

Abstract

The major facilitator superfamily (MFS) is the largest known superfamily of secondary active transporters. MFS transporters are responsible for transporting a broad spectrum of substrates, either down their concentration gradient or uphill using the energy stored in the electrochemical gradients. Over the last 10 years, more than a hundred different MFS transporter structures covering close to 40 members have provided an atomic framework for piecing together the molecular basis of their transport cycles. Here, we summarize the remarkable promiscuity of MFS members in terms of substrate recognition and proton coupling as well as the intricate gating mechanisms undergone in achieving substrate translocation. We outline studies that show how residues far from the substrate binding site can be just as important for fine-tuning substrate recognition and specificity as those residues directly coordinating the substrate, and how a number of MFS transporters have evolved to form unique complexes with chaperone and signaling functions. Through a deeper mechanistic description of glucose (GLUT) transporters and multidrug resistance (MDR) antiporters, we outline novel refinements to the rocker-switch alternating-access model, such as a latch mechanism for proton-coupled monosaccharide transport. We emphasize that a full understanding of transport requires an elucidation of MFS transporter dynamics, energy landscapes, and the determination of how rate transitions are modulated by lipids.
1. Introduction
The transport of small molecules across cell membranes is essential for the healthy life of a cell. As small molecules diffuse poorly across membranes, nature has evolved a diverse set of membrane-integrated transporters to act as gatekeepers. In essence, these doors open in a controlled fashion to catalyze either the import or export of small molecules across biological membranes to maintain cell homeostasis and communicate information. Small-molecule transporters, commonly referred to as solute carrier (SLC) transporters, represent the second-largest fraction of the human membrane proteome after the G-protein coupled receptors (GPCRs).1,2 They are distinct from primary active transporters, which typically use ATP to translocate molecules across the membrane and against their concentration gradient and channels that catalyze the high flux of molecules down their electrochemical gradient.
In humans, there are currently ∼550 recognized SLC transporters which have to undergo multiple conformational states to translocate a single molecule across the cell membrane.3 While transporters can be structurally similar to channels, they are functionally more similar to enzymes, with activities described by Michaelis–Menten kinetics. Like enzymes, their activities become saturated at high substrate concentrations and they form the equivalent of a transition state, which is an intermediate (occluded) state, whereby access to the substrate binding site is simultaneously blocked off from either side of the membrane. The formation of the occluded state is of critical importance, and its formation ensures that the electrochemical gradients established by the cell are maintained.4 By coupling substrate translocation together with the movement of ions down their electrochemical gradient, substrates can be transported uphill. This process is referred to as secondary active transport, which can be further subclassified as either symport or antiport, depending on if the ion and substrate are moving in the same or in opposite directions, respectively.
Regardless of the source of energy, the foundation of the transport mechanism is that the substrate-binding site is alternatively exposed to only one side of the membrane at a time. The transporter’s mode-of-action is referred to as the “alternating-access model”,5 and so far, molecular details have shown that the alternating-access mechanism can be generally described by three types of models: the “rocker-switch”, the “rocking-bundle,” or the “elevator” mechanism (Figure 1).3 During the past decade, a combination of transporter structures in multiple functional states together with biochemical analysis, biophysical analysis, and in silico molecular dynamics (MD) calculations, have enabled us to interpret more specific details of their transport mechanisms. In this review, we explore the structure, function, regulation, dynamics, oligomerization, and complexes of the major facilitator superfamily (MFS). In particular, we use sugar porters (SP) and multidrug resistance (MDR) transport members, as model systems for describing alternating-access mechanisms at a deeper level. We also highlight the importance of lipids and recent research that breaks the paradigm that all MFS transporters are “transporters” but can have dedicated signaling functions.
Figure 1.
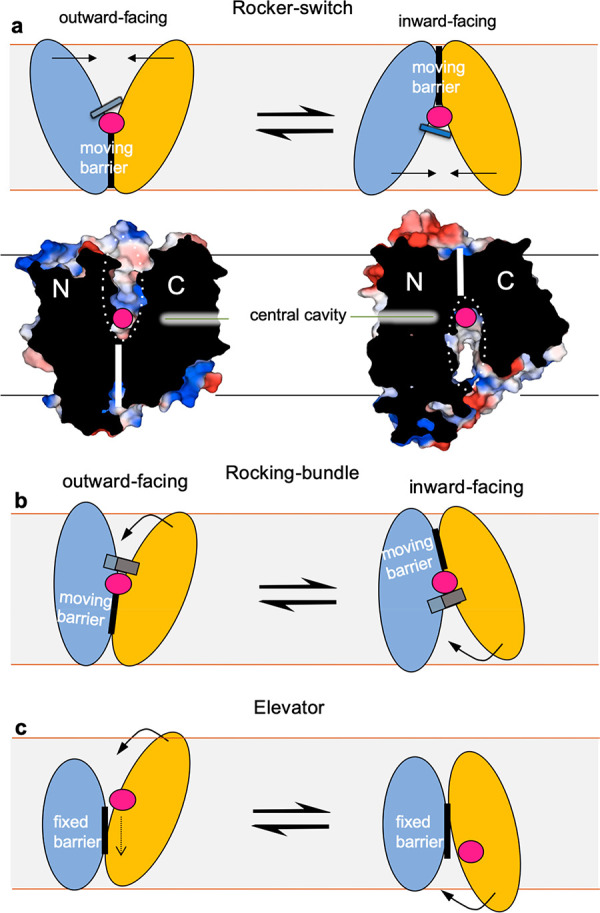
The three different SLC transporter models for alternating-access. (a) In the rocker-switch mechanism, the structurally similar bundles (N(light-blue)- and C(light-orange)-terminal bundle) rearrange symmetrically around the centrally located substrate-binding site (substrate shown as pink sphere) to alternate access to the other side of the membrane. This is the alternating access mechanism used by MFS transporters, as depicted here below in the surface representation of the fructose transporter GLUT5 in the apo outward- and inward-facing conformations (PDB 4YBQ, 4YB9). (b) In the rocking-bundle mechanism, the structurally dissimilar bundles rearrange asymmetrically around the centrally located substrate-binding site to alternate access to either side of the membrane. An example of a rocking-bundle transporter is the sodium-coupled neurotransmitter symporters harboring the LeuT-fold,6 wherein the C-terminal bundle undergoes large, local gating rearrangements to coordinate sodium and substrate to rock around the less labile N-terminal bundle. The less labile N-terminal bundle is typically referred to as the “scaffold” and the C-terminal bundle as the “transport” domain. Both rocker-switch and rocking-bundle models can further be referred to as operating by a moving barrier mechanism as originally postulated by Peter Mitchell,7,8 where the barrier between the two bundles (thick vertical lines) moves from the inside to outside during substrate translocation. The two end states shown here are further connected by intermediate conformations that involve local gating rearranges around the substrate, as illustrated here by a gray thin line for rocker-switch proteins and a gray thick line for rocking-bundle proteins. (c) In the elevator mechanism, the two bundles are highly divergent and the substrate (pink sphere) is transported across the membrane by only the C-terminal bundle, whereas the N-terminal remains fixed, typically due to oligomerization. An example of an elevator transporter is the sodium-coupled glutamate transporter harboring the GltPh-fold,9 wherein the C-terminal bundle carries the substrate by a vertical distance of approximately 18 Å across the membrane against the immobile N-terminal bundle. The immobile N-terminal bundle is typically referred to as the “scaffold” and the C-terminal bundle the “transport” domain. In reference to the moving-barrier mechanism, the elevator mechanism has also been referred to as a fixed-barrier mechanism,3 as the protein does not rearrange around the substrate. Instead, the N-terminal bundle forming the barrier stays fixed. Substrate binding and release in each state are likely facilitated by local gating transitions in the transporter domain (thick gray line). Adapted from Drew and Boudker.3
2. The Major Facilitator Superfamily (MFS) Topology
MFS is the largest and most diverse superfamily of secondary active transporters found ubiquitously in all living organisms.10 MFS members are thought to be one of the oldest protein families on Earth, being present more than 3 billion years ago.10 The Transporter Classification Database (TCDB, http://www.tcdb.org), which includes recognized and hypothetical membrane transport proteins, classifies the MFS based on phylogeny and function into 16 different families (Table 1), with 89 subfamilies and 1244 annotated proteins. With the exception of three integral membrane proteins that were recently classified as MFS members but are not currently recognized as transporters,11 all of the proteins are known or assumed transport proteins. The Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC) has curated human genes into larger families based on their function, homology, or phenotype.12 To date, genes coding for human membrane transport SLC systems represent 65 families (http://slc.bioparadigms.org/), and 16 of these belong to the MFS (Table 1). In humans, this is the largest cluster of phylogenetically related SLCs.13 The MFS transporters are somewhat easier to identify than other SLC members as they have a very distinct topology. The canonical MFS-fold has 12 transmembrane (TM) segments organized from two 6-TM bundles connected by a long and flexible intracellular loop (Figure 2).10,14,15
Table 1. MFS Family Classifications.
| TCDB | ||
|---|---|---|
| TCDB ID | family description | proteins |
| 2.A.1 | the major facilitator superfamily (MFS)a | 942 |
| 2.A.2 | the glycoside-pentoside-hexuronide (GPH):cation symporter family | 45 |
| 2.A.12 | the ATP:ADP antiporter (AAA) family | 21 |
| 2.A.17 | the proton-dependent oligopeptide transporter (POT/PTR) family | 46 |
| 2.A.48 | the reduced folate carrier (RFC) family | 6 |
| 2.A.57 | the equilibrative nucleoside transporter (ENT) family | 42 |
| 2.A.60 | the organo anion transporter (OAT) family | 29 |
| 2.A.71 | the folate-biopterin transporter (FBT) family | 12 |
| 2.A.85 | the aromatic acid exporter (ArAE) family | 33 |
| 2.A.100 | the ferroportin (Fpn) family | 9 |
| 2.A.125 | the eukaryotic riboflavin transporter (E-RFT) family | 5 |
| 4.H.1 | the lysyl-phosphatidylglycerol synthase/flippase (MprF) family | 14 |
| 5.B.2 | The Eukaryotic Cytochrome b561 (Cytb561) Family | 18 |
| 9.B.57 | the conidiation and conidial germination protein (CCGP) family | 5 |
| 9.B.111 | the 6 TMS lysyl tRNA synthetase (LysS) family | 5 |
| 9.B.143 | the 6 TMS DUF1275/Pf06912 (DUF1275) family | 22 |
| SLC
tables | ||
|---|---|---|
| SLC ID | family description | membersb |
| SLC2 | facilitative GLUT transporter family | 14 |
| SLC15 | protein oligopeptide cotransporter family | 4 |
| SLC16 | monocarboxylate transporter family | 14 |
| SLC17 | vesicular glutamate transporter family | 9 |
| SLC18 | vesicular amine transporter family | 4 |
| SLC19 | folate/thiamine transporter family | 3 |
| SLCO/SLC21 | organic anion transporter family | 11 |
| SLC22 | organic cation/anion/zwitterion transporter family | 23 |
| SLC29 | facilitative nucleoside transporter family | 4 |
| SLC33 | acetyl-CoA transporter family | 1 |
| SLC37 | sugar–phosphate/phosphate exchanger family | 4 |
| SLC40 | basolateral iron transporter family | 1 |
| SLC43 | Na+-independent, system-L like amino acid transporter family | 3 |
| SLC45 | proton/sugar cotransporter family | 4 |
| SLC46 | folate transporter family | 3 |
| SLC49 | FLVCR-related transporter family | 4 |
Contains 89 subfamilies.
Not including pseudogenes: the classification according to the Transporter Classification Database (TCDB) and the human solute carrier (SLC) gene tables (SLC tables).30
Figure 2.

Schematic representation of the canonical MFS-fold transporter topology. The canonical MFS topology is comprised of 12 TMs with a Nin and Cin orientation, forming two structurally similar six-helix bundles. The N-terminal bundle (TM1–6; light-blue/blue) and the C-terminal bundle (TMs 7–12; light-orange/red) are connected together by a cytosolic loop, which can sometimes contain structural elements (gray). Each of the bundles are made up from 3-TM structural-inverted repeats. The first TM in each of the 3-TM repeats (TM1, TM4, TM7, and TM10) form the central cavity helices and often undergo local changes to bind and release the substrate (yellow pentagon) during alternating access.
The first crystal structures describing the MFS-fold were that of the proton (H+)-coupled lactose symporter LacY16 and the glycerol-3-phosphate–phosphate antiporter GlpT from Escherichia coli.17 The crystal structures confirmed that the two 6-TM segments adopt separate structural entities with the substrate binding pocket located between the two bundles as seen in earlier low resolution projections from 2D crystal structures of OxlT18 and MelB19 and reminiscent of the transport schematics by Peter Mitchell in the 1950s (Figure 1a).7 Indeed, prior to the availability of solved structures, it has been shown that it was possible to reconstruct a functional LacY transporter by expression of the N- and C-terminal 6-TM bundles separately.20
Although the two structurally similar 6-TM bundles are related by symmetry perpendicular to the plane of the membrane, it is not thought that MFS members have evolved from gene fusions of two 6-TM segments.14 Rather, each 6-TM bundle is itself made up of two 3-TM segments related by a 180° rotation running parallel to the plane of the membrane (Figure 2a).21,22 Indeed, structural-inverted repeats are found in all transporter folds.23 Even simple non-MFS transporters, such as the 4-TM multidrug transporter EmrE, inserts itself into the membrane in two different orientations (Nin, Cin, and Nout, Cout) to form functional homodimers.24,25 Such “dual-topology” membrane proteins have homologues that have fused genes with opposite topologies and therefore provide evidence of how larger transporter folds could have evolved from smaller subunits.26 Most likely, the evolutionary pressure to evolve gene fusions was to increase substrate diversity3 and the complexity of coupling substrate binding and transport. Although it is generally accepted that three-helix bundles were likely the evolutionary origin of the MFS, a 3-TM ancestor has not yet been identified.27 Intriguingly, it was predicted that a small fraction of MFS members have only 11 TMs,14 and this was most recently confirmed by the crystal structure of the equilibrative nucleoside transporter ENT1.28 The 11-TM topology has mostly arisen from a 12-TM ancestor that has lost the last TM segment.29 The evolutionary and functional reason for this structural divergence in unclear.
3. MFS Transporter Physiology
Secondary-active MFS symporters and antiporters energize transport by (typically) utilizing the proton motive force (PMF), also known as the proton electrochemical gradient (ΔμH+), which is made up of the proton concentration difference (ΔpH) and the membrane potential (Δψ). If net transport results in a charge difference across the membrane, then the transporter is considered “electrogenic”; if the net charge difference is zero, then the transport is “electroneutral” and driven solely by ΔpH.31 Although the MFS-fold appears fairly simple at first glance, this robustness has nevertheless arisen to be the most popular secondary active transporter fold used by the cell and is widely acknowledged for its ability to transport highly diverse substrates.10 This includes, but is not limited to, a variety of sugars, polyols, drugs, neurotransmitters, amino acids, peptides, lipids, organic and inorganic ions, vitamins, nucleobases, nucleosides, and nucleotides.10,15,32 Although it appears that macromolecules such as polysaccharides, nucleic acids, and proteins are not transported by MFS proteins,29 a recent study suggests an MFS transporter from Drosophila melanogaster was responsible for the presentation of the truncated core1 O-glycan T-antigen.33
Most MFS members consist of 400–600 amino acid residues and share low sequence conservation of 12–18%.34 As expected, MFS members from the different subfamilies show greater sequence divergence.10 MFS transporters are pivotal at the cellular level for growth, metabolism, and homeostasis across all kingdoms of life. A large number of MFS members (>25) are present across all kingdoms for sugars (sugar porters, TC no. 2A.1.1; glycoside-pentoside-hexuronide:cation symporters, TC no. 2.A.2), drugs and other hydrophobic substrates (drug:proton (H+) antiporters 1 and 2, TC nos. 2.A.1.2 and 2.A.1.3, respectively), inorganic or organic anions or cations (anion:cation symporters, TC no. 2.A.1.14; organic cation transporters, TC no. 2.A.1.19; organo anion transporters, TC no. 2.A.60), peptides (proton-dependent oligopeptide transporters, TC no. 2.A.17), nucleosides (equilibrative nucleoside transporters, TC no. 2.A.57), and aromatic acids (aromatic acid exporters, TC no. 2.A.85). Although, the TCDB lists more than 80 MFS subfamilies, only a number of MFS subfamilies, in particular, sugar porters (SP), drug:proton (H+) antiporter-1 and -2 (DHA1, 2), anion:cation symporter (ACS), organic cation transporter (OCT), and the proton-dependent oligopeptide transporter (POT/PTR) family are present across bacteria, yeast, human, and plants.35,36 Thus, many MFS transporter subfamilies have evolved specific functions in their respective domains.
MFS transporters are pivotal at the cellular level for growth, metabolism, and homeostasis in all organisms. There are over 100 human MFS transporter genes classified in the HGNC (Table 1).12 In human physiology, the SLC transporters of the MFS superfamily are involved in the transport of nutrients, metabolites, and other substrates between both cells and their intracellular compartments (Figure 3). This is important in a multitude of critical processes such as development, neurotransmission, signaling, nutrient absorption, and renal and hepatic clearance.10,37 Thus, it is not surprising that defects in MFS transporters are associated with a plethora of serious diseases such as cancers and metabolic diseases.38,39
Figure 3.

The distribution of human MFS-type SLCs and atypical SLCs. The proportion of MFS transporters have been grouped according to their SLC designation (https://www.bioparadigms.org) (left). Each wedge is labeled with its respective short-hand TCDB classification (http://www.tcdb.org). The atypical SLCs include proteins that are described as SV2 (synaptic vesicle protein 2), SVOP (synaptic vesicle-2 related protein), MFSD (MFS domain containing proteins), SPNS (spinster homologue), Unc93 (Unc93 homologue) and CLN3 (ceroid lipofuscinosis, neuronal 3). The atypical SLCs have been classified or are currently unclassified into a number of TCDB groups (right). The TCDB abbreviations are as follows: the sugar porter (SP) family, the proton-dependent oligopeptide transporter (POT) family, the monocarboxylate transporter (MCT) family, the anion:cation symporter (ACS) family, the drug:H+ antiporter-1 (DHA1) family, the reduced folate carrier (RFC) family, the organo anion transporter (OAT) family, the organic cation transporter (OCT) family, the equilibrative nucleoside transporter (ENT) family, the peptide/acetyl-coenzyme A/drug transporter (PAT) family, the organophosphate:Pi antiporter (OPA) family, the ferroportin (FPN) family, the l-amino acid transporter-3 (LAT3) family, the glycoside-pentoside-hexuronide (GPH):cation symporter family, the proton coupled folate transporter/heme carrier protein (PCFT) family, the feline leukemia virus subgroup C receptor (FLVCR)/heme importer family, the unidentified major facilitator-14 (UMF14) family, the plant copper uptake porter (PI-Cu-UP) family, the endosomal spinster (Spinster) family, and the N-acetylglucosamine transporter (NAG-T) family.
In mammals, the physiological roles of MFS transporters are very diverse. For example, ferropotin (FPN1, also referred to as SLC40A1) is considered to play a major role in cellular iron homeostasis (Figure 3).40 After dietary iron has been absorbed into the cells of the small intestine, FPN1 transports the iron from the cells of the small intestine into the bloodstream. FPN1 also functions in macrophages, allowing the iron to be recovered from the broken-down cells to be released back into the bloodstream for reuse. Because FPN1 is ubiquitously expressed but is the only known iron exporter in mammals, the absorption and recycling of iron can be centrally regulated by its expression. A number of mutations in FPN1 are associated with ferroportin diseases.41 The peptide hormone hepcidin, secreted by the liver, binds to FPN1 and directs it to intracellular lysosomes, leading to its degradation, thus hepcidin regulates iron absorption and utilization by regulating the expression of ferroportin,42 indicating FPN1 and its modulation by hepcidin has been an attractive therapeutic target for treating ferroportin diseases.43
Another example of the physiological role of an MFS transporter is the mammalian vesicular monoamine transporters (VMAT) belonging to the SLC18 family, which have broad substrate specificities and uptake all the monoamine neurotransmitters, e.g., dopamine, serotonin, norepinephrine, epinephrine, and histamine, into presynaptic neuronal vesicles (Figure 3).44,45 Notably, the monoamine neurotransmitters are first taken from the neurosynaptic cleft and into the presynaptic neurons by highly specific non-MFS SLC6 transporters belonging to the sodium-coupled neurotransmitter superfamily, e.g., serotonin transporter (SERT), dopamine transporter (DAT), and norepinephrine transporter (NET).46 The active transport of cytosolic monoamines into storage vesicles, against a high concentration gradient is driven by a transmembrane pH and electrochemical gradient generated by the vesicular H+-ATPase (V-ATPase) in the granule membrane. A number of studies suggest VMATs have a critical role in neuronal and endocrine informational output by fine-tuning sorting, storing, and releasing of neurotransmitters.47,48
The MFS vesicular glutamate transporter (VGLUT) belonging to SLC17 family is also playing a critical role in neurotransmission. VGLUT is responsible for loading of glutamate into synaptic vesicles (Figure 3).49 Three isoforms of VGLUT (VGLUT1–3) exist. The two major isoforms, VGLUT1 and VGLUT2, exhibit complementary expression in the cortex and diencephalon, respectively.50 Glutamate is the main excitatory neurotransmitter in the central nervous system and also in various peripheral tissues. VGLUTs utilize the positive-inside membrane potential established by the V-ATPase to concentrate the negatively charged glutamate about 10-fold against the H+-gradient and is also nonstoichiometrically linked to a channel-like anion (Cl–) conductance.51,52 Interestingly, the recent cryo-EM structure of VGLUT2 together with previous biochemical analyses,53,54 indicates that positively charged glutamate binding residues intersects with a potential Cl– channel. From a functional perspective, VGLUT differs greatly from the VMAT, which are antiporters that utilize the outwardly directed H+ to uptake monoamines against their concentration gradient by up to 10 000-fold.55
In plants, MFS members dominate the transport of carbon and nitrogen. In Arabidopsis thaliana, for example, sugar porter (SP) and the nitrate transporter 1/peptide transporter (NRT1/PTR) members make up 60% of all MFS transporters.56 Just like in mammals, sugar porter isoforms are likely to have differences in sugar preferences, kinetics, and tissue localizations, e.g., four out of the 47 sugar porter members are specific to pollen.56 Interestingly, some of the NRT1 members have evolved to preferentially transport compounds very different from nitrate and peptides, such as phytohormones or glucosinolates.57 Phosphate is also a major substrate of the plant MFS, where there are currently three families comprising 18 members in total, involved in phosphate uptake/translocation.56 In bacteria, MFS transporters are important for the uptake of nutrients, and the extrusion of harmful compounds such as antibiotics and heavy metals.58 For example, 19 out of the 37 putative multidrug resistance (MDR) transporter genes in E. coli belong to the MFS.59 As antimicrobial resistance poses an enormous threat to public health, they have been targeted by antibacterial approaches in clinically relevant bacteria.
3.1. MFS Transporters As Drug Targets
In human, a number of MFS members are drug targets or already have FDA-approved drugs targeting them.60,61 A subset of MFS transporters belonging to the SLC22 family are further classified as drug transporters, as they effect the pharmacokinetics of many orally administrated drugs (Figure 3).62 More specifically, the MFS drug transporters belong to two separate clades of the SLC22 family; the organic anion transporters (OATs) and the organic cation transporters (OCTs), which are expressed in the intestine, liver, brain, and kidney.62 Roughly 30 out of the 32 SLC22 transporters have broad substrate specificity, ranging from organic anions to organic zwitterions and organic cations, as well as other molecules.62 This promiscuity enables them to transport a diverse range of compounds, including bile acids, steroid conjugates, thyroid hormones, anionic peptides, as well as numerous drugs, e.g., statins, nonsteroidal anti-inflammatory (NSAID) drugs, and other xenobiotic substances.63 Their importance to the pharmaceutical industry is highlighted by the fact that the FDA recommends several of these MFS transporters are to be tested for the transport of new drugs.63
Other MFS proteins that are well-known to be important to human health and disease are the oligopeptide transporters (PepT1 and PepT2; SLC15),64,65 the monocarboxylate transporters (MCT; SLC16),66,67 and the glucose (GLUT; SLC2)68,69 transporters (Figure 3). The oligopeptide transporters are highly expressed in the intestine and have been shown to be able to aid the absorption of orally administrated drugs.64 Indeed, drugs such as antivirals valacyclovir and valganciclovir have been modified into so-called “pro-drugs” to improve their adsorption by the intestinal oligopeptide transporter PepT1.64,70,71 MCTs are required in the H+-dependent transport of l-lactate, pyruvate, and monocarboxylate drugs.66,72 Many cancer cells have increased glucose consumption and derive ATP primarily by aerobic glycolysis, which is referred to as the Warburg effect.73,74 The increased lactate is exported by the low-affinity lactate transporter MCT4 and taken up again into other cells by the high-affinity lactate transporter MCT1.67,75 Thus, both MCT1 and MCT4 are targets for anticancer drugs.75,76 The passive glucose (GLUT) transporters have an essential role in maintaining whole-body glucose homeostasis and are also highly expressed in many tumors and metastases.74,77 In addition to cancer, aberrant functioning of various GLUTs is also linked to many other diseases such as type 2 diabetes, obesity,78 GLUT1 deficiency syndrome, referred to as De Vivo disease,79,80 and Fanconi–Bickel syndrome.81
3.2. Orphan MFS Transporters
Despite the importance of MFS transporters to cellular physiology and drug development, up to 30% of SLC transporters in the human genome are still considered orphans in that their function remains unknown (Figure 3).60 Of the orphan SLCs, there is a small subset of transporters that are referred to as “atypical SLCs”. These transporters have sequences similar to SLCs,30,82 but they deviate to such an extent that they were “missed” during annotation of the SLC family. The majority of atypical SLCs belong to the MFS (currently 28 out of 30) and are sometimes found annotated as MFS domain (MFSD) containing proteins. A number of these MFSD proteins still cannot be clearly classified (Figure 3). SLCs have well-established roles in the etiology and treatment of several human diseases,1,2,60,83,84 however, the importance of these MFSD proteins to human health is unknown. Indeed, deorphanization of MFS transporters remains a large challenge. Furthermore, even if a transporter has been shown to transport a particular substrate, it is still unclear if this is the physiological substrate and/or if there are others. For example, human have 14 different glucose (GLUT) transporter isoforms GLUT1–GLUT14, and although most are thought to transport d-glucose, it is unclear why there are so many different isoforms that appear to have overlapping substrate preferences and kinetics.68 For example, the isoform GLUT5 is thought to be the only isoform specific in the transport of d-fructose, but it is also unclear why GLUT5 is expressed in the brain where the levels of circulating fructose are very low.85
4. MFS Transporter Methods and Characterization Approaches
The difficulty in characterizing substrates of MFS transporters is several-fold. In general, their intrinsic dynamics and large conformational changes undergone during transport86 means that they are often unstable in detergent solubilized solution,87 making them difficult to purify. Furthermore, unlike other highly dynamic membrane proteins, such as GPCRs that typically bind ligands with high affinity (nM range) and specificity,88,89 MFS transporters often bind their substrates weakly (high μM to mM range) so that they can be transported across membranes at physiologically relevant concentrations,68,70,90−92 e.g., blood d-glucose levels needs to be maintained around 5–7 mM.68 The low affinity of the transporter for their substrates means binding assays are difficult to carry out (see ref (93)) and often requires that the protein is first purified to remove interference from endogenously expressed transporters where binding can be assessed, e.g., using scintillation proximity assays (SPA).94−96 Even if it is possible to measure the substrate binding, transport needs to be further assessed as binding does not guarantee transport.3 If the cell has additional transporters with overlapping activities and/or the substrate being analyzed is metabolized, it is not always possible to test transport activities in cell-based assays.97 In this regard, the yeast Saccharomyces cerevisiae is often a useful expression host as its relatively easy to genetically manipulate in the removal of competing transporters.98 The use of Xenopus oocytes is also a useful expression host due to its limited competition with endogenous transport activity in the oocyte plasma membrane, e.g., glucose (GLUT) transporters,99,100 but like yeast, transporters may not be functional in Xenopus oocytes as they may require mammalian cells due to the requirement for certain lipids and/or complex N-linked glycosylation for folding.101 Cell-based assays further have the limitation that it is not possible to control the internal environment, which might be critical for functional characterization of antiporters, for example. This is particularly an issue if the MFS transporters is localized to an internal compartment. Moreover, while inhibitors are often used to validate the transport catalyzed by a specific transporter, it can never be excluded that inhibitors may have off-target effects.102
The reconstitution of a purified MFS transporter into liposomes and the measurement of solute uptake into proteoliposomes is considered the gold standard for validation of a substrate transporter pairing (Figure 4).103,104 A proteoliposome transport assay also makes it possible to analyze the energetics and kinetics of vectorial transport in a controlled fashion and is therefore necessary for detailed mechanistic analysis. For instance, it becomes possible to apply different driving forces to catalyze substrate transport, such as either the application of Δp (PMF), ΔpH, Δψ, or under counterflow or exchange conditions (Figure 4a).105,106 Under Δp-driven conditions, substrate accumulation of a H+-coupled symporter, for example, will require intact H+ translocation as it does in vivo. Yet mutations that abolish H+ translocation might still be able to carry out some of the steps in the transport cycle and are still active in counterflow transport.90,106 To measure counterflow transport, proteoliposomes are preloaded with a high concentration of the unlabeled substrate and then diluted into a transport buffer containing the radiolabeled substrate.90 The radiolabeled substrate will only accumulate during resetting of the transporter on the outside if a successful substrate-binding event has first taken place on the inside, i.e., transport is able to be driven by passive efflux of the unlabeled substrate. However, residues that need to be protonated, in order for substrate to bind, will be defective under both Δp-driven and counterflow conditions. As such, one can use these transport methods, for example, to help establish which residues are likely required for H+ translocation only versus what residue(s) are required for both H+ translocation and substrate binding.107−109 One can further determine substrate: ion stoichiometries and apply separate ΔpH or Δψ gradients to establish if the overall net transport is either electroneutral or electrogenic, as only the later can transport be driven solely by a membrane potential (Figure 4a). As will be discussed later, guided by structural details and by the manipulation of different driving forces and mutagenesis, one can then begin to establish meaningful mechanistic mechanisms that are not readily tractable in most cell-based assays, if at all. At a pragmatic level, proteoliposome-based assays are further amenable to inhibitors that might otherwise be toxic to cells and is further of critical importance to confirm transport of drugs that may have off-target recognition and are difficult to validate in vivo,102 such as those transported by promiscuous organic-anion transporters (OATs), for example.
Figure 4.

Proteoliposome transport assays. (a) Schematic representations of different proteoliposme transport assays. (top left) Uptake of a radiolabeled substrate (green pentagon) is driven by an inwardly directed pH gradient for a H+-coupled symporter or without for a passive uniporter (zero trans). (top right) As described in the left panel except for H+-coupled transporters, a nonlabeled substrates can alternatively be used and H+ transport is measured by pH sensitive fluorescent dyes (e.g., either 9-amino-6-chloro-2-methoxyacridine (ACMA) (star) or pyranine). (bottom left) The F-type ATPase can be co-reconstituted with the transporter into liposomes to create a proton motive force. Such a setup has been used to measure the uptake of l-glutamate by VGLUT, which requires a positive inside membrane potential and natively colocalizes in synaptic vesicles with the V-type ATPase.121 Alternatively, K+ and valinomycin can be added to dissipate the electrical potential generated by the ATPase to meaure transport activity with an outwardly directed pH gradient only. (bottom right) Proteoliposomes can be loaded with KCl, and the addition of valinomycin establishes a negative inside potassium-diffusion potential, which can be used to drive either antiporters or symporters that are electrogenic. (b) Detergent mediated-reconstitution method. The detergent purified transporter (blue, orange) is mixed together with liposomes (blue), which are typically small unilamellar vesicles premade by rehydration of multilamellar stacks of crude membranes by freeze–thawing and sonication. To facilitate reconstitution, the liposomes are destabilized with a low concentration of detergent such as Triton X-100 or cholate. The detergent is subsequently removed from the mixture, leading to the incorporation of the transporter into the liposomes. The detergent can be removed by a number of different methods as shown here. For detergents with a high critical micelle concentration (CMC) such as sodium cholate or octyl-β-glucoside (OG), methods such as rapid dilution, dialysis or size exclusion chromatography are typically used. For detergents with a low CMC, adsorption to polystyrene biobeads in combination with dialysis is more favored. However, rapid-dilution methods have also been successfully applied to GLUT transporters purified in low CMC detergents like DDM.121 Multiple rounds of freezing in liquid nitrogen and thawing helps to make uniform proteoliposomes and is most often later combined with extrusion through filters of an appropriate size (ca. 200–400 nm).
Historically, the freeze–thaw sonication procedure has been used, which consists of freezing a mixture of liposomes and a transport protein solubilized in a nonionic detergent and then slowly thawing these samples to enable reconstitution (Figure 4b).102,110 The addition of detergent such as Triton X-100 destabilizes the liposome and would often support efficient reconstitution (Figure 4b).105,111 However, such detergent medicated reconstitution procedures involve “co-micellization” of the purified membrane protein in an excess of phospholipids and detergent to form a solution of mixed lipid–protein–detergent and lipid–detergent micelles. Therefore four methods, i.e., dialysis, dilution, size exclusion chromatography (SEC), and biobeads methods have been widely used to remove detergents for the formation of closed lipid bilayers in which the proteins eventually incorporate.104 Many examples of proteoliposome-based assays have been carried out with bacterial MFS transporters, which has provided a wealth of mechanistic insights.90,105,112 Unfortunately, there are not as many examples of mammalian MFS transporters reconstituted into proteoliposomes.113−118 The reason for this discrepancy is that it is still challenging to purify mammalian MFS transporters, as they are often unstable in detergent solution.119 Even if one can isolate functional material, it may take many months or even years to optimize the proteoliposome transport assay so that it is robust enough for comparing the effect of mutations.120 Anecdotally, it seems that the mammalian MFS transporters might be more sensitive to the lipid composition than their bacterial homologues,119 adding a further layer of complexity in the development of a robust functional assay in proteoliposomes (Figure 5a).
Figure 5.

