

Abstract
Recent studies suggested a beneficial role of lymphatics in restoring heart function after cardiac injury1–6. Here we report that in mice lymphatics promote cardiac growth, repair and cardio-protection. We show that a lymphoangiocrine signal produced by lymphatic endothelial cells (LECs) controls cardiomyocyte (CM) proliferation and survival during heart development, improves neonatal cardiac regeneration and is cardioprotective after myocardial infarction (MI). Embryos devoid of LECs develop smaller hearts as a consequence of reduced CM proliferation and increased CM apoptosis. Culturing primary mouse CMs in LEC-conditioned media increases CM proliferation and survival, indicating that LECs produce lymphoangiocrine signals controlling CM homeostasis. Characterization of the LEC secretome identified Reelin as a key player responsible for such function. Moreover, we report that LEC-specific Reln-null embryos also develop smaller hearts, that Reelin is required for efficient heart repair and function following neonatal MI, and that cardiac delivery of REELIN using collagen patches improves adult heart function after MI through a cardioprotective effect. These results identify a lymphoangiocrine role of LECs during cardiac development and injury response, and Reelin as an important mediator of this function.
Keywords: Lymphatics, heart, Reelin, mouse, myocardial infarction
The molecular and functional characterization of the lymphatic vasculature has greatly improved1. Recent data suggest that natural or therapeutic formation of new lymphatics (lymphangiogenesis) correlates with improved systolic function after experimental MI; it delays atherosclerotic plaque formation, facilitates the healing process after MI, and can be a natural response to fluid accumulation into the myocardium during cardiac edema2,3,4. These new findings argue that stimulation of lymphangiogenesis in the infarcted heart could improve cardiac function and prevent adverse cardiac remodeling3. Studies in mouse and zebrafish suggested that newly formed lymphatics provided a route for the clearance of immune cells in the injured heart, and therefore promote cardiac repair5,6. However, whether lymphatics have additional functional roles during heart development and cardiac repair is not known.
Lymphatics regulate heart growth
As previously reported2, at around E14.5 cardiac lymphatics become evident, particularly over the dorsal side of the heart (Fig. 1a). As development progresses, lymphatics expand over the dorsal and ventral surfaces, and into the myocardium during embryonic and postnatal stages (Fig. 1a). To evaluate a possible developmental role of cardiac-associated lymphatics we took advantage of Prox1 floxed mice7. Prox1 is a master regulator required to promote and maintain LEC fate identity8,9 and germ-line deletion of Prox1 in mice results in complete lack of LECs and embryonic lethality at around E14.58. To conditionally delete Prox1 from LECs, we crossed Cad5(PAC)-CreERT2 mice10 with Prox1 floxed mice and injected pregnant females with tamoxifen at E13.5 and E14.5. Analysis of E17.5 Cad5(PAC)-CreERT2;Prox1f/f null embryos (Prox1ΔLEC/ΔLEC) revealed appreciably edema (Fig. 1b, arrow); a phenotype associated with defective lymphatics and they die soon after birth. Surprisingly, these mutant embryos have significantly smaller hearts than control littermates (approximately 1/3 smaller, Fig. 1c, j). Most, if not all cardiac lymphatics were missing (Fig. 1d, g and Extended Data Fig. 1a) and the blood vasculature was not affected in Prox1ΔLEC/ΔLEC embryos (Fig. 1e, f, h, i).
Figure 1. Lymphatics are required for embryonic heart growth.
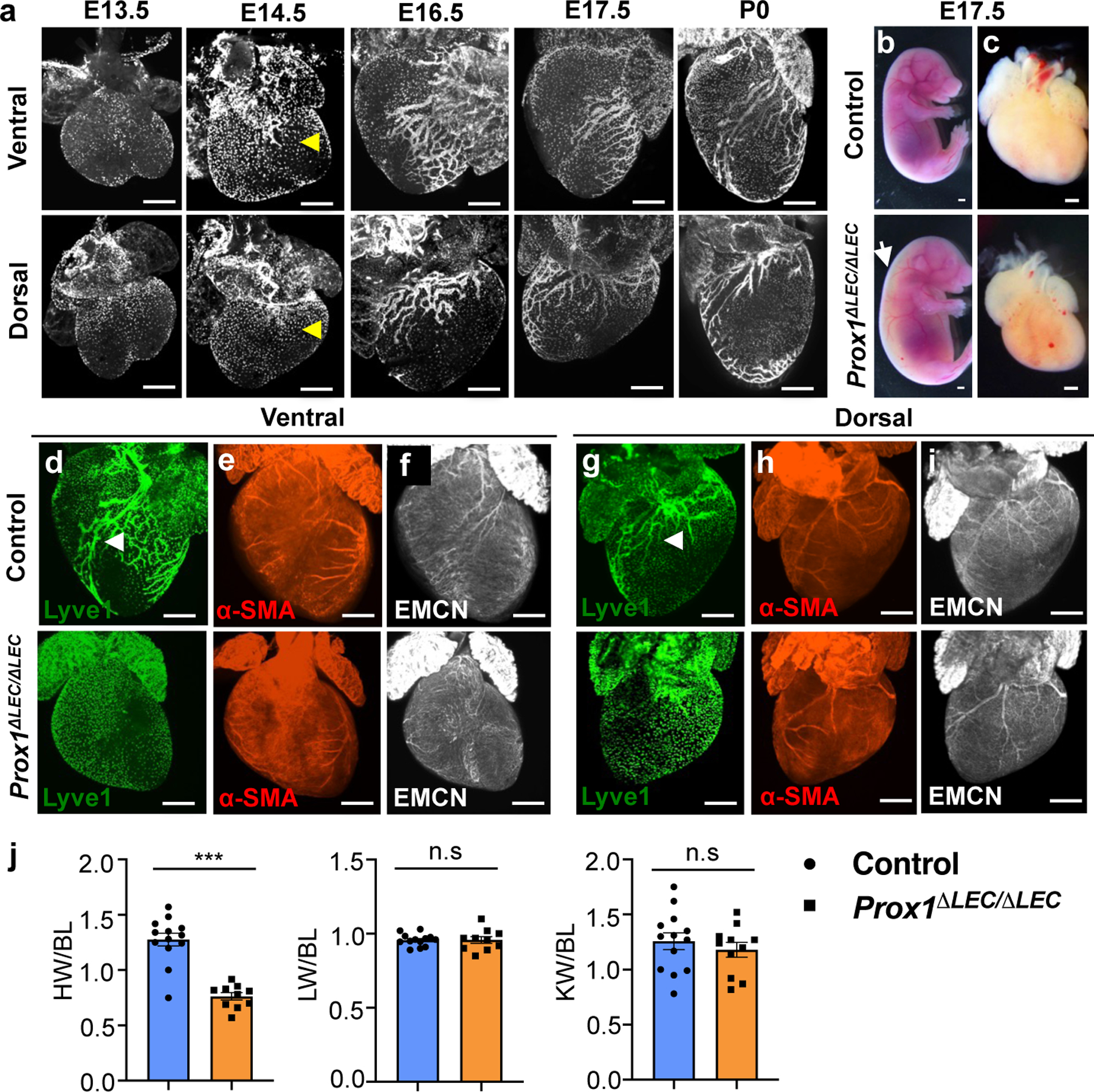
a, Wild type mouse cardiac lymphatic vasculature development as depicted by anti-Lyve1 whole mount immunostaining. Yellow arrowheads indicate cardiac lymphatics at E14.5. b-c, Bright field images of E17.5 control and Prox1ΔLEC/ΔLEC embryos and hearts. White arrow indicates edema in Prox1ΔLEC/ΔLEC embryos. d-i, Whole mount immunostaining shows that E17.5 Prox1ΔLEC/ΔLEC hearts lack Lyve1+ cardiac lymphatics and have normal major coronary arteries and veins, as indicated by α-SMA and endomucin (EMCN) staining. Arrowheads indicate developing lymphatics in control hearts. j, Quantification of organ weight relative to body length (BL) shows reduced heart size and normal liver and kidney sizes in E17.5 Prox1ΔLEC/ΔLEC embryos (N=13 controls and N=10 Prox1ΔLEC/ΔLEC embryos; 3 different litters). Data is presented as mean ± S.E.M. ***p=3.19062E-06 by unpaired two-tailed Student’s t test. n.s, not significant. Control embryos are TAM treated Cre- and Cre+;Prox1+/+ littermates. HW, heart weight; LW, liver weight; KW, kidney weight. N = 3 embryos/genotype (a, d-i). Scale bars, 500 μm (a, c-i), 2 mm (b).
Decreased CM mass causes heart size reduction
H&E staining confirmed that the overall size of the ventricles in Prox1ΔLEC/ΔLEC embryos is smaller; however, cardiac valves appear normal (Fig. 2a, arrows). Immunostaining of heart sections against α-actinin and F-actin show that overall, cardiac muscle structure and arrangement are not disrupted in Prox1ΔLEC/ΔLEC hearts (Extended Data Fig. 1b). Flow cytometry analysis (FACS) indicated that the percentage of CMs is significantly reduced (approximately 1/3 reduction) (Extended Data Fig. 1c), a result suggesting that a decrease in CM mass underlies the reduction in heart size. Hoechst 33342 labeling showed no differences in CMs ploidy in Prox1ΔLEC/ΔLEC hearts (Extended Data Fig. 1d). However, an increased percentage of multinucleated CMs was observed in these E17.5 mutant hearts after CM dissociation and o/n plating (Extended Data Fig. 1e, f), but no overall differences in CM size were detected (Extended Data Fig. 1e, g). Similarly, α-Laminin staining showed that the overall CM size was not affected in the mutant hearts (Fig. 2b). Next, we evaluated possible alterations in CM proliferation and survival. Indeed, CM proliferation is greatly reduced in E17.5 Prox1ΔLEC/ΔLEC embryos, as indicated by EdU labeling (Fig. 2c, g, Extended Data Fig. 2a–e), and phospho-histone H3 (pH3), Ki67 and aurora kinase B (AuroraB) immunostainings (Fig. 2d–g). This reduction in proliferation is seen in different regions of the E17.5 mutant heart (Extended Data Fig. 2f). In addition, CM apoptosis was significantly increased in Prox1ΔLEC/ΔLEC hearts (Fig. 2h). These alterations in CM proliferation and apoptosis were not seen in other cardiac cell types (blood endothelial cells, fibroblasts or macrophages) or in other organs (nephron progenitors and hepatocytes) in these mutant embryos (Extended Data Fig. 2g).
Figure 2. Lymphatics are required for CM proliferation and survival.
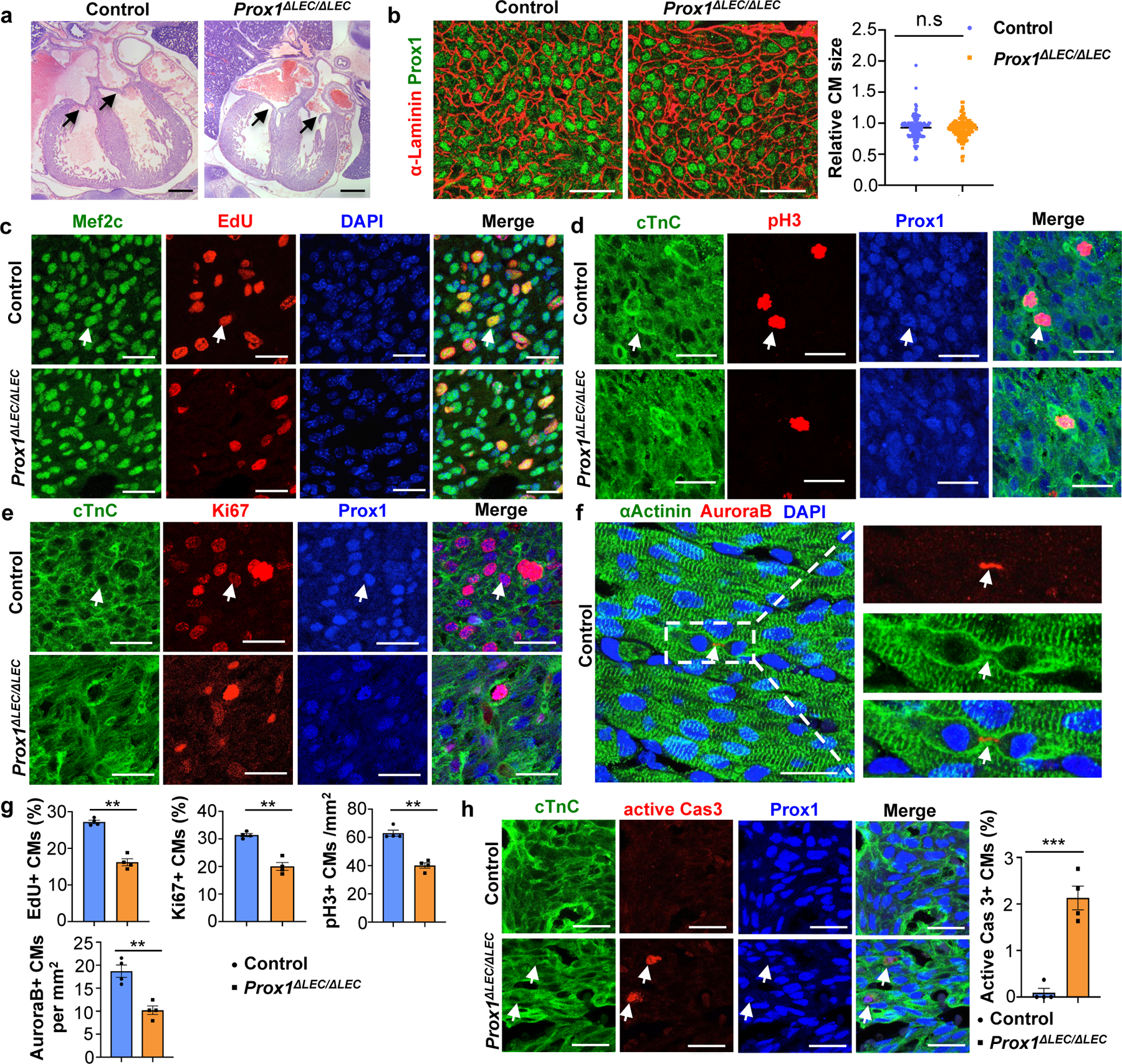
a, H&E staining shows no obvious defects in cardiac valves (arrows) or ventricular wall compaction in E17.5 Prox1ΔLEC/ΔLEC hearts (TAM injected at E13.5 and E14.5). N = 4 embryos/genotype. b, α-Laminin staining shows no differences in Prox1+ CM size between E17.5 controls and Prox1ΔLEC/ΔLEC hearts. Right panel shows quantification of Prox1+ CM size (α-Laminin+ area). Average cell size was measured from five fields/ventricle, 8–10 Prox1+ CMs/field, 3 embryos per genotype; N = 152 (control) and 155 (Prox1ΔLEC/ΔLEC). c-f, Immunostaining with proliferation markers (EdU, pH3, Ki67 and AuroraB) together with CM markers (cardiac Troponin C [cTnC], Prox1, αActinin and/or Mef2c). In all images, arrows indicate the double positive CMs selected for counting. N = 4 embryos/genotype from 3 separate litters. g, Quantification of the immunostaining in c-f shows reduced number of EdU+, Ki67+, AuroraB+ and pH3+ CMs in E17.5 Prox1ΔLEC/ΔLEC hearts. N = 4 embryos/genotype from 3 separate litters. **p=0.003 (EdU, Ki67 and AuroraB), **p=0.002 (pH3). h, Active Caspase-3 immunostaining shows increased CM apoptosis in Prox1+ CMs in E17.5 Prox1ΔLEC/ΔLEC hearts. Arrows indicate apoptotic CMs. N = 4 embryos/genotype from 3 separate litters. ***p=0.0003. Control embryos are TAM treated Cre− and Cre+;Prox1+/+ littermates. Data are presented as mean ± S.E.M. p values were calculated by unpaired two-tailed Student’s t test. n.s, not significant. Scale bars, 1 mm (a), 25 μm (b, c-f, h). Lower magnification of panels c-e and h are included in Supplementary Fig 1.
To support these findings, we performed similar analysis using another mouse model without lymphatics. Accordingly, we used Vegfr3kd/kd, a natural occurring mouse strain with a point mutation in the kinase domain of VEGFR3 that impacts Vegfr3 signaling and therefore, lymphatic development11. As seen in Extended Data Fig. 3a, b, E17.5 Vegfr3kd/kd embryos lacking cardiac-associated lymphatics also have smaller hearts. Similar to Prox1ΔLEC/ΔLEC embryos (Fig. 1j), although no significant size differences were seen in E17.5 Vegfr3kd/kd livers or kidneys, some individual samples show a size reduction trend (Extended Data Fig. 3a). Also, similar to Prox1ΔLEC/ΔLEC embryos, CM proliferation is significantly reduced in E17.5 Vegfr3kd/kd hearts (Extended Data Fig. 3c–g), CM apoptosis is significantly increased (Extended Data Fig. 3h), and proliferation in other cardiac cell types or in nephron progenitors and hepatocytes is not affected (Extended Data Fig. 3i). Because E17.5 Prox1ΔLEC/ΔLEC embryos develop edema, their reduced heart size could be secondary to hemodynamic defects consequence of their lack of lymphatics and therefore, of lymphatic flow. However, E17.5 Cad5(PAC)-CreERT2;Prox1f/+ embryos (Prox1ΔLEC/+) also exhibit severe edema, their cardiac lymphatics show reduced branching, but their heart size and CM proliferation is normal (Extended Data Fig. 4a–e). Similarly, E14.5 Prox1ΔLEC/ΔLEC null embryos (a stage when cardiac lymphatics will just start to grow into the heart, Fig 1a; tamoxifen injection at E10.5 and E11.5) also lack LECs and exhibit severe edema, but their heart size and CM proliferation are normal (Extended Data Fig. 4f–h).
