IV | Decreased risk of obesity. | - | [86, 92] |
| Haplogroup J | Non-synonymous | Complex I Complex III |
Decreased ROS production, mtDNA damage, blood pressure, NO production and expression of IL33 and NF-κB; Longer telomere length and increased longevity; Decreased risk of morbid obesity. | Decreased OA incidence and prevalence | [93–95] |
| Haplogroup T | Non-synonymous | Complex I | Increased production of ROS. Increased risk of morbid obesity and diabetic retinopathy. Decreased risk of Parkinson’s disease. | Decreased OA prevalence and slows progression | [93] [96] [98] |
| Haplogroup K | Non-synonymous | Complex I Complex V |
Decreased ROS production. Decreased risk of Parkinson’s disease. | - | [86, 92] |
| Haplogroup U | Non-coding | Complex I | Decreased ROS production Decreased risk of Parkinson’s disease. | - | [86, 92] |
| Haplogroup V | Synonymous | Complex I | - | - | [86, 92] |
| Haplogroup L | - | Complexes I, III, IV and V | Decreased mtDNA copy numbers, ROS production and ATP turnover rates; Increased inflammation-related genes. | - | [86, 92] |
| Haplogroup G | Non-synonymous | Complex I Complex III |
Increased OXPHOS and lower glycolysis Increased risk of myocardial infarction. | Increased OA prevalence | [93, 98] |
| Haplogroup B | - | Complex I | Increased risk of type 2 diabetes. | Decreased OA prevalence | [90] |
1.4. Other mitochondrial changes observed in osteoarthritic chondrocytes
Activities of various sirtuins are found to decline in OA chondrocytes, and that reduced sirtuin activity has many detrimental effects on cartilage metabolism and health [101]. For example, SIRT3 regulates mitochondrial dynamics and deacetylates many central mitochondrial antioxidant enzymes such as isocitrate dehydrogenase (IDH2) and superoxide dismutase 2 (SOD2). Therefore, SIRT3 participates in regulating the mitochondrial MRC to maintain normal oxidative mechanisms [102]. The decline in SIRT3 activity and levels impairs antioxidant protein function, leading to the accumulation of ROS and RNS [103]. Increased levels of ROS and RNS cause damage to mtDNA, decrease mitochondrial membrane potential (Δψm), reduce the activity of complex IV, and eventually lead to cell death [104, 105]. The increase in mitochondrial mass observed in OA chondrocytes therefore can be compared to a compensatory mechanism to offset the loss of mitochondria due to mitophagy, deficiency in electron transfer, and resultant low ATP production [106].
Matrix-vesicle-mediated microcalcification is another feature of OA cartilage, and mitochondria play important roles in this process as well. Particularly, calcium stored in mitochondria participates in regulating intracellular Ca2+ homeostasis [107]. When mitochondrial respiration is being suppressed, mineralization in both extracellular matrix (ECM) vesicles and mitochondria themselves is promoted [27, 79]. The movement of mitochondrial Ca2+ also induces cyclooxygenase-2 (COX-2) expression and prostaglandin E2 (PGE2) production, resulting in increased generation of pro-inflammatory cytokines, including IL-1β, IL-33, IL-8, TNFα, NF-κB (nuclear factor kappa-light-chain-enhancer of activated B), COX2, PEG2, and matrix metallopeptidases (MMPs) including MMP1,3 and 13 [108, 109]. Moreover, the release of Ca2+ from mitochondria can also trigger a cascade of caspases, leading to apoptosis and cell death [110, 111].
2. Mitochondrial function in cell responses to mechanical loading
Mechanical loading has complex and diverse effects on regulating the signaling pathway in chondrocytes. Moderate exercise shows protective effects, increasing cartilage thickness, proteoglycan content, and mechanical stiffness [112]. This protective effect may partially be achieved by supplying chondrocytes with more nutrients. As we know, the cartilage matrix restricts the travel of molecules by size, charge, and molecular configuration. Additionally, the average pore size within the ECM is approximately 6.0 nm [19]. Such a dense structure forms the basis for low permeability, which prevents fluid from being quickly perfused into or squeezed out of the matrix [113]. Under moderate dynamic mechanical loading, however, the compression of articular cartilage augments the fluid flow, promoting the transport of nutrients into and waste products out of the tissue [113]. Regular joint motion, even once per day, is helpful to enhance fluid flow in a joint effusion [114]. This also partly explains why static loading does not have a protective role. While loading above or below the suitable range is believed to cause atrophy of the cartilage, characterized by cartilage thinning and a decrease in proteoglycan content [115].Despite extensive research efforts in the past, mechanical stress sensing, and transduction in chondrocytes, known as mechanotransduction, is still poorly understood. Here we summarize several molecular mechanisms and discuss the role of mitochondria in this process.
2.1. Mechanotransduction in chondrocytes
Mechanotransduction refers to the processes through which cells sense mechanical stimuli and convert these stimuli into biochemical signals that elicit specific cellular responses [116]. There are six major types of stress: compression, tension, shear, bending, fatigue, and torsion. During development, the location of the subchondral growth front is influenced by intermittent shear stress and hydrostatic pressure within the deep zone cartilage [117]. In mature cartilage, compression and tension are more common for chondrocytes as they are encased by ECM in cartilage [118, 119]. Unlike sensory cells which develop special receptors, chondrocytes percept and respond to mechanical loading with non-specialized cell structures or molecules, such as pericellular matrix, cilia, integrins, microfilaments, and the focal adhesion complex [120]. Downstream pathways of each signal receptor are different, which ultimately leads to different resultant responses.
Primary cilia are solitary, non-motile extensions of the centriole that consist of an axoneme of nine doublet microtubules that extend from a basal body surrounded by the ciliary membrane [121]. First named by Sergei Sorokin in 1968, primary cilium has been identified as a cellular sensory organelle and can be found on the cell surface of nearly all nucleated eukaryotic cells during growth arrest [122, 123]. Incidence and length of chondrocyte primary cilia change in response to varying intensity and action time of mechanical stress during joint activity, either “stretched” by mechanical intervention, or “cut” by the excessive intensity of a mechanical stimulus [124]. The unique hair-like structure enables primary cilia to act as mechanosensory organelles that transduce mechanical forces and initiate specific biological signals through regulating Ca2+, receptor tyrosine kinases (RTKs), Hedgehog (Hh), Wingless-INT (WNT), neuronal and purinergic receptors, as well as through communication with the ECM [125]. For instance, Indian Hedgehog (Ihh) is a key component of the regulatory apparatus that regulates chondrocyte proliferation and differentiation [126]. All the proteins required for Hh signal transduction are enriched in primary cilia and change their distribution in response to different mechanical stimuli [127]. As such, the mechanical signals are then converted into biochemical signals through cilia and Ihh. Under cyclic tensile strain (10% strain), hedgehog signaling transduction was activated, which increased the expression of ADAM metallopeptidase with thrombospondin Type 1 Motif 5 (ADAMTS- 5) [128]. However, this response was not seen in the condition of 20% strain, which is partially due to significantly reduced cilia length at high strains. Interestingly, this study also indicated that suppressing histone deacetylase (HDAC)6 prevented cilia disassembly and preserved their capacity in sensing mechanical overloading. Primary cilia’s function is also under the regulation of polycystic kidney disease protein 1-like 1(PKD1L1), a heteromeric transient receptor potential (TRP) channel anchored on the surface of the primary cilia. PKD1L1 controls ciliary calcium concentration and thereby modifies smoothened (SMO)-activated GLI2 translocation and GLI1 expression [129]. It is still unclear how exactly primary cilia recruit Ihh and activate downstream pathways under mechanical stress. Given that protein kinase A (PKA) represses the expression of Hh target genes through phosphorylating sites on GLI3, and PKA activity is closely related to intracellular Ca2+ concentration, we hypothesize that cilia regulate the Ihh pathway under mechanical stress via regulating Ca2+ level.
Mechanosensitive ion channels are another group of force sensors and regulators in chondrocyte mechanotransduction. Activated by mechanical inputs, these transmembrane protein channels react rapidly, leading to the localized flux of ions across the membrane [130]. Mechanosensitive ion channels that play a role in chondrocytes’ perception of stress include transient receptor potential vanilloid 4 (TRPV4), piezo type mechanosensitive ion channel component (PIEZO) 1, and PIEZO2 [131]. TRPV4, a non-selective but Ca2+ preferred membrane cation channel, can be activated by a wide range of stimuli including physical (cell swelling, heat, and mechanical stimulation) and chemical (endocannabinoids, arachidonic acid, and, surprisingly, 4α-phorbol esters) stimuli [132]. In cartilage development, this ion channel is involved in chondrocyte differentiation and loading-induced ECM biosynthesis [133, 134]. GSK205, a TRPV4 antagonist, blocked the biosynthetic response of chondrocytes to dynamic mechanical loading [134]. Similar results were reported that silencing the TRPV4 gene with siRNA abolished the effects of mechanical strain [135]. In contrast, chemical activation of TRPV4 in the absence of loading mimicked the effects of moderate loading, which enhanced chondrocyte anabolic activity and suppressed catabolic expression [134]. An in vivo study showed that global Trpv4 knockout increased the susceptibility to both age-related and obesity-induced OA [136, 137]. However, a conflicting result was reported as well, in which cartilage-specific knockout of TRPV4 in adult mice demonstrated a protective effect and reduced the severity of aging-associated OA [138]. Given that increased intracellular Ca2+ concentration and mitochondrial ROS generation have a close mutual promotion effect in several mammalian cells, TRPV4-mediated Ca2+ intracellular accumulation may play an important role in mediating the loading-induced release of mitochondrial ROS [139, 140].