GFP-TS assay and comparing the lipid stabilization of bacterial versus eukaryotic transporters. (a) Box-and-whisker plots show the distribution of thermostabilities for eukaryotic transporter (red bars) and bacterial transporters (black bars) before and after purification as assessed by the GFP-TS assay; the median is shown as a line in the box, while bottom and top boundaries represent the lower and upper quartile, respectively. Whiskers indicate the minimum and maximum apparent Tm. (b) Schematic representation of the GFP-TS assay for monitoring ligand interactions, including lipids. (c) The GFP-TS melting curves for the bacterial monosaccharide transporter XylE (left) and rat GLUT5 (right) in crude detergent solubilized membranes (black circles) and as a purified fusion (cyan squares). Error bars show the range of two technical replicates, and the values reported for the apparent Tm are the mean ± SEM of the fit. (d) Supernatant fluorescence of detergent purified rat GLUT5-GFP before heating at apparent Tm +5 °C (nonfilled bars) and that remaining after heating and centrifugation (black bars) in the presence of listed lipids solubilized in the same detergent or detergent only (control); the asterisk indicates the most stabilizing lipid (bars show the range of two technical replicates). (e) GFP-TS melting curves for purified rat GLUT5 in the absence (black) and presence of brain lipids (cyan); apparent Tm were calculated as described in (b), and the values reported are the mean ± SEM of the fit. Reproduced with permission from ref (119). Copyright 2018 Nature.
In addition to the difficulties in optimizing a robust proteoliposome assay, one cannot further control the orientation of the transporter during reconstitution into liposomes, which can even be influenced by many factors including the detergent used during reconstitution and its rate of removal.122,123 Because substrate binding affinities can be different on either side of the transporter a biased orientation, a mixed orientation could lead to differences in transport kinetic parameters (KM, Vmax) or in the case of competitive inhibitors, Ki and IC50 estimates, i.e., the substrate binds with higher affinity on the outside versus the inside.124 This is not always the case, even for similar types of MFS transporters and has to be experimentally tested. For example, while the facilitative glucose transporter GLUT1 binds its substrate with 10-fold higher affinity on the outside,125,126 for the H+-coupled lactose sympoter LacY, in the absence of a H+ electrochemical gradient, galactoside affinity is essentially identical on both sides of the symporter.127 Typically, the ratio of inside:outside in proteoliposomes is estimated based on cysteine or protease accessibility and, in many cases, the orientations are fairly even.123 In E. coli at least, it is also straightforward to directly isolate vesicles with preferred right-side or inside-out orientations and, if there are no endogenous competing systems, they can also be used for transport measurements with the same driving forces applied.128
A major limitation to the deorphanization of MFS and SLC transporters in general is that the direct measurement of vectorial transport typically requires a labeled substrate.129 Radiolabeled or fluorescently labeled substrates are often prohibitory expensive or commercially unavailable, making it practically infeasible to screen for a large collection of “potential” labeled substrate candidates using proteoliposome assays.129 For known or suspected H+-coupled MFS transporters, indirect methods, such as using pH sensitive fluorescent dyes like 9-amino-6-chloro-2-methoxyacridine (ACMA), can be utilized to follow transport in replace of an labeled substrate (Figure 4a), however, these dyes still lack adequate sensitivity for low turnover transporters to be useful in a medium- to high-throughput setup.123 Nevertheless, if pH sensitive dyes are developed with improved specificity and sensitivity, it should be possible to use proteoliposome based assays for H+-coupled MFS transporters in high-throughput setting. For instance, ∼100 000 compound libraries have been screened for K+ channel inhibitors using the pH sensitive dye ACMA, wherein K+ efflux was converted into H+ influx by the addition of a H+ ionophore in proteoliposomes.130 Alternatively, a more sensitive approach is to monitor transport indirectly by following the charge displacement of an unlabeled substrate across proteoliposomes, which is capacitively coupled to a gold electrode by adsorption to a lipid monolayer using solid support membrane (SSM)-based electrophysiology.131 SSM-based electrophysiology has been effectively applied to a number of MFS transporters to determine kinetic parameters and to dissect differences in H+-coupling mechanisms for a number of sugar symporters132,133 and is now commercially available as SURFE2R N1.131 Because the technology can monitor the half-reaction, presteady-state kinetics is also tractable to SSM-based electrophysiology, which opens up the possibility to probe conformational dynamics134 and to estimate rate constants linking the different conformational states, for example.131,132 The SURFE2R N1 technology has been applied as a rapid method to screen for potential substrates of a non-MFS rocker-switch transporter,135 and the 96-well version of the system holds promise as a medium- to high-throughput platform for substrate screening.131
One main drawback with proteoliposome-based assays is that in the absence of known substrate for use as a positive control, failure to show transport activity might be a problem with the experimental setup itself rather than the substrate, meaning that false negatives are a real concern,129 e.g., the transporter might be too unstable to be incorporated efficiently into liposomes and/or the lipid composition is suboptimal. One avenue is to utilize information from large-scale screening approaches and bioinformatic analysis to limit the number of possible substrates to be biochemically tested. Functional approaches, such as genome-wide RNA interference (RNAi) screens, have been used to identify genes whose loss affected the cellular function’s homeostasis.136,137 More recently, CRISPR knockout and knockin experiments are being carried out for phenotype mapping in the presence and absence of compounds, like drugs, for example.138,139 If the MFS transporter can be expressed in the yeast S. cerevisiae, then this also opens the possibility of screening for substrates by complementation, as there are many engineered yeast strains where essential transporters have been knocked out, e.g., hexose deletion strains.140 Indeed, the Yeast Knockout (YKO) Collection contains over 6000 gene-disruption mutants, covering 96% of the yeast genes.141
An attractive complementary approach to find potential substrates is to instead screen for binders. Specific binders could turn out to be bona fide substrates or if not, they could be inhibitors that might turn out to provide useful tool compounds for characterization of a potential substrate in vivo. Clearly, knowledge that a molecule binds will greatly facilitate the deorphanization of an MFS transporter in vivo as well as in proteoliposome-based transport assays. Arguably the most efficacious high-throughput label-free binding assays are based on monitoring the change in the thermostability of the target in the presence of a ligand.129 The premise of the thermal-shift assay (TSA) is to measure a change in its thermal denaturation temperature by calculating its melting temperature (Tm) in the absence and presence of a ligand.142,143 The most common TSA is differential scanning fluorimetry (DSF), otherwise commonly referred to as thermofluor assays, which monitors protein unfolding upon heating by including an environmentally sensitive fluorescent dye that increases binding and fluorescence as the protein unfolds.144 For membrane proteins, the hydrophobic sulfhydryl-binding dye N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM) has been successfully developed for monitoring unfolding and ligand binding to many different types of membrane proteins.87,129,145 Recently, the CPM assay was successfully applied to demonstrate that it was possible to detect specific substrate binding in a library of many different compounds to mitochondrial carriers and also the MFS d-galactose transporter GalP.129 Alternatively, following the change in intrinsic tryptophan fluorescence upon thermal denaturation, it should be possible to use nanoDSF to screen for potential substrates in a high-throughput format.146
One major limitation of DSF is its demand for large amounts of purified proteins, which can be difficult to obtain in some cases and can sometimes suffer from a high background originating from fluorescent compounds or hydrophobic proteins.144 An alternative approach is to overexpress the transporter with a C-terminal green fluorescent protein (GFP)-fusion tag, which makes it possible to use less material and monitor the melting temperature (TM) in either crude membranes or as purified fusion by fluorescence-detection size exclusion chromatography (FSEC);119,147 at least up to 76 °C as above this temperature GFP is no longer fluorescent. Alternatively, if a generally “harsh” detergent like octyl-glucoside (OG) is added after solubilization in a mild-detergent (e.g., dodecyl-maltopyranoside (DDM), it is possible to centrifuge the heat-induced aggregates and remove the requirement for SEC, which has been termed the GFP-TS assay (Figure 5b).119 Like the CPM assay, the GFP-TS is also amenable in a 96-well format and offers an attractive alternative for screening substrates using unpurified samples to deorphanize SLC transporters.148 Indeed, using a test set of nine different transporters, the melting temperatures by GFP-TS correlated well with the unfolding estimates monitored using the CPM assay.119 The GFP-TS assay has also been utilized to study structural-functional relationships of SLC transporters, in addition to deorphanization of several SLC transporters.17,20 Moreover, the binding affinities (Kd) of an ligand to the human SLC35A1 transporter could be calculated from crude detergent-solubilized membranes and were equivalent to the binding affinities estimated by isothermal titration calorimetry (ITC) measurements using purified transporter.119 As purification is not required, the GFP-TS assay facilitates substrate binding measurements of many mutants,149 an obvious advantage compared to the CPM assay for poorly producing transporters, which would otherwise need to be purified to monitor substrate binding. A disadvantage of the TSA is that in order to thermostabilize the transporter, the assay requires the concentration of the ligand to be at least several-fold higher than its binding affinity to the transporter.129 Current small-molecule libraries are often in the μM concentration range, and therefore the screening of substrates may not be feasible, although it might be possible to detect for inhibitors. Screening for substrates using TSA approaches may instead require the construction of in-house targeted screens, as previously demonstrated.129
Complementary to binding and transport assays is the attainment of structural information. To date, there are around 110 structures of MFS transporters, including 24 unique bacterial structures and 12 unique eukaryotic structures, of which seven are mammalian, two are from plants, one is fungal, and another one is protozoan (Table 2 and Figure 6). The low number of mammalian MFS crystal structures reflects the difficulties in isolating large amounts of detergent stable protein.3,87 With the development of direct electron counting detectors, single-particle cryoelectron microscopy (cryo-EM) has proven to be a revolution in the determination of membrane protein structures.150,151 Although we have seen an explosion in single-particle cryo-EM structures of respiratory complexes, ion channels, and GPCRs, the number of MFS and SLC single-particle transporter structures is still lagging behind.150 To date, there are only six single-particle cryo-EM structures of novel MFS transporters: MCT1 and MCT2 from Homo sapiens (PDBs 7BP3, 7CKR), vesicular glutamate transporter VGLUT2 from Rattus norvegicus (PDB 6V4D), atypical MFS UNC93B1 from Homo sapiens and Mus musculus (PDBs 7C76, 7C77),152 and ferroportin FPN transporters from Homo sapiens (PDBs 6W4S, 6WBV) and primates (PDB 6VYH). The problem is that assuming modest amounts of ∼0.5 mg of the transporter can be purified, most MFS transporters are monomers in detergent and are only between 40 and 80 kDa in size, which is currently considered still very challenging for structure determination by single-particle cryo-EM on their own. They are also highly dynamic, which means that even if it is possible to stabilize oligomers in lipid-mimetics such as nanodiscs, it is still challenging to determine their structures by single-particle cryo-EM. In particular, MFS transporters often have small nonmembranous domains, making it difficult to achieve accurate image alignment due to the high contrast from either noisy detergent micelles or lipid mimetics.150 Similar to crystallography, conformational thermostabilization approaches and/or the use of scaffolds, such as single-chain antibodies or fiducial tags to aid alignment,153 might be essential for obtaining structures of MFS transporters in many cases. Indeed, three out of the five single-particle cryo-EM MFS transporter structures were obtained in complex with Fab antibodies. In the remaining examples, human MCT1 and human and mouse UNC93B1 were in a complex together with single-TM containing proteins harboring large soluble domains152,154 and human MCT2 could be isolated as a stable homodimer.155−157
Table 2. Representative List of the Known MFS Transporter Structuresa.
| MFS family | TCDB | protein | organism | conformation | ligand bound | resolution | PDB | ref |
|---|---|---|---|---|---|---|---|---|
| sugar porter | 2.A.1.1 | XylE | E. coli | outward-occluded | d-glucose | 1.50 Å | 4GBZ | Sun et al., 2012177 |
| drug:H+ antiporter | 2.A.1.2 | EmrD | E. coli | occluded | 3.50 Å | 2GFP | Yin et al., 2006178 | |
| organophosphate:Pi antiporter | 2.A.1.4 | GlpT | E. coli | inward-open | 3.30 Å | 1PW4 | Huang et al., 200317 | |
| oligosaccharide:H+ symporter | 2.A.1.5 | LacY | E. coli | inward-open | TDG | 3.60 Å | 1PV7 | Abramson et al., 200316 |
| fucose:H+ symporter | 2.A.1.7 | FucP | E. coli | outward-open | n-nonyl-β-d-glucopyranoside | 3.14 Å | 3O7Q | Dang et al., 2010179 |
| nitrate/nitrite porter | 2.A.1.8 | NarU | E. coli | partially inward-open occluded | 2.80 Å | 4IUP | Yan et al., 2013180 | |
| phosphate:H+ symporter | 2.A.1.9 | PipT | Piriformospora indica | inward-occluded | phosphate | 2.90 Å | 4J05 | Pedersen et al., 2013181 |
| monocarboxylate transporter | 2.A.1.13 | SfMCT | S. fumaroxidans | outward-open | l-lactate | 2.69 Å | 6HCL | Bosshart et al., 2019182 |
| organic anion:cation symporter | 2.A.1.14 | DgoT | E. coli | inward-open | d-gluconic acid | 2.91 Å | 6E9N | Leano et al., 2019183 |
| proton-dependent oligopeptide transporter | 2.A.17 | PepTSo | S. oneidensis | inward-occluded | 3.62 Å | 2XUT | Newstead et al., 2011159 | |
| glycoside-pentoside-hexuronide:cation symporter | 2.A.2.1 | MelB | S. typhimurium | outward-partially occluded | 3.35 Å | 4M64 | Ethayathulla et al., 2014184 | |
| equilibrative nucleoside transporter | 2.A.57 | ENT1 | H. sapiens | outward-open | dilazep | 2.30 Å | 6OB7 | Wright and Lee, 201928 |
| ferroportin | 2.A.100 | ferroportin | Bdellovibrio bacteriovorus | outward-open | potassium | 2.20 Å | 5AYN | Taniguchi et al., 2015185 |
| atypical SLC | 2.A.1.2 | MFSD10 (TETRAN) | H. sapiens | outward-open | 2.40 Å | 6S4M | unpublished |
The first determined structure in each family is shown.
Figure 6.

One representative from each one of MFS subfamilies where a structure has been determined. The canonical MFS fold is comprised of 12 TMs, made up of two six-helix bundles that are connected by a cytosolic loop. The N-terminal bundle is colored in pale-blue, while the C-terminal bundle is colored in pale-yellow. TMs 1, 4, 7, and 10 are labeled and colored in deep-blue and red, respectively. The PDBs for each representative structure are in parentheses as follows: GLUT3 (4ZW9), MdfA (4ZOW), GlpT (1PW4), LacY (1PV7), FucP (3O7Q), NarK (4JRE), PipT (4J05), SfMCT(6HCL), VGLUT2 (6V4D), PepTSt (5OXN), MelB (4M64), human ENT1 (6OB7), BbFPN (5AYN), and MFSD10 (6S4M). The first structure of a human atypical SLC, MFSD10 (TETRAN), is shown. Similar to MdfA, this is currently classified as a drug:H+ antiporter 1 in the TCDB. Some MFS members deviate from the canonical 12 TMs and instead have 14 TMs with the two extra helices located between the two six-helix bundles such as in PepTSt or have one less TM at the C-terminus and only 11 TMs as for human ENT1. Bound ligands are shown as gray spheres.
A static structure of an MFS transporter is an important first step, but many different structures are required to build up a full transport cycle. Because there are currently no examples of a single MFS transporter with structures that have been determined in all conformations of its transport cycle, in the best case, one must rely on comparing structures of different homologues and the obvious drawbacks associated in doing so. In most cases, however, only one or two of the three major conformations have been determined, such as the oligopeptide transporter family, where a representative outward-facing state is still “missing” despite numerous inward-facing crystal structures.70,107,158−164 Moreover, the fully occluded conformation is rarely captured yet is an important intermediate for establishing how substrate binding and gating are coupled.165 Even if a transport cycle can be reconstructed at the molecular level, we further require methods such as fluorescence resonance energy transfer (FRET),166−168 nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) spectroscopy,169−171 and hydrogen–deuterium exchange mass spectrometry (HDX-MS)172 to assess how these conformations are connected together and their population distributions under various driving forces. In this regard, computational methods such as MD simulations and direct coupling analysis (DCA) based on evolutionary-based sequence contacts, are powerful tools to deepen mechanistic understanding.86,173−176 With these caveats in place, we first describe the generic structural basis for a “rocker-switch” alternating-access mechanism used by MFS transporters before focusing on particular transport systems that have developed mechanistic models by using a combination of these different methods.
5. A Generic Overview of the Rocker-Switch Alternating Access Mechanism
Rocker-switch proteins are made up of two helical bundles that are related by a pseudo-2-fold symmetry axis that runs through the center of the transporter and perpendicular to the plane of the membrane.16,17 At the most basic, the rocker-switch mechanism involves nearly symmetrical movements of two symmetrically related bundles around a centrally located substrate-binding site (Figure 1a).3 In essence, the protein moves around the substrate, alternately exposing the binding site to each side of the membrane.3 The term “rocker-switch” depicts the symmetrical rocking of the two structurally similar bundles as would be expected by global transitions from outward- to inward-facing states.3,16,17,186,187 This is in contrast to “rocking-bundle” transporters, wherein the two bundles are structurally different and the domains are not thought to move symmetrically, but large conformational changes predominantly occur in only one-half of the transporter (Figure 1b). Inevitably, however, a pure “rocker-switch” model breaks down when structures of intermediate, occluded conformations are also included. This is most easily observed by structures of semi-SWEETs, which are parallel homodimers of just 3-TM segments each.188−190 The “V” and “Λ” shaped conformations are easily recognizable in the outward- and inward-facing structures, but in the occluded SWEET structure, an “O”-shaped conformation is observed.3 The reason is that rocker-switch transitions are further coupled with local, gating rearrangements from each of the two symmetrical bundles during formation of the occluded conformation. Simplistically, even rocker-switch transporters with the most basic architecture use rearrangements of nonrigid bodies.
During global, rocker-switch structural transitions in MFS transporters, cavity-closing contacts are predominantly formed by TMs lining the central cavity (Figures 6, 7a,b), particularly between TM4 and TM10 in the outward-facing conformation and between TM1 and TM7 in the inward-facing conformation (Figure 7a,b).3,186,191 While these global, rocker-switch transitions are structurally conserved, the local gating events appear to be fine-tuned to the substrate being transported. In contrast to the symmetrical semi-SWEET proteins, where gating is acquired by symmetrical, local rearrangements from both bundles,3 gating in the MFS transporters appears to be asymmetrical in many cases.3,78,171,191−193 In some cases, it is clear that this asymmetry is established by the asymmetry in the substrate binding site, such as the glucose transporter GLUT3, where the sugar is only coordinated by a single residue from the N-terminal bundle.192 Indeed, in GLUT3 and related sugar porters, sugar binding and substrate gating is primarily driven by rearrangements in the C-terminal bundle.78,193,194 In many cases, however, the substrate appears to bind evenly to both domains, yet either the C-terminal bundle171,195 or the N-terminal bundle is thought to contribute more to the opening dynamics, such as in LacY196,197 or in the phosphate transporter PipT.181 As a consequence, rather than only three conformations of outward-facing, occluded, and inward-facing MFS transporters have at least five distinct structural conformations: outward-facing, outward-occluded, occluded, inward-occluded, and inward-facing (Figure 8a). These partially occluded states represent local changes by TMs that are referred to as “gating helices”, which occlude the substrate from exiting, but the MFS transporter is yet to undergo the global rocker-switch conformations to its opposite-facing conformation.193 In many cases, the substrate gating helices are made up from one or two of the central cavity helices of either TM1 or TM4 in the N-terminal bundle or TM7 and TM10 in the C-terminal bundle,3,78,107,173,181,191,192 which are often broken or highly flexible in the middle (Figure 6, Figure 7c). The central cavity helices that are also substrate gating helices are not always easy to ascertain from apo outward- or inward-facing crystal structures, as they might only contain a well-conserved glycine or proline residues that will eventually bend in the middle to accommodate substrate binding in partially or fully occluded states, such as in LacY (Figure 6).107 In some cases, the substrate gating helices are clearer as they are fully broken and contain unwound regions that connect the two half-helices (Figure 7c).
Figure 7.

General structural architectures of MFS transporters. (a) Ribbon representation of open outward-facing rat GLUT5 (left) (PDB 4YBQ) and open inward-facing bovine GLUT5 (right) structures (PDB 4YB9), viewed in the plane of the membrane. TMs 1 and 4 and TMs 2, 3, 5, and 6 in the N-terminal TM bundle are colored in blue and light-blue, respectively. TMs 7 and 10 and TMs 8, 9, 11, and 12 in the C-terminal TM bundle are colored in red and light-orange, respectively. Cavity-closing contacts in the outward-facing conformation and predominantly formed between TM4 and TM10 (dotted ellipse) and in the inward-facing conformation between TM1 and TM7 (dotted ellipse). Neighboring TMs 5 and 11 and TMs 2 and 8 in the outward- and inward-facing states, respectively, also contribute to cavity closing by a varying extent. The intracellular domain helices (ICH) unique to the sugar porters are shown in gray. (b) As in (a), viewed from the extracellular side. (c) MFS transporter structures of the glucose sugar porter STP10 (left) (PDB 6H7D), the nucleoside transporter ENT1 (PDB 6OB7) (middle), and the drug:H+ antiporter transporter YajR (PDB 3WDO) (right), where substrate-gating helices have been proposed as apparent by highly bent of broken helices in either TM1, TM4, TM7, or TM10. These structures also highlight extra non-TM domains that can be located in an extracellular loop and/or in the cytoplasmic loop located between the N- and C-terminal bundle and/or at the C-terminus or N-terminus (not shown).
Figure 8.

The major conformations in the transport cycle of an MFS transporter. (a) Schematic illustrating the six major conformations of an MFS transporter cycle: outward-open, outward-occluded with bound substrate (pink sphere), occluded with substrate, inward-occluded with substrate, inward-open and occluded with no substrate. (b) A structural based example of all the major conformations of the MFS transporter cycle as illustrated here by monosaccharide sugar porters, which have a highly conserved fold and for which structures are available in all the major conformations. The “fully-occluded” conformation of the hexose transporter from the malarial parasite Plasmodium falciparum was the last remaining state to be observed within the rocker-switch alternating access mechanism. The observed structural states shown as surface transversal cross sections and clockwise from the top left: “outward-open” rat GLUT5 (PDB 4YBQ), “outward-occluded” human GLUT3 (PDB 4ZW9), PfHT1 “fully occluded” (PDB 6RW3), “inward-occluded” XylE (PDB 4JA3), and “inward-open” bovine GLUT5 (PDB 4YB9). In either the forward or reverse direction, the attainment of the occluded intermediate is required. Below the structures is the principal component analysis from the conserved MFS ensemble core (n = 17 structures from 16 PDB codes), which yields a major principle component one (∼65% of the total structural variance) that tracks the 16° global rocker-switch motion between the bundles. Projections were colored according to their trajectory angle. (b) Adapted from Qureshi et al.193
The fully occluded conformation is thought to be metastable and only transiently occupied during structural isomerization between outward-occluded and inward-occluded states or vice versa.45 Consistent with this line of reasoning, structures of fully occluded conformations of MFS transporters are rare, and out more than 110 determined MFS transporter structures, the occluded conformation has clearly only been observed in three cases: the multidrug transporter EmrD,178 the nitrate/nitrite antiporter NarK,195 and the hexose transporter from the malarial parasite P. falciparumPf HT1(Figure 8b).193,198 Structural details from all of these different conformational states have led to general themes of how substrate binding catalyzes global rearrangements. Although the molecular details are different for every MFS transporter, a clear requirement is the breakage and reformation of salt bridges that hold the N- and C-terminal bundles together, as first seen in crystal structures of LacY16 and GlpT.17 Interbundle salt bridges have consistently been observed in MFS transporter structures and are often found proximal to the substrate binding site.78,107,184,191,195 Indeed, on the basis of bioinformatic analysis across all MFS subfamilies, a clear sequence consensus emerges in many members, which has been called the “A-motif”. The A-motif is located between the ends of TM2 and TM3 and/or between the ends of TM8 and TM9, and structures have shown these charged residues form salt bridges that are often connected to the interbundle salt-bridges, e.g., as seen in the sugar porter subfamily (Figure 9a).10,78,191,199 Notably, salt bridges are formed and broken in both passive as well active MFS transporters, and therefore ionic interactions are thought to establish the energetic barriers to be overcome by substrate binding in most, if not all, MFS transporters.
Figure 9.

Salt bridges are often formed within and between the N- and C-terminal bundles in MFS transporters. (a) Cartoon representation of the fructose transporter GLUT5 as viewed from the cytoplasm in the outward- (PDB 4YBQ) (left) and inward-facing (PDB 4YB9) (right) conformations. ICHs are not shown for clarity. The residues forming salt bridges are shown as sticks, and interbundle salt bridges are only formed in the outward-facing conformation for the monosaccaharide sugar porters. Note, in most other MFS transporters, interbundle salt bridges are formed in both outward- and inward-facing conformations. (b) The salt bridge forming residues are highly conserved and pseudosymmetrically related. These residues make up the sugar porter motif together with ICH1, which was used before structures became available to identify sugar porters. (c) Unique to the sugar porter structures is an intracellular helical bundle that can either have three or four intracellular helices between the N- and C-terminal bundles and an intracellular helix at the C-terminal bundle. In the outward-facing GLUT5 structure ICH1–3 are linked together by several salt bridges (side chains are labeled and shown as sticks in yellow). In contrast, no polar interactions are formed between ICH5 and either ICH1–3 or cytoplasmic ends of N-terminal TM bundle helices. A salt bridge forms (dotted line in magenta), however, between E225 in ICH3 and R407 in TM 11, which also forms part of the interbundle salt bridge network (side chains are labeled and shown as sticks in cyan). The ICH domain functional role is proposed to act as a scaffold domain that further helps to stabilize the outward-facing conformation. Adapted from Nomura et al.78
For active H+-coupled transport, the local and global conformational changes must be coupled to avoid “forbidden” transport of either the substrate or H+ on its own.200 Indeed, most of the key residues for H+ translocation are located in the central cavity helices, including TM1, TM4, TM7, and TM10.201 As such, both the binding of a H+ and a substrate is coupled and required for energized transport. Notably, as shown for the melibose transporter MelB, the driving ion can also be Na+ or Li+.202,203 To ensure concomitant binding, often ionizable groups, such as aspartic acid and histidine, close to or part of the substrate-binding site, must first be protonated for the substrate to bind with measurable affinity.90,108,204 Typically, residues upstream of the substrate binding site, however, are first protonated, which elicits a change in the local electrostatic network, leading to the subsequent protonation of the substrate binding residue.34,90 In some cases, these H+ binding residues reform salt bridges between the N- and C-terminal bundles together,108 providing a clear explanation of how substrate binding triggers larger, global conformational changes. In other cases, substrate binding catalyzes salt bridge breakage between the N- and C-terminal bundles distant from the substrate-binding site.78,205 Most often, however, even with structural details on hand, H+-coupling pathways are mechanistically challenging to detangle. In the bacterial MCT homologue SfMCT for example, a histidine residue far from the substrate-binding site and located at the end of a helix facing the extracellular space, was found to be essential for H+-coupled transport.182 In some MFS members, the H+-coupling pathway is stringent, and the neutralization of just a single acidic residue can convert an H+-coupled symporter into a passive transporter.194,206−210 For example, in the H+/galactoside symporter LacY, a glutamate residue (Glu325) in TM10 is the primary H+-binding site.90,211 Although the glutamate residue does not interact with galactoside directly, it is connected to the sugar binding site via a histidine (His322), which itself is salt-bridged to an aspartate residue.16,90 If either the glutamate or histidine is substituted to alanine, LacY can still carry out passive downhill transport of sugar, but cannot perform active H+-coupled sugar transport.90,211 In LacY, it has been shown that the Glu325 alanine mutant is still able to carry out counterflow and exchange with rates similar to wild-type.211 These findings demonstrate that substrate binding drives the conformational rearrangements in both passive and active H+-coupled transporters.68,106 In the latter case, however, the presence of an ionizable group in the wild-type transporter ensures that the substrate does not bind until key residue(s) are first protonated.212
In some active MFS members, however, there appears to be some flexibility in the H+ coupling pathway and is not thought to be dependent on the protonation state of any one key residue. For example, while three acidic residues in the yeast sucrose sugar porter Mal11 were required for maximal active transport, individual mutations to a neutral amino acid were still able to utilize a H+ gradient to drive maltose uptake.109 It was only when all three residues were neutralized that H+ and substrate cotransport uncoupled, with no impairment to the substrate binding site because the system is fully active in downhill (counterflow) transport,109 i.e., mutation of all three residues completely abolished H+-coupled transport, but still enabled facilitated diffusion of the substrate. It was shown recently that it was possible to evolve yeast strains to grow in a medium only containing sucrose as a carbon source complemented with the MalII triple mutant.213 Remarkably, two of the suppressor mutants regained H+-coupled sucrose transport through acidic residues now located in TM7 and TM11, which are positioned parallel to the naturally occuring position of the acidic residues that were mutated in TM1 and TM4.213 In the melibose transporter MelB, both Na+ and H+ compete for binding to the same ion-binding site, but due to their respective affinities and physiological concentrations, Na+ is thought to be the primary driving ion.214 Interestingly, however, some sugar epimers can be coupled to either Na+ or H+, whereas others can only use Na+.215 Ion-coupled promiscuity has further been observed for oligopeptide symporters and multidrug antiporters, where even the H+:substrate stoichiometry can vary depending on the substrate being transported.216−219 Such consequences can change the energetics, as the net overall transport process can then be either electroneutral or electrogenic.218 In the multidrug exporter MdfA and LmrP, the H+-coupling residues can be shifted to a different helix and still retain substrate-H+-coupling.220,221 The ability of MFS transporters to alter energy transduction through modifying vectoral H+-coupling pathways is remarkable. Such adaptability has not been observed, so far, in other transporter folds and could explain why MFS members in higher eukaryotes remain H+-coupled even when sodium gradients are otherwise available.
In reality, a complete description of substrate translocation requires the construction of a multidimensional energy landscape that defines the relative probabilities of each of the conformational states and the energetic barriers between them. Simplistically, the equilibrium is thought to be shifted by the binding of either substrates and/or ion-coupling. The substrate binding and gating interactions in the different conformational states are all subtly different, which is fine-tuned to the substrates that are being transported. Rather than describing the molecular details of the alternating-access mechanisms for many different MFS transporters, we have limited our focus to the sugar porter (SP) and MFS multidrug resistance (MDR) proteins belonging to the drug proton (H+) antiporter (DHA) subfamilies for several different reasons: (i) both SPs and DHA proteins represent large MFS subfamilies, (ii) crystal structures are available in all (for SP) or multiple (for DHA) major conformations of their transport cycle, (iii) SPs are either uniporters or symporters, whereas DHA members are antiporters, and (iv) SPs are highly specific for their substrates, in comparison to DHA proteins that typically show substrate promiscuity. Thus, the similarities and differences offered between these two large families covers most of the deeper mechanistic principles of the alternating-access mechanisms found in most MFS transporters. Notably, recent reviews have outlined detailed alternating-access mechanisms for oligopeptide transport,71 lactose permease LacY,90 and in this themed issue for nucleoside transport.222
6. The Alternating-access Mechanism of Glucose (GLUT) Transport
Typically, sugar transporters must be highly specific but bind their substrates weakly to facilitate high sugar flux. One of the most well-described model systems are the “passive” glucose (GLUT) transporters, which work to maintain blood glucose levels at ∼4–12 mM and do so with turnover rates (kcat) of up to 6500 molecules/s reported.68 At these physiological glucose concentrations, the GLUT transporters have high μM to low mM Michaelis constants (KM) for the sugars, yet surprisingly they maintain high specificity, i.e., not even stereoisomers of the transported sugars are recognized. Human has 14 different GLUT isoforms, and each isoform shows a distinct pattern of tissue distribution, gene regulation, kinetic properties, and substrate selectivity.68 For example, GLUT1 is distributed in a wide range of tissues, including the blood–brain barrier, and is essential for glucose transport into the brain, whereas GLUT4 is mostly localized to skeletal muscles and adipose tissue and is the major insulin-stimulated glucose transporter.223,224 GLUT5 is the only member specific to fructose and together with GLUT2 are the major fructose transporters in the body.225
The GLUT transporters belong to a subset of the MFS sugar porter (SP) family (TC no. 2.A.1.1), called the facilitative sugar transporter family SLC2A, which is responsible for the majority of organism-wide sugar transport in mammals.226 The GLUT proteins were one of the first transporters to be functionally characterized and the substrate coordination mapped, owing to their importance and the natural abundance of GLUT1 in red blood cells.227,228 From a structural perspective, they are the only MFS subfamily to date where structures have clearly been determined in all of the major conformations of the transport cycle (Table 2 and Figure 8b), albeit from different organisms. Nevertheless, the high degree of structural conservation, as observed when overlaying crystal structures determined in the same conformation, demonstrates that the approach of combining these different structural states is robust enough to describe the major structural transitions.193 In addition to the canonical MFS-fold, these sugar porters have an additional intracellular helical domain between the six-helix bundles that is comprised of three to four intracellular helices (ICH) in the N-terminal bundle and a short intracellular helix at the C-terminus (Figure 7a).177,186,229 Interestingly, the sugar porter motif used to classify MFS members as sugar porters is an intracellular salt bridge network located at the ends of the TM segments that connects the N-and C-terminal bundles together in the outward-facing conformation (Figure 9a).10,199,230 The salt bridge residues are related by pseudosymmetry as can be seen by superimposition of the N- and C-terminal bundles (Figure 9b). The sugar porter motif also includes ICH1, which further forms salt bridges with other ICH’s (Figure 9c).78 Because this motif is separated from the residues coordinating sugar,78 it remains unclear how many of the members currently annotated as sugar porters are actually sugar transporters.
Crystal structures of apo GLUT5 have been determined in both outward- and inward-facing conformations (Figure 7a,b and Figure 8b).78 GLUT3 was solved in outward-open and outward-occluded states, with and without maltose/glucose bound,192 and GLUT1 in an inward-facing state (Figure 8b).231,232 These GLUT structures are highly homologous to the structure of the H+-coupled xylose transporter XylE from E. coli determined in several conformational states177,194,205 as well as the hexose transporter PfHT1 determined in an occluded conformation (Figure 8b).193,233 Other monosaccharide sugar porter structures include Staphylococcus epidermidis (GlcPSe) in an inward-facing confromation206 and the STP10 from A. thaliana in a glucose-bound outward-occluded conformation (Figure 7c).234 Recently, 17 structures belonging to the monosaccharide sugar porter family were aligned and assigned across all conformations by statistically based principle component analysis,193 which provided further support that these structures could be assembled to reconstruct a reliable structural basis for their alternating-access mechanism (Figure 8b).
6.1. The Sugar Binding Site
The best structural understanding of d-glucose recognition is apparent from the structure of human GLUT3 with d-glucose bound at 1.5 Å resolution.235 In GLUT3, residues located in TM7 and TM10 of the C-terminal bundle predominantly bind d-glucose. Indeed, only a single residue in TM5 of the N-terminal bundle contributes to d-glucose coordination (Figure 10a). Inverted-symmetry-related TM7 and TM10 make up highly conserved sugar transporter signatures,199 and their predominant role in sugar recognition is in agreement with previous functional data.236−241 Indeed, prior to crystal structures, the end of TM7 of GLUT1 was proposed to be important in both the coordination of d-glucose and the closing of the outside gate during translocation.242 The d-glucose binding mode with the C1-OH and C2-OH groups facing the inside, and the C4-OH and C6-OH groups facing the exterior is perfectly consistent with the extensive biochemical analysis of GLUT1 transporters, as concluded in the 1970s (Figure 10a).69,228,243 As one might expect, the sugar binding site in GLUT3 is very polar, with extensive hydrogen bonding to all six oxygen atoms (Figure 10a). The sugar binding site is very conserved, with only small side chain differences between the high-affinity glucose transporter GLUT3 and the fructose-specific transporter GLUT578 (Figure 10a). In fact, the sugar binding site is highly similar to the xylose transporter XylE,177 the promiscuous sugar transporter from the malarial parasite PfHT1193 or even the bacterial GlcPSe and plant STP10 transporters, which are specific to d-glucose and bind with very high affinities at ∼3–30 μM (Figure 10b).206,234,244 From the structure of the sugar binding pocket itself, it is thus unclear how monosaccharide sugar porters recognize sugars with different binding affinities and preferences.
Figure 10.