To investigate the molecular basis of this lymphatics-dependent defects, we performed RNA sequencing (RNA-seq) of the ventricular portions of E17.5 control and Prox1ΔLEC/ΔLEC hearts. Gene set expression analysis (GSEA) revealed that genes and pathways related to cell cycle were greatly reduced; instead, expression of genes and pathways involved in apoptosis were enriched (Extended Data Fig. 5a). These results were validated by qPCR showing that expression of pro-apoptotic genes is significantly upregulated, but that of cell cycle related genes is significantly downregulated (Extended Data Fig. 5b).
LEC media promote CM proliferation
Signaling between blood endothelial cells (BECs) and CMs is important during cardiac growth and repair12,13. To evaluate whether LECs produce lymphoangiocrine signals promoting CM proliferation and survival we first cultured human iPSCs-derived CMs (hiPSC-CMs) with LECs-conditioned media obtained from culturing commercially available human dermal LECs. We then examined AKT and ERK signaling since phosphorylated AKT and ERK (p-AKT and p-ERK) are frequently used as readouts of proliferative signaling. As seen in Fig. 3a, compared with DMEM control media, LEC-conditioned media significantly increases p-AKT and p-ERK signaling in the cultured hiPSC-CMs. Similar results were seen using mouse primary CMs isolated from wild-type E14.5 to E17.5 hearts (Fig. 3b). Furthermore, treatment of mouse primary CMs with LEC-conditioned media significantly increases cell proliferation as indicated by Ki67 staining (Extended Data Fig. 5c), and protects CM from apoptosis when cultured under CoCl2-induced hypoxia conditions (Extended Data Fig. 5d). Together, these results argue that LECs-conditioned medium promotes CM proliferation and survival in vitro, and that lymphangiocrine factor/s present in that conditioned media play an important in vivo role during heart development.
Figure 3. LECs-secreted Reelin promotes CM proliferation and survival.
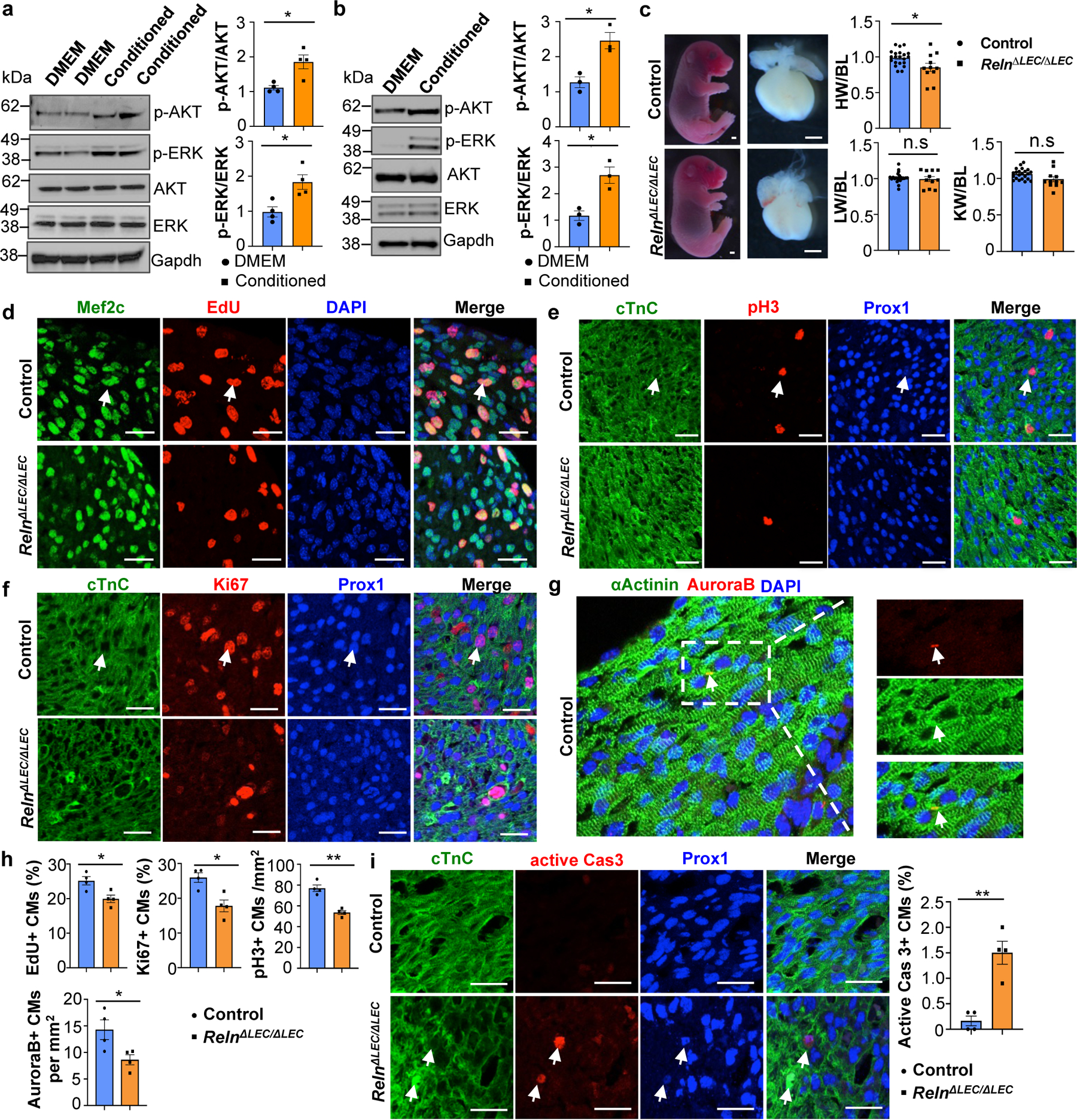
a, b, Quantitative Western blot results show increased p-AKT and p-ERK in LEC-conditioned media-treated hiPSC-CMs (a) and mouse primary CMs (b). *p=0.012 (p-AKT, a), *p=0.015 (p-ERK, a), *p=0.013 (p-AKT and p-ERK, b). N=4 (a) and N=3 (b). c, Bright field images of E17.5 RelnΔLEC/ΔLEC and control embryos and hearts (TAM injected at E13.5 and E14.5). Quantification of organ weight [heart (HW), liver (LW) and kidney (KW)] relative to body length (BL) indicates that hearts are smaller in RelnΔLEC/ΔLEC embryos. N=22 (controls) and N=11 (RelnΔLEC/ΔLEC) from 5 litters. *p=0.016. Controls are TAM treated Cre- embryos and Cre+;Reln+/+littermates. d-g, Double immunostaining using proliferation (EdU, pH3, Ki67 and AuroraB) and CM (cardiac Troponin C [cTnC], Prox1, αActinin and/or Mef2c) markers shows reduced CM proliferation in E17.5 RelnΔLEC/ΔLEC hearts. Arrows indicate proliferating CMs. h, Quantification of the immunostaining in d-g shows reduced number of EdU+, Ki67+, AuroraB+ and pH3+ CMs in E17.5 RelnΔLEC/ΔLEC hearts. N = 4 embryos/genotype from 3 separate litters. *p=0.02 (EdU), *p=0.01 (Ki67), **p=0.001 (pH3) and *p=0.035 (AuroraB). i, Active Caspase-3 immunostaining shows increased CM apoptosis (arrows) in E17.5 RelnΔLEC/ΔLEC hearts. Right panel shows quantification of the percentage of active caspase-3+ CMs in E17.5 control and RelnΔLEC/ΔLEC hearts. N = 4 embryos/genotype from 3 separate litters. **p=0.002. Control embryos are TAM treated Cre- embryos and Cre+;Reln+/+ littermates. Data are presented as Mean ± S.E.M. p values were calculated by unpaired two-tailed Student’s t test. n.s, not significant. Scale bars, 1 mm (c), 25 μm (d-g, i). Lower magnification for panels d-f and i are included in Supplementary Fig 2. For western blot source data, see Supplementary Fig 6 and 7.
Reelin is required for heart growth
To identify such secreted factor/s we performed mass spectrometry of the LEC conditioned media and identified 317 unique proteins. From that list, we initially focused on all secreted proteins by comparing changes in their expression levels in the RNAseq dataset described above. Among those candidates, Reelin is greatly reduced in Prox1ΔLEC/ΔLEC hearts (log2 fold change: -0.6098 compared to control). Indeed, qPCR analysis confirmed about 80% reduction in Reln expression in Prox1ΔLEC/ΔLEC hearts (Extended Data Fig. 6a). We then validated by qPCR the gene expression levels of Reln, as well as of several other enriched proteins identified in the LECs secretome (Extended Data Fig. 6b). Quantification of Reelin secretion in 3 separate LEC preparations by ELISA revealed similar concentrations of this protein in their supernatants (average OD450 is 0.453±0.065) (Extended Data Fig. 6c). Reelin is an extracellular matrix protein widely known for its roles during neuronal development and migration, and Reln mutant mice are ataxic14,15. Reelin is also expressed in LECs and regulates collecting lymphatic vessel maturation16. In agreement with those results16, in the heart Reelin is mainly expressed in LECs (Extended Data Fig. 6d), although some cardiac blood vessels also express low levels of Reelin (Extended Data Fig. 6e). Accordingly, the observed qPCR reduction in Reln expression in Prox1ΔLEC/ΔLEC hearts is consequence of their lack of lymphatics. Indeed, Reelin is almost undetected in E17.5 Prox1ΔLEC/ΔLEC hearts (Extended Data Fig. 6f).
Importantly, the heart size of E17.5 Reln−/− embryos17 was also significantly reduced, but cardiac lymphatics appear normal (Extended Data Fig. 6g–i). To further demonstrate that the smaller heart phenotype is a consequence of Reln loss in LECs, we deleted Reelin from LECs (RelnΔLEC/ΔLEC) by crossing Reln floxed mice18 with Prox1CreERT2 mice19 (tamoxifen injections at E13.5 and E14.5). Immunostaining confirmed efficient Reelin deletion in cardiac lymphatics at E17.5 (Extended Data Fig. 7a). Significantly, E17.5 RelnΔLEC/ΔLEC embryos also developed smaller hearts (although no significant differences are seen in the size of other organs such as kidneys and livers) (Fig. 3c). In addition, CM proliferation was also reduced in RelnΔLEC/ΔLEC embryos as indicated by EdU, pH3, Ki67 and AuroraB labeling (Fig. 3d–h), and CM apoptosis was increased as indicated by active Caspase 3 (Fig. 3i). No changes in proliferation and apoptosis were detected in other cardiac cell types or in kidney and liver (Extended Data Fig. 7b). These results agree with those seen in Reln−/− embryos, arguing that LEC-derived Reelin plays a critical role during heart development and growth by regulating CM proliferation and apoptosis. To validate this statement, we collected LEC-conditioned media from Reln siRNA and control siRNA treated LECs. Analysis by qPCR showed that Reln expression is efficiently silenced in Reln siRNA treated LECs (Extended Data Fig. 8a). Western analysis showed that the identified increase in p-AKT and p-ERK signaling induced by the LEC conditioned media was greatly reduced when using the Reln deficient LEC conditioned media (Extended Data Fig. 8b).
Reelin signaling requires Integrinβ1
Previous studies about the role of Reelin during neuronal development, neuronal migration and in tumor cells identified VLDLR15, ApoER220,21 and integrinβ122,23 as Reelin receptors. Upon its binding to those receptors, Reelin stimulates intracellular signaling transduction through the phosphorylation of the intracellular protein Disabled-1 (Dab1) and the activation of PI3K/AKT/GSK3β24 and mToR25 signaling cascades. Integrinβ1 has been shown to play important roles in heart development, as its deletion in embryonic CMs results in smaller hearts with reduced CM proliferation26. Therefore, we investigated whether LEC-derived Reelin regulates CM proliferation and survival by regulating Integrinβ1 signaling. Western analysis confirmed that LEC conditioned media treated CMs increased the activity of Integrinβ1 and Reelin downstream signals such as FAK, Dab1, AKT and ERK; in contrast, LEC conditioned media from Reln deficient LECs failed to induce Integrinβ1 signaling activity (Extended Data Fig. 8b). More importantly, blocking Integrinβ1 signaling in CMs by adding Integrinβ1 blocking antibodies to the LEC conditioned media partially abolished the pro-survival effects of the intact LEC conditioned media (Extended Data Fig. 8b). Furthermore, LEC conditioned media from Reln deficient LECs or media containing Integrinβ1 blocking antibodies also failed to promote CM proliferation or to protect against CM apoptosis (Extended Data Fig. 8c, d). These data further support our proposal that LEC secreted Reelin regulates CM proliferation and survival mainly by activating the Integrinβ1 signaling pathway. Furthermore, we also observed an increase in Reelin/Integrinβ1 signaling activity in mouse primary CMs upon Reelin stimulation (addition of supernatant from Reln transfected 293T cells), and this signaling was greatly inhibited by adding Integrinβ1 blocking antibodies (Extended Data Fig. 8e). Moreover, E17.5 Integrinβ1fl/+,MhcCre;Reln+/− (β1ΔCM/+;Reln+/−) double heterozygous embryos generated by crossing Integrinβ1fl/+,MhcCre (β1ΔCM/+) with Reln+/− mice, also developed smaller hearts without significant size differences in livers or kidneys (Extended Data Fig. 8f). Reln+/− and β1ΔCM/+ littermates exhibited no differences in heart size compared to WT (Extended Data Fig. 8f), and embryo size and cardiac lymphatics appear normal in all 3 genotypes resulting from those crosses (Extended Data Fig. 8g). Consistently, CM proliferation was also significantly reduced and apoptosis increased in E17.5 β1ΔCM/+;Reln+/− embryos (Extended Data Fig. 9a, b). No changes in proliferation and apoptosis were detected in other cardiac cell types or in kidney and liver (Extended Data Fig. 9c). Together, these data support our proposal that LECs-secreted Reelin regulates CM proliferation and survival through the Integrinβ1 signaling pathway.
Neonatal heart repair requires Reelin
At E17.5, Reelin is highly expressed in cardiac lymphatics nearby the epicardium, as well as in the base of the myocardium; however, its expression levels get steadily reduced from P2 to P14, such that at P14 it is barely detected (Extended Data Fig. 10a). This reduction in the levels of Reelin is accompanied by a similar change in the levels of Reln mRNA (Extended Data Fig. 10b), suggesting that Reln expression levels are temporally regulated in cardiac lymphatics.
Since this reduction in Reelin expression coincides with the loss of cardiac regenerative potential in mice27, we first examined Reln role in WT mouse neonatal cardiac regeneration. We performed neonatal MI at P2 and the analysis of P7 pups showed Lyve1-expressing lymphatics in both, the infarcted and the nearby non-infarcted cardiac tissue. Reelin expression was re-activated in the infarcted hearts, with higher levels in the infarcted area and low in the non-infarcted tissue (Extended Data Fig. 10c). A similar analysis in P7 Reelin null pups showed that similar to WT controls, Lyve1-expressing lymphatics were present in the infarcted and non-infarcted tissues, although in both cases Reelin expression was not detected (Extended Data Fig. 10c). Compared to WT controls, although cardiac function was not affected in Reln−/− hearts at P7, it was reduced at P14 and P21, as determined by echocardiography (Fig. 4a). In line with this reduced cardiac function, Masson’s trichrome staining showed increased fibrosis in P21 Reln−/− hearts (Fig. 4b). Immunostaining revealed that neither lymphatic density nor LEC proliferation is affected in P21 Reln−/− hearts after injury (Extended Data Fig. 10d, e). Similar to the results observed in E17.5 Reln null embryos, CM proliferation was reduced and CM apoptosis was increased in the infarcted area of P7 Reln−/− hearts (Fig. 4c, d, Extended Data Fig 11a–e). Importantly, no alterations in cardiac function or increased fibrosis were seen in Reln+/− mice (Fig. 4a–b). Immunostaining revealed that neither lymphatic density nor LEC proliferation is affected in Reln−/− hearts after injury (Extended Data Fig. 10d, e). These results demonstrate that Reelin re-expression in cardiac-associated lymphatics of the injured neonatal heart improves cardiac regeneration and function after MI.
Figure 4. Reelin improves neonatal and adult cardiac function after myocardial infarction.