PIEZOs are large-transmembrane proteins identified as pore-forming ion channels directly gated by membrane stretch and are conserved among various species [141]. PIEZO2 is mainly involved in mediating gentle touch sensation, while PIEZO1 has broad roles in multiple physiological processes, including sensing mechanical stress, regulating cell function, and controlling cell migration and differentiation [142, 143]. Hydrostatic pressure sensed by PIEZO1 in bone marrow-derived mesenchymal stem cells (BMSCs) promoted the expression of bone morphogenetic protein-2 expression via the activation of the ERK1/2 and p38 MAPK signaling pathway [144]. In chondrocytes, compressive overloading induces PIEZO-mediated accumulation of intracellular Ca2+, resulting in caspase-12 and ER-stress-mediated cell apoptosis, potentially contributing to the development of OA [145]. Blocking PIEZO1 with PIEZO-blocking peptide, or Piezo-specific siRNA, protected chondrocytes from apoptosis [146, 147]. It should be noted that TRPV4 and PIEZOs extensively interact with each other. High-speed pressure clamps and elastomeric pillar arrays revealed that both TRPV4 and PIEZO1 channels contributed to stimuli-activated currents in primary murine chondrocytes [148]. Recently, PIEZO1-induced TRPV4 channel openings were found in pancreatic acinar cells [149, 150]. These findings indicate the possibility that these two ion channels might regulate through separate, but overlapping mechanoelectrical transduction pathways to regulate intracellular Ca2+ levels and mitochondrial function [148, 151].
The cytoskeleton and structurally connected proteins, such as integrins and lamin A, are also involved in mechanotransduction. Mechanical stimulation of chondrocytes leads to cytoskeleton deformation and associated distortion of intracellular organelles [152, 153]. Subsequent cytoskeleton reorganization is related to lethal mitochondrial ROS and mitochondrial oxidant release [154]. For example, Brouillette et al. identified that the release of lethal amounts of ROS from mitochondria increased linearly relative to the magnitude of cartilage deformation. Significant cell death was observed at strains > 40%. By contrast, hydrostatic stress, which causes minimal tissue strain, had no significant effect [153]. More evidence was found by using agents that promote the dissolution of major cytoskeleton protein, microfilaments (cytochalasin B), or microtubules (nocodazole). Both agents significantly reduced impact-induced oxidant release and cell death [155]. Integrins are heterodimeric transmembrane glycoproteins composed of a large extracellular domain, a single transmembrane region, and a short cytoplasmic tail [156]. Extracellular domains bind a host of cartilage extracellular matrix (ECM) proteins, most notably fibronectin and collagen types II and VI [157]. Excessive mechanical signals are sensed and transmitted to the intracellular tail of the integrin, which regulates different kinases, phosphatases, and other signaling molecules via adapter proteins such as paxillin, Shc, and tensin [158, 159]. Interestingly, the number and subunit composition of integrins change with age, suggesting the impact of aging on chondrocyte response to mechanical loading [157].
Lamin A is an architectural protein of the cell nucleus which lines the interior of the nuclear membrane to provide binding sites and confer mechanical stability [160]. Nuclear deformation caused by either substrate stiffness or applied forces have the potential to induce the redistribution of lamin A from the nuclear membrane to the nucleoplasm [161]. The translocation of lamin A then directly promotes the transportation of Yes-associated Protein (YAP) to the nucleus, further activating its paralog, the transcriptional co-activator with PDZ-binding motif (TAZ) [162]. Nuclear YAP activity controls the transcription of genes involved in cell cycle control, typically driving proliferation and survival, blocking apoptosis and differentiation [163, 164]. YAP also has crosstalk with other mechanosensitive pathways such as Akt/mTOR and TGFß/SMAD, together regulating mechanotransduction and cell reactions [163]. The mechanistic role of lamin A in YAP transport remains unclear, however, it is likely that lamin A interacts with nuclear pore complexes (NPCs) to regulate YAP translocation as a correlation between lamin A distribution and NPC density, which has been observed in the nuclear membrane [165]. Moreover, studies on other mechanosensors such as microRNA-365, and voltage-dependent Cl- channels are limited and need further exploration [166, 167].
2.2. Mechanical loading, mitochondria, and OA
It has been long acknowledged that exposure of articular cartilage to supraphysiological mechanical loading can directly evoke cartilage degeneration and the onset of osteoarthritis [168]. Dr. David Felson actually claimed that most or almost all osteoarthritis is caused, in part, by mechanically induced injury to joint tissues [169]. The possible reasons may include inducing apoptosis and cell death, evoking inflammation due to the breakdown of cartilage matrix, and activating the stresses-associated pathways that generate detrimental molecules [170]. In fact, the term “mechanoflammation” has been proposed to reflect the fact that mechanical stress can drive inflammatory signaling [171]. However, little is known about the molecular mechanism underneath. Given demonstrated effects of mechanical stress on mitochondrial ATP production, membrane potential change, and the increased generation and release of oxytoxins, a causal link between mitochondrial dysfunction and OA pathogenesis, has been implied [16]. In Figure 3, we summarize the known molecules that are involved in this relationship. It should be noted, however, that a significant knowledge gap still remains.
Figure 3. The pathways that may participate in regulating the responses of mitochondria to mechanical stimuli.
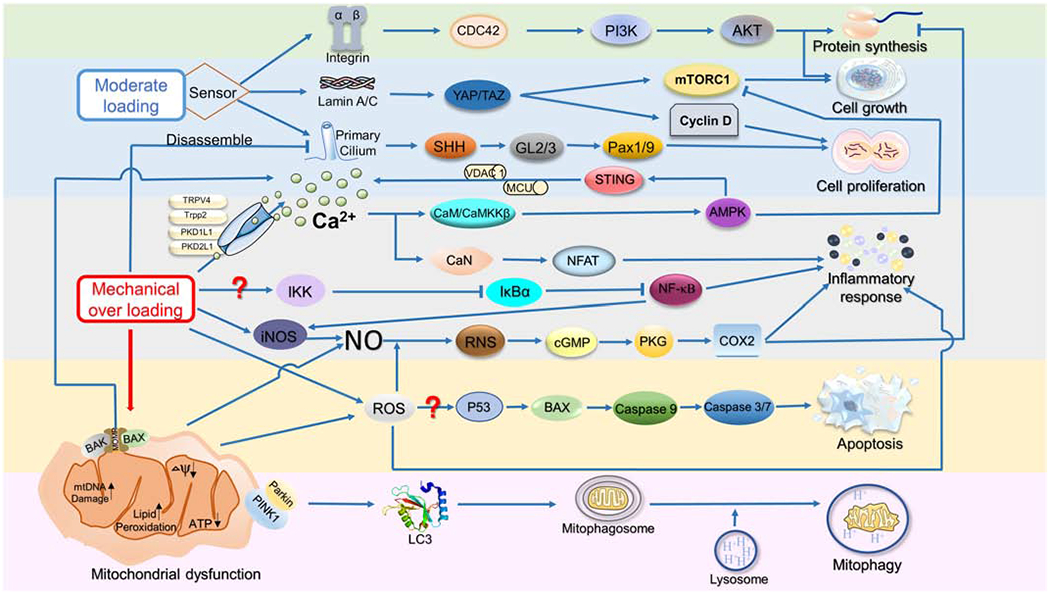
Mechanotransduction in chondrocytes starts from the perception of mechanical stimuli, and then cilia, integrins, microfilaments, and focal adhesion complex activate different downstream resultant responses, eventually affecting molecular synthesis, cell growth, proliferation, and cells death. ADAMTS-5: A Disintegrin And Metalloproteinase with Thrombospondin Motifs 5; AKT: RAC-alpha serine/threonine-protein kinase; AMPK: 5′ AMP-activated protein kinase; CaMKKβ: Calcium/calmodulin-dependent protein kinase kinase 2; CaN: Calmodulin; Cdc42: Cell division control protein 42 homolog; COX: cyclooxygenase; eIFs: Eukaryotic initiation factors; FAK: Focal adhesion kinase; MAPK: Mitogen-activated protein kinase; MCU: Mitochondrial calcium uniporter; NAD: Nicotinamide adenine dinucleotide; NFAT: Nuclear factor of activated T-cells; NRF1/2: Nuclear respiratory factor1/2; PI3K: Phosphatidylinositol 3-kinase ; RNS: Reactive nitrogen species; SIRT1: Silent information regulator 1; SOD: Superoxide dismutase; TNF-α: Tumor necrosis factor β; UPR: Unfolded protein response; VDAC1: Voltage Dependent Anion Channel 1.
ATP production and ATP/ADP ratio decrease under cyclic compression accompanied by suppressed respiratory activity, increased proton leakage, decreased mitochondrial membrane potential, as well as increased ROS formation in bovine cartilage explants [172]. These changes have a significant impact on chondrocytes. For example, mitochondrial damage and depolarization lead to the accumulation of PTEN-induced putative kinase protein 1 (PINK1) on the mitochondrial surface, which normally is imported into healthy mitochondria and cleaved by mitochondrial proteases [173]. If the importing process fails due to mitochondrial dysfunction, PINK1 autophosphorylates, dimerizes, accumulates on the outer mitochondrial membrane, and then phosphorylates Parkin and ubiquitin to trigger mitophagy [174]. Studies have found that Pink1-mediated mitophagy leads to chondrocyte death, while deficiency in Pink1 expression was associated with decreased cartilage damage and pain behaviors in the Monoiodoacetate (MIA)-induced OA model, suggesting that targeting the Pink1 pathway may provide a therapeutic avenue for OA treatment [175]. Also, this oxidant-dependent mitochondrial dysfunction, caused by mechanical stimuli, may contribute to the destruction of cartilage during various stages of OA by disrupting chondrocyte anabolic responses[176]. Activation of the AMPK-SIRT1-PGC1α pathway then increases mitochondrial biogenesis in chondrocytes. Conversely, the activation of AMPK-SIRT-3 pathway induced Parkin expression, SOD2 activation, and mitophagy, together reducing mitochondrial stress [104]. The activity of AMPK and its regulatory upstream kinase, serine/threonine-protein kinase (STK11), is reduced in bovine chondrocytes after dynamic compression-induced biomechanical injury [177]. This reduced activity has been associated with substantially reduced mitochondrial biogenesis and ATP production [178].
The mitochondrial membrane potential is generated by proton pumps (Complexes I, III, and IV), and forms the transmembrane potential of hydrogen ions in the process of energy storage during oxidative phosphorylation (OXPHOS) [179]. Under mechanical stress, the endoplasmic reticulum releases Ca2+ to the cytoplasm via the ryanodine receptor, which is then taken up by the mitochondria to maintain cellular Ca2+ balance. The accumulation of excessive calcium in the mitochondrial matrix leads to the formation of the permeability transition pore (PTP), in turn causing the collapse of the mitochondrial transmembrane potential, resulting in the release of Ca2+, and mitochondrial proteins, such as cytochromes [180, 181]. Such mitochondrial depolarization and loss of mitochondrial proteins causes a series of cascade reactions, constituting a positive feedback regulation, and eventually leads to caspase-9 dependent apoptosis and cartilage degradation [182].
Expression level and activity SOD2 constitute an important part of mechanical-overloading-induced mitochondrial dysfunction and aging-related cartilage degeneration. In a surgically induced destabilization of the medial meniscus (DMM) OA mice model, mechanical overloading resulted in decreased Sod2 expression and SOD2 levels in knee chondrocytes. SOD2 deficiency resulted in mitochondrial superoxide overproduction, thus leading to mitochondrial dysfunction and cartilage damage [34]. This mitochondrial superoxide imbalance and mitochondrial dysfunction could be eliminated by the supplement of small-molecular-weight superoxide dismutase mimetics in a dose-dependent effect [35].
Conclusion and Future Perspectives
Despite tremendous efforts spent in OA research in last decades, few breakthroughs have been made in the prevention or treatment of OA. For example, pharmacological benefits for OA development using MMP and ADAMTS5 inhibitors in humans were not observed, and the benefits of anticytokine therapies in OA clinical trials are so far uncertain [183, 184]. With no effective disease-modifying osteoarthritis drugs (DMOADs) approved by regulatory bodies, OA is still a worldwide disease that affects the health of millions of people. For symptomatic end-stage OA patients, total joint replacement is an effective treatment with high success rates. But potential side effects, such as infection, and fibrosis, and poor functional outcome are also observed in ~2% OA patients undergoing total joint replacement [185]. Additionally, in spite of progress in the generatio of artificial joints, the lifespan of prostheses is limited and variable [2]. Given that more than 2 million total joint replacements are performed globally each year [186], the number of OA patients that don’t benefit from this major surgery is remarkable. Thus, identifying and treating OA before it causes structural changes and functional disability are of fundamental importance [187].
OA is known as a multifactorial disease. Although mechanical stress is the discussing topic in this review, other risk factors are also involved, such as aging and obesity. Interestingly, both aging and obesity accompany the changes of mitochondrial function as well [188, 189]. Autophagy has been identified as a protective mechanism in normal cartilage, and aging-related decline of autophagy is linked with cell death and osteoarthritis [190]. As a selective degradation of mitochondria via autophagy, mitophagy is decreased in Parkin-depleted chondrocytes, resulting in increased production of ROS, accumulation of dysfunctional mitochondria, and apoptosis [191]. Therefore, instead of targeting the individual upstream signals/molecules, we propose modulating the common downstream hub of multiple regulatory pathways, such as mitochondria, may provide a more efficient strategy to find therapeutic treatments.
There are several strategies to mitigate the impact of mitochondrial dysfunction on cell functionality. The most straightforward method is removing malfunctional mitochondria via enhancing autophagy or mitophagy, which have shown ameliorating effects upon OA development, pointing to the notion that enhancing/restoring normal mitophagy might be a good OA treatment approach [192, 193]. Other methods aim to restore the normal function of mitochondria through targeting key molecules. For example, a novel mTOR-independent autophagic inducer, Trehalose, displays a protective effect in OA mice models by ameliorating oxidative stress-mediated mitochondrial membrane potential collapse, ATP level decrease and ER stress, via selective autophagy stimulation and autophagic flux restoration [194]. Other mitochondrial protective drugs that have been tested in OA animal models include quercetin [195], SS peptides [196], mitochondria-specific antioxidant MitoTempo [197] and metformin [198]. Among the many changes resulted from the loss of mitochondrial homeostasis, ion imbalance, especially calcium imbalance, seem to be at the core. As discussed above, Ca2+ released from the mitochondria leads to mitochondrial membrane depolarization, oxidative phosphorylation suppression, reduced ATP production, as well as increased ROS production that may cause mtDNA damage and denaturation of mitochondrial proteins. These harmful changes, once started, influence each other to form a positive feedback regulation path. Therefore, modulation Ca2+ flux to break this circle might represent a therapeutic strategy for the prevention and treatment of OA. Specifically, reducing mitochondrial permeability and transition pore, or modulating the function of key factors such as calcium/calmodulin-regulated kinase II (CaMKII) and calpains, are other potential options to treat OA.
As one of the major risk factors, the effects of mechanical stress in OA pathogenesis is specially focused on in this review. Through summarizing the relevant findings, we propose the connective role of mitochondria in the relationship between mechanical stress and OA. Mechanical overloading-induced SOD reduction led to swollen mitochondria with disrupted cristae [34], which resulted in increased proton leakage, decreased mitochondrial membrane potential, as well as suppressed respiratory activity, and ATP levels, eventually contributing to OA [172]. Furthermore, ECM turnover was also influenced by mechanical loading through an ATP-dependent mitochondrion-regulated way [199]. Taken together, these molecules mediate mitochondrial dysfunction induced by mechanical stress, which can serve as the therapeutic targets of post-traumatic OA.
In future studies, non-invasive methods should be generated to monitor the health of mitochondria in cartilage-based chondrocytes, which will inform us of the intervention timing. For example, the protoporphyrin IX-triplet state lifetime technique (PpIX-TSLT) has been used to noninvasively measure the mitochondrial oxygen tension (mitoPO2) in rats [200]. On the other hand, safe, specific and efficient treatment is needed to restore the normal mitochondrial function. Zhang et al. recently reviewed the drug therapy for mitochondrial diseases [103] and many of them haven’t been examined in the context of osteoarthritis. In addition, since animal models generally don’t allow for high throughput screening, the newly emerged microphysiological tissue chips that simulate OA pathology in humans [201, 202] will be powerful tools to quickly identify potential mitochondria-restoring drugs for pre-clinical studies and clinical trials.