High structural conservation and coordination of d-glucose in distantly related sugar porter monosaccharide homologues harboring different substrate preferences, kinetics, and binding affinities. (a) Cartoon representation of the outward-occluded human GLUT3 (PDB 4ZW9) sugar binding site with d-glucose bound (yellow sticks), and the residues forming hydrogen bonds are labeled. The corresponding residues in the outward-open fructose transporter GLUT5 (PDB 4YBQ) are also shown (pink sticks) and are labeled where different. (b) The sugar binding site comparison between the d-glucose bound sugar porter structures for the glucose specific human GLUT3, glucose specific plant STP10 (PDB 6H7D), the xylose specific E. coli XylE (PDB 4GBZ), and the pan-specific P. falciparumPfHT1 (PDB 6RW3). While GLUT3 (KM = 1.3 mM) and STP10 (0.005 mM) transport d-glucose, PfHT1 (KM = 0.9 mM) can transport many different sugars including d-fructose and d-xylose in addition to d-glucose, and XylE cannot transport d-glucose but binds it with the same affinity as its preferred substrate d-xylose. All residues hydrogen bonding to bound d-glucose are identical apart from the TM10a residue corresponding to TM10 residue, which is in an alanine in all structures except GLUT3, where it is a glutamate. Most residues in the N-terminal bundle surrounding the d-glucose but not coordinating the sugar are also highly conserved (not shown). (c) The substrate gating helices TM7b and TM10b control access of the sugar to the outside and inside, respectively, in GLUT5. Interactions between hydrophobic residues between TM7b and TM10b in the outward-facing conformation (PDB 4YBQ) (left) are lost in the inward-facing conformation (PDB 4YB9) (right) and may help to increase mobility of TM10b to catalyze interbundle salt bridge breakage. Note that the residue corresponding to I295 has been reported as the key residue for fine-tuning d-glucose affinities as single-point mutations can have 10-fold differences in KM values.241 (d) Schematic highlighting the local substrate gating rearrangements by TM7b from the outward-open to the outward-occluded state and by symmetry-related TM10b from the inward-occluded to the inward-open state. (c,d) Adapted from Qureshi et al.193
6.2. The Determinants for Evolving Sugar Recognition and Transport
It is becoming increasingly clearer that in monosaccharide sugar porters, the environment around the central binding pocket and the substrate gating helices themselves appear to play a larger role in shaping substrate recognition than previously anticipated. The residues in the N-terminal bundle juxtaposed to the d-glucose binding site in GLUTs are hydrophobic, which could push d-glucose toward the binding site and increase its apparent affinity to the sugar binding pocket in the C-terminal bundle. Consistently, it has been proposed that the hydrophobic surface of the N-terminal bundle in STP10 is important for d-glucose binding affinity.234 However, hydrophobicity in the N-terminal bundle is also a general structural feature across a number of determined sugar porter structures, and this feature alone does not adequately explain these large differences.
One of the most insightful sets of biochemical studies to tackle this question was carried out with the hexose transporters from S. cerevisiae, wherein a comprehensive chimeric screen was performed between the high-affinity glucose transporter HXT2 (KM = 3 mM) and a low-affinity glucose transporter HXT1 (KM = 46 mM). In brief, each TM segment of HXT2 was replaced with the corresponding TM segment in HXT1.245 It was revealed that TM1, TM5, TM7, and TM8 of HXT2 were essential for high-affinity glucose transport.245 Subsequently, these TMs were systematically and individually replaced between HXT1 and HXT2, and with further mutagenesis it was concluded that the asparagine residue in TM7 (Asn331) was the most important residue for the determining the affinity for d-glucose.246,247 Remarkably, the replacement of this residue (Asn331) with each of the other 19 amino acids yielded transporters with KM values for d-glucose ranging from 0.87 to 54 mM, compared with a KM of 3.3 mM for the wild-type protein. To show that this is a general site, it was further possible to engineer a TM7 mutant with higher binding affinity than wild-type for a different yeast hexose transporter, HXT7.248 Moreover, the corresponding TM7 residue in GLUT1 (Ile287) was further substituted to every other amino acid and analyzed by in-depth kinetics.241 Somewhat surprisingly, despite their sequence divergence, a remarkably clear correlation between all 19 mutations was observed.241 Substitution of this single TM7 residue could drastically alter d-glucose binding affinities in GLUT1. It was proposed that the TM7 residue likely “interacts with surrounding residues within van der Waals distance that directly communicates with the substrate and thereby contributes to the fine-tuning of the binding reaction at the presumed exofacial site”.241 Consistently, the GLUT1 and other GLUT structures have confirmed that the Ile287 residue is not coordinating the substrate sugar but is sandwiched between a strictly conserved TM7b asparagine that coordinates d-glucose and a glycine residue that breaks TM7 into the half-helices TM7a and TM7b (Figure 10c).78,235 It thus seems likely that the Ile287 has an indirect influence on substrate binding by somehow affecting the coupling between sugar binding and TM7b gating. Because the equivalent residue in GLUT5 was found to only interact with the gating helix TM10 in the outward-facing conformation, it was also postulated that perhaps this region might also be important for the communication between TM7 and TM10 gating helices (Figure 10c).78
6.3. The Extracellular Substrate Gating Helix TM7b
The asymmetry of sugar binding, predominantly by the C-terminal bundle, is consistent with proposed asymmetric local rearrangements of the substrate gating helices TM7b and TM10b in the C-terminal bundle as first proposed in GLUT3 and GLUT5 (Figure 10a,d).3,78,191−194 As seen in MD simulations,193 the extracellular gating helix TM7b is very mobile and consistently in several of the outward-facing sugar porter structures, lipids, detergents, or nonphysiological substrates appear to interact to stabilize the TM7b gate.177,231 TM7b gating dynamics is further consistent with the fact that, even in the presence of maltose, GLUT3 crystallizes in both outward-open and outward-occluded conformations.235 Although the exact coupling mechanism between sugar binding and extracellular gating remains unclear, the repositioning of an asparagine in TM7b to hydrogen bond to the bound sugar is clearly an important first step (Figure 11a). In detail, between the outward and outward-occluded conformations a strictly conserved asparagine residue (Asn288 in GLUT1), located at the beginning of TM7b, moves inward to coordinate the most critical hydroxyl groups, 3-OH and 4-OH, in d-glucose.235 Consistently, the TM7b asparagine residue directly precedes the TM7a-7b breakpoint residue (Ile287) that, as just discussed, is a key determinant for tuning d-glucose binding affinities. During transition from an outward-open into an outward-occluded state, a conserved tyrosine residue (Tyr293 in GLUT1) located one helical turn from the TM7b asparagine, moves inward to obstruct sugar exit, although it does not interact with the substrate sugar itself (Figure 11a).177,235 Extraordinarily, biochemical studies some 27 years ago had proposed that Tyr293 was required for closing the exofacial site around C4-OH and C6-OH groups of d-glucose.242 Notably, a crystallization lipid blocks the tyrosine residue in TM7b of human GLUT3 to adopt a more occluded state as seen in the outward-occluded structure of XylE (Figure 11a).
Figure 11.

The extracellular gate TM7b forms the occluded state and is coupled to sugar binding by an asparagine residue. (a) The extracellular substrate-gating helix TM7b. (left) A cartoon representation of TM7b of human GLUT3 in the outward-open conformation (PDB 4ZWC, red) and the outward-occluded d-glucose bound conformation of XylE (PDB 4GBZ. salmon). During transition into the outward-occluded conformation, a strictly conserved asparagine coordinates the C3- and C4- hydroxyl groups of d-glucose to stabilize the inward movement of TM7b, as shown in the upper panel. Conserved tyrosine residues in TM7b form the occlusion. (left middle) Comparison between the outward-occluded XylE (salmon) and outward-occluded human GLUT3. The crystallization lipid in human GLUT3 blocks the inward movement of the occlusion-forming tyrosine, and TM7b does not transition into a full outward-occlusion. (right middle) Comparison between the outward-occluded XylE (PDB 4GBZ, salmon) and the inward-open conformation of GLUT5 (blue). During transition into the inward-open conformation, the TM7b asparagine and tyrosine move closer to the sugar, and TM7b breaks into an elbow-shaped conformation as shown in the upper panel. (right) A cartoon representation of TM7b of GLUT5 (PDB 4YB9, blue) in the inward-open conformation, and the occluded-occluded d-glucose bound conformation of PfHT1 (PDB 6RW3, red). TM7b forms the same elbow shaped structure as in the inward-facing conformation, demonstrating that the full inward movement of TM7b and its breakage is a transition that occurs prior to the rocker-switch transition. (b) Cartoon representation of the sugar binding site of occluded PfHT1 (green sticks) superimposed with outward GLUT5 (PDB 4YBQ, blue sticks), outward-occluded GLUT3 (PDB 4ZW9 gray sticks), and inward GLUT5 (PDB 4YB9, orange sticks). Asn311 (dotted ellipsoid) is the only residue clearly repositioning during the entire transport cycle. Adapted from Qureshi et al.193 (c) The highly conserved tyrosine residues that occlude the substrate from exiting in the outward-occluded conformations of GLUTs are replaced by serine (S315) and asparagine (N316) in PfHT1. (d) Cartoon representation of PfHT1 extracellular gating interactions between TM7b (magenta) and TM1 (blue). Potential hydrogen bond interactions are indicated by dotted lines and prominent residue side chains labeled. (e) Transport activity for PfHT1 TM1–TM7b interacting residue mutants for [14C]-d-glucose (black bars) and [14C]-d-fructose (white bars), respectively, residues in bold facing toward TM1 were determined to be just as essential for sugar transport as residues coordinating d-glucose directly. Stabilization of cavity-closing contacts between TM1 and TM7 are essential for stabilization of the occluded state. Data are mean ± SEM of N = 3 biologically independent experiments. Adapted from Qureshi et al.193
Upon superimposition of all the major conformation of the GLUT transport cycle, the TM7b asparagine is the only sugar-coordinating residue significantly changing its position during the whole transport cycle (Figure 11b). It has been proposed that GLUT transport must therefore be (primarily) driven by conformational selection. Because the TM7b asparagine residue is strictly conserved in all GLUT transporters and related sugar porters, regardless of their sugar preference and affinities, it appears this is a key and generic interaction required for coupling sugar binding, extracellular gating, and transport (Figure 11a).193 Of importance, in the occluded structure of PfHT1, TM7b further breaks in the middle to adopt an elbow-shaped helix (Figure 11a). In all inward-facing states, the gating helix TM7b always adopts the same elbow-shaped helix (Figure 11a).78,193,231 The structural transition of TM7b takes place before transitioning into the inward-facing state. Presumably, TM7b breakage is not spontaneous and only a correctly bound sugar will induce formation of the occluded state to achieve transport.
Intriguingly, the TM7b tyrosine residues that are important for substrate occlusion in GLUT proteins have polar residue counterparts of asparagine and serine residues in PfHT1 (Figure 11a,c). The TM7b residues were found to form contacts with TM1, consistent with the cavity closing contacts eventually formed between TM1 and TM7 in the inward-facing state (Figure 7a).193 It is likely that TM7b sequence differences have made it possible to capture the occluded conformation of PfHT1 by crystallography. Consistently, MD simulations have confirmed that the occluded state of PfHT1 was stable, whereas outward-occluded GLUT3 spontaneously opens back to an open state even when d-glucose is present.193 However, there is no evidence that the transition intermediate in PfHT1 is rate limiting; in fact, the malarial sugar transporter PfHT1 is uniquely capable of transporting both d-glucose and d-fructose with equal efficiency (KM, kcat) as the dedicated high-affinity d-glucose GLUT3 and d-fructose GLUT5 transporters, respectively.68,193,249,250 The PfHT1 sugar binding site and the coordination of d-glucose is very similar to that seen in human GLUT3192 and also with the d-fructose binding site observed in rat GLUT5 (Figure 10a,b).78 Mutation of sugar binding residues in PfHT1 made to match either GLUT3 or GLUT5 binding sites failed to rationally shift sugar preferences in PfHT1.193 For example, the mutation of the TM10 tryptophan to alanine to match GLUT5 did not shift the sugar preference of PfHT1 to d-fructose but instead abolished d-fructose transport, while d-glucose transport was unaffected. This result was further unexpected, because in all GLUT transporters harboring the TM10 tryptophan, it has been critical for d-glucose transport.68
In contrast to the high conservation of the sugar binding site, as discussed, TM7b tyrosine residues in PfHT1 have been replacedby more polar residues (Figure 11c). Strikingly, in PfHT1, the mutation of most TM7b residues to alanine completely abolished transport and were found to be just as critical for transport as the residues directly coordinating d-glucose (Figure 11d,e).193 This data illustrates that the gating helix TM7b in PfHT1 can be considered as an extension of the sugar binding site. The polar TM1 residues pointing toward the TM7b breakpoint were also found to be important, consistent with the fact that TM7b is required to break and move closer to TM1 in forming the occluded state.193 A number of TM7b mutants retaining transport showed a reduced kinetic preference for d-glucose but not for d-fructose. By making modifications to the TM7b gate, it therefore seems possible to alter the coupling with the sugar binding site. Rather than evolving the sugar binding site to be able to transport so many different sugars effectively, it was proposed that PfHT1 has evolved the extracellular gating helix TM7b instead. Simply put, imply, it was concluded that PfHT1 has an outside gate that shuts more easily and has therefore become less stringent as to which sugars catalyze transport.193 Although TM7b gating dynamics might be exaggerated in PfHT1, the conceptual framework is consistent with the importance of the TM7a–TM7b breakpoint residues that have found to be important for fine-tuning d-glucose binding affinities in GLUT transporters. Furthermore, in an exhaustive forward-evolution approach of the yeast glucose transporter (HXT14) in S. cerevisiae, only five residues with improved d-xylose uptake could be identified.251 The only single mutation shifting sugar preference from d-glucose to d-xylose in HXT14 was not found in the sugar binding site, but in the middle of TM7b, the residue equivalent to Asn318 in TM7b of PfHT1 (Figure 11d).251,252 Moreover, in several patients with GLUT1 deficiency syndrome, a missense mutation in TM7b, equivalent to Asn218 in PfHT1, was also found to have shifted the KM for d-glucose.253
The required and evolved coupling between the sugar binding site and the extracellular gate TM7b offers a rational explanation as to why XylE binds d-glucose in the same position and with the same affinity as in human GLUT3, but is incapable of transporting the sugar,34,177 i.e., the binding of d-glucose in XylE is unable to induce the breakage of the TM7b gate so that it can transition into the occluded state. Consistently, the mutation of a sugar binding site residue and a TM7b residue can be combined in XylE to enable the transport of d-glucose. Specifically, while single-point mutations of Gln175Leu and Leu297Phe in XylE were incapable of d-glucose transport, combining both mutations enabled transported of d-glucose while retaining 75% of wild-type d-xylose transport.34 The Gln175 residue is located in the sugar binding pocket, and the Leu297 residue is located in TM7b, one helical turn from the TM7b asparagine (Figure 11d). Lastly, mutation of the TM7a–TM7b breakpoint isoleucine to a valine in GLUT7 abolishes d-fructose transport, while leaving d-glucose transport unaffected.240
6.4. The Lid Domain of the Plant Homologue STP10 Controls Outside Occlusion
The plant homologue STP10 is a very high-affinity glucose transporter (low μM affinity), yet the coordination of d-glucose is almost identical to that seen in other d-glucose bound sugar porter structures (Figure 10b). The most notable structural differences between STP10 and the other sugar porter structures for d-glucose is that STP10 has an additional helical domain located between TM1 and TM2, which has been referred to as the “lid” domain (Figure 12a). Surprisingly, the lid domain harbors a cysteine residue that forms a disulfide to a cysteine residue in TM11 in the C-terminal bundle and, thus, covalently links the two bundles together in the outward-facing conformation (Figure 12a).234 The disulfide linkage is not essential for sugar binding and transport, however, as a lid domain cysteine-to-alanine mutant has similar sugar binding and kinetics as the wild-type protein; the cysteine mutant does however alter STP10 activity at more neutral pH values, as will be later discussed. Instead of TM7b, the lid domain now contributes to the outward-occlusion through a different tyrosine residue, whilst TM7 adopts a straighter helix (Figure 12b).234 While the STP10 structure could indicate that extracellular TM7b gating is not a conserved mechanism, in the more recently determined inward-open conformation of STP10,254 TM7b has fully broken into the same elbow-shaped configuration as seen in all other inward-facing sugar porter structures.78,193,194,205,231 In the inward-facing state of STP10, the lid domain has been pushed away and the lid domain tyrosine has been replaced by the TM7b occlusion-forming tyrosine.254 This result implies that STP10 will go through a similar occluded intermediate. Consistently, mutation of either the lid domain tyrosine or the TM7b tyrosine to alanine shifts the KM for d-glucose from 20 to 300 μM.254 Therefore, the TM7b gate is also contributing to the high sugar binding affinities in STP10, and the lid domain located at the end of TM1 has further evolved a unique coupling with TM7b gating.
Figure 12.

Extracellular TM7b gating occlusion opens a potential allosteric pocket in sugar porters and TM7b and the lid domain in the plant homologue STP10. (a) The high-affinity d-glucose transporter STP10 has an additional, extracellular helical loop domain between TM1 and TM2 that forms a covalent disulfide bond to the C-terminal bundle (dotted eclipse). A tyrosine residue in the loop domain contributes to the outward-occlusion and a phenylalanine in TM7b. (b) In the outward-occluded conformation of STP10 (PDB 6H7D), TM7b adopts a position more similar to an outward-open position (red) as can be compared to the outward-occluded state of XylE (salmon) (PDB 4GBZ). (c) Slab through the electrostatic surface representation of PfHT1 in the occluded conformation (PDB 6RW3) opens an extracellular side vestibule that could be a site for potential regulation by other ligands. (d) In the PfHT1 structure crystallized in the short-chain detergent n-nonyl β-d-glucopyranoside (PDB 6M20), the glucose headgroup of the detergent can bind in the open vestibule or between TM1 and TM7b and likely restrict TM7b dynamics (yellow sticks). In other GLUT structures, crystallization lipids have also been modeled between the extracellular cavities, as shown here for human GLUT3 in the outward-occluded conformation (pink sticks) (PDB 4ZW9). One of these crystallization lipids even forms direct hydrogen bonding to d-glucose in the high-resolution human GLUT3 structure.192,193
6.5. Interbundle Salt Bridges and the Intracellular Gating Helix TM10b
In GLUT and related sugar porters, no salt bridges are observed near the central cavity, in either outward- or inward-facing conformations,78,177,193,231,235 perhaps to avoid inadvertent H+ coupling. Instead, interbundle salt bridges are formed far from the central cavity and only in the outward-facing conformation, linking the cytoplasmic ends of TM3, TM4, and TM5 in the N-terminal bundle to those of TM9, TM10, and TM11 in the C-terminal bundle (Figure 9a).78,235 These charged residues are the most conserved and make up the well-described sugar porter signature motifs.10,199 The charged residues are also structurally related by a pseudo-2-fold symmetry axis that runs through the center of the transporter and perpendicular to the membrane plane (Figure 9b).78 In the inward-facing conformation, the interbundle salt bridges are broken and are located far apart.3
In addition to the intracellular salt bridge network located between the end of TM helices, there are further salt bridges that latch intracellular helices on the N-terminal bundle with an intracellular helix on the C-terminal bundle (Figure 9b,c).78,177,192 In the occluded PfHT1 structures, only a portion of the C-terminal intracellular helix could be modeled or not at all.193,233 As was the case for previously determined inward-facing structures of human GLUT1, bovine GLUT5, and E. coli XylE.194,205,231 Because the interbundle salt bridge network was still intact in the occluded PfHT1 structure, it seems that the ICH5 latch may move first to further weaken salt bridge stability and enable subsequent transition into the inward-occluded conformation. Consistently, long MD simulations of GLUT1 found that the ICH5 interaction to ICH1–4 is a critical step for the transition between the inward- to outward-facing states.255 Moreover, removal of the C-terminal tail arrests GLUT1 in an inward-facing conformation.256 In GLUT1, the return of the empty carrier from the inside to the outside is reported to be the slowest step the GLUT transport cycle.257,258 As such, one might expect that the rate of forming the outward-conformation is fine-tuned by the intracellular salt bridge network.259 Consistent with this rationale, recent mutagenesis between GLUT1 and GLUT3 in Xenopous oocytes have shown that their turnover rates are changed by selective mutations to the intracellular salt bridge forming residues.259 Moreover, a bound chloride ion was found to coordinate residues between N-terminal salt bridge interactions of GLUT1 and STP10 in the inward-facing structures,254,259 implying a potential allosteric site. In a similar manner, GLUT activities are thought to be dependent on anionic lipids because their negatively charged headgroups can compete for interbundle salt bridge formation to facilitate salt bridge breakage.113 Connected to the interbundle salt bridge network is a strictly conserved glutamate residue in the intracellular gating helix TM10b.3,78,235 Comparison of inward-occluded and inward-open structures reveals that the largest movement is TM10b,78,194,205,231 the symmetry related helix to TM7b (Figure 10c,d).78 It has been proposed that during formation of the occluded conformation, TM7b weakens its interactions with TM10b, which becomes more mobile and catalyzes interbundle salt bridge breakage.78
6.6. Asymmetrical Sugar Binding and Substrate Gating
Unlike the extracellular gate TM7b, the intracellular TM10b gate itself appears to have a more passive role in regard to sugar binding affinities or preferences, i.e., there is no equivalent residue to the TM7b asparagine that dramatically changes its positioning upon sugar binding. Furthermore, unlike the strictly conserved TM7b asparagine, the highly conserved tryptophan in TM10b is not always essential for transport.193 Here, for the first time, we propose that the different roles between the outside and inside gates are correlated with the fact that sugar binding affinities are (typically) asymmetric. As first demonstrated in the late 1970s,125 GLUT1 and related sugar porters have a ∼10-fold higher affinity for d-glucose on the outside as compared to the inside.120,260 Because the structure of the sugar binding site is identical in either conformation, we propose that the higher binding affinities from the outside are caused by differences between the TM7b and TM10b gates. We propose that the substrate sugar binds with higher affinity when it coordinates to the coupling asparagine in TM7b and facilitates transition of TM7b from a bent to an elbow-shaped helix in the occluded state. If one considers the occluded state of the transporter as analogous to the transition state of a soluble enzyme, then this reasoning fits the classical description of enzyme catalysis, in which there is relatively weak binding of the substrate to the enzyme but tight binding of the transition state.261 This comparison is consistent with the induced transition fit theory, which introduced the important concept that the energy barrier for conversion between states is lowered by substrate binding (Figure 13).165,262 The main fundamental difference is that in transporters the transition state is the activation energy barrier for global conformational changes, whereas in enzymes the barrier is imposed by substrate remodeling in the transition state.165
Figure 13.

An energy diagram for GLUT transport based on the induced transition fit (ITF) model for transport catalysis. Klingenberg introduced the ITF model165 that stipulates, similarly to enzymes, the substrate has a higher affinity for the transition state. The difference to enzymes is that substrate binding provides the catalytic energy for the rearrangements of the transporter, in contrast to providing the transition strain or destabilization of the substrate in an enzymatic reaction; to date, the induced transition fit theory model has largely been used to described the alternating-access mechanism of ADP/ATP exchangers.271 The energy barriers are low between empty (Cout, Cin) and bound outward- or inward-substrate-bound complexes (CoutSout or CinSin) because of the poorer fit of the substrate but are larger going into the transition state, and substrate binding largely compensates for the catalytic barrier (ΔG°). In more descriptive terms, between the outward- and outward-occluded conformations (PDB 4YB9, 4ZW9), the extracellular substrate-gating helix TM7b is mobile and samples either state (magenta and transparent). Substrate binding via an asparagine reside in TM7b conformationally stabilizes the outward-occluded state, thus increasing the likelihood for TM7b to break in the middle, completely close over the substrate, and interact more tightly with the substrate (see Figure 11). In the occluded state, the salt bridge interactions between ICH5 in the C-terminal bundle and ICH1–4 are weakened, which indirectly destabilizes the highly conserved intrabundle salt bridge network. Breakage of the intrabundle salt bridge network catalyzes the 16° global rocker-switch rearrangements of the N- and C-terminal bundles (Figure 8b). In the inward-occluded conformation (PDB 4JA3), the intracellular gating helix TM10b (cyan), which is related by inverted symmetry to TM7b, spontaneously moves from the inward-occluded to the inward-open conformation (PDB 4YB9). After sugar release, the sugar porter spontaneously resets itself to the outward-facing conformation through an “empty” occluded-state, but TM10b does not need to break. Spontaneous resetting means that the energetic barriers separating opposite-facing states must be low enough that the occluded state can form in the absence of sugar binding. Nevertheless, substrate binding catalyzes transport rates are significantly faster through “substrate-bound” versus “empty” occluded-state transitions. Adapted from Qureshi et al.193
Tighter binding of the substrate sugar in the outward-facing state might be required to ensure the intracellular salt bridge network is broken, which is also asymmetric as it is only formed in the outward-facing conformation.3,78,186,191 It is possible that the negatively charged lipid interactions to interbundle salt bridges may further ensure that the occluded state is not rate-limiting.263 However, TM10b only needs to spontaneously close to transition through the empty-occluded state. Consistent with sugar binding asymmetries, GLUT1 transport rates are asymmetric, with rates ∼100 times faster through a substrate-bound occluded conformation than through the empty occluded conformation,68,264 i.e., lacking energy from substrate binding to catalyze structural transitions through an empty occluded state (Figure 13). Moreover, the KM of GLUT1 for d-glucose is 10-fold lower at 7 °C than at 47 °C,265 and whilst enzyme rates increase to an optimum temperature,266 in GLUT1 the lower temperature selectively widens the differences between influx and efflux kinetics.264,267,268 In other words, at a lower temperature, the reduced GLUT1 dynamics selectively increases d-glucose binding affinities to the outside. Consistently, principle component analysis places the occluded state structurally closer to the open outward-facing conformation than the open inward-facing conformation (Figure 8b).193 Remarkably, the disease causing TM7b mutant in the GLUT1 deficiency syndrome (Thr294Met) was found to enlarge the differences between influx and efflux kinetic asymmetries 10-fold in comparison to the wild-type protein at room temperature.253 Taken together, the most likely answer to the some 50-year-old conundrum of how GLUT1 can have a single binding site when it binds d-glucose with much higher affinity on the outside than the inside, is because the gates are not symmetric. During influx, TM7b must break and close to accommodate the substrate in formation of its transition state, whereas during efflux, TM10b does not rearrange but simply closes. The GLUT kinetics is consistent with the proposed role of the TM7b extracellular gate for fine-tuning sugar affinities and kinetics via controlling the height of the energy barrier in the formation of the occluded state.193
Although sugar porters have a single binding site that the protein rearranges around, we cannot rule out the possibility that TM7b gating might be further sensitive to weak interactions between sugars and the external cavity during influx. In formation of the occluded state, the inward movement of TM7b opens an extracellular vestibule on the surface, which likely opens a hotspot for allosteric regulation (Figure 12c).193,233 In one of the occluded crystal structures of PfHT1, in addition to d-glucose binding to the canonical sugar-binding site, several d-glucose headgroups from the detergent nonyl-β-d-glucoside could further be modeled on either side of TM7b (Figure 12d).233 Thus, it seems entirely plausible that TM7b gating dynamics could further be modulated by low affinity sugar interactions in either the cavity or the extracellular vestibule and would not be inconsistent with the concept of the “multisite” model.269 Although sugar asymmetry appears to be a common theme in sugar porters, GLUT4 has reported to bind sugar symmetrically, and the influx and efflux kinetics are equivalent.270 Chimera studies between GLUT1 and GLUT4 mapped the differences in these kinetic barriers to TM6 in the N-terminal bundle.270 While substrate binding and gating appear to have predominantly evolved in the C-terminal bundle, the N-terminal bundle could nonetheless influence overall dynamics to influence how the transporter spontaneously resets, or relaxes itself, through the empty occluded conformation.
6.7. A “Latch” Mechanism for Proton-Coupled Monosaccharide Sugar Porter Symporters
While the substrate sugar is sufficient to catalyze the energy barriers between opposite-facing states, a proton-coupled mechanism ensures this process is coupled. The presence of an acidic residue in TM1 of proton-coupled sugar porters is key for H+ coupling.34,206,207 The mutation of the aspartic residue to asparagine in TM1 of XylE (Asp27) or GlcPSe (Asp22) converts the transporter into a variant capable of only downhill sugar transport.34,206 Most GLUT transporters have an uncharged alanine or asparagine in the equivalent position. In the outward-occluded structure of XylE, Asp27 in TM1 forms a salt bridge to Arg133 in TM4 (Figure 14a).177 The Asp27 residue is located 12 Å from the bound d-xylose. Given its salt bridge interaction with Arg133, the Asp27 must be in a deprotonated state, consistent with MD simulations.272 Yet, the fact that the substrate can bind in this conformation implies that protonation of Asp27 is not a prerequisite for substrate binding in XylE, unlike in LacY and other H+-coupled symporters, where protonation of a key residue in the substrate binding site is required in the wild-type situation.90 In contrast to LacY, the transporter XylE, like many of the monosaccharide sugar porters, lacks an ionizable residue in the sugar binding pocket (Figure 10b). Consistently, an Asp27Asn mutant in XylE and an Asp22Asn mutant in GlcPSe binds d-xylose and d-glucose with the same affinity as wild-type, respectively.34,273 The structure of the H+-coupled glucose symporter STP10 was determined from crystals grown at low pH.234 At low pH, the equivalent aspartate (Asp42) is thought to be in the protonated state and no longer forming a salt bridge to the equivalent arginine (Arg142), and TM1 has moved in toward the bound d-glucose (Figure 14a). Indeed, MD simulations of STP10 have calculated an pKa of 8 for Asp42, and a salt bridge was only formed and maintained with Arg142 when Asp42 was deprotonated and in the negatively charged state.254 Because STP10 is required to be functional under low pH conditions, it is thought the lid domain controls its solvation to the outside so that Asp42 can still be efficiently protonated.234 It was proposed that protonation of the aspartic acid of STP10 and the inward movement of TM1 favors d-glucose binding as TM1 hydrophobic residues (Phe39 and Leu43) stabilizes the sugar binding pocket.234 This model, however, does imply the protonation of Asp42 was a prerequite for d-glucose binding; unfortunately, as far as we are aware, the binding affinities of the Asp42 to asparagine mutant are not known.
Figure 14.