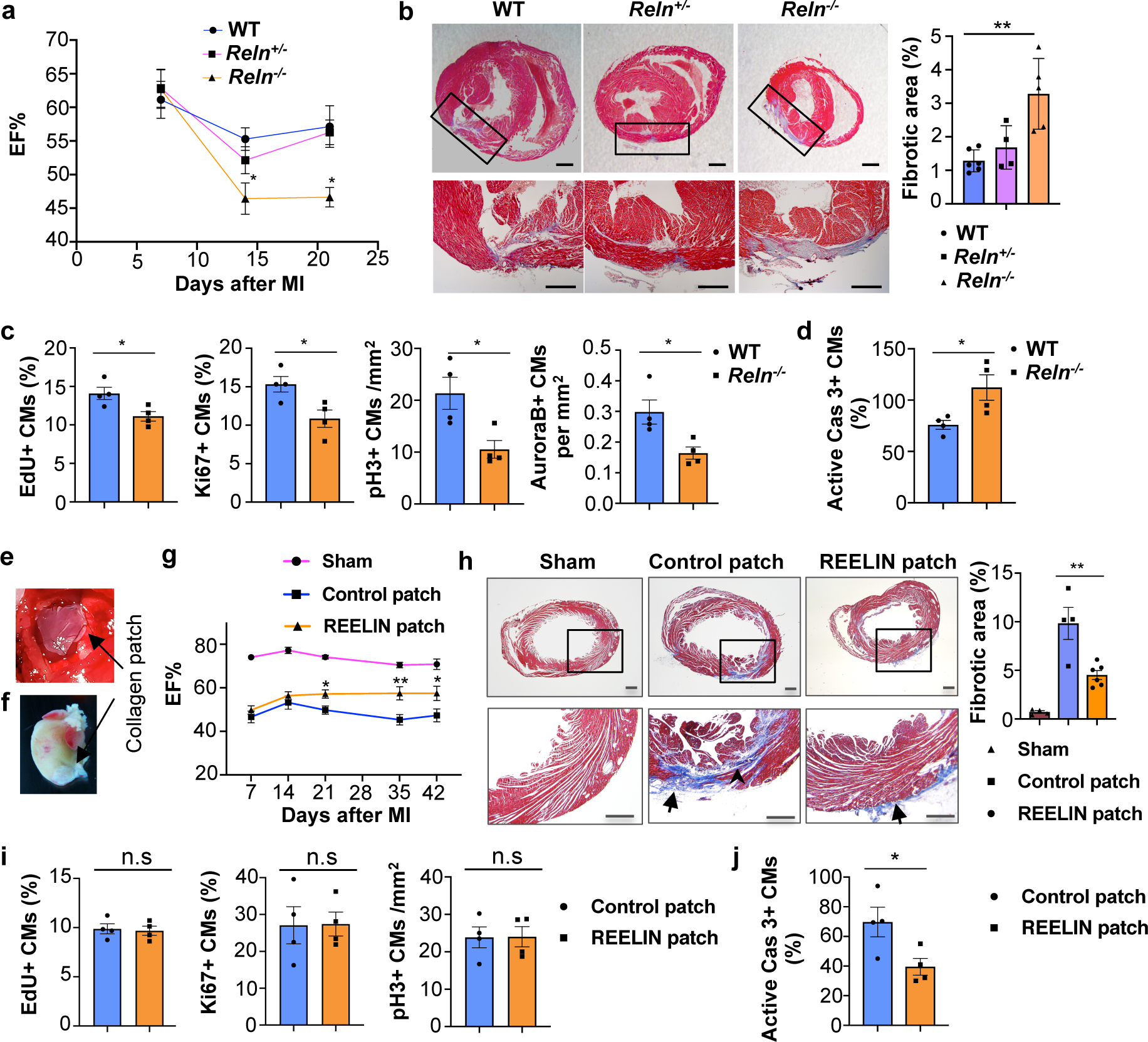
a, Echocardiography reveals relatively normal cardiac function at P7 and reduced at P14 and P21 in Reln−/− mice after MI at P2. P7: N=9 (WT), N=9 (Reln+/−), N=10 (Reln−/−); P14: N=10 (WT), N=8 (Reln+/−), N=7 (Reln−/−); P21: N=9 (WT), N=13 (Reln+/−), N=8 (Reln−/−). *p=0.04 (P14) and *p=0.012 (P21) by two-way ANOVA followed by Bonferroni test. b, Masson’s trichrome staining shows increased fibrosis in P21 Reln−/− hearts (MI at P2). Right panel, quantification of the percentage of fibrotic area. N=6 (WT), N=4 (Reln+/−), N=5 (Reln−/−). **p = 0.002 by one-way ANOVA followed by Tukey’s test. c, CM proliferation is decreased in the border of the infarcted area of P7 Reln−/− hearts (N= 4 mice/genotype). *p=0.026 (EdU), *p=0.025 (Ki67), *p=0.022 (pH3) and *p=0.023 (AuroraB) by unpaired two-tailed Student’s t test. d, CM apoptosis increases significantly in the infarcted area of P7 Reln−/− hearts (N= 4 mice/genotype). *p =0.032 by unpaired two-tailed Student’s t test. e, Sutured collagen patch onto the adult mouse heart following MI. f, Residual collagen patch remains up to 42 days after MI. g, Echocardiography reveals significantly improved cardiac function (EF%) in adult mice with REELIN patches starting at around 21 days after MI and up to 42 days post-MI. N=6 (sham), N=6 (control patch) and N=7 (REELIN patch). *p=0.04 (P21), **p=0.001 (P35) and *p=0.01 (P42) by two-way ANOVA followed by Bonferroni test. h, Masson’s trichrome staining shows reduced cardiac fibrotic area in REELIN patch-treated mice 42 days post-MI. Arrowhead shows fibrotic area; arrows indicate residual collagen patch. Right panel, quantification of the percentage of fibrotic area. N=4 (sham), N=4 (control patch), N=6 (REELIN patch). **p = 0.003 by one-way ANOVA followed by Tukey’s test. i, No differences in CM proliferation between control patch and REELIN patch treated hearts were observed in the infarcted areas 7 days post-MI (N = 4 hearts/group). n.s. not significant difference by unpaired two-tailed Student’s t test. j, CM apoptosis is reduced in the infarcted area of REELIN patch treated hearts (N=4 mice/group). *p = 0.039 by unpaired two-tailed Student’s t test. Data are presented as Mean ± S.E.M. Scale bars, 500 μm (b, h). Representative images of c, d, i and j are in Extended Data Fig. 11. Functional parameters for the neonatal and adult MI echocardiography are in Source Data.
Reelin improves adult MI recovery
We next assessed whether delivery of Reelin directly into the heart could improve cardiac repair in adult WT mice after MI. We took advantage of well-established bioengineered collagen patches28,29 as a scaffold to deliver recombinant REELIN protein into the heart. REELIN-containing patches and control patches were surgically sutured onto approximately 2-month old injured hearts immediately following acute MI (Fig. 4e, f). Cardiac function was evaluated weekly (1–6 weeks after MI) and as shown in Fig. 4g, 21 days after MI ejection fraction (EF) was significantly improved in mice with REELIN patches. Consistent with this improved heart function, 42 days after MI the size of the fibrotic scar in the infarcted area was remarkably reduced in REELIN-patched mice (Fig. 4h). To evaluate whether this improved cardiac function and reduced fibrotic tissue was consequence of increased CM proliferation and/or reduced CM cell death, we performed immunostaining seven days after MI, a stage when increased CM proliferation is normally detected after injury. As seen in Fig. 4i and Extended Data Fig 11f, no differences in CM proliferation were observed in the infarcted area between mice with control or REELIN patches as indicated by EdU labeling and Ki67 or pH3 immunostaining. Importantly, CM apoptosis was greatly reduced in the infarcted area of REELIN patched mice (Fig. 4j, Extended Data Fig 11g). These data indicate that following adult cardiac injury, REELIN protects CMs from apoptosis, which correlates with a reduced scar and improved heart function.
Discussion
Using mouse embryos devoid of LECs or Reelin-producing LECs we demonstrate that their hearts are smaller as a consequence of increased CM apoptosis and reduced CM proliferation. We showed that the percentage of CMs is significantly reduced in E17.5 Prox1ΔLEC/ΔLEC and RelnΔLEC/ΔLEC hearts, suggesting that communication between LECs and CMs is required for CM survival during cardiac development. We also found that LEC-conditioned medium increases CM survival and prevents CM apoptosis consequence of hypoxia; a result suggesting potent LEC lymphoangiocrine cardioprotective effects. We identified Reelin as a factor performing such functional role likely via the Integrinβ1 signaling pathway, both in vivo and in vitro. Finally, we provide some additional insight about the proposed beneficial roles of lymphatics on cardiac repair by showing that at least partially, is mediated by Reelin activity. We demonstrate Reelin relevance in the endogenous cardiac regenerative ability by showing that following MI at P2, Reelin expression in LECs is particularly reactivated in the MI area of wild-type mice, and that Reln−/− mice do not fully regenerate. We found that Reelin is required for CM proliferative activity at P7, although proliferation was not completely abolished in Reln−/− pups, indicating that other factors contribute to CM proliferation. Cardiomyocytes apoptosis was also increased in Reln−/− mice during an extended period after MI (up to P21), suggesting that in addition to the reduced proliferation, loss of CM protection underlies the inability of Reln−/− postnatal hearts to fully regenerate.
We also demonstrate that exogenously applied Reelin is useful for cardiac repair in the adult heart after MI. Whereas during cardiac growth and in neonatal cardiac regeneration Reelin promotes both, CM proliferation and survival, in the adult heart Reelin beneficial activity on cardiac function seems to be mostly consequence of reduced CM cell death and a smaller scarred myocardial area, both features indicative of a cardioprotective effect.
Although our results argue that Reelin regulation of Integrin mediated signaling is specifically critical for CM proliferation and survival, it is likely that alternative signals or receptors mask similar effects on other cardiac cell types such as fibroblasts and BECs. Furthermore, it is also likely that Reelin and/or other lymphoangiocrine signals play similar homeostatic roles in other organs.
In summary, our study highlights the importance of LECs and REELIN during heart growth and repair, and provides some ideas about possible paths to improve cardiac regeneration and cardio-protection in mammals. Our results suggest that the use of REELIN could be a valuable therapeutic approach to improve cardiac function in humans.
Methods
Mouse models
LEC-specific Prox1 deficient mice were generated by crossing Prox1f/f mice7 with Cad5(PAC)-CreERT2 mice10. These mice are maintained in a mixed C57B6 and NMRI background. LEC-specific Reln deficient mice were generated by crossing Relnf/f mice18 with Prox1CreERT2 mice19. These mice are in a mixed 129, FVB and C57B6 background. Reln+/− mice were kindly provided by Dr. Bianka Brunne and are originally from the Jackson laboratory and are maintained in a mixed Balb/c and C57B6 background. For induction of Cre mediated recombination in Prox1ΔLEC/ΔLEC and RelnΔLEC/ΔLEC embryos, two consecutive intraperitoneal tamoxifen (TAM) injections of 5 mg/40g were administered to pregnant dams. Integrin β1f/f mice and MhcCre mice were obtained from the Jackson laboratory and are in a mixed C57B6 and NMRI background. These strains were bred to generate MhcCre;Integrin β1f/f+ mice that were crossed with Reln+/− mice to obtain MhcCre;Integrinβ1f/f+;Reln+/− (β1ΔCM/+;Reln+/−) embryos. Heterozygous mice carrying the kinase-dead Flt4Chy allele (Vegfr3kd) (MRC Harwell) were described previously30 and are maintained in the NMRI background. Twelve-weeks-6 month old mice of both sexes were used for breeding and experiments. Mice were not randomized into experimental groups, but were age and sex-matched and littermates were used whenever possible.
All animal husbandry was performed in accordance with protocols approved by Northwestern University and UT Southwestern Medical Center Institutional Animal Care and Use Committee, as well as Animal Experimentation Review Board of the Semmelweis University. Animal facilities are equipped with a 14:10 or 12:12 light cycle. Temperatures are maintained between 18–23°C with 40–60% humidity.
Mouse embryonic CM isolation
CMs were isolated from E15.5–17.5 mouse embryos using the Pierce Primary Cardiomyocyte Isolation Kit (Thermo Fisher). Briefly, ventricles were isolated from embryonic hearts and minced and washed with cold HBSS and further digested according to the manufacture instructions. To examine the relative CM cell size, dissociated cells were cultured in DMEM containing 10% FBS o/n and then cells were fixed in 4% PFA for immunostaining. For any other experiments, primary cells were cultured in DMEM containing 10% FBS and cardiomyocyte growth supplements for 3–4 days before experiments.
Human iPSC derived-CMs (hiPSC-CMs)
Cardiac differentiation was performed using the CDM3 (chemically defined medium, three components) system as described with slight modifications31,32. hiPSCs are split at 1:15 ratios and grown in B8 medium for 4 days reaching ~80% confluence. On day 0, B8 medium is changed to CDM331, consisting of RPMI 1640 (Corning, 10-040-CM), 500 μg/ml fatty acid-free bovine serum albumin (GenDEPOT), and 200 μg/ml l-ascorbic acid 2-phosphate (Wako), supplemented with 6 μM of CHIR99021 (LC Labs, C-6556). After 24 hours (day 1), medium is changed to CDM3. On day 2, medium is changed to CDM3 supplemented with 2 μM of Wnt-C59 (Biorbyt, orb181132). Medium is then changed every other day for CDM3 starting on day 4. Contracting cells are noted from day 7. On day 16 of differentiation, CMs are dissociated using DPBS for 20 min at 37 °C followed by 1:200 Liberase TH (Roche) diluted in DPBS for 20 min at 37 °C, centrifuged at 300 g for 5 min, and filtered through a 100 μm cell strainer (Falcon). The purity of the differentiated cells was determined by expression of CM cell marker TNNT2 using flow cytometry. Only cell lines that show over 85% are TNNT2+ were used for experiments.
LEC conditioned medium
Human dermal LECs were purchased from Lonza and cultured with endothelial basal medium (EBM) complemented with supplement mix (Lonza). Passages 4 or 5 were cultured in 10 cm dishes until confluent, washed with cold PBS three times and then 8 ml of serum free DMEM (without phenol red) with penicillin/streptomycin was added. Cells were then cultured o/n before collecting the conditioned media that was filtered through a 0.22 μm pore membrane (Millipore). Control conditioned media (DMEM) was prepared in the same way but without LECs.
siRNA knockdown
Human LECs were transfected as described previously33. Briefly, P4 human LECs were transfected with scrambled or Reln siRNA (Santa Cruz) with Lipofectamine 2000 (Invitrogen), according to the manufacture’s instruction. After 48 h, cells were washed and replaced with DMEM and further cultured o/n to collect the conditioned media. LECs were collected and qPCR was performed to check transfection efficiency.
LEC conditioned media treatment
To examine the effects of the LECs conditioned media, mouse primary CM or human iPSC-CM were cultured in 12 well plates (about 80% confluence), and cells were treated either with DMEM, conditioned media, conditioned media from scrambled siRNA treated LECs (siCtrl-conditioned), conditioned media from siReln treated LECs (siReln-conditioned) or conditioned media with Integrinβ1 blocking antibodies (10 μg/ml, BD Biosciences) o/n. Cells were either fixed in 4%PFA for immunofluorescent staining, or lysed in RIPA buffer for Western blot analysis.
Reelin conditioned media and treatment
HEK-293T cells (ATCC) were cultured in DMEM with 10% fetal bovine serum and transfected with the Reelin cDNA construct pCrl, kindly provided by Dr. Gabriella D’Arcangelo using Lipofectamine 2000 (Invitrogen). Control cells were mock transfected in the same way without adding the vector. Twenty-four hours after transfection, the medium was changed to serum free DMEM and Reelin conditioned medium and mock conditioned media (control) were collected two days after the medium change. The conditioned medium was filtered through a 0.22 μm pore membrane. To examine the effects of the Reelin conditioned media, mouse primary CM were starved o/n with DMEM and stimulated for 30 min with Reelin conditioned media (supernatant from transfected cells) or control media (supernatant from mock-transfected cells). To examine the Reelin/Integrinβ1 pathway, primary CM were treated in the presence or absence of Integrinβ1 blocking antibodies (10 μg/ml, BD Biosciences) for 3 h prior to Reelin conditioned media treatment.
Western blot analysis
To examine signaling changes in primary CMs or iPSC-CMs, cells were lysed in RIPA buffer and subject to Western blot analysis. The following primary antibodies were used: p-AKT (Rabbit, Cell Signaling, 4060, 1:500), p-ERK (Rabbit, Cell Signaling, 4370, 1:1,000), total AKT (Rabbit, Cell Signaling, 4691, 1:500), total ERK (Rabbit, Cell Signaling, 4695, 1:500), p-Dab1 (Rabbit, Cell Signaling, 3327S, 1:100), p-FAK (Rabbit, Cell Signaling, 3284, 1:200), integrin β1 (Mouse, BD, 610467, 1:100), Gapdh (Rabbit, Santa Cruz, sc32233, 1:5,000). Blots were imaged using a ChemiDock imaging system (Bio-Rad) and bands were acquired using Quantity One 1-D software. Quantification of Western blot was analyzed using ImageJ 1.51. Included images are representative blots. All raw data used for the quantifications is included in the Supplementary information.
