

In this study, Villa et al. compared the targeting mechanisms of the dosage compensation complex in two Drosophila species, D. melanogaster and D. virilis, that are separated by >40 million years of evolution. Using biochemical and genomic approaches, they found that the main determinants of X-chromosome recognition are conserved, but their relative contributions diverged, illustrating how components of an ancient dosage compensation system can be refined along distinct, species-specific evolutionary trajectories.
Keywords: DNA binding, chromatin, dosage compensation, evolution, long noncoding RNA
Abstract
The dosage compensation complex (DCC) of Drosophila identifies its X-chromosomal binding sites with exquisite selectivity. The principles that assure this vital targeting are known from the D. melanogaster model: DCC-intrinsic specificity of DNA binding, cooperativity with the CLAMP protein, and noncoding roX2 RNA transcribed from the X chromosome. We found that in D. virilis, a species separated from melanogaster by 40 million years of evolution, all principles are active but contribute differently to X specificity. In melanogaster, the DCC subunit MSL2 evolved intrinsic DNA-binding selectivity for rare PionX sites, which mark the X chromosome. In virilis, PionX motifs are abundant and not X-enriched. Accordingly, MSL2 lacks specific recognition. Here, roX2 RNA plays a more instructive role, counteracting a nonproductive interaction of CLAMP and modulating DCC binding selectivity. Remarkably, roX2 triggers a stable chromatin binding mode characteristic of DCC. Evidently, X-specific regulation is achieved by divergent evolution of protein, DNA, and RNA components.
Regulated gene expression largely relies on binding of transcription factors (TFs) to distinct DNA sequences. Robust and specific binding of TFs often not only depends on the intrinsic properties of their DNA binding domain (DBD) but requires cooperative interactions of two or more proteins with complex DNA elements (Morgunova and Taipale 2017; Lambert et al. 2018). The diversification and refinement of binding selectivity during evolution therefore involved coevolution of DBDs along with their targeting sites as well as of cooperative protein interactions. Given the multifactorial nature of transcription regulation, individual contributions are often difficult to tease apart. A better knowledge of overarching principles and detailed mechanisms is required to understand how phenotypic variation results from protein–DNA interactions.
The process of dosage compensation in Drosophila provides a powerful experimental system to dissect the determinants of specific DNA binding sites. Dosage compensation enhances the transcription on the male X chromosome to approximate the combined levels of the two female X chromosomes (Samata and Akhtar 2018). All functional binding sites for the transcription activator, the male-specific-lethal dosage compensation complex (MSL-DCC, or DCC for short), the “high-affinity sites” (HASs), are located on the X chromosome and can be easily distinguished from similar-looking, nonfunctional DNA elements on the autosomes. The DCC contains five proteins and at least one of the two long noncoding (lnc) roX RNAs. The MSL2 subunit is the only component that confers specific DNA binding (Villa et al. 2016). It is connected via the scaffold protein MSL1 to an epigenetic reader–writer module, consisting of MSL3 and MOF (Scott et al. 2000). MSL3 recognizes actively transcribed chromatin marked by methylation of histone H3 at lysine 36 (H3K36me3), where the acetyltransferase MOF then specifically acetylates H4K16 to boost transcription through chromatin decompaction (Akhtar and Becker 2000; Smith et al. 2000; Sural et al. 2008). In addition, the DNA/RNA helicase activity of the subunit MLE is required to incorporate the roX RNA into the complex (Ilik et al. 2013; Maenner et al. 2013; Müller et al. 2020).
MSL2 is the fundamental component of the DCC, as it is the only male-specific protein subunit (Bashaw and Baker 1995; Kelley et al. 1995; Zhou et al. 1995). Ectopic expression of MSL2 in female cells leads to DCC assembly and X-chromosome binding (Kelley et al. 1995; Villa et al. 2016). Selective X-chromosome targeting requires carefully tuned MSL2 levels, which is assured by S-phase-specific transcription (Lim and Kelley 2012) and an intrinsic E3 ubiquitin ligase activity (Villa et al. 2012).
The principle that initiates the unambiguous, exclusive targeting of the X chromosome has remained controversial. We reported earlier that MSL2 of D. melanogaster has the intrinsic ability to recognize specific sequence elements. The CXC-type DBD of MSL2 binds a subset of HASs, the so-called “PionX sites,” defined by a distinct motif (the PionX motif) and a DNA shape signature. These sites are enriched on the X chromosome, discriminate between the X chromosome and autosomes, and are recognized early in the series of events that leads to coating of the X with DCC (Villa et al. 2016).
Others suggested that the selective interaction of the DCC requires tethering by the CLAMP (chromatin-linked adaptor for MSL proteins) zinc finger protein (Soruco et al. 2013). Indeed, binding of DCC to many non-PionX HASs relies on cooperation of MSL2 with CLAMP (Albig et al. 2019; Tikhonova et al. 2019). More recently, Valsecchi et al. argued a radically different principle for initiating the binding of MSL2 to the X chromosome, which does not involve MSL2-HAS recognition. Rather, they suggest that interactions of MSL2 via an intrinsically disordered domain with roX2 RNA lead to nucleation of an X-chromosomal “condensate” (Valsecchi et al. 2021). In such a model, the fact that both roX genes lie on the X chromosome is of fundamental importance, an interesting analogy to the initiation of mammalian X-chromosome inactivation through XIST RNA (Cerase et al. 2019; Żylicz and Heard 2020). However, unlike XIST, roX RNA also functions in trans if transcribed from an autosomal location, arguing against the nucleation model (Meller et al. 1997; Ramírez et al. 2015; Ilik et al. 2017).
In the absence of the helicase MLE (and hence the roX RNAs, which require MLE for stability), the DCC does not assemble properly and the DNA-binding module consisting of MSL2/MSL1 binds to PionX sites, thus clearly demonstrating the potential of MSL2 to recognize the X in the absence of roX (Villa et al. 2016). These conflicting observations raise questions about the contributions of MSL, CLAMP, and roX for the initial identification of the X through PionX sites for further binding to the majority of HASs and, finally, for the “spreading” of the DCC to actively transcribed genes.
The MSL-dependent dosage compensation arose 60 million years ago in the Drosophila genus, along with the evolution of heteromorphic sex chromosomes and its main components, including roX RNA, have been conserved among the different Drosophila species (Bone and Kuroda 1996; Marín et al. 1996; Quinn et al. 2016; Ellison and Bachtrog 2019). “Chromatin isolation by RNA purification” (ChIRP) experiments performed with roX-specific probes in several Drosophila species identified a GA-rich motif as a common feature of the MSL-DCC binding sites (Quinn et al. 2016; Ellison and Bachtrog 2019). These MSL recognition elements (MREs) (Alekseyenko et al. 2008; Straub et al. 2008) apparently originated through point mutation of presites, microexpansion of GA repeats or transposons bearing precursor sequences (Ellison and Bachtrog 2019). However, the GA-rich consensus motifs differ in detail in different species, suggesting variation in MSL-MRE recognition (Quinn et al. 2016; Ellison and Bachtrog 2019). The corresponding species-specific variation of the DNA-binding properties of MSL2, and of the DCC in general, were never investigated. In particular, it is not known whether the initial recognition of PionX motifs by CXC domains is a conserved feature of dosage compensation in different Drosophila species.
Here, we compared the targeting mechanisms of the DCC in two Drosophila species, D. melanogaster and D. virilis, that are separated by >40 million years of evolution (referred to here as “virilis” and “melanogaster”). We found that the main determinants of X-chromosome recognition (MSL2, CLAMP, and roX RNA) are conserved, but their relative contributions diverged. In virilis, the X chromosome is not enriched in PionX motifs and, correspondingly, the CXC domain of MSL2vir does not show the PionX specificity of its melanogaster homolog. We found that the intrinsic specificity of MSL2 for the X chromosome correlates with the enrichment of PionX motifs on the X, suggesting a coevolution of proteins and genomes. In virilis, where this mechanism is not active, the roX2 RNA is required to refine MSL2 selectivity for HASs. The comparison of both systems also revealed that roX2 changes the mode of DCC–chromosome interaction, suggesting a topological linkage between the DCC and DNA that may “lock in” the complex after the successful identification of the X chromosome. Our data show that the initial selection of the X chromosome by the MSL-DCC is achieved in a species-specific manner via the modulation of conserved cofactors.