Lastly, in this review we only summarize the mitochondrial function and alterations in chondrocytes/cartilage. As mentioned above, OA is a whole joint disease that affects all joint elements. Therefore, understating the mitochondrial alterations in other cells/tissues is also necessary, which, however, has not been well investigated. Particularly, whether the methods to enhance the mitochondrial function in chondrocytes work on other cell types needs special attention, which is critical to assess the efficacy of mitochondria-targeting strategy in treating osteoarthritis.
Acknowledgement
This work was supported in part by the National Institutes of Health (UG3TR003090) and Department of Orthopaedic Surgery at the University of Pittsburgh.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Availability of data and materials
Not applicable.
References:
- 1.Dequeker J, Luyten FP: The history of osteoarthritis-osteoarthrosis. Ann Rheum Dis 2008, 67(1):5. [DOI] [PubMed] [Google Scholar]
- 2.Glyn-Jones S, Palmer AJR, Agricola R, Price AJ, Vincent TL, Weinans H, Carr AJ: Osteoarthritis. Lancet 2015, 386(9991):376–387. [DOI] [PubMed] [Google Scholar]
- 3.Hootman JM, Helmick CG, Barbour KE, Theis KA, Boring MA: Updated projected prevalence of self-reported doctor-diagnosed arthritis and arthritis-attributable activity limitation among US adults, 2015-2040. Arthritis Rheum 2016, 68(7):1582–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace IJ, Worthington S, Felson DT, Jurmain RD, Wren KT, Maijanen H, Woods RJ, Lieberman DE: Knee osteoarthritis has doubled in prevalence since the mid-20th century. Proc Natl Acad Sci U S A 2017, 114(35):9332–9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia B, Di C, Zhang J, Hu S, Jin H, Tong P: Osteoarthritis pathogenesis: a review of molecular mechanisms. Calcif Tissue Int, 2014, 95(6):495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldring SR, Goldring MB: Changes in the osteochondral unit during osteoarthritis: structure, function and cartilage-bone crosstalk. Nat Rev Rheumatol 2016, 12(11):632. [DOI] [PubMed] [Google Scholar]
- 7.Chen D, Shen J, Zhao W, Wang T, Han L, Hamilton JL, Im H-J: Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res 2017, 5(1):16044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, Goldring SR, Jones G, Teichtahl AJ, Pelletier JP: Osteoarthritis. Nat Rev Dis Primers 2016, 2:16072. [DOI] [PubMed] [Google Scholar]
- 9.Sacitharan PK: Ageing and osteoarthritis. Subcell Biochem 2019, 91:123–159. [DOI] [PubMed] [Google Scholar]
- 10.Litwic A, Edwards MH, Dennison EM, Cooper C: Epidemiology and burden of osteoarthritis. Br Med Bull 2013, 105(1):185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun HB: Mechanical loading, cartilage degradation, and arthritis. Ann NY Acad Sci 2010, 1211(l):37–50. [DOI] [PubMed] [Google Scholar]
- 12.Albro MB, Cigan AD, Nims RJ, Yeroushalmi KJ, Oungoulian SR, Hung CT, Ateshian GA: Shearing of synovial fluid activates latent TGF-β. Osteoarthr Cartil 2012, 20(11):1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madej W, van Caam A, Davidson EB, Buma P, van der Kraan PJO, cartilage: Unloading results in rapid loss of TGFβ signaling in articular cartilage: role of loading-induced TGFβ signaling in maintenance of articular chondrocyte phenotype? Osteoarthr Cartil 2016, 24(10):1807–1815. [DOI] [PubMed] [Google Scholar]
- 14.Vanwanseele B, Eckstein F, Knecht H, Stiissi E, Spaepen A: Knee cartilage of spinal cord-injured patients displays progressive thinning in the absence of normal joint loading and movement. Arthritis Rheum 2002, 46(8):2073–2078. [DOI] [PubMed] [Google Scholar]
- 15.Griffin TM, Guilak F: The role of mechanical loading in the onset and progression of osteoarthritis. Exerc Sport Sci Rev 2005, 33(4):195–200. [DOI] [PubMed] [Google Scholar]
- 16.Chang SH, Mori D, Kobayashi H, Mori Y, Nakamoto H, Okada K, Taniguchi Y, Sugita S, Yano F, Chung U-I et al. : Excessive mechanical loading promotes osteoarthritis through the gremlin-1-NF-κB pathway. Nat Commun 2019, 10(1):1442–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Musumeci G: The effect of mechanical loading on articular cartilage. J Fund Morphol Kinesiol 2016. [Google Scholar]
- 18.Li KW, Williamson AK, Wang AS, Sah RL: Growth responses of cartilage to static and dynamic compression. Clin Orthop Relat Res 2001(391 Suppl):S34–48. [DOI] [PubMed] [Google Scholar]
- 19.Sophia Fox AJ, Bedi A, Rodeo SA: The basic science of articular cartilage: structure, composition, and function. Sports Health 2009, l(6):461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holyoak DT, Chlebek C, Kim MJ, Wright TM, Otero M, van der Meulen MCH: Low-level cyclic tibial compression attenuates early osteoarthritis progression after joint injury in mice. Osteoarthr Cartil 2019, 27(10):1526–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolduc JA, Collins JA, Loeser RF: Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med 2019, 132:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palade GE: An electron microscope study of the mitochondrial structure. J Histochem Cytochem 1953, 1(4):188–211. [DOI] [PubMed] [Google Scholar]
- 23.Anderson S, Bankier AT, Barrell BG, de Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F et al. : Sequence and organization of the human mitochondrial genome. Nature 1981, 290(5806):457–465. [DOI] [PubMed] [Google Scholar]
- 24.Zheng J, Li L, Jiang H: Molecular pathways of mitochondrial outer membrane protein degradation. Biochem Soc Trans 2019, 47(5):1437–1447. [DOI] [PubMed] [Google Scholar]
- 25.Suhaili SH, Karimian H, Stellato M, Lee T-H, Aguilar M-DBr: Mitochondrial outer membrane permeabilization: a focus on the role of mitochondrial membrane structural organization. Biophys Rev 2017, 9(4):443–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munn EA: The structure of mitochondria: Academic Press; 1974. [Google Scholar]
- 27.Blanco FJ, Rego I, Ruiz-Romero C: The role of mitochondria in osteoarthritis. Nat Rev Rheumatol 2011, 7(3):161. [DOI] [PubMed] [Google Scholar]
- 28.Waypa GB, Smith KA, Schumacker PT: 02 sensing, mitochondria and ROS signaling: the fog is lifting. Mol Asp Med 2016, 47:76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ighodaro O, Akinloye O: First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alexandria J Med 2018, 54(4):287–293. [Google Scholar]
- 30.Azadmanesh J, Borgstahl GE: A review of the catalytic mechanism of human manganese superoxide dismutase. Antioxidants (Basel, Switzerland) 2018, 7(2):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lane RS, Fu Y, Matsuzaki S, Kinter M, Humphries KM, Griffin TM: Mitochondrial respiration and redox coupling in articular chondrocytes. Arthritis Res Ther 2015, 17(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grishko VI, Ho R, Wilson GL, Pearsall IV AW: Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthr Cartil 2009, 17(1):107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Picard M, Wallace DC, Burelle Y: The rise of mitochondria in medicine. Mitochondrion 2016, 30:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koike M, Nojiri H, Ozawa Y, Watanabe K, Muramatsu Y, Kaneko H, Morikawa D, Kobayashi K, Saita Y, Sasho T et al. : Mechanical overloading causes mitochondrial superoxide and SOD2 imbalance in chondrocytes resulting in cartilage degeneration. Sci Rep 2015, 5:11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coleman MC, Brouillette MJ, Andresen NS, Oberley-Deegan RE, Martin JM: Differential effects of superoxide dismutase mimetics after mechanical overload of articular cartilage. Antioxidants (Basel, Switzerland) 2017, 6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman JR, Nunnari J: Mitochondrial form and function. Nature 2014, 505(7483):335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phaniendra A, Jestadi DB, Periyasamy L: Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem 2015, 30(1):11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu H, Li Z, Cao Y, Cui Y, Yang X, Meng Z, Wang R: Effect of chondrocyte mitochondrial dysfunction on cartilage degeneration: A possible pathway for osteoarthritis pathology at the subcellular level. Mol Med Rep 2019, 20(4):3308–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chandel NS, Jasper H, Ho TT, Passegue E: Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat Cell Biol 2016, 18(8):823–832. [DOI] [PubMed] [Google Scholar]