Mass spectrometry analysis of LEC conditioned-media
50 ml of LEC-conditioned was collected from five 10-cm dishes of cultured LECs and filtered through a 0.22μm pore membrane as mentioned above. LEC-conditioned media was further concentrated into 500uls using the Protein-Concentrate Kit (Millipore) according to the manufacture’s instruction. Protein concentration was then measured by the BCA protein assay (Thermo Fisher). Experiments were repeated three times and three biological samples were submitted to Northwestern Proteomics Core for untargeted quantitative proteomics analyses by Label-free Quantitative Proteomics: Briefly, samples were analyzed using an UltiMate™ 3000 RSLCnano system (ThemoFisher Scientific, CA) that is coupled with electrospray ionization (ESI) to a linear ion trap (LTQ) Orbitrap mass spectrometer (iLTQ-Orbitrap, ThermoFisher, CA). The resulting raw mass spectra from all three replicates were analyzed by the MaxQuant search engine (version 1.6.0.16) using UniprotKB human database with the allowance of up to 2 missed cleavages and precursor mass tolerance of 20 p.p.m. The secretome was acquired using software Scaffold 4 and annotated using Gene Ontology (GO), which assigns putative cellular compartmentalization, biological process and molecular functions.
ELISA
To validate the presence of Reelin in the LECs conditioned media, 3 different batches of commercial LECs were cultured and their conditioned media was collected as described. Sandwich enzyme-linked immunosorbent assay (ELISA) was performed to examine the relative levels of Reelin in the 3 different batches of LECs conditioned media. Briefly, conditioned media were pre-coated to Nunc MaxiSorp™ Flat-Bottom 96-well plates (Invitrogen) o/n and blocked with 5% milk in TBST. Plates were then incubated with Reelin primary antibody (R&D, AF3820, 1:100) and followed by incubation with HRP conjugated Donkey anti-goat antibody (Jackson ImmunoResearch, 705-035-003, 1:1000). Subsequently, plates were washed and the substrate solution (3,3,5,5-tetramethylbenzidine liquid substrate system for ELISA, Abcam) was added. The reaction was stopped by adding 2N H2SO4, and plates were measured at 450 nm using the Opsys Mr microplate reader (Dynex Technologies). Relative Reelin levels in different batches of conditioned media were quantified by OD intensity.
FACS analyses and sorting
For analysis of percentages of CMs in the heart, whole E17.5 ventricles were dissociated from control and Prox1ΔLEC/ΔLEC hearts using the Pierce Primary Cardiomyocyte Isolation Kit (Thermo Fisher). Cells were fixed and permeabilized using a Permeabilization kit for intracellular staining (eBioscience) following the manufacturer’s instruction. Cells were then incubated with Cy3-conjugated mouse anti-cardiac Troponin C antibody (Abcam, ab45931, 1:100) and Hoechst 33342 (Invitrogen, 1:1000) at room temperature for 1 h. Cells were washed and percentage of cTnC+ CMs was determined after 20,000 total cell counts by flow cytometry. Percentage of polyploidy CMs was determined by Hoechst 33342 intensity. Flow data was collected using the flow software BD FACS Diva 8.0.3 and analyzed by FlowJo v10.
For analysis of the purity of differentiated hiPSC-CM, dissociated CMs were fixed with 4% PFA and permeabilized using 0.5% saponin. Cells were then incubated with 647-conjugated mouse anti-cardiac TNNT2 antibody (BD Biosciences, clone 13-11, 1:200) for 1 h. Cells were washed and percentage of TNNT2+ CM was determined after 10,000 total cell counts by flow cytometry.
Neonate myocardial infarction
Neonatal myocardial infarction was performed in P2 pups. Briefly, P2 pups were anaesthetized under isoflurane anesthesia (1–2%). Once pups did not respond to toe pinch, they were moved to a cold platform to undergo hypothermia anesthesia. Each neonate undergoes acute myocardial infarction by ligation of the left anterior descending coronary artery. Thoracic wall incisions were sutured and the wound closed using skin adhesive. Pups were warmed on a warm pad. After confirmation of spontaneous movement pups received a dose of subcutaneous buprenorphine (0.05mg/kg). Once neonate recovered from hypothermia, they are moved back to its fostering mother’s cage.
Compressed collagen patches
Compressed acellular collagen patches were prepared as described previously. Briefly, control collagen patches were prepared by adding 1.1 ml DMEM to 0.9 ml of sterile rat tail type I collagen solution in acetic acid (3.84 mg/ml, Millipore) and neutralized with 0.1 M NaOH (~50 μl). REELIN collagen patches were prepared by adding 20 μg of recombinant human REELIN protein (R&D) into the collagen mix. Then, 0.9 ml of the collagen solution was added into one well of 24-well plates and placed into a tissue culture incubator for 30 min at 37 °C for polymerization. Polymerized collagen gel was then compressed by application of a static compressive stress of ~1,400 Pa for 5 min as described28 . Each collagen patch was then trimmed to 3 even pieces for application in vivo.
Myocardial infarction and insertion of collagen patches in adult mice
Nine-11 week-old NMRI female mice were anaesthetized using an isoflurane inhalational chamber, endotracheally intubated using a 22-gauge angiocatheter and connected to a small animal volume-control ventilator (Harvard Apparatus, Holliston, MA). All mice underwent acute myocardial infarction by ligation of the left anterior descending coronary artery and ligation was considered successful when the LV wall turned pale. Immediately after ligation, prepared collagen patches (with and without REELIN) were sutured (at two points) onto the surface of the ischaemic myocardium (Fig. 4e). The patch size used was ~ one-third of the 15.6 mm-diameter collagen gel. Animals were kept on a heating pad until they recovered. After confirmation of spontaneous movement, pups received a dose of subcutaneous buprenorphine (0.05mg/kg).
Echocardiography
Two-dimensional echocardiograms were measured on a 55 MHz probe using Vevo 3100 micro-ultrasound imaging system (VisualSonics), short axis views of the left ventricles were taken at the level of papillary muscles and used to calculate end-diastolic and -systolic dimensions using Vevo LAB 3.2.6 software (VisualSonics). All echocardiography measurements were performed in a blinded manner.
Histology, immunohistochemistry and immunofluorescent staining
For H&E staining, samples were embedded in paraffin and sectioned longitudinally at 6um thickness and staining was performed according to standard protocols.
For whole mount heart staining, isolated hearts were fixed in 4% PFA o/n and blocked. Antibodies were used as followed: Lyve1 (Goat, R&D, AF2125, 1:200), Endomucin (Rat, Invitrogen, 14-5851-82, 1:500), Reelin (Goat, R&D, AF3820, 1:50), Prox1 (Goat, R&D, AF2727, 1:100) and Cy3-conjugated α-SMA (Mouse, Sigma, C6198, 1:300). Cy3-conjugated donkey anti-goat (Jackson ImmunoResearch, 705-165-147, 1:300) and Cy5-conjugated donkey anti-rat (Jackson ImmunoResearch, 712-175-150, 1:300) were used for immunofluorescent staining.
For cryosections, embryos or isolated hearts were fixed in 4% PFA o/n and dehydrated in 30% sucrose. Samples were embedded in OCT compound and frontal sectioned at 10-um thickness to show four chambers. Primary antibodies were used as follows: α-Actinin (Mouse, Sigma, A7811, 1:500), cardiac Troponin C (Mouse, Abcam, ab8295, 1:1000), Ki67 (Rabbit, Invitrogen, SP6, MA5-14520, 1:200), active Caspase-3 (Rabbit, BD Pharmingen, C92- 605, 559565, 1:200), Lyve1 (Goat, R&D, AF2125, 1:200), Reelin (Goat, R&D, AF3820, 1:50), Lyve1 (Rabbit, AngioBio, 11-034, 1:500), Prox1 (Rabbit, AngioBio, 11002, 1:500), Prox1 (Goat, R&D, AF2727, 1:100) and Mef2c (Rabbit, LSBio, LSC356188, 1:1000). Secondary antibodies were used as follows: Alexa 488-conjugated donkey anti-rabbit (Invitrogen, A21206, 1:300); Alexa 488-conjugated donkey anti-goat (Invitrogen, A11055, 1:300); Cy3-conjugated donkey anti-rabbit (Jackson ImmunoResearch, 711-165-152, 1:300); Alexa 488-conjugated donkey anti-mouse (Invitrogen, A21202, 1:300); Cy3-conjugated donkey anti-goat (Jackson ImmunoResearch, 705-165-147, 1:300) and Cy5-conjugated donkey anti-goat (Jackson ImmunoResearch, 705-495-147, 1:300).
For cell staining, cells were fixed in 4% PFA for 30 min on ice, blocked, and incubated with primary antibody against α-Actinin (Mouse, Sigma, A7811, 1:500), cardiac Troponin C (Mouse, Abcam, ab8295, 1:1000), Ki67 (Rabbit, Invitrogen, SP6, MA5-14520, 1:200), Prox1 (Goat, R&D, AF2727, 1:100) and active Caspase-3 (Rabbit, BD Pharmingen, C92- 605, 559565, 1:200). Secondary antibodies were used as follows: Alexa 488-conjugated donkey anti-mouse (Invitrogen, A21202, 1:300) and Cy3-conjugated donkey anti-rabbit (Jackson ImmunoResearch, 711-165-152, 1:300). At least 3 heart samples per genotype were used for whole mount staining and 3 sections per heart per staining for immunohistochemistry and immunofluorescent staining, respectively. Cell staining was repeated at least 3 times.
For Masson’s Trichrome staining, mouse hearts were harvested and fixed in 4% PFA and embedded. Paraffin sections were cut from apex to base into serial sections at 0.8μm thickness. Masson’s trichrome staining was performed according to standard procedures (Sigma) and used for detection of fibrosis. Scar size was quantified using NIH ImageJ 1.51 software and the percentage of the fibrosis area was calculated relative to left ventricle area.
EdU administration
To examine the EdU incorporation in Prox1ΔLEC/ΔLEC, Vegfr3kd/kd, β1ΔCM/+;Reln+/− or Prox1ΔLEC/ΔLEC strains, 5-ethynyl-2′-deoxyuridine (EdU, 3mg/mouse) was administrated into pregnant females by intraperitoneal injections. 2 h after injections, mice were euthanized and hearts, livers and kidney collected and cryosectioned as described above. To examine EdU incorporation in control or REELIN patched treated mice after MI, EdU (3mg/mouse) was injected intraperitoneally for 3 days starting 4 days after MI. Hearts were collected at day 7 and subjected to EdU immunohistochemistry using Click-iT® EdU Alexa Fluor® 488 Imaging Kit (Life Technologies) according to the manufacture’s instruction.
Quantitative reverse transcription PCR (qRT-PCR)
Total RNAs was extracted using RNeasy Mini Kit (Qiagen). cDNA was generated (Clontech Laboratories) and 20 ng used for qRT-PCR using Power SYBR Green PCR Master Mix (Life Technologies) on a StepOnePlus Real-Time PCR system (Applied Biosystems). At least three individual samples per group were performed for each run of qPCR. Primer sequences used in this study are listed below.
For mouse qPCR:
Bcl2l11: GAGATACGGATTGCACAGGA, ATTTGAGGGTGGTCTTCAGC; P21 GAAAGAAGCGGAAGATCCTCC, GGGCCTCAGGGATTGTTTGG; Pdcd4 GAAATTGGATTTCCGCATCT, TAACCGCTTCACTTCCATT; Stat1: AGGGGCCATCACATTCACAT, AGATACTTCAGGGGATTCTC; Trp53ip1: TCCTCAGCAGAGCACACTTC, TCCATTGGACAGGACTCAAA; Cdc6: AGGGTGACTTTGAGCCAAGA, ATGAAGATTCTGGGGGCTCT; E2f1: TGCAGAAACGGCGCATCTAT, CCGCTTACCAATCCCCACC; Pcna: TTGCACGTATATGCCGAGACC, GGTGAACAGGCTCA TTCA TCTCT ;Mcm5: GGAGGCTATTGTGCGCATTG, CTGGTCCTCCTGGGTAGTGA; Ccne2: TCTGTGCATTCTAGCCATCG, ACAAAAGGCACCATCCAGTC; Reelin: GGACTAAGAATGCTTATTTCC, GGAAGTAGAATTCATCCATCAG; Rlp32: GCCTCTGGTGAAGCCCAAG, TTGTTGCTCCCATAACCGATGT,
For human qPCR:
Reelin: CAATCTGAATGGCGAAACC, CTTTCGCTATAAATCGGAGAGAGA; Gapdh: TGACCACAGTCCATGCCATC, GACGGACACATTGGGGGTAG; MMRN1: TTGGATTGGAGGTGCTGTC, GCCTGGTTGGTGTGTATCA; THBS1: CACCAACCGCATTCCAGAG, TCAGGGATGCCAGAAGGAG; HSPG2: CTCCATCGTCATCTCCGTCT, GTCTGCCCTTCTGCCACTC; FN1: CCATCGCAAACCGCTGCCAT; AACACTTCTCAGCTATGGGCT T; FSTL1: CGATGGACACTGCAAAGAGA, CCAGCCATCTGGAATGATCT; LAMA4: GCGGCCGAGAAATGCA, AGTCGCAGGGCACACATTC; SERPINE1: ACAAGTTCAACTATACTGAGTTCACCACGCCC, TGAAACTGTCTGAACATGTCGGTCATTCCC.
All sequences are included forward and reversed and are annotated from 5’to 3.
RNAseq
Total RNA was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Extracted total RNAs were quantitated by NanoDrop and RNA integrity number value measured with an Agilent Bioanalyzer. In all RNAseq samples, quality control was performed using the 2100 Bioanalyzer (Agilent). RNA library was prepared using the TruSeq mRNA-Seq Library Prep and sequenced using the HiSEQ Next-generation Sequencing System at the NUSeq Core.
Imaging acquisition and quantification
Confocal images in Fig. 1a, d–i, Extended data Figures 1a, 3b, 4b, 6i and 8g were acquired using a Nikon W1 Dual CAM Spinning Disk confocal microscope. All other confocal images were acquired using Zeiss LSM510 laser-scanning confocal microscope. All confocal images represent maximum projection images of z stacks. For quantifications of CM proliferation and apoptosis, images were taken from 3 myocardium regions of frontal heart sections: myocardium nearby left ventricle, myocardium nearby right ventricle and septum. At least 9 images were taken from each heart (at least 3 images/region) and at least 3 hearts from each genotype were quantified. Bright field images were taken using a Leica stereomicroscope. Embryo body length was measured from head to tail (crown-rump) using Image J 1.51 with all the images under the same magnification. CM cell size, as well as fibrosis area were also measured by Image J 1.51 software with all the images under the same magnification.
Statistical analysis
No statistical analysis was used to predetermine sample size. Statistical analysis was performed using GraphPad Prism 7 and Microsoft Excel 2016. Differences between two groups were determined by two-tailed unpaired t-test, and differences between multiple groups were calculated using one-way ANOVA or two-way ANOVA. Differences with p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) were considered statistically significant.
Extended Data
Extended Data Figure 1. E17.5 Prox1ΔLEC/ΔLEC hearts lack LECs and have a reduced number of CMs.