A latch mechanism for proton-coupled monosaccharide sugar porter symport. (a) Superimposition of the d-glucose bound structures of E. coli XylE (PDB 4GBZ) and plant STP10 (PDB 6H7D) showing the difference in the position of TM1 when the critical TM1 residue for H+ coupling (D27 in XylE) is protonated due to crystallization at a low pH of 4 and no longer interacts with the TM4 arginine. (b) A latch mechanism for H+-coupling for monosaccharide sugar porters. Protonation of TM1 aspartic acid in the outward-open state allows it to move closer to TM7b. If TM7b has bound a substrate, then the interactions with TM1 will catalyze formation of the occluded state, which is a prerequisite for alternating-access. In this model, both intrinsic TM1 and TM7b dynamics and interactions are also important to achieve H+-coupled symport, and therefore protonated symporters are not the equivalent of uniporters.
Here, we suggest a “latch” mechanism that considers the occluded PfHT1 structure193 represents a common transition state structure that is formed by all the monosaccharide sugar porters. In the occluded conformation, the extracellular TM7b gate is broken and has moved closer to TM1 (Figures 11a, 12a, and 14b). It seems likely that TM1 needs to be mobile enough to interact fully with TM7b to facilitate its transition from a bent to elbow-shaped helix. Indeed, in PfHT1, point mutations of polar residues in TM1 interacting with TM7b abolishes transport (Figure 11d,e).193 Simply put, a deprotonated TM1 aspartic acid forming a salt bridge to the TM4 arginine, restricts the movement of TM1 so that it cannot come close enough to TM7b and facilitate its transition into the fully occluded conformation (Figure 14b). Protonation of the aspartic acid, however, removes this “latch”, enabling TM1 to come close enough to TM7b. If TM7b interacts with a bound substrate and has moved to an outward-occluded state, then TM1 can facilitate TM7b into the occluded state. Consistent with this rationale, recent HDX-MS and MD simulations of XylE concluded that (i) when Asp27 is negatively charged, the binding of d-xylose stabilizes the outward-facing state and is stably bound, (ii) upon Asp27 protonation, TM1 become more dynamic and increased water solvation that leads to d-xylose adopting multiple states, and (iii) that Asp27 protonation is a critical switch for conformational transition into the inward-facing state.272
What is appealing with this simple “latch” mechanistic concept is that it also means the intrinsic dynamics of both TM1 and TM7b helices are important in achieving H+ coupling. Indeed, the Asp27Asn mutant in XylE is not capable of sugar influx (so-called zero trans transport) but can only uptake radiolabeled d-glucose when driven from the inside (counterflow transport)34 and, as such, the Asp27Asn mutant is nonfunctional in E. coli.274 In other words, the Asp27Asn mutant creates a defective outside gate, as the TM1 mutant cannot facilitate TM7b occlusion. On the other hand, the intracellular TM10b gate is unaffected by the TM1 Asp27Asn mutation and can still be used to drive sugar uptake by counterflow. Although the TM1 mutant is defective, it still retains an ability to enable spontaneous closing of TM7b and uptake low levels of radiolabeled sugar from the outside. Consistently, MD simulations show that the energetic landscape in XylE and GLUT1 are different beyond the presence or absence of an acidic residue in TM1.274 Interestingly, a seven amino acid mutation was constructed in XylE based on differences in conformational contacts in GLUT1 versus XylE. This XylE mutant was now able to transport low levels of D-xylose in E. coli.274 Three out of the seven mutants identified map onto the TM1 and TM7b interface, further providing support for importance of TM1 and TM7b interactions and dynamics.
On a more general level, evolved TM7b gating dynamics and the interaction with TM1 further provide an explanation as to why other GLUT isoforms, such as GLUT2, harbor an aspartic acid in TM1 but are not H+-coupled.34 Nevertheless, a more quantitative description of the free energy sugar porter landscape and the role of dynamics in H+ coupling in sugar porters are required to develop deeper mechanistic models. For example, SSM-based electrophysiology measurements of GlcPSe have shown that, unlike XylE, H+ and sugar transport may not always be strictly coupled.132 The potential explanation for this behavior is that GlcPSe lacks a nearby glutamate residue that can further fine-tune the pKa of the TM1 aspartate as it does in XylE.132,272 Though unclear, the data nonetheless implies that the energetic differences between H+ bound and unbound events can be low enough that H+ and sugar translocation are not always strictly coupled. Indeed, just as mutants can be found that abolish the requirement for H+-coupling, other mutants can also lead to H+-leaks as shown in LacY.275,276 H+-leak pathways are best characterized by comparing presteady-state and steady-state kinetics.200 Uncoupled substrate and H+ transport is unlikely under most normal conditions as it would create a futile transport cycle and dissipate the electrochemical gradients established by the cell.4 However, it has been put forward that “slippage” could be important under conditions of transitory high intracellular accumulation or large transmembrane ion gradients by acting as a sort of safety valve.4
6.8. Summary of the Rocker-Switch Mechanism in Monosaccharide Sugar Porters
Most MFS transporters, with the exception of antiporters, must spontaneously reset themselves from an inward-facing conformation to an outward-facing conformation. This requirement means that both local- and global-conformational rearrangements have to take place in the absence of substrate binding. This requirement implies that the energetic barriers separating opposite-facing conformations must be fairly low, as if they were not, the transporter would be arrested in an inward-facing state after releasing the substrate. One can start to understand how weakly binding substrates can drive seemingly large conformational changes. The binding of a substrate is just required to conformationally stabilize a state that can already be spontaneously populated. However, for a sugar to be a substrate, it not only has to bind but must also induce formation of the transition state.
To summarize the rocker-switch alternating-access mechanism for GLUTs and related monosaccharide sugar porters, in the outward-open and outward-occluded conformations, the substrate-gating helix TM7b is mobile and samples either state. Substrate binding on the outside conformationally stabilizes the outward-occluded state by recruitment of the TM7b sugar-coordinating-asparagine, thus increasing the likelihood for TM7b to break in the middle as fine-tuned between interactions with TM1 and possibly other transient sugar binding interactions to TM7b. A bound sugar, that is also a transported substrate, enables TM7b to break and completely close to fully accommodate the sugar. If the sugar porter is H+-coupled, then the TM1 aspartate first needs to be protonated to remove its salt bridge “latch” so that TM1 is mobile enough to interact with TM7b and facilitate its transition into the fully occluded state. In the occluded conformation, the salt bridge interactions between the C-terminal bundle intracellular helices and the N-terminal intracellular helices are broken, removing one of the restraints between the N- and C-terminal bundles. Further, with weakened contacts between the TM7b and TM10b gates, a more mobile TM10b triggers destabilization of the interbundle salt bridge network. Breakage of the interbundle salt bridge network catalyzes the global rocker-switch rearrangements of the N- and C-terminal bundles. In the inward-occluded conformation, the intracellular gating helix TM10b spontaneously moves to the inward-open conformation. After sugar release, the sugar porter spontaneously resets itself to the outward-facing conformation through an “empty” occluded state that is typically the rate-limiting step of the transport cycle, which is further fine-tuned by the rate of interbundle salt bridge reformation and specific lipid–protein interactions (Figure 14).
7. The Rocker-Switch Mechanisms of MFS Multidrug Resistance (MDR) Transporters
The most widespread and understood microbial MFS-MDR transport systems belong to the 12-TM drug:H+ antiporter 1 (DHA1) and 14-TM drug:H+ antiporter 2 (DHA2) families32,277 (TC nos. 2.A.1.2 and 2.A.1.3, respectively). MFS-MDR transporters are antiporters, which recognize and extrude a large range of structurally unrelated drugs from the cell using the free energy released from the downhill flux of ions along their electrochemical gradient.91,217,278 MFS-MDR transporters are found across all kingdoms of life, but are most highly represented among microbial genomes, where they render cells resistant to multiple drugs.32,58 These transporters contribute significantly to drug resistance in Gram-positive bacteria in particular. Efflux pump-mediated antibiotic resistance is a growing problem for the treatment of pathogenic bacterial infections. The first characterized microbial MFS-MDR transporter was NorA, which was discovered in quinolone and methicillin resistant Staphylococcus aureus clinical isolates.279 Elevated expression levels in plasmid borne copies of the 14-TM QacA and QacB MFS-MDR pumps have further shown a high degree of drug resistance in clinically relevant isolates of S. aureus.280,281
Bacterial genomes harbor MFS-MDR transporters that are also capable of exporting native cellular constituents, and therefore MFS-MDR activities are not always contributing to drug resistance.282 For example, a putative Gram-positive MFS-MDR transporter known as Blt exports a number of toxic molecules from the cell, but it is encoded in a polycistronic operon with an enzyme that catalyzes the acetylation of a cellular polyamine.283 It has been shown that Blt can remove polyamine from the cell and, based on its genetic arrangement, it is likely that polyamine is its physiological substrate.284 Another example is the bacterial homologue of mammalian VMAT, which in mammals transports monoamine neurotransmitters into presynaptic neuronal vesicles.285 The Brevibacillus brevis monoamine transporter (BbMAT) transports not only monoamines but also multiple drugs.286 While MFS-MDR transporters show substrate promiscuity, the drug:H+ antiporter MdtM in E. coli is only thought to catalyze Na+(K+)/H+ exchange in order to regulate intracellular pH under external alkalization conditions.287 Thus, MFS-MDR transporters do not only expel drugs but can have other, specific and physiologically relevant substrates.
Putative MFS-MDR transporters are widely distributed in pathogenic fungi such as Candida, Cryptococcus, and Aspergillus,288,289 but many of the analyses regarding MFS-MDR transporters come only from studies in the nonpathogenic baker’s yeast S. cerevisiae, as it is tractable to genetic engineering and knock-in/-out studies.290,291 In total, 22 putative MFS drug transporters have been reported to be present in the genome of S. cerevisiae.292 ATP-binding cassette (ABC) transporter and MFS transporter are two major families potentially contributing to pleiotropic drug resistance in fungi. However, compared with the ABC transporters, the role of the MFS transporters in MDR is still poorly understood. One possible reason is that MFS transporters often share overlapping drug specificity with ABC transporters.289 For example, Flr1p (MFS) contributes to resistance to the antifungal agent fluconazole and cycloheximide,293 but Pdr5p (ABCG) also mediates tolerance to both of these compounds and is clearly a major determinant in the resistance phenotype to these drugs.294 In fungi, the physiological roles of many of MDRs are well investigated, e.g., review for S. cerevisiae.295 For example, Dtr1 from S. cerevisiae was shown to be essential for biosynthesis of the spore wall by facilitating the translocation of bisformyl dityrosine through the prospore membrane during spore wall maturation.291 Another example is polyamines transporters. Four DHA1 family proteins (Tpo1–4) are involved in polyamine transport among the DHA1 family, which are essential organic cations for regulating nucleic acid and protein synthesis.296 Tpo1–4 confers resistance to toxic concentrations of the polyamines spermine, spermidine, and putrescine.296 As such examples shown, due to the relative ease of handling yeast genetics, most of the understanding of physiological functions has been identified in S. cerevisiae. Contrarily, only a few DHA transporters have been linked to antifungal drug resistance in pathogenic fungi.288,297 For example, two MFS transporters, CaMDR1(298) and Flu1,299 in C. albicans are involved in azole and fluconazole resistance, respectively, but the native function of these transporters is not known, most likely due to a lack of characterization efforts.295
In higher eukaryotes, the roles of MFS-MDR transporters are even less well understood. In plants, MFS-MDR transporters make a primary contribution to cellular detoxification processes but also contribute to numerous processes essential for optimal plant growth and development.56,300 The multiantimicrobial extrusion (MATE), the ATP-binding cassette (ABC), and the major facilitator superfamily (MFS) are the main plant transporter families.56,300 A number of MFS-MDR homologues are present in plants, and they transport a variety of nutrients, such as phosphate, nitrate, and amines as original function,301 but only a few have “MDR-like” functions reported. One example is the ZIF1 and ZIFL2 proteins that protect cells from toxic effects: ZIF1 from noxious concentrations of zinc and ZIFL2 from cesium and excess potassium.302 Some transporters might unexpectedly confer MDR activity. The tetracycline transporter-like protein (TETRAN) is an example of this. In the process of searching for resistant genes for nonsteroidal anti-inflammatory drugs (NSAID), the DHA1 family Tpo1 member was identified in yeast. Overexpression of TPO1 enabled NSAID resistance. A possible human orthologue of Tpo1, referred to as TETRAN, has also been identified and in cultured human cells caused resistance to some NSAIDs.303 In mammals, MDR has been focused on the ABC transporters, such as P-glycoprotein,304 while MFS-MDR members belonging to the DHA family are less understood. While monocarboxylate transporter (MCT),72 oligopeptide transporters (PepT),70 and organic anion (OAT) and organic cation (OCT)62 are all MFS members capable of transporting drugs, the MFS-MDR members belonging to the DHA1 and DHA2 families specialize in drug export. Here we have focused on MFS-MDR proteins belonging to these families only, but it is worth mentioning that the mechanistic themes are likely to be applicable to other promiscuous MFS transporters.
MFS-MDR transporters use the proton motive force to drive the efflux of chemically distinct substrates. MFS-MDR transporters are able to expel either charged or neutral substrates, meaning that they are also capable of electrically distinct transport reactions. The characteristics of the bacterial inner membrane (Δψ, inside negative, and ΔpH, inside alkaline) and the degree to which these features contribute to the movement of a given solute, explains the versatility of efflux pumps in handling electrically distinct substrates. A prominent characteristic of MFS-MDR transporters is that many have overlapping substrate profiles, and most appear to have promiscuous substrate-binding sites. To achieve flexible recognition of structurally divergent compounds, the substrate binding pockets are typically large and flexible and bind substrates through a number of nonspecific electrostatic and hydrophobic interactions.148 A shared feature of compounds recognized by MFS-MDR members is that they are highly hydrophobic and harbor at least one aromatic moiety.217 Much of understanding of the molecular mechanisms of drug transport in MFS-MDR transporters have been developed on several bacterial transporters. Several MFS-MDR antiporter structures belong to the DHA1 family have been determined from E. coli, namely EmrD,178 YajR,305 MdfA,306−311 SotB,312 and LmrP from Lactococcus lactis.221
More recently, the first DHA2 family MDR transporter NorC from S. aureus was determined, which in contrast to DHA1 members have 14-TMs.313 MdfA and LmrP are biochemically well-characterized, while the functional analysis of YajR, EmrD, SotB, and NorC is less well-known.312 Structures of MdfA have been captured in multiple states over the past few years, including an apo-form of the outward-open conformation and several mutant structures in the inward- and inward-occluded conformations with either cationic, neutral, or detergent molecules bound (Figure 15).306−311 Very recently the sugar efflux member SotB from E. coli was determined in multiple inward-open states and an occluded conformation with substrate bound.312 For the other MFS-MDR proteins, single or single-facing conformational states have been determined in the occluded conformation (EmrD178), or outward-open conformation (YajR305 and LmrP221). The NorC structure was determined in an outward-open conformation in complex with a single-domain camelid antibody bound at the extracellular cleft of the NorC. The complementarity determining region (CDR) of the antibody was deeply inserted into the outside cavity and was thus blocking substrate accessibility.
Figure 15.

The major conformations in the transport cycle of an MFS-MDR DHA family transporter. Schematic illustrating major conformations of an H+-coupled MDR-MFS transporter cycle: outward-open, outward-occluded with bound substrate (light-yellow hexagon), occluded with substrate, inward-occluded with substrate, and inward-open and occluded with no substrate. N-terminal (light-blue) and C-terminal bundles (yellow) are shown. The order of protonation and substrate bind/release is not fully confirmed yet. The abbreviations are as follows: chloramphenicol (CLM), deoxycholate (DXC), n-dodecyl-N,N-dimethylamine-N-oxide (LDAO), isopropyl β-d-thiogalactoside (IPTG), methyl viologen (MV), phosphatidylglycerol (PG), and Indian Camelid antibody (ICab).
7.1. The Drug Binding Pocket and Promiscuous Substrate Recognition
In MdfA, the highly conserved TM1 acidic residues Glu26 and Asp34 are both required to achieve H+-coupled efflux of positively charged substrates, whereas only one out of these two residues are required to expel noncharged substrates.217,220,314 While the substrate binding residues vary, all substrates directly form electrostatic interactions with Asp34 in TM1, which also serves as the principal H+-coupling residue (Figure 16). Despite the variation of substrate-binding modes, mutation of substrate coordinating residues significantly reduces the efflux of monocationic compounds and neutrally charged drugs. Remarkably, however, it is possible to remove both acidic residues in TM1 (Glu26Thr and Asp34Met) and then add back to an acidic residue to TM5 (Ala150Glu) to recover H+-coupled drug efflux.181 Interestingly TM5MdfA mutant structure shows that the substrate reorients ∼180° in the binding pocket to fulfill H+ coupling with electrostatic interactions to Glu150.311 Moreover, with the addition of a third acidic residue in the wild-type substrate binding site (as Ile239Thr/Gly354Glu), MdfA is now able to efflux the doubly charged compound methyl viologen (MV).218 The crystal structure of this gain-of-function mutant shows that it might be capable of transporting two zwitterionic LDAO detergent molecules by binding at two distinct sites. Specifically, one LDAO molecule interacts to the principle H+ binding site (Asp34), whereas the other binds to the newly introduced glutamate (Glu354) (Figure 16b). Collectively, biochemical and crystal structures highlight the remarkable and inherent plasticity of the MdfA substrate binding pocket to expand both its drug profile and energetics to do so.311 Consistently, it has been shown that shifting the H+ coupling site in LmrP also preserves H+-coupled drug efflux.315
Figure 16.

Promiscuity of substrate binding in MdfA. (a) The central substrate binding cavity of MdfA viewed from the membrane side. Bound substrates (sticks) observed in crystal structures are superimposed in a single structure (PDB 4ZP2). Depending on the substrate and MdfA mutant, a varying subset of residues are involved in substrate binding. The residues from both N- and C-terminal domain directly make contact to the substrate. N-terminal and C-terminal bundles were shown as blue and light-yellow, respectively, with substrate (LDAO (orange), chloramphenicol (CLM-yellow), deoxycholate (light-green), and methyl viologen (MV) as black sticks). The acidic residues interacting with the substrate via electrostatics interaction are shown in yellow. (b) The LDAO binding site in MdfA mutants, Q131R (PDB 4ZP2), E26T/D34M/A150E (PDB 6VS2), and I239T/G354E (PDB 6OOM) reveals that the substrate is hydrogen-bonded to an acidic residue. LDAO1 interacts with D34 via long charge–charge interactions due to rearrangement of interaction caused by the mutation I239T and G354E. The LDAO2 molecule occupies the site 5.5 Å away from the LDAO1 molecule toward the cytoplasmic side of the binding pocket and is hydrogen-bonded to Q357. The residue G354E interacts via a long charge–charge interaction. (c) The CLM binding site in Q131R (PDB 4ZOW) and E26T/D34M/A150E (PDB 6VRZ) mutant. (d) The MV, a dicationic compound, binding site in a I239T/G354E (PDB 6OOP) mutant. MV interacts via long charge–charge interaction with the D34 and G354E.
The flexible nature of the substrate binding site was apparent in computational simulations of EmrD, whereby fluctuations of side chains were modeled to have a considerable effect on the cavity’s flexibility, even in the absence of substrate.316 Intrinsic EmrD dynamics reveals states in which the cavity is wider than necessary for the substrate CCCP. This could indicate that EmrD is able to simultaneously accommodate multiple substrates in the central cavity.317 In the recently determined LmrP structure,221 a lipid molecule was bound to the central cavity together with the substrate Hoechst 33342, which is a cell-permeable DNA staining dye. It is not fully understood but is plausible that specific lipids may control the substrate binding pocket itself, contributing further to substrate promiscuity. The flexibility of H+ coupling and substrate binding appears to be a conserved feature of MFS-MDR transporters, with obvious physiological benefits. As previously mentioned, substrate promiscuity is a clear feature of other MFS transporter members, such as the oligopeptide transporters that appear to be able to accommodate peptides with different binding modes318−320 and H+:substrate stoichiometries.216 Indeed, like MdfA, the prototypical E. coli oligopeptide transporter YdgR has been also shown to be able to transport chloramphenicol,321 which is analogous to the ability of human PepT1 and PepT2 homologues to transport β-lactam containing antibacterial and antiviral compounds.64,71,322
7.2. Proton–Substrate Coupling
MdfA is a promiscuous drug transporter as it couples the extrusion of cationic, neutral, and zwitterionic compounds using the proton gradient with a drug/H+ stoichiometry of 1:1.217,220,314 However, how and when proton coupling takes place is still unclear. The triple MdfA mutant Glu26Thr, Asp34Met, Ala150Glu, sheds some light into proton coupling pathways, as the introduced Glu150 becomes the only acidic residue in the substrate-binding pocket. Titration experiments have demonstrated that while the MdfA Gly354Glu mutant binds two zwitterionic detergents and releases two proton’s (1:1 stoichiometry), the triple mutant only triggered the release of a single proton. The addition of an acidic residue to wild-type MdfA in a different location to Glu150 (position Gly354), however, handles the H+/substrate coupling differently, as Asp34 and the newly added acidic residue are functionally not equivalent, being located at opposite ends of the binding site (Figure 16).218,310,323 Collectively, Asp34 in TM1 has a central role in the wild-type MdfA,310 but if the acidic residue is removed and placed elsewhere in the binding cavity (e.g., Glu150 in TM5), the substrate can adapt how it binds.311 In other words, the same substrate is coordinated by different residues so that it maintains an interaction with at least one acidic residue, which is essential for substrate binding (Figure 16). In this case, the change of overall structure is subtle with only small substrate-induced differences.
In contrast to MdfA, an electrogenic drug/nH+ (with n ≥ 2) transport mode is the default coupling stoichiometry in the MFS-MDR antiporter members LmrP, NorA, and QacA.324 In LmrP from L. lactis, the proton motive force drives the extrusion of propidium with an apparent propidium2+/3H+ antiport stoichiometry.325 There are three negatively charged residues, Asp142TM5, Asp235TM7, and Glu327TM10, that can take part in H+/substrate coupling between the N- and C-terminal bundles. Replacement of these carboxyl groups alters the drug/nH+ stoichiometry, i.e., 3H+/propidium2+ to electroneutral mode, 2H+/propidium2+, 2H+/ethidium1+ to 1H+/ethidium1+.325 While the two carboxyl groups are required for propidium binding, loss of one of these carboxylates in the C-terminal domain was not required for ethidium transport. This data suggests that a dedicated H+ binding site, as well as a flexible H+/ligand binding site is required, depending on the charged status of the exported substrated.325,326 In LmrP, the addition of an acidic group in the central cavity had wild-type-like electrogenic transport for both propidium and ethidium, whereas its placement in a different location could only recover electrogenic transport for ethidium but not for propidium.315,325 These findings further highlight the functional redundancy in the H+ binding sites and, given the large distances to the substrate binding site at ∼15 Å, highlights the plasticity of proton coupling.315
7.3. Conformational Switching
How does proton and/or drug binding catalyze conformational rearrangements? The MFS-MDR transporters harbor an “antiporter motif” localized to TM5.280,327,328 The antiporter motif “XPXXXP” containing the interspersed proline residues is highly conserved and is expected to have an essential role in antiport activity.277 Studies have shown that the antiporter motif acts as a molecular hinge for the N- and C-terminal bundles to rock against each other.327,329 In the outward-facing conformation, TM5 in MdfA has a 15° kink that is accompanied by a ca. 45° clockwise twist that terminates with the two proline residues in the antiporter motif (Figure 17a,b,e). In the inward-facing conformation, TM5 in no longer bent (Figure 17c,d).306,308 How TM5 bends in the outward-facing conformation is unclear, but the bending motion could be a general mechanism for conformational switching in the MFS-MDR. In addition, the hydrophobic cluster in TM4 has slightly shifted its position between outward- and inward-facing conformations. In the outward-facing structure of MdfA, the cytoplasmic side is closed off by numerous interactions between the N- and C-terminal bundles. In particular, TM5 juxtaposes against the ends of TM8 and TM10, and TM11 nestles between the ends of TM2 and TM4. Hydrophobic side chains from each of these helices pack against each other to form a hydrophobic seal to the cytoplasmic face, supported by further electrostatic interactions.
Figure 17.

Proposed transport cycle of MdfA. (a) Exposure to the low pH periplasmic space in the outward-open state allows acidic residue D34 protonation. (b) Upon protonation of D34, the periplasmic region of MdfA begins to close, leading to an occluded state. (c) A H+ is likely to be released from Asp34 to the cytosol in a Glu26-mediated manner (this state is not confirmed yet) upon substrate binding. (d) Substrate bound inward-facing form. Substrate binding triggers a conformational switch that exposes the substrate binding pocket to the extracellular side (periplasm). (e) Substrate is released to the periplasmic side. The helix bending and twisted motion of TM5 (black arrow) and the expansion of hydrophobic cluster in the periplasmic facing domain between TM1 and TM2 are likely to be important for driving the conformational cycle. This process possibly occurs the state between (b) and (d). Key functionally important residues (Glu26, Asp34, and Arg112) are shown. The schematic representation of transport cycle of MdfA (center) with functionally important TM1 that has a protonation site shown in blue (Asp34, star; cyan, deprotonated; red, protonated state) and the gating helix TM5 in either green (bent helix) or pink (straight). The substrate is shown as a yellow hexagon. The order of protonation/deprotonation and the connection with substrate binding and release has not yet been established in detail.
MD simulations of MdfA have concluded that the protonation of Asp34 in the outward conformation immediately leads to an occluded state of MdfA with an energetically favorable direction,308 but TM5 remains kinked (Figure 17b). The protonation of Asp34 in the inward-facing conformation also leads to an occluded state with a kinked TM5. This implies that the protonation of Asp34 in both the outward and apo-inward facing conformations drives occlusion; indeed, in EmrD, an acidic residue (Glu227) was also proposed to be important for the transition between the occluded to the outward-open state.316 In MdfA, the conformational rearrangements between the occluded to the inward-facing conformations may result in the untwisting of TM5. Notably, the orientation of an TM4 residue (Tyr127) could be coupled with movement of an arginine residue (Arg112), which is essential for transport.220 The buried guanidinium moiety is part of an elaborate hydrogen bonding network involving a buried glutamine, a glycine carbonyl, an asparagine, and Asp34. Changes in the bulk solvent (e.g., substrate binding and/or protonation) could drive the observed reorganization of the hydrophobic cluster immediately adjacent to the arginine. In turn, communication of this change coupled with H+ or substrate binding dictates the position of TM5 and repositioning of the hydrophobic clusters associating with large conformational changes. Indeed, in MdfA there are tyrosine residues (Tyr127) located in TM4, which are rearranged by the hydrogen bonding network that also includes a glutamic acid (Glu26).
What is the preferred conformation in MdfA? The answer to this question is unclear, but MD analysis308 cross-linking experiments309 have indicated that the inward-facing (most likely inward-occluded) conformation was preferred upon protonation of Asp34 under physiological conditions. In contrast, electron paramagnetic resonance (EPR) data in detergent has shown that MdfA was a predominantly in an outward-open conformation.330 More recent EPR data for MdfA embedded into nanodiscs, have rather concluded that MdfA samples both outward- and inward-occluded conformations.331 These results probably reflect the low energetic barriers between opposite facing conformations, which are almost certainly shaped by the lipid bilayer composition. Interestingly, either the monocationic substrate TPP or protonation of Asp34 did not induce large conformational changes in MdfA-embedded nanodiscs.331 These results suggest that MdfA in lipids may not open up to “the conventional wide-open cavity” observed in crystal structures but rather that it may use smaller rearrangements to achieve alternating access. Indeed, it is possible that hydrophobic substrates could enter into the substrate binding cavity through lateral diffusion from the lipid bilayer itself.331
As in most MFS transporters, interbundle salt bridges are broken and reformed during transport. The initial structure of MdfA was determined for a Gln131Arg mutant in the N-terminal bundle, which is nonfunctional as it confers almost no resistance against either chloramphenicol or cationic compounds.309 It was, however, possible to restore some activity for the MdfA mutant (Gln131Arg) by removing a positively charged residue in the C-terminal bundle. The double mutant Gln131Arg/Arg336Gln, could transport some but not all of the different compounds of the wild-type protein and was therefore less promiscuous.309 Interestingly, a single positive charge inserted into this location also inhibited transport activity.309 The impaired transport might be caused by electrostatic repulsion between the two positively charged residues in each of the bundles and could partially limit the drug-induced conformational response. Although the turnover rates of the different mutants are not known, a likely explanation is that the loss of extra charged interactions makes the barriers between inward and outward-facing states somewhat lower, and so the crippled transporter is only capable of transporting substrates that require less catalytic energy.
7.4. Local Gating Mechanisms in MFS-MDR Transporters
Owing to larger structural differences between different MFS-MDR members, it is difficult to reach a consensus for the conserved conformational changes by comparing multiple states of different homologues. MdfA is the only MFS-MDR member where structures have been determined in both outward- and inward-facing conformations. The most obvious local rearrangements are centered on TM5 in the N-terminal bundle, which changes between a bent and straight configuration during rocker-switch global rearrangements.308 In SotB, there are two structures in the inward-facing conformation that differ by the width of the distance between the N- and C-terminal bundles, highlighting the inherent flexibility between the two bundles.312 SotB is able to efflux IPTG and arabinose in E. coli.332 Interestingly, SotB has also been determined in an inward-occluded state with IPTG bound.312 Although the physiological function of SotB is poorly understood, between the inward-open and the inward-occluded states, most of the rearrangements can be described as rigid-body rearrangements showing classic rocker-switch transitions. Comparing just the inward-facing conformation that has a narrow intracellular cavity with the inward-occluded conformation, shows that the N-terminal helices TM4 and TM5, however, may need to move more than their symmetry related helices in the C-terminal bundle.312 Nevertheless, it was concluded that nonlinear rigid-body rearrangements were apparent in both the N- and C-terminal bundles and, indeed, the substrate is evenly coordinated by TMs in both N- and C-terminal bundles.312
Overall, there are no obvious gating helices driving substrate occlusion observed for MFS-MDR members to date. Yet, given the large structural diversity of this family, it seems likely that some members will show large asymmetry during transport. Indeed, in the MDR antiporter YajR,305 the structure clearly has a broken N-terminal bundle TM4 helix (Figure 7c), which indicates that substrate occlusion could be achieved by local gating rearrangements in the N-terminal bundle; currently, the substrate preferences for YajR are unknown. Moreover, YajR has been used for modeling the structure of the vesicular monoamine transporters (VMAT), which belong to the DHA1 family. Indeed, three gain-of-function mutations in rat VMAT enable the transport of drugs,333 and the bacterial homologue BbMAT is capable of transporting multiple drugs.286 Using the homology model based on YajR,305 a cytoplasmic gate has been proposed between TM5 and TM1145 yet is unclear if the N-terminal bundle will contribute more than the C-terminal bundle to gating. In EPR studies of LmrP, flexible motions of TM8 was observed in the presence of substrate and this helix is thought to act as a substrate-gating helix.334 Molecular dynamics analysis of EmrD have also suggested that TM8 might act as a gating helix316 but lacks experimental evidence.
7.5. Comparison of Rocker-switch Mechanisms between Sugar Porters (SP) and MDR Antiporters
Well-established models of monosaccharide sugar porters and MDR transporters are likely to represent opposite ends of an MFS alternating-access spectrum. MDR transporters, as typified by MdfA, have large substrate binding cavities that are used to select compounds based on their overall size and charge through long-range electrostatic interactions. This flexibility enables them to recognize many different compounds as well as enabling flexibility in H+ coupling in order to energize transport, presumably to enable them to transport multiple-charged substrates if needed. Monosaccharide sugar porters, as typified by the GLUTs, on the other hand, have narrow polar cavities with a highly conserved sugar binding site, which is highly specific for certain sugars. When H+ coupled, such as in the bacterial homologues XylE and GlcPSe, a single residue is strictly required to energize transport and, to date, there is no evidence that the acidic residue can be shifted to a different helix and retain active transport. Rather than changes to the chemistry of the substrate-binding site, they appear to have evolved the extracellular gate TM7b to fine-tune their substrate preferences and kinetics. Indeed, sugars are only coordinated directly by a single residue in the N-terminal bundle, while all other residues are located in the C-terminal bundle, predominantly on the TM7b and TM10b gating helices. In contrast, substrate binding residues in MDR transporters appear to be evenly distributed in both N- and C-terminal bundles and local gating rearrangements are thought to be either minimal or spread evenly across both bundles.
Taken together, while the MFS-MDR crystal structures determined to date appear to operate more closely with a classical rocker-switch definition, the GLUT transporters almost blur the lines between “rocker-switch” and “rocking-bundle” alternating-access definitions, in which conformational asymmetry is immediately apparent by their structures. If one considers the GLUT transporters as analogous to rocking-bundle proteins (also referred to as gated-pores), then their ability to allosterically modulated by their extracellular gate becomes an even more obvious characteristic, e.g., in the rocking-bundle transporter SERT the extracellular half-helix TM1a is a well-established site for allosteric modulation by ligands.335 In the recent structure of the malarial parasite transporter PfHT1, an antimalarial compound was also found to enable selective inhibition by blocking TM7b gate closure.233 It should be stressed, however, that we retain using separate “rocker-switch” and “rocking-bundles” definitions and, although partly historical-based, it is not intended that they represent absolutes but are rather useful starting schematics for which mechanistic details can be compared with and built upon. Owing to the large variety of MFS-folds and substrate gating rearrangements, within the large family sugar porter and DHA subfamilies, we expect other MFS members will demonstrate conformational changes and transport behavior that are more similar to one another than these examples highlighted here. For example, similar to DHA1 members, in the disaccharide sugar porter Mal11, it was possible to retain H+ coupling by relocating the acidic residues to a different helix.109,213 Furthermore, the structure of the DHA1 member YajR,305 in particular, indicates that substrate occlusion might still be achieved by local gating helices from one of the two bundles in some cases.
Sugar porter and MFS-MDR rocker-switch mechanisms highlight the remarkably adaptability of the MFS fold for accomplishing substrate translocation. At a more general level, it seems that as the structural asymmetry between the two bundles increases, the substrate promiscuity decreases.3,336 For example, while there is highly specific Na+-coupled rocking-bundle and elevator proteins to reuptake individual neurotransmitters from the synaptic cleft after neurotransmitter release, the transporters exporting the neurotransmitters into synaptic vesicles are the lower-affinity H+-coupled transporters VMAT and VGLUT. As discussed, VMAT is able to transport all monoamines, and while VGLUT shows specificity for glutamate, it binds glutamate with much lower affinity than in the elevator-protein homologues (EAATs) and is also capable of transporting inorganic phosphate.115
8. MFS Transporter Complexes and Regulation by Lipids
8.1. Regulation of MFS Transport by the Lipid Composition
Membrane bilayers are assembled from different lipid classes with varying biophysical properties.337 Lipids regulate the functional activity, stability, and oligomerization state of many membrane proteins,337,338 including MFS transporters.172 Despite this prevailing view, due to technical hurdles, it is often difficult to dissect if it is the lipid bilayer properties themselves that are indirectly influencing functional activity and/or specific lipid–protein interaction(s) to the protein of interest.119,339 Furthermore, the majority of studies have analyzed the effect of different lipids to bacterial MFS function,172,340−342 in part due to the technical challenges in developing robust proteoliposome assays for eukaryotic MFS transporters.
The inner membrane of E. coli is made up of ∼70% phosphatidylethanolamine (PE), ∼25% phosphatidylglycerol (PG), and ∼5% cardiolipin (CL).343 From the few studies analyzing the influence of the lipid composition in MFS transporters, we know that the folding of LacY in E. coli requires the presence of PE,340 which can be restored upon the induction of a gene required for PE synthesis.344 PE is a nonbilayer forming lipid, and its presence increases membrane fluidity as well as introducing packing defects in the membrane.337 The endoplasmic reticulum membrane is highly enriched in PE lipids, and this is where eukaryotic membrane protein’s fold via the Sec61 translocon.337 However, not all MFS transporters require PE for obtaining the correct topology, as shown for the melibose transporter MelB, which obtains its correct fold in an E. coli PE deletion strain.342
Assuming proper MFS transporter folding, how do lipids influence activity? As previously discussed, MFS transporters harbor salt bridges within and between N-and C-terminal bundles, which are broken and reformed during the transport cycle.191 It is well-established that lipid headgroups can interact with charged groups to fine-tune energetic barriers imposed by salt bridge interactions. In LacY, for example, mutant cells lacking PE dramatically effects its turnover with a Vmax that is 5–10-fold lower relative to cells containing PE, with an KM that was unaffected.345 In LacY, after the release of sugar on the inside, deprotonation of Glu325 in TM10 is thought to trigger the resetting between inward- and outward-facing conformations.90 MD simulations of LacY have shown that the PE lipid headgroup interacts with Glu325 and facilitates its deprotonated and subsequent reformation of a salt bridge to Arg302.86 In contrast, when MD simulations were carried out in phosphatidylcholine (DMPC) lipids, the lipid headgroups were unable to interact in a similar manner to Glu325 and LacY remained too static.86 These results imply that the PE lipid is required to facilitate dynamics to enable correct proton-coupling rather than influence substrate binding (Figure 18a). Indeed, in the absence of PE lipid, LacY is still able to carry out passive downhill sugar transport.345
Figure 18.