- 40.Herzig S, Shaw RJ: AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol 2018, 19(2):121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia D, Shaw RJ: AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell 2017, 66(6):789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zoncu R, Efeyan A, Sabatini DM: mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol 2011, 12(l):21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunlop EA, Tee AR: mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 2014, 36:121–129. [DOI] [PubMed] [Google Scholar]
- 44.Li X: SIRT1 and energy metabolism. Acta Biochim Biophys Sin 2013, 45(1):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang H-C, Guarente L: SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab 2014, 25(3):138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Femandez-Marcos PJ, Auwerx J: Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 2011, 93(4):884S–890S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu C, Li S, Liu T, Boijigin J, Lin JD: Transcriptional coactivator PGC-1α integrates the mammalian clock and energy metabolism. Nature 2007, 447(7143):477–481. [DOI] [PubMed] [Google Scholar]
- 48.Schlafli P, Borter E, Spielmann P, Wenger RH: The PAS-domain kinase PASKIN: a new sensor in energy homeostasis. Cell Mol Life Sci 2009, 66(5):876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeMille D, Grose JH: PAS kinase: A nutrient sensing regulator of glucose homeostasis. IUBMB Life 2013, 65(11):921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rossetti L: Perspective: hexosamines and nutrient sensing. Endocrinology 2000, 141(6):1922–1925. [DOI] [PubMed] [Google Scholar]
- 51.Hanover JA, Krause MW, Love DC: The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta Gen Snbj 2010, 1800(2):80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L: A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature 1998, 393(6686):684–688. [DOI] [PubMed] [Google Scholar]
- 53.Grimaldi PA: Peroxisome proliferator-activated receptors as sensors of fatty acids and derivatives. Cell Mol Life Sci 2007, 64(19-20):2459–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poulsen LC, Siersbsek M, Mandrup S: PPARs: Fatty acid sensors controlling metabolism. Semin Cell Dev Biol 2012, 23(6):631–639. [DOI] [PubMed] [Google Scholar]
- 55.Massafra V, van Mil SWC: Farnesoid X receptor: A “homeostat” for hepatic nutrient metabolism. Biochim Biophys Acta Mol Basis Dis 2018, 1864(1):45–59. [DOI] [PubMed] [Google Scholar]
- 56.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD: Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516(7529):112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma K, Saha PK, Chan L, Moore DD: Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest 2006, 116(4):1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hetz C: The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol 2012, 13(2):89–102. [DOI] [PubMed] [Google Scholar]
- 59.Kaufman RJ, Scheuner D, Schroder M, Shen X, Lee K, Liu CY, Arnold SM: The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol 2002, 3(6):411–421. [DOI] [PubMed] [Google Scholar]
- 60.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J: AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458(7241):1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hurtado-Cameiro V, Roncero I, Egger SS, Wenger RH, Blazquez E, Sanz C, Alvarez E: PAS kinase is a nutrient and energy sensor in hypothalamic areas required for the normal function of AMPK and mTOR/S6K1. Mol Neurobiol 2014, 50(2):314–326. [DOI] [PubMed] [Google Scholar]
- 62.Canto C, Auwerx J: PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Carr Opin Lipidol 2009, 20(2):98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye W, Zhu S, Liao C, Xiao J, Wu Q, Lin Z, Chen J: Advanced oxidation protein products induce apoptosis of human chondrocyte through reactive oxygen species-mediated mitochondrial dysfunction and endoplasmic reticulum stress pathways. Fundam Clin Pharmacol 2017, 31(l):64–74. [DOI] [PubMed] [Google Scholar]
- 64.Zhou S, Lu W, Chen L, Ge Q, Chen D, Xu Z, Shi D, Dai J, Li J, Ju H et al. : AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci Rep 2017, 7:43245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alvarez-Garcia O, Matsuzaki T, Olmer M, Plate L, Kelly JW, Lotz MK: Regulated in development and DNA damage response 1 deficiency impairs autophagy and mitochondrial biogenesis in articular cartilage and increases the severity of experimental osteoarthritis. Arthritis & rheumatology 2017, 69(7):1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feng K, Chen Z, Pengcheng L, Zhang S, Wang X: Quercetin attenuates oxidative stress-induced apoptosis via SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and prevents the progression of osteoarthritis in a rat model. J Cell Physiol 2019, 234(10):18192–18205. [DOI] [PubMed] [Google Scholar]
- 67.Li J, Zhang B, Liu WX, Lu K, Pan H, Wang T, Oh CD, Yi D, Huang J, Zhao L et al. : Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann Rheum Dis 2020, 79(5):635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen LY, Wang Y, Terkeltaub R, Liu-Bryan R: Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthr Cartil 2018, 26(11):1539–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao X, Petursson F, Viollet B, Lotz M, Terkeltaub R, Liu-Bryan R: Peroxisome proliferator–activated receptor γ coactivator 1α and FoxQ3A mediate chondroprotection by AMP- activated protein kinase. Arthritis Rheumatol 2014, 66(11):3073–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y, Vasheghani F, Li YH, Blati M, Simeone K, Fahmi H, Lussier B, Roughley P, Lagares D, Pelletier JP et ah Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann Rheum Dis 2015, 74(7):1432–1440. [DOI] [PubMed] [Google Scholar]
- 71.Cai C, Min S, Yan B, Liu W, Yang X, Li L, Wang T, Jin A: MiR-27a promotes the autophagy and apoptosis of IL-1β treated-articular chondrocytes in osteoarthritis through PI3K/AKT/mTOR signaling. Aging 2019, 11(16) 6371–6384 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Xue JF, Shi ZM, Zou J, Li XL: Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed Pharmacother 2017, 89:1252–1261. [DOI] [PubMed] [Google Scholar]
- 73.Wu H, Meng Z, Jiao Y, Ren Y, Yang X, Liu H, Wang R, Cui Y, Pan L, Cao Y: The endoplasmic reticulum stress induced by tunicamycin affects the viability and autophagy activity of chondrocytes. J Clin Lab Anal 2020:e23437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Wei L, Zeng L, He D, Wei X: Nutrition and degeneration of articular cartilage. Knee Surg Sports Traumatol Arthrosc 2013, 21(8):1751–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou S, Cui Z, Urban JPG: Factors influencing the oxygen concentration gradient from the synovial surface of articular cartilage to the cartilage–bone interface: A modeling study. Arthritis Rheum 2004, 50(12):3915–3924. [DOI] [PubMed] [Google Scholar]
- 76.Lund-Olesen K: Oxygen tension in synovial fluids. Arthritis Rheum 1970, 13(6):769–776. [DOI] [PubMed] [Google Scholar]
- 77.Stockwell RA: Biology of cartilage cells. Arthritis Rheum 1979, 7.103559 [Google Scholar]
- 78.Johnson K, Jung A, Murphy A, Andreyev A, Dykens J, Terkeltaub R: Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum 2000, 43(7):1560–1570. [DOI] [PubMed] [Google Scholar]
- 79.Terkeltaub R, Johnson K, Murphy A, Ghosh S: Invited review: the mitochondrion in osteoarthritis. Mitochondrion 2002, l(4):301–319. [DOI] [PubMed] [Google Scholar]
- 80.Hayami T, Pickarski M, Zhuo Y, Wesolowski GA, Rodan GA, Duong LT: Characterization of articular cartilage and subchondral bone changes in the rat anterior cruciate ligament transection and meniscectomized models of osteoarthritis. Bone 2006, 38(2):234–243. [DOI] [PubMed] [Google Scholar]
- 81.Lane LB, Villacin A, Bullough PJTJob, volume jsB: The vascularity and remodelling of subchondrial bone and calcified cartilage in adult human femoral and humeral heads. An age-and stress-related phenomenon. J Bone Joint Surg Br 1977, 59(3):272–278. [DOI] [PubMed] [Google Scholar]
- 82.Mahjoub M, Berenbaum F, Houard X: Why subchondral bone in osteoarthritis? The importance of the cartilage bone interface in osteoarthritis. Osteoporosis Int 2012, 23 Suppl 8:S841–846. [DOI] [PubMed] [Google Scholar]
- 83.Lepetsos P, Papavassiliou AG: ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta Mol Basis Dis 2016, 1862(4):576–591. [DOI] [PubMed] [Google Scholar]