Results
MSL2vir does not have intrinsic specificity for the X chromosome
MSL2 in melanogaster, MSL2mel, targets the DCC to the X chromosome via the intrinsic ability of the CXC domain to recognize the PionX sites (Villa et al. 2016). To explore whether this mechanism was conserved across the Drosophila genus, we searched the genome of 13 Drosophila species for PionX sequence motifs and plotted their enrichments on the X chromosome (Fig. 1A). Only strong matches to the PionX motifs (score >22) that proved to be a good discriminator of X specificity were considered (Villa et al. 2016). Interestingly, the analysis revealed an inverse relationship between number of PionX motifs and their enrichment on the X chromosome: The lower the number, the more they are enriched on the X (Fig. 1A). To investigate the functional meaning of these findings, we focused on virilis, a species that diverged from melanogaster 40 million years ago. Virilis has an MSL-based dosage compensation system but does not show a strong enrichment of PionX motifs on the X chromosome (Fig. 1A; Quinn et al. 2016). Additional computational analyses highlighted that the virilis genome contains numerous PionX motifs. At a low statistical threshold (q = 0.2), we identified >70,000 motifs, compared with 2667 motifs in melanogaster. A higher threshold (q = 0.05) yielded 3770 motifs in the virilis genome compared with 34 motifs in melanogaster (Fig. 1B; Villa et al. 2016). Intriguingly, the abundant PionX motif was not enriched on the X chromosome in virilis, independently of the score applied (Fig. 1B). Assuming that MSL2 is responsible for the targeting of the DCC also in virilis, we then compared the amino acid sequences of MSL2vir and MSL2mel. The sequence alignment of the two proteins showed conservation in the RING, the CXC, and the CLAMP binding (CBD) domains. The unstructured parts of MSL2mel, like the region between the RING and the CXC domains and the proline/basic amino acid stretch (PB domain) in the C terminus are poorly conserved (Supplemental Fig. S1A). To test how the level of structural conservation of the CXC domains affects their DNA binding specificity, we performed DNA immunoprecipitation assays followed by sequencing (DIP-seq) on fragmented genomic DNA from melanogaster. Analogous experiments had previously led to the identification of PionX sites in melanogaster (Villa et al. 2016). Cataloging the preferred binding sites of recombinant MSL2vir in vitro using melanogaster genomic DNA, we observed that indeed MSl2vir did not select PionX sites but rather bound thousands of genomic loci with no intrinsic specificity for the X chromosome (Fig. 1C,D). We concluded that the PionX motif does not mark the X chromosome in all Drosophila species and that, in virilis, this is in line with the inability of the CXC domain of MSL2vir to recognize this motif specifically.
Figure 1.

PionX sites are not universal X-chromosome determinants. (A) Number (top panel) and X-chromosomal enrichment (bottom panel) of PionX motif hits (score >22) in different Drosophila species. Species are in ascending order with respect to X-enrichment. (B) Number (top) and X-chromosomal enrichment (bottom) of PionX motif hits at different q-value cutoffs for D. melanogaster (left) and D. virilis (right) (see also Villa et al. 2016). (C) Chromosomal distribution of in vitro binding sites of MSL2mel (MEL) and MSL2vir (VIR). The chromosomal size distribution (genome) is provided for reference. Note the difference in absolute numbers of DIP-seq peaks (MSL2mel: 246, MSL2vir: 1850). Considered are only common peaks in N = 2 and N = 5 independent experiments for D. melanogaster and D. virilis, respectively. (D) Representative DIP-seq profiles of MSL2mel and MSL2vir illustrating a region on chromosome X. MSL2mel in vitro (DIP) binding sites are marked at the bottom.
The X chromosome of virilis may recruit MSL2vir through other DNA determinants. To test this hypothesis, we performed DIP assays with genomic DNA from virilis male flies. MSL2mel mildly enriched the X chromosomal sequences from the virilis genome, in agreement with the enrichment of high-score PionX motifs on the X (Fig. 1A,B; Supplemental Fig. S1B), but MSL2vir did not show any selectivity (Supplemental Fig. S1B). We conclude that MSL2vir can bind to DNA in vitro but, in contrast to MSL2mel, does not possess an intrinsic specificity for the X chromosome. Evidently, the targeting mechanism for the DCC must be different in the two species.
DNA binding of MSL2vir depends on the CXC domain
We previously showed that MSL2mel can bind DNA with at least two structures in vitro: The CXC domain determines PionX selectivity and the C terminus (C-ter) of the protein contributes to general DNA binding to GA copolymers (Villa et al. 2016). The C-ter contains an intrinsically disordered region (IDR) rich in prolines and basic amino acid residues (PB) that is also involved in roX RNA binding (Li et al. 2008; Müller et al. 2020; Valsecchi et al. 2021). To explore whether the MSL2vir DNA-binding properties depend on the CXC domain, we swapped CXC domains between MSL2mel and MSL2vir and tested the chimeric proteins in DIP-seq assays (Fig. 2A,B; Supplemental Fig. S2A).
Figure 2.

The DNA binding of both MSL2mel and MSL2vir depends on their CXC domains. (A, top) Representation of MSL2 swap constructs used in DIP assays. (Bottom) Representative DIP-seq profiles of a region on the X chromosome of the indicated MSL2 swap constructs: MSL2mel (MEL), MSL2vir (VIR), MSL2virCXCmel (VIRCXCmel), MSL2melCXCvir (MELCXCvir), MSL2virCTmel (VIRCTmel), MSL2virPBmel (VIRPBmel). The MSL2mel in vitro (DIP) binding sites are marked at the bottom. (B) Hierarchically clustered heat map of DIP log2 (read counts) at MSL2mel in vitro (DIP) binding sites using the indicated swap constructs. Reads at peak sites were counted after subsampling to the same library size. Note that the signal of the DIP assays cluster in two main groups indicated as “MSL2mel” (MEL) and “MSL2vir”(VIR). (C) Superposition of 1H,15N HSQC NMR spectra, of Drosophila virilis CXC domain free (black) and bound to S12 (red) and S12 mutant (green) DNA. (D) NMR titration curves globally fitted to a three-site binding model along with the binding affinity for the binding of S12 and S12 mutant DNA to the virilis CXC domain.
A cluster analysis of the DIP-seq data confirmed the visual impression gained from browser screenshots (Fig. 2A): The substitution of the CXC domain of MSL2vir with the one of MSL2mel (MSL2virCXCmel) was sufficient to partially restore the in vitro binding pattern of MSL2mel. The data were obtained with the MSL2virCXCmel cluster in the “MSL2mel group” and not in the “MSL2vir group” (Fig. 2B). Conversely, the CXC domain of MSL2vir conferred to MSL2mel (MSL2melCXCvir) the nonspecific binding mode characteristic of MSL2vir (Fig. 2A,B).
Because IDRs sometimes contribute to binding specificity (Brodsky et al. 2020) and the C-terminal IDR of MSL2mel is also involved in DNA binding, we tested the DNA-binding potential of this region in MSL2vir. Substitution of the C-ter of MSL2vir with the IDR of MSL2mel did not affect the binding properties of the protein, that is, MSL2virPBmel still clusters with MSL2vir while MSL2virCTmel (in which the entire melanogaster C-ter, including the CXC domain, is transplanted onto MSL2vir) clusters with MSL2mel (like MSL2virCXCmel) (Fig. 2A,B). We conclude that the CXC domain is the main determinant for DNA binding in both proteins.
The CXC domain of MSL2vir lacks MRE specificity
The isolated CXC domain of melanogaster (CXCmel) binds a GA-rich DNA derived from the MRE motif (S12) 15-fold better than a mutated sequence, in which GA is replaced by an A tract (Zheng et al. 2014). To explore the DNA-binding properties of the CXC domain of MSL2vir (CXCvir) in an analogous setting, we monitored the binding of the isolated domain to S12 DNA using 1H-15N HSQC NMR titrations (Fig. 2C,D; Supplemental Fig. S2B–D). Upon titration with S12 DNA, 1H-15N HSQC peaks of the CXCvir domain exhibited chemical shift perturbations (CSPs) in the fast to intermediate exchange, suggesting affinity in the low micromolar to high nanomolar range (Fig. 2C; Supplemental Fig. S2C). In agreement with this, quantitative analysis of CSPs revealed an apparent dissociation constant (KD) of 2.8 µM for the binding of S12 to CXCvir, which was in the same range as that for CXCmel (2.7 µM) (Fig. 2D; Zheng et al. 2014). Similar to S12, titration of the S12 mutant with CXCvir showed fast to intermediate exchange in the NMR titration, suggesting a similar affinity (Fig. 2D; Supplemental Fig. S2C). CXCvir showed no decrease in affinity for the S12 mutant (KD = 2.4 µM), in contrast to the 15-fold drop of affinity for this nonconsensus site observed with CXCmel (KD = 42.8 µM) (Fig. 2C,D; Zheng et al. 2014). Comparison of CSPs suggests that the binding mode of the S12 and S12 mutant DNA to the CXCvir remains similar upon titrations (Supplemental Fig. S2C,D). We conclude that the CXCvir can bind DNA in vitro but lacks sequence specificity.
A male-specific factor confers X-chromosome specificity to MSL2vir in vivo
So far, the results suggest that MSL2vir binds DNA in vitro via its CXC domain, but it lacks the intrinsic selectivity for the X chromosome of MSL2mel. However, virilis DCC binds specifically to the X chromosome in male flies (Marín and Baker 1998; Chlamydas et al. 2016; Quinn et al. 2016). What, then, provides this specificity in vivo? To address this question, we expressed MSL2vir and MSL2mel as GFP fusion proteins in male (S2) or in female (Kc167) melanogaster cells.
Male cells are a good model system to study dosage compensation in the steady state. Female cells, instead, offer the opportunity to investigate the capability of the two MSL2 proteins to initiate the de novo assembly of the DCC and its first-time targeting to the X chromosome. Immunofluorescence (IF) staining confirmed that MSL2mel binds specifically to the X chromosome in male cells, visualized by staining of the characteristic X-chromosome territory. Importantly, MSL2mel was mainly localized to the compact territory in female cells. MSL2vir, in contrast, was able to associate with the X chromosome in male cells but failed to mark the territory in female cells (Fig. 3A). In female cells, MSl2vir displayed a distributed nuclear staining with some aggregates, suggesting inappropriate targeting (Fig. 3A). Remarkably, MSL2vir colocalized with MSL3 and H4K16ac also in female cells, arguing that it formed an acetylation-competent complex with endogenous proteins, which was, however, not properly targeted to the X chromosome (Fig. 3A).
Figure 3.