Lipids and MFS transporter complexes. (a) Annular lipids (left) can indirectly influence transporter function by modulating lipid bilayer fluidity and packing; (left middle) lipids can directly interact with the transporter to regulate activity; (right middle) lipids can promote oligomerization such as cholesterol or cardiolipin; (right) lipids can behave like ligands and form part of the substrate binding site to promote or potentially inhibit transport. (b) Oligomeric stability scale (green) with buried surface area shown in parentheses, highlighting the weaker oligomeric surfaces become more dependent on lipids as measured by native MS (PDB 2A65 (LeuT), 4QND (Semi-SWEET), 4AU5 (NhaA), 4BWZ (NapA), 4GPO (β1-adrenergic receptor), 4DKL (μ-opioid receptor), and 4DJH (κ-opioid receptor)). (c) Glycoprotein targeting chaperone complexes between Basigin (green) and human MCT1 (blue and light-orange; PDB 6LZ0 and 7DA5) in the outward- and inward-open conformations (left, middle left), CDC98hc (red), and the LAT1 transporter (blue and light-orange; PDB 6JMQ) (middle right), and TLR3 (cyan) and the UNC93B1 (blue and light-orange; PDB 7C76). (b) Adapted from Gupta et al.350
For the melibose transporter MelB, it was shown that in an E. coli mutant strain lacking PE, both Na+ and H+-driven uptake were greatly impaired,342 despite controls showing that PE-lacking cells still establish the appropriate electrochemical gradient.345 In contrast, an E. coli strain lacking PG and CL dramatically diminished H+-coupled symport, but not the Na+-coupled symport of melibose. Because the presence of these lipids was not associated with sugar binding, it is likely the lipid composition has an indirect effect on MelB conformational dynamics and ion coupling,342 perhaps through interactions with ionic networks that are connected to the ion-coupled pathways.
In agreement with the results obtained for LacY and MelB, EPR data of LmrP346 and HDX-MS measurements combined with MD simulations for XylE, GlpT, and LacY, respectively, have also concluded that the headgroup of the PE lipid competes for ionic interactions to modulate preferred conformational states.172 In particular, the interbundle salt bridges that stabilize the outward-facing conformation.172 It appears that direct hydrogen bond(s) interactions between the PE headgroup and the transporter influences conformational dynamics, because if the amino headgroup in PE is methylated it affects both activity and conformational dynamics.346 Nevertheless, in all these cases, no clear PE binding sites have been uncovered, and so it is also plausible that the influences of PE are indirect through membrane bilayer properties.342,346 Given that 70% of the E. coli inner membrane is composed of PE lipids, although its presence is clearly required for function, it seems unlikely that the PE lipid is acting as a regulatory fashion through specific lipid-binding sites. In fact, there are only a few MFS transporters structures with clearly defined bound lipids. As previously mentioned, in the crystal structure of LmrP, a PG lipid was found bound to the central cavity, forming contacts with the cocrystallized substrate (Figure 18a).221 MD simulations indicate that the negatively charged lipid stabilizes the N-terminal bundle through ionic interactions that favor substrate binding.221 It will be interesting to ascertain if LmrP has evolved specific lipid-binding sites to expand substrate promiscuity.
Given the diversity and complexity of lipids in eukaryotic membrane bilayers, one might expect that eukaryotic MFS transporters might even be more finely tuned to their lipid composition. Indeed, in a large study monitoring the thermostability of transporters including MFS members, it was found that bacterial detergent-solubilized transporters were just as stable before and after purification in a mild detergent, whereas the eukaryotic transporters become significantly more unstable (Figure 5b,c).119 While the mammalian transporters like GLUT5 could be stabilized by the addition of brain lipids to the detergent purified samples, the addition of E. coli lipids had no stabilizing effect (Figure 5d,e).119 Given that dynamics and function are intimately entwined, this study implies that the greater sensitivity of the eukaryotic proteins to their environmental lipid composition might be connected with their clearer ability to be allosterically regulated by them. The greater sensitivity to their lipid composition could anecdotally explain why there are so few successful examples of eukaryotic transporters reconstituted into liposomes.120 From the few studies analyzing the role of lipids in eukaryotic transporters work on GLUT transporters, for example, it was concluded that functional activity requires the presence of anionic lipids as well as the inverted-cone lipid PE.113 Furthermore, it is thought that anionic lipids, such as phosphatidylserine (PS), are required to compete with the interactions between the interbundle salt bridges (Figure 9).172 Consistently, in both GLUT3 and GLUT4, an increase in anionic or conical lipids PE and DAG lipids increased turnover number linearly but had no measurable change on the Michaelis constant KM.113 This data highlights how sensitive GLUT transporters are to their lipid composition and how lipids can directly fine-tune the rate-determining step in their transport cycle. Interestingly, FRET studies analyzing the population dynamics of the oligopeptide transporter DtpA in detergent and lipid mimetics have also found the protein highly sensitive to its lipid environment, where negatively charged lipids POPA and POPS stabilize inward-open states.168 Given the intrinsic dynamics of MFS transporters167 and the high-frequency of salt bridges in MFS transporters between the ends TM2 and TM3 and/or between TM8 and TM9 (i.e., motif A),14,191 it is likely that turnover rates of many MFS transporters are likely to be fine-tuned by a composition of negatively charged lipids and lipids affecting membrane packing and fluidity.
8.2. Lipids, Oligomerization, and Complexes
The lactose transporter LacS is a dimer and coexpression of WT with a nonfunctional mutant abolishes H+-active transport, demonstrating cooperativity.347 Blue-native page and negative stain electron microscopy of the human H+-coupled folate MFS transporter PCFT demonstrates it is also a dimer.348,349 Interestingly, coexpression of wild-type PCFT with an inactive mutant, dramatically increases transport activity.348 By using native mass spectrometry approaches, it has been proposed that membrane protein’s with weak oligomerization interfaces are more dependent on lipids to enable oligomerization (Figure 18b).350 Certainly, this analysis could explain why many MFS transporters, such as GLUT transporters, have been reported to be oligomers in membranes351,352 but nonetheless have been extracted, purified and crystallized in detergent as monomers.78,192,231 Cholesterol was also found to be stabilizing to GLUT5 (Figure 5d), and given cholesterol is such a high fraction of the plasmamembrane,337 it is possible that this lipid may modulate oligomerization. A number of non-MFS neurotransmitter sodium symporters (NSSs) members have been found to have cholesterol binding sites that appear to promote homodimerization through TM11 and TM12 of their scaffold domains.350,353 Although the functional consequences of oligomerization and the role of lipids to influence higher oligomeric states is currently unclear, it is tempting to speculate that oligomerization could stabilize either the N- or C-terminal bundles during transport. In the rocking-bundle and elevator-alternating-access mechanisms, it becomes increasingly clear that lipids and oligomerization are important for stabilization of the less mobile domain elements during transport.3,119,354 Using this rationale, one might expect that oligomerization might be more important in MFS transporters, where one of the domains participates less in substrate-gating and transport and could therefore be less labile, e.g., the N-terminal bundle of sugar porters. Along these lines, the sugar porter PfHT1 purifies and crystallizes in a mild detergent as a dimer though interactions in the N-terminal bundle.193
Although dimer formation by interactions between the N-terminal bundles intuitively makes the most sense in sugar porters, there are examples of structural oligomers that have been found in the bacterial oligopeptide transporters that are mediated instead via the C-terminal bundles. In particular, orthologues sharing 23% sequence identity from Shewanella oneidensis (PepTSo and PepTSo2) are found to exist as either a monomer or a dimer and tetramer, respectively.159,355 A recent single particle cryo-EM structure of the 200 kDa PepTSo2 tetramer at 4.1 Å in the lipid mimetic saposin356 confirms the earlier crystal structures355 and the lower resolution cryo-EM reconstruction.357 Tetrameric organization of PepTSo2 is mediated through a small extracellular loop domain in the C-terminal bundle of one protomer and two asparagine residues in TM12 of the C-terminal bundle of another;356 the loop domain sequence is not found in PepTSo. It has been put forward that such an assembly might be regulated by lipids,356 yet the rationale and physiological significance of a tetramer remains unclear. Furthermore, because intracellular gating helices in PepTSo and PepTSo2 both involve the local movement of TM10 and TM11 in the C-terminal bundle in the transition from inward-occluded to inward-open states,107,355 it further makes such oligomeric assembles difficult to rationalize as this could potentially restrict local gating. Interestingly, bacterial oligopeptide transporters often have two additional helices to the canonical 12 TM fold (referred to as HA and HB), which are inserted after TM6 with the HA helix following the N-terminal bundle and HB the C-terminal bundle.107,159,355,358 At a structural level, the HA and HB helices appear separated from the rest of the transporter, and while these two helices would seem the most obvious regions to mediate homodimerization, this has not been observed to date and their functional role is also unclear.
As well as oligomeric contacts between either the N- or C-terminal domains, in the recent cryo-EM structure of the human monocarboxylate transporter 2 (MCT2), an extensive dimer interface of 5100 Å2 involving 4 TMs from both the N- and C-terminal bundles was observed.156 Because N- and C-terminal rocker-switch movements would be restricted by side-by-side dimerization of both bundles, the alternating-access mechanism of transport has been postulated to occur predominantly by only gating rearrangements.156 MCT2 has an unusually high number of broken helices in that TM1, TM2, TM5, TM7, TM8, and TM10 are all discontinuous; in contrast, GLUTs and related sugar porters typically have only two broken helices at most. It is plausible that these half-helices could support gating rearrangements, and biochemical evidence supports cooperativity between the two substrates.156 However, a more recent cryo-EM structure of human MCT1 in complex with Basigin-2 was found to be highly similar to MCT2.154 In MCT1, the comparison of outward- and inward-facing structures of the monomer were instead consistent with a conventional rocker-switch mechanism, as initially deduced from the monomeric bacterial homologue structure from Syntrophobacter fumaroxidans (SfMCT)154,182 (Figure 18c). Taken together, while structural information appears to support the biochemical analysis of MFS transporter oligomers, in most cases the physiological assembly and significance is still unclear.
Basigin is a transmembrane glycoprotein with two immunoglobulin-like domains.359 Basigin has also been given a CD name, CD147. The cell surface glycoprotein Basigin is thought to be connected to a number of biological functions, and it is associated with a number of human diseases.360 Basigin interacts tightly with MCT1 and MCT4 and acts as a trafficking chaperone.361 In detail, a loss of Basigin in knockout mice reduces MCT localization, leading to an impairment of lactate excretion in photoreceptor cells and impairs their activity.362 MCT4 and Basigin are both commonly expressed in glioblastoma363 and inhibiting their interaction may be an effective approach to inhibit tumor progression.364 In the recent human MCT1-Basigin-2 cryo-EM structure, the extracellular domain harboring the flexible immunoglobulin-like domains was found to extend over the extracellular surface of MCT1 (Figure 18c).154 The single TM segment of Basigin-2 interacts with the peripheral helix TM6 in MCT1, mostly through hydrophobic interactions and likely also through polar interactions through a glutamate residue in Basigin and an TM6 asparagine residue in MCT1.154 Despite the complex structure, the molecular basis for the trafficking function of Basigin-2 is unclear and whether the complex affects the function of MCT1 in any other way, such as helping to recruit lactate to the transporter and oligomerization. Interestingly, analogous to the MCT1-Basigin-2 complex structure, the non-MFS transporter belonging to the SLC7 family, LAT1 (l-type amino acid transporter (1) forms a similar complex with CD98 heavy chain (Figure 18c).365,366 Similar to Basigin, the glycoprotein CD98hc acts as a trafficking chaperone for LAT1.367 The main difference is that CD98hc forms a disulfide bridge to LAT1, which enables a closer association to the extracellular side of the transporter as compared to the Basigin-2 with MCT1.365 It has been proposed that this association may directly affect extracellular gating of LAT1,365 but again the functional role of the ancillary subunit remains unclear. One interesting proposal is that the lipid cholesterol was found at the complex interface and cholesterol binding could promote homodimerization.368 In support for this notion, the polar headgroups of phosphatidic acids interact with a conserved arginine in CD98hc, and the mutation of this arginine abolishes the transport function of LAT.366 Clearly, the role of lipids and further their association between MFS transporter complexes is likely to be important, but mechanistic details are lacking.368 In regard to MCT, it is further unclear if the differences between MCT1 and MCT2 transport models are based on nonphysiological complexes or reflect adaptions that can take place.
One of the most extensive system analyzed for the role of oligomerization in MFS transporters is for the plant nitrate transporter NRT1.1, which physiologically is in a dynamic equilibrium between low and high affinity states.369 Under high nitrate levels (>1 mM) NRT1.1 is a low-affinity transporter (KM ∼ 4 mM), but when nitrate levels drop to less than 1 mM, NRT1.1 is turned into a high-affinity transporter (KM ∼ 40 μM).370 The transformation between a low and high affinity nitrate transporter is due to phosphorylation of a threonine residue (Thr101) by the CIPK kinase.369 In the crystal structures, the threonine residue is not located near the substrate binding site but at the membrane interface on the cytoplasmic side. A Thr101Asp mutant mimicking phosphorylation in NTR1.1 destabilizes the protein.371,372 It has been proposed that destabilization is a result of the protein switching between its dimeric (low affinity) and monomeric (high affinity) states.372,373 However, the Thr101Asp mutant does not shift the binding affinity for nitrate.371 Thus, the monomer vs dimer kinetic differences appears to be result of changes of overall transporter dynamics, highlighting their importance to elucidate, i.e., just like GLUT1, in which the KM for glucose changes 10-fold between high and low temperatures.
In addition to specific lipid binding sites and intrinsic regulation by direct phosphorylation of the MFS transporter, extrinsic factors can also regulate MFS transport activity indirectly. Bacteria import sugars by a predetermined preference.374 Glucose is the most preferred sugar as it can directly enter glycolysis. The phosphoenolpyruvate (PEP):carbohydrate phosphotransferase system (PTS) controls the uptake of non-PTS sugars.375,376 The phosphotransferase protein IIAGlc of PTS, plays a key role for the regulation of carbohydrate metabolism by binding to sugar transporters and other proteins to modulate their activities.374 LacY and MelB are examples of sugar transporters regulated by IIAGlc. Binding analysis by ITC experiments have shown a direct interaction between the IIAGlc protein to LacY and MelB in a 1:1 ratio.377−379 IIAGlc binding to LacY and MelB is thought to restrict conformational dynamics and decrease the binding affinity for sugar.379 In the presence of glucose IIAGlc is unphosphorylated, which binds to LacY and inhibits galactoside binding. As such, the PTS is induced in the presence of glucose, and alternative carbon sources are not taken up, a process referred to as inducer exclusion.374
Although the same components for regulation take place in LacS from Streptococcus thermophilus, the regulation mechanism is thought to be different. LacS consists of a membrane-embedded carrier domain and a hydrophilic IIA domain, which is homologous to IIAGlc-like domains of the PEP–PTS system.380 To ensure efficient metabolism of lactose, the phosphoryl transfer protein HPr plays a central role in regulating LacS expression and activity. The histidine-phosphorylated form of HPr phosphorylates the IIA domain of LacS and thereby affects transport.381 The unphosphorylated LacS-IIA does not functionally interact with the carrier domain. Instead, only the phosphorylated form of LacS-IIA interacts with the carrier and stimulates lactose counterflow transport.382 The regulation differs from “inducer exclusion” mechanism in E. coli, i.e., the function of LacY and MelB are inhibited by the unphosphorylated form of IIAGlc, whereas LacS is inhibited by phosphorylated form of intramolecularly LacS-IIA. These results are consistent with the dimeric form of LacS.347,383,384 Despite the clear effect of IIAGlc and HPr complex interactions on sugar transporter kinetics, the structural basis for their extrinsic regulation it still poorly understood. Compared to bacterial MFS transporters, additional and longer loops and tails are more frequent in eukaryotic MFS transporters. These nonmembrane regions are likely interaction sites for extrinsic regulation of MFS transporters by the binding of allosteric modulators. For example, the oligopeptide transporter PepT1 has a 19 kDa extracellular loop domain between TM9 and TM10 that has been proposed to be the binding site for trypsin, which would be a means to couple the generation of peptides directly with their H+-coupled symport.385
9. Novel Functions of MFS Transporters
Although the majority of MFS transporters are assumed to be single proteins such as glucose transporters (Figure 19a), examples of fusions with enzymatic, regulatory, or signaling domains have also emerged. In bacteria, various MFS-fusion proteins have been identified, including some that are not yet characterized.386,387 A lysophospholipid transporter LplT was the first MFS member shown to use a lipid substrate in the transbilayer movement of lysophospholipid 2-acylglycerophosphoethanolamine (2-acyl-GPE). Interestingly, 2-acyl-GPE is thought to be acylated by a fused bifunctional enzyme known as acyltransferase/acyl-ACP synthetase to form phosphatidylethanolamine (Figure 19b).388
Figure 19.