- 84.Murphy Michael P: How mitochondria produce reactive oxygen species. Biochemical Journal 2008, 417(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ruiz-Romero C, Calamia V, Mateos J, Carreira V, Martinez-Gomariz M, Fernandez M, Blanco FJ: Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics 2009, 8(1):172–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taylor RW, Turnbull DM: Mitochondrial DNA mutations in human disease. Nat Rev Genet 2005, 6(5):389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stefano GB, Bjenning C, Wang F, Wang N, Kream RM: Mitochondrial heteroplasmy. Adv Exp Med Biol 2017, 982:577–594. [DOI] [PubMed] [Google Scholar]
- 88.Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, Hosseini S, Brandon M, Easley K, Chen E, Brown MD et al. : Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci U S A 2003, 100(1):171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Torroni A, Huoponen K, Francalacci P, Petrozzi M, Morelli L, Scozzari R, Obinu D, Savontaus ML, Wallace DC: Classification of European mtDNAs from an analysis of three European populations. Genetics 1996, 144(4):1835–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stewart JB, Chinnery PF: The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet 2015, 16(9):530–542. [DOI] [PubMed] [Google Scholar]
- 91.Martinez-Redondo D, Marcuello A, Casajus JA, Ara I, Dahmani Y, Montoya J, Ruiz-Pesini E, Lopez-Perez MJ, Diez-Sanchez C: Human mitochondrial haplogroup H: the highest VO2max consumer--is it a paradox? Mitochondrion 2010, 10(2):102–107. [DOI] [PubMed] [Google Scholar]
- 92.Blanco FJ, Valdes AM, Rego-Perez I: Mitochondrial DNA variation and the pathogenesis of osteoarthritis phenotypes. Nat Rev Rheumatol 2018, 14(6):327–340. [DOI] [PubMed] [Google Scholar]
- 93.Blanco FJ, June RK: Cartilage metabolism, mitochondria, and osteoarthritis. J Am Acad Orthop Surg 2020, 28(6):e242–e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fernández-Moreno M, Soto-Hermida A, Vázquez-Mosquera ME, Cortés-Pereira E, Relaño S, Hermida-Gómez T, Pértega S, Oreiro-Villar N, Fernández-López C, Garesse R et al. : Mitochondrial DNA haplogroups influence the risk of incident knee osteoarthritis in OAI and CHECK cohorts. A meta-analysis and functional study. Ann Rheum Dis 2017, 76(6):1114. [DOI] [PubMed] [Google Scholar]
- 95.Soto-Hermida A, Fernandez-Moreno M, Oreiro N, Fernandez-Lopez C, Rego-Perez I, Blanco FJ: mtDNA haplogroups and osteoarthritis in different geographic populations. Mitochondrion 2014, 15:18–23. [DOI] [PubMed] [Google Scholar]
- 96.Rego I, Fernandez-Moreno M, Fernandez-Lopez C, Gomez-Reino JJ, Gonzalez A, Arenas J, Blanco FJ: Role of European mitochondrial DNA haplogroups in the prevalence of hip osteoarthritis in Galicia, Northern Spain. Ann Rheum Dis 2010, 69(1):210–213. [DOI] [PubMed] [Google Scholar]
- 97.Fang H, Liu X, Shen L, Li F, Liu Y, Chi H, Miao H, Lu J, Bai Y: Role of mtDNA haplogroups in the prevalence of knee osteoarthritis in a southern Chinese population. Int J Mol Sci 2014, 15(2):2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao Z, Li Y, Wang M, Jin Y, Liao W, Zhao Z, Fang J: Mitochondrial DNA haplogroups participate in osteoarthritis: current evidence based on a meta-analysis. Clin Rheumatol 2020, 39(4):1027–1037. [DOI] [PubMed] [Google Scholar]
- 99.Fernández-Moreno M, Soto-Hermida A, Vázquez-Mosquera ME, Cortés-Pereira E, Pértega S, Relaño S, Oreiro-Villar N, Fernñndez-López C, Blanco FJ, Rego-Pérez I: A replication study and meta-analysis of mitochondrial DNA variants in the radiographic progression of knee osteoarthritis. Rheumatology 2016, 56(2):263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kenney MC, Chwa M, Atilano SR, Falatoonzadeh P, Ramirez C, Malik D, Tarek M, Cáceres-del-Carpio J, Nesburn AB, Boyer DS et al. : Inherited mitochondrial DNA variants can affect complement, inflammation and apoptosis pathways: insights into mitochondrial–nuclear interactions. Ham Mol Genet 2014, 23(13):3537–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baeza J, Smallegan MJ, Denu JM: Mechanisms and dynamics of protein acetylation in mitochondria. Trends Biochem Sci 2016, 41(3):231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu W, Dittenhafer-Reed KE, Denu JM: SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem 2012, 287(17):14078–14086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang L, Zhang Z, Khan A, Zheng H, Yuan C, Jiang H: Advances in drug therapy for mitochondrial diseases. Ann TranslMed 2020, 8(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 104.Blanco FJ, Rego-Perez I: Mitochondria and mitophagy: biosensors for cartilage degradation and osteoarthritis. Osteoarthr Cartil 2018, 26(8):989–991. [DOI] [PubMed] [Google Scholar]
- 105.Davies CM, Guilak F, Weinberg JB, Fermor B: Reactive nitrogen and oxygen species in interleukin-l-mediated DNA damage associated with osteoarthritis. Osteoarthr Cartil, 2008, 16(5):624–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mobasheri A, Rayman MP, Gualillo O, Sellam J, van der Kraan P, Fearon U: The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol 2017, 13(5):302–311. [DOI] [PubMed] [Google Scholar]
- 107.Shapiro IM, Golub EE, Kakuta S, Hazelgrove J, Havery J, Chance B, Frasca P: Initiation of endochondral calcification is related to changes in the redox state of hypertrophic chondrocytes. Science 1982, 217(4563):950–952. [DOI] [PubMed] [Google Scholar]
- 108.Vaamonde-Garcia C, Riveiro-Naveira RR, Valcarcel-Ares MN, Hermida-Carballo L, Blanco FJ, Lopez-Armada MJ: Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum 2012, 64(9):2927–2936. [DOI] [PubMed] [Google Scholar]
- 109.Cillero-Pastor B, Rego-Perez I, Oreiro N, Fernandez-Lopez C, Blanco FJ: Mitochondrial respiratory chain dysfunction modulates metalloproteases -1, -3 and -13 in human normal chondrocytes in culture. BMC Musculoskel Disord 2013, 14(1):235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, Giorgi C, Pinton P: Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell calcium 2018, 69:62–72. [DOI] [PubMed] [Google Scholar]
- 111.Orrenius S, Gogvadze V, Zhivotovsky B: Calcium and mitochondria in the regulation of cell death. Biochem Biophys Res Comntun 2015, 460(1):72–81. [DOI] [PubMed] [Google Scholar]
- 112.Yokota H, Leong DJ, Sun HB: Mechanical loading: bone remodeling and cartilage maintenance. Carr Osteoporos Rep 2011, 9(4):237. [DOI] [PubMed] [Google Scholar]
- 113.Mow VC, Holmes MH, Michael Lai W: Fluid transport and mechanical properties of articular cartilage: A review. J Biomech 1984, 17(5):377–394. [DOI] [PubMed] [Google Scholar]
- 114.Brody LT: Knee osteoarthritis: Clinical connections to articular cartilage structure and function. Rhys Ther Sport 2015, 16(4):301–316. [DOI] [PubMed] [Google Scholar]
- 115.Kurz B, Lemke AK, Fay J, Pufe T, Grodzinsky AJ, Schiinke M: Pathomechanisms of cartilage destruction by mechanical injury. Ann Anat 2005, 187(5–6):473–485. [DOI] [PubMed] [Google Scholar]
- 116.Paluch EK, Nelson CM, Biais N, Fabry B, Moeller J, Pruitt BL, Wollnik C, Kudryasheva G, Rehfeldt F, Federle W: Mechanotransduction: use the force(s). BMC Biol 2015, 13:47–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carter DR, Orr TE, Fyhrie DP, Schurman D: Influences of mechanical stress on prenatal and postnatal skeletal development. Clin Orthop Relat Res 1987(219):237–250. [PubMed] [Google Scholar]
- 118.Wolff KJ, Ramakrishnan PS, Brouillette MJ, Joumot BJ, McKinley TO, Buckwalter JA, Martin JA: Mechanical stress and ATP synthesis are coupled by mitochondrial oxidants in articular cartilage. J Orthop Res 2013, 31(2):191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wilkins RJ, Browning JA, Urban JPG: Chondrocyte regulation by mechanical load. Biorheology 2000, 37:67–74. [PubMed] [Google Scholar]
- 120.Zhao Z, Li Y, Wang M, Zhao S, Zhao Z, Fang J: Mechanotransduction pathways in the regulation of cartilage chondrocyte homoeostasis. J Cell Mol Med 2020, 24(10):5408–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Satir P, Christensen ST: Overview of structure and function of mammalian cilia. Annu Rev Physiol 2007, 69:377–400. [DOI] [PubMed] [Google Scholar]
- 122.Sorokin SP: Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J Cell Sci 1968, 3(2):207. [DOI] [PubMed] [Google Scholar]
- 123.Wheatley DN, Wang Am, Strugnell GEJCbiE: Expression of primary cilia in mammalian cells. Cell Biol Int 1996, 20(1):73–81. [DOI] [PubMed] [Google Scholar]