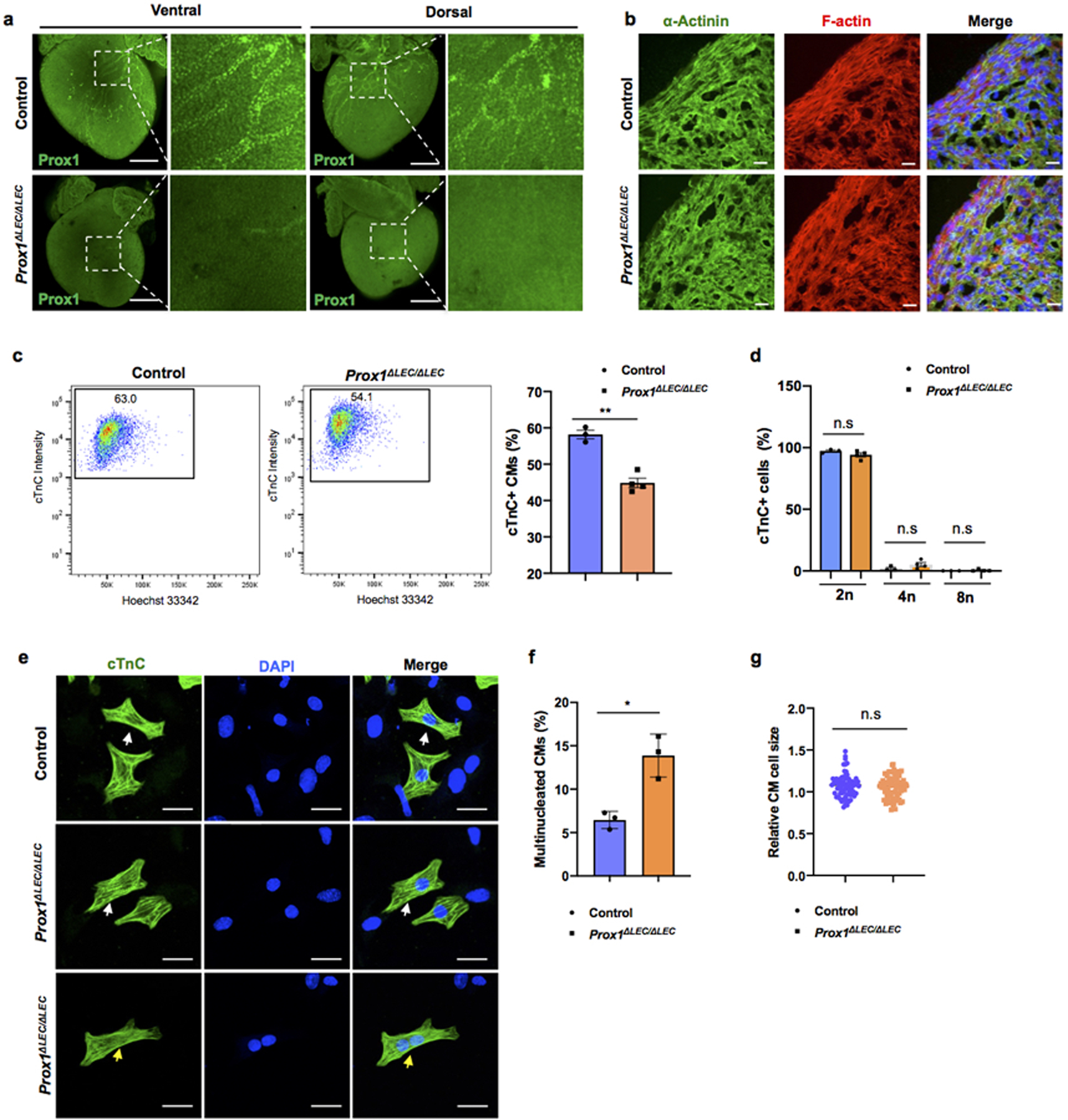
a, Whole mount immunostaining with anti-Prox1 antibody shows that cardiac lymphatics are missing in E17.5 Prox1ΔLEC/ΔLEC hearts (TAM injected at E13.5 and E14.5). Squared areas are shown in larger magnification in the adjacent images. N = 3 embryos/group from two litters. b, Co-immunostaining of E17.5 control and Prox1ΔLEC/ΔLEC heart sections with anti-α-Actinin and F-actin antibodies show that cardiac muscle is not affected in Prox1ΔLEC/ΔLEC embryos (TAM injected at E13.5 and E14.5). N = 3/group. c, Flow cytometry analysis shows reduced CM numbers in E17.5 Prox1ΔLEC/ΔLEC hearts (TAM injected at E13.5 and E14.5). d, Hoechst 33342 labeling shows no significant differences in CMs ploidy between control and Prox1ΔLEC/ΔLEC hearts. N = 3 (control) and N=4 (Prox1ΔLEC/ΔLEC) embryos from the same litter used in c and d. Data are presented as mean ± S.E.M. **p = 0.001 and n.s (not significant) were calculated by unpaired two-tailed Student’s t test. e-g, The percentage of multinucleated CMs in E17.5 mutant hearts is increased (e, f), and no global differences in CM size were detected (e, g) after CM dissociation and o/n plating. N=3 embryo/genotype from the same litter. White arrows indicate CMs and yellow arrows indicate a bi-nucleated CM (g). The average cell size was calculated from 25 cTnC+ CMs /culture (1 whole heart/culture; 3 cultures per genotype). N = 75 (control CMs) and N=75 (Prox1ΔLEC/ΔLEC CMs). Data are presented as mean ± S.E.M. *p=0.023 and n.s (not significant) as calculate by unpaired two-tailed Student’s t test. Scale bars, 500 μm (a), 25μm (b, e). Flow cytometry gating strategy is included in Supplementary Fig 10. Source data.
Extended Data Figure 2. CM proliferation is reduced in E17.5 Prox1ΔLEC/ΔLEC hearts.
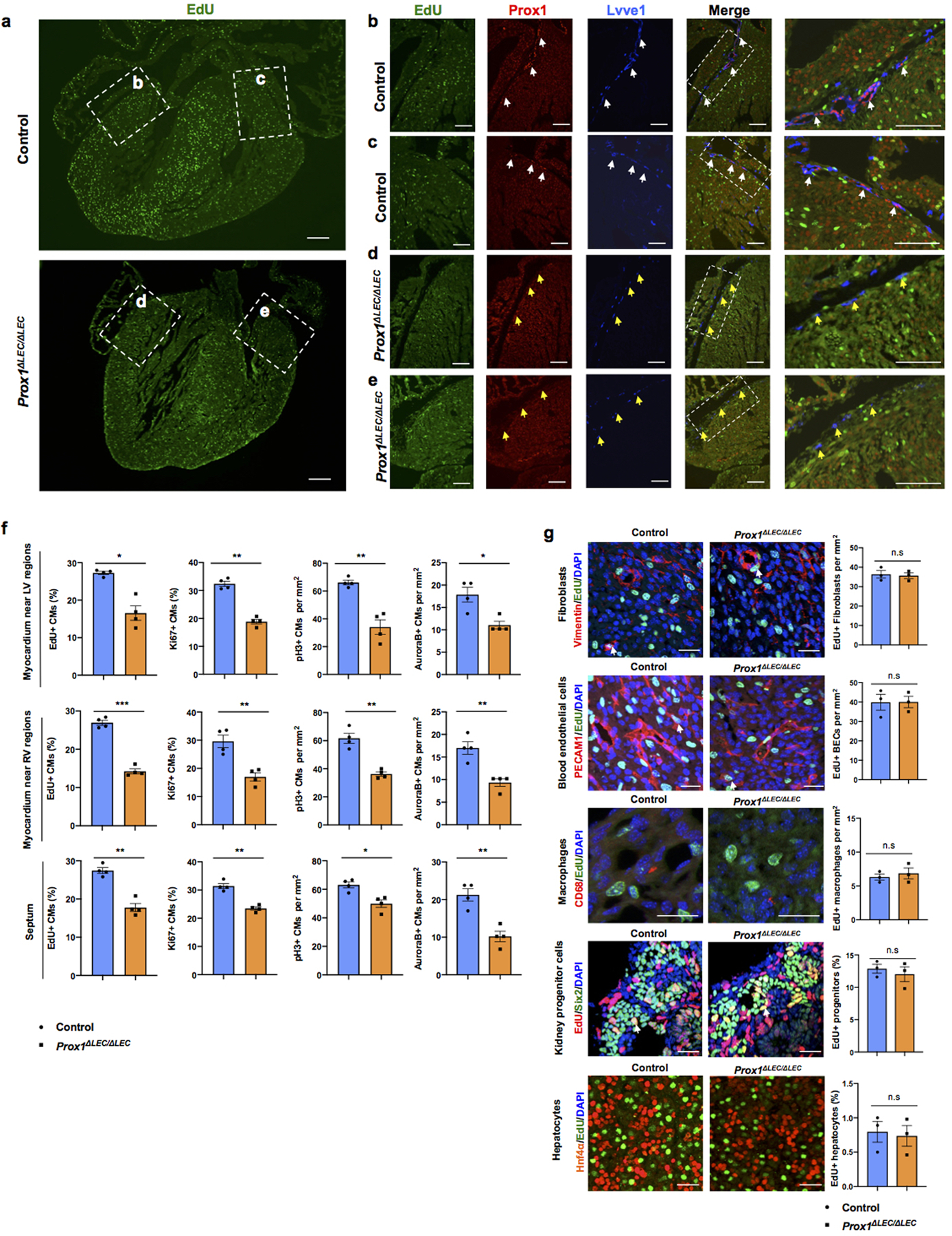
a. EdU labeling shows an overall reduction in the number of EdU+ cells in sections of E17.5 Prox1ΔLEC/ΔLEC hearts. Dashed boxes indicate the corresponding areas of the heart that are shown at higher magnification in panels b-e. b-e, Immunostaining results show the presence of Prox1+Lyve1+ cardiac lymphatics (white arrows) in sections of control hearts (b, c), and lack of lymphatics in Prox1ΔLEC/ΔLEC hearts (d, e). Yellow arrows indicate Lyve1+ Prox1- macrophages. N = 3 embryos/genotype from 3 separate litters. f, CM proliferation is reduced in the myocardium of the left ventricle area (LV), the right ventricle area (RV) and the septum. N = 4 embryos/genotype from 3 separate litters. At least 3 images/region and 3 separate regions/heart were quantified. Data are presented as mean ± S.E.M. *p= 0.01, **p =0.003, 0.006 and *p=0.02 (upper panel); ***p= 0.0001, **p= 0.004, 0.002 and 0.005 (middle panel); **p= 0.001, 0.001, *p= 0.01 and **p =0.002 (bottom panel) as calculated by unpaired two-tailed Student’s t test. g, Immunostaining with antibodies against Vimentin (fibroblasts), PECAM1 (blood endothelial cells), CD68 (macrophages), Six2 (nephron progenitors) or Hnf4α (hepatocytes) together with EdU labeling (white arrows) shows no differences in proliferation in those cell types between E17.5 Prox1ΔLEC/ΔLEC and control hearts (TAM injected at E13.5 and E14.5). n.s. no significant differences by unpaired two-tailed Student’s t test. N = 3 embryos/genotype from 3 separate litters. Control are TAM treated Cre- embryos and Cre+;Prox1+/+ littermates. Data are presented as mean ± S.E.M. Scale bars, 200 μm (a), 100 μm (b-e), 25μm (g).
Extended Data Figure 3. Vegfr3kd/kd embryos lack cardiac lymphatics and have smaller hearts.
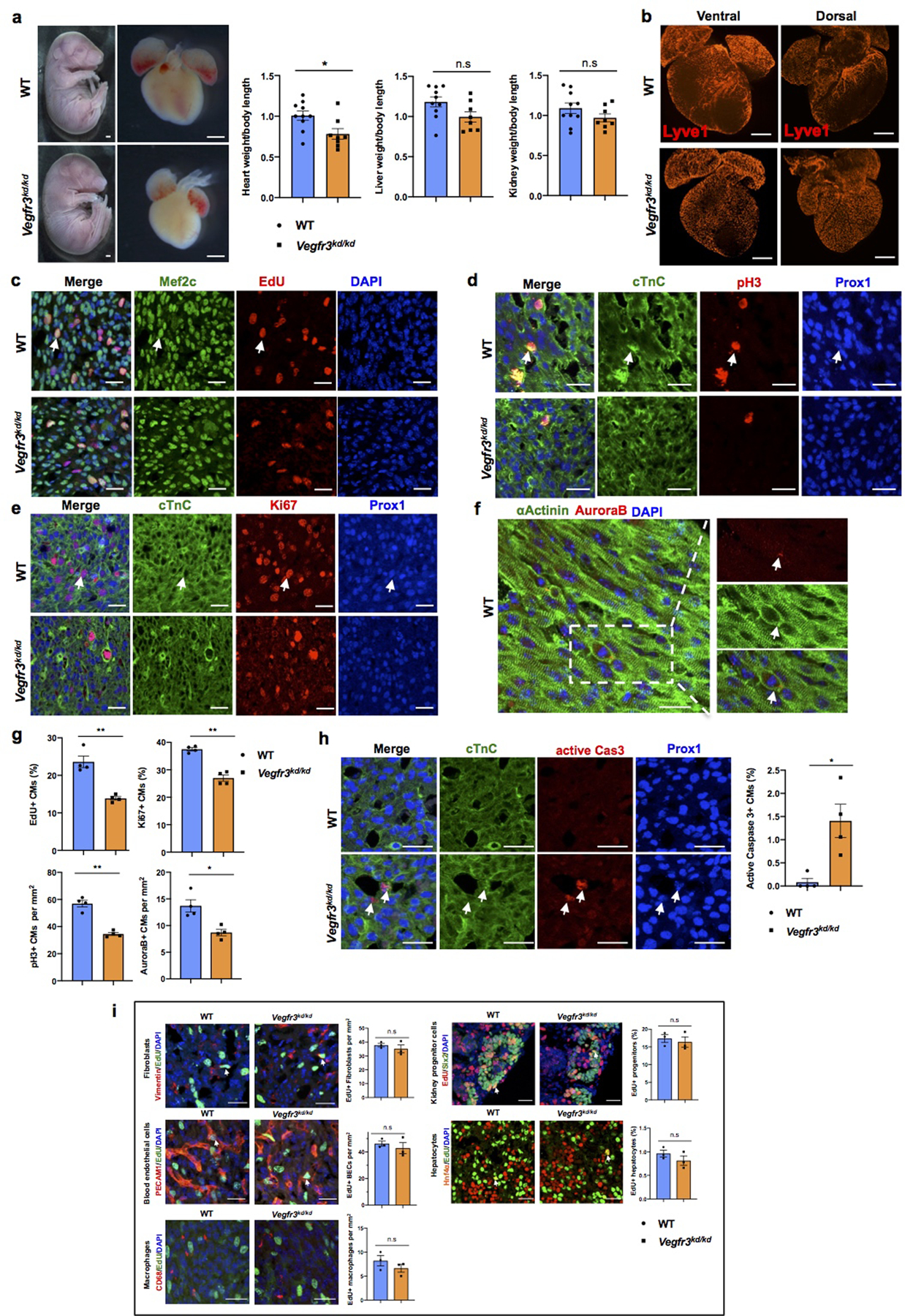
a, Bright field images of whole E17.5 Vegfr3kd/kd and WT embryos and hearts. Quantification of organ weight (heart, liver and kidney) relative to body length indicates that the heart is smaller and the liver and kidney have comparable sizes between Vegfr3kd/kd and control embryos. N=10 (WT) and N=8 (Vegfr3kd/kd). Embryos are from 3 different litters. *p=0.019. b, Lyve1 whole mount immunostaining shows that ventral and dorsal sides of the heart are devoid of lymphatics in Vegfr3kd/kd embryos. N = 3/genotype. c-f, Co-immunostaining using antibodies against cell proliferation markers (EdU, pH3, Ki67 and AuroraB) and antibodies against CM markers (cardiac Troponin C [cTnC], Prox1, αActinin and/or Mef2c) shows reduced CM proliferation in Vegfr3kd/kd hearts compared to wild-type hearts at E17.5. Arrows indicate representative proliferating CMs. g, Quantification shows significantly reduced percentage of EdU+ and Ki67+ CMs and significantly reduce number of pH3+ and AuroraB+ CMs in Vegfr3kd/kd hearts compared to controls. N = 4 embryos/genotype from 3 separate litters. **p=0.005 (EdU), 0.001(Ki67, pH3), *p =0.02 (AuroraB). h, Active Caspase-3 immunostaining shows increased CM apoptosis (white arrows) in Vegfr3kd/kd hearts compared to wild-type hearts at E17.5. Right panel is the quantitative data showing significantly increased percentage of active caspase-3+ CMs (Prox1+) in Vegfr3kd/kd hearts compared to wild-types. N = 4 embryos/genotype from 3 separate litters. *p=0.032. i, Co-immunostaining with antibodies against Vimentin, PECAM1, CD68, Six2 and Hnf4α, together with EdU labeling shows comparable proliferation of cardiac fibroblasts, blood endothelial cells and macrophages, and of nephron progenitors and hepatocytes between wild-type and Vegfr3kd/kd embryos at E17.5. White arrows indicate EdU+ proliferating cells. Quantification of the proliferation for each of those cell types is shown on the right panels. n.s. not significant. N = 3 embryos/genotype from 3 separate litters. Data are presented as mean ± S.E.M. p values were calculated by unpaired two-tailed Student’s t test. Scale bars, 1 mm (a), 500 μm (b), 25 μm (c-f, h), 25μm (i). Lower magnification images for panels c-e and h are included in Supplementary Fig 4.
Extended Data Figure 4. Heart size and CM proliferation is normal in E17.5 Prox1ΔLEC/+ embryos and E14.5 Prox1ΔLEC/ΔLEC embryos.
