

Abstract
A physiologically based pharmacokinetic (PBPK) model was developed to evaluate and predict (1) the effect of concomitant cytochrome P450 3A (CYP3A) inhibitors or inducers on the exposures of zanubrutinib, (2) the effect of zanubrutinib on the exposures of CYP3A4, CYP2C8, and CYP2B6 substrates, and (3) the impact of gastric pH changes on the pharmacokinetics of zanubrutinib. The model was developed based on physicochemical and in vitro parameters, as well as clinical data, including pharmacokinetic data in patients with B‐cell malignancies and in healthy volunteers from two clinical drug‐drug interaction (DDI) studies of zanubrutinib as a victim of CYP modulators (itraconazole, rifampicin) or a perpetrator (midazolam). This PBPK model was successfully validated to describe the observed plasma concentrations and clinical DDIs of zanubrutinib. Model predictions were generally within 1.5‐fold of the observed clinical data. The PBPK model was used to predict untested clinical scenarios; these simulations indicated that strong, moderate, and mild CYP3A inhibitors may increase zanubrutinib exposures by approximately four‐fold, two‐ to three‐fold, and <1.5‐fold, respectively. Strong and moderate CYP3A inducers may decrease zanubrutinib exposures by two‐ to three‐fold or greater. The PBPK simulations showed that clinically relevant concentrations of zanubrutinib, as a DDI perpetrator, would have no or limited impact on the enzyme activity of CYP2B6 and CYP2C8. Simulations indicated that zanubrutinib exposures are not impacted by acid‐reducing agents. Development of a PBPK model for zanubrutinib as a DDI victim and perpetrator in parallel can increase confidence in PBPK models supporting zanubrutinib label dose recommendations.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Drug interactions between zanubrutinib and cytochrome P450 3A4 (CYP3A4) modulators are expected to be clinically significant, as seen in clinical studies of zanubrutinib with itraconazole and rifampicin.
WHAT QUESTION DID THIS STUDY ADDRESS?
The study explored whether PBPK models can be used to assess zanubrutinib's clinical drug‐drug interaction (DDI), including its potential as a victim of CYP3A modulators or PPIs and as a perpetrator for CYP3A, CYP2C9, and CYP2C19 substrates. The simulation work supported dosing recommendations in the product label of the drug in DDI situations not clinically tested.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Development of a PBPK model for zanubrutinib as a DDI victim and perpetrator in parallel can increase the confidence of PBPK models.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT AND/OR THERAPEUTICS?
Specific dosing guidance on the use of zanubrutinib with CYP3A4 perpetrators or other CYP victims can be given to prescribers in the absence of observed clinical data.
INTRODUCTION
Zanubrutinib (BGB‐3111, BRUKINSA™) is a potent and highly selective Bruton's tyrosine kinase (BTK) inhibitor that is being developed for the treatment of a variety of B‐cell malignancies. Zanubrutinib is differentiated from other medications in its class in a number of ways, including enhanced selectivity for BTK, and higher therapeutic exposure. 1 These differentiating factors have further translated into clinically meaningful benefits of zanubrutinib with respect to safety and efficacy compared with ibrutinib, as demonstrated in its clinical program. 2 Zanubrutinib received accelerated approval by regulatory authorities in the United States for relapsed/refractory mantle cell lymphoma. The recommended total daily dose of zanubrutinib is 320 mg, administered as 160 mg twice daily (b.i.d.) or 320 mg once daily (q.d.).
Clinical pharmacokinetics (PK) data have shown that zanubrutinib is rapidly absorbed and eliminated after oral administration in patients with B‐cell malignancies. The median time to reach peak plasma concentration for zanubrutinib was 2 hours, with a mean apparent terminal elimination half‐life of approximately 2 to 4 hours. The maximum plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC) for zanubrutinib increased proportionally over a dose range of 40 mg to 320 mg. 3 There were limited systemic accumulations following multiple‐dose administrations with accumulation ratios on Day 8/Day 1 of approximately 1. 3 Following administration of a single radiolabeled dose of zanubrutinib 320 mg in healthy subjects, approximately 87% of the dose was recovered in feces, and 8% was recovered in urine. Unchanged zanubrutinib was the predominant drug‐related entity in feces, accounting for 38% of the dose, which indicated that 60–70% of zanubrutinib was absorbed. Renal excretion of unchanged zanubrutinib was minor, with 0.4% of the dose excreted as the parent drug. CYP3A‐mediated metabolism is the major clearance pathway with additional contribution from direct cysteine conjugation (~11% of dose was recovered in excreta as cysteine and N‐acetyl cysteine adducts). There are no major active metabolites in circulation.
The results of a clinical drug‐drug interaction (DDI) study (BGB–3111–104) 4 showed that co‐administration of itraconazole increased zanubrutinib Cmax by 2.6‐fold and increased AUC from time 0 to last observation (AUC0‐t) by 3.9‐fold. Co‐administration of rifampicin decreased zanubrutinib Cmax and AUC0‐t by 12.6‐fold and 13.5‐fold, respectively. Another clinical DDI study (BGB–3111–108)5 was conducted to assess the effect of zanubrutinib on the PK of substrates of CYP3A (midazolam), CYP2C9 (warfarin), CYP2C19 (omeprazole), P‐glycoprotein (digoxin), and breast cancer resistance protein, BCRP (rosuvastatin) using a cocktail approach. The results showed that the zanubrutinib 320‐mg total daily dose had minimal or no effect on the activity of CYP2C9, BCRP and P‐gp, but decreased the systemic exposure of CYP3A and CYP2C19 substrates (mean reduction <50%). Zanubrutinib is likely to be a substrate of P‐gp, but is not a substrate or inhibitor of OAT1, OAT3, OCT2, OATP1B1, or OATP1B3.
Physiologically based PK (PBPK) modeling has been successfully used to facilitate the design of DDI studies, extrapolated to untested drug interactions, and to inform drug labeling. 6 , 7 To support dosing recommendations for zanubrutinib, the primary objective of this study was to develop a PBPK model for a comprehensive assessment of its clinical DDI, including: (1) the effect of CYP3A4 modulators on the PK of zanubrutinib; (2) the effect of zanubrutinib on exposures to substrates of CYP3A, CYP2C8, and CYP2B6 and (3) the potential impact of acid‐reducing agents (ARAs) on the PK of zanubrutinib. Of note, there are currently no available sensitive and validated CYP2C8 and CYP2B6 probes for a cocktail DDI study. Since the half‐maximal inhibitory concentration (IC50) is comparable for these three CYP2C isoenzymes (CYP2C8, CYP2C9, and CYP2C19), and in view of the coupled induction potential of CYP3A and CYP2B6, 8 the PBPK model was developed based on clinical data for CYP2C9, CYP2C19 and CYP3A, and was used to explore the impact of zanubrutinib on CYP2C8 and CYP2B6 substrates.
METHODS
Overview of modeling strategy
The zanubrutinib PBPK model was developed using a population‐based human absorption, metabolism, and excretion simulator (Simcyp version 16 release 1; Simcyp, Sheffield, UK). The default virtual North European Caucasian population was used for the simulations, 9 with the exception of demographic data noted in each clinical study. Clinical PK data from healthy volunteers and patients with B‐cell malignancies, as well as data from clinical DDI and human absorption, distribution, metabolism, and excretion (ADME) studies (Table 1), physicochemical parameters, and in vitro data (Table 2), were used for model development and verification. The overview of the PBPK model development process consisted of three parts: model development, model verification, and model simulation/application (Figure 1). The prespecified acceptance criteria (1.5‐fold of observed values) was used as a guide during model development and verification.
Table 1.
Zanubrutinib clinical study data used in PBPK model development and verification.
| Clinical study | Population | Study description | Dose regimen | PK data used in PBPK model | |
|---|---|---|---|---|---|
|
BGB−3111–104 4 |
Healthy volunteer | Clinical DDI study with strong CYP3A inhibitor/inducer | 20 mg q.d. (DDI with itraconazole, n = 18) | Development | |
| 320 mg (DDI with rifampicin, n = 20) | Verification | ||||
|
BGB−3111–108 5 |
Healthy volunteer |
Effects of zanubrutinib on substrates of CYP3A, CYP2C9, CYP2C19, P‐gp and BCRP |
2 mg midazolam (n = 18) | Development | |
|
160 mg b.i.d. dose (n = 17) b.i.d. Days 1–7 |
Verification | ||||
|
BGB−3111–106 38 |
Healthy volunteer |
A thorough QTc study |
160 mg (n = 28) Single dose |
Verification | |
|
BGB−3111–107 39 |
Healthy volunteer |
A hepatic impairment study |
80 mg (n = 11) Single dose |
Verification | |
|
BGB−3111–AU−003 3 |
B‐cell malignancy |
Phase 1/2 dose escalation and expansion study |
40 mg q.d. (n = 4) 80 mg q.d. (n = 5) 160 mg q.d. (n = 6) 320 mg q.d. (n = 22) 160 mg b.i.d. (n = 72) q.d. Days 1–8 b.i.d. Days 1–8 |
Verification |
Abbreviations: b.i.d., twice daily; DDI, drug‐drug interaction; PBPK, physiologically based pharmacokinetics; q.d., once daily.
Table 2.
Final input parameters for the zanubrutinib PBPK model
| Parameter | Value | Source |
|---|---|---|
| Physicochemical properties | ||
| MW (g/mol) | 471.55 | Internal data |
| logP | 4.2 | Monoprotic base, experimental data a |
| pKa | 3.3 | Monoprotic base, experimental data a |
| B/P | 0.804 | Experimental data a |
| f u | 0.0582 | Experimental data a |
| Human absorption, dissolution, metabolism, and elimination | ||
| Peff,man (10−4 cm/s) | 0.9 |
Experimental data a Adjusted to match Fa~0.7 based on clinical data |
| Qgut | 5.9 | Predicted by Simcyp |
| Solubility (mg/mL) | 0.247, 0.073, 0.054, and 0.052 at pH 1.2, 4.5, 6.8, and 7.4, respectively | Experimental data a |
| CLin (L/h) | 188.19 | Adjusted by comparing time‐concentration profile of zanubrutinib 20 mg |
| CLout (L/h) | 142.15 | Adjusted by comparing time‐concentration profile of zanubrutinib 20 mg |
| Vss (L/kg) | 9.4 | Predicted by Simcyp Method 1 |
| Vsac (L/kg) | 9.2 | Adjusted by comparing time‐concentration profile of zanubrutinib 20 mg |
| CLint (μL/min/mg) | 120 |
Adjusted; A well‐stirred liver model for IVIVE CLint from HLM is 109 μL/min/mg a Estimated fmCYP3A4 is 81.6% |
| Additional clearance HLM (μL/min/mg) | 60 | Adjusted by comparing with the observed DDI data with itraconazole in study BGB−3111–104 4 |
| fu,mic | 0.407 | Predicted (pH=7.4, microsomal protein 0.5 mg/mL) |
| CLR (L/h) | 0.5 | Based on human absorption, metabolism, and excretion study (BGB−3111–105); Estimated renal contribution: 1.6% of total CL |
| Drug interaction: induction/inhibition | ||
| Induction/suppression |
CYP3A4: Indmax=6.27, IndC50=0.47 CYP2B6: Indmax=2.21, IndC50=0.73 CYP2C8: Indmax=4.172, IndC50=0.53 CYP2C9: Indmax=1.694, IndC50=0.119 CYP2C19: Indmax=2.02, IndC50=0.155 |
Estimated by the Emax model based on experimental data a Zanubrutinib at 0.3, 3, and 30 μM increased CYP3A4 activity by 2.02, 6.27, and 2.51‐fold, respectively Zanubrutinib at 0.3, 3, and 30 μM increased CYP2B6 mRNA levels by 1.6, 3.6, and 2.6‐fold, respectively Zanubrutinib at 0.3, 3, and 30 μM increased CYP2C8 activity by 1.39, 3.78, and 3.94‐fold, respectively Zanubrutinib at 0.3, 3, and 30 μM increased CYP2C9 activity by 1.21, 1.65, and 1.67‐fold, respectively. Zanubrutinib at 0.3, 3, and 30 μM increased CYC2C19 activity by 1.31, 2.04, and 1.91‐fold, respectively |
| Competitive inhibition |
KiCYP1A2=60.5 μM KiCYP2B6=60.5 μM KiCYP2C8=2.015 μM KiCYP2C9=2.845 μM KiCYP2C19=3.790 μM KiCYP2D6=36.45 μM KiCYP3A4=7.15 μM |
Experimental data a For a competitive enzyme inhibition, Ki calculated by Ki=IC50/2 Fraction unbound in microsomes, fu, mic=0.774 (predicted by microsomal protein: 0.1 mg/mL) |
Abbreviations: B/P, blood/plasma partition ratio; CLin and CLout, clearance from the systemic compartment to the single‐adjusted compartment and from the single‐adjusted compartment to the systemic compartment, respectively; CLint, intrinsic clearance; CLR, renal clearance; CYP, cytochrome P450; fa, fraction absorbed; fmCYP3A4, fraction of drug metabolized by CYP3A4; fu, fraction of unbound drug in plasma; fu,mic, microsomal protein binding; HLM, human liver microsomes; IndC50, calibrated inducer concentration that supports half maximal induction (μM); Indmax, calibrated maximal fold induction over vehicle (1= no induction); IVIVE, in vitro–in vivo extrapolation; ka, absorption rate constant; Ki, enzyme inhibition constant (concentration of inhibitor associated with half maximal inhibition); logP, Log of the octanol‐water partition coefficient for the neutral compound; MW, molecular weight; PBPK, physiologically based pharmacokinetics; Peff, man, effective human jejunum permeability; PK, pharmacokinetics; pKa, acid dissociation constant; Qgut, flow rate for overall delivery of drug to the gut (drug dependent); SAC, single adjusting compartment; Vmax maximum velocity; Vsac, volume of the single adjusted compartment; Vss volume of distribution at steady state.
Internal data.
Figure 1.

The overall model development, verification process, and simulation flow chart. Abbreviations: b.i.d., twice daily; CLint, intrinsic clearance; CYP, cytochrome P450; DDI, drug‐drug interaction; Fa, fraction absorbed; fmcyp3A4,fraction of drug metabolized by CYP3A4; PBPK, physiologically based pharmacokinetics; q.d., once daily; Vsac, volume of the single adjusted compartment.
Model development and model parameters
A summary of model parameters used in the PBPK model and the corresponding references are shown in Table 2. For absorption, the advanced dissolution absorption metabolism (ADAM) model was used. 10 , 11 The apparent permeability (A‐B) values for zanubrutinib were based on the in vitro permeability study (Caco‐2; Table 2) and adjusted further based on results of the ADME study. As zanubrutinib has pH‐dependent solubility profiles, measured solubility data at different pH levels were used in the model.
Distribution models evaluated during model development included a full PBPK model, a minimal PBPK model, and a minimal PBPK model with a single adjusting compartment (SAC), which represents a lump of tissues, 12 and considers metabolism in the liver and intestine. Since the PBPK model with a SAC provided a better description of the observed clinical data (zanubrutinib PK at 20 mg4 and DDI data with itraconazole), this model was selected for further model development and validation.
The intrinsic clearance value of 120 μL/min/mg was assigned for CYP3A based on the human liver microsome study (internal data); an additional clearance value of 60 µL/min/mg was included to account for non–CYP3A‐mediated clearance and to better describe clinical data associated with DDIs with itraconazole. A well‐stirred liver model was used in the in vitro–in vivo extrapolation (IVIVE). Renal clearance with a value of 0.5 L/h was used in all simulations.
CYP3A induction parameters in the zanubrutinib PBPK model were calibrated based on a series of simulations using in vitro induction data in human hepatocytes (CYP3A4 mRNA or activity) and compared with observed midazolam concentration data during coadministration of zanubrutinib in the clinical DDI study (BGB–3111–108). The maximum fold induction (Indmax) and half maximum‐fold induction concentration (IndC50) values were obtained by fitting a maximum drug effect (Emax) model based on CYP3A4 mRNA or activity induction data. The CYP3A induction parameters based on the activity data were used in the model due to better agreement with clinical data. The Indmax and IndC50 values for CYP2C8, CYP2C9, and CYP2C19 were obtained by fitting an Emax model based on in vitro induction activity data (Table 2). The IC50 values for zanubrutinib reversible CYP inhibition are comparable for CYP2C8, CYP2C9 and CYP2C19 (Table 2).
Model verification
The developed PBPK model was further verified by comparing the model against PK parameters and/or plasma concentration profiles from studies in healthy volunteers and a study in patients with B‐cell malignancies (Table 1). The PBPK model was also verified against clinical DDI data with rifampicin (BGB–3111–104). 4 Model‐predicted effects of zanubrutinib on CYP2C9 and CYP2C19 activities were compared with clinical DDI data in the cocktail DDI study (BGB–3111–108).
Model application
After the verification step, the PBPK model was used to simulate untested clinical DDI scenarios for zanubrutinib as a victim. To maximize the impact of the CYP modulators on steady‐state zanubrutinib exposure, both CYP inducers and inhibitors were administered for 14 days and 7 days, respectively, prior to starting zanubrutinib. The model was also used to simulate the DDI potential for zanubrutinib as a perpetrator when co‐administered with CYP3A (midazolam), CYP2B6 (bupropion), and CYP2C8 (repaglinide and rosiglitazone) substrates. Plasma concentration profiles of these substrate drugs with or without multiple‐dose administration of zanubrutinib 160 mg were simulated. Geometric mean ratios (with/without perpetrator) for Cmax and AUC were provided.
To simulate the impact of gastric pH changes on the PK of zanubrutinib, the impact of administration of esomeprazole on zanubrutinib absorption was modeled by increasing the gastric pH from 1.5 to 4.5 in the standard Simcyp healthy volunteer population (fasted model). Plasma concentrations of zanubrutinib under these two conditions (gastric pH of 1.5 or 4.5) were compared.
PBPK models of other compounds used in the study
Except for itraconazole and rifampicin, compound files in Simcyp V.16 were used. Additional model verification and in‐house simulations were conducted for itraconazole and rifampin models. The PBPK models for itraconazole and hydroxy‐itraconazole were based on the work of Chen et al. 13 The rifampicin PBPK model was modified to include CYP3A induction parameters based on publications by Fahmi and Yamashita. 8 , 14
Simulation setup
For model development and verification, the virtual trials used in each of the simulations were based on the corresponding doses used in each of the clinical studies (Table 1). The virtual population (100 subjects [10 trials]) used in the simulations mimicked the populations used in these studies. For model application, all simulations were conducted with the “Sim‐Healthy volunteer” population that was built into the software. The CYP modulators were administered from Day 1 to Day 14, and zanubrutinib 160 mg b.i.d. was administered from Day 7 to Day 14. Steady‐state Cmax and AUC of zanubrutinib were simulated on Day 14 with and without modulators.
RESULTS
Model development
The performance of the PBPK model was tested by comparing predicted and observed PK parameters and plasma concentration–time profiles following the 20‐mg dose of zanubrutinib from the BGB–3111–104 study 4 (Figure 2). The predicted plasma concentration–time profiles and PK parameters were generally in agreement with observed clinical data.
Figure 2.
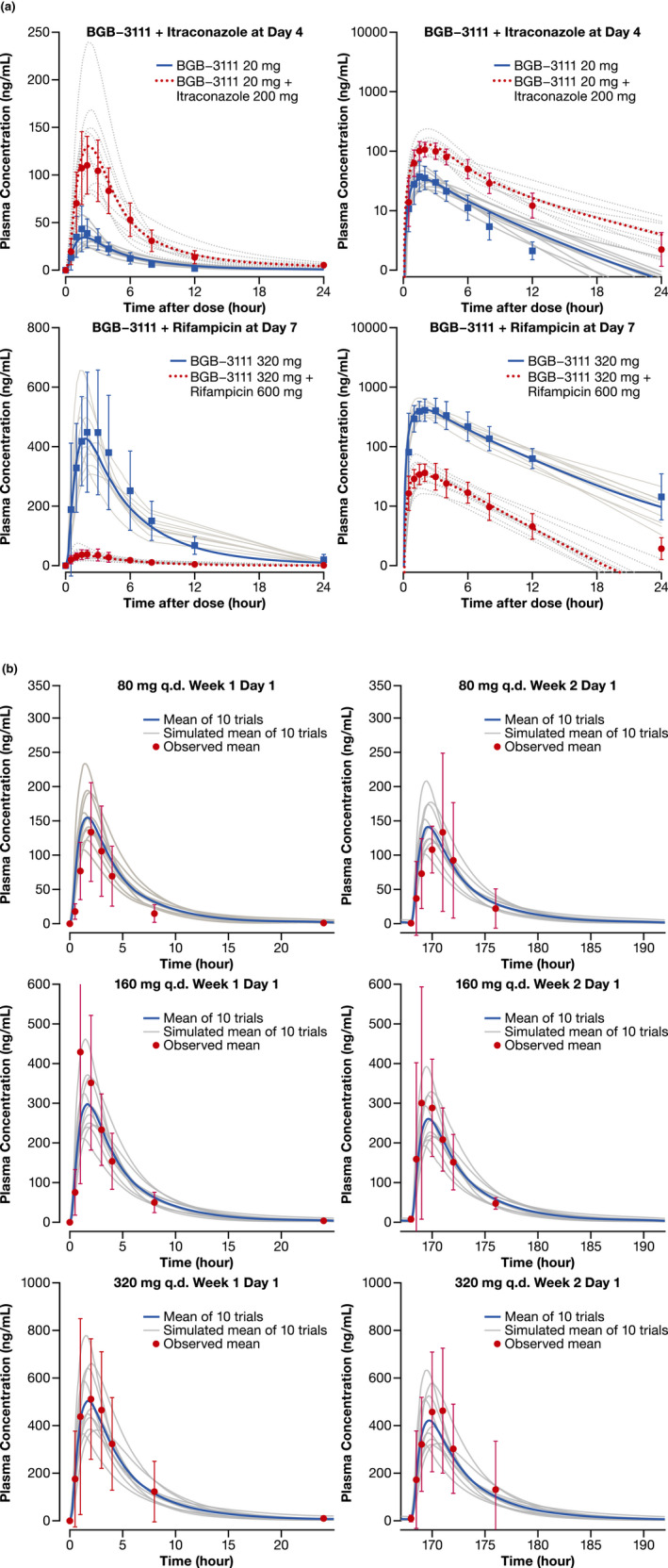
Simulated and observed plasma concentration‐time profiles of zanubrutinib in healthy subjects and in patients with B‐cell malignancies. (a) Healthy subjects with or without co‐administration of itraconazole or rifampicin (Left = Linear Scale; Right = Semi‐Log Scale) (b) Patients with B‐cell malignancies following single and repeated dose of zanubrutinib 80 mg, 160 mg, and 320 mg. The grey lines represent individual trials (n = 10×10) and the solid blue lines or red dashed lines are the mean of the simulated population (n = 100). Observed data shown are mean (the dotted points) and standard deviation. Age range is 20–50 years old and ratio of females is 0.16 (healthy subjects). Age range is 20–90 years old and ratio of females is 0.3 (patients). Abbreviations: b.i.d., twice daily; q.d., once daily.
Zanubrutinib as a DDI victim with a strong CYP3A4 inhibitor (itraconazole)
A modified itraconazole model was developed and verified in‐house. Model verification was conducted by the ability of the itraconazole models to describe the concentration–time profiles of itraconazole and hydroxy‐itraconazole following single‐ and multiple‐dose administration (Figure S1). Furthermore, model verification was conducted by comparing the model with the observed DDI effects of itraconazole co‐administered with midazolam (Table S1). The optimized itraconazole model (with parameters summarized in Table S2) was then used to simulate the impact of itraconazole on the PK of zanubrutinib.
The sensitivity analysis showed that the fraction of drug metabolized by CYP3A4 (fmCYP3A4) appears to be the most sensitive parameter for simulating DDI and that changes in fmCYP3A4 from 98.2% to 76.7% would decrease the Cmax and AUC ratio (with/without itraconazole) from 4.0‐ to 2.9‐fold and 4.9‐ to 3.1‐fold, respectively. An estimated fmCYP3A4 of 81.6% was used in the final model.
We assessed the ability of the model to describe concentration–time profiles of 20‐mg zanubrutinib co‐administered with itraconazole in the clinical DDI study 4 (Figure 2A). The observed geometric mean ratios (in the presence/absence of itraconazole) of the Cmax and AUC from time 0 to infinity (AUC0‐inf) for zanubrutinib were 2.57‐ and 3.86‐fold, respectively. The corresponding PBPK model‐predicted geometric mean Cmax and AUC ratios were consistent with observed values in the clinical DDI study (Table 3).
Table 3.
PBPK model‐predicted geometric mean ratios of Cmax and AUC for zanubrutinib as a DDI victim or perpetrator compared with observed data from clinical DDI studies.
| Parameter | Ratio | |||
|---|---|---|---|---|
| Observed a (90% CI) | Prediction a (90% PI) | Observed/predicted b | ||
| Inhibitor (itraconazole) | AUC | 3.86 (3.49–4.22) | 3.47 (3.32–3.63) | 1.11 |
| Cmax | 2.57 (2.26–2.91) | 3.20 (3.03–3.38) | 0.80 | |
| Inducer (rifampicin) | AUC | 0.071 (0.062–0.090) (↓14.1‐fold) | 0.060 (0.053–0.068) (↓16.6‐fold) | 1.18 |
| Cmax | 0.079 (0.068–0.095) (↓12.6‐fold) | 0.062 (0.054–0.072) (↓16.0‐fold) | 1.27 | |
| Substrate (midazolam) c | AUC | 0.53 (0.48–0.57) | 0.51 (0.49–0.053) | 1.02 |
| Cmax | 0.70 (0.63–0.78) | 0.53 (0.50–0.055) | 0.74 | |
| Substrate (omeprazole) d | AUC | 0.63 (0.5–0.70) | 0.94 (0.93–0.95) | 0.67 |
| Cmax | 0.79 (0.65–0.97) | 0.97 (0.96–0.98) | 0.81 | |
| Substrate (warfarin) e | AUC | 1.00 (0.98–1.03) | 0.97 (0.96–0.97) | 1.03 |
| Cmax | 0.95 (0.87–1.04) | 1.00 (1.00–1.00) | 0.95 | |
Abbreviations; AUC, area under the plasma concentration–time curve; AUC0‐24 h, area under the plasma concentration–time curve from time 0 to 24 hours; AUC0‐inf, area under the plasma concentration–time curve from time 0 to infinity; AUC0‐t, area under the plasma concentration–time curve from time 0 to last observation; CI, confidence interval; Cmax, maximum plasma concentration; DDI, drug‐drug interaction; PBPK, physiologically based pharmacokinetics; PI, prediction interval.
Expressed by ratio = (substrate +interaction)/substrate.
AUC0‐t was used to calculate the observed ratio; AUC0‐24 h was used to calculate the predicted ratio on Day 4 for zanubrutinib 20 mg used with itraconazole. Observed clinical data were from a clinical DDI study of Mu S, et al. 38
Predicted AUC0‐24 h on Day 7 for midazolam (2 mg) with/without zanubrutinib co‐administration. AUC0‐inf was used to calculate the predicted ratio.
Predicted AUC0‐24 h on Day 12 for omeprazole (20 mg) with/without zanubrutinib co‐administration. AUC0‐t was used to calculate the predicted ratio.
Predicted AUC0‐24 h on Day 8 for warfarin (10 mg) with/without zanubrutinib co‐administration. AUC0‐inf was used to calculate the predicted ratio.
Zanubrutinib as a DDI perpetrator with a sensitive CYP3A substrate (midazolam)
The CYP3A induction parameters of zanubrutinib as a DDI perpetrator were tested by comparing the simulated midazolam plasma concentration–time profiles (Figure S2) and PK parameters with the observed clinical data with or without co‐administration of zanubrutinib. Model‐predicted geometric mean Cmax and AUC ratios (Table 3) of midazolam 2 mg co‐administered with zanubrutinib 160 mg were consistent with observed clinical data (BGB–3111–108).
Model verification
To assess the robustness of the developed model, simulations of different clinical scenarios were compared with the observed clinical data not included in the original model development. As shown in Figure 2B, the predicted plasma concentration profiles after single and repeated oral doses (40, 80, 160, 320 mg) were consistent with the observed profiles in patients with B‐cell malignancies in the BGB–3111–AU–003 study. 2 Additionally, the Cmax and AUC values were successfully predicted within 1.5‐fold of observed values (Table 3). Similarly, the predicted concentration profiles after a single oral dose of 80, 160, or 320 mg were consistent with the observed data in studies of healthy volunteers (Table 1). Model‐predicted concentrations after repeated dosing appeared to be trending lower compared with those after a single dose (Figure 2B). Nevertheless, the predicted steady‐state concentrations were within 1.5‐fold of observed data (Table S3), and consistent with minimal presence of auto‐induction from the observed clinical PK data. Thus, despite moderate to high PK variability, the developed PBPK model was able to capture plasma concentration–time profiles and exposure levels of zanubrutinib after single and multiple oral doses in four healthy volunteer studies and in patients with B‐cell malignancies.
Zanubrutinib as a DDI victim with a strong CYP3A4 inducer (rifampicin)
As was the case for itraconazole, additional simulations were performed to verify the rifampicin PBPK models used in our PBPK analyses. The final rifampicin model retained the parameters of the rifampicin PBPK model from the default compound file in Simcyp while parameters governing the CYP3A induction potential of rifampicin were modified. As shown in Figure S3, simulations based on modified Indmax and IndC50 values of 37.1 and 0.28 µM, respectively, provided a better representation of observed data when compared with the rifampicin default compound file in the Simcyp library.
Good agreement was observed between simulated and observed plasma concentration–time profiles of zanubrutinib in healthy subjects with or without co‐administration with rifampicin (Figure 2A). The PBPK model‐predicted geometric mean Cmax and AUC ratios (with/without rifampicin) of zanubrutinib 320 mg were <1.5 times the observed values (Table 3). Thus, the optimized fmCYP3A value, based on data for itraconazole, were subsequently verified by clinical DDI data for rifampicin clinical DDI.
Zanubrutinib as a DDI perpetrator with the substrates of CYP2C9 and CYP2C19 (warfarin and omeprazole)
Because zanubrutinib showed CYP2C9 and CYP2C19 induction potential in vitro, PBPK models incorporating in vitro CYP2C9 and CYP2C19 induction parameters were developed. The model simulations were compared with the results from the cocktail DDI study, in which zanubrutinib was co‐administered with the sensitive substrates of CYP2C9 (warfarin) and CYP2C19 (omeprazole). The results indicated that the induction parameters of CYP2C9 enabled successful description of observed data, whereas the induction parameters of CYP2C19 slightly underestimated the DDI effect (Table 3). Nevertheless, the ratios (observed/predicted) were all within the acceptable criteria of 1.5‐fold.
Simulation of zanubrutinib as a DDI victim
The developed PBPK model was used to simulate other untested clinical DDI scenarios for zanubrutinib during co‐administration with CYP3A inhibitors and inducers. Simulated zanubrutinib PK parameters and corresponding ratios with and without co‐administration of CYP3A4 modulators are presented in Table 4.
Table 4.
Simulated Cmax and AUC0‐24 h ratios of zanubrutinib at steady state in the presence and absence of CYP modulators.
| Inhibitor/inducer | Dose (mg) | Treatment (days) | No. of subjects (trials) | Dose regimen | Cmax ratio | AUC0‐24 h ratio |
|---|---|---|---|---|---|---|
| Strong CYP3A inhibitors | ||||||
| Ritonavir | 100 | 14 | 100 (10) | b.i.d. | 6.68 | 8.32 |
| Itraconazole | 200 | 14 | 100 (10) | q.d. | 3.95 | 2.97 |
| Clarithromycin | 250 | 14 | 100 (10) | b.i.d. | 2.75 | 2.83 |
| Moderate CYP3A inhibitors | ||||||
| Erythromycin | 500 | 14 | 100 (10) | q.6.h. | 3.84 | 4.17 |
| Fluconazole | 200 | 14 | 100 (10) | q.d. | 2.79 | 2.77 |
| Fluconazole | 400 | 14 | 100 (10) | q.d. | 3.70 | 3.84 |
| Diltiazem | 60 | 14 | 100 (10) | q.8.h. | 2.51 | 2.57 |
| Ciprofloxacin | 500 | 14 | 100 (10) | q.8.h. | 1.00 | 1.00 |
| Mild CYP3A inhibitors | ||||||
| Fluvoxamine | 50 | 14 | 100 (10) | q.d. | 1.12 | 1.09 |
| Cyclosporine | 200 | 14 | 100 (10) | q.d. | 1.19 | 1.11 |
| Cimetidine | 400 | 14 | 100 (10) | b.i.d. | 1.00 | 1.00 |
| Strong CYP3A inducers | ||||||
| Rifampicin | 600 | 14 | 100 (10) | q.d. | 0.07 | 0.07 |
| Carbamazepine | 400 | 14 | 100 (10) | b.i.d. | 0.39 | 0.42 |
| Moderate CYP3A inducer | ||||||
| Efavirenz | 600 | 14 | 100 (10) | q.d. | 0.42 | 0.40 |
Simulation conditions: 10 virtual trials, each trial included 10 subjects (aged 20–50 years and 50% female). Each subject received an inhibitor or inducer for 14 days; zanubrutinib 160 mg b.i.d. was administered from Day 7 to Day 14. Geometric mean ratios (with/without perpetrator) for AUC and Cmax are provided ([substrate +interaction]/substrate). For an inducer, %decrease = (1‐ ratio)*100% is also shown.
Abbreviations: AUC, area under the plasma concentration–time curve; AUC0‐24 h, area under the plasma concentration–time curve from time 0 to 24 hours; b.i.d., twice daily; Cmax, maximum plasma concentration; q.6.h., every 6 hours; q.8.h., every 8 hours; q.d., once daily.
PBPK simulations indicate that steady‐state zanubrutinib exposures may increase by approximately four‐fold and by two‐ to three‐fold during co‐administration with the strong and moderate CYP3A inhibitors, respectively; the exceptions were erythromycin 500 mg and ritonavir 400 mg. Simulations suggest that a moderate CYP3A inducer may decrease the AUC of zanubrutinib by two‐ to three‐fold. Of note, while carbamazepine is considered a strong CYP3A inducer, the magnitude of the zanubrutinib exposure decrease was similar to that of a moderate CYP3A inducer, efavirenz.
The magnitude of zanubrutinib exposure increases by time‐dependent inhibitors, such as ritonavir and erythromycin, appears to be larger compared with those of the strong CYP3A inhibitor, itraconazole. The PBPK model was also used to explore changes in zanubrutinib exposure at different doses of fluconazole. The model showed that the simulated AUC ratio of zanubrutinib (with/without inhibitor) following 400‐mg and 200‐mg doses of fluconazole was 3.84 to 2.77, respectively.
A summary of model simulations for CYP3A modulators along with the observed clinical DDI data is visually displayed in Figure S4.
Simulation of zanubrutinib as a DDI perpetrator
To assess the potential effects of zanubrutinib on the PK of substrate drugs, the PBPK model was also used to explore clinical DDI scenarios beyond those reported in the cocktail DDI study. Predicted PK parameters and the mean Cmax and AUC ratios (with/without zanubrutinib) of substrate drugs for CYP3A, CYP2C8, and CYP2B6 were simulated in Table S4. Findings from the simulation show that concomitant use of zanubrutinib has no impact or a limited impact on the exposure of both rosiglitazone, a CYP2C8 substrate, and bupropion, a CYP2B6 substrate. For repaglinide, a CYP2C8 and CYP3A4 substrate, simulations predicted a reduction in exposure of approximately 23% (on Day 10) following multiple doses of zanubrutinib (160 mg b.i.d.) when considering combined effects from CYP3A4 induction and CYP2C8 inhibition. The simulations predicted no impact on exposure when only CYP2C8 inhibition potential was considered. Additional simulations were also conducted at zanubrutinib 320 mg q.d.; Cmax and AUC ratios (with/without zanubrutinib) of midazolam were similar to those following the 160‐mg b.i.d. dose (~0.7 for 320 mg q.d.).
Simulation of effect of acid‐reducing agents on zanubrutinib exposure
To mimic physiologic changes following dosing with ARAs, simulated zanubrutinib Cmax and AUC0‐24 at gastric pH values of 1.5 and 4.5 were calculated (Figure 3). The predicted solubility of zanubrutinib in the stomach decreases from 0.25 to 0.13 mg/mL, with gastric pH value changing from 1.5 to 4.5; the corresponding predicted Cmax and AUC changes are minimal. Based on the PBPK simulations, ARAs such as proton pump inhibitors (PPIs) do not significantly impact the PK of zanubrutinib.
Figure 3.
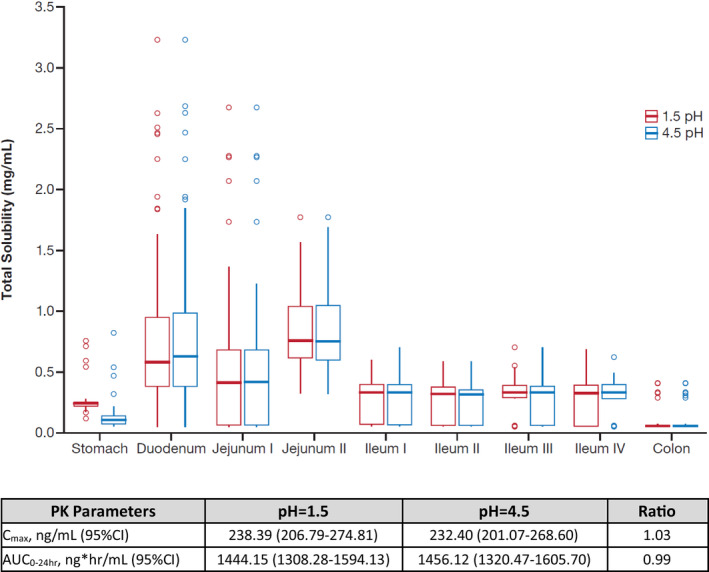
Predicted effect of gastric pH values (pH=1.5 and 4.5) on solubility and PK parameters of zanubrutinib. Abbreviations: AUC0‐24 hr, area under the plasma concentration–time curve from time 0 to 24 hours; CI, 95% confidence interval; Cmax, maximum plasma concentration; PK, pharmacokinetic; ratio, calculated by the ratio of pH=1.5 and pH=4.5.
DISCUSSION
The current work describes a model‐based approach for comprehensive evaluation of DDI profiles for zanubrutinib, including evaluation of its potential as a victim of CYP3A modulators or PPIs and as a perpetrator for CYP3A, CYP2C9, and CYP2C19 substrates. The PBPK approach adopted an example of the learning and confirming paradigm to enhance the efficiency of clinical development proposed by Sheiner et al. 15 The robustness of the current PBPK model was demonstrated by comparing model predictions with the observed clinical data following single and multiple doses of zanubrutinib ranging from 20 to 320 mg and from various drug interaction pathways. Results of the two clinical DDI studies, together with the PBPK model, support DDI dose recommendations for zanubrutinib. 16
In addition to supporting dose recommendations for clinical DDI scenarios, the developed PBPK model, which integrates all available in vitro and clinical data, continues to guide further understanding of zanubrutinib ADME profiles. Sensitivity analyses indicated that fmcyp3A4 appears to be the most sensitive parameter. An estimated fmCYP3A4 of 81.6% was used in the model, which is comparable to estimates based on experimental data from the in vitro phenotyping study (internal data). Based on the PBPK model, the estimated bioavailability of oral zanubrutinib is approximately 15%, with corresponding estimated values for Fa, Fg, and Fh of 0.7, 0.44, and 0.5, respectively. While a clinical absolute bioavailability study for zanubrutinib has not been conducted, these estimates appear to be reasonable approximations, as the current PBPK model was validated based on all available preclinical and clinical data for zanubrutinib.
Higher drug exposure has been reported in cancer patients relative to healthy subjects due to altered drug binding or reduced CYP3A4 abundance in the liver and gut. 17 , 18 , 19 In contrast to reported findings, zanubrutinib exposure appears to be 26.8% (Cmax) and 43.7% (AUC) lower in patients compared with that in healthy volunteers based on population PK analysis. 20 Thus, reported characteristics in cancer patients 17 , 18 , 19 were not included in the PBPK model. The median age in patient studies was 66 years, whereas the median age in healthy volunteer studies was 43 years. It is plausible that the absorption of zanubrutinib could be impacted by age‐related physiological changes in elderly cancer patients, such as reduced gastrointestinal motility and splanchnic blood flow. 21 , 22 Alternatively, the difference in zanubrutinib exposure could be partly due to the high PK variability in elderly patients with comorbidities who often receive multiple concomitant medications. Since model simulation was able to describe observed PK data in healthy volunteers and patients within pre‐defined criteria of 1.5‐fold of the observed values, the PK difference is expected to have a limited impact on DDI predictions.
The current simulation showed that effects of carbamazepine on the PK of zanubrutinib is modest and is similar to that of a moderate CYP3A inducer, efavirenz. Similar observations have also been reported. The simulated ribociclib AUC ratios (with/without inducer) from efavirenz, carbamazepine, and rifampicin were 0.40, 0.48, and 0.16, respectively. 23 Although rifampicin and carbamazepine are both characterized as strong inducers of CYP3A4, rifampicin is a much stronger CYP3A inducer than carbamazepine based on in vitro 24 and clinical data for certain CYP3A substrates. 25 , 26 , 27 , 28 Another observation based on the current simulation is that the magnitude of zanubrutinib exposure increases with fluconazole is approaching those of itraconazole, especially at the high dose of 400 mg. Of note, while itraconazole has a much lower Ki compared with that of fluconazole, the magnitude of interaction is a function of inhibitor concentrations at the target site relative to Ki [I/Ki] and fmCYP3A4. The fmCYP3A4 of zanubrutinib is about 0.8, so the fold increase in exposure (AUC ratio) as a function of [I/Ki] would be relatively flat compared with compounds that have fmCYP3A4 values of 0.9. All of these factors could account for the small difference in AUC ratio between fluconazole and itraconazole.
Following validation, the power of a PBPK model lies in its ability to inform other untested clinical scenarios. Based on results from clinical DDI studies that are supported by PBPK model simulations, the proposed dose reduction for zanubrutinib in the US Prescribing Information 16 is 80 mg q.d. and 80 mg b.i.d. when co‐administered with a strong or moderate CYP3A inhibitor, respectively. Zanubrutinib administration is not recommended with strong/moderate CYP3A inducers, and no dose reductions are required with weak CYP3A inhibitors/inducers. The proposed dose modification considers the extent of DDI within each inhibitor category and is intended to provide simplified dosing guidelines for each of these categories: a four‐fold reduction in dose with strong CYP3A inhibitors and a two‐fold reduction with moderate CYP3A inhibitors. Simulations showed that erythromycin (500 mg every 6 hours [q.6.h.]), a time‐dependent CYP inhibitor (moderate CYP3A inhibitor category), had a higher DDI impact on the steady‐state concentration of zanubrutinib compared with itraconazole (strong CYP3A inhibitor category). Similar results have been observed for other CYP3A substrates. 29 , 30 Effects of erythromycin on substrate drugs appear to be highly dependent on erythromycin dose and frequency. A 2.47‐ and 6.53‐fold increase in AUC of substrate drugs was observed following erythromycin 400 mg and 500 mg three times daily, respectively. 30 The current study used a more frequent dose of 500 mg q.6.h. In light of these model predictions, additional DDI and safety data for zanubrutinib during concomitant use of strong and moderate CYP inhibitors are being generated in an ongoing study in patients with B‐cell malignancies. Nevertheless, the proposed dose reduction appears to be supported by the exposure‐response analysis for zanubrutinib, which shows no apparent exposure‐response relationships for efficacy and safety endpoints over a wide range of zanubrutinib concentrations. 31
Zanubrutinib exhibits pH‐dependent solubility, although the change in solubility from pH 1.2 to pH 4.5 or pH 7.4 was only about three‐fold. Given that PPIs and other ARAs were allowed in clinical studies, the potential effects of ARAs on the PK of zanubrutinib were also assessed using PBPK analyses. Parrott et al 32 showed that PPIs can increase the gastric pH from 1 or 2, as reported in untreated individuals, to approximately 4.5. 32 , 33 Consistent with the approach used by other investigators, 34 the impact of ARAs (including PPIs) on zanubrutinib absorption was modeled by increasing the gastric pH from 1.5 to 4.5 in healthy volunteers (fasted model). The current model did not incorporate solubility data in biorelevant media, which would further increase the accuracy of PBPK simulations for ARAs. 35 Another approach would be to incorporate effects from dynamic pH changes as demonstrated by Pepin et al. 36 However, current results show minimal differences in zanubrutinib PK at the static pH value of 4.5 and 1.5. Thus, no clinically meaningful changes in zanubrutinib PK would be expected from dynamic changes within this pH range. PBPK simulations indicate that ARAs such as PPIs do not significantly impact the PK of zanubrutinib. This bottom‐up approach further corroborated results of the top‐down population PK analysis, 31 which showed that zanubrutinib exposure was not significantly impacted by concurrent ARA treatment. Together, results from these model‐based approaches were used to inform the language regarding the use of ARAs and PPIs in the zanubrutinib drug label.
Given the lack of available sensitive and validated probes for CYP2B6 for cocktail DDI studies, the PBPK simulations were used to predict interactions between zanubrutinib and CYP2B6 substrates. Of note, a dedicated clinical study with bupropion, a CYP2B6 substrate, is likely confounded by co‐induction of CYP3A by zanubrutinib. 8 A PBPK model was previously described for CYP2B6 clinical DDI prediction. 37 After adopting this compound file, the PBPK simulation indicated that the clinical CYP2B6 induction potential of zanubrutinib is predicted to be low. Based on in vitro data, assessment of R3 values according to FDA DDI guidance indicated that the induction potential for CYP2B6 (R3=0.55) is relatively weak compared with that for CYP3A (R3=0.23). A clinical study (BGB–3111–108) with the CYP3A‐sensitive substrate midazolam confirmed that zanubrutinib is a weak CYP3A inducer in vivo. 5 Apart from the PBPK modeling, an additional assessment was conducted based on the Consortium Working Group framework. 8 It has been noted that if new molecular entities exhibit CYP2B6 and CYP3A4 induction in vitro, a clinical CYP3A DDI study could serve as a surrogate for identifying the potential risk of CYP2B6 induction in clinical settings. If the CYP3A4 clinical induction study is negative or mild, it can be concluded that the likelihood of CYP2B6 clinical induction is low. Thus, the predicted low CYP2B6 DDI potential of zanubrutinib by PBPK appears to be consistent with available mechanistic drug interaction data.
The current work describes the development and validation process for a PBPK model integrating all available in vitro and clinical data for a comprehensive assessment of clinical DDI for zanubrutinib. Development of a PBPK model for zanubrutinib as a DDI victim and perpetrator in parallel synergistically supported the model verification process and can increase confidence in PBPK models. As the clinical development of zanubrutinib continues to expand, this validated PBPK model may be used to support future clinical programs, including studies related to the DDI potential of various drug combinations, dose regimens, and formulations, as well as studies in pediatric populations.
CONFLICT OF INTEREST
K.W. and Y.G. are employees of Shanghai Qiangshi Information Technology Co., Ltd. X.Y., M.Z., and D.L. declare no conflicts of interest. S.S. and Y.C.O. are employees and own stock in BeiGene, Inc.
AUTHOR CONTRIBUTIONS
Y.C.O. and K.W. wrote the manuscript. Y.C.O, and S.S. designed the research; K.W., X.Y., M.Z., D.L, Y.G., and Y.C.O. performed the research and analyzed the data. K.W., X.Y., M.Z., D.L contributed new analytical tools.
Supporting information
Table S1‐S4
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to acknowledge the clinical center study staff, the study patients, and their families. Special thanks to Bilal Tariq, PharmD, MS; Tristin Tang, PhD, and Nageshwar Budha, PhD, and Heather Zhang, PhD, for their assistance and input during model development. BeiGene, Ltd. provided financial support for this manuscript, including editorial assistance by OPEN Health Medical Communications, Chicago, IL.
Authors Kun Wang and Xueting Yao contributed equally to the work.
Funding information
The analysis plan was developed by BeiGene, Ltd. in conjunction with the collaborators in this study. BeiGene, Ltd. was also involved in data collection, model analysis, and interpretation of results.
DATA AVAILABILITY STATEMENT
Upon request, and subject to certain criteria, conditions, and exceptions, BeiGene will provide access to individual de‐identified participant data from BeiGene‐sponsored global interventional clinical studies conducted for medicines (1) with indications that have been approved or (2) in programs that have been terminated. BeiGene will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data requests may be submitted to medicalinformation@beigene.com.
REFERENCES
- 1. Guo Y, Liu Y, Hu N, et al. Discovery of zanubrutinib (BGB‐3111), a novel, potent, and selective covalent inhibitor of Bruton's tyrosine kinase. J Med Chem. 2019;62:7923‐7940. [DOI] [PubMed] [Google Scholar]
- 2. Tam CS, Opat S, D'Sa S, et al. A randomized phase 3 trial of zanubrutinib vs. ibrutinib in symptomatic Waldenström macroglobulinemia: the Aspen study. Blood. 2020;136:2038‐2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tam CS, Trotman J, Opat S, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B‐cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134:851‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mu S, Tang Z, Novotny W, et al. Effect of rifampin and itraconazole on the pharmacokinetics of zanubrutinib (a Bruton's tyrosine kinase inhibitor) in Asian and non‐Asian healthy subjects. Cancer Chemother Pharmacol. 2020;85:391‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ou YC, Tang Z, Novotny W, et al. Evaluation of drug interaction potential of zanubrutinib with cocktail probes representative of CYP3A4, CYP2C9, CYP2C19, P‐gp and BCRP. Br J Clin Pharmacol. 2020. 10.1111/bcp.14707. [Online ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shepard T, Scott G, Cole S, Nordmark A, Bouzom F. Physiologically based models in regulatory submissions: output from the ABPI/MHRA forum on physiologically based modeling and simulation. CPT Pharmacometrics Syst Pharmacol. 2015;4:221‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao P, Zhang L, Grillo JA. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011;89:259‐267. [DOI] [PubMed] [Google Scholar]
- 8. Fahmi OA, Shebley M, Palamanda J, et al. Evaluation of CYP2B6 induction and prediction of clinical drug–drug interactions: considerations from the IQ Consortium Induction Working Group—an industry perspective. Drug Metab Dispos. 2016;44:1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Howgate EM, Rowland YK, Proctor NJ, Tucker GT, Rostami‐Hodjegan A. Prediction of in vivo drug clearance from in vitro data. I: Impact of inter‐individual variability. Xenobiotica. 2006;36:473‐497. [DOI] [PubMed] [Google Scholar]
- 10. Darwich AS, Neuhoff S, Jamei M, Rostami‐Hodjegan A. Interplay of metabolism and transport in determining oral drug absorption and gut wall metabolism: a simulation assessment using the advanced dissolution, absorption, metabolism (ADAM; model. Curr Drug Metab. 2010;11:716‐729. [DOI] [PubMed] [Google Scholar]
- 11. Jamei M, Turner D, Jiansong Y, et al. Population‐based mechanistic prediction of oral drug absorption. AAPS J. 2009;11:225‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou D, Podoll T, Xu Y, et al. Evaluation of the drug‐drug interaction potential of acalabrutinib and its active metabolite, ACP‐5862, using a physiologically‐based pharmacokinetic modeling approach. CPT Pharmacometrics Syst Pharmacol. 2019;8:489‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y, Ma F, Lu T, et al. Development of a physiologically based pharmacokinetic model for itraconazole pharmacokinetics and drug–drug interaction prediction. Clin Pharmacokinetics. 2016;55:735‐749. [DOI] [PubMed] [Google Scholar]
- 14. Yamashita F, Sasa Y, Yoshida S, et al. Modeling of rifampicin‐induced CYP3A4 activation dynamics for the prediction of clinical drug‐drug interactions from in vitro data. PLoS One. 2013;8:e70330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sheiner LB. Learning versus confirming in clinical drug development. Clin Pharmacol Ther. 1997;61:275‐291. [DOI] [PubMed] [Google Scholar]
- 16. BeiGene USA, Inc . Brukinsa [package insert]. San Mateo, CA: BeiGene USA, Inc; 2019. https://www.brukinsa.com/prescribing‐information.pdf. Accessed December 1, 2020. [Google Scholar]
- 17. Cheeti S, Budhe NR, Dresser MJ, Jin KY. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm Drug Dispos. 2013;34:141‐154. [DOI] [PubMed] [Google Scholar]
- 18. Coutant DE, Kulanthaivel P, Turner PK, et al. Understanding disease‐drug interactions in cancer patients: implications for dosing within the therapeutic window. Clin Pharmacol Ther. 2015;98:76‐86. [DOI] [PubMed] [Google Scholar]
- 19. Schwenger E, Reddy VP, Moorthy G, et al. Harnessing meta‐analysis to refine an oncology patient population for physiology‐based pharmacokinetic modeling of drugs. Clin Pharmacol Ther. 2018;103:271‐280. [DOI] [PubMed] [Google Scholar]
- 20. Ou YC, Liu L, Tariq B, et al. Population pharmacokinetic analysis of the BTK inhibitor zanubrutinib in healthy volunteers and patients with B‐cell malignancies. Clin Transl Sci. 10.1111/cts.12948. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cusack BJ. Pharmacokinetics in older persons. Am J Geriatr Pharmacother. 2004;2:274‐302. [DOI] [PubMed] [Google Scholar]
- 22. Klotz U. Pharmacokinetics and drug metabolism in the elderly. Drug Metab Rev. 2009;41:67‐76. [DOI] [PubMed] [Google Scholar]
- 23. US Food and Drug Administration Center for Drug Evaluation and Research . Approval package for KISQALI. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/209092Orig1s001.pdf. Accessed 1 December, 2020.
- 24. Almond LM, Mukadem S, Gardner I, et al. Prediction of drug‐drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metab Dispos. 2016;44:821‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chung E, Nafziger AN, Kazierad DJ, Bertino JS Jr. Comparison of midazolam and simvastatin as cytochrome P450 3A probes. Clin Pharmacol Ther. 2006;79:350‐361. [DOI] [PubMed] [Google Scholar]
- 26. Ucar M, Neuvonen M, Luurila H, Dahlqvist R, Neuvonen PJ, Mjörndal T, et al. Carbamazepine markedly reduces serum concentrations of simvastatin and simvastatin acid. Eur J Clin Pharmacol. 2004;59:879‐882. [DOI] [PubMed] [Google Scholar]
- 27. Vlase L, Popa A, Neag M, Muntean D, Bâldea I, Leucuţa SE, et al. Pharmacokinetic interaction between zolpidem and carbamazepine in healthy volunteers. J Clin Pharmacol. 2011;51:1233‐1236. [DOI] [PubMed] [Google Scholar]
- 28. Villikka K, Kivisto KT, Luurila H, Neuvonen PJ. Rifampin reduces plasma concentrations and effects of zolpidem. Clin Pharmacol Ther. 1997;62:629‐634. [DOI] [PubMed] [Google Scholar]
- 29. de Kanter R, Sidharta PN, Delahaye S, et al. Physiologically‐based pharmacokinetic modeling of macitentan: prediction of drug–drug interactions. Clin Pharmacokinetics. 2016;55:369‐380. [DOI] [PubMed] [Google Scholar]
- 30. Wagner C, Pan Y, Hsu V, et al. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin Pharmacokinetics. 2015;54:117‐127. [DOI] [PubMed] [Google Scholar]
- 31. FDA summary basis of approval. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/213217Orig1s000MultidisciplineR.pdf. Accessed September 15. 2020.
- 32. Parrott NJ, Yu LJ, Takano R, Nakamura M, Morcos PN. Physiologically based absorption modeling to explore the impact of food and gastric pH changes on the pharmacokinetics of alectinib. AAPS J. 2016;18:1464‐1474. [DOI] [PubMed] [Google Scholar]
- 33. Rasmussen L, Oster‐Jørgensen E, Qvist N, Pedersen SA. The effects of omeprazole on intragastric pH, intestinal motility, and gastric emptying rate. Scand J Gastroenterol. 1999;34:671‐675. [DOI] [PubMed] [Google Scholar]
- 34. Einolf HJ, Lin W, Won CS, et al. Physiologically based pharmacokinetic model predictions of panobinostat (LBH589) as a victim and perpetrator of drug‐drug interactions. Drug Metab Dispos. 2017;45:1304. [DOI] [PubMed] [Google Scholar]
- 35. Dodd S, Kollipara S, Sanchez‐Felix M, et al. Prediction of ARA/PPI drug‐drug interactions at the drug discovery and development interface. J Pharmaceutical Sci. 2019;108:87‐101. [DOI] [PubMed] [Google Scholar]
- 36. Pepin XJH, Moir AJ, Mann JC, et al. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part II. A mechanistic PBPK model for IR formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. Eur J Pharm Biopharm. 2019;142:435‐448. [DOI] [PubMed] [Google Scholar]
- 37. Ke A, Barter Z, Rowland‐Yeo K, Almond L. Towards a best practice approach in PBPK modeling: case example of developing a unified efavirenz model accounting for induction of CYPs 3A4 and 2B6. CPT Pharmacometrics Syst Pharmacol. 2016;5:367‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mu S, Darpo B, Tang Z, et al. No QTc prolongation with zanubrutinib: results of concentration‐QTc analysis from a thorough QT study in healthy subjects. Clin Transl Sci. 2020;13:923‐931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ou YC, Preston RA, Marbury TC, et al. A phase 1, open‐label, single‐dose study of the pharmacokinetics of zanubrutinib in subjects with varying degrees of hepatic impairment. Leukemia Lymphoma. 2020;61:1355‐1363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S4
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material
Supplementary Material
Data Availability Statement
Upon request, and subject to certain criteria, conditions, and exceptions, BeiGene will provide access to individual de‐identified participant data from BeiGene‐sponsored global interventional clinical studies conducted for medicines (1) with indications that have been approved or (2) in programs that have been terminated. BeiGene will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data requests may be submitted to medicalinformation@beigene.com.