Schematic representations of an MFS transporter and MFS transporter complexes. (a) A mammalian glucose (GLUT) MFS transporter protein in the outward open conformation, which facilitatively binds and transports glucose across the membrane bilayer. (b) A bacterial lysophospholipid (LplT, TC no. 2.A.1.42) MFS transporter that facilitatively translocates 2-acyl-GPE into the cytoplasm. Following translocation, 2-acyl-GPE is then acylated by a fused bifunctional enzyme comprised of two domains: a putative 2 TM lysophospholipid acyltransferase (PlsC) domain and an acyl-CoA synthetase (ACS) domain. (c) An MFS transceptor and signaling complex, using Rgt2 or Snf3 from yeast as an example. An intracellular signal for the upregulation of hexose transporters is generated upon glucose binding to Rgt2/Snf3. Glucose binding is proposed to activate phosphorylation of the repressor proteins Mth1 and Std1 (by Yck1/2), which further subjects them to ubiquitination (by the SCFGrr1 ubiquitin–protein ligase) and then degradation at the proteasome.
Some MFS transporters have been classified as “transceptors”,389,390 which are essentially transporters with a receptor-like function for detecting substrates. Tranceptors have been identified in many organisms ranging from bacteria to mammals389 and can either be substrate sensors, which transport their own substrates, or substrate sensors that bind their substrate but are incapable of transport. The simplest example of a transporter with a signaling function is the H+-coupled nitrate symporter NRT1.1 in plants, as discussed.391,392 Another example is the glucose-6-phosphate (G6P) sensor protein UhpC from E. coli that induces expression of the G6P transport protein UhpT via the UhpABC signaling cascade but has also been shown to possess G6P transport activity itself.393,394 UhpT and UhpC are both members of the MFS and share 32% sequence identity.
Maintaining glucose homeostasis is an essential, but complicated cellular process. In yeast, Rgt2 and Snf3 are thought to act as energy sensors that bind d-glucose, but are not capable of transport.395−397 Sugar binding elicits a myriad of intracellular responses that up-regulate the expression of several hexose transporters.395−397 Specifically, Rgt2 functions as a low-affinity glucose sensor and Snf3 functions as a high-affinity glucose sensor. There are at least 20 known or predicted sugar porters in yeast that are highly conserved, sharing between 50 and 100% sequence identity.395 Rgt2 and Snf3 only share ∼25% sequence similarity to these related proteins,97,398,399 but should nonetheless adopt a fold similar to that seen in GLUT transporters.395 A major feature distinguishing Rgt2 and Snf3 from their relatives is the unusually long 337 residue C-terminal tail on the cytoplasmic side.400 The C-terminal tail is thought to be a hub for the recruitment of signaling factors.396 It is postulated that the intracellular signal generated by these proteins activates the membrane-bound casein kinase I (Yck1/2), which then phosphorylates proteins Mth1 and Std1 bound to the C-terminal tail of the respective sensor, rendering them as substrates for the SCFGrr1 ubiquitin–protein ligase that further targets them to the proteasome (see schematic, Figure 19c).401−404 Mth1 and Std1 are required for repression of hexose transporter gene expression,397 and when glucose binds to the transceptors, activation of these proteins by phosphorylation is believed to be the rate-limiting signaling event.396 Thus, it is not the extended C-termini of Rgt2 and Snf3 that are directly responsible for glucose signaling, they instead serve to enhance signaling in a receptor-mediated fashion.396,397 This occurs without any apparent glucose transport,405 but the molecular details of glucose activation are unclear. What is known is that disruption of the predicted salt bridge network in Rgt2 as seen in the GLUTs (Figure 9), actually increases the upregulation of hexose transporters. This indirectly confirms that the Rgt2 activity is unlikely linked to d-glucose transport because the mutations should only form the inward-facing conformation.396 It is possible that these “transceptors” have evolved from former glucose transporters, and sequence changes have hindered their ability to switch between the conventional outward-facing and inward facing conformation upon glucose binding.3,186,396 As yet, however, these complexes and models for allosteric regulation and signaling are yet to be experimentally validated using purified components.
Rgt2/Snf3 represent examples of MFS proteins that are thought to be incapable of substrate transport. Another is the atypical MFS mammalian transporter UNC93B1,152 which is not thought to have a transporter function but instead acts as a dedicated trafficking chaperone for Toll-like receptors (TLRs).406,407 Nucleic acid-sensing TLRs are important to recognize microbial DNA and RNA and activate the innate immune response.408 TLRs are single-spanning TM glycoproteins, which are made up an extracellular leucine-rich repeat domain and a cytosolic Toll/interleukin-1 receptor domain.152,408 The atypical MFS transporter UNC93B1 is essential for the trafficking nucleic acid-sensing TLRs from the endoplasmic reticulum to the endosome.406,407 Recently, cryo-EM structures of human and mouse UNC93B1 were determined in complex with TLR3 and TLR7.152 Interestingly, the TLR3 protein forms a similar complex with UNC93B1 as Basigin does with MCT1,154 with protein–protein interactions also formed between the transmembrane domain of the glycoprotein and TM6 of the MFS transporter (Figure 18c).152 The functional roles, however, are reversed with the MFS transporter UNC93B1 now having the role as the trafficking chaperone rather than the glycoprotein. Although there might still be an unidentified transport function for UNC93B1, these studies nevertheless highlight how the MFS-fold has been adopted for physiological roles outside of substrate transport.
10. Concluding Remarks
During the past decade, MFS transporter structures and their biochemical, biophysical, and computational characterization has provided a richer insight into the molecular basis for their alternating-access mechanisms. Although their large global rearrangements resemble the early sketches of rigid rocking-bodies, molecular movies of “simple” transporters, such as the passive GLUT transporters, have rather demonstrated the complexity and the intricate coupling of the numerous conformational states formed to achieve substrate translocation. Elucidating the energetic barriers between each of these conformational states and how these barriers are modulated by substrates, proton(ion)-coupling, and the membrane bilayer itself, is an overarching goal that needs to be addressed in developing more meaningful mechanistic models. These goals will require the development of methods that can monitor transporter dynamics, not just in detergent, but in lipid bilayers, such as NMR, EPR, and also population dynamics, such as FRET and HDX-MS. First, however, considerable effort must be placed on the development of proteoliposome assays for MFS transporters, particularly for the mammalian MFS transporters. We would argue this is important first step for developing deeper kinetic models and for monitoring conformational dynamics in an environment that will be meaningful to the physiological situation and for a better understanding of the role of lipids in fine-tuning transport. At the other end of the spectrum, however, considerable effort is still required to determine the physiological substrates for many of the orphan MFS transporters. The molecular basis for how MFS transporters are allosterically regulated by the binding of extrinsic factors is also unclear, as is how they could contribute to signaling cascades. With improvements in single-particle cryo-EM, it is likely that in the next 10 years, we see an even larger expansion in structural data. We envisage that these molecular details will be the driver for structure–function analysis of these intricate machines to reveal novel, conceptual insights in MFS transporter biology.
Acknowledgments
We thank the Drew lab for feedback and scientific discussions and Michael Landreh for Figure 18a. This work was supported by the Knut and Alice Wallenberg Foundation (D.D), the Novo Nordisk foundation (no. 34188 D.D), the EMBO long-term fellowship (ALTF 33-2019) (R.A.N), the Grant-in-Aid for Scientific Research (no. 19H03186 to M.T) from the Japan Society for the Promotion of Science (JSPS) and partially supported by Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS) from the Japan Agency for Medical Research and Development (AMED) under grant numbers JP20am0101071 (to M.T).
Biographies
David Drew is a professor of Biochemistry at Stockholm University. Dr. Drew completed his university studies in New Zealand and moved to Stockholm University in Sweden for a doctoral position, where he developed the use of GFP as a tool for monitoring production and purification of membrane proteins. In 2006, he continued method development and structural studies of membrane transporters as an EMBO postdoctoral fellow with Dr. So Iwata at Imperial College London. In 2009, Dr. Drew started his independent lab at Imperial College as a Royal Society University Research fellow on the structural elucidation of SLC transporters and later returned to Sweden as a Wallenberg Academy Fellow. Dr. Drew has established a team focusing on developing mechanistic models of SLC transporters, with a particular emphasis on glucose (GLUT) transporters and Na+/H+ exchangers. Dr. Drew is a reviewing editor for eLife (2020–present) and has been recognized with several awards and distinctions for his work on small molecule transporters, including the Arrhenius medal from The Swedish Chemical Society and Göran Gustafsson Prize in Chemistry from the Royal Swedish Academy of Science.
Rachel A. North is an EMBO Research Fellow working with David Drew in the Department of Biochemistry and Biophysics, Stockholm University, Sweden. Rachel was previously a Research Fellow in the School of Biological Sciences and Biomolecular Interaction Centre at the University of Canterbury, New Zealand. She graduated with a Ph.D. in Biochemistry in 2017, also at the University of Canterbury. Her research focuses on understanding the function and structure of membrane transporters at a molecular level.
Kumar Nagarathinam is a postdoctoral researcher at the Institute of Biochemistry, University of Lübeck, Germany. He did his doctoral training under the cosupervision of Mikio Tanabe and Milton T. Stubbs at the HALOmem Membrane Protein Biochemistry group, Martin-Luther-University Halle-Wittenberg, Germany, and received his Ph.D. in 2020. He studied the mechanism of antibiotic efflux mediated by the major facilitator superfamily multidrug resistant transporters during his doctoral study. Currently, his research work focuses on the development of vaccine against hepatitis-C virus (HCV) and trying to understand the molecular mechanism of HCV entry into hepatocytes by interacting with the host receptors.
Mikio Tanabe is an Associate Professor in Structural Biology Research Center, Institute of Materials Structure Science in the KEK/High Energy Accelerator Research Organization. He completed his Ph.D. in biochemistry in 2006 at Imperial College London, where he worked under Bernadette Byrne and Katy Brown’s direction. Following studies as a Postdoctoral Fellow in the Department of Pharmacology at Vanderbilt University Medical Center with Tina M Iverson, he was appointed as a junior research group leader of HALOmem Membrane Protein and Dynamic at Martin-Luther-University Halle-Wittenberg in 2009, joined to Structural Biology Research Center in 2015. He served as a topic editor of Antibiotics from (2020–present). His research focuses on understanding multidrug resistance transporter and potential antimicrobial drug target proteins using biochemical and biophysical approaches to elucidate the molecular mechanisms by which these proteins contribute to infectious diseases.
The authors declare no competing financial interest.
Dedication
This review is dedicated to the memory of Michihiro Kasahara.
References
- Hediger M. A.; Clemencon B.; Burrier R. E.; Bruford E. A. The ABCs of membrane transporters in health and disease (SLC series): introduction. Mol. Aspects Med. 2013, 34, 95–107. 10.1016/j.mam.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger M. A.; Romero M. F.; Peng J. B.; Rolfs A.; Takanaga H.; Bruford E. A. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteinsIntroduction. Pfluegers Arch. 2004, 447, 465–468. 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- Drew D.; Boudker O. Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 2016, 85, 543–572. 10.1146/annurev-biochem-060815-014520. [DOI] [PubMed] [Google Scholar]
- Henderson R. K.; Fendler K.; Poolman B. Coupling efficiency of secondary active transporters. Curr. Opin. Biotechnol. 2019, 58, 62–71. 10.1016/j.copbio.2018.11.005. [DOI] [PubMed] [Google Scholar]
- Jardetzky O. Simple allosteric model for membrane pumps. Nature 1966, 211, 969–970. 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- Kazmier K.; Claxton D. P.; McHaourab H. S. Alternating access mechanisms of LeuT-fold transporters: trailblazing towards the promised energy landscapes. Curr. Opin. Struct. Biol. 2017, 45, 100–108. 10.1016/j.sbi.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. A general theory of membrane transport from studies of bacteria. Nature 1957, 180, 134–136. 10.1038/180134a0. [DOI] [PubMed] [Google Scholar]
- Mitchell P. Foundations of vectorial metabolism and osmochemistry. Biosci. Rep. 1991, 11, 297–344. discussion 345–346. 10.1007/BF01130212. [DOI] [PubMed] [Google Scholar]
- Reyes N.; Ginter C.; Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature 2009, 462, 880–885. 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao S. S.; Paulsen I. T.; Saier M. H. Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. 10.1128/MMBR.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Sands Z. A.; Biggin P. C. A numbering system for MFS transporter proteins. Front Mol. Biosci 2016, 3, 21. 10.3389/fmolb.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray K. A.; Seal R. L.; Tweedie S.; Wright M. W.; Bruford E. A. A review of the new HGNC gene family resource. Hum. Genomics 2016, 10, 6. 10.1186/s40246-016-0062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglund P. J.; Nordstrom K. J.; Schioth H. B.; Fredriksson R. The solute carrier families have a remarkably long evolutionary history with the majority of the human families present before divergence of Bilaterian species. Mol. Biol. Evol. 2011, 28, 1531–1541. 10.1093/molbev/msq350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy V. S.; Shlykov M. A.; Castillo R.; Sun E. I.; Saier M. H. Jr. The major facilitator superfamily (MFS) revisited. FEBS J. 2012, 279, 2022–2035. 10.1111/j.1742-4658.2012.08588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier M. H. Jr.; Beatty J. T.; Goffeau A.; Harley K. T.; Heijne W. H.; Huang S. C.; Jack D. L.; Jahn P. S.; Lew K.; Liu J.; et al. The major facilitator superfamily. J. Mol. Microbiol. Biotechnol. 1999, 1, 257–279. [PubMed] [Google Scholar]
- Abramson J.; Smirnova I.; Kasho V.; Verner G.; Kaback H. R.; Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science 2003, 301, 610–615. 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Lemieux M. J.; Song J.; Auer M.; Wang D. N. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 2003, 301, 616–620. 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- Heymann J. A.; Sarker R.; Hirai T.; Shi D.; Milne J. L.; Maloney P. C.; Subramaniam S. Projection structure and molecular architecture of OxlT, a bacterial membrane transporter. EMBO J. 2001, 20, 4408–4413. 10.1093/emboj/20.16.4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacksell I.; Rigaud J. L.; Purhonen P.; Pourcher T.; Hebert H.; Leblanc G. Projection structure at 8 A resolution of the melibiose permease, an Na-sugar co-transporter from Escherichia coli. EMBO J. 2002, 21, 3569–3574. 10.1093/emboj/cdf378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibi E.; Kaback H. R. In vivo expression of the lacY gene in two segments leads to functional lac permease. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 4325–4329. 10.1073/pnas.87.11.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej M. G.; Kaback H. R. Evolutionary mix-and-match with MFS transporters II. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E4831–4838. 10.1073/pnas.1319754110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radestock S.; Forrest L. R. The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J. Mol. Biol. 2011, 407, 698–715. 10.1016/j.jmb.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Forrest L. R. Structural symmetry in membrane proteins. Annu. Rev. Biophys. 2015, 44, 311–337. 10.1146/annurev-biophys-051013-023008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloris-Garcera P.; Bianchi F.; Slusky J. S.; Seppala S.; Daley D. O.; von Heijne G. Antiparallel dimers of the small multidrug resistance protein EmrE are more stable than parallel dimers. J. Biol. Chem. 2012, 287, 26052–26059. 10.1074/jbc.M112.357590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubarretxena-Belandia I.; Baldwin J. M.; Schuldiner S.; Tate C. G. Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. EMBO J. 2003, 22, 6175–6181. 10.1093/emboj/cdg611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp M.; Seppala S.; Granseth E.; von Heijne G. Emulating membrane protein evolution by rational design. Science 2007, 315, 1282–1284. 10.1126/science.1135406. [DOI] [PubMed] [Google Scholar]
- Keller R.; Ziegler C.; Schneider D. When two turn into one: evolution of membrane transporters from half modules. Biol. Chem. 2014, 395, 1379–1388. 10.1515/hsz-2014-0224. [DOI] [PubMed] [Google Scholar]
- Wright N. J.; Lee S. Y. Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat. Struct. Mol. Biol. 2019, 26, 599–606. 10.1038/s41594-019-0245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. C.; Davejan P.; Hendargo K. J.; Javadi-Razaz I.; Chou A.; Yee D. C.; Ghazi F.; Lam K. J. K.; Conn A. M.; Madrigal A.; et al. Expansion of the major facilitator superfamily (MFS) to include novel transporters as well as transmembrane-acting enzymes. Biochim. Biophys. Acta, Biomembr. 2020, 1862, 183277. 10.1016/j.bbamem.2020.183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perland E.; Fredriksson R. Classification systems of secondary active transporters. Trends Pharmacol. Sci. 2017, 38, 305–315. 10.1016/j.tips.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Poolman B.; Konings W. N. Secondary solute transport in bacteria. Biochim. Biophys. Acta, Bioenerg. 1993, 1183, 5–39. 10.1016/0005-2728(93)90003-X. [DOI] [PubMed] [Google Scholar]
- Saier M. H. Jr.; Paulsen I. T. Phylogeny of multidrug transporters. Semin. Cell Dev. Biol. 2001, 12, 205–213. 10.1006/scdb.2000.0246. [DOI] [PubMed] [Google Scholar]
- Valoskova K.; Biebl J.; Roblek M.; Emtenani S.; Gyoergy A.; Misova M.; Ratheesh A.; Reis-Rodrigues P.; Shkarina K.; Larsen I. S. B.; Vakhrushev S. Y.; Clausen H.; Siekhaus D. E. A conserved major facilitator superfamily member orchestrates a subset of O-glycosylation to aid macrophage tissue invasion. eLife 2019, 8, e41801 10.7554/eLife.41801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej M. G.; Sun L.; Yan N.; Kaback H. R. Functional architecture of MFS D-glucose transporters. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E719–727. 10.1073/pnas.1400336111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S. S.; Prashant R.; Kadoo N. Y.; Upadhyay A.; Gupta V. S. Global study of MFS superfamily transporters in arabidopsis and grapes reveals their functional diversity in plants. Plant Gene 2019, 18, 100179. 10.1016/j.plgene.2019.100179. [DOI] [Google Scholar]
- Steiner H. Y.; Naider F.; Becker J. M. The PTR family: a new group of peptide transporters. Mol. Microbiol. 1995, 16, 825–834. 10.1111/j.1365-2958.1995.tb02310.x. [DOI] [PubMed] [Google Scholar]
- Marger M. D.; Saier M. H. Jr. A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biochem. Sci. 1993, 18, 13–20. 10.1016/0968-0004(93)90081-W. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhang Y.; Sun K.; Meng Z.; Chen L. The SLC transporter in nutrient and metabolic sensing, regulation, and drug development. J. Mol. Cell Biol. 2019, 11, 1–13. 10.1093/jmcb/mjy052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. W.; Gallo L.; Jadhav A.; Hawkins R.; Parker C. G. The druggability of solute carriers. J. Med. Chem. 2020, 63, 3834–3867. 10.1021/acs.jmedchem.9b01237. [DOI] [PubMed] [Google Scholar]
- Vlasveld L. T.; Janssen R.; Bardou-Jacquet E.; Venselaar H.; Hamdi-Roze H.; Drakesmith H.; Swinkels D. W. Twenty years of ferroportin disease: a review or an update of published clinical, biochemical, molecular, and functional features. Pharmaceuticals 2019, 12, 132. 10.3390/ph12030132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrangelo A. Ferroportin disease: pathogenesis, diagnosis and treatment. Haematologica 2017, 102, 1972–1984. 10.3324/haematol.2017.170720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E.; Tuttle M. S.; Powelson J.; Vaughn M. B.; Donovan A.; Ward D. M.; Ganz T.; Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- Sebastiani G.; Wilkinson N.; Pantopoulos K. Pharmacological targeting of the hepcidin/ferroportin axis. Front. Pharmacol. 2016, 7, 160. 10.3389/fphar.2016.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D.; Vergara-Jaque A.; Forrest L. R.; Schuldiner S. Emulating proton-induced conformational changes in the vesicular monoamine transporter VMAT2 by mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E7390–E7398. 10.1073/pnas.1605162113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D.; Forrest L. R.; Schuldiner S. The ins and outs of vesicular monoamine transporters. J. Gen. Physiol. 2018, 150, 671–682. 10.1085/jgp.201711980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S.; Mortensen O. V. Overview of monoamine transporters. Curr. Protoc Pharmacol 2017, 79, 12.16.1–12.16.17. 10.1002/cpph.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner S. A molecular glimpse of vesicular monoamine transporters. J. Neurochem. 1994, 62, 2067–2078. 10.1046/j.1471-4159.1994.62062067.x. [DOI] [PubMed] [Google Scholar]
- Wimalasena K. Vesicular monoamine transporters: structure-function, pharmacology, and medicinal chemistry. Med. Res. Rev. 2011, 31, 483–519. 10.1002/med.20187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer R. J.; Edwards R. H. Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pfluegers Arch. 2004, 447, 629–635. 10.1007/s00424-003-1087-y. [DOI] [PubMed] [Google Scholar]
- Fremeau R. T. Jr.; Troyer M. D.; Pahner I.; Nygaard G. O.; Tran C. H.; Reimer R. J.; Bellocchio E. E.; Fortin D.; Storm-Mathisen J.; Edwards R. H. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 2001, 31, 247–260. 10.1016/S0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- Burger P. M.; Mehl E.; Cameron P. L.; Maycox P. R.; Baumert M.; Lottspeich F.; De Camilli P.; Jahn R. Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron 1989, 3, 715–720. 10.1016/0896-6273(89)90240-7. [DOI] [PubMed] [Google Scholar]
- Martineau M.; Guzman R. E.; Fahlke C.; Klingauf J. VGLUT1 functions as a glutamate/proton exchanger with chloride channel activity in hippocampal glutamatergic synapses. Nat. Commun. 2017, 8, 2279. 10.1038/s41467-017-02367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen J.; Chang R.; McGregor M.; Silm K.; Suzuki T.; Edwards R. H. Protons regulate vesicular glutamate transporters through an allosteric mechanism. Neuron 2016, 90, 768–780. 10.1016/j.neuron.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang R.; Eriksen J.; Edwards R. H. The dual role of chloride in synaptic vesicle glutamate transport. eLife 2018, 7, e34896 10.7554/eLife.34896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons S. M. Transport mechanisms in acetylcholine and monoamine storage. FASEB J. 2000, 14, 2423–2434. 10.1096/fj.00-0203rev. [DOI] [PubMed] [Google Scholar]
- Nino-Gonzalez M.; Novo-Uzal E.; Richardson D. N.; Barros P. M.; Duque P. More transporters, more substrates: the arabidopsis major facilitator superfamily revisited. Mol. Plant 2019, 12, 1182–1202. 10.1016/j.molp.2019.07.003. [DOI] [PubMed] [Google Scholar]
- Corratge-Faillie C.; Lacombe B. Substrate (un)specificity of arabidopsis NRT1/PTR Family (NPF) proteins. J. Exp. Bot. 2017, 68, 3107–3113. 10.1093/jxb/erw499. [DOI] [PubMed] [Google Scholar]
- Pasqua M.; Grossi M.; Zennaro A.; Fanelli G.; Micheli G.; Barras F.; Colonna B.; Prosseda G. The varied role of efflux pumps of the MFS family in the interplay of bacteria with animal and plant cells. Microorganisms 2019, 7, 285. 10.3390/microorganisms7090285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K.; Yamaguchi A. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 2001, 183, 5803–5812. 10.1128/JB.183.20.5803-5812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesar-Razquin A.; Snijder B.; Frappier-Brinton T.; Isserlin R.; Gyimesi G.; Bai X.; Reithmeier R. A.; Hepworth D.; Hediger M. A.; Edwards A. M.; et al. A call for systematic research on solute carriers. Cell 2015, 162, 478–487. 10.1016/j.cell.2015.07.022. [DOI] [PubMed] [Google Scholar]
- Lin L.; Yee S. W.; Kim R. B.; Giacomini K. M. SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug Discovery 2015, 14, 543–560. 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam S. K. The SLC22 transporter family: a paradigm for the impact of drug transporters on metabolic pathways, signaling, and disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 663–687. 10.1146/annurev-pharmtox-010617-052713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini K. M.; Huang S. M.; Tweedie D. J.; Benet L. Z.; Brouwer K. L.; Chu X.; Dahlin A.; Evers R.; Fischer V.; Hillgren K. M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discovery 2010, 9, 215–236. 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandsch M. Drug transport via the intestinal peptide transporter PepT1. Curr. Opin. Pharmacol. 2013, 13, 881–887. 10.1016/j.coph.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Daniel H.; Spanier B.; Kottra G.; Weitz D. From bacteria to man: archaic proton-dependent peptide transporters at work. Physiology 2006, 21, 93–102. 10.1152/physiol.00054.2005. [DOI] [PubMed] [Google Scholar]
- Jones R. S.; Morris M. E. Monocarboxylate transporters: therapeutic targets and prognostic factors in disease. Clin. Pharmacol. Ther. 2016, 100, 454–463. 10.1002/cpt.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap A. P. The SLC16 gene family - structure, role and regulation in health and disease. Mol. Aspects Med. 2013, 34, 337–349. 10.1016/j.mam.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Mueckler M.; Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 2013, 34, 121–138. 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman G. D. Chemical biology probes of mammalian GLUT structure and function. Biochem. J. 2018, 475, 3511–3534. 10.1042/BCJ20170677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minhas G. S.; Newstead S. Structural basis for prodrug recognition by the SLC15 family of proton-coupled peptide transporters. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 804–809. 10.1073/pnas.1813715116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minhas G. S.; Newstead S. Recent advances in understanding prodrug transport through the SLC15 family of proton-coupled transporters. Biochem. Soc. Trans. 2020, 48, 337–346. 10.1042/BST20180302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmlee M. A.; Jones R. S.; Rodriguez-Cruz V.; Follman K. E.; Morris M. E. Monocarboxylate transporters (SLC16): function, regulation, and role in health and disease. Pharmacol. Rev. 2020, 72, 466–485. 10.1124/pr.119.018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhouse S.; Warburg O.; Burk D.; Schade A. L. On respiratory impairment in cancer cells. Science 1956, 124, 267–272. 10.1126/science.124.3215.267. [DOI] [PubMed] [Google Scholar]
- Adekola K.; Rosen S. T.; Shanmugam M. Glucose transporters in cancer metabolism. Curr. Opin. Oncol. 2012, 24, 650–654. 10.1097/CCO.0b013e328356da72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin D.; Robay D.; Hindupur S. K.; Pohlmann J.; Colombi M.; El-Shemerly M. Y.; Maira S. M.; Moroni C.; Lane H. A.; Hall M. N. Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep. 2018, 25, 3047–3058. 10.1016/j.celrep.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen V. L.; Mina E.; Van Hee V. F.; Porporato P. E.; Sonveaux P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. 10.1016/j.molmet.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancey P. B.; Contat C.; Meylan E. Glucose transporters in cancer - from tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. 10.1111/febs.14577. [DOI] [PubMed] [Google Scholar]
- Nomura N.; Verdon G.; Kang H. J.; Shimamura T.; Nomura Y.; Sonoda Y.; Hussien S. A.; Qureshi A. A.; Coincon M.; Sato Y.; et al. Structure and mechanism of the mammalian fructose transporter GLUT5. Nature 2015, 526, 397–401. 10.1038/nature14909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer I. E. GLUT1 deficiency: a glut of epilepsy phenotypes. Neurology 2012, 78, 524–525. 10.1212/WNL.0b013e318248a245. [DOI] [PubMed] [Google Scholar]
- Brockmann K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev 2009, 31, 545–552. 10.1016/j.braindev.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Santer R.; Schneppenheim R.; Dombrowski A.; Gotze H.; Steinmann B.; Schaub J. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nat. Genet. 1997, 17, 324–326. 10.1038/ng1197-324. [DOI] [PubMed] [Google Scholar]
- Perland E.; Bagchi S.; Klaesson A.; Fredriksson R. Characteristics of 29 novel atypical solute carriers of major facilitator superfamily type: evolutionary conservation, predicted structure and neuronal co-expression. Open Biol. 2017, 7, 170142. 10.1098/rsob.170142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S.; Bentz S.; Hediger M. A.; Anderle P. Solute carriers (SLCs) in cancer. Mol. Aspects Med. 2013, 34, 719–734. 10.1016/j.mam.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Bhutia Y. D.; Babu E.; Ramachandran S.; Yang S.; Thangaraju M.; Ganapathy V. SLC transporters as a novel class of tumour suppressors: identity, function and molecular mechanisms. Biochem. J. 2016, 473, 1113–1124. 10.1042/BJ20150751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douard V.; Ferraris R. P. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol Endocrinol Metabol 2008, 295, E227–237. 10.1152/ajpendo.90245.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M.; Bondar A. N.; Freites J. A.; Tobias D. J.; Kaback H. R.; White S. H. Proton-coupled dynamics in lactose permease. Structure 2012, 20, 1893–1904. 10.1016/j.str.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda Y.; Newstead S.; Hu N. J.; Alguel Y.; Nji E.; Beis K.; Yashiro S.; Lee C.; Leung J.; Cameron A. D.; et al. Benchmarking membrane protein detergent stability for improving throughput of high-resolution X-ray structures. Structure 2011, 19, 17–25. 10.1016/j.str.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis W. I.; Kobilka B. K. The molecular basis of G protein-coupled receptor activation. Annu. Rev. Biochem. 2018, 87, 897–919. 10.1146/annurev-biochem-060614-033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Stevens R. C.; Xu F. The importance of ligands for G protein-coupled receptor stability. Trends Biochem. Sci. 2015, 40, 79–87. 10.1016/j.tibs.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Kaback H. R.; Guan L. It takes two to tango: The dance of the permease. J. Gen. Physiol. 2019, 151, 878–886. 10.1085/jgp.201912377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewinson O.; Adler J.; Sigal N.; Bibi E. Promiscuity in multidrug recognition and transport: the bacterial MFS Mdr transporters. Mol. Microbiol. 2006, 61, 277–284. 10.1111/j.1365-2958.2006.05254.x. [DOI] [PubMed] [Google Scholar]
- Fluman N.; Cohen-Karni D.; Weiss T.; Bibi E. A promiscuous conformational switch in the secondary multidrug transporter MdfA. J. Biol. Chem. 2009, 284, 32296–32304. 10.1074/jbc.M109.050658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani F.; Serrano-Vega M. J.; Shibata Y.; Abdul-Hussein S.; Lebon G.; Miller-Gallacher J.; Singhal A.; Strege A.; Thomas J. A.; Tate C. G. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat. Protoc. 2016, 11, 1554–1571. 10.1038/nprot.2016.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr M. L.; Singh S. K. Radioligand binding to nanodisc-reconstituted membrane transporters assessed by the scintillation proximity assay. Biochemistry 2014, 53, 4–6. 10.1021/bi401412e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M.; Javitch J. A. Monitoring the function of membrane transport proteins in detergent-solubilized form. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 3603–3608. 10.1073/pnas.0609573104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder D.; Fotiadis D. Measuring substrate binding and affinity of purified membrane transport proteins using the scintillation proximity assay. Nat. Protoc. 2012, 7, 1569–1578. 10.1038/nprot.2012.090. [DOI] [PubMed] [Google Scholar]
- Boles E.; Hollenberg C. P. The molecular genetics of hexose transport in yeasts. FEMS Microbiol Rev. 1997, 21, 85–111. 10.1111/j.1574-6976.1997.tb00346.x. [DOI] [PubMed] [Google Scholar]
- Wieczorke R.; Dlugai S.; Krampe S.; Boles E. Characterisation of mammalian GLUT glucose transporters in a heterologous yeast expression system. Cell. Physiol. Biochem. 2003, 13, 123–134. 10.1159/000071863. [DOI] [PubMed] [Google Scholar]
- Long W.; O’Neill D.; Cheeseman C. I. GLUT characterization using frog Xenopus laevis oocytes. Methods Mol. Biol. 2018, 1713, 45–55. 10.1007/978-1-4939-7507-5_4. [DOI] [PubMed] [Google Scholar]
- Feng H.; Xia X.; Fan X.; Xu G.; Miller A. J. Optimizing plant transporter expression in Xenopus oocytes. Plant Methods 2013, 9, 48. 10.1186/1746-4811-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrell J.; Tate C. G. Overexpression of membrane proteins in mammalian cells for structural studies. Mol. Membr. Biol. 2013, 30, 52–63. 10.3109/09687688.2012.703703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalise M.; Pochini L.; Giangregorio N.; Tonazzi A.; Indiveri C. Proteoliposomes as tool for assaying membrane transporter functions and interactions with xenobiotics. Pharmaceutics 2013, 5, 472–497. 10.3390/pharmaceutics5030472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson Z. L.; Lee S. Y. Liposome reconstitution and transport assay for recombinant transporters. Methods Enzymol. 2015, 556, 373–383. 10.1016/bs.mie.2014.11.048. [DOI] [PubMed] [Google Scholar]
- Rigaud J. L.; Levy D. Reconstitution of membrane proteins into liposomes. Methods Enzymol. 2003, 372, 65–86. 10.1016/S0076-6879(03)72004-7. [DOI] [PubMed] [Google Scholar]
- Knol J.; Veenhoff L.; Liang W. J.; Henderson P. J.; Leblanc G.; Poolman B. Unidirectional reconstitution into detergent-destabilized liposomes of the purified lactose transport system of Streptococcus thermophilus. J. Biol. Chem. 1996, 271, 15358–15366. 10.1074/jbc.271.26.15358. [DOI] [PubMed] [Google Scholar]
- Kaback H. R. A chemiosmotic mechanism of symport. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 1259–1264. 10.1073/pnas.1419325112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solcan N.; Kwok J.; Fowler P. W.; Cameron A. D.; Drew D.; Iwata S.; Newstead S. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 2012, 31, 3411–3421. 10.1038/emboj.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J. L.; Li C.; Brinth A.; Wang Z.; Vogeley L.; Solcan N.; Ledderboge-Vucinic G.; Swanson J. M. J.; Caffrey M.; Voth G. A.; et al. Proton movement and coupling in the POT family of peptide transporters. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 13182–13187. 10.1073/pnas.1710727114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R.; Poolman B. Proton-solute coupling mechanism of the maltose transporter from Saccharomyces cerevisiae. Sci. Rep. 2017, 7, 14375. 10.1038/s41598-017-14438-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara M.; Hinkle P. C. Reconstitution and purification of the D-glucose transporter from human erythrocytes. J. Biol. Chem. 1977, 252, 7384–7390. 10.1016/S0021-9258(19)66976-0. [DOI] [PubMed] [Google Scholar]
- Holloway P. W. A simple procedure for removal of Triton X-100 from protein samples. Anal. Biochem. 1973, 53, 304–308. 10.1016/0003-2697(73)90436-3. [DOI] [PubMed] [Google Scholar]
- Henderson P. J.; Kagawa Y.; Hirata H. Reconstitution of the GalP galactose transport activity of Escherichia coli into liposomes made from soybean phospholipids. Biochim. Biophys. Acta, Biomembr. 1983, 732, 204–209. 10.1016/0005-2736(83)90204-3. [DOI] [PubMed] [Google Scholar]