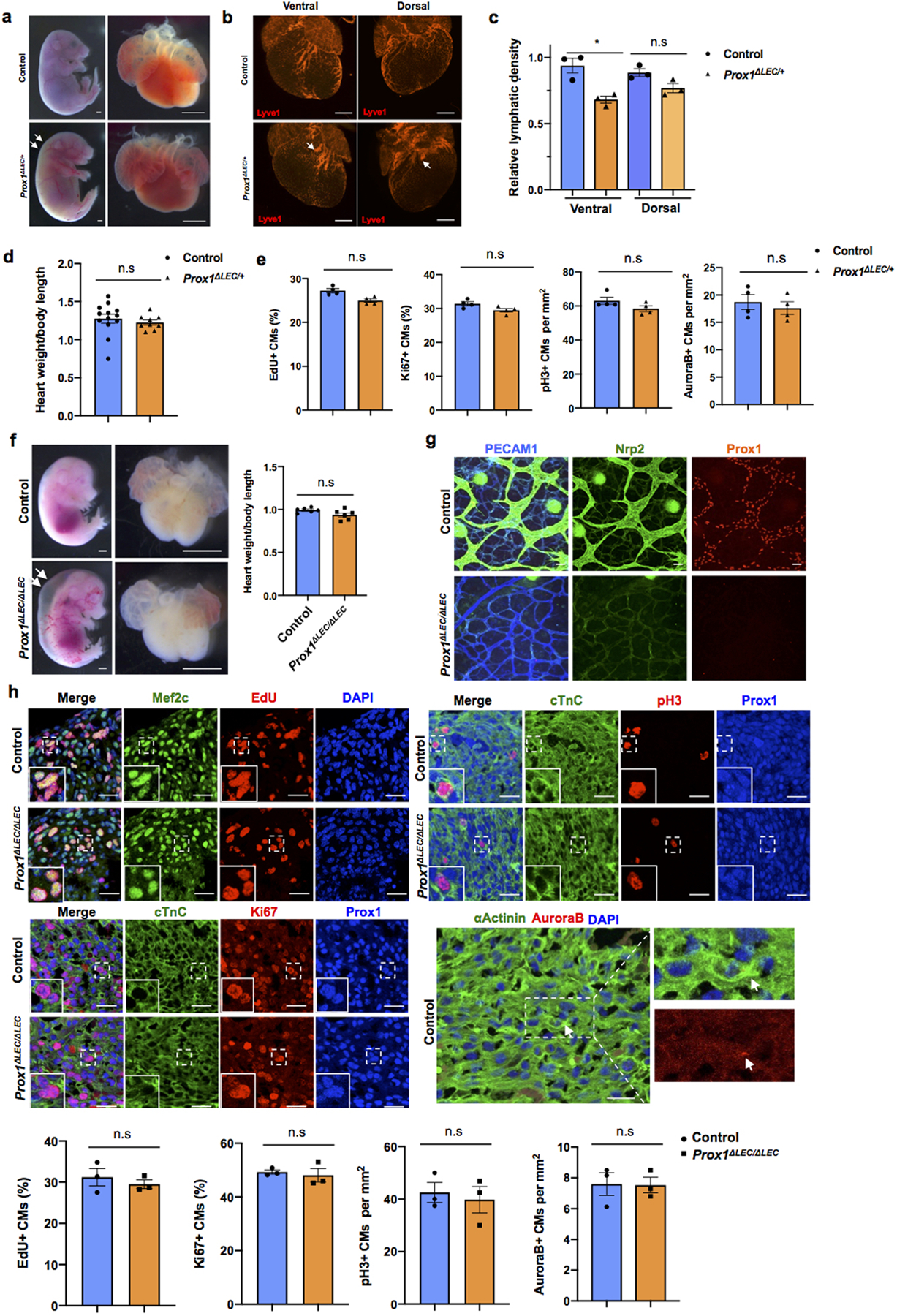
a, Bright field images of whole embryos and hearts show no difference in heart size in E17.5 Prox1ΔLEC/+ embryos (TAM injected at E13.5 and E14.5). White arrows indicate edema in the Prox1ΔLEC/+ embryo. b, Whole mount immunostaining shows that cardiac lymphatics are present in both dorsal and ventral sides of Prox1ΔLEC/+ hearts. Lymphatics are less branched (arrows). c, Cardiac lymphatic density is significantly reduced on the ventral surface of the heart but not on the dorsal one in Prox1ΔLEC/+ embryos. This difference may be because cardiac lymphatics on the dorsal side and the ventral side originate from two different lineages during embryonic development. N=3 embryos/genotype from 3 separate litters. *p=0.027. d, Heart size is normal in E17.5 Prox1ΔLEC/+ embryos. N = 13 (controls) and N=9 (Prox1ΔLEC/+) embryos from 3 separate litters. e, Quantification of the immunostaining analysis shows no significant differences in CM proliferation between E17.5 Prox1ΔLEC/+ hearts and controls, as indicated by the percentage of EdU+ and Ki67+ CMs and the number of pH3+ and AuroraB+ CMs. N = 4 embryos/genotype from 3 separate litters. Controls are TAM treated Cre- embryos and Cre+;Prox1+/+ littermates. f, Bright field images of whole embryos and hearts show no difference in cardiac size in between E14.5 wild-type and Prox1ΔLEC/ΔLEC embryos (TAM injected at E10.5 and E11.5). White arrows indicate severe edema. N=6 embryos/genotype from 2 separate litters. Control embryos are TAM treated Cre- embryos and Cre+;Prox1+/+ littermates. g, Whole mount staining of skin shows efficient Prox1 deletion as indicated by the lack of Prox1+ or Nrp2+ lymphatics at E14.5 in Prox1ΔLEC/ΔLEC embryos. N=3 embryos/genotype from same litter. h, Co-immunostaining against cell proliferation markers (EdU, Ki67, pH3 and AuroraB) together with CM markers (cardiac Troponin C [cTnC], Prox1, αActinin and/or Mef2c). Quantification of those immunostainings shows no differences in CM proliferation between wild-type and Prox1ΔLEC/ΔLEC hearts at E14.5. Squares indicate proliferating CMs. N = 3 embryos/genotype from the same litter. Data are presented as mean ± S.E.M. n.s. not significant difference by unpaired two-tailed Student’s t test. Scale bars, 1mm (a, f), 500 μm (b), 25μm (g, h).
Extended Data Figure 5. Pathways related to cell cycle are downregulated in E17.5 Prox1ΔLEC/ΔLEC embryos and LECs-conditioned media promotes CM proliferation and survival in vitro.
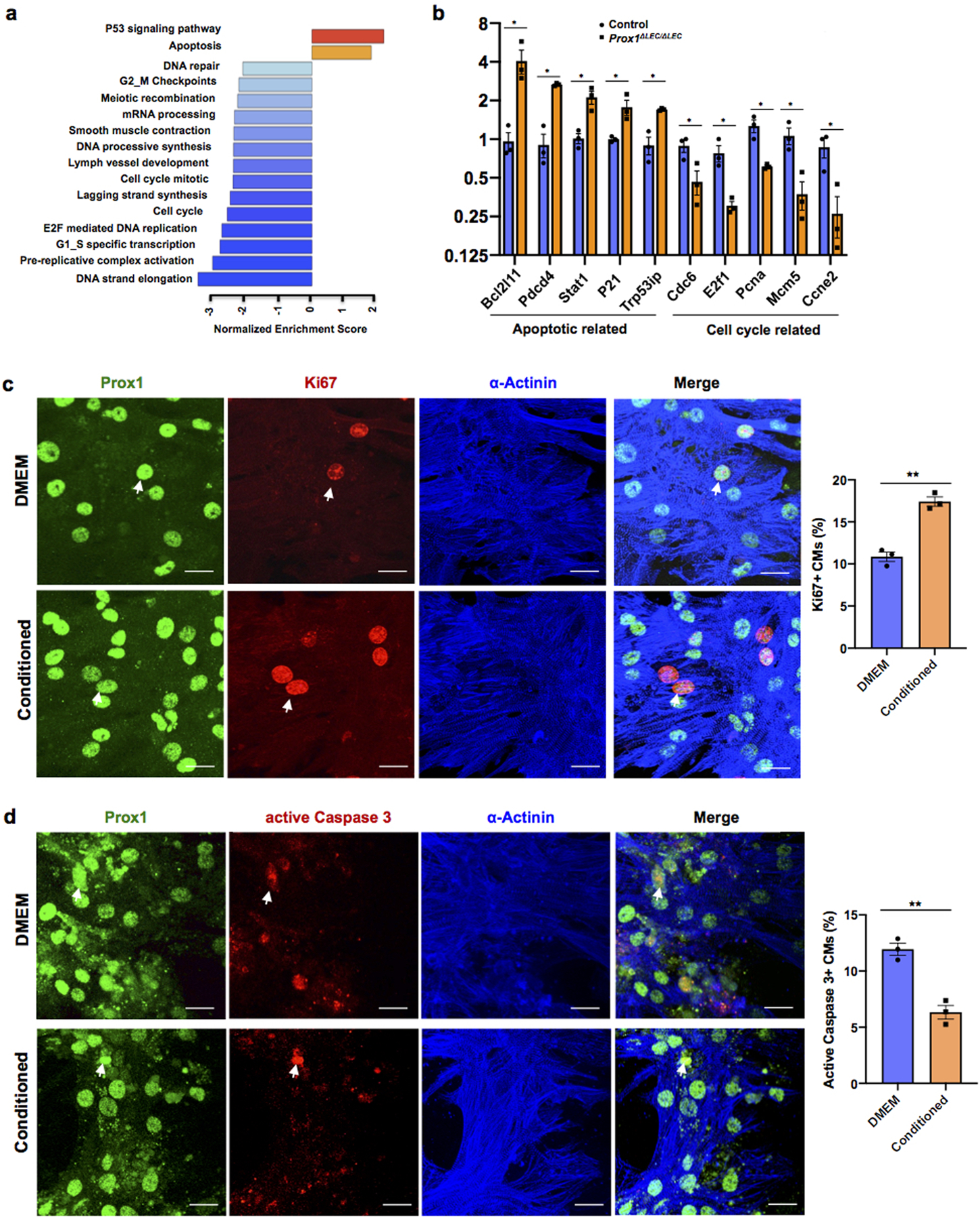
a, GSEA shows downregulation of cell cycle pathways and upregulation of cell death pathways in Prox1ΔLEC/ΔLEC hearts. N=4/genotype from the same litter. b, qPCR analysis confirmed the upregulation of pro-apoptotic genes (Bcl12ll, Pdcd4, Trp53ip, Stat1 and P21) and downregulation of cell cycle related genes (Cdc6, E2f1, Pcna, Mcm5 and Ccne2) in Prox1ΔLEC/ΔLEC hearts. N=3/genotype from the same litter. TAM was injected at E13.5 and E14.5. Control embryos are TAM treated Cre- embryos and Cre+;Prox1+/+ littermates. *p=0.02 (Bcl12ll), **p =0.001 (Pdcd4), 0.005 (Trp53ip), *p =0.01 (Stat1), 0.03 (P21), 0.04 (Cdc6), 0.02 (E2f1), 0.01 (Pcna), 0.02 (Mcm5) and 0.03 (Ccne2). c, Co-immunostaining against the proliferation marker Ki67 and the CM markers α-Actinin and Prox1 shows that LECs-conditioned media increases primary CM proliferation. Arrows indicate proliferating CMs. Percentage of CM proliferation was quantified by the number of Ki67+ Prox1+ CMs relative to total number of Prox1+CMs. N=3. **p=0.001. d, Co-immunostaining against the apoptotic marker active Caspase-3 and the CM markers α-Actinin and Prox1 shows reduced primary CM apoptosis upon LEC-conditioned media treatment under CoCl2 induced hypoxia. Arrows indicate apoptotic CMs. Percentage of apoptotic CMs was quantified by the number of active Caspase 3+ CMs relative to Prox1+ CMs. N=3. **p=0.003. Data are presented as mean ± S.E.M. p values were calculated by unpaired two-tailed Student’s t test. n.s, not significant. Scale bar, 25 μm (c,d).
Extended Data Figure 6. E17.5 Reln−/− embryos develop smaller hearts.
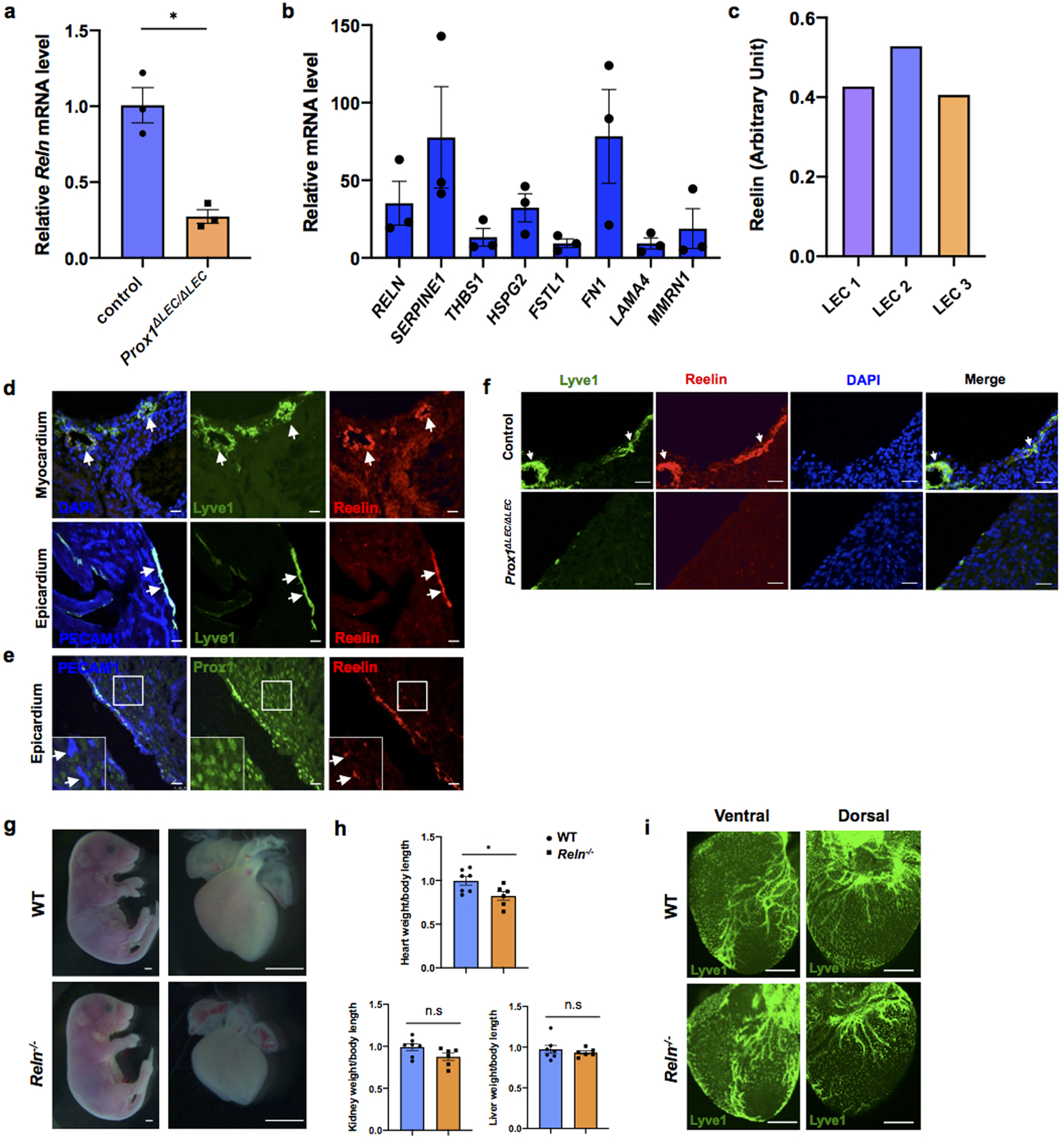
a, qPCR analysis shows reduced Reln expression in E17.5 Prox1ΔLEC/ΔLEC hearts (TAM injected at E13.5 and E14.5). N = 3 embryos/genotype from the same litter. Control embryos are TAM treated Cre- embryos and Cre+;Prox1+/+littermates. *p=0.014. b, qPCR analysis validates the expression of candidates from the LECs secretome (SERPINE1, FN1, RELN, HSPG2, MMRN1, LAMA4, FSTL1 and THBS1). Experiments were repeated 3 times using different batches of LECs. Gene expression is normalized as a fold change relative to 100x Gapdh. c, Reelin protein can be detected in 3 different batches of LEC conditioned media and the relative Reelin level is quantified by ELISA according to the OD intensity. d-e, Immunostaining of sections of E17.5 WT hearts shows Reelin is highly expressed in cardiac lymphatics of the epicardium and myocardium. Some blood vessels in the heart express low levels of Reelin (e, arrows). N = 3 WT embryos. f, Immunostaining of E17.5 control and Prox1ΔLEC/ΔLEC heart sections with antibodies against Reelin and Lyve1 shows that cardiac lymphatics and Reelin are absent in Prox1ΔLEC/ΔLEC hearts (TAM injected at E13.5 and E14.5). N = 3 embryos/genotype from the same litter. Control embryos are TAM treated littermate Cre- and Cre+; Prox1+/+embryos. g, Representative bright field images show smaller hearts in E17.5 Reln−/− embryos. h, Quantifications of organ weight (heart, liver and kidney) relative to body length indicate that hearts are smaller in E17.5 Reln−/− embryos compared to controls. N = 7 (WT) and N=6 (Reln−/−) embryos from 3 separate litters. *p =0.03. i, Whole mount immunostaining shows that cardiac lymphatic development is normal in Reln−/− embryos. N = 3 embryos/genotype from 2 separate litters. Data are presented as mean ± S.E.M. p values were calculated by unpaired two-tailed Student’s t test. n.s, not significant. Scale bar, Scale bar, 25 μm (d, e, f), 1mm (g), 500 μm (i).
Extended Data Figure 7. Reelin is efficiently deleted in RelnΔLEC/ΔLEC cardiac-associated lymphatics.
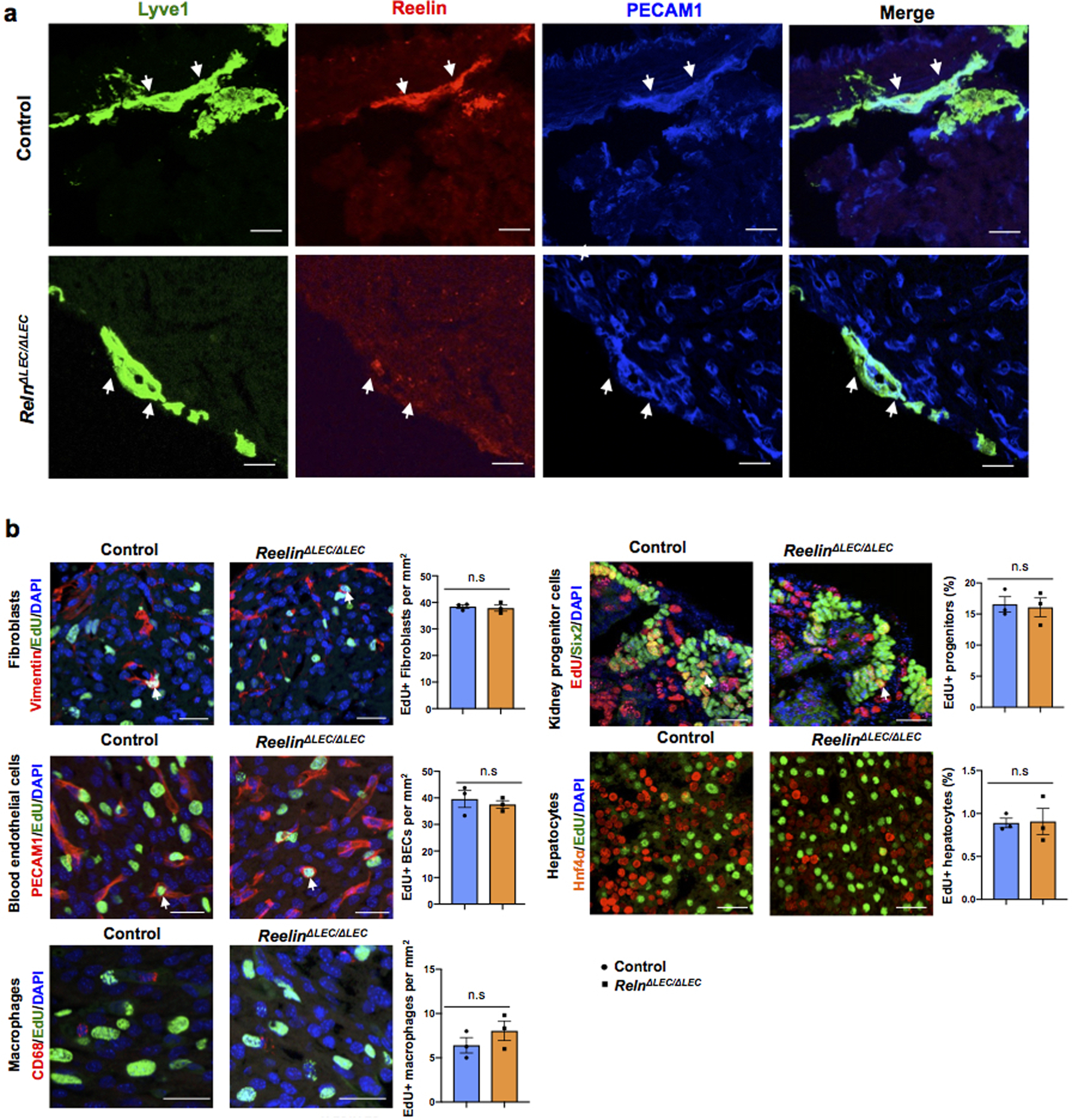
a, Immunostaining of E17.5 control and RelnΔLEC/ΔLEC heart sections with antibodies against Reelin and Lyve1 confirms that Reelin is deleted from cardiac lymphatics in RelnΔLEC/ΔLEC hearts (TAM injected at E13.5 and E14.5). N = 3 embryos/genotype from 2 separate litters. Control embryos are TAM treated Cre- embryos and Cre+; Reln+/+embryos. b, Co-immunostaining with antibodies against Vimentin, PECAM1, CD68, Six2 and Hnf4α, together with EdU labeling shows comparable proliferation of cardiac fibroblasts, blood endothelial cells and macrophages, and of nephron progenitors and hepatocytes between controls and E17.5 RelnΔLEC/ΔLEC hearts (TAM injected at E13.5 and E14.5). White arrows indicate EdU+ proliferating cells. Quantification of the proliferation for each of those cell types is shown on the right panels. N = 3 embryos/genotype from 2 separate litters. Control embryos are TAM treated Cre- and Cre+;Reln+/+ littermates. Data are presented as mean ± S.E.M. n.s. not significant difference by unpaired two-tailed Student’s t test. Scale bar, 25 μm.
Extended Data Figure 8. Cardiac size is reduced in E17.5 β1ΔCM/+;Reln+/− embryos.
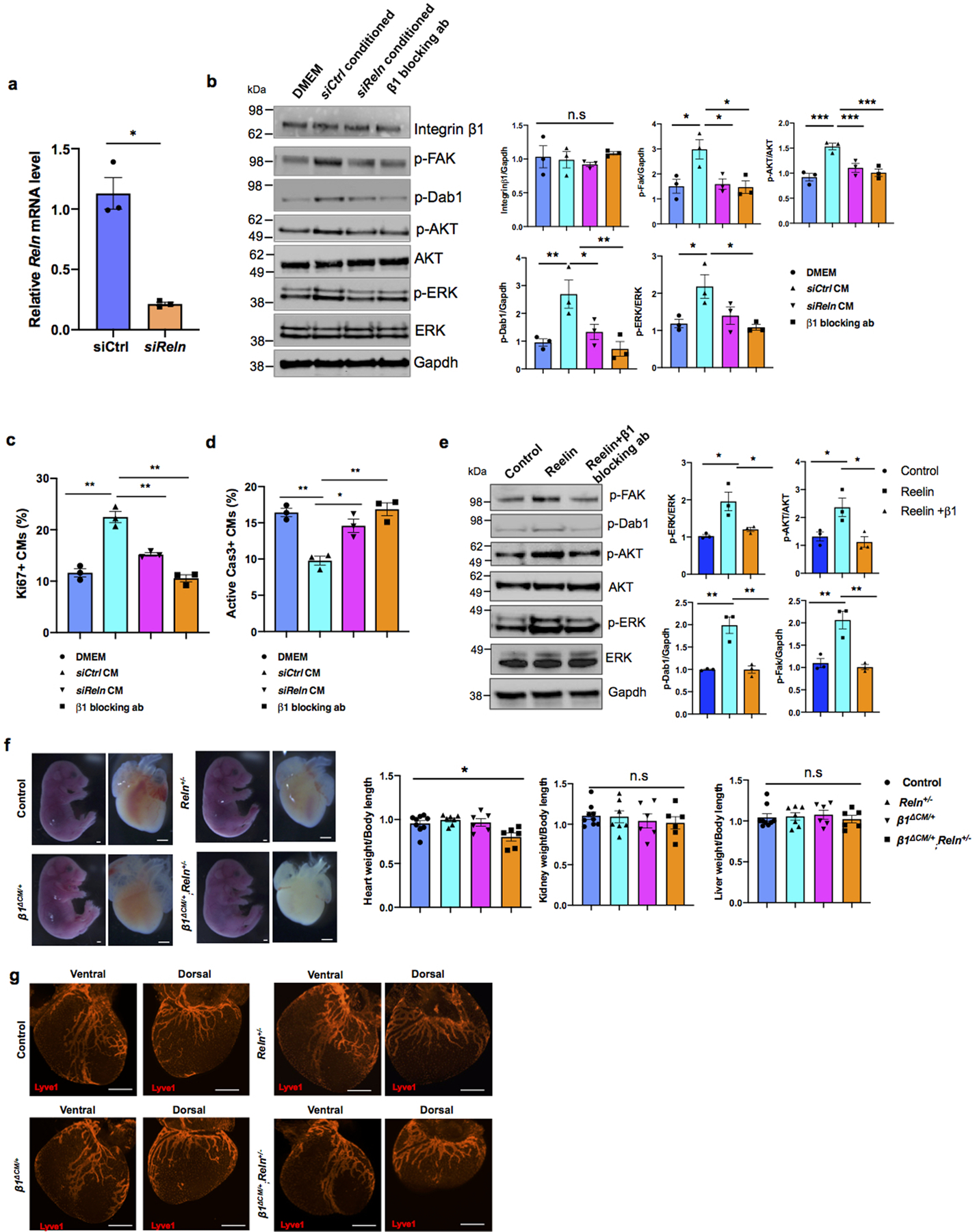
a, qPCR analysis shows efficient Reln knockdown in LECs after siRNA treatment. N=3. Mean ± S.E.M. *p < 0.05 by unpaired two-tailed Student’s t test. b, Representative Western blot of primary CMs cultured with DMEM, siCtrl and siReln treated conditioned media, or with conditioned media + Integrinβ1 blocking antibody o/n. Addition of the LEC conditioned media (siCtrl group) to primary CMs increases Dab1, FAK, AKT and ERK activities. These activities are reduced when cultured CMs are treated with Reelin deficient LECs conditioned media or with LECs conditioned media with β1 blocking antibody. Experiments were repeated 3 times. Data are presented as mean ± S.E.M. *p < 0.05; **p <0.01; ***p<0.001 by two-way ANOVA followed by Bonferroni test. c, Ki67 quantification of immunostained cultured cells (similar to Extended Data Fig. 5c) shows that addition of the LEC conditioned media (siCtrl group) to cultured primary CMs improves CM proliferation and this effect is partially abolished in CMs treated with Reln (siReln) deficient LECs conditioned media or with LECs conditioned media containing β1 blocking antibody. Percentage of CM proliferation was quantified by the number of Ki67+ Prox1+ CMs relative to total numbers of Prox1+CMs. N=3. Mean ± S.E.M. **p < 0.01 by two-way ANOVA followed by Bonferroni test. d, Quantification of active Caspase 3 immunostained cultured CMs shows that addition of the LEC conditioned media (siCtrl group) to primary CMs protect them from apoptosis and this effect is partially abolished in CMs treated with Reln deficient LECs conditioned media or with LECs conditioned media with β1 blocking antibodies. Percentage of apoptotic CMs was quantified by the number of active Caspase 3+ CMs relative to Prox1+ CMs. N=3. Mean ± S.E.M. *p < 0.05; **p <0.01 by two-way ANOVA followed by Bonferroni test. e, Representative Western blot of primary CMs after treatment with Reelin conditioned media from Reelin transfected cells (Reelin), or conditioned media from mock-transfected cells (control) or Reelin conditioned media with Integrin β1 blocking antibody (Reelin + β1 blocking ab) shows that Reelin treatment increases Dab1, FAK, AKT and ERK activities in primary CMs, and these activities are reduced by adding the Integrinβ1 blocking antibody. N=3. Data are presented as mean ± S.E.M. *p < 0.05; **p <0.01 by one-way ANOVA followed by Tukey’s test. f, Bright field images show no difference in embryo size at E17.5 among control, Reln+/−, β1ΔCM/+ and β1ΔCM/+;Reln+/− embryos. Quantification of organ weight (heart, liver and kidney) relative to body length indicates that hearts are smaller in E17.5 β1ΔCM/+;Reln+/− embryos. N=9 (control), N=7 (Reln+/−), N=6 (β1ΔCM/+) and N=6 (β1ΔCM/+;Reln+/−) embryos from 3 separate litters. Data are presented as mean ± S.E.M. *p =0.015 by one-way ANOVA followed by Tukey’s test. n.s, not significant. g, Whole mount immunostaining using Lyve1 antibodies shows normal cardiac lymphatic development in control, β1ΔCM/+, β1ΔCM/+;Reln+/− and Reln+/− embryos. N = 3 embryos/genotype from 3 separate litters. Scale bars, 1 mm (f), 500 μm (g). For western blot source data, see Supplementary Fig 8 and 9. Exact p values included in Source Data.
Extended Data Figure 9. Reelin promotes CM proliferation and survival through Integrin β1 signaling.
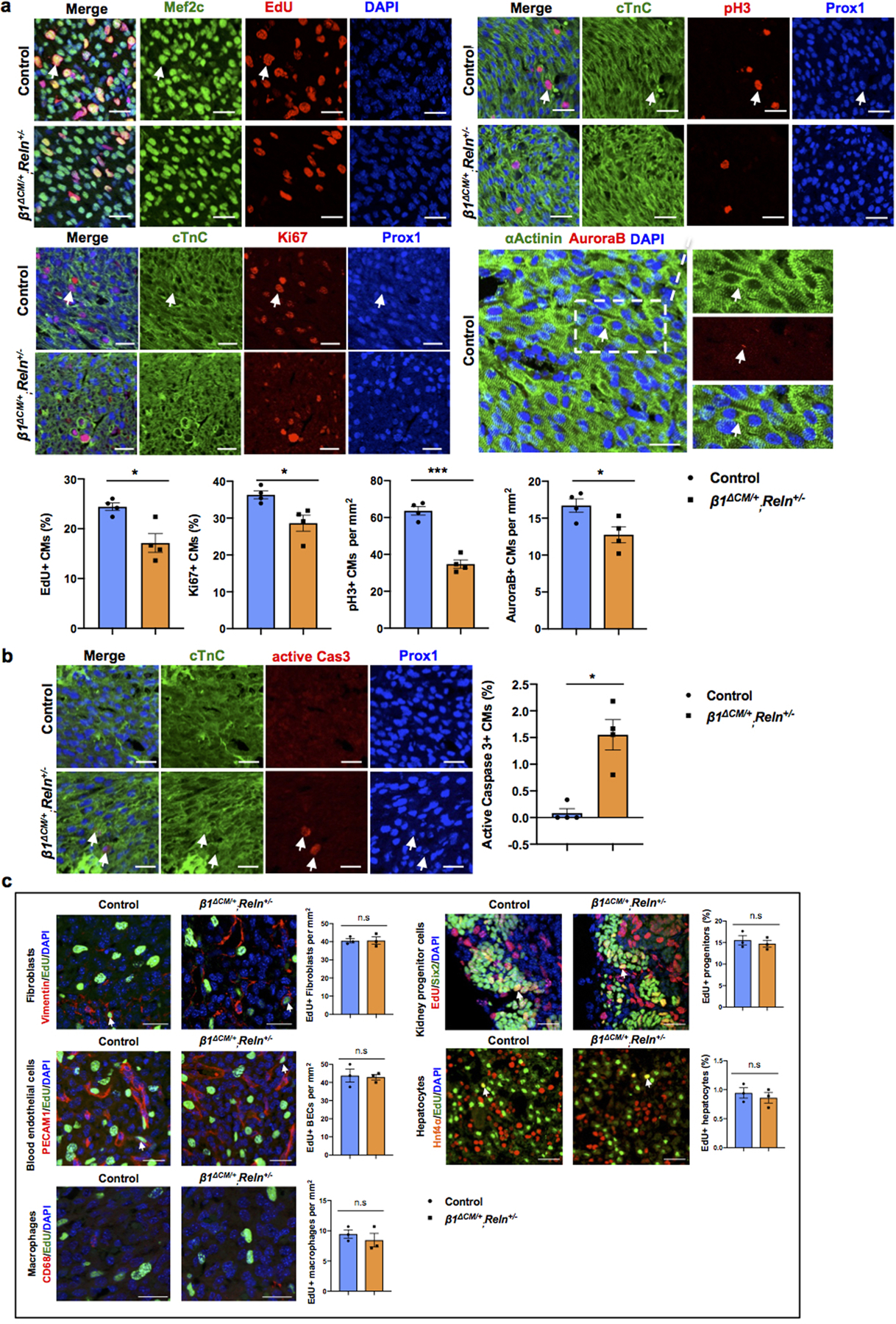
a, Co-immunostaining using cell proliferation markers (EdU, Ki67, pH3 and AuroraB) together with CM markers (cardiac Troponin C [cTnC], Prox1, αActinin and/or Mef2c) shows reduced CM proliferation in β1ΔCM/+;Reln+/− hearts at E17.5. Arrows indicate proliferating CMs. Quantification in the lower panel shows reduced proliferation in E17.5 β1ΔCM/+;Reln+/− hearts, as indicated by the percentage of EdU+ and Ki67+ CMs and the number of pH3+ and AuroraB+ CMs. N = 4 embryos/genotype from 3 separate litters. *p=0.022 (EdU), 0.029 (Ki67), ***p =0.0001 (pH3) and *p= 0.033 (AuroraB). b, Active Caspase-3 immunostaining shows increased CM apoptosis in β1ΔCM/+;Reln+/− hearts at E17.5, as quantified by the percentage of active caspase-3+ CMs relative to Prox1+ CMs. Arrows indicate apoptotic CMs. N = 4 embryos/genotype from 3 separate litters. Control embryos are Cre- embryos and Cre+;β1+/+ littermates. *p=0.01. c, Co-immunostaining with antibodies against Vimentin, PECAM1, CD68, Six2 and Hnf4α, together with EdU labeling shows comparable proliferation of cardiac fibroblasts, blood endothelial cells and macrophages, and of nephron progenitors and hepatocytes between controls and E17.5 β1ΔCM/+;Reln+/− embryos. White arrows indicate EdU+ proliferating cells. Quantification of the proliferation analysis for each of those cell types is shown on the right panels. N = 3 embryos/genotype from 3 separate litters. Control are Cre- embryos and Cre+;β1+/+ littermates. Data are presented as mean ± S.E.M. n.s. not significant difference by unpaired two-tailed Student’s t test. Scale bars, 25μm. Lower magnification images for panels a and b are included in Supplementary Fig 5.
Extended Data Figure 10. Reelin expression is developmentally downregulated, but is upregulated in newly formed cardiac lymphatics after myocardial infarction.