- 124.McGlashan SR, Knight MM, Chowdhury TT, Joshi P, Jensen CG, Kennedy S, Poole CA: Mechanical loading modulates chondrocyte primary cilia incidence and length. Cell Biol Int 2010, 34(5):441–446. [DOI] [PubMed] [Google Scholar]
- 125.Satir P, Pedersen LB, Christensen ST: The primary cilium at a glance. J Cell Sci 2010, 123(4):499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shao YY, Wang L, Welter JF, Ballock RT: Primary cilia modulate Ihh signal transduction in response to hydrostatic loading of growth plate chondrocytes. Bone 2012, 50(1):79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bangs F, Anderson KV: Primary cilia and mammalian hedgehog signaling. Cold Spring Harb Perspect Biol 2017, 9(5):a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thompson CL, Chappie JP, Knight MM: Primary cilia disassembly down-regulates mechanosensitive hedgehog signalling: a feedback mechanism controlling ADAMTS-5 expression in chondrocytes. Osteocirthr Cartil 2014, 22(3):490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE: Primary cilia are specialized calcium signalling organelles. Nature 2013, 504(7479):311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Martinac B: Mechanosensitive ion channels: molecules of mechanotransduction. J Cell Sci 2004, 117(12):2449–2460. [DOI] [PubMed] [Google Scholar]
- 131.Servin-Vences MR, Richardson J, Lewin GR, Poole K: Mechanoelectrical transduction in chondrocytes. Clin Exp Pharmacol Physiol 2018, 45(5):481–488. [DOI] [PubMed] [Google Scholar]
- 132.Garcia-Elias A, Mrkonjić S, Jung C, Pardo-Pastor C, Vicente R, Valverde MA: The TRPV4 channel. In: Mammalian Transient Receptor Potential (TRP) Cation Channels. Springer; 2014:293–319. [DOI] [PubMed] [Google Scholar]
- 133.Muramatsu S, Wakabayashi M, Ohno T, Amano K, Ooishi R, Sugahara T, Shiojiri S, Tashiro K, Suzuki Y, Nishimura R: Functional gene screening system identified TRPV4 as a regulator of chondrogenic differentiation. J Biol Chem 2007, 282(44):32158–32167. [DOI] [PubMed] [Google Scholar]
- 134.O’Conor CJ, Leddy HA, Benefield HC, Liedtke WB, Guilak F: TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc Natl Acad Sci U S A 2014, 111(4):1316–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ohtsuki T, Shinaoka A, Kumagishi-Shinaoka K, Asano K, Hatipoglu OF, Inagaki J, Takahashi K, Oohashi T, Nishida K, Naruse K et al. : Mechanical strain attenuates cytokine-induced ADAMTS9 expression via transient receptor potential vanilloid type 1. Exp Cell Res 2019, 383(2):111556. [DOI] [PubMed] [Google Scholar]
- 136.Clark AL, Votta BJ, Kumar S, Liedtke W, Guilak F: Chondroprotective role of the osmotically sensitive ion channel transient receptor potential vanilloid 4: Age- and sex- dependent progression of osteoarthritis in Trpv4- deficient mice. Arthritis Rheum 2010, 62(10):2973–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.O’Conor CJ, Griffin TM, Liedtke W, Guilak F: Increased susceptibility of Trpv4- deficient mice to obesity and obesity-induced osteoarthritis with very high-fat diet. Arm Rheum Dis 2013, 72(2):300–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.O’Conor CJ, Ramalingam S, Zelenski NA, Benefield HC, Rigo I, Little D, Wu C-L, Chen D, Liedtke W, McNulty AL et al. : Cartilage-specific knockout of the mechanosensory ion channel TRPV4 decreases age-related osteoarthritis. Sci Rep 2016, 6:29053–29053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Knighten J, Phillips V, Taylor M, Townsley M, Francis CM: Mitochondria amplify TRPV4-dependent calcium dynamics within the pulmonary microvascular endothelium. FASEB J 2020, 34(S1):1–1. [Google Scholar]
- 140.Suresh K, Servinsky L, Philip N, Jiang H, Huetsch J, Kirsch B, Hoang G, Le A, Damarla M, Shimoda L: Increased fatty acid oxidation promotes mitochondrial ROS (mtROS) production and Ca2+ entry via TRPV4 in microvascular endothelial cells in PAH. FASEB J 2020, 34(S1):1–1. [Google Scholar]
- 141.Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A: Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 2010, 330(6000):55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.He L, Si G, Huang J, Samuel ADT, Perrimon N: Mechanical regulation of stem-cell differentiation by the stretch-activated Piezo channel. Nature 2018, 555(7694):103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Volkers L, Mechioukhi Y, Coste B: Piezo channels: from structure to function. Pflugers Arch 2015, 467(l):95–99. [DOI] [PubMed] [Google Scholar]
- 144.Sugimoto A, Miyazaki A, Kawarabayashi K, Shono M, Akazawa Y, Hasegawa T, Ueda-Yamaguchi K, Kitamura T, Yoshizaki K, Fukumoto S et al. : Piezo type mechanosensitive ion channel component 1 functions as a regulator of the cell fate determination of mesenchymal stem cells. Sci Rep 2017, 7(1):17696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li X-F, Zhang Z, Chen Z-K, Cui Z-W, Zhang H-N: Piezo1 protein induces the apoptosis of human osteoarthritis-derived chondrocytes by activating caspase-12, the signaling marker of ER stress. Int J Mol Med 2017, 40(3):845–853. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 146.Lawrence KM, Jones RC, Jackson TR, Baylie RL, Abbott B, Bruhn-Olszewska B, Board TN, Locke IC, Richardson SM, Townsend PA: Chondroprotection by urocortin involves blockade of the mechanosensitive ion channel Piezo1. Sci Rep 2017, 7(1):5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lee W, Leddy HA, Chen Y, Lee SH, Zelenski NA, McNulty AL, Wu J, Beicker KN, Coles J, Zauscher S et al. : Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. Proc Nath Acad Sci U S A 2014, 111(47):E5114–E5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Servin-Vences MR, Moroni M, Lewin GR, Poole K: Direct measurement of TRPV4 and PIEZO 1 activity reveals multiple mechanotransduction pathways in chondrocytes. Elife 2017, 6:e21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Swain SM, Romac JMJ, Shahid RA, Pandol SJ, Liedtke W, Vigna SR, Liddle RA: TRPV4 channel opening mediates pressure-induced pancreatitis initiated by Piezo1 activation. J Clin Invest 2020, 130(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Borbiro I, Badheka D, Rohacs T: Activation of TRPV1 channels inhibits mechanosensitive Piezo channel activity by depleting membrane phosphoinositides. Sci Signal 2015, 8(363):ral5–ral5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yoneda M, Suzuki H, Hatano N, Nakano S, Muraki Y, Miyazawa K, Goto S, Muraki K: PIEZO1 and TRPV4, which are distinct mechano-sensors in the osteoblastic MC3T3-E1 cells, modify cell-proliferation. Int J Mol Sci 2019, 20(19):4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Knight MM, Bomzon Z, Kimmel E, Sharma AM, Lee DA, Bader DL: Chondrocyte deformation induces mitochondrial distortion and heterogeneous intracellular strain fields. Biomech Model Mechanobiol 2006, 5(2):180. [DOI] [PubMed] [Google Scholar]
- 153.Brouillette MJ, Ramakrishnan PS, Wagner VM, Sauter EE, Joumot BJ, McKinley TO, Martin JA: Strain-dependent oxidant release in articular cartilage originates from mitochondria. Biomech Model Mechanobiol 2014, 13(3):565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Durrant LA, Archer CW, Benjamin M, Ralphs JR: Organisation of the chondrocyte cytoskeleton and its response to changing mechanical conditions in organ culture. J Anat 1999, 194 (Pt 3):343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sauter E, Buckwalter JA, McKinley TO, Martin JA: Cytoskeletal dissolution blocks oxidant release and cell death in injured cartilage. J Orthop Res 2012, 30(4):593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hynes RO: Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69(1):11–25. [DOI] [PubMed] [Google Scholar]
- 157.Loeser RF: Integrins and chondrocyte–matrix interactions in articular cartilage. Matrix Biol 2014, 39:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Millward-Sadler SJ, Salter DM: Integrin-dependent signal cascades in chondrocyte mechanotransduction. Ann Biomed Eng 2004, 32(3):435–446. [DOI] [PubMed] [Google Scholar]
- 159.Song H, Liang W, Xu S, Li Z, Chen Z, Cui W, Zhou J, Wang Q, Liu F, Fan W: A novel role for integrin-linked kinase in periodic mechanical stress-mediated ERK1/2 mitogenic signaling in rat chondrocytes. Cell Biol Int 2016, 40(7):832–839. [DOI] [PubMed] [Google Scholar]
- 160.Dittmer TA, Misteli T: The lamin protein family. Genome Biol 2011, 12(5):222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Athirasala A, Hirsch N, Buxboim A: Nuclear mechanotransduction: sensing the force from within. Carr Opin Cell Biol 2017, 46:119–127. [DOI] [PubMed] [Google Scholar]
- 162.Koushki N, Ghagre A, Srivastava LK, Sitaras C, Yoshie H, Molter C, Ehrlicher AJ: Lamin A redistribution mediated by nuclear deformation determines dynamic localization of YAP. bioRxiv 2020:2020.2003.2019.998708. [Google Scholar]
- 163.Fischer M, Rikeit P, Knaus P, Coirault C: YAP-mediated mechanotransduction in skeletal muscle. Front Physiol 2016, 7(41). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S et al. : Role of YAP/TAZ in mechanotransduction. Nature 2011, 474(7350):179–183. [DOI] [PubMed] [Google Scholar]
- 165.Maeshima K, Yahata K, Sasaki Y, Nakatomi R, Tachibana T, Hashikawa T, Imamoto F, Imamoto N: Cell-cycle-dependent dynamics of nuclear pores: pore-free islands and lamins. J Cell Sci 2006, 119(Pt 21):4442–4451. [DOI] [PubMed] [Google Scholar]
- 166.Zheng Q, Li X-x, Xiao L, Shao S, Jiang H, Zhang X-l, Sun L-y, Xu H-g: MicroRNA-365 functions as a mechanosensitive microRNA to inhibit end plate chondrocyte degeneration by targeting histone deacetylase 4. Bone 2019, 128:115052. [DOI] [PubMed] [Google Scholar]
- 167.Yamamura H, Suzuki Y, Imaizumi Y: Physiological and pathological functions of Cl - channels in chondrocytes. Biol Pharm Bull 2018, 41(8):1145–1151. [DOI] [PubMed] [Google Scholar]
- 168.Bader DL, Salter DM, Chowdhury TT: Biomechanical influence of cartilage homeostasis in health and disease. Arthritis 2011, 2011:979032–979032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Felson DT: Osteoarthritis as a disease of mechanics. Osteoarthr Cartil 2013, 21(1):10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Buckwalter JA, Anderson DD, Brown TD, Tochigi Y, Martin JA: The roles of mechanical stresses in the pathogenesis of osteoarthritis: implications for treatment of joint injuries. Cartilage 2013, 4(4):286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Vincent TL: Mechanoflammation in osteoarthritis pathogenesis. Semin Arthritis Rheum 2019, 49(3s):S36–s38. [DOI] [PubMed] [Google Scholar]
- 172.Coleman MC, Ramakrishnan PS, Brouillette MJ, Martin JA: Injurious loading of articular cartilage compromises chondrocyte respiratory function. Arthritis Rheumatol 2016, 68(3):662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bingol B, Sheng M: Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radical Biol Med 2016, 100:210–222. [DOI] [PubMed] [Google Scholar]
- 174.Wang X, Winter D, Ashrafi G, Schlehe J, Wong Yao L, Selkoe D, Rice S, Steen J, LaVoie Matthew J, Schwarz Thomas L: PINK1 and Parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147(4):893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shin HJ, Park H, Shin N, Kwon HH, Yin Y, Hwang JA, Song HJ, Kim J, Kim DW, Beom J: Pink1-mediated chondrocytic mitophagy contributes to cartilage degeneration in osteoarthritis. J Clin Med 2019, 8(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hardie DG, development: AMP-activated protein kinase—an energy sensor that regulates all aspects of cell function. Genes Dev 2011, 25(18):1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Petursson F, Husa M, June R, Lotz M, Terkeltaub R, Liu-Bryan R: Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res Ther 2013, 15(4):R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang Y, Zhao X, Lotz M, Terkeltaub R, Liu-Bryan R: Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol 2015, 67(8):2141–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M et al. : Mitochondrial membrane potential. Anal Biochem 2018, 552:50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Duchen MR: Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. J Physiol 1999, 516 (Pt 1)(Pt 1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Borutaite V, Morkuniene R, Brown GC: Release of cytochrome c from heart mitochondria is induced by high Ca2+ and peroxynitrite and is responsible for Ca(2+)-induced inhibition of substrate oxidation. Biochimica et biophysica acta 1999, 1453(l):41–48. [DOI] [PubMed] [Google Scholar]
- 182.Lv M, Zhou Y, Chen X, Han L, Wang L, Lu XL: Calcium signaling of in situ chondrocytes in articular cartilage under compressive loading: Roles of calcium sources and cell membrane ion channels. J Orthop Res 2018, 36(2):730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Martel-Pelletier J, Wildi LM, Pelletier J-P: Future therapeutics for osteoarthritis. Bone 2012, 51(2):297–311. [DOI] [PubMed] [Google Scholar]
- 184.Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier J-P, Fahmi H: Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 2011, 7(1):33–42. [DOI] [PubMed] [Google Scholar]
- 185.Kurtz SM, Lau E, Watson H, Schmier JK, Parvizi J: Economic burden of periprosthetic joint infection in the United States. J Arthroplasty 2012, 27(8 Suppl):61–65.e61. [DOI] [PubMed] [Google Scholar]
- 186.Pabinger C, Lothaller H, Geissler A: Utilization rates of knee-arthroplasty in OECD countries. Osteoarthr Cartil 2015, 23(10):1664–1673. [DOI] [PubMed] [Google Scholar]
- 187.Kraus VB, Hsueh M-F: Chapter 22 - Biomarkers and osteoarthritis In: Genomic and Precision Medicine (Third Edition). Edited by Ginsburg GS, Willard HF, Tsalik EL, Woods CW. Boston: Academic Press; 2019:429–444. [Google Scholar]
- 188.Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV: Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res Int 2014, 2014:238463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.de Mello AH, Costa AB, Engel JDG, Rezin GT: Mitochondrial dysfunction in obesity. Life Sci 2018, 192:26–32. [DOI] [PubMed] [Google Scholar]
- 190.Caramés B, Taniguchi N, Otsuki S, Blanco FJ, Lotz MJA, Rheumatism: Autophagy is a protective mechanism in normal cartilage, and its aging- related loss is linked with cell death and osteoarthritis. Arthritis Rheum 2010, 62(3):791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ansari MY, Khan NM, Ahmad I, Haqqi TM: Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthr Cartil 2018, 26(8):1087–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M: Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis 2012, 71(4):575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Carames B, de Figueroa PL, Lotz M, Blanco FJ, Cartilage: Autophagy activation protects from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol 2014, 22:S135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tang Q, Zheng G, Feng Z, Chen Y, Lou Y, Wang C, Zhang X, Zhang Y, Xu H, Shang P et al. : Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis 2017, 8(10):e3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Qiu L, Luo Y, Chen X: Quercetin attenuates mitochondrial dysfunction and biogenesis via upregulated AMPK/SIRT1 signaling pathway in OA rats. Biomed Pharmacother 2018, 103:1585–1591. [DOI] [PubMed] [Google Scholar]
- 196.Bartell LR, Fortier LA, Bonassar LJ, Szeto HH, Cohen I, Delco ML: Mitoprotective therapy prevents rapid, strain-dependent mitochondrial dysfunction after articular cartilage injury. J Orthop Res 2020, 38(6):1257–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Famaghi S, Prasadam I, Cai G, Friis T, Du Z, Crawford R, Mao X, Xiao Y: Protective effects of mitochondria-targeted antioxidants and statins on cholesterolinduced osteoarthritis. FASEB J 2017, 31(l):356–367. [DOI] [PubMed] [Google Scholar]
- 198.Wang C, Yang Y, Zhang Y, Liu J, Yao Z, Zhang C: Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci Trends 2019, 12(6):605–612. [DOI] [PubMed] [Google Scholar]
- 199.Lee R, Wilkins R, Razaq S, Urban J: The effect of mechanical stress on cartilage energy metabolism. Biorheology 2002, 39(1, 2):133–143. [PubMed] [Google Scholar]
- 200.Harms FA, Bodmer SI, Raat NJ, Mik EG: Non-invasive monitoring of mitochondrial oxygenation and respiration in critical illness using a novel technique. Crit Care 2015, 19(1):343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lin Z, Li Z, Li EN, Li X, Del Duke CJ, Shen H, Hao T, O’Donnell B, Bunnell BA, Goodman SB et al. : Osteochondral tissue chip derived from iPSCs: modeling OA pathologies and testing drugs. Front Bioeng Biotechnol 2019, 7(411). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Occhetta P, Mainardi A, Votta E, Vallmajo-Martin Q, Ehrbar M, Martin I, Barbero A, Rasponi M: Hyperphysiological compression of articular cartilage induces an osteoarthritic phenotype in a cartilage-on-a-chip model. Nat Biomed Eng 2019, 3(7):545–557. [DOI] [PubMed] [Google Scholar]