- Hresko R. C.; Kraft T. E.; Quigley A.; Carpenter E. P.; Hruz P. W. Mammalian glucose transporter activity is dependent upon anionic and conical phospholipids. J. Biol. Chem. 2016, 291, 17271–17282. 10.1074/jbc.M116.730168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller T.; Egenberger B.; Gorboulev V.; Bernhard F.; Uzelac Z.; Gorbunov D.; Wirth C.; Koppatz S.; Dotsch V.; Hunte C.; et al. The large extracellular loop of organic cation transporter 1 influences substrate affinity and is pivotal for oligomerization. J. Biol. Chem. 2011, 286, 37874–37886. 10.1074/jbc.M111.289330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juge N.; Yoshida Y.; Yatsushiro S.; Omote H.; Moriyama Y. Vesicular glutamate transporter contains two independent transport machineries. J. Biol. Chem. 2006, 281, 39499–39506. 10.1074/jbc.M607670200. [DOI] [PubMed] [Google Scholar]
- Scalise M.; Galluccio M.; Pochini L.; Indiveri C. Over-expression in Escherichia coli, purification and reconstitution in liposomes of the third member of the OCTN sub-family: the mouse carnitine transporter OCTN3. Biochem. Biophys. Res. Commun. 2012, 422, 59–63. 10.1016/j.bbrc.2012.04.105. [DOI] [PubMed] [Google Scholar]
- Hiasa M.; Matsumoto T.; Komatsu T.; Omote H.; Moriyama Y. Functional characterization of testis-specific rodent multidrug and toxic compound extrusion 2, a class III MATE-type polyspecific H+/organic cation exporter. Am. J. Physiol Cell Physiol 2007, 293, C1437–1444. 10.1152/ajpcell.00280.2007. [DOI] [PubMed] [Google Scholar]
- Komatsu T.; Hiasa M.; Miyaji T.; Kanamoto T.; Matsumoto T.; Otsuka M.; Moriyama Y.; Omote H. Characterization of the human MATE2 proton-coupled polyspecific organic cation exporter. Int. J. Biochem. Cell Biol. 2011, 43, 913–918. 10.1016/j.biocel.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Nji E.; Chatzikyriakidou Y.; Landreh M.; Drew D. An engineered thermal-shift screen reveals specific lipid preferences of eukaryotic and prokaryotic membrane proteins. Nat. Commun. 2018, 9, 4253. 10.1038/s41467-018-06702-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Wu J.; Ke M.; Zhang S.; Yuan Y.; Lin J. Y.; Yan N. Engineered XylE as a tool for mechanistic investigation and ligand discovery of the glucose transporters GLUTs. Cell Discovery 2019, 5, 14. 10.1038/s41421-019-0082-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda Y.; Cameron A.; Newstead S.; Omote H.; Moriyama Y.; Kasahara M.; Iwata S.; Drew D. Tricks of the trade used to accelerate high-resolution structure determination of membrane proteins. FEBS Lett. 2010, 584, 2539–2547. 10.1016/j.febslet.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Knol J.; Sjollema K.; Poolman B. Detergent-mediated reconstitution of membrane proteins. Biochemistry 1998, 37, 16410–16415. 10.1021/bi981596u. [DOI] [PubMed] [Google Scholar]
- Amati A. M.; Graf S.; Deutschmann S.; Dolder N.; von Ballmoos C. Current problems and future avenues in proteoliposome research. Biochem. Soc. Trans. 2020, 48, 1473–1492. 10.1042/BST20190966. [DOI] [PubMed] [Google Scholar]
- Tsai M. F.; Fang Y.; Miller C. Sided functions of an arginine-agmatine antiporter oriented in liposomes. Biochemistry 2012, 51, 1577–1585. 10.1021/bi201897t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker G. F.; Widdas W. F. The asymmetry of the facilitated transfer system for hexoses in human red cells and the simple kinetics of a two component model. J. Physiol. 1973, 231, 143–165. 10.1113/jphysiol.1973.sp010225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker G. F.; Naftalin R. J. Evidence of multiple operational affinities for D-glucose inside the human erythrocyte membrane. Biochim. Biophys. Acta, Biomembr. 1979, 550, 474–484. 10.1016/0005-2736(79)90150-0. [DOI] [PubMed] [Google Scholar]
- Guan L.; Kaback H. R. Binding affinity of lactose permease is not altered by the H+ electrochemical gradient. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12148–12152. 10.1073/pnas.0404936101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos S.; Kaback H. R. The electrochemical proton gradient in Escherichia coli membrane vesicles. Biochemistry 1977, 16, 848–854. 10.1021/bi00624a006. [DOI] [PubMed] [Google Scholar]
- Majd H.; King M. S.; Palmer S. M.; Smith A. C.; Elbourne L. D.; Paulsen I. T.; Sharples D.; Henderson P. J.; Kunji E. R. Screening of candidate substrates and coupling ions of transporters by thermostability shift assays. eLife 2018, 7, e38821 10.7554/eLife.38821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z.; Brown E. C.; Wang W.; MacKinnon R. Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 5748–5753. 10.1073/pnas.1602815113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzone A.; Barthmes M.; Fendler K. SSM-Based electrophysiology for transporter research. Methods Enzymol. 2017, 594, 31–83. 10.1016/bs.mie.2017.05.008. [DOI] [PubMed] [Google Scholar]
- Bazzone A.; Zabadne A. J.; Salisowski A.; Madej M. G.; Fendler K. A Loose relationship: incomplete H(+)/sugar coupling in the MFS sugar transporter GlcP. Biophys. J. 2017, 113, 2736–2749. 10.1016/j.bpj.2017.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzone A.; Madej M. G.; Kaback H. R.; Fendler K. pH Regulation of electrogenic sugar/H+ symport in MFS sugar permeases. PLoS One 2016, 11, e0156392 10.1371/journal.pone.0156392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganea C.; Meyer-Lipp K.; Lemonnier R.; Krah A.; Leblanc G.; Fendler K. G117C MelB, a mutant melibiose permease with a changed conformational equilibrium. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 2508–2516. 10.1016/j.bbamem.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Kermani A. A.; Macdonald C. B.; Burata O. E.; Ben Koff B.; Koide A.; Denbaum E.; Koide S.; Stockbridge R. B. The structural basis of promiscuity in small multidrug resistance transporters. Nat. Commun. 2020, 11, 6064. 10.1038/s41467-020-19820-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagan J. D.; Kitaoka M.; Anthony R. M. Engineered sialylation of pathogenic antibodies in vivo attenuates autoimmune disease. Cell 2018, 172, 564–577. 10.1016/j.cell.2017.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njiaju U. O.; Gamazon E. R.; Gorsic L. K.; Delaney S. M.; Wheeler H. E.; Im H. K.; Dolan M. E. Whole-genome studies identify solute carrier transporters in cellular susceptibility to paclitaxel. Pharmacogenet. Genomics 2012, 22, 498–507. 10.1097/FPC.0b013e328352f436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardi E.; Cesar-Razquin A.; Lindinger S.; Papakostas K.; Konecka J.; Hemmerich J.; Kickinger S.; Kartnig F.; Gurtl B.; Klavins K.; et al. A widespread role for SLC transmembrane transporters in resistance to cytotoxic drugs. Nat. Chem. Biol. 2020, 16, 469–478. 10.1038/s41589-020-0483-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovskich A.; Goldmann U.; Kartnig F.; Lindinger S.; Konecka J.; Fiume G.; Girardi E.; Superti-Furga G. The transporters SLC35A1 and SLC30A1 play opposite roles in cell survival upon VSV virus infection. Sci. Rep. 2019, 9, 10471. 10.1038/s41598-019-46952-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorke R.; Krampe S.; Weierstall T.; Freidel K.; Hollenberg C. P.; Boles E. Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in Saccharomyces cerevisiae. FEBS Lett. 1999, 464, 123–128. 10.1016/S0014-5793(99)01698-1. [DOI] [PubMed] [Google Scholar]
- Giaever G.; Nislow C. The yeast deletion collection: a decade of functional genomics. Genetics 2014, 197, 451–465. 10.1534/genetics.114.161620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Molina D.; Nordlund P. The cellular thermal shift assay: a novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 141–161. 10.1146/annurev-pharmtox-010715-103715. [DOI] [PubMed] [Google Scholar]
- Alexandrov A. I.; Mileni M.; Chien E. Y.; Hanson M. A.; Stevens R. C. Microscale fluorescent thermal stability assay for membrane proteins. Structure 2008, 16, 351–359. 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Lo M. C.; Aulabaugh A.; Jin G.; Cowling R.; Bard J.; Malamas M.; Ellestad G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- World Malaria Report; World Health Organization, 2018.
- Alexander C. G.; Wanner R.; Johnson C. M.; Breitsprecher D.; Winter G.; Duhr S.; Baaske P.; Ferguson N. Novel microscale approaches for easy, rapid determination of protein stability in academic and commercial settings. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844, 2241–2250. 10.1016/j.bbapap.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M.; Hibbs R. E.; Gouaux E. A fluorescence-detection size-exclusion chromatography-based thermostability assay for membrane protein precrystallization screening. Structure 2012, 20, 1293–1299. 10.1016/j.str.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyfakh A. A. Mystery of multidrug transporters: the answer can be simple. Mol. Microbiol. 2002, 44, 1123–1130. 10.1046/j.1365-2958.2002.02965.x. [DOI] [PubMed] [Google Scholar]
- Nji E.; Gulati A.; Qureshi A. A.; Coincon M.; Drew D. Structural basis for the delivery of activated sialic acid into Golgi for sialyation. Nat. Struct. Mol. Biol. 2019, 26, 415–423. 10.1038/s41594-019-0225-y. [DOI] [PubMed] [Google Scholar]
- Cheng Y. Membrane protein structural biology in the era of single particle cryo-EM. Curr. Opin. Struct. Biol. 2018, 52, 58–63. 10.1016/j.sbi.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y. Single-particle cryo-EM-How did it get here and where will it go. Science 2018, 361, 876–880. 10.1126/science.aat4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida H.; Asami J.; Zhang Z.; Nishizawa T.; Shigematsu H.; Ohto U.; Shimizu T. Cryo-EM structures of Toll-like receptors in complex with UNC93B1. Nat. Struct. Mol. Biol. 2021, 28, 173–180. 10.1038/s41594-020-00542-w. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Erramilli S. K.; Ammirati M.; Alvarez F. J. D.; Fennell K. F.; Purdy M. D.; Skrobek B. M.; Radziwon K.; Coukos J.; Kang Y.; et al. Synthetic antibodies against BRIL as universal fiducial marks for single-particle cryoEM structure determination of membrane proteins. Nat. Commun. 2020, 11, 1598. 10.1038/s41467-020-15363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Jiang X.; Zhang S.; Zhu A.; Yuan Y.; Xu H.; Lei J.; Yan C. Structural basis of human monocarboxylate transporter 1 inhibition by anti-cancer drug candidates. Cell 2021, 184, 370–383. 10.1016/j.cell.2020.11.043. [DOI] [PubMed] [Google Scholar]
- Li F.; Eriksen J.; Finer-Moore J.; Chang R.; Nguyen P.; Bowen A.; Myasnikov A.; Yu Z.; Bulkley D.; Cheng Y.; et al. Ion transport and regulation in a synaptic vesicle glutamate transporter. Science 2020, 368, 893–897. 10.1126/science.aba9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Jin Q.; Xu L.; Li N.; Meng Y.; Chang S.; Zheng X.; Wang J.; Chen Y.; Neculai D.; et al. Cooperative transport mechanism of human monocarboxylate transporter 2. Nat. Commun. 2020, 11, 2429. 10.1038/s41467-020-16334-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billesbolle C. B.; Azumaya C. M.; Kretsch R. C.; Powers A. S.; Gonen S.; Schneider S.; Arvedson T.; Dror R. O.; Cheng Y.; Manglik A. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 2020, 586, 807–811. 10.1038/s41586-020-2668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doki S.; Kato H. E.; Solcan N.; Iwaki M.; Koyama M.; Hattori M.; Iwase N.; Tsukazaki T.; Sugita Y.; Kandori H.; et al. Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter POT. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11343–11348. 10.1073/pnas.1301079110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newstead S.; Drew D.; Cameron A. D.; Postis V. L.; Xia X.; Fowler P. W.; Ingram J. C.; Carpenter E. P.; Sansom M. S.; McPherson M. J.; et al. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 2011, 30, 417–426. 10.1038/emboj.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ural-Blimke Y.; Flayhan A.; Strauss J.; Rantos V.; Bartels K.; Nielsen R.; Pardon E.; Steyaert J.; Kosinski J.; Quistgaard E. M.; et al. Structure of prototypic peptide transporter DtpA from E. coli in complex with valganciclovir provides insights into drug binding of human PepT1. J. Am. Chem. Soc. 2019, 141, 2404–2412. 10.1021/jacs.8b11343. [DOI] [PubMed] [Google Scholar]
- Guettou F.; Quistgaard E. M.; Raba M.; Moberg P.; Low C.; Nordlund P. Selectivity mechanism of a bacterial homolog of the human drug-peptide transporters PepT1 and PepT2. Nat. Struct. Mol. Biol. 2014, 21, 728–731. 10.1038/nsmb.2860. [DOI] [PubMed] [Google Scholar]
- Boggavarapu R.; Jeckelmann J. M.; Harder D.; Ucurum Z.; Fotiadis D. Role of electrostatic interactions for ligand recognition and specificity of peptide transporters. BMC Biol. 2015, 13, 58. 10.1186/s12915-015-0167-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quistgaard E. M.; Martinez Molledo M.; Low C. Structure determination of a major facilitator peptide transporter: Inward facing PepTSt from Streptococcus thermophilus crystallized in space group P3121. PLoS One 2017, 12, e0173126 10.1371/journal.pone.0173126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Mao G.; Liu M.; Zhang L.; Wang X.; Zhang X. C. Crystal structure of the E. coli peptide transporter YbgH. Structure 2014, 22, 1152–1160. 10.1016/j.str.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Klingenberg M. Ligand-protein interaction in biomembrane carriers. The induced transition fit of transport catalysis. Biochemistry 2005, 44, 8563–8570. 10.1021/bi050543r. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; He L.; Liu Y.; Zhao Y.; Zhang X. C. smFRET probing reveals substrate-dependent conformational dynamics of E. coli multidrug MdfA. Biophys. J. 2019, 116, 2296–2303. 10.1016/j.bpj.2019.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar D. S.; Smirnova I.; Kasho V.; Nir E.; Kong X.; Weiss S.; Kaback H. R. Single-molecule FRET reveals sugar-induced conformational dynamics in LacY. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12640–12645. 10.1073/pnas.0700969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasitza-Male T.; Bartels K.; Jungwirth J.; Wiggers F.; Rosenblum G.; Hofmann H.; Low C. Membrane chemistry tunes the structure of a peptide transporter. Angew. Chem., Int. Ed. 2020, 59, 19121–19128. 10.1002/anie.202008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich U. A.; Glaubitz C. NMR and EPR studies of membrane transporters. Biol. Chem. 2009, 390, 815–834. 10.1515/BC.2009.084. [DOI] [PubMed] [Google Scholar]
- Chiaramonte M.; Koviach J. L.; Moore C.; Iyer V. V.; Wagner C. R.; Halcomb R. L.; Miller W.; Melancon P.; Kuchta R. D. Inhibition of CMP-sialic acid transport into Golgi vesicles by nucleoside monophosphates. Biochemistry 2001, 40, 14260–14267. 10.1021/bi011262w. [DOI] [PubMed] [Google Scholar]
- Fowler P. W.; Orwick-Rydmark M.; Radestock S.; Solcan N.; Dijkman P. M.; Lyons J. A.; Kwok J.; Caffrey M.; Watts A.; Forrest L. R.; et al. Gating topology of the proton-coupled oligopeptide symporters. Structure 2015, 23, 290–301. 10.1016/j.str.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens C.; Shekhar M.; Borysik A. J.; Lau A. M.; Reading E.; Tajkhorshid E.; Booth P. J.; Politis A. Direct protein-lipid interactions shape the conformational landscape of secondary transporters. Nat. Commun. 2018, 9, 4151. 10.1038/s41467-018-06704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl L. S.; Fowler P. W.; Sansom M. S.; Beckstein O. Flexible gates generate occluded intermediates in the transport cycle of LacY. J. Mol. Biol. 2014, 426, 735–751. 10.1016/j.jmb.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpole T. J.; Delemotte L. Conformational landscapes of membrane proteins delineated by enhanced sampling molecular dynamics simulations. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 909–926. 10.1016/j.bbamem.2017.10.033. [DOI] [PubMed] [Google Scholar]
- Nicoludis J. M.; Gaudet R. Applications of sequence coevolution in membrane protein biochemistry. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 895–908. 10.1016/j.bbamem.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf T. A.; Colwell L. J.; Sheridan R.; Rost B.; Sander C.; Marks D. S. Three-dimensional structures of membrane proteins from genomic sequencing. Cell 2012, 149, 1607–1621. 10.1016/j.cell.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.; Zeng X.; Yan C.; Sun X.; Gong X.; Rao Y.; Yan N. Crystal structure of a bacterial homologue of glucose transporters GLUT1–4. Nature 2012, 490, 361–366. 10.1038/nature11524. [DOI] [PubMed] [Google Scholar]
- Yin Y.; He X.; Szewczyk P.; Nguyen T.; Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science 2006, 312, 741–744. 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang S.; Sun L.; Huang Y.; Lu F.; Liu Y.; Gong H.; Wang J.; Yan N. Structure of a fucose transporter in an outward-open conformation. Nature 2010, 467, 734–738. 10.1038/nature09406. [DOI] [PubMed] [Google Scholar]
- Yan H.; Huang W.; Yan C.; Gong X.; Jiang S.; Zhao Y.; Wang J.; Shi Y. Structure and mechanism of a nitrate transporter. Cell Rep. 2013, 3, 716–723. 10.1016/j.celrep.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Pedersen B. P.; Kumar H.; Waight A. B.; Risenmay A. J.; Roe-Zurz Z.; Chau B. H.; Schlessinger A.; Bonomi M.; Harries W.; Sali A.; et al. Crystal structure of a eukaryotic phosphate transporter. Nature 2013, 496, 533–536. 10.1038/nature12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosshart P. D.; Kalbermatter D.; Bonetti S.; Fotiadis D. Mechanistic basis of L-lactate transport in the SLC16 solute carrier family. Nat. Commun. 2019, 10, 2649. 10.1038/s41467-019-10566-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leano J. B.; Batarni S.; Eriksen J.; Juge N.; Pak J. E.; Kimura-Someya T.; Robles-Colmenares Y.; Moriyama Y.; Stroud R. M.; Edwards R. H. Structures suggest a mechanism for energy coupling by a family of organic anion transporters. PLoS Biol. 2019, 17, e3000260 10.1371/journal.pbio.3000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethayathulla A. S.; Yousef M. S.; Amin A.; Leblanc G.; Kaback H. R.; Guan L. Structure-based mechanism for Na(+)/melibiose symport by MelB. Nat. Commun. 2014, 5, 3009. 10.1038/ncomms4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi R.; Kato H. E.; Font J.; Deshpande C. N.; Wada M.; Ito K.; Ishitani R.; Jormakka M.; Nureki O. Outward- and inward-facing structures of a putative bacterial transition-metal transporter with homology to ferroportin. Nat. Commun. 2015, 6, 8545. 10.1038/ncomms9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N. Structural biology of the major facilitator superfamily transporters. Annu. Rev. Biophys. 2015, 44, 257–283. 10.1146/annurev-biophys-060414-033901. [DOI] [PubMed] [Google Scholar]
- Shi Y. Common folds and transport mechanisms of secondary active transporters. Annu. Rev. Biophys. 2013, 42, 51–72. 10.1146/annurev-biophys-083012-130429. [DOI] [PubMed] [Google Scholar]
- Wang J.; Yan C.; Li Y.; Hirata K.; Yamamoto M.; Yan N.; Hu Q. Crystal structure of a bacterial homologue of SWEET transporters. Cell Res. 2014, 24, 1486–1489. 10.1038/cr.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Tao Y.; Cheung L. S.; Fan C.; Chen L. Q.; Xu S.; Perry K.; Frommer W. B.; Feng L. Structures of bacterial homologues of SWEET transporters in two distinct conformations. Nature 2014, 515, 448–452. 10.1038/nature13670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Nishizawa T.; Yamashita K.; Ishitani R.; Nureki O. Structural basis for the facilitative diffusion mechanism by SemiSWEET transporter. Nat. Commun. 2015, 6, 6112. 10.1038/ncomms7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quistgaard E. M.; Low C.; Guettou F.; Nordlund P. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat. Rev. Mol. Cell Biol. 2016, 17, 123–132. 10.1038/nrm.2015.25. [DOI] [PubMed] [Google Scholar]
- Deng D.; Sun P.; Yan C.; Ke M.; Jiang X.; Xiong L.; Ren W.; Hirata K.; Yamamoto M.; Fan S.; et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature 2015, 526, 391–396. 10.1038/nature14655. [DOI] [PubMed] [Google Scholar]
- Qureshi A. A.; Suades A.; Matsuoka R.; Brock J.; McComas S. E.; Nji E.; Orellana L.; Claesson M.; Delemotte L.; Drew D. The molecular basis for sugar import in malaria parasites. Nature 2020, 578, 321–325. 10.1038/s41586-020-1963-z. [DOI] [PubMed] [Google Scholar]
- Wisedchaisri G.; Park M. S.; Iadanza M. G.; Zheng H.; Gonen T. Proton-coupled sugar transport in the prototypical major facilitator superfamily protein XylE. Nat. Commun. 2014, 5, 4521. 10.1038/ncomms5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M.; Takeda H.; Kato H. E.; Doki S.; Ito K.; Maturana A. D.; Ishitani R.; Nureki O. Structural basis for dynamic mechanism of nitrate/nitrite antiport by NarK. Nat. Commun. 2015, 6, 7097. 10.1038/ncomms8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D.; Lindahl E.; Andersson M. Uptake dynamics in the Lactose permease (LacY) membrane protein transporter. Sci. Rep. 2018, 8, 14324. 10.1038/s41598-018-32624-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Smirnova I.; Kasho V.; Wu J.; Hirata K.; Ke M.; Pardon E.; Steyaert J.; Yan N.; Kaback H. R. Crystal structure of a LacY-nanobody complex in a periplasmic-open conformation. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 12420–12425. 10.1073/pnas.1615414113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althoff T.; Abramson J. Protein structure reveals how a malaria parasite imports a wide range of sugars. Nature 2020, 578, 220–221. 10.1038/d41586-020-00148-8. [DOI] [PubMed] [Google Scholar]
- Henderson P. J. The homologous glucose transport proteins of prokaryotes and eukaryotes. Res. Microbiol. 1990, 141, 316–328. 10.1016/0923-2508(90)90005-B. [DOI] [PubMed] [Google Scholar]
- Lolkema J. S.; Poolman B. Uncoupling in secondary transport proteins. A mechanistic explanation for mutants of lac permease with an uncoupled phenotype. J. Biol. Chem. 1995, 270, 12670–12676. 10.1074/jbc.270.21.12670. [DOI] [PubMed] [Google Scholar]
- Yan N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013, 38, 151–159. 10.1016/j.tibs.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Pourcher T.; Bassilana M.; Sarkar H. K.; Kaback H. R.; Leblanc G. The melibiose/Na+ symporter of Escherichia coli: kinetic and molecular properties. Philos. Trans R Soc. Lond B Biol. Sci. 1990, 326, 411–423. 10.1098/rstb.1990.0021. [DOI] [PubMed] [Google Scholar]
- Pourcher T.; Zani M. L.; Leblanc G. Mutagenesis of acidic residues in putative membrane-spanning segments of the melibiose permease of Escherichia coli. I. Effect on Na(+)-dependent transport and binding properties. J. Biol. Chem. 1993, 268, 3209–3215. 10.1016/S0021-9258(18)53679-6. [DOI] [PubMed] [Google Scholar]
- Smirnova I. N.; Kasho V.; Kaback H. R. Protonation and sugar binding to LacY. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8896–8901. 10.1073/pnas.0803577105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quistgaard E. M.; Low C.; Moberg P.; Tresaugues L.; Nordlund P. Structural basis for substrate transport in the GLUT-homology family of monosaccharide transporters. Nat. Struct. Mol. Biol. 2013, 20, 766–768. 10.1038/nsmb.2569. [DOI] [PubMed] [Google Scholar]
- Iancu C. V.; Zamoon J.; Woo S. B.; Aleshin A.; Choe J. Y. Crystal structure of a glucose/H+ symporter and its mechanism of action. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17862–17867. 10.1073/pnas.1311485110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspari T.; Stadler R.; Sauer N.; Tanner W. Structure/function relationship of the Chlorella glucose/H+ symporter. J. Biol. Chem. 1994, 269, 3498–3502. 10.1016/S0021-9258(17)41890-4. [DOI] [PubMed] [Google Scholar]
- Hama H.; Wilson T. H. Replacement of alanine 58 by asparagine enables the melibiose carrier of Klebsiella pneumoniae to couple sugar transport to Na+. J. Biol. Chem. 1994, 269, 1063–1067. 10.1016/S0021-9258(17)42221-6. [DOI] [PubMed] [Google Scholar]
- Smirnova I.; Kasho V.; Sugihara J.; Vazquez-Ibar J. L.; Kaback H. R. Role of protons in sugar binding to LacY. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16835–16840. 10.1073/pnas.1214890109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolman B.; Knol J.; Lolkema J. S. Kinetic analysis of lactose and proton coupling in Glu379 mutants of the lactose transport protein of Streptococcus thermophilus. J. Biol. Chem. 1995, 270, 12995–13003. 10.1074/jbc.270.22.12995. [DOI] [PubMed] [Google Scholar]
- Carrasco N.; Antes L. M.; Poonian M. S.; Kaback H. R. lac permease of Escherichia coli: histidine-322 and glutamic acid-325 may be components of a charge-relay system. Biochemistry 1986, 25, 4486–4488. 10.1021/bi00364a004. [DOI] [PubMed] [Google Scholar]
- Sahin-Toth M.; Karlin A.; Kaback H. R. Unraveling the mechanism of the lactose permease of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 10729–10732. 10.1073/pnas.200351797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R. K.; de Valk S. C.; Poolman B.; Mans R. Energy coupling of membrane transport and efficiency of sucrose dissimilation in yeast. Metab. Eng. 2021, 65, 243–254. 10.1016/j.ymben.2020.11.014. [DOI] [PubMed] [Google Scholar]
- Poolman B.; Knol J.; van der Does C.; Henderson P. J.; Liang W. J.; Leblanc G.; Pourcher T.; Mus-Veteau I. Cation and sugar selectivity determinants in a novel family of transport proteins. Mol. Microbiol. 1996, 19, 911–922. 10.1046/j.1365-2958.1996.397949.x. [DOI] [PubMed] [Google Scholar]
- Wilson T. H.; Ding P. Z. Sodium-substrate cotransport in bacteria. Biochim. Biophys. Acta, Bioenerg. 2001, 1505, 121–130. 10.1016/S0005-2728(00)00282-6. [DOI] [PubMed] [Google Scholar]
- Parker J. L.; Mindell J. A.; Newstead S. Thermodynamic evidence for a dual transport mechanism in a POT peptide transporter. eLife 2014, 3, e04273 10.7554/eLife.04273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluman N.; Bibi E. Bacterial multidrug transport through the lens of the major facilitator superfamily. Biochim. Biophys. Acta, Proteins Proteomics 2009, 1794, 738–747. 10.1016/j.bbapap.2008.11.020. [DOI] [PubMed] [Google Scholar]
- Tirosh O.; Sigal N.; Gelman A.; Sahar N.; Fluman N.; Siemion S.; Bibi E. Manipulating the drug/proton antiport stoichiometry of the secondary multidrug transporter MdfA. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 12473–12478. 10.1073/pnas.1203632109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.; Bibi E. A single membrane-embedded negative charge is critical for recognizing positively charged drugs by the Escherichia coli multidrug resistance protein MdfA. EMBO J. 1999, 18, 822–832. 10.1093/emboj/18.4.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal N.; Fluman N.; Siemion S.; Bibi E. The secondary multidrug/proton antiporter MdfA tolerates displacements of an essential negatively charged side chain. J. Biol. Chem. 2009, 284, 6966–6971. 10.1074/jbc.M808877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debruycker V.; Hutchin A.; Masureel M.; Ficici E.; Martens C.; Legrand P.; Stein R. A.; McHaourab H. S.; Faraldo-Gomez J. D.; Remaut H.; et al. An embedded lipid in the multidrug transporter LmrP suggests a mechanism for polyspecificity. Nat. Struct. Mol. Biol. 2020, 27, 829–835. 10.1038/s41594-020-0464-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright N. J.; Lee S. Y.. Toward a molecular basis of cellular nucleoside transport in humans. Chem. Rev. 2021 10.1021/acs.chemrev.0c00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson I. A.; Vannucci S. J.; Maher F. Glucose transporters in mammalian brain. Biochem. Soc. Trans. 1994, 22, 671–675. 10.1042/bst0220671. [DOI] [PubMed] [Google Scholar]
- James D. E.; Strube M.; Muecdler M. Molecular cloning and characterization of an insulin-regulatable glucose transporter. Nature 1989, 338, 83–87. 10.1038/338083a0. [DOI] [PubMed] [Google Scholar]
- Kayano T.; Burant C. F.; Fukumoto H.; Gould G. W.; Fan Y. S.; Eddy R. L.; Byers M. G.; Shows T. B.; Seino S.; Bell G. I. Human facilitative glucose transporters. Isolation, functional characterization, and gene localization of cDNAs encoding an isoform (GLUT5) expressed in small intestine, kidney, muscle, and adipose tissue and an unusual glucose transporter pseudogene-like sequence (GLUT6). J. Biol. Chem. 1990, 265, 13276–13282. 10.1016/S0021-9258(19)38295-X. [DOI] [PubMed] [Google Scholar]
- Cura A. J.; Carruthers A. Role of monosaccharide transport proteins in carbohydrate assimilation, distribution, metabolism, and homeostasis. Comprehensive Physiology 2012, 2, 863–914. 10.1002/cphy.c110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre P. G. Sugar transport in the red blood cell: structure-activity relationships in substrates and antagonists. Pharmacol. Rev. 1961, 13, 39–70. [PubMed] [Google Scholar]
- Barnett J. E.; Holman G. D.; Munday K. A. Structural requirements for binding to the sugar-transport system of the human erythrocyte. Biochem. J. 1973, 131, 211–221. 10.1042/bj1310211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng D.; Yan N. GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci. 2016, 25, 546–558. 10.1002/pro.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden M. C.; Davis E. O.; Baldwin S. A.; Moore D. C.; Henderson P. J. Mammalian and bacterial sugar transport proteins are homologous. Nature 1987, 325, 641–643. 10.1038/325641a0. [DOI] [PubMed] [Google Scholar]
- Deng D.; Xu C.; Sun P.; Wu J.; Yan C.; Hu M.; Yan N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- Kapoor K.; Finer-Moore J. S.; Pedersen B. P.; Caboni L.; Waight A.; Hillig R. C.; Bringmann P.; Heisler I.; Muller T.; Siebeneicher H.; et al. Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 4711–4716. 10.1073/pnas.1603735113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Yuan Y.; Huang J.; Zhang S.; Luo S.; Wang N.; Pu D.; Zhao N.; Tang Q.; Hirata K.; et al. Structural aasis for blocking sugar uptake into the malaria parasite Plasmodium falciparum. Cell 2020, 183, 258–268. 10.1016/j.cell.2020.08.015. [DOI] [PubMed] [Google Scholar]
- Paulsen P. A.; Custodio T. F.; Pedersen B. P. Crystal structure of the plant symporter STP10 illuminates sugar uptake mechanism in monosaccharide transporter superfamily. Nat. Commun. 2019, 10, 407. 10.1038/s41467-018-08176-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng D.; Sun P.; Yan C.; Ke M.; Jiang X.; Xiong L.; Ren W.; Hirata K.; Yamamoto M.; Fan S.; et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature 2015, 526, 391–396. 10.1038/nature14655. [DOI] [PubMed] [Google Scholar]
- Mueckler M.; Makepeace C. Analysis of transmembrane segment 10 of the Glut1 glucose transporter by cysteine-scanning mutagenesis and substituted cysteine accessibility. J. Biol. Chem. 2002, 277, 3498–3503. 10.1074/jbc.M109157200. [DOI] [PubMed] [Google Scholar]
- Seatter M. J.; De la Rue S. A.; Porter L. M.; Gould G. W. QLS motif in transmembrane helix VII of the glucose transporter family interacts with the C-1 position of D-glucose and is involved in substrate selection at the exofacial binding site. Biochemistry 1998, 37, 1322–1326. 10.1021/bi972322u. [DOI] [PubMed] [Google Scholar]
- Hruz P. W.; Mueckler M. M. Cysteine-scanning mutagenesis of transmembrane segment 7 of the GLUT1 glucose transporter. J. Biol. Chem. 1999, 274, 36176–36180. 10.1074/jbc.274.51.36176. [DOI] [PubMed] [Google Scholar]
- Hashiramoto M.; Kadowaki T.; Clark A. E.; Muraoka A.; Momomura K.; Sakura H.; Tobe K.; Akanuma Y.; Yazaki Y.; Holman G. D.; et al. Site-directed mutagenesis of GLUT1 in helix 7 residue 282 results in perturbation of exofacial ligand binding. J. Biol. Chem. 1992, 267, 17502–17507. 10.1016/S0021-9258(19)37070-X. [DOI] [PubMed] [Google Scholar]
- Manolescu A.; Salas-Burgos A. M.; Fischbarg J.; Cheeseman C. I. Identification of a hydrophobic residue as a key determinant of fructose transport by the facilitative hexose transporter SLC2A7 (GLUT7). J. Biol. Chem. 2005, 280, 42978–42983. 10.1074/jbc.M508678200. [DOI] [PubMed] [Google Scholar]
- Kasahara T.; Maeda M.; Boles E.; Kasahara M. Identification of a key residue determining substrate affinity in the human glucose transporter GLUT1. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 1051–1055. 10.1016/j.bbamem.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Mori H.; Hashiramoto M.; Clark A. E.; Yang J.; Muraoka A.; Tamori Y.; Kasuga M.; Holman G. D. Substitution of tyrosine 293 of GLUT1 locks the transporter into an outward facing conformation. J. Biol. Chem. 1994, 269, 11578–11583. 10.1016/S0021-9258(19)78163-0. [DOI] [PubMed] [Google Scholar]
- Barnett J. E.; Holman G. D.; Chalkley R. A.; Munday K. A. Evidence for two asymmetric conformational states in the human erythrocyte sugar-transport system. Biochem. J. 1975, 145, 417–429. 10.1042/bj1450417a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottmann T.; Zierer W.; Subert C.; Sauer N.; Stadler R. STP10 encodes a high-affinity monosaccharide transporter and is induced under low-glucose conditions in pollen tubes of Arabidopsis. J. Exp. Bot. 2016, 67, 2387–2399. 10.1093/jxb/erw048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara T.; Kasahara M. Transmembrane segments 1, 5, 7 and 8 are required for high-affinity glucose transport by Saccharomyces cerevisiae Hxt2 transporter. Biochem. J. 2003, 372, 247–252. 10.1042/bj20030044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara T.; Ishiguro M.; Kasahara M. Eight amino acid residues in transmembrane segments of yeast glucose transporter Hxt2 are required for high affinity transport. J. Biol. Chem. 2006, 281, 18532–18538. 10.1074/jbc.M602123200. [DOI] [PubMed] [Google Scholar]
- Kasahara T.; Maeda M.; Ishiguro M.; Kasahara M. Identification by comprehensive chimeric analysis of a key residue responsible for high affinity glucose transport by yeast HXT2. J. Biol. Chem. 2007, 282, 13146–13150. 10.1074/jbc.C700041200. [DOI] [PubMed] [Google Scholar]
- Kasahara T.; Kasahara M. Identification of a key residue determining substrate affinity in the yeast glucose transporter Hxt7: a two-dimensional comprehensive study. J. Biol. Chem. 2010, 285, 26263–26268. 10.1074/jbc.M110.149716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodrow C. J.; Penny J. I.; Krishna S. Intraerythrocytic Plasmodium falciparum expresses a high affinity facilitative hexose transporter. J. Biol. Chem. 1999, 274, 7272–7277. 10.1074/jbc.274.11.7272. [DOI] [PubMed] [Google Scholar]
- Woodrow C. J.; Burchmore R. J.; Krishna S. Hexose permeation pathways in Plasmodium falciparum-infected erythrocytes. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 9931–9936. 10.1073/pnas.170153097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Li S.; Zhao H. Design and engineering of intracellular-metabolite-sensing/regulation gene circuits in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2016, 113, 206–215. 10.1002/bit.25676. [DOI] [PubMed] [Google Scholar]
- Wang M.; Yu C.; Zhao H. Identification of an important motif that controls the activity and specificity of sugar transporters. Biotechnol. Bioeng. 2016, 113, 1460–1467. 10.1002/bit.25926. [DOI] [PubMed] [Google Scholar]
- Wang D.; Yang H.; Shi L.; Ma L.; Fujii T.; Engelstad K.; Pascual J. M.; De Vivo D. C. Functional studies of the T295M mutation causing Glut1 deficiency: glucose efflux preferentially affected by T295M. Pediatr. Res. 2008, 64, 538–543. 10.1203/PDR.0b013e318184d2b5. [DOI] [PubMed] [Google Scholar]
- Bavnhøj L; Paulsen P. A.; Flores-Canales J. C.; Schiøtt B.; Pedersen B. P.. Molecular mechanism of high affinity sugar transport in plants unveiled by structures of glucose/H+ symporter STP10. bioRxiv 2020 10.1101/2020.11.05.369397. [DOI] [PubMed] [Google Scholar]
- Galochkina T.; Chong M. N. F.; Challali L.; Abbar S.; Etchebest C. New insights into GluT1 mechanics during glucose transfer. Sci. Rep. 2019, 9, 998. 10.1038/s41598-018-37367-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka Y.; Asano T.; Shibasaki Y.; Lin J. L.; Tsukuda K.; Katagiri H.; Akanuma Y.; Takaku F. C-terminal truncated glucose transporter is locked into an inward-facing form without transport activity. Nature 1990, 345, 550–553. 10.1038/345550a0. [DOI] [PubMed] [Google Scholar]
- Vollers S. S.; Carruthers A. Sequence determinants of GLUT1-mediated accelerated-exchange transport: analysis by homology-scanning mutagenesis. J. Biol. Chem. 2012, 287, 42533–42544. 10.1074/jbc.M112.369587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo N.; Griffin L. D.; Elsas L. J. Influx and efflux of 3-O-methyl-D-glucose by cultured human fibroblasts. Am. J. physiol 1988, 254, C628–633. 10.1152/ajpcell.1988.254.5.C628. [DOI] [PubMed] [Google Scholar]
- Custódio T. P.; Paulsen P. A.; Frain K. M.; Pedersen B. P. Structural comparison of GLUT1 to GLUT3 reveal transport regulation mechanism in sugar porter family. Life Sci. Alliance 2021, 4, e202000858 10.26508/lsa.202000858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloherty E. K.; Heard K. S.; Carruthers A. Human erythrocyte sugar transport is incompatible with available carrier models. Biochemistry 1996, 35, 10411–10421. 10.1021/bi953077m. [DOI] [PubMed] [Google Scholar]
- Arcus V. L.; Prentice E. J.; Hobbs J. K.; Mulholland A. J.; Van der Kamp M. W.; Pudney C. R.; Parker E. J.; Schipper L. A. On the temperature dependence of enzyme-catalyzed rates. Biochemistry 2016, 55, 1681–1688. 10.1021/acs.biochem.5b01094. [DOI] [PubMed] [Google Scholar]
- Kunji E. R.; Aleksandrova A.; King M. S.; Majd H.; Ashton V. L.; Cerson E.; Springett R.; Kibalchenko M.; Tavoulari S.; Crichton P. G.; et al. The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta, Mol. Cell Res. 2016, 1863, 2379–2393. 10.1016/j.bbamcr.2016.03.015. [DOI] [PubMed] [Google Scholar]
- Zhang X. C.; Han L. Uniporter substrate binding and transport: reformulating mechanistic questions. Biophys Rep 2016, 2, 45–54. 10.1007/s41048-016-0030-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe A. G. The kinetics and thermodynamics of glucose transport in human erythrocytes: indications for the molecular mechanism of transport. Biochem. Soc. Trans. 1989, 17, 435–438. 10.1042/bst0170435. [DOI] [PubMed] [Google Scholar]
- Sen A. K.; Widdas W. F. Determination of the temperature and pH dependence of glucose transfer across the human erythrocyte membrane measured by glucose exit. J. Physiol. 1962, 160, 392–403. 10.1113/jphysiol.1962.sp006854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs J. K.; Jiao W.; Easter A. D.; Parker E. J.; Schipper L. A.; Arcus V. L. Change in heat capacity for enzyme catalysis determines temperature dependence of enzyme catalyzed rates. ACS Chem. Biol. 2013, 8, 2388–2393. 10.1021/cb4005029. [DOI] [PubMed] [Google Scholar]
- Lowe A. G.; Walmsley A. R. The kinetics of glucose transport in human red blood cells. Biochim. Biophys. Acta, Biomembr. 1986, 857, 146–154. 10.1016/0005-2736(86)90342-1. [DOI] [PubMed] [Google Scholar]
- Carruthers A.; DeZutter J.; Ganguly A.; Devaskar S. U. Will the original glucose transporter isoform please stand up!. Am. J. Physiol Endocrinol Metabol 2009, 297, E836–848. 10.1152/ajpendo.00496.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naftalin R. J. A critique of the alternating access transporter model of uniport glucose transport. Biophys Rep 2018, 4, 287–299. 10.1007/s41048-018-0076-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor L. P.; Holman G. D. Symmetrical kinetic parameters for 3-O-methyl-D-glucose transport in adipocytes in the presence and in the absence of insulin. Biochim. Biophys. Acta, Biomembr. 1981, 642, 325–335. 10.1016/0005-2736(81)90449-1. [DOI] [PubMed] [Google Scholar]
- Ruprecht J. J.; Kunji E. R. S. The SLC25 mitochondrial carrier family: structure and mechanism. Trends Biochem. Sci. 2020, 45, 244–258. 10.1016/j.tibs.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia R.; Martens C.; Shekhar M.; Pant S.; Pellowe G. A.; Lau A. M.; Findlay H. E.; Harris N. J.; Tajkhorshid E.; Booth P. J.; Politis A. Hydrogen-deuterium exchange mass spectrometry captures distinct dynamics upon substrate and inhibitor binding to a transporter. Nat. Commun. 2020, 11, 6162. 10.1038/s41467-020-20032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos Seica A. F.; Iancu C. V.; Pfeilschifter B.; Madej M. G.; Choe J. Y.; Hellwig P. Asp22 drives the protonation state of the Staphylococcus epidermidis glucose/H(+) symporter. J. Biol. Chem. 2020, 295, 15253–15261. 10.1074/jbc.RA120.014069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke M.; Yuan Y.; Jiang X.; Yan N.; Gong H. Molecular determinants for the thermodynamic and functional divergence of uniporter GLUT1 and proton symporter XylE. PLoS Comput. Biol. 2017, 13, e1005603 10.1371/journal.pcbi.1005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooker R. J. An analysis of lactose permease “sugar specificity” mutations which also affect the coupling between proton and lactose transport. I. Val177 and Val177/Asn319 permeases facilitate proton uniport and sugar uniport. J. Biol. Chem. 1991, 266, 4131–4138. 10.1016/S0021-9258(20)64296-X. [DOI] [PubMed] [Google Scholar]
- King S. C.; Wilson T. H. Towards an understanding of the structural basis of ‘forbidden’ transport pathways in the Escherichia coli lactose carrier: mutations probing the energy barriers to uncoupled transport. Mol. Microbiol. 1990, 4, 1433–1438. 10.1111/j.1365-2958.1990.tb02053.x. [DOI] [PubMed] [Google Scholar]
- Paulsen I. T.; Skurray R. A. Topology, structure and evolution of two families of proteins involved in antibiotic and antiseptic resistance in eukaryotes and prokaryotes--an analysis. Gene 1993, 124, 1–11. 10.1016/0378-1119(93)90755-R. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Lekshmi M.; Parvathi A.; Ojha M.; Wenzel N.; Varela M. F. Functional and structural roles of the major facilitator superfamily bacterial multidrug efflux pumps. Microorganisms 2020, 8, 266. 10.3390/microorganisms8020266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H.; Bogaki M.; Nakamura S.; Ubukata K.; Konno M. Nucleotide sequence and characterization of the Staphylococcus aureus norA gene, which confers resistance to quinolones. J. Bacteriol. 1990, 172, 6942–6949. 10.1128/JB.172.12.6942-6949.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen I. T.; Brown M. H.; Littlejohn T. G.; Mitchell B. A.; Skurray R. A. Multidrug resistance proteins QacA and QacB from Staphylococcus aureus: membrane topology and identification of residues involved in substrate specificity. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 3630–3635. 10.1073/pnas.93.8.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. H.; Skurray R. A. Staphylococcal multidrug efflux protein QacA. J. Mol. Microbiol. Biotechnol. 2001, 3, 163–170. [PubMed] [Google Scholar]
- Neyfakh A. A. Natural functions of bacterial multidrug transporters. Trends Microbiol. 1997, 5, 309–313. 10.1016/S0966-842X(97)01064-0. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Lyass L.; Markham P. N.; Taylor S. S.; Vazquez-Laslop N.; Neyfakh A. A. Two highly similar multidrug transporters of Bacillus subtilis whose expression is differentially regulated. J. Bacteriol. 1995, 177, 3904–3910. 10.1128/JB.177.14.3904-3910.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolridge D. P.; Vazquez-Laslop N.; Markham P. N.; Chevalier M. S.; Gerner E. W.; Neyfakh A. A. Efflux of the natural polyamine spermidine facilitated by the Bacillus subtilis multidrug transporter Blt. J. Biol. Chem. 1997, 272, 8864–8866. 10.1074/jbc.272.14.8864. [DOI] [PubMed] [Google Scholar]
- Yelin R.; Schuldiner S. The pharmacological profile of the vesicular monoamine transporter resembles that of multidrug transporters. FEBS Lett. 1995, 377, 201–207. 10.1016/0014-5793(95)01346-6. [DOI] [PubMed] [Google Scholar]
- Yaffe D.; Vergara-Jaque A.; Shuster Y.; Listov D.; Meena S.; Singh S. K.; Forrest L. R.; Schuldiner S. Functionally important carboxyls in a bacterial homologue of the vesicular monoamine transporter (VMAT). J. Biol. Chem. 2014, 289, 34229–34240. 10.1074/jbc.M114.607366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth S. R.; Law C. J. Multidrug resistance protein MdtM adds to the repertoire of antiporters involved in alkaline pH homeostasis in Escherichia coli. BMC Microbiol. 2013, 13, 1987. 10.1186/1471-2180-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalheiro M.; Pais P.; Galocha M.; Teixeira M. C. Host-pathogen interactions mediated by MDR transporters in fungi: as pleiotropic as it gets!. Genes 2018, 9, 332. 10.3390/genes9070332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulshan K.; Moye-Rowley W. S. Multidrug resistance in fungi. Eukaryotic Cell 2007, 6, 1933–1942. 10.1128/EC.00254-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzi E.; Goffeau A. Yeast multidrug resistance: the PDR network. J. Bioenerg. Biomembr. 1995, 27, 71–76. 10.1007/BF02110333. [DOI] [PubMed] [Google Scholar]
- Felder T.; Bogengruber E.; Tenreiro S.; Ellinger A.; Sa-Correia I.; Briza P. Dtrlp, a multidrug resistance transporter of the major facilitator superfamily, plays an essential role in spore wall maturation in Saccharomyces cerevisiae. Eukaryotic Cell 2002, 1, 799–810. 10.1128/EC.1.5.799-810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C.; Dias P. J.; Sa-Correia I.; Teixeira M. C. MFS multidrug transporters in pathogenic fungi: do they have real clinical impact?. Front. Physiol. 2014, 5, 197. 10.3389/fphys.2014.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarco A. M.; Balan I.; Talibi D.; Mainville N.; Raymond M. AP1-mediated multidrug resistance in Saccharomyces cerevisiae requires FLR1 encoding a transporter of the major facilitator superfamily. J. Biol. Chem. 1997, 272, 19304–19313. 10.1074/jbc.272.31.19304. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis D. P.; Sagar N.; Hirschi K. D. Overexpression of Erg11p by the regulatable GAL1 promoter confers fluconazole resistance in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 1999, 43, 2798–2800. 10.1128/AAC.43.11.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos S. C.; Teixeira M. C.; Dias P. J.; Sa-Correia I. MFS transporters required for multidrug/multixenobiotic (MD/MX) resistance in the model yeast: understanding their physiological function through post-genomic approaches. Front. Physiol. 2014, 5, 180. 10.3389/fphys.2014.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomitori H.; Kashiwagi K.; Sakata K.; Kakinuma Y.; Igarashi K. Identification of a gene for a polyamine transport protein in yeast. J. Biol. Chem. 1999, 274, 3265–3267. 10.1074/jbc.274.6.3265. [DOI] [PubMed] [Google Scholar]
- Redhu A. K.; Shah A. H.; Prasad R. MFS transporters of Candida species and their role in clinical drug resistance. FEMS Yeast Res. 2016, 16, fow043 10.1093/femsyr/fow043. [DOI] [PubMed] [Google Scholar]
- Pasrija R.; Panwar S. L.; Prasad R. Multidrug transporters CaCdr1p and CaMdr1p of Candida albicans display different lipid specificities: both ergosterol and sphingolipids are essential for targeting of CaCdr1p to membrane rafts. Antimicrob. Agents Chemother. 2008, 52, 694–704. 10.1128/AAC.00861-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese D.; Bille J.; Sanglard D. A novel multidrug efflux transporter gene of the major facilitator superfamily from Candida albicans (FLU1) conferring resistance to fluconazole. Microbiology (London, U. K.) 2000, 146, 2743–2754. 10.1099/00221287-146-11-2743. [DOI] [PubMed] [Google Scholar]
- Juliao M. H. M.; Silva S. R.; Ferro J. A.; Varani A. M. A genomic and transcriptomic overview of MATE, ABC, and MFS Transporters in Citrus sinensis interaction with Xanthomonas citri subsp. citri. Plants 2020, 9, 794. 10.3390/plants9060794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy E.; Duque P. Beyond cellular detoxification: a plethora of physiological roles for MDR transporter homologs in plants. Front. Physiol. 2014, 5, 201. 10.3389/fphys.2014.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon M. J.; Cobbett C. S. A novel major facilitator superfamily protein at the tonoplast influences zinc tolerance and accumulation in Arabidopsis. Plant Physiol. 2007, 143, 1705–1719. 10.1104/pp.106.092015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima S.; Ushijima H.; Hwang H. J.; Tsutsumi S.; Makise M.; Yamaguchi Y.; Tsuchiya T.; Mizushima H.; Mizushima T. Identification of the TPO1 gene in yeast, and its human orthologue TETRAN, which cause resistance to NSAIDs. FEBS Lett. 2007, 581, 1457–1463. 10.1016/j.febslet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Breier A.; Gibalova L.; Seres M.; Barancik M.; Sulova Z. New insight into p-glycoprotein as a drug target. Anti-Cancer Agents Med. Chem. 2013, 13, 159–170. 10.2174/1871520611307010159. [DOI] [PubMed] [Google Scholar]
- Jiang D.; Zhao Y.; Wang X.; Fan J.; Heng J.; Liu X.; Feng W.; Kang X.; Huang B.; Liu J.; et al. Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 14664–14669. 10.1073/pnas.1308127110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng J.; Zhao Y.; Liu M.; Liu Y.; Fan J.; Wang X.; Zhao Y.; Zhang X. C. Substrate-bound structure of the E. coli multidrug resistance transporter MdfA. Cell Res. 2015, 25, 1060–1073. 10.1038/cr.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Heng J.; Gao Y.; Wang X. Crystal structures of MdfA complexed with acetylcholine and inhibitor reserpine. Biophys Rep 2016, 2, 78–85. 10.1007/s41048-016-0028-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarathinam K.; Nakada-Nakura Y.; Parthier C.; Terada T.; Juge N.; Jaenecke F.; Liu K.; Hotta Y.; Miyaji T.; Omote H.; et al. Outward open conformation of a Major Facilitator Superfamily multidrug/H(+) antiporter provides insights into switching mechanism. Nat. Commun. 2018, 9, 4005. 10.1038/s41467-018-06306-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomot E.; Yardeni E. H.; Vargiu A. V.; Tam H. K.; Malloci G.; Ramaswamy V. K.; Perach M.; Ruggerone P.; Pos K. M.; Bibi E. A new critical conformational determinant of multidrug efflux by an MFS transporter. J. Mol. Biol. 2018, 430, 1368–1385. 10.1016/j.jmb.2018.02.026. [DOI] [PubMed] [Google Scholar]
- Wu H. H.; Symersky J.; Lu M. Structure of an engineered multidrug transporter MdfA reveals the molecular basis for substrate recognition. Commun. Biol. 2019, 2, 210. 10.1038/s42003-019-0446-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H. H.; Symersky J.; Lu M. Structure and mechanism of a redesigned multidrug transporter from the Major Facilitator Superfamily. Sci. Rep. 2020, 10, 3949. 10.1038/s41598-020-60332-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q.; Sun B.; Zhou Y.; Wang C.; Guo L.; He J.; Deng D. Visualizing the nonlinear changes of a drug-proton antiporter from inward-open to occluded state. Biochem. Biophys. Res. Commun. 2021, 534, 272–278. 10.1016/j.bbrc.2020.11.096. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Mahendran I.; Athreya A.; Ranjan R.; Penmatsa A. Isolation and structural characterization of a Zn(2+)-bound single-domain antibody against NorC, a putative multidrug efflux transporter in bacteria. J. Biol. Chem. 2020, 295, 55–68. 10.1074/jbc.RA119.010902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluman N.; Ryan C. M.; Whitelegge J. P.; Bibi E. Dissection of mechanistic principles of a secondary multidrug efflux protein. Mol. Cell 2012, 47, 777–787. 10.1016/j.molcel.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair A. V.; Singh H.; Raturi S.; Neuberger A.; Tong Z.; Ding N.; Agboh K.; van Veen H. W. Relocation of active site carboxylates in major facilitator superfamily multidrug transporter LmrP reveals plasticity in proton interactions. Sci. Rep. 2016, 6, 38052. 10.1038/srep38052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J.; Wright S. H.; Tama F. Simulations of substrate transport in the multidrug transporter EmrD. Proteins: Struct., Funct., Genet. 2012, 80, 1620–1632. 10.1002/prot.24056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X.; Wang B. Inward open characterization of EmrD transporter with molecular dynamics simulation. Biochem. Biophys. Res. Commun. 2016, 474, 640–645. 10.1016/j.bbrc.2016.04.006. [DOI] [PubMed] [Google Scholar]
- Martinez Molledo M.; Quistgaard E. M.; Low C. Tripeptide binding in a proton-dependent oligopeptide transporter. FEBS Lett. 2018, 592, 3239–3247. 10.1002/1873-3468.13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Molledo M.; Quistgaard E. M.; Flayhan A.; Pieprzyk J.; Low C. Multispecific substrate recognition in a proton-dependent oligopeptide transporter. Structure 2018, 26, 467–476. 10.1016/j.str.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons J. A.; Parker J. L.; Solcan N.; Brinth A.; Li D.; Shah S. T.; Caffrey M.; Newstead S. Structural basis for polyspecificity in the POT family of proton-coupled oligopeptide transporters. EMBO Rep. 2014, 15, 886–893. 10.15252/embr.201338403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhala B. K.; Aduri N. G.; Sharma N.; Shaheen A.; Sharma A.; Iqbal M.; Hansen P. R.; Brasen C.; Gajhede M.; Rahman M.; et al. The prototypical proton-coupled oligopeptide transporter YdgR from Escherichia coli facilitates chloramphenicol uptake into bacterial cells. J. Biol. Chem. 2018, 293, 1007–1017. 10.1074/jbc.M117.805960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckner P.; Brandsch M. Interaction of 31 beta-lactam antibiotics with the H+/peptide symporter PEPT2: analysis of affinity constants and comparison with PEPT1. Eur. J. Pharm. Biopharm. 2005, 59, 17–24. 10.1016/j.ejpb.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Lewinson O.; Adler J.; Poelarends G. J.; Mazurkiewicz P.; Driessen A. J.; Bibi E. The Escherichia coli multidrug transporter MdfA catalyzes both electrogenic and electroneutral transport reactions. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 1667–1672. 10.1073/pnas.0435544100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putman M.; van Veen H. W.; Konings W. N. Molecular properties of bacterial multidrug transporters. Microbiol. Mol. Biol. Rev. 2000, 64, 672–693. 10.1128/MMBR.64.4.672-693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaedler T. A.; van Veen H. W. A flexible cation binding site in the multidrug major facilitator superfamily transporter LmrP is associated with variable proton coupling. FASEB J. 2010, 24, 3653–3661. 10.1096/fj.10-156927. [DOI] [PubMed] [Google Scholar]
- Mazurkiewicz P.; Konings W. N.; Poelarends G. J. Acidic residues in the lactococcal multidrug efflux pump LmrP play critical roles in transport of lipophilic cationic compounds. J. Biol. Chem. 2002, 277, 26081–26088. 10.1074/jbc.M203141200. [DOI] [PubMed] [Google Scholar]
- De Jesus M.; Jin J.; Guffanti A. A.; Krulwich T. A. Importance of the GP dipeptide of the antiporter motif and other membrane-embedded proline and glycine residues in tetracycline efflux protein Tet(L). Biochemistry 2005, 44, 12896–12904. 10.1021/bi050762c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. C.; Zhao Y.; Heng J.; Jiang D. Energy coupling mechanisms of MFS transporters. Protein Sci. 2015, 24, 1560–1579. 10.1002/pro.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D.; Radestock S.; Shuster Y.; Forrest L. R.; Schuldiner S. Identification of molecular hinge points mediating alternating access in the vesicular monoamine transporter VMAT2. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E1332–1341. 10.1073/pnas.1220497110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardeni E. H.; Bahrenberg T.; Stein R. A.; Mishra S.; Zomot E.; Graham B.; Tuck K. L.; Huber T.; Bibi E.; McHaourab H. S.; Goldfarb D. Probing the solution structure of the E. coli multidrug transporter MdfA using DEER distance measurements with nitroxide and Gd(III) spin labels. Sci. Rep. 2019, 9, 12528. 10.1038/s41598-019-48694-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardeni E. H.; Mishra S.; Stein R. A.; Bibi E.; McHaourab H. S. The multidrug transporter MdfA deviates from the canonical model of alternating access of MFS Transporters. J. Mol. Biol. 2020, 432, 5665–5680. 10.1016/j.jmb.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carole S.; Pichoff S.; Bouche J. P. Escherichia coli gene ydeA encodes a major facilitator pump which exports L-arabinose and isopropyl-beta-D-thiogalactopyranoside. J. Bacteriol. 1999, 181, 5123–5125. 10.1128/JB.181.16.5123-5125.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros Y.; Schuldiner S. Directed evolution reveals hidden properties of VMAT, a neurotransmitter transporter. J. Biol. Chem. 2010, 285, 5076–5084. 10.1074/jbc.M109.081216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masureel M.; Martens C.; Stein R. A.; Mishra S.; Ruysschaert J. M.; McHaourab H. S.; Govaerts C. Protonation drives the conformational switch in the multidrug transporter LmrP. Nat. Chem. Biol. 2014, 10, 149–155. 10.1038/nchembio.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge P.; Abramyan A. M.; Sorensen G.; Mork A.; Weikop P.; Gether U.; Bang-Andersen B.; Shi L.; Loland C. J. The mechanism of a high-affinity allosteric inhibitor of the serotonin transporter. Nat. Commun. 2020, 11, 1491. 10.1038/s41467-020-15292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaeva A. A.; Slotboom D. J. Elevator-type mechanisms of membrane transport. Biochem. Soc. Trans. 2020, 48, 1227–1241. 10.1042/BST20200290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harayama T.; Riezman H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- Phillips R.; Ursell T.; Wiggins P.; Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature 2009, 459, 379–385. 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning E. J.; Beckstein O. Influence of lipids on protein-mediated transmembrane transport. Chem. Phys. Lipids 2013, 169, 57–71. 10.1016/j.chemphyslip.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Bogdanov M.; Dowhan W. Phospholipid-assisted protein folding: phosphatidylethanolamine is required at a late step of the conformational maturation of the polytopic membrane protein lactose permease. EMBO J. 1998, 17, 5255–5264. 10.1093/emboj/17.18.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas F.; Tocanne J. F.; Leblanc G.; Lebrun M. C. Consequences of hydrophobic mismatch between lipids and melibiose permease on melibiose transport. Biochemistry 2000, 39, 4846–4854. 10.1021/bi992634s. [DOI] [PubMed] [Google Scholar]
- Hariharan P.; Tikhonova E.; Medeiros-Silva J.; Jeucken A.; Bogdanov M. V.; Dowhan W.; Brouwers J. F.; Weingarth M.; Guan L. Structural and functional characterization of protein-lipid interactions of the Salmonella typhimurium melibiose transporter MelB. BMC Biol. 2018, 16, 85. 10.1186/s12915-018-0553-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlett V. W.; Mallampalli V.; Karlstaedt A.; Dowhan W.; Taegtmeyer H.; Margolin W.; Vitrac H. Impact of membrane phospholipid alterations in Escherichia coli on cellular function and bacterial stress adaptation. J. Bacteriol. 2017, 199, e00849-16 10.1128/JB.00849-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M.; Heacock P. N.; Dowhan W. A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J. 2002, 21, 2107–2116. 10.1093/emboj/21.9.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M.; Dowhan W. Phosphatidylethanolamine is required for in vivo function of the membrane-associated lactose permease of Escherichia coli. J. Biol. Chem. 1995, 270, 732–739. 10.1074/jbc.270.2.732. [DOI] [PubMed] [Google Scholar]
- Martens C.; Stein R. A.; Masureel M.; Roth A.; Mishra S.; Dawaliby R.; Konijnenberg A.; Sobott F.; Govaerts C.; McHaourab H. S. Lipids modulate the conformational dynamics of a secondary multidrug transporter. Nat. Struct. Mol. Biol. 2016, 23, 744–751. 10.1038/nsmb.3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenhoff L. M.; Heuberger E. H.; Poolman B. The lactose transport protein is a cooperative dimer with two sugar translocation pathways. EMBO J. 2001, 20, 3056–3062. 10.1093/emboj/20.12.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z.; Kugel Desmoulin S.; Etnyre E.; Olive M.; Hsiung B.; Cherian C.; Wloszczynski P. A.; Moin K.; Matherly L. H. Identification and functional impact of homo-oligomers of the human proton-coupled folate transporter. J. Biol. Chem. 2012, 287, 4982–4995. 10.1074/jbc.M111.306860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aduri N. G.; Ernst H. A.; Prabhala B. K.; Bhatt S.; Boesen T.; Gajhede M.; Mirza O. Human proton coupled folic acid transporter is a monodisperse oligomer in the lauryl maltose neopentyl glycol solubilized state. Biochem. Biophys. Res. Commun. 2018, 495, 1738–1743. 10.1016/j.bbrc.2017.12.008. [DOI] [PubMed] [Google Scholar]
- Gupta K.; Donlan J. A. C.; Hopper J. T. S.; Uzdavinys P.; Landreh M.; Struwe W. B.; Drew D.; Baldwin A. J.; Stansfeld P. J.; Robinson C. V. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 2017, 541, 421–424. 10.1038/nature20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert D. N.; Carruthers A. Cholate-solubilized erythrocyte glucose transporters exist as a mixture of homodimers and homotetramers. Biochemistry 1991, 30, 4654–4658. 10.1021/bi00233a003. [DOI] [PubMed] [Google Scholar]
- Zottola R. J.; Cloherty E. K.; Coderre P. E.; Hansen A.; Hebert D. N.; Carruthers A. Glucose transporter function is controlled by transporter oligomeric structure. A single, intramolecular disulfide promotes GLUT1 tetramerization. Biochemistry 1995, 34, 9734–9747. 10.1021/bi00030a011. [DOI] [PubMed] [Google Scholar]
- Ponzoni L.; Zhang S.; Cheng M. H.; Bahar I. Shared dynamics of LeuT superfamily members and allosteric differentiation by structural irregularities and multimerization. Philos. Trans. R. Soc., B 2018, 373, 20170177. 10.1098/rstb.2017.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alguel Y.; Cameron A. D.; Diallinas G.; Byrne B. Transporter oligomerization: form and function. Biochem. Soc. Trans. 2016, 44, 1737–1744. 10.1042/BST20160217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guettou F.; Quistgaard E. M.; Tresaugues L.; Moberg P.; Jegerschold C.; Zhu L.; Jong A. J.; Nordlund P.; Low C. Structural insights into substrate recognition in proton-dependent oligopeptide transporters. EMBO Rep. 2013, 14, 804–810. 10.1038/embor.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamura R.; Fukuda M.; Kawamoto A.; Matoba K.; Dohmae N.; Ishitani R.; Takagi J.; Nureki O. Structural basis for oligomerization of the prokaryotic peptide transporter PepTSo2. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2019, 75, 348–358. 10.1107/S2053230X19003546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfeld J.; Loving R.; Armache J. P.; Sonnen A. F.; Guettou F.; Moberg P.; Zhu L.; Jegerschold C.; Flayhan A.; Briggs J. A.; et al. A saposin-lipoprotein nanoparticle system for membrane proteins. Nat. Methods 2016, 13, 345–351. 10.1038/nmeth.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newstead S. Recent advances in understanding proton coupled peptide transport via the POT family. Curr. Opin. Struct. Biol. 2017, 45, 17–24. 10.1016/j.sbi.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu T.; Miyauchi T. Basigin (CD147): a multifunctional transmembrane protein involved in reproduction, neural function, inflammation and tumor invasion. Histol Histopathol 2003, 18, 981–987. 10.14670/HH-18.981. [DOI] [PubMed] [Google Scholar]
- Iacono K. T.; Brown A. L.; Greene M. I.; Saouaf S. J. CD147 immunoglobulin superfamily receptor function and role in pathology. Exp. Mol. Pathol. 2007, 83, 283–295. 10.1016/j.yexmp.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk P.; Wilson M. C.; Heddle C.; Brown M. H.; Barclay A. N.; Halestrap A. P. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000, 19, 3896–3904. 10.1093/emboj/19.15.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philp N. J.; Ochrietor J. D.; Rudoy C.; Muramatsu T.; Linser P. J. Loss of MCT1, MCT3, and MCT4 expression in the retinal pigment epithelium and neural retina of the 5A11/basigin-null mouse. Invest. Ophthalmol. Visual Sci. 2003, 44, 1305–1311. 10.1167/iovs.02-0552. [DOI] [PubMed] [Google Scholar]
- Miranda-Goncalves V.; Honavar M.; Pinheiro C.; Martinho O.; Pires M. M.; Pinheiro C.; Cordeiro M.; Bebiano G.; Costa P.; Palmeirim I.; et al. Monocarboxylate transporters (MCTs) in gliomas: expression and exploitation as therapeutic targets. Neuro Oncol 2013, 15, 172–188. 10.1093/neuonc/nos298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss D. M.; Spina R.; Carter D. L.; Lim K. S.; Jeffery C. J.; Bar E. E. Disruption of the monocarboxylate transporter-4-basigin interaction inhibits the hypoxic response, proliferation, and tumor progression. Sci. Rep. 2017, 7, 4292. 10.1038/s41598-017-04612-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Wiriyasermkul P.; Jin C.; Quan L.; Ohgaki R.; Okuda S.; Kusakizako T.; Nishizawa T.; Oda K.; Ishitani R.; et al. Cryo-EM structure of the human L-type amino acid transporter 1 in complex with glycoprotein CD98hc. Nat. Struct. Mol. Biol. 2019, 26, 510–517. 10.1038/s41594-019-0237-7. [DOI] [PubMed] [Google Scholar]
- Yan R.; Zhao X.; Lei J.; Zhou Q. Structure of the human LAT1–4F2hc heteromeric amino acid transporter complex. Nature 2019, 568, 127–130. 10.1038/s41586-019-1011-z. [DOI] [PubMed] [Google Scholar]
- Palacin M.; Kanai Y. The ancillary proteins of HATs: SLC3 family of amino acid transporters. Pfluegers Arch. 2004, 447, 490–494. 10.1007/s00424-003-1062-7. [DOI] [PubMed] [Google Scholar]
- Newstead S. Insights into L-type heteromeric amino acid transporters. Nat. Struct. Mol. Biol. 2019, 26, 395–396. 10.1038/s41594-019-0240-z. [DOI] [PubMed] [Google Scholar]
- Liu K. H.; Tsay Y. F. Switching between the two action modes of the dual-affinity nitrate transporter CHL1 by phosphorylation. EMBO J. 2003, 22, 1005–1013. 10.1093/emboj/cdg118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K. H.; Huang C. Y.; Tsay Y. F. CHL1 is a dual-affinity nitrate transporter of arabidopsis involved in multiple phases of nitrate uptake. Plant Cell 1999, 11, 865–874. 10.2307/3870820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J. L.; Newstead S. Molecular basis of nitrate uptake by the plant nitrate transporter NRT1.1. Nature 2014, 507, 68–72. 10.1038/nature13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Bankston J. R.; Payandeh J.; Hinds T. R.; Zagotta W. N.; Zheng N. Crystal structure of the plant dual-affinity nitrate transporter NRT1.1. Nature 2014, 507, 73–77. 10.1038/nature13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid M.; Bera S.; Banerjee M.; Medvinsky A. B.; Sun G. Q.; Li B. L.; Sljoka A.; Chakraborty A. Feedforward control of plant nitrate transporter NRT1.1 biphasic adaptive activity. Biophys. J. 2020, 118, 898–908. 10.1016/j.bpj.2019.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeckelmann J. M.; Erni B. Transporters of glucose and other carbohydrates in bacteria. Pfluegers Arch. 2020, 472, 1129–1153. 10.1007/s00424-020-02379-0. [DOI] [PubMed] [Google Scholar]
- Postma P. W.; Lengeler J. W.; Jacobson G. R. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 1993, 57, 543–594. 10.1128/MR.57.3.543-594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher J.; Ake F. M.; Derkaoui M.; Zebre A. C.; Cao T. N.; Bouraoui H.; Kentache T.; Mokhtari A.; Milohanic E.; Joyet P. The bacterial phosphoenolpyruvate:carbohydrate phosphotransferase system: regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microbiol. Mol. Biol. Rev. 2014, 78, 231–256. 10.1128/MMBR.00001-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan P.; Guan L. Insights into the inhibitory mechanisms of the regulatory protein IIAGlc on melibiose permease activity. J. Biol. Chem. 2015, 290, 6752. 10.1074/jbc.A114.609255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan P.; Guan L. Insights into the inhibitory mechanisms of the regulatory protein IIA(Glc) on melibiose permease activity. J. Biol. Chem. 2014, 289, 33012–33019. 10.1074/jbc.M114.609255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan P.; Balasubramaniam D.; Peterkofsky A.; Kaback H. R.; Guan L. Thermodynamic mechanism for inhibition of lactose permease by the phosphotransferase protein IIAGlc. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 2407–2412. 10.1073/pnas.1500891112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolman B.; Royer T. J.; Mainzer S. E.; Schmidt B. F. Lactose transport system of Streptococcus thermophilus: a hybrid protein with homology to the melibiose carrier and enzyme III of phosphoenolpyruvate-dependent phosphotransferase systems. J. Bacteriol. 1989, 171, 244–253. 10.1128/JB.171.1.244-253.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnewijk M. G.; Poolman B. HPr(His approximately P)-mediated phosphorylation differently affects counterflow and proton motive force-driven uptake via the lactose transport protein of Streptococcus thermophilus. J. Biol. Chem. 2000, 275, 34080–34085. 10.1074/jbc.M003513200. [DOI] [PubMed] [Google Scholar]
- Geertsma E. R.; Duurkens R. H.; Poolman B. The activity of the lactose transporter from Streptococcus thermophilus is increased by phosphorylated IIA and the action of beta-galactosidase. Biochemistry 2005, 44, 15889–15897. 10.1021/bi051638w. [DOI] [PubMed] [Google Scholar]
- Friesen R. H.; Knol J.; Poolman B. Quaternary structure of the lactose transport protein of Streptococcus thermophilus in the detergent-solubilized and membrane-reconstituted state. J. Biol. Chem. 2000, 275, 33527–33535. 10.1074/jbc.M004066200. [DOI] [PubMed] [Google Scholar]
- Geertsma E. R.; Duurkens R. H.; Poolman B. Identification of the dimer interface of the lactose transport protein from Streptococcus thermophilus. J. Mol. Biol. 2003, 332, 1165–1174. 10.1016/j.jmb.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Beale J. H.; Parker J. L.; Samsudin F.; Barrett A. L.; Senan A.; Bird L. E.; Scott D.; Owens R. J.; Sansom M. S. P.; Tucker S. J.; et al. Crystal structures of the extracellular domain from PepT1 and PepT2 provide novel insights into mammalian peptide transport. Structure 2015, 23, 1889–1899. 10.1016/j.str.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willson B. J.; Dalzell L.; Chapman L. N. M.; Thomas G. H. Enhanced functionalisation of major facilitator superfamily transporters via fusion of C-terminal protein domains is both extensive and varied in bacteria. Microbiology (London, U. K.) 2019, 165, 419–424. 10.1099/mic.0.000771. [DOI] [PubMed] [Google Scholar]
- Willson B. J.; Chapman L. N.; Thomas G. H. Evolutionary dynamics of membrane transporters and channels: enhancing function through fusion. Curr. Opin. Genet. Dev. 2019, 58–59, 76–86. 10.1016/j.gde.2019.07.017. [DOI] [PubMed] [Google Scholar]
- Harvat E. M.; Zhang Y. M.; Tran C. V.; Zhang Z.; Frank M. W.; Rock C. O.; Saier M. H. Jr. Lysophospholipid flipping across the Escherichia coli inner membrane catalyzed by a transporter (LplT) belonging to the major facilitator superfamily. J. Biol. Chem. 2005, 280, 12028–12034. 10.1074/jbc.M414368200. [DOI] [PubMed] [Google Scholar]
- Thevelein J. M.; Voordeckers K. Functioning and evolutionary significance of nutrient transceptors. Mol. Biol. Evol. 2009, 26, 2407–2414. 10.1093/molbev/msp168. [DOI] [PubMed] [Google Scholar]
- Diallinas G. Transceptors as a functional link of transporters and receptors. Microb Cell 2017, 4, 69–73. 10.15698/mic2017.03.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gojon A.; Krouk G.; Perrine-Walker F.; Laugier E. Nitrate transceptor(s) in plants. J. Exp. Bot. 2011, 62, 2299–2308. 10.1093/jxb/erq419. [DOI] [PubMed] [Google Scholar]
- Ho C. H.; Lin S. H.; Hu H. C.; Tsay Y. F. CHL1 functions as a nitrate sensor in plants. Cell 2009, 138, 1184–1194. 10.1016/j.cell.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Schwoppe C.; Winkler H. H.; Neuhaus H. E. Connection of transport and sensing by UhpC, the sensor for external glucose-6-phosphate in Escherichia coli. Eur. J. Biochem. 2003, 270, 1450–1457. 10.1046/j.1432-1033.2003.03507.x. [DOI] [PubMed] [Google Scholar]
- Schwoppe C.; Winkler H. H.; Neuhaus H. E. Properties of the glucose-6-phosphate transporter from Chlamydia pneumoniae (HPTcp) and the glucose-6-phosphate sensor from Escherichia coli (UhpC). J. Bacteriol. 2002, 184, 2108–2115. 10.1128/JB.184.8.2108-2115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan S.; Dover J.; Johnston M. Glucose sensing and signaling by two glucose receptors in the yeast Saccharomyces cerevisiae. EMBO J. 1998, 17, 2566–2573. 10.1093/emboj/17.9.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharff-Poulsen P.; Moriya H.; Johnston M. Genetic analysis of signal generation by the Rgt2 glucose sensor of Saccharomyces cerevisiae. G3: Genes, Genomes, Genet. 2018, 8, 2685–2696. 10.1534/g3.118.200338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya H.; Johnston M. Glucose sensing and signaling in Saccharomyces cerevisiae through the Rgt2 glucose sensor and casein kinase I. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 1572–1577. 10.1073/pnas.0305901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson L. F.; Coons D. M.; Kruckeberg A. L.; Lewis D. A. Yeast sugar transporters. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 259–308. 10.3109/10409239309078437. [DOI] [PubMed] [Google Scholar]
- Kruckeberg A. L. The hexose transporter family of Saccharomyces cerevisiae. Arch. Microbiol. 1996, 166, 283–292. 10.1007/s002030050385. [DOI] [PubMed] [Google Scholar]
- Marshall-Carlson L.; Celenza J. L.; Laurent B. C.; Carlson M. Mutational analysis of the SNF3 glucose transporter of Saccharomyces cerevisiae. Mol. Cell. Biol. 1990, 10, 1105–1115. 10.1128/MCB.10.3.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick K. M.; Spielewoy N.; Kalashnikova T. I.; Guaderrama M.; Zhu Q.; Chang H. C.; Wittenberg C. Grr1-dependent inactivation of Mth1 mediates glucose-induced dissociation of Rgt1 from HXT gene promoters. Mol. Biol. Cell 2003, 14, 3230–3241. 10.1091/mbc.e03-03-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H.; Polish J.; Johnston M. Specificity and regulation of DNA binding by the yeast glucose transporter gene repressor Rgt1. Mol. Cell. Biol. 2003, 23, 5208–5216. 10.1128/MCB.23.15.5208-5216.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan J.; Mosley A. L.; Ozcan S. Repression of transcription by Rgt1 in the absence of glucose requires Std1 and Mth1. Curr. Genet. 2003, 44, 19–25. 10.1007/s00294-003-0423-2. [DOI] [PubMed] [Google Scholar]
- Mosley A. L.; Lakshmanan J.; Aryal B. K.; Ozcan S. Glucose-mediated phosphorylation converts the transcription factor Rgt1 from a repressor to an activator. J. Biol. Chem. 2003, 278, 10322–10327. 10.1074/jbc.M212802200. [DOI] [PubMed] [Google Scholar]
- Ozcan S.; Dover J.; Rosenwald A. G.; Wolfl S.; Johnston M. Two glucose transporters in Saccharomyces cerevisiae are glucose sensors that generate a signal for induction of gene expression. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 12428–12432. 10.1073/pnas.93.22.12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. M.; Brinkmann M. M.; Paquet M. E.; Ploegh H. L. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 2008, 452, 234–238. 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- Tabeta K.; Hoebe K.; Janssen E. M.; Du X.; Georgel P.; Crozat K.; Mudd S.; Mann N.; Sovath S.; Goode J.; et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- Takeuchi O.; Akira S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]