MSL2vir does not support de novo X-chromosome targeting in female melanogaster cells. (A) Representative immunofluorescence pictures of either male (top panels) or female (bottom panels) D. melanogaster cells transfected with the indicated MSL2-GFP constructs, MSL2mel-GFP (MEL-GFP) and MSL2vir-GFP (VIR-GFP). Cells were stained using antibodies against GFP, MSL3, H4K16ac, and DAPI to stain DNA. Scale bar, 3 µm. (B) Chromosomal distribution of GFP-ChIP peaks obtained from two independent experiments using female melanogaster cells transfected with the indicated constructs, MSL2mel (MEL) and MSL2vir (VIR). The relative size of the chromosomes (genome) serves as a reference for uniform distribution. (C) Representative ChIP-seq profiles of a region on chromosome X (GFP or H4K16ac) obtained using female melanogaster cells expressing MSL2mel (MEL) or MSL2vir (VIR). MSL2mel in vivo binding sites (HASs) are marked in the lowest track. The colors represent the different cells used in the experiments. Untransfected cells are labeled as Ctrl. (D) X-chromosomal H4K16ac coverage relative to total genome coverage for two independent experiments. Extent of domains equals the sum of widths of called peaks. H4K16ac ChIP-seq profiles obtained using female melanogaster cells expressing the indicated MSL2-GFP constructs (MSL2mel [MEL] and MSL2vir [VIR]). Ctrl indicates untransfected cells.
Failure to assemble a properly targeted DCC in female cells upon MSL2 expression may be due to limiting amounts of the other complex members: MSL1 and MSL3 express at low levels in Kc cells and the roX RNAs are almost undetectable. Western blots confirmed that expression of MSL2mel and MSL2vir led to increased expression of MSL1 and MSL3 in female cells, albeit to different degrees (Supplemental Fig. S3B). MSL2 is responsible for inducing roX2 transcription by binding to a 3′ enhancer element that contains MREs, an early event in dosage compensation (Rattner and Meller 2004; Lim and Kelley 2012). We explored whether expression of MSL2 proteins from different Drosophila species can induce roX2 in melanogaster cells, with the idea in mind that the quantitative readout may inform about the ability to interact with the roX2 MREs. Expression of MSL2 proteins from melanogaster, virilis, willistoni, and busckii all induced roX2 transcription in female cells, albeit to different degrees (Supplemental Fig. S3C). The result confirms the functionality of the proteins in the heterologous system. Not surprisingly, homologous MSL2mel induced roX2 transcription most. The fact that expression of MSL2vir led to the lowest induction of roX2 is in line with its poor ability to recognize MRE sequences.
To understand the different nuclear localization of the virilis and the melanogaster MSL2-GFP proteins in female cells, we mapped their chromosomal localization by ChIP-seq using antibodies against GFP, MSL3, and H4K16ac. While GFP and MSL3 antibodies inform about the genomic localization of MSL2 and the recruitment of the MSL-DCC, respectively, H4K16ac antibodies report on the functionality of the chromatin-bound complex. In agreement with the IF data, the ChIP-seq results confirmed that MSL2mel was able to initiate dosage compensation in female cells. MSL2mel was found almost exclusively bound to the X chromosome (Fig. 3B) along with a functional DCC complex, visualized by MSL3 and H4K16ac at gene bodies (Fig. 3C,D). In contrast, MSL2vir was bound all over the genome, with just modest enrichment for the X chromosome, in line with the IF staining (Fig. 3B). Of note, the fraction of MSL2vir bound to the X chromosome also recruited a functional DCC, witnessed by MSL3 and H4K16ac ChIP (Fig. 3C,D; Supplemental Fig. S3A). We conclude that MSL2vir, when expressed in melanogaster cells, can initiate the assembly of a functional DCC despite low roX2 levels in female cells. In male cells, MSL2vir binds specifically to the X chromosome, but in female cells it cannot, suggesting that there must be a male-specific factor that provides this selectivity to MSL2vir.
CLAMP is an exceptionally strong interactor of MSL2vir in vivo
We considered that the male-specific factor might be a protein that interacts with MSL2vir in male cells. In an unbiased search for such an interactor, recombinant FLAG-tagged MSL2vir was incubated in soluble nuclear extracts (devoid of DNA) from male virilis cells and immunoprecipitated with an αFLAG antibody under stringent conditions. We used a male diploid embryonic cell line from D. virilis for this experiment to be sure to also detect species-specific interactors. Mass spectrometry analyses revealed some unique interactors of MSL2vir in comparison with the control (Fig. 4A). Among the strongest interactors were, as expected, the virilis MSL proteins (MSL1, MSL3, and MOF), but not MLE, which is known not to interact with the other MSLs under stringent conditions (Smith et al. 2000). We also found some nucleoporins, an RNA helicase (BAM1G4), the DNA replication-related element factor (DREF), and two zinc finger proteins (A0A0Q9W5D2 and CLAMP) (Supplemental Table 1). Surprisingly, CLAMP was the most highly enriched protein apart from bystander chaperones (Fig. 4A).
Figure 4.

The zinc finger protein CLAMP is a strong interactor of MSL2vir in vivo. (A) Volcano plot showing log10 P-value in relation to the average log2 fold change (n = 4 biological replicates) comparing the FLAG-MSL2vir (MSL2vir) AP-MS versus the control purification with FLAG-only beads (Ctrl). MSL2vir is represented in red, other DCC subunits are represented in blue, and CLAMP is in orange. Heat shock proteins (green) are considered irrelevant bystander proteins. (B) Western blot analyses of coimmunoprecipitation (“pull-down”) experiments using the indicated MSL2-FLAG recombinant construct MSL2mel (MEL) or MSL2vir (VIR) as bait to retrieve proteins from extracts of either melanogaster or virilis cells. Five percent of the extracts was loaded as a reference (Input). “M” indicates the protein marker. (C) Western blot analyses of pull-down experiments as in B. Five percent of the extracts was loaded as a reference (Input). “M” indicates the protein marker. (D) Venn diagram showing overlapping peaks of MSL2vir (VIR; GFP ChIP N = 2), MSL2mel (MEL; GFP ChIP N = 2), and CLAMP (CLAMP ChIP from Kc cells N = 2) (from Soruco et al. 2013). Only peaks common to all replicates are considered.
CLAMP is a highly conserved DNA-binding protein that was previously described to bind to melanogaster MSL2 and synergize with it for HAS binding (Larschan et al. 2012; Albig et al. 2019). Although CLAMP promotes the interaction of MSL2 with HASs, it also has thousands of binding sites scattered throughout the genome and thus, per se, does not confer X-chromosome selectivity. Unfortunately, neither CLAMP nor any of the other interactors are specifically expressed in male cells, making their involvement in targeting MSL2vir to the X chromosome in male cells unlikely. However, intrigued by the finding that, in virilis MSL2 interacts with CLAMP even better than with MSL1, we compared this interaction in virilis and melanogaster. To this end, we performed “pull-down” experiments as described above with recombinant MSL2vir-FLAG and MSL2mel-FLAG and nuclear extracts from male virilis or melanogaster cells. Interestingly, while both MSL2vir and MSL2mel precipitated CLAMP from the two extracts, MSL2vir always interacted stronger with CLAMP than MSL2mel (Fig. 4B). We observed the opposite for MSL1: Both MSL1mel and MSL1vir interact better with MSL2mel than MSL2vir (Fig. 4B). Likewise, CLAMP in extracts of male or female melanogaster cells always interacted stronger with MSl2vir than with MSL2mel (Fig. 4C). These results suggest that the different strength of CLAMP interaction is an intrinsic property of the two MSL2 proteins. Recently, the CLAMP binding domain on MSL2 was mapped to amino acids 620–685 (Albig et al. 2019; Tikhonova et al. 2019). This region is highly conserved between MSL2mel and MSL2vir (Supplemental Fig. S4A), and it is likely that CLAMP interacts with the same region in both MSL2 proteins. Indeed, pull-down experiments using the MSL2 swap constructs (Fig. 2A) are in agreement with this assumption. Replacing half of the CBD in MSL2vir with the conserved one of MSL2mel (MSL2virPBmel) reduced the affinity for CLAMP, and substituting the CXC of MSL2mel with the CXC of MSL2vir (MSL2melCXCvir) in some experiments improved the interaction with CLAMP (Supplemental Fig. S4B).
The observation that MSL2vir binds tighter to CLAMP raises the idea that CLAMP might have a more prominent role in targeting the DCC in virilis than in melanogaster. Accordingly, MSL2vir may be “derouted” by CLAMP in the melanogaster context. Support for this idea comes from reinspection of the ChIP-seq binding profile of ectopic MSL2vir in female cells. Remarkably, 85% of MSL2vir in vivo binding sites overlap with CLAMP binding sites, 65% of which are not shared with MSL2mel (Fig. 4D). Likewise, the binding profile of MSL2virCXCmel-GFP also resembled the CLAMP binding pattern in female cells. Apparently, the strong virilis CBD overrules the binding properties of the CXCmel. This is in contrast to its DNA selectivity in vitro, which resembles more that of MSL2mel, since in the absence of CLAMP, the intrinsic CXC properties dominate (Fig. 2A,B; Supplemental Fig. S4C ). We conclude that the strong interaction between MSL2vir and CLAMP may indeed deroute the former away from the X to nonfunctional CLAMP binding sites. In summary, our affinity purification approach did not identify any protein with a male-specific role in targeting MSL2vir to the X chromosome but suggested an explanation for the observed autosomal binding of MSL2vir.
Long, noncoding roX2 RNA confers specificity to MSL2vir X-chromosome binding
Because we could not identify a protein that provides X-binding specificity to MSL2vir, we considered that the missing, male-specific factor might be roX2 RNA, which is expressed in male cells and a subunit of the DCC. Nascent roX2 RNA can concentrate MSL proteins at chromosomal loci (Kelley et al. 1999; Oh et al. 2003; Valsecchi et al. 2021). Our previous studies had ruled out a role for roX in the initial selection of the X chromosome by MSL2mel (Villa et al. 2016), but this may be different in other species, like virilis. To test this possibility, we cotransfected the roX2 gene together with MSL2vir-GFP into female cells and identified the transfected cells by immunostaining. Remarkably, we observed that, in the presence of roX2, most GFP-positive cells showed coherent X-territories defined by colocalization of MSL2-GFP, MSL3, and H4K16ac (Fig. 5A). In addition, the total number of ChIP-seq peaks for MSL3 doubled, indicating an improved binding of the DCC (Fig. 5B). Notably, this was not just due to an overall gain of ChIP efficiency but reflected an improved specificity, since the signals for MSL3 and H4K16ac, the functional readout of targeting, shifted from autosomes to the X chromosome in the presence of roX2 (Fig. 5B,C). ChIP-seq signals for MSL3 and H4K16ac shifted from autosomes to the X chromosome in the presence of roX2 (Fig. 5B,C). Similar effects were observed using roX2 genes from melanogaster or virilis, confirming their functional conservation (Supplemental Fig. S5; Quinn et al. 2016). We conclude that roX2 is the male-specific factor that provides targeting selectivity to MSL2vir.
Figure 5.

The long, noncoding roX2 RNA provides X specificity to MSL2vir. (A) Representative immunofluorescence pictures of female melanogaster cells transfected with the MSL2vir-GFP (VIR) construct with (+roX2) or without the roX2 gene. Cells were stained using antibodies against GFP, MSL3, H4K16ac, and DAPI to visualize DNA. Scale bar, 3 µm. (B) Chromosomal distribution of MSL3 ChIP peaks obtained from three independent experiments using female melanogaster cells transfected with MSL2vir-GFP (VIR) construct with (+roX2) or without the roX2 gene. Only peaks common to all replicates are considered. The relative size of the chromosomes (genome) serves as a reference for uniform distribution. (C) Distributions of mean signal intensity changes for MSL3 (left) and H4K16ac (right) on their corresponding autosomal (A) and X-chromosomal (X) domains upon expression of roX2 in female melanogaster cells transfected with the MSL2vir-GFP construct. (N = 4 for each ChIP target).
Diverse roles for roX2 in MSL2 targeting and DCC assembly
The increased signal of MSL3 and H4K16ac ChIP-seq on the X chromosome in the presence of roX2 could mean that (1) roX2 improves the DNA binding specificity of MSL2vir, which binds to additional HASs, or (2) roX2 promotes the assembly of DCC at chromatin-bound MSL2vir. To test for these possibilities, we compared MSL3 ChIP-seq peaks in the presence or absence of roX2 with HASs, the physiological MSL2 binding sites. MSL2vir alone was able to bind ∼14% of HASs (conceivably due to the modest activation of the endogenous roX2 gene) (Supplemental Fig. S3C), but upon cotransfection of roX2, the number of bound HASs doubled (Fig. 6A). This indicates that the RNA triggers the binding of MSL2vir to new HASs.
Figure 6.

RoX2 affects the DNA binding of MSL2vir in different ways. (A) Venn diagram showing overlapping peaks of three independent MSL3 ChIP experiments using female melanogaster cells transfected with the MSL2vir-GFP (VIR) construct with (+roX2) or without the roX2 gene and MSL2mel in vivo binding sites (HASs). Only peaks common to all replicates are considered. (B) Western blot analyses of pull-down experiments using the recombinant MSL2mel-FLAG (MEL) or MSL2vir-FLAG (VIR) constructs as bait to retrieve interacting proteins from extracts of male melanogaster cells, untreated or treated with RNase A. (C) Representative DIP-seq profiles of MSL2vir (VIR) in the presence of no RNA (VIR), of a control RNA (CTRL), or of the roX box RNA (RB4). (D) Read counts on 9534 ensemble binding sites from DIP-seq experiments of MSL2vir in the presence of no RNA (VIR), of control RNA (CTRL), or of the roX box RNA (RB4). Two independent experiments are shown. (E) FRAP analyses of female melanogaster cells expressing either MSL2vir-GFP (nucleoplasm) or MSL2vir-GFP + roX2 (X). Relative fluorescence intensity measured in the bleached area was plotted against recovery time. Shade curves represent the standard error of the measurements. N = 9.
Comparing the roX2-dependent changes in H4K16ac and MSL3 signals at HASs constitutively bound by MSL2vir versus HASs just bound in the presence of roX2, we found that both increased in the presence of roX2 (Supplemental Fig. S6A,B). This shows that more active DCC was recruited to these loci. In summary, the presence of roX2 instructs MSL2vir to bind to more HASs and also promotes the assembly of functional DCC.
RoX2 binds to the C terminus of MSL2 (Li et al. 2008; Müller et al. 2020; Valsecchi et al. 2021). Given that CLAMP also contacts this region, both factors may mutually affect each other's interactions. We explored this possibility in pull-down experiments monitoring the MSL2-CLAMP interaction in the presence or absence of RNA. MSL2vir-FLAG was incubated in nuclear extracts from male cells under low-salt conditions compatible with RNA binding. Endogenous RNA was removed by RNase or benzonase treatment (enzymes were omitted from appropriate controls). In the absence of RNA, the interaction between CLAMP and MSL2vir was remarkably enhanced (Fig. 6B; Supplemental Fig. S6C). As a control, the interaction between MSL2vir and MLE was much reduced upon RNA degradation (Fig. 6B; Ilik et al. 2013). These findings are consistent with the idea that roX2 and CLAMP compete for the binding to the C terminus of MSL2vir. Accordingly, in male cells expressing roX2, MSL2vir will not be rerouted by CLAMP to autosomal sites and will bind better to HASs on the X (Fig. 3A).
MSL2 binds roX2 specifically at the roX box, once this motif is exposed by MLE-dependent secondary structure remodeling (Ilik et al. 2017; Müller et al. 2020). It is conceivable that roX2 RNA has an additional, instructive effect modulating the conformation of MSL2. To explore this possibility, we carried out DIP-seq assays in the presence of RNA oligonucleotides containing either a roX box sequence or an unrelated control sequence. We observed that MSL2vir retrieved more DNA fragments in a standardized DIP with a genomic DNA substrate in the presence of the specific roX2 oligonucleotide (but not the control), albeit the specificity of binding was unaltered. These data suggest that roX improves the affinity of MSL2vir for DNA (Fig. 6C,D).
roX2 switches MSL2vir to a stable X-chromosome binding mode
In contrast to other chromatin complexes, the DCC associates unusually tightly with the X chromosome. “Fluorescence recovery after photobleaching” (FRAP) studies had shown earlier that MSL2 binds the X chromosome in such a stable manner that is incompatible with canonical protein–DNA interactions but only observed with factors that engage chromatin with a topological linkage (Straub et al. 2005). These early studies also identified a minor, nucleoplasmic pool of MSL2 with fast exchange rates. Intrigued by our observation that roX2 RNA was required for MSL2vir to associate with a coherent chromosomal territory, we examined the mobility of MSL2vir-GFP expressed in female cells. In the presence of roX2, MSL2-GFP localized to the X chromosome seen as a coherent territory with very little nucleoplasmic staining (Fig. 5A). In the absence of the RNA, MSL2 was diffusely distributed throughout the nucleus, with occasional condensates, but without an identifiable X-chromosome territory (Figs. 3A, 5A). We now tested by FRAP whether these different nuclear localizations also differed in binding mode. Remarkably, MSL2vir bound to the X chromosome (in the presence of roX2) had a low mobility similar to the one of MSL2mel (Fig. 6E; Supplemental Fig. S6D; Straub et al. 2005). In contrast, in the absence of roX2, MSL2vir was localized more diffusely and exhibited highly dynamic chromatin binding (Fig. 6E; Supplemental Fig. S6D). The analogous experiment with MSL2mel could not be done since expression of MSL2mel rapidly induces roX RNA expression, so that the RNA-dependency cannot be tested in this setting. We conclude that the tight binding of MSL2 to the X chromosome is a feature conserved during the evolution of the Drosophila genus. We show that the tight binding of MSL2vir depends on the presence of roX2 and speculate that this may also be the case for MSL2mel.
Discussion
Understanding the mechanism underlying the selective targeting of the MSL-DCC to the X chromosome in Drosophila is instructive for many similar problems of transcription regulation. The binding to the X chromosome appears as a multistep process, involving an initial recognition of a limited number of sequence-defined “high-affinity sites” (HASs), followed by distribution to sites of lower affinity and, finally, the association with transcribed chromatin via epigenetic marks (Lucchesi and Kuroda 2015). Our study mainly addresses the initial recognition of the X chromosome, where several targeting principles, all involving the DNA binding subunit of the DCC, MSL2, had been described: the specific binding of its CXC domain to PionX sites (Villa et al. 2016), cooperative interactions with the zinc finger protein CLAMP at GA-rich MREs (Soruco et al. 2013; Albig et al. 2019), and, most recently, a tethering of MSL2 through roX2 (Valsecchi et al. 2021). We explored the evolutionary conservation of these principles, hoping to evaluate their broader relevance. Focusing on D. virilis, a species that diverged from D. melanogaster 40 million years ago, we found that the involvement in X-chromosome targeting of the three components, MSL2, CLAMP, and roX2 RNA, have been conserved, but their precise contributions differ. Evidently, starting from an ancestral set of components, evolution diverged and achieved the goal of X-specific DCC association through different strategies. These findings impressively illustrate the nondirectional nature of evolution and the emergence of different, viable solutions to the overarching problem of attaining X-chromosome selectivity (Fig. 7).
Figure 7.
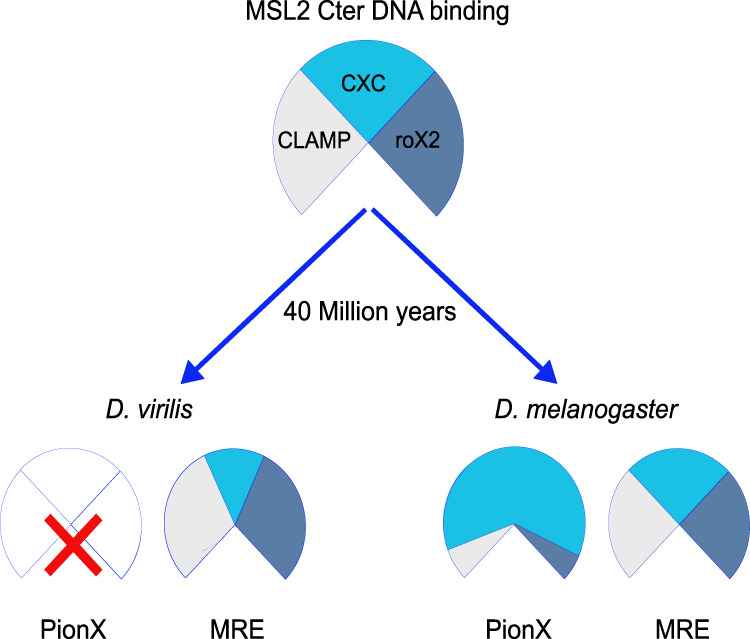
Model. Species-specific ways to achieve X-chromosome specificity in dosage compensation. The CXC domain of MSL2, the CLAMP protein, and the roX2 lnc RNA are conserved factors involved in the targeting of the DCC to the X chromosome both in D. virilis and in D. melanogaster, two species that diverged 40 million years ago. The X chromosome of D. virilis is not marked by PionX motifs, and the binding of the CXC domain of MSL2 to DNA (MREs) relies on CLAMP cooperativity and on roX2 modulation to achieve X specificity. In D. melanogaster, the coevolution of PionX sites enriched on the X chromosome, together with the ability of the CXC domain of MSL2 to specifically recognize them, endows MSL2 with direct X-chromosome selectivity. The cooperative interaction with CLAMP and roX2 RNA contributes to recognition of non-PionX MREs.
Coevolution of PionX sites and CXC domain specificity
Recently, we demonstrated that the melanogaster X chromosome is marked by a distinct class of MREs, termed PionX sites, which serve as first X-specific DNA determinants (Villa et al. 2016). These sites are distinguished by a 5′ sequence extension of the GA-rich MRE (the PionX motif). The CXC domain of MSL2 specifically recognizes a PionX DNA shape signature. Such sites are found to mark dosage-compensated X chromosomes in other Drosophila species as well. For example, in D. miranda, PionX sites apparently derive from a transposon-borne precursor sequence and are fixed on the neo-X chromosome (Ellison and Bachtrog 2013; Villa et al. 2016). To explore whether PionX sites play a similarly important role in other Drosophila species, we computationally identified high-confidence PionX motifs and monitored their X-chromosome enrichment (Fig. 1A). The results show that, in many species, including virilis, PionX motifs cannot be involved in dosage compensation because they are too numerous and not enriched on the X. The comparison of melanogaster and virilis leads us to hypothesize that along with the enrichment of the PionX motif on the X, the CXC domain of MSL2 evolved a specific recognition mode, as was observed for transcription factors and their binding sites (Yang et al. 2011). The general applicability of this hypothesis remains to be tested in light of the inverse relationship between the number of PionX motifs and their X-chromosome enrichment in different Drosophila species. However, our findings that activation of roX2 in female cells (as a proxy of X-chromosome binding) expressing MSL2 constructs from different species correlate with the enrichment of PionX motifs on the X provides support for this idea (Fig. 1A; Supplemental Fig. S3C).
roX RNA antagonizes the nonproductive side effects of CLAMP cooperation
The specialized CXC domain of MSL2 in melanogaster autonomously discriminates the PionX signature. The majority of HASs lack PionX features and rather contain GA-rich MREs. MSL2 can bind to them but cannot distinguish these sequences from nonfunctional GA-rich sites on autosomes (Villa et al. 2016). MSL2 binding to GA-rich sequences is promoted by cooperation with the ubiquitous CLAMP protein, which also binds GA with a subset of its zinc fingers. However, CLAMP not only binds to functional MREs but to thousands of GA-rich sites throughout the genome (Kuzu et al. 2016). If both proteins are analyzed together in genome-wide DIP, a relatively low X-chromosome enrichment is scored, since CLAMP stabilizes the binding of MSL2 to GA-rich sequences everywhere in the genome. In cells, however, the cooperativity between CLAMP and MSL2 is clearly focused on MREs (Soruco et al. 2013; Albig et al. 2019). The reason for this has been enigmatic.
Our results now suggest that roX2 binding to MSL2, which we and others mapped to the C terminus in the direct vicinity of CLAMP binding (Müller et al. 2020; Valsecchi et al. 2021), counteracts the derouting effect of CLAMP either by weakening the CLAMP interaction or by allosteric modulation of the DNA-binding properties of MSL2. This mechanism was observed following MSL2vir chromosome association in a DCC-naïve situation in the presence and absence of roX2. The analogous experiment with MSL2mel is not possible, because expression of MSL2 in females induces roX2 (Villa et al. 2016). Nevertheless, modulation of DNA binding of melanogaster MSL2 by roX is clearly compatible with the known requirement of roX RNA and MLE for non-PionX HAS recognition in vivo (Oh et al. 2003; Li et al. 2008; Figueiredo et al. 2014; Villa et al. 2016), the localization of MLE at most HASs in cells (Straub et al. 2013), and the massive derouting activity of CLAMP in the absence of RNA in vitro (Albig et al. 2019).
Most interestingly, we found that a gain of PionX specificity of the CXC domain in melanogaster went along with a decreased affinity for CLAMP (relative to virilis). We speculate that the evolution of intrinsic PionX specificity of the CXC domain (along with PionX enrichment on the X) permitted relaxation of the CLAMP cooperativity and hence the danger of rerouting, further improving the X specificity. In the absence of the PionX sites (virilis), MSL2 relies more on CLAMP cooperativity, which necessitated modulation by roX2 to counteract rerouting to autosomes.
Potential roles for roX RNA
Long, noncoding RNA bears enormous potential to tune molecular interactions, since their fast evolution (Pang et al. 2006; Quinn et al. 2016) allows accumulation of different functions. The fact that MSL2vir binds more HASs in the presence of a transfected roX2 gene demonstrates that roX2 can act in trans and must not be transcribed from the X chromosome, in agreement with earlier findings (Meller et al. 1997; Ramírez et al. 2015; Ilik et al. 2017). So far, there is no evidence for a direct base-pairing mechanism, either through triple helices or R-loops, between roX and HAS DNA.
Our data rather suggest that roX2 affects DCC–X chromosome interactions and functionality in several ways: (1) Small roX box oligonucleotides improve the affinity of MSL2 for DNA without changing the specificity (Fig. 6C,D). (2) roX2 counteracts the rerouting activity of CLAMP (Figs. 5, 6). (3) RoX2 increases the assembly of functional DCC at HASs (Supplemental Fig. S6A,B). The roX RNA may be sufficiently long to allosterically affect several protein subunits of the DCC, all of which have RNA-binding potential but can form complexes in the absence of RNA (Müller et al. 2020). (4) roX2 triggers a tight chromosome binding mode of MSL2 (see below; Fig. 6E). All effects are consistent with the speculative idea that roX RNA may combine various RNA aptamers that allosterically modulate the conformation of DCC subunits in a species-specific manner. Future experiments will test this idea.
roX RNA triggers a switch to a tight chromatin binding mode
We earlier reported FRAP experiments showing that the association of MSL2 and MSL1 with the X chromosome is characterized by an unusually long residence time (Straub et al. 2005). In those experiments, the heterochromatin protein HP1 was much more mobile than MSL2. Such immobile behavior is not explained by classical, even multivalent protein–DNA interaction but is reminiscent of the topological linkages of cohesin or the MCM complexes to the chromatin fiber (Kalfalah et al. 2015; Rhodes et al. 2017).
The distinct chromosome binding of MSL2vir in the presence and absence of roX2 provided a unique opportunity to assess the effect of the RNA on chromosomal residence time. This showed that the tight interaction of MSL2 was conserved through 40 million years of evolution and, excitingly, is not an intrinsic property of the protein but induced by roX2 expression. Tight binding is only observed when MSL2 stains a coherent X-chromosomal territory after successful chromosome recognition and complex assembly. The nature of this switch is unclear at present, but we speculate that the involvement of roX2 in the initial selection of the X chromosome (tuning protein–DNA interactions) and the final switch of the DCC to a “locked-in” conformation reflect distinct functions that diverged or were conserved, respectively, during evolution of the melanogaster and virilis species.
The tight interaction we present appears unrelated to an “RNA tethering” phenomenon recently described, where MSL2, through IDR–RNA interactions, is concentrated around nascent roX RNA (Valsecchi et al. 2021). The fact that MSL2 binds nascent roX RNA had been suggested previously (Kelley et al. 1999; Meller et al. 2000; Oh et al. 2003). It is plausible that the binding of MSL proteins to nascent roX RNA triggers first steps in DCC assembly and, because both roX genes reside on the X, facilitates the search for X chromosomal HASs. However, X-chromosome targeting and dosage compensation still works if the only roX gene is integrated on an autosome (Meller et al. 1997; Ramírez et al. 2015; Ilik et al. 2017) or in our transient transfection experiments. Therefore, MSL2–RNA interactions per se bear only very limited potential for X/autosome discrimination.
Conclusion
MSL2 is the only male-specific protein component of the dosage compensation system in Drosophila; it is the crucial activator of roX2 transcription and central to X-chromosome recognition and DCC assembly (Rattner and Meller 2004; Lim and Kelley 2012; Villa et al. 2016). DCC assembly and homeostasis are integrated in the N-terminal RING finger domain, which mediates MSL1 interaction and E3 ubiquitin ligase activity (Villa et al. 2012). All known components of the X-chromosome targeting system, the CXC domain, CLAMP, and roX binding surfaces, are integrated in the C terminus. A full appreciation of their mutual interactions will require structural knowledge of the complex bound to DNA, RNA, and CLAMP. The assembly of a functional DCC involved coopting available epigenetic reader and writer components and adapting them for a new function. It is tempting to speculate that this was possible through the action of fast-evolving roX lncRNA that bears the potential for allosteric regulation.
Our study illustrates how components of an ancient dosage compensation system can be refined along distinct, species-specific evolutionary trajectories (Fig. 7). The coevolution of protein domains, DNA sequence, and lncRNA beautifully illustrates the existence of alternative, equivalent solutions to the problem of balancing the genome along with diversifying sex chromosomes.
Materials and methods
Plasmids and antibodies
MSL2 cDNAs from D. virilis, D. busckii, and D. willistoni were synthetized by Genewiz and cloned in a phsp70-GFP vector and in a pFastBac-FLAG for expression of recombinant proteins. The CXC domains of MSL2vir (523–558 amino acids) and MSL2mel (525–561 amino acids) were swapped using Gibson cloning. In the MSL2virCTmel construct, the C terminus of MSL2vir (from amino acid 523) was substituted with the C terminus of MSL2mel (from amino acid 525) using Gibson cloning. In MSL2virPBmel, the region from amino acid 644 of MSL2vir was substituted with the region from amino acid 646 of MSL2mel via Gibson cloning. The recombinant proteins MSL2virCXCmel and MSL2virCTmel present a deletion between amino acids 234–274 and amino acids 130–182, respectively. This deletion is important for the proper expression and purification of the recombinant proteins. The functionality of these deleted constructs was verified in vivo (transfection in S2 cells), where we could not detect any difference in comparison with the full-length constructs. The roX2 virilis gene was synthetized by Gene Universal and cloned in a phsp70-GFP vector. The roX2 melanogaster gene was amplified from the D. melanogaster genome and cloned in a phsp70-GFP vector using Gibson cloning. All constructs were verified by sequencing.
Rat monoclonal anti-MLE 6E11, rabbit anti-MSL1, rabbit anti-MSL2, rabbit anti-MOF, rat monoclonal anti-MSL3 1C9, and guinea pig anti-MSL3 antibodies were previously described in Akhtar and Becker (2000), Izzo et al. (2008), Straub et al. (2008), and Albig et al. (2019). Rabbit anti-CLAMP antibody was a kind gift of Erica Larschan (Brown University). Rabbit anti-MSL1vir was a kind gift of Asifa Akhtar (MPI). Mouse anti-FLAG M2 affinity gel and mouse monoclonal anti-FLAG M2 antibody were from Sigma-Aldrich (F3165). Mouse monoclonal anti-Lamin antibody (T40) was provided by Professor H. Saumweber. Mouse monoclonal anti-GFP antibody was from Roche (11814460001).
Cell lines and culture conditions
The D. melanogaster embryonic Kc167 cell line was obtained from the Drosophila Genomic Resource Center (https://dgrc.bio.indiana.edu/home). The D. melanogaster S2 (subclone L2-4) cell line was a kind gift of P. Heun (Villa et al. 2016). The D. virilis 79f7Dv3 cell line was a kind gift of B.V. Adrianov (Albig et al. 2019). The identity of cell lines was verified by high-throughput sequencing. Cells were subjected to mycoplasma testing. Cells were maintained at 26°C in Schneider's Drosophila medium (Thermo Fisher 21720024) supplemented with 10% FBS (Kc167 and S2) or 5% FBS (79f7Dv3) and 1% penicillin-streptomycin solution (Sigma-Aldrich P-4333). Spodoptera frugiperda 21 (SF21) cells (Gibco) were used for amplification of recombinant baculoviruses and baculovirus-driven expression of recombinant proteins. SF21 cells were cultured at 26°C in Sf-900 II medium (Gibco) supplemented with 10% fetal calf serum and gentamycin.
Cells were transfected with the plasmid of interest plus 1:20 of pCoBlast (Thermo Fisher K5150-01) using Effectene transfection reagent (Qiagen 301425). Seventy-two hours after transfection, cells were diluted two to three times, and Blasticidin was added to the medium at a final concentration of 50 ng/µL for 7 or 10 d.
Nuclear extraction and pull-down
For nuclear extraction, 80 million to 120 million cells were collected by centrifugation at 1500 rpm for 5 min. Cells were washed with 10 mL of PBS, resuspended in 2 mL of ice-cold NBT-10 buffer (15 mM HEPES at pH 7.6, 15 mM NaCl, 60 mM KCl, 0.5 mM EGTA at pH 8, 10% sucrose, 0.15% Triton X-100, 0.5 mM DTT, 1× cOmplete EDTA-free protease inhibitor [Roche 5056489001]), and rotated for 10 min at 4°C. Lysed cells were gently overlaid on 8 mL of ice-cold NB-1.2 buffer (15 mM HEPES at pH 7.5, 15 mM NaCl, 60 mM KCl, 0.5 mM EGTA at pH 8, 1.2 M sucrose, 0.5 mM DTT, 1× cOmplete EDTA-free protease inhibitor) and spun at 4000 rpm for 20 min. Nuclei were resuspended in 500 μL of nuclear solubilization buffer (10 mM HEPES, 12.5 mM MgCl2, 400 mM NaCl, 0,05% NP40) and rotated for 45 min at 4°C. Extracts were cleared by centrifugation at top speed for 30 min and used for Western blot or pull-down experiments.
For pull-down experiments, 0.5–1 µg of recombinant proteins was incubated with 600–800 µg of extracts diluted to the right salt concentration (300 nM for high-salt conditions or 100 nM for low-salt conditions) for 2 h. Thirty microliters of FLAG beads were used to recover the pulled-down material and analyze it. For the RNase and benzonase treatment, we used 50 µg and 1 µL (Millipore 103773), respectively.
Immunofluorescence
For immunofluorescence, 0.2 million to 0.4 million cells in 200 µL of complete Schneider's Drosophila medium were seeded onto round 12-mm coverslips (Paul Marienfeld GmbH and Co. 0117520) placed separately inside wells of 12-well plates. Cells were allowed to attach for 30 min and the coverslips were gently rinsed with 500 µL of PBS. Cells were fixed in 500 µL of ice-cold PBS + 2% formaldehyde for 7.5 min. After removal of fixative, cells were permeabilized by adding 500 µL of ice-cold PBS + 0.25% Triton X-100 + 1% formaldehyde and incubated for 7.5 min. Coverslips were washed two times with 1 mL of PBS and blocked with PBS + 3% BSA for 1 h at room temperature. Coverslips were transferred onto a piece of parafilm, placed into a wet chamber, and 40 µL of primary antibody solution were gently added onto the coverslip. After overnight incubation at 4°C, coverslips were transferred back to 12-well plates and washed twice with 1 mL of PBS. Coverslips were transferred again onto a piece of parafilm, placed into a wet chamber, and 40 µL of secondary antibody were gently added onto the coverslip. After 1 h incubation at room temperature, coverslips were transferred back to 12-well plates and washed twice with 1 mL of PBS. Cells were incubated with 1 mL of 0.2 µg/mL DAPI (Sigma-Aldrich 10236276001) for 2 min at room temperature. Coverslips were washed with PBS and with deionized water, mounted on slides with 9 µL of VectaShield (Vector Laboratories H-1000), and sealed with nail polish. Images were acquired on a Zeiss Axiovert 200M equipped with a 63× objective and processed and analyzed using Fiji. Confocal microscopy was performed at the Bioimaging Core Facility of the Biomedical Center with either upright or inverted Leica SP8X WLL microscopes, upgraded with Klondike linear scan electronics, equipped with 405-nm laser, WLL2 laser (470–670 nm), Argon laser (inverted microscope), and acusto-optical beam splitter (see the Supplemental Material).
Protein expression and purification
The MSL2 recombinant proteins were purified according to Fauth et al. (2010). See also the Supplemental Material.
NMR data acquisition, backbone assignment, and titrations
All spectra were acquired using Avance III Bruker NMR spectrometers with 700 or 800 MHz proton Larmor frequencies and equipped with cryogenic (800 MHz) or room temperature (700 MHz) triple-resonance gradient probe heads. All experiments were performed at 298 K. Spectra for backbone experiments were acquired with 0.33 mM protein in 50 mM potassium phosphate buffer, pH 6.0 supplemented with 10% D2O for the deuterium lock. For backbone resonance assignments, HNCA, CBCA(CO)NH, and HNCACB experiments were recorded (Sattler et al. 1995).
1H,15N HSQC NMR titrations were performed with 0.2 mM 15N-labeled protein that was titrated with increasing amounts of HPLC purified double-stranded DNA (S12: ATGAGCGAGATG, or S12 mutant: ATGAAAAAAATG) purchased from Integrated DNA Technologies. The titrations were performed with molar ratios ranging from 0.025, 0.05, 0.1, 0.15, 0.2, 0.3, 0.4, 0.5 and 0.6 equivalents of DNA to protein. All spectra were processed using NMRPipe and analyzed using CARA and CCPNmr (Delaglio et al. 1995; Keller 2004; Morin et al. 2013; Skinner et al. 2016). Chemical shift perturbations were calculated using the equation
where ΔH and ΔN are the difference of the protein chemical shifts between the DNA bound and free state in the proton and nitrogen dimension, respectively. Fitting of chemical shift perturbations for calculating dissociation constants was performed in GraphPad Prism (version 6) using a multisite global fit model described previously (Fielding 2003). When the number of binding sites was allowed to float during fitting of the equation, a value between 3 and 4 was obtained for both DNAs. To be consistent with the previously published data, the number of binding sites was fixed at three sites (Zheng et al. 2014).
Mass spectrometry
For mass spectrometry experiments, after pull-down, beads were washed three times with 50 mM NH4HCO3 and incubated with 10 ng/µL trypsin in 1 M urea/50 mM NH4HCO3 for 30 min, and washed with 50 mM NH4HCO3. The supernatant was digested overnight in the presence of 1 mM DTT. Digested peptides were alkylated and desalted prior to LC-MS analysis (see the Supplemental Material).
Genomic DNA preparation and DIP
The pellet from 6 × 107 S2 or virilis cells was suspended in 1.2 mL of lysis buffer (10 mM Tris at pH 8, 100 mM NaCl, 25 mM EDTA at pH 8, 0.5% SDS, 0.15 mg/mL of proteinase K) and incubated overnight at 56°C. After addition of sodium acetate to a final concentration of 0.3 M, nucleic acids were extracted with phenol–chloroform and precipitated with an equal volume of isopropanol for 1 h at −20°C. Precipitated nucleic acids were centrifuged and washed with 70% ethanol. Dried pellets were resuspended in TE buffer and sonicated with Covaris AFA S220 (microTUBEs, peak incident power 175 W, duty factor 10%, cycles per burst 200, 430 sec) to generate 200-bp fragments. After RNase digestion (0.1 mg/mL, 1 h at 37°C), DNA was purified with the GenElute kit (Sigma-Aldrich).
DNA immunoprecipitation (DIP) experiments were performed as in Villa et al. (2016). See also the Supplemental Material.
ChIP-seq
Fifty million to 100 million D. melanogaster Kc167 cells (untransfected or transfected with the indicated constructs), resuspended in 20 mL of complete Schneider's Drosophila medium, were crosslinked by adding 1:10 of the volume of fixing solution (100 mM NaCl, 50 mM HEPES at pH 8, 1 mM EDTA, 0.5 mM EGTA, 10% methanol-free formaldehyde) and rotated for 8 min at room temperature. Freshly prepared 2.5 M glycine (1.17 mL) was added to stop the fixation (final concentration 125 mM). Cells were pelleted at 500g for 10 min at 4°C and resuspended in 10 mL of ice-cold PBS. We added 3.5 million fixed virilis cells (fixed as described for D. melanogaster cells) for every 70 million melanogaster cells. Cells were pelleted at 526g for 10 min at 4°C and resuspended in 1 mL of PBS + 0.5% Triton X-100 + 1× cOmplete EDTA-free protease inhibitor for every 70 million melanogaster cells and rotated for 15 min at 4°C to release nuclei. Nuclei were pelleted at 2000g for 10 min and washed once with 10 mL of ice-cold PBS. Nuclei were suspended in 1 mL of RIPA buffer (10 mM Tris-HCl at pH 8, 140 mM NaCl, 1 mM EDTA, 0.1% Na-deoxycholate, 1% Triton X-100, 0.1% SDS, 1 mM PMSF, 1× cOmplete EDTA-free protease inhibitor) + 2 mM CaCl2 for every 70 million melanogaster cells, divided into 1-mL aliquots, and flash-frozen in liquid N2. One milliliter of fixed nuclei was quickly thawed and 1 µL of MNase (to 0.6 U; Sigma-Aldrich N5386) added. Chromatin was digested for 35 min at 37°C. MNase digestion was stopped by transferring the samples on ice and adding 22 µL of 0.5 M EGTA. Samples were mildly sonicated using a Covaris S220 instrument with the following settings: 50-W peak power, 20% duty factor, 200 cycles/burst, 8 min total time. Insoluble chromatin was removed by centrifugation at 16,000g for 30 min at 4°C. ChIPs were performed as described in the Supplemental Material.
Library preparation and sequencing
Libraries were prepared using the UltraII NEBNext Ultra II DNA library preparation kit or the MicroPlex library preparation kit v12 from Diagenode. Libraries were sequenced on an Illumina HiSeq 1500 instrument at the Laboratory of Functional Genomic Analysis (LAFUGA, Gene Center Munich, Ludwig-Maximilians-University).
Processing of sequencing reads
Sequencing reads were aligned to the dm6 version of the D. melanogaster genome or the caf1 version of virilis using bowtie2 (version 2.3.5) allowing only unique matches. In the case of paired-end sequencing (ChIP of GFP-tagged MSL2 constructs), only fragments <135 bp were kept for further analyses. Peak calling against the corresponding input controls was performed with Homer (version 4.10). For quantitative comparisons between conditions, sequence read libraries were subsampled to the size of the smallest library.
Sequence motif searches
Genomes of Drosophila species (dm6 for melanogaster, caf1 for virilis, willistoni, sechellia, yakuba, ananassae, erecta, persimilis, mojavensis, and grimshawi; Dpse_3.0 for pseudoobscura; ASM75419v3 for simulans; and GSE69208 for busckii) were searched for hits of the PionX PWM using fimo (version 5.1.1)
FRAP
For the FRAP experiment, Kc cells were transfected with the MSL2vir-GFP construct with or without roX2 as explained in “transfection.” After one week of selection, cells were seeded for 30 min on cell culture dishes (CELLview PS, 35/10 mm, glass bottom) pretreated with concavalin A (0.5 mg/mL in water) and treated with nocodazole (25µg/mL) to try to prevent nuclear movements during the FRAP experiment.
FRAP was performed at the Bioimaging Core Facility of the Biomedical Center with an inverted Leica SP8X WLL microscope, upgraded with Klondike linear scan electronics, equipped with a 405-nm laser, WLL2 laser (470–670 nm), Argon laser, and acusto-optical beam splitter. Live cells were recorded at 25°C (see the Supplemental Material).
Data availability
NMR chemical shift assignments of the MSL2 CXC domain from D. virilis were deposited to the BMRB under accession number 50724. The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD023747. Sequencing data were deposited to GEO with accession number GSE165833.
Supplementary Material
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft (Be1140/8-1). P.K.A.J. acknowledges EMBL and the EU Marie Curie Actions Cofund for the EIPOD fellowship. J.H. gratefully acknowledges EMBL for funding. We thank C. Margulies for sharing her antibody against GFP, S. Krause and A. Zabel for technical assistance, S. Baldi and M. Müller for critical reading the manuscript, all members of the Becker laboratory for discussion, and S. Krebs and H. Blum for their sequencing service.
Author contributions: R.V. conceived the study and performed experiments. P.K.A.J. performed all the NMR experiments and the related analyses. A.W.T. performed the FRAP experiments and acquired and processed microscope images. A.C.S. performed experiments. I.F. processed and analyzed the mass spectrometry samples. J.H. secured funding and supervised the NMR experiments. T.S. performed all bioinformatic analyses and analyzed the FRAP experiments. P.B.B. secured funding, supervised the study, and provided intellectual support for the interpretation of the results. R.V. and P.B.B. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348411.121.
Competing interest statement
The authors declare no competing interests.
References
- Akhtar A, Becker PB. 2000. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol Cell 5: 367–375. 10.1016/S1097-2765(00)80431-1 [DOI] [PubMed] [Google Scholar]
- Albig C, Tikhonova E, Krause S, Maksimenko O, Regnard C, Becker PB. 2019. Factor cooperation for chromosome discrimination in Drosophila. Nucleic Acids Res 47: 1706–1724. 10.1093/nar/gky1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, et al. 2008. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 134: 599–609. 10.1016/j.cell.2008.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashaw GJ, Baker BS. 1995. The msl-2 dosage compensation gene of Drosophila encodes a putative DNA-binding protein whose expression is sex specifically regulated by Sex-lethal. Development 121: 3245–3258. 10.1242/dev.121.10.3245 [DOI] [PubMed] [Google Scholar]
- Bone JR, Kuroda MI. 1996. Dosage compensation regulatory proteins and the evolution of sex chromosomes in Drosophila. Genetics 144: 705–713. 10.1093/genetics/144.2.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky S, Jana T, Mittelman K, Chapal M, Kumar DK, Carmi M, Barkai N. 2020. Intrinsically disordered regions direct transcription factor in vivo binding specificity. Mol Cell 79: 459–471.e4. 10.1016/j.molcel.2020.05.032 [DOI] [PubMed] [Google Scholar]
- Cerase A, Armaos A, Neumayer C, Avner P, Guttman M, Tartaglia GG. 2019. Phase separation drives X-chromosome inactivation: a hypothesis. Nat Struct Mol Biol 26: 331–334. 10.1038/s41594-019-0223-0 [DOI] [PubMed] [Google Scholar]
- Chlamydas S, Holz H, Samata M, Chelmicki T, Georgiev P, Pelechano V, Dündar F, Dasmeh P, Mittler G, Cadete FT, et al. 2016. Functional interplay between MSL1 and CDK7 controls RNA polymerase II Ser5 phosphorylation. Nat Struct Mol Biol 23: 580–589. 10.1038/nsmb.3233 [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293. 10.1007/BF00197809 [DOI] [PubMed] [Google Scholar]
- Ellison CE, Bachtrog D. 2013. Dosage compensation via transposable element mediated rewiring of a regulatory network. Science 342: 846–850. 10.1126/science.1239552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison C, Bachtrog D. 2019. Contingency in the convergent evolution of a regulatory network: dosage compensation in Drosophila. PLoS Biol 17: e3000094. 10.1371/journal.pbio.3000094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauth T, Müller-Planitz F, König C, Straub T, Becker PB. 2010. The DNA binding CXC domain of MSL2 is required for faithful targeting the Dosage Compensation Complex to the X chromosome. Nucleic Acids Res 38: 3209–3221. 10.1093/nar/gkq026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding L. 2003. NMR methods for the determination of protein-ligand dissociation constants. Curr Top Med Chem 3: 39–53. 10.2174/1568026033392705 [DOI] [PubMed] [Google Scholar]
- Figueiredo ML, Kim M, Philip P, Allgardsson A, Stenberg P, Larsson J. 2014. Non-coding roX RNAs prevent the binding of the MSL-complex to heterochromatic regions. PLoS Genet 10: e1004865. 10.1371/journal.pgen.1004865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilik IA, Quinn JJ, Georgiev P, Tavares-Cadete F, Maticzka D, Toscano S, Wan Y, Spitale RC, Luscombe N, Backofen R, et al. 2013. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Mol Cell 51: 156–173. 10.1016/j.molcel.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilik IA, Maticzka D, Georgiev P, Gutierrez NM, Backofen R, Akhtar A. 2017. A mutually exclusive stem-loop arrangement in roX2 RNA is essential for X-chromosome regulation in Drosophila. Genes Dev 31: 1973–1987. 10.1101/gad.304600.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo A, Regnard C, Morales V, Kremmer E, Becker PB. 2008. Structure–function analysis of the RNA helicase maleless. Nucleic Acids Res 36: 950–962. 10.1093/nar/gkm1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalfalah FM, Berg E, Christensen MO, Linka RM, Dirks WG, Boege F, Mielke C. 2015. Spatio-temporal regulation of the human licensing factor Cdc6 in replication and mitosis. Cell Cycle 14: 1704–1715. 10.1080/15384101.2014.1000182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R. 2004. The computer aided resonance assignment tutorial. Cantina Verlag, Goldau, Switzerland. [Google Scholar]
- Kelley RL, Solovyeva I, Lyman LM, Richman R, Solovyev V, Kuroda MI. 1995. Expression of msl-2 causes assembly of dosage compensation regulators on the X chromosomes and female lethality in Drosophila. Cell 81: 867–877. 10.1016/0092-8674(95)90007-1 [DOI] [PubMed] [Google Scholar]
- Kelley RL, Meller VH, Gordadze PR, Roman G, Davis RL, Kuroda MI. 1999. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell 98: 513–522. 10.1016/S0092-8674(00)81979-0 [DOI] [PubMed] [Google Scholar]
- Kuzu G, Kaye EG, Chery J, Siggers T, Yang L, Dobson JR, Boor S, Bliss J, Liu W, Jogl G, et al. 2016. Expansion of GA dinucleotide repeats increases the density of CLAMP binding sites on the X-chromosome to promote Drosophila dosage compensation. PLoS Genet 12: e1006120. 10.1371/journal.pgen.1006120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT. 2018. The human transcription factors. Cell 175: 598–599. 10.1016/j.cell.2018.09.045 [DOI] [PubMed] [Google Scholar]
- Larschan E, Soruco MM, Lee OK, Peng S, Bishop E, Chery J, Goebel K, Feng J, Park PJ, Kuroda MI. 2012. Identification of chromatin-associated regulators of MSL complex targeting in Drosophila dosage compensation. PLoS Genet 8: e1002830. 10.1371/journal.pgen.1002830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Schiemann AH, Scott MJ. 2008. Incorporation of the noncoding roX RNAs alters the chromatin-binding specificity of the Drosophila MSL1/MSL2 complex. Mol Cell Biol 28: 1252–1264. 10.1128/MCB.00910-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CK, Kelley RL. 2012. Autoregulation of the Drosophila noncoding roX1 RNA gene. PLoS Genet 8: e1002564. 10.1371/journal.pgen.1002564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi JC, Kuroda MI. 2015. Dosage compensation in Drosophila. Cold Spring Harb Perspect Biol 7: a019398. 10.1101/cshperspect.a019398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maenner S, Müller M, Fröhlich J, Langer D, Becker PB. 2013. ATP-dependent roX RNA remodeling by the helicase maleless enables specific association of MSL proteins. Mol Cell 51: 174–184. 10.1016/j.molcel.2013.06.011 [DOI] [PubMed] [Google Scholar]
- Marín I, Baker BS. 1998. The evolutionary dynamics of sex determination. Science 281: 1990–1994. 10.1126/science.281.5385.1990 [DOI] [PubMed] [Google Scholar]
- Marín I, Franke A, Bashaw GJ, Baker BS. 1996. The dosage compensation system of Drosophila is co-opted by newly evolved X chromosomes. Nature 383: 160–163. 10.1038/383160a0 [DOI] [PubMed] [Google Scholar]
- Meller VH, Wu KH, Roman G, Kuroda MI, Davis RL. 1997. roX1 RNA paints the X chromosome of male Drosophila and is regulated by the dosage compensation system. Cell 88: 445–457. 10.1016/S0092-8674(00)81885-1 [DOI] [PubMed] [Google Scholar]
- Meller VH, Gordadze PR, Park Y, Chu X, Stuckenholz C, Kelley RL, Kuroda MI. 2000. Ordered assembly of roX RNAs into MSL complexes on the dosage-compensated X chromosome in Drosophila. Curr Biol 10: 136–143. 10.1016/S0960-9822(00)00311-0 [DOI] [PubMed] [Google Scholar]
- Morgunova E, Taipale J. 2017. Structural perspective of cooperative transcription factor binding. Curr Opin Struct Biol 47: 1–8. 10.1016/j.sbi.2017.03.006 [DOI] [PubMed] [Google Scholar]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P. 2013. Collaboration gets the most out of software. Elife 2: e01456. 10.7554/eLife.01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Schauer T, Krause S, Villa R, Thomae AW, Becker PB. 2020. Two-step mechanism for selective incorporation of lncRNA into a chromatin modifier. Nucleic Acids Res 48: 7483–7501. 10.1093/nar/gkaa492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Park Y, Kuroda MI. 2003. Local spreading of MSL complexes from roX genes on the Drosophila X chromosome. Genes Dev 17: 1334–1339. 10.1101/gad.1082003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang KC, Frith MC, Mattick JS. 2006. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet 22: 1–5. 10.1016/j.tig.2005.10.003 [DOI] [PubMed] [Google Scholar]
- Quinn JJ, Zhang QC, Georgiev P, Ilik IA, Akhtar A, Chang HY. 2016. Rapid evolutionary turnover underlies conserved lncRNA-genome interactions. Genes Dev 30: 191–207. 10.1101/gad.272187.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Lingg T, Toscano S, Lam KC, Georgiev P, Chung HR, Lajoie BR, de Wit E, Zhan Y, de Laat W, et al. 2015. High-affinity sites form an interaction network to facilitate spreading of the MSL complex across the X chromosome in Drosophila. Mol Cell 60: 146–162. 10.1016/j.molcel.2015.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner BP, Meller VH. 2004. Drosophila male-specific lethal 2 protein controls sex-specific expression of the roX genes. Genetics 166: 1825–1832. 10.1093/genetics/166.4.1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JDP, Haarhuis JHI, Grimm JB, Rowland BD, Lavis LD, Nasmyth KA. 2017. Cohesin can remain associated with chromosomes during DNA replication. Cell Rep 20: 2749–2755. 10.1016/j.celrep.2017.08.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samata M, Akhtar A. 2018. Dosage compensation of the X chromosome: a complex epigenetic assignment involving chromatin regulators and long noncoding RNAs. Annu Rev Biochem 87: 323–350. 10.1146/annurev-biochem-062917-011816 [DOI] [PubMed] [Google Scholar]
- Sattler M, Schwendinger MG, Schleucher J, Griesinger C. 1995. Novel strategies for sensitivity enhancement in heteronuclear multi-dimensional NMR experiments employing pulsed field gradients. J Biomol NMR 6: 11–22. 10.1007/BF00417487 [DOI] [PubMed] [Google Scholar]
- Scott MJ, Pan LL, Cleland SB, Knox AL, Heinrich J. 2000. MSL1 plays a central role in assembly of the MSL complex, essential for dosage compensation in Drosophila. Embo J 19: 144–155. 10.1093/emboj/19.1.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner SP, Fogh RH, Boucher W, Ragan TJ, Mureddu LG, Vuister GW. 2016. CcpNmr AnