

Abstract
Bile acids (BAs) are diverse molecules that are synthesized from cholesterol in the liver. The synthesis of BAs has traditionally been shown to occur via two pathways. Cholesterol 7α-hydroxylase (CYP7A1) performs the initial and rate-limiting step in the classical pathway and sterol 27-hydroxylase (CYP27A1) initiates the hydroxylation of cholesterol in the alternative pathway. While the role of individual BA species as physiological detergents is relatively ubiquitous, their endocrine functions as signaling molecules and roles in disease pathogenesis have been emerging to be BA species specific. In order to better understand the pharmacologic and toxicologic roles of individual BA species in an in vivo model, we created Cyp7a1 and Cyp27a1 double knockout mice (DKO) by cross-breeding single KO mice (Cyp7a1−/− and Cyp27a1−/−). BA profiling and quantification by LC-MS of serum, gallbladder, liver, small intestine and colon of wild type, Cyp7a1−/−, Cyp27a1−/−, and DKO mice showed that DKO mice exhibited a reduction of BAs in the plasma (45.9%), liver (60.2%), gallbladder (76.3%), small intestine (88.7%) and colon (93.6%), while maintaining a similar BA pool composition as compared to WT mice. The function of the farnesoid X receptor (FXR) in DKO mice was lower, revealed by decreased mRNA expression of well-known FXR target genes, hepatic small heterodimer partner (Shp) and ileal fibroblast growth factor 15 (Fgf15). However, response to FXR synthetic ligands was maintained in DKO mice as treatment with GW4064 resulted in similar changes in gene expression in all strains of mice.
Conclusion:
We provide a useful tool for studying the role of individual BAs in vivo. DKO mice have a significantly reduced BA pool, similar BA profile, and maintained response to FXR activation.
Keywords: farnesoid X receptor, cholic acid, chenodeoxycholic acid, small heterodimer partner, fibroblast growth factor 15
INTRODUCTION
Bile acids (BAs) are physiological detergents synthesized through the enzymatic oxidation of cholesterol in the liver. Most BAs are conjugated to either glycine or taurine in the liver to form negatively charged bile salts, which increases their solubility (1). Conjugated BAs are effluxed by the bile salt export pump (BSEP) into bile canaliculi for transport and storage in the gallbladder. The postprandial release of cholecystokinin stimulates contraction of the gallbladder and the release of BAs into the duodenum (2). Inside the small intestine, BAs facilitate the emulsification, digestion, and absorption of dietary fats, cholesterol, and lipid soluble vitamins through the formation of mixed micelles (3). BAs are efficiently reabsorbed in the distal small intestine (ileum) through the apical sodium-dependent bile acid transporter (ASBT) and organic solute transporter alpha and beta (OSTa/b) where they are returned to the liver through portal circulation. Upon returning to the liver, BAs are taken up by hepatocytes through sodium taurocholate transporter (NTCP) and organic anion transporting polypeptides (OATPs)(4–8).
BA synthesis in the liver is a complex process involving at least 17 different enzymes and is predominantly accomplished through two distinct pathways (9). The classical (or neutral) pathway is initiated by the rate-limiting enzyme cholesterol 7α-hydroxylase (CYP7A1) and results in the formation of the primary BAs, cholic acid (CA) and chenodeoxycholic acid (CDCA). The differential formation of CA and CDCA in the classical pathway is determined by cholesterol 12α-hydroxylase (CYP8B1) with CDCA being formed in the absence of CYP8B1 activity (10). The alternative (or acidic) pathway is initiated with the oxidation of the cholesterol side chain by the mitochondrial cytochrome p450 sterol 27-hydroxylase (CYP27A1) followed by 25-hydroxycholesterol 7-alpha-hydroxylase (CYP7B1). Mice utilize an additional enzyme, cytochrome p450 2c70 (CYP2C70), to rapidly convert CDCA to β-muricholic acid (βMCA) (11). Primary BAs are conjugated in the liver before undergoing biliary excretion. Luminal bacteria in the intestine de-conjugate and then metabolize a portion of primary bile acids to form more hydrophobic and cytotoxic secondary BAs, including lithocholic acid (LCA) and deoxycholic acid (DCA) (12, 13).
Previous research using mice deficient in Cyp7a1 (Cyp7a1−/− mice) has shown over 75% reductions in the BA pool size of three-month old, male mice. While the BA synthesis in these mice was markedly reduced, there were no changes in cholesterol levels, likely due to significantly reduced absorption of dietary cholesterol (14). The concentration of primary BAs displayed a trend to shift from more CA in male wild type (WT) mice to more MCA in Cyp7a1−/− mice (15). Similarly, in mice deficient in Cyp27a1 (Cyp27a1−/− mice), there was a significant reduction in BAs with minimal changes in cholesterol levels. The expression of the Cyp7a1 gene is regulated by BAs in a negative feedback manner, therefore the reduction in BAs in Cyp27a1−/− mice results in an induction of Cyp7a1 gene expression. The side chain hydroxylation required for side chain cleavage in these mice was likely carried out by microsomal cholesterol 25-hydroxylase (CH25H) and/or CYP3A11 (16).
In addition to their role as physiological detergents, BAs act as signaling molecules by activating the farnesoid X receptor (FXR), vitamin D receptor, pregnane X receptor, and G protein-coupled BA receptors (TGR5 and S1PR2), in multiple organs (17). BAs have been found to regulate a multitude of biological processes including lipid and glucose homeostasis, energy expenditure, inflammation, bacterial proliferation and gastrointestinal motility (18–22). Increased understanding of the pleiotropic role BAs play in human health has led to the implication of BA dysregulation in a number of disease states, including cancer as well as an array of metabolic and liver diseases (23–25). As such, BAs, their receptors, and downstream pathways have provided novel targets for drug intervention (26).
To date, over 30 unique BAs have been identified in both humans and rodents (27, 28). Due to the diversity of BA species and potential toxicity associated with BA feeding, studying the role of individual BAs in vivo can prove difficult. Through the development of a mouse model deficient in Cyp7a1 and Cyp27a1, we hope to attain a better model for studying the effects of individual BAs in vivo. In the current study, we aimed to characterize the BA profile and regulation of gene expressions involved in BA homeostasis in Cyp7a1/Cyp27a1 double knockout mice (DKO).
MATERIALS AND METHODS
Animals and treatments
Cholesterol 7α-hydroxylase null mice (Cyp7a1−/−) and sterol 27-hydroxylase null mice (Cyp27a1−/−) were purchased from The Jackson Laboratory (Bar Harbor, ME). Cyp7a1−/− and Cyp27a1−/− mice were crossbred resulting in progeny that were heterozygous for Cyp7a1 and Cyp27a1 (Cyp7a1−/+ and Cyp27a1−/+). The double heterozygous mice were crossed to produce Cyp7a1−/−/Cyp27a1−/− double knockout mice (DKO) that are deficient in both enzymes. Cyp7a1−/− mice were maintained on a mixed strain background (C57BL/6J:129Sv). Cyp27a1−/− mice were developed on a 129Sv background and have been backcrossed to C57BL/6J inbred mice for over 13 generations. Wild-type (WT) mice derived from heterozygous Cyp7a1−/+ and Cyp27a1−/+ breeding pairs were used as controls. All mice in this study were genotyped according to The Jackson Laboratory protocols. A representative agarose gel used to genotype mice and a table including all primers used can be found in Suppl. Fig. 1. For BA profiling, 3 to 6-month-old, non-fasted WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice were euthanized. Blood, liver, gallbladder, small intestine and colon were collected and frozen in liquid nitrogen. To achieve in vivo activation of FXR, mice were treated via oral gavage with a synthetic FXR agonist, GW4064. In detail, 3 to 5-month old, male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice were treated with either GW4064 suspended in 1% Tween-20 and 1% methylcellulose at 150mg/kg or vehicle at 6:00pm and 8:00am. The mice were fasted overnight and euthanized 2 hrs after the second treatment. Blood, liver, gallbladder, and intestine samples were collected and frozen in liquid nitrogen. All mice were group-housed and maintained under standard 12-hr light/dark cycles. Food and water was provided ad libitum unless otherwise noted. Additional animal information can be found in suppl. Table 1. The experiments performed in this study were approved by the Rutgers Institutional Animal Care and Use Committee.
Serum biochemistry
Activities of serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP), and levels of serum triglycerides and total cholesterol were measured with the use of commercially available kits (Pointe Scientific, Canton MI).
Gene expression
Total RNA was extracted from frozen liver and ileum using the TRIzol reagent (Thermo Fisher Scientific; Waltham, MA) and reverse transcription was performed to attain cDNA. Relative gene expression was determined by real-time polymerase chain reaction (RT q-PCR) by SYBR green chemistry using the ViiA7 Real Time PCR machine (Life Technologies, Grand Island, NY) in a 384-well plate. All Ct values were converted to delta delta Ct values and were normalized to β-actin mRNA levels. Primer sequences can be found in suppl. Table 2.
BA extraction and profiling
Total BAs were extracted and purified from serum, liver, gallbladder, small intestine and colon samples. Liver BA extraction was performed using previously described methods (29). BA extraction from plasma was performed using 90μL of plasma. 900μL of acetonitrile was added to the samples for protein precipitation. The samples were incubated for 1 hr at room temperature on a shaker table and then spun at 12,000g for 10 mins; the supernatant was collected and dried in a speed-vac. Dried samples were reconstituted in 400μL of 50% methanol, filtered through a 0.22μm Costar Spin-X centrifuge tube, and used for analysis. Gallbladder samples were suspended in 1.5mL of 1x PBS. An aliquot of the gallbladder suspension was diluted 50x in PBS. BA extraction from the diluted gallbladder samples was then performed in the same manner as plasma samples. Intact frozen small intestine samples (including luminal content) were homogenized in 18mL of HPLC grade H2O and centrifuged. Intact frozen large intestine samples (including luminal content) were homogenized in 3mL of HPLC grade H2O and centrifuged. For both small and large intestines, a 300μL aliquot of the pooled supernatant was used for BA extraction following previously described methods (Zhang and Klaassen 2010). All BA extracts were analyzed using a Thermo Accela Ultra Performance Liquid Chromatography System (Thermo Fisher Scientific, Waltham, MA) coupled to a Thermo Finnigan LTQ XL Ion Trap Mass Spectrometer (Thermo Fisher Scientific, Waltham, MA). Chromatography was performed on a reverse phase 1.3μ 2.1 × 50 mm C18 Kinetex column (Phenomenex, Torrance, CA).
Immunohistochemistry
Intrahepatic ductal mass was measured using previously described methodologies (30). Frozen liver sections (n=3–6) from WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice were stained using an anti-cytokeratin 19 (CK-19) antibody (ab52625, Abcam, Cambridge, MA). Stained sections were scanned with Olympus VS120 slide scanner. At least 20 random fields were taken from each liver section. The stained area was quantified using ImageJ software. Data are shown as stained area relative to WT.
Statistical analysis
Data are represented as mean ± standard deviation (SD) (n=3–8/group). Comparison of groups was performed using one-way ANOVA followed by Tukey post-hoc test unless otherwise noted. Comparison of GW4064 treatment groups was performed using two-way ANOVA followed by Tukey post-hoc test. The results of statistical analysis were considered significant with P-values < 0.05.
RESULTS
Serum lipids, liver injury markers, and histology
Liver histology was examined by a pathologist and showed no evidence of liver injury (Suppl. Fig 2A.). Serum assays showed no significant alterations to AST, ALP, triglycerides, or total cholesterol. DKO mice displayed an elevation in ALT but still within the normal range (Suppl. Fig 2B.). Biliary mass was assessed with cytokeratin 19 (CK19) immunohistochemistry staining. The DKO mice trended toward a reduction in CK19 staining with no significant alterations between groups (Suppl. Fig 2C & 2D)
The effects of Cyp7a1 and Cyp27a1 deficiencies on BA concentration and composition
In order to assess the effect of Cyp7a1 and/or Cyp27a1 deficiencies on BA concentrations in the serum, liver, gallbladder, small intestine and large intestine, 3–6 month old, male mice maintained on chow diet for at least 2 months were used for measuring tissue concentrations of 23 BAs using LC-MS. First, we determined plasma BA concentrations and profile. Shown in Fig. 1, WT mice had an average concentration of 1280 ± 542.8 ng/ml with unconjugated BAs representing 83.9% of plasma BAs in WT mice and specifically, ωMCA (42%), βMCA (14%), and CA (14%) were most abundant. Cyp7a1−/− mice displayed an increase in total plasma BAs as compared to WT mice with βMCA (19%), TβMCA (17%), CA (17%), TCA (16%), and ωMCA (16%) being most abundant. Cyp27a1−/− mice had significantly less plasma BAs than WT mice with TCA (35%), CA (18%), ωMCA (16%) and HDCA (10%) being most abundant. DKO mice had an average BA concentration of 692.3 ± 348.8 ng/ml, representing a 45.9% reduction when compared to WT mice. 66.7% of BAs in DKO mice were conjugated to taurine, with TβMCA (30%), ωMCA (23%), TωMCA (18%) and TCA (15%) being abundant.
Fig. 1. Plasma BA pool size and composition of male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice.
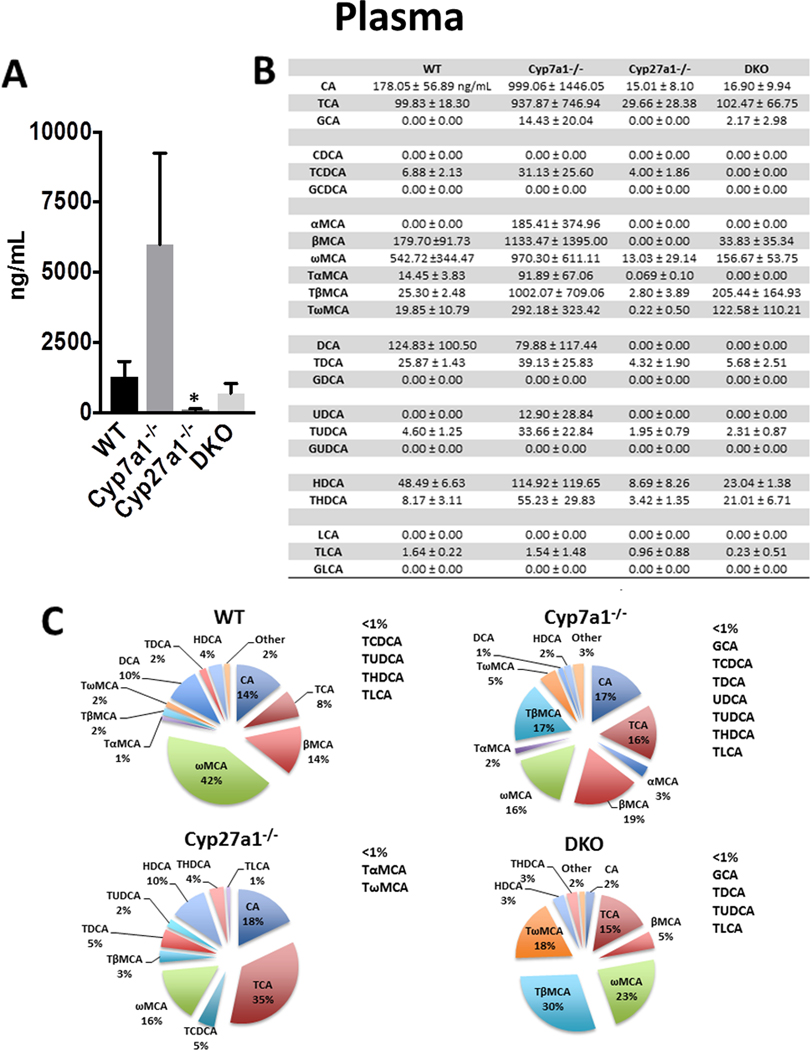
(A) Plasma BA pool size was measured with 90μL aliquots of plasma using UPLC-ITMS. Values are displayed in ng/mL plasma ± 1 SD. These data failed Levene’s test therefore Kruskal-Wallis was used for analysis. An asterisk denotes a significant difference from WT (P < 0.05). (B) Plasma concentration of individual BA species ± 1 SD. (C) Percent composition of BA species in plasma. BAs that represent <1% of total BAs in plasma are represented as “other” and denoted alongside the pie charts.
In the liver (Fig 2), the average BA concentration in WT mice was 139.9 ± 88.8 μg/g liver and the predominant BAs were TCA (67%) and TMCA (24%). The Cyp7a1−/− mice had no significant change in total BAs but had higher concentrations of TMCA (66%) than TCA (28%), suggesting BAs were largely produced via the alternative pathway. The Cyp27a1−/− and DKO mice had significant reductions of BAs in the liver, 75.0% and 60.2%, respectively. The hepatic BA content of Cyp27a1−/− mice was composed mainly of TCA (69%) and TMCA (23%). The DKO mice consisted principally of TMCA (53%) and TCA (37%).
Fig. 2. Liver BA pool size and composition of male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice.

(A) Liver BA pool size was measured using UPLC-ITMS. Values are displayed in ng/mg liver ± 1 SD. These data failed Levene’s test therefore Kruskal-Wallis was used for analysis. An asterisk denotes a significant difference from WT (P < 0.05). (B) Liver concentration of individual BA species ± 1 SD. (C) Percent composition of BA species in plasma. BAs that represent <1% of total BAs in the liver are represented as “other” and denoted alongside the pie charts.
In the gallbladder (Fig 3), WT mice had an average BA concentration of 5,443 ± 1640 μg/100g body weight. Conjugated BAs constituted 97% of gallbladder BAs in WT mice with TCA (55%), TβMCA (24%), TαMCA (7%), and TDCA (4%) being most abundant (Fig. 3B and 3C). All knockout groups had significantly reduced BA levels when compared to WT mice with no significant difference between knockout groups. The reduction of total BAs in Cyp7a1−/−, Cyp27a1−/−, and DKO mice was 81.4%, 83.5% and 76.3% respectively. All groups had significantly reduced CA, TCA, MCA, TMCA and TDCA compared to WT mice (Fig. 3B and 3C).
Fig. 3. Gallbladder BA pool size and composition of male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice.

(A) Gallbladder BA pool size was measured using UPLC-ITMS. Values are displayed in μg/100g body weight (x1000) ± 1 SD. An asterisk denotes a significant difference from WT (P < 0.05). (B) Gallbladder concentration of individual BA species (μg/100g body weight) ± 1 SD. (C) Percent composition of BA species in the gallbladder. BAs that represent <1% of total BAs in the gallbladder are represented as “other” and denoted alongside the pie charts.
In the small intestine (Fig 4), analysis of homogenized whole small bowel of WT mice (including luminal content) showed an average BA concentration of 58,763 ± 11,686 μg/100g body weight (Fig. 4A). Over 73% of BAs in WT mice were primary BAs and the predominant BAs were CA (29%), TCA (15%), ωMCA (14%), and βMCA (13%) (Fig. 4B and 4C). All KO mice had significantly reduced BA levels as compared to WT with no significant difference in total BAs among KO groups. Cyp7a1−/− mice had the greatest reduction at 93.5% and consisted largely of ωMCA (55%), βMCA (19%), and CA (16%). Cyp27a1−/− mice showed a decrease of 78.7% in total BAs with CA (41%), βMCA (15%), TCA (14%), and ωMCA (14%) being most abundant. The DKO group had a reduction of 88.7% in the small bowel with the predominant BAs being TCA (31%), CA (18%), TβMCA (13%) and βMCA (13%)..
Fig. 4. Small intestine BA pool size and composition of male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice.

(A) Small intestine BA pool size was measured using UPLC-ITMS and includes luminal content. Values are displayed in μg/100g body weight (x1000) ± 1 SD. An asterisk denotes a significant difference from WT (P < 0.05). (B) Small intestine concentration of individual BA species (μg/100g body weight) ± 1 SD. (C) Percent composition of BA species in the small intestine. BAs that represent <1% of total BAs in the small intestine are represented as “other” and denoted alongside the pie charts.
In the large intestine (Fig. 5), the BA analysis of homogenized whole colon (including luminal content) was determined. WT mice had an average large intestine BA concentration of 1,615 ± 1,008 μg/100g body weight. The BAs in the large intestine of WT, Cyp7a1−/−. Cyp27a1−/−, and DKO mice were largely secondary (70.9%, 77.1%, 62.1%, and 80.1% respectively) and unconjugated BAs (84.75%, 85.21%, 84.9%, and 93.0% respectively). Cyp7a1−/− and Cyp27a1−/− mice trended towards a decrease in BA concentration compared to WT mice with reductions of 57.1% and 59.3%, respectively. DKO mice showed a significant reduction of 93.6% total BAs, compared to WT.
Fig. 5. Large intestine BA pool size and composition of male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice.

(A) Large intestine BA pool size was measured using UPLC-ITMS and includes luminal content. Values are displayed in μg/100g body weight (x1000) ± 1 SD. An asterisk denotes a significant difference from WT (P < 0.05). (B) Large intestine concentration of individual BA species (μg/100g body weight) ± 1 SD. (C) Percent composition of BA species in the large intestine. BAs that represent <1% of total BAs in the large intestine are represented as “other” and denoted alongside the pie charts.
Gene expression of BA synthetic enzymes and transporters
The hepatic expression at the mRNA level of 17 genes (Cyp7a1, Cyp27a1, Cyp46a1, Ch25h, Cyp7b1, Cyp8b1, Hsd17b4, Hsd3b7, Baat, Amacr, Scp2, Slc27a5, FXRα, Shp, Fgfr4, Ntcp, and Bsep) involved in BA synthesis, conjugation, transport, and regulation are shown in Fig 6. In Fig. 6A, the relative mRNA levels of Cyp7a1 and Cyp27a1 were below detection limit for their corresponding KO groups. Interestingly, the mRNA levels of Cyp7a1 were increased 11.3 fold in Cyp27a1−/− mice while Cyp27a1 showed no induction in Cyp7a1−/− mice. The expression of Cyp8b1, which performs 12-alpha hydroxylation in the production of CA, was increased over 2 fold in all groups but failed to reach significance in the DKO mice. Cyp46a1, which initiates the 24-hydroxylase pathway of BA synthesis and Ch25h, which initiates the 25-hydroxylase pathway of BA synthesis, showed no significant changes in mRNA expression among groups. Fig. 6B shows the mRNA expression for intermediate genes in BA synthesis and conjugation. Cyp7a1−/− mice had a significant reduction in the mRNA levels of alpha-methyacyl-CoA racemase (Amacr), which plays a role in the peroxisomal cleavage of C27 precursors into mature C24 BAs. There were no other significant alterations to the mRNA expression of intermediate BA synthesizing or BA conjugating genes examined. All groups displayed similar levels of genes involved in BA transport (Ntcp, Bsep, Ibabp, OSTb) in the liver and ileum (Fig. 6C & D). The hepatic gene expression of Shp was significantly reduced in all knockout groups (Fig. 6C), which is in line with reduced BA levels and reduced ileal Fgf15 expression (Fig. 6D).
Fig. 6. Relative mRNA levels of BA related genes for male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice.

Genes were measured using RT q-PCR, normalized to β-actin mRNA levels, and graphed as relative mRNA ± 1 SD. An asterisk denotes a significant difference from WT (P < 0.05). (A) Relative mRNA of hepatic genes involved in classic, alternative, and minor BA synthetic pathways. (B) Relative mRNA of hepatic genes for BA conjugation. (C) Relative mRNA of hepatic FXR, Shp, Fgfr4, Ntcp, and Bsep. (D) Relative mRNA of ileal genes involved in BA regulation and transport.
The mRNA expression of 7 additional hepatic BA transporters has been quantified and are shown in suppl. Fig. 3. Genes of Osta, Ostb, Mrp3 and Mrp4 could encode transporter to efflux BAs to the sinusoidal side of hepatocytes, and Mrp2 could efflux anion conjugated substrates to canalicular side of hepatocytes. Genes of Oatp1a1 and Oatp1a4 encode transporters that have been shown to uptake unconjugated BAs into hepatocytes. Compared to WT mice, Cyp7a1−/− mice tended to have lower expression of Ostb and Oatp1a4, but higher Mrp4, Cyp27a1−/− mice tended to have lower expression of Ostb and Oatp1a1, but higher expression of Mrp2, Mrp3, Mrp4, and Oatp1a4. The DKO mice tend to have lowed expression of Osta and Ostb.
Hepatic and ileal BA related gene expression following GW4064 treatment
FXR is a BA-activated nuclear receptor and essential for regulating BA synthesis and transport. The extent of FXR agonism or antagonism by BAs is dependent upon the BA concentration and composition (31, 32). In order to assess the effect of FXR activation on gene expression in mice with deficiencies in major BA synthetic enzymes, we treated 3–5 month old male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice with GW4064, a synthetic FXR agonist.
Shown in Fig. 7, the activation of FXR by GW4064 significantly reduced hepatic Cyp7a1 mRNA levels in WT and Cyp27a1−/− mice to 18.3% and 12.6% of vehicle-treated WT mice, respectively. Cyp27a1−/− mice had a significant decrease of 69.7% in mRNA levels of Cyp8b1 in response to GW4064 treatment. There were no other significant changes in response to GW4064 treatment for the genes examined relating to BA synthesis. Cyp27a1−/− mice were the most susceptible group to alteration by FXR activation likely due to their reliance on the classical pathway for BA metabolism.
Fig. 7. Relative mRNA levels of hepatic enzymes involved in BA synthesis for male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice with FXR activation.

Mice were treated with vehicle or a synthetic FXR agonist, GW4064. The expression of genes at mRNA levels was measured using RT q-PCR and normalized to β-actin mRNA levels, and graphed as relative mRNA ± 1 SD. An asterisk denotes a significant difference from WT vehicle treated mice, an octothorpe denotes a significant difference between vehicle and GW4064 treatments (P < 0.05).
Ileal mRNA levels of Fxr were significantly reduced in WT and Cyp27a1−/− mice following treatment with GW4064, as shown in Fig. 8. DKO mice showed a similar trend to WT mice in response to GW4064 treatment with 53.9% reduction of Fxr mRNA. All groups responded to Fxr activation by inducing the transcription of Shp and Fgf15 mRNA over 16 fold and Ibabp mRNA roughly 4 fold as compared to vehicle-treated WT mice.
Fig. 8. Relative mRNA levels of ileal enzymes involved in BA transport and regulation for male WT, Cyp7a1−/−. Cyp27a1−/−, and DKO mice with FXR activation.

Mice were treated with vehicle or a synthetic FXR agonist, GW4064. The expression of genes at mRNA levels was measured using RT q-PCR and normalized to β-actin mRNA levels, and graphed as relative mRNA ± 1 SD. An asterisk denotes a significant difference from WT vehicle treated mice, an octothorpe denotes a significant difference between vehicle and GW4064 treatments (P < 0.05).
DISCUSSION
In the present study, we characterized the BA profile and hepatic/ileal gene expression of mice with gene deletion of Cyp7a1, Cyp27a1, and both. These two genes encode the initial enzymes in the conversion of cholesterol to BAs in the classical and alternative pathway, respectively. DKO mice presented with significantly lower BAs with 45.9%, 60.2%, 76.3%, 88.7%, and 93.6% reduction in plasma, liver, gallbladder, small intestine, and large intestine, respectively, as compared to WT mice. DKO mice displayed few alterations in the mRNA expression of genes involved in BA synthesis, conjugation, or transport aside from the intentional knockout of Cyp7a1 and Cyp27a1. Furthermore, DKO mice maintained a BA composition similar to WT mice amongst the 23 BA species profiled in the study.
Our findings on Cyp7a1 and Cyp27a1 single KO mice are in line with previous studies that have shown BA reductions of 66.7% and 73% in Cyp7a1−/− and Cyp27a1−/− mice, respectively, with Cyp7a1−/− mice shifting their BA composition from CA to MCA (15, 33). While these DKO mice showed significantly reduced BAs as compared to WT mice, their BA levels were somewhat altered when compared to Cyp7a1−/− or Cyp27a1−/− mice. For example, the plasma BA concentration of DKO mice were much lower than Cyp7a1−/− but slightly higher than Cyp27a1−/− mice. The liver BA concentrations of DKO mice were comparable to Cyp27a1−/− mice and significantly lower than the other two strains. The BA concentrations in the gallbladder and small intestine were significantly reduced in the single and double KO mice, with little difference between KO groups. DKO mice displayed the largest reduction of BAs in the colon. These large intestine BA data suggest that DKO mice may reabsorb primary conjugated BAs more efficiently than other groups despite showing no significant changes in the expression of Asbt mRNA.
Expression of the genes in minor BA synthetic pathways catalyzed by CYP46A1 and CH25H showed no significant changes at hepatic mRNA levels. This is in agreement with a previous work that has shown no changes in BA metabolism in the Cyp46a1−/− mice, as measured through the fecal excretion of acidic sterols (34). Taken together these findings suggest there is a possibly novel minor pathway of BA synthesis or route of oxysterol intermediate production for the metabolism of cholesterol to BAs.
In order to assess the potential of differential FXR activation in these genetically modified mice with lower BAs, we treated mice with a synthetic FXR agonist, GW4064. Following FXR activation, mice expressing Cyp7a1 had significantly reduced Cyp7a1 gene expression. The expression of Cyp8b1 displayed a trend for reduction in the livers of all groups, with only Cyp27a1−/− mice reaching significance. The hepatic mRNA levels of genes involved in the alternative pathway of BA synthesis (Cyp27a1, Cyp7b1) showed no significant changes following GW4064 treatment. In the ileum, all groups responded similarly to FXR activation with strong inductions of Shp, Fgf15, and Ibabp. These data suggest that the loss of Cyp7a1 and Cyp27a1 does not affect the transcriptional response following FXR activation. Increasing interest in the modification of FXR and BA signaling pathways for the treatment of hepatic and metabolic diseases provide a need for tools to dissect the mechanistic consequences of these alterations in vivo. In addition to currently approved BA therapies, basic research continues to elucidate potential applications of individual BA species for the intervention of disease pathologies. While these investigations prove promising, they are complicated by the diversity of BA species in vivo as well as the potential for BA induced toxicity at higher dosages. By maintaining homeostatic levels of genes involved in BA synthesis while not affecting FXR signaling, Cyp7a1/Cyp27a1 DKO mice may serve as a potential model to investigate the mechanistic alterations which may be induced by individual BA species in vivo.
In conclusion, the creation of Cyp7a1 and Cyp27a1 DKO mice resulted in a significantly reduced BA pool size while maintaining a similar BA profile and homeostatic expression of genes involved in BA synthesis, conjugation, and transport as compared to WT mice. While we work to extrapolate these studies into female DKO mice, we feel this model may provide a useful tool to researchers for investigations into the mechanistic consequences of individual BA therapies as well as BA and FXR signaling pathways.
Supplementary Material
Agarose gel depicting results of genotyping in Cyp7a1 & Cyp27a1DKO mice and a table including primer sequences. Cyp7a1 WT allele ~450bp, Cyp7a1 mutant allele ~500bp, Cyp27a1 WT allele= 359bp and Cyp27a1 mutant allele= 223bp. Genotyping is depicted for 12 animals with lanes 1–12 detecting Cyp7a1 alleles and lanes 13–24 detecting Cyp27a1 respectively. Lanes 1–5 and 13–17 show 5 DKO mice. Lanes 6–8, 11–12, 17–19 and 23–24 show Cyp7a1 heterozygous and Cyp27a1 WT mice. Lanes 9–10 and 21–22 show 2 WT mice.
(A) Representative images of H&E stained liver sections. (B) Serum assay of triglycerides and cholesterol (mg/dL) and liver injury biomarkers AST, ALP, and ALT (IU/L). (C) Representative images of CK19 stained liver sections. (D) Quantification of relative area stained. An asterisk denotes a significant difference from WT (P < 0.05).
Relative mRNA levels of BA transporter genes in the liver for male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice.
Gene expression was measured using RT q-PCR, normalized to β-actin mRNA levels, and graphed as relative mRNA ± 1 SD. An asterisk denotes a significant difference from WT (P < 0.05). Genes of Osta, Ostb, Mrp3 and Mrp4 could encode transporter to efflux BAs to the sinusoidal side of hepatocytes, and of Mrp2 could efflux anion conjugated substrates to canalicular side of hepatocytes. Genes of Oatp1a1 and Oatp1a4 encode transporters that have been shown to uptake unconjugated BAs into hepatocytes.
List of primer sequences used in this study.
Animal numbers and average body weights for all groups used in this study.
Acknowledgments
Financial Support
Support for this study was provided by National Institute of Health and Department of Veterans Affairs grants (R21ES029258, BX002741, T32ES007148, R01GM104037, and P3-ES005022).
Abbreviations
- αMCA
α-muricholic acid
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- Amacr
alpha-methylacyl-CoA racemase
- ASBT
apical sodium-dependent bile acid transporter
- AST
aspartate aminotransferase
- βMCA
β-muricholic acid
- BA
bile acid
- Baat
bile acid-Coenzyme A
- amino
acid N-acyltransferase
- BSEP
bile salt export pump
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- Ch25h
cholesterol 25-hydroxylase
- CK-19
cytokeratin 19
- Cyp27a1
sterol 27-hydroxylase
- Cyp46a1
cytochrome p450 46a1
- Cyp7a1
cholesterol 7α-hydroxylase
- Cyp7b1
25-hydroxycholesterol 7-alpha-hydroxylase
- Cyp8b1
cytochome p450 8b1
- DCA
deoxycholic acid
- DKO
Cyp7a1/Cyp27a1 double knock out
- Fgf15
fibroblast growth factor 15
- FXR
farnesoid X receptor
- GCA
glycocholic acid
- GCDCA
glycochenodeoxycholic acid
- GDCA
glycodeoxycholic acid
- GLCA
glycolithocholic acid
- HDCA
hyodeoxycholic acid
- Hsd17b4
hydroxysteroid 17-beta dehydrogenase 4
- Hsd3b7
3β-hydroxy-Δ5-C27-steroid oxidoreductase
- Ibabp
ileal bile acid-binding protein
- LCA
lithocholic acid
- NTCP
sodium taurocholate transporter
- OATPs
organic anion-transporting polypeptides
- OSTa/b
organic solute transporter alpha and beta
- S1PR2
sphingosine 1-phosphate receptor 2
- Scp2
sterol carrier protein 2
- Shp
small heterodimer partner
- Slc27a5
solute carrier family 27, member 5
- TCA
taurocholic acid
- TCDCA
taurochenodeoxycholic acid
- TDCA
taurodeoxycholic acid
- TGR5
G protein-coupled BA receptor
- THDCA
taurohyodeoxycholic acid
- TLCA
taurolithocholic acid
- TUDCA
tauroursodeoxycholic acid
- TαMCA
tauro-α-muricholic acid
- TβMCA
tauro-β-muricholic acid
- TωMCA
tauro-ω-muricholic acid
- UDCA
ursodeoxycholic acid
- ωMCA
ω-muricholic acid
Reference
- 1.Li T, Chiang JYL. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacological Reviews. 2014;66(4):948–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hundt M, John S. Physiology, Bile Secretion. StatPearls. Treasure Island (FL): StatPearls Publishing StatPearls Publishing LLC; 2018. [PubMed] [Google Scholar]
- 3.Hofmann A The continuing importance of bile acids in liver and intestinal disease. Archives of Internal Medicine. 1999;159(22):2647–58. [DOI] [PubMed] [Google Scholar]
- 4.Hagenbuch B, Meier PJ. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. Journal of Clinical Investigation. 1994;93(3):1326–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacquemin E, Hagenbuch B, Stieger B, Wolkoff AW, Meier PJ. Expression cloning of a rat liver Na(+)-independent organic anion transporter. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(1):133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong MH, Oelkers P, Craddock AL, Dawson PA. Expression cloning and characterization of the hamster ileal sodium-dependent bile acid transporter. The Journal of biological chemistry. 1994;269(2):1340–7. [PubMed] [Google Scholar]
- 7.Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, et al. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. The Journal of biological chemistry. 2005;280(8):6960–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Csanaky IL, Lu H, Zhang Y, Ogura K, Choudhuri S, Klaassen CD. Organic anion-transporting polypeptide 1b2 (Oatp1b2) is important for the hepatic uptake of unconjugated bile acids: Studies in Oatp1b2-null mice. Hepatology (Baltimore, Md). 2011;53(1):272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russell DW. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annual Review of Biochemistry. 2003;72(1):137–74. [DOI] [PubMed] [Google Scholar]
- 10.Chiang JYL. Bile Acid Metabolism and Signaling. Comprehensive Physiology. 2013;3(3):1191–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res. 2016;57(12):2130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile Acids and the Gut Microbiome. Current opinion in gastroenterology. 2014;30(3):332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuno T, Hirayama-Kurogi M, Ito S, Ohtsuki S. Reduction in hepatic secondary bile acids caused by short-term antibiotic-induced dysbiosis decreases mouse serum glucose and triglyceride levels. Scientific Reports. 2018;8(1):1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwarz M, Russell DW, Dietschy JM, Turley SD. Marked reduction in bile acid synthesis in cholesterol 7α-hydroxylase-deficient mice does not lead to diminished tissue cholesterol turnover or to hypercholesterolemia. Journal of Lipid Research. 1998;39(9):1833–43. [PubMed] [Google Scholar]
- 15.Schwarz M, Russell DW, Dietschy JM, Turley SD. Alternate pathways of bile acid synthesis in the cholesterol 7α-hydroxylase knockout mouse are not upregulated by either cholesterol or cholestyramine feeding. Journal of Lipid Research. 2001;42(10):1594–603. [PubMed] [Google Scholar]
- 16.Rosen H, Reshef A, Maeda N, Lippoldt A, Shpizen S, Triger L, et al. Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27-hydroxylase gene. The Journal of biological chemistry. 1998;273(24):14805–12. [DOI] [PubMed] [Google Scholar]
- 17.Chow MD, Lee Y-H, Guo GL. The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Molecular Aspects of Medicine. 2017;56:34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harach T, Pols TWH, Nomura M, Maida A, Watanabe M, Auwerx J, et al. TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Scientific Reports. 2012;2:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vítek L, Haluzík M. The role of bile acids in metabolic regulation. Journal of Endocrinology. 2016;228(3):R85–R96. [DOI] [PubMed] [Google Scholar]
- 20.Sipka S, Bruckner G. The Immunomodulatory Role of Bile Acids. International Archives of Allergy and Immunology. 2014;165(1):1–8. [DOI] [PubMed] [Google Scholar]
- 21.Hellstrom PM, Nilsson I, Svenberg T. Role of bile in regulation of gut motility. Journal of internal medicine. 1995;237(4):395–402. [DOI] [PubMed] [Google Scholar]
- 22.Kong B, Sun R, Huang M, Chow MD, Zhong XB, Xie W, et al. A Novel Fibroblast Growth Factor 15 Dependent- and Bile Acid Independent-Promotion of Liver Regeneration in Mice. Hepatology (Baltimore, Md). 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuipers F, Stroeve JH, Caron S, Staels B. Bile acids, farnesoid X receptor, atherosclerosis and metabolic control. Current opinion in lipidology. 2007;18(3):289–97. [DOI] [PubMed] [Google Scholar]
- 24.Narisawa T, Magadia NE, Weisburger JH, Wynder EL. Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-methyl-N’-nitro-N-nitrosoguanidine in rats. Journal of the National Cancer Institute. 1974;53(4):1093–7. [DOI] [PubMed] [Google Scholar]
- 25.Bechmann LP, Kocabayoglu P, Sowa JP, Sydor S, Best J, Schlattjan M, et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology (Baltimore, Md). 2013;57(4):1394–406. [DOI] [PubMed] [Google Scholar]
- 26.Schaap FG, Trauner M, Jansen PLM. Bile acid receptors as targets for drug development. Nature Reviews Gastroenterology & Amp; Hepatology. 2013;11:55. [DOI] [PubMed] [Google Scholar]
- 27.Hofmann A Bile acids: Trying to understand their chemistry and biology with the hope of helping patients. Hepatology (Baltimore, Md). 2009;49(5):1403–18. [DOI] [PubMed] [Google Scholar]
- 28.Han J, Liu Y, Wang R, Yang J, Ling V, Borchers CH. Metabolic Profiling of Bile Acids in Human and Mouse Blood by LC–MS/MS in Combination with Phospholipid-Depletion Solid-Phase Extraction. Analytical Chemistry. 2015;87(2):1127–36. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Klaassen CD. Effects of feeding bile acids and a bile acid sequestrant on hepatic bile acid composition in mice. Journal of Lipid Research. 2010;51(11):3230–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong B, Sun R, Huang M, Chow MD, Zhong XB, Xie W, et al. Fibroblast Growth Factor 15-Dependent and Bile Acid-Independent Promotion of Liver Regeneration in Mice. Hepatology (Baltimore, Md). 2018;68(5):1961–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lew JL, Zhao A, Yu J, Huang L, De Pedro N, Pelaez F, et al. The farnesoid X receptor controls gene expression in a ligand- and promoter-selective fashion. The Journal of biological chemistry. 2004;279(10):8856–61. [DOI] [PubMed] [Google Scholar]
- 32.Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell metabolism. 2013;17(2):225–35. [DOI] [PubMed] [Google Scholar]
- 33.Repa JJ, Lund EG, Horton JD, Leitersdorf E, Russell DW, Dietschy JM, et al. Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia. Reversal by cholic acid feeding. The Journal of biological chemistry. 2000;275(50):39685–92. [DOI] [PubMed] [Google Scholar]
- 34.Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW. Knockout of the Cholesterol 24-Hydroxylase Gene in Mice Reveals a Brain-specific Mechanism of Cholesterol Turnover. Journal of Biological Chemistry. 2003;278(25):22980–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Agarose gel depicting results of genotyping in Cyp7a1 & Cyp27a1DKO mice and a table including primer sequences. Cyp7a1 WT allele ~450bp, Cyp7a1 mutant allele ~500bp, Cyp27a1 WT allele= 359bp and Cyp27a1 mutant allele= 223bp. Genotyping is depicted for 12 animals with lanes 1–12 detecting Cyp7a1 alleles and lanes 13–24 detecting Cyp27a1 respectively. Lanes 1–5 and 13–17 show 5 DKO mice. Lanes 6–8, 11–12, 17–19 and 23–24 show Cyp7a1 heterozygous and Cyp27a1 WT mice. Lanes 9–10 and 21–22 show 2 WT mice.
(A) Representative images of H&E stained liver sections. (B) Serum assay of triglycerides and cholesterol (mg/dL) and liver injury biomarkers AST, ALP, and ALT (IU/L). (C) Representative images of CK19 stained liver sections. (D) Quantification of relative area stained. An asterisk denotes a significant difference from WT (P < 0.05).
Relative mRNA levels of BA transporter genes in the liver for male WT, Cyp7a1−/−, Cyp27a1−/−, and DKO mice.
Gene expression was measured using RT q-PCR, normalized to β-actin mRNA levels, and graphed as relative mRNA ± 1 SD. An asterisk denotes a significant difference from WT (P < 0.05). Genes of Osta, Ostb, Mrp3 and Mrp4 could encode transporter to efflux BAs to the sinusoidal side of hepatocytes, and of Mrp2 could efflux anion conjugated substrates to canalicular side of hepatocytes. Genes of Oatp1a1 and Oatp1a4 encode transporters that have been shown to uptake unconjugated BAs into hepatocytes.
List of primer sequences used in this study.
Animal numbers and average body weights for all groups used in this study.