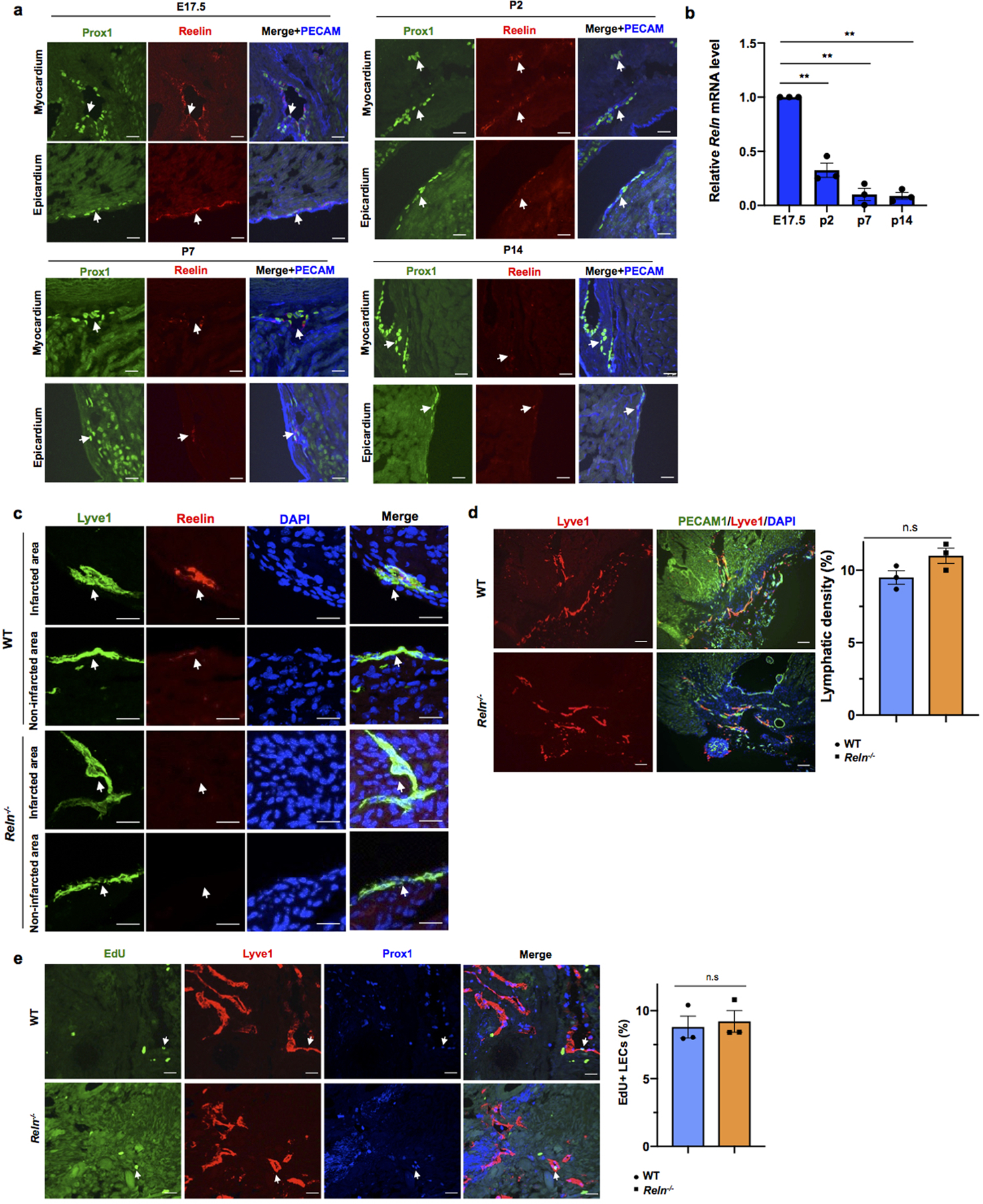
a. Immunostaining with Reelin, Prox1 and PECAM shows Reelin is highly expressed in cardiac lymphatics in the epicardium and myocardium nearby the base of the heart at E17.5. Reelin expression level is gradually downregulated during development from P2 to P14. N = 3 hearts/stage. Arrows indicate Prox1+ cardiac lymphatics. b, qPCR analysis using sorted cardiac lymphatics shows Reln levels are drastically downregulated in cardiac LECs during development. N=3. Reln relative level from each experiment is presented as fold changes relative to E17.5. Data are presented as mean ± S.E.M. **p=0.009 (P2 vs E17.5), 0.004 (P7 vs E17.5), 0.001 (P14 vs E17.5) by one-way ANOVA followed by Tukey’s test. c, Immunostaining shows Reelin expression is highly upregulated in the newly formed cardiac lymphatics in WT P7 pups (myocardial infarction was performed at P2). Notably, the pre-existing cardiac lymphatics in the non-infarcted area express low levels of Reelin. Reln−/− hearts are completely devoid of Reelin expression in both, newly formed cardiac lymphatics and pre-existing lymphatics. Arrows indicate cardiac lymphatics. N = 3 hearts/group. d, Immunostaining against the pan-endothelial marker PECAM1 and the lymphatic marker Lyve1 shows normal lymphangiogenesis in WT and Reln−/− hearts 21 days after MI (MI performed at P2). N = 3 hearts/group. Data are presented as mean ± S.E.M. n.s. not significant difference by unpaired two-tailed Student’s t test. e, EdU labeling shows no differences in LECs proliferation in WT and Reln−/− hearts 21 days after MI (MI performed at P2). N = 3 hearts/group. Data are presented as mean ± S.E.M. n.s. not significant difference by unpaired two-tailed Student’s t test. Arrow indicates EdU+ LECs. Scale bars, 100μm (d), 25μm (a,c,e).
Extended Data Figure 11. Reelin improves cardio-protection in neonates and adult mice after MI.
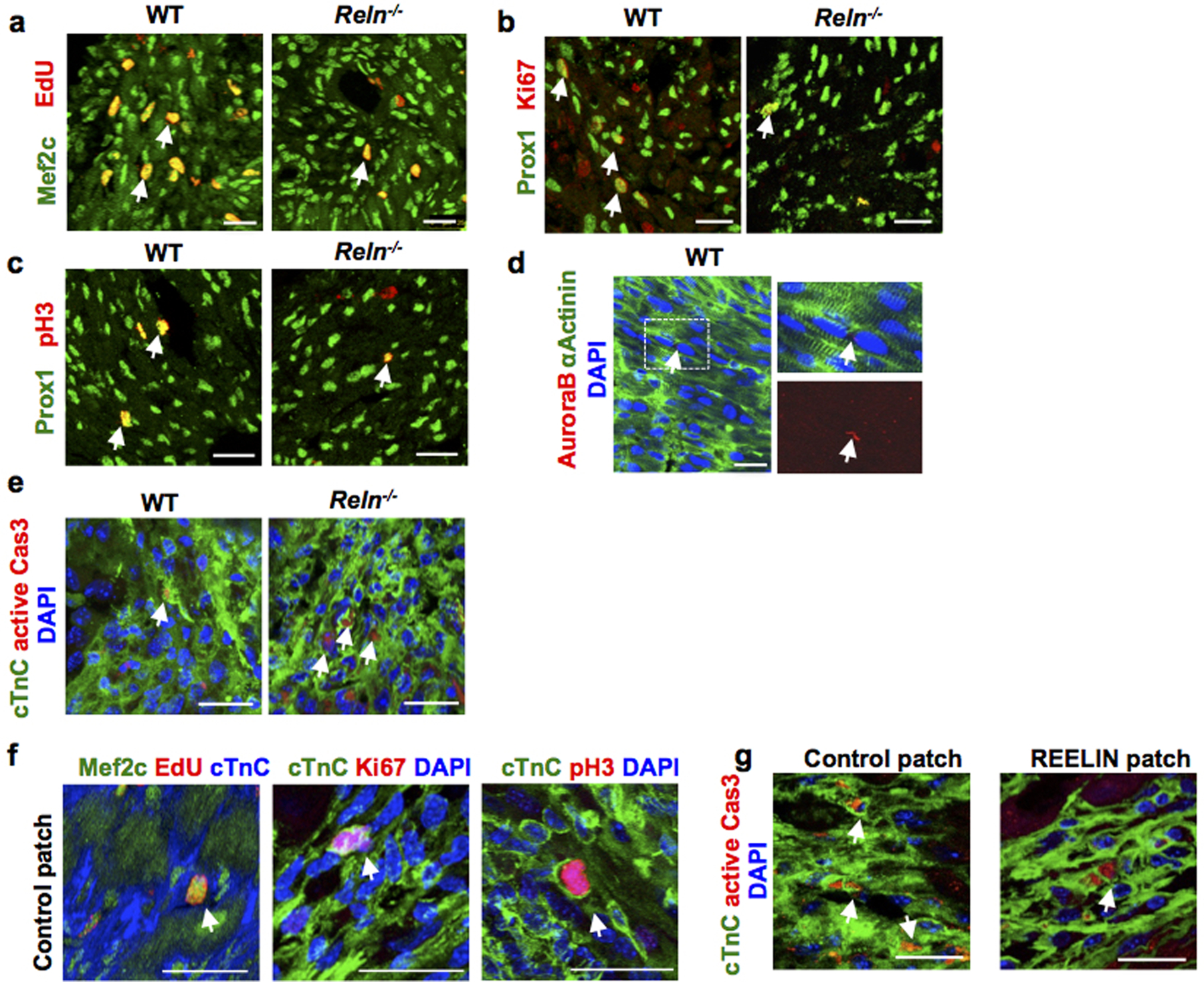
a-d, Co-immunostaining using cell proliferation markers (EdU, Ki67, pH3 and AuroraB) together with the CM markers Prox1, αActinin or Mef2c shows decreased CM proliferation in the border of the infarcted area of Reln−/− hearts at P7. Arrows indicate proliferating CMs. N= 4 mice/group. e, Immunostaining using active Caspase-3 shows increased CM apoptosis in the infarcted area of Reln−/− hearts at P7. Arrows indicate apoptotic CMs in the section. N=4 mice/group. f, Immunostaining against the cell proliferation markers EdU, Ki67 and pH3 together with the CM markers Mef2c or cTnC shows no differences in CM proliferation in the infarcted areas between control patch or REELIN patch treated hearts 7 days after MI. Arrows indicate proliferating CMs. N = 4 hearts/group. g, Immunostaining using active Caspase-3 shows reduced CM apoptosis in the infarcted area of REELIN patch treated hearts. Arrows indicate apoptotic CMs. N=4 mice/group. Arrows indicate apoptotic CMs. Scale bars, 25 μm. Lower magnification for panels a-c, e and g are included in Supplementary Fig 3.
Supplementary Material
Acknowledgments
This work was supported by NIH grant (RO1HL073402-16) to G.O, AHA grant (18CDA34110356) to X.L, 5T32HL134633 to W.M, FPU grant from the Spanish Ministry of Education, Culture and Sports and EMBO Short-Term Fellowship to E.C.C, Leducq TNE-17CVD and RD16/0011/0019 (ISCIII) from the Spanish Ministry of Science, Innovation, and Universities to M.T, NIH T32 GM008061 to C.L, HL63762, and NS093382 to J.H. We thank Dr. Gabriele M. Rune and Dr. Bianka Brunne for the Reln +/− strain. RNAseq work was supported by the Northwestern University NUSeq Core Facility. We thank the Robert H. Lurie Cancer Center Flow Cytometry facility supported by NCI CCSG P30 CA060553 for their invaluable assistance. Flow Cytometry Cell Sorting was performed on a BD FACSAria SORP system and BD FACSymphony S6 SORP system, purchased through the support of NIH 1S10OD011996-01 and 1S10OD026814-01. Imaging work was performed at the Northwestern University Center for Advanced Microscopy supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center. Spinning disk confocal microscopy was performed on an Andor XDI Revolution microscope, purchased through the support of NCRR 1S10 RR031680-01. Proteomics services were performed by the Northwestern Proteomics Core Facility supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center, instrumentation award (S10OD025194) from NIH Office of Director, and the National Resource for Translational and Developmental Proteomics supported by P41 GM108569. We thank the George M. O’Brien Kidney Research Core Center (NU GoKidney, supported by a P30 DK114857 award from NIDDK) for the use of the Echocardiography machine. The myocardial infarction surgeries were performed by the comprehensive Transplant Center Microsurgery Core, partially supported by NIH NIAID P01AI112522. We thank Dr. Jing Jin and Dr. Pan Liu for help with the ELISA reagents and data analysis, Dr. Ruihua Ma for help with DNA polyploidy analysis, Ao Shi for the Mef2c antibodies, Dr. Michael Dellinger for the Prox1CreERT2 mice and Dr. Hossein Ardehali for the MhcCre mice. We specially thank Dr. Beatriz Sosa-Pineda for advice and valuable suggestions and Dr. Pilar Ruiz-Lozano for shearing her expertise in the preparation of the collagen patches.
Footnotes
Competing Interests
JH is a shareholder of Reelin Therapeutics and a coinventor on a pending US patent application filed by his institution (UT Southwestern; application number 15/763,047 and publication number 20180273637, title “Methods and Compositions for Treatment of Atherosclerosis”; Inventors: Joachim Herz, Yinyuan Ding, Xunde Xian, Linzhang Huang, Chieko Mineo, Philip Shaul, Laurent Calvier). This patent application covers no aspects of the current manuscript. Findings regarding the potential applications and methods for using Reelin to treat cardiac diseases are the subject of provisional patent application US 63/091,558 owned by Northwestern University and list X.L. and G.O. as inventors. The rest of the authors declares no competing interests.
Data availability
All data from the manuscript are available from the corresponding author on request. RNAseq raw data have been deposited to the Gene Expression Omnibus (GEO) repository with accession number GSE158504. Source data are provided with this paper.
Supplementary information
This file contains Supplementary Figures 1–5 (lower magnification images for CM proliferation and apoptosis), Supplementary Figures 6–9 (source data for western blots), Supplementary Figure 10 (the gating strategy for flow cytometry).
References
- 1.Oliver G, Kipnis J, Randolph GJ & Harvey NL The Lymphatic Vasculature in the 21st Century: Novel Functional Roles in Homeostasis and Disease. Cell 182, 270–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klotz L et al. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature 522, 62–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vuorio T, Tirronen A & Ylä-Herttuala S Cardiac Lymphatics – A New Avenue for Therapeutics? Trends in Endocrinology and Metabolism 28, 285–296 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Henri O et al. Selective Stimulation of Cardiac Lymphangiogenesis Reduces Myocardial Edema and Fibrosis Leading to Improved Cardiac Function Following Myocardial Infarction. Circulation 133, 1484–97; discussion 1497 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Vieira JM et al. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J. Clin. Invest 128, 3402–3412 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrison MR et al. Late developing cardiac lymphatic vasculature supports adult zebrafish heart function and regeneration. Elife 8, pii: e42762 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harvey NL et al. Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nat. Genet 37, 1072–1081 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Wigle JT & Oliver G Prox1 function is required for the development of the murine lymphatic system. Cell 98, 769–778 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Johnson NC et al. Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev 22, 3282–3291 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sörensen I, Adams RH & Gossler A DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 113, 5680–8 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Zhang L et al. VEGFR-3 ligand-binding and kinase activity are required for lymphangiogenesis but not for angiogenesis. Cell Res. 20, 1319–1331 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Ross ON, Moore CM, Suggett DJ, Macintyre HL & Geider RJ Endothelial - Cardiomyocyte Interactions in Cardiac Development and Repair. Annu. Rev Physiol 68:1–736, 1835–1852 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brutsaert DL & Cotran RS Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev 83, 59–115 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Jossin Y Neuronal Migration and the Role of Reelin During Early Development of the Cerebral Cortex. Mol. Neurobiol 30, 225–252 (2004). [DOI] [PubMed] [Google Scholar]
- 15.D’Arcangelo G Reelin in the Years: Controlling Neuronal Migration and Maturation in the Mammalian Brain. Adv. Neurosci 2014, 1–19 (2014). [Google Scholar]
- 16.Lutter S, Xie S, Tatin F & Makinen T Smooth muscle–endothelial cell communication activates Reelin signaling and regulates lymphatic vessel formation. J. Cell Biol 197, 837–849 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Arcangelo G et al. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723 (1995). [DOI] [PubMed] [Google Scholar]
- 18.Lane-Donovan C et al. Reelin protects against amyloid β toxicity in vivo. Sci. Signal 8, ra67–ra67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srinivasan RS et al. Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes and Development 21, 2422–2432 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trommsdorff M et al. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97, 689–701 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Hiesberger T et al. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24, 481–9 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Lin L et al. Reelin promotes the adhesion and drug resistance of multiple myeloma cells via integrin β1 signaling signaling and STAT3. Oncotarget 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dulabon L et al. Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron 27, 33–44 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Beffert U et al. Reelin-mediated Signaling Locally Regulates Protein Kinase B/Akt and Glycogen Synthase Kinase 3β. J. Biol. Chem 277, 49958–49964 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Jossin Y & Goffinet AM Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol. Cell. Biol 27, 7113–24 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ieda M et al. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev. Cell 16, 233–44 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porrello ER et al. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 331, 1078–1080 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei K et al. Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature 525, 479–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Serpooshan V et al. The effect of bioengineered acellular collagen patch on cardiac remodeling and ventricular function post myocardial infarction. Biomaterials 34, 9048–9055 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karkkainen MJ et al. A model for gene therapy of human hereditary lymphedema. Proc. Natl. Acad. Sci 98, 12677–82. (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burridge PW et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 11, 855–860 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burridge PW, Holmström A & Wu JC Chemically Defined Culture and Cardiomyocyte Differentiation of Human Pluripotent Stem Cells in Current Protocols in Human Genetics 87, 21.3.1–21.3.15 (John Wiley & Sons, Inc., 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X et al. Rasip1 controls lymphatic vessel lumen maintenance by regulating endothelial cell junctions. Development 145, dev165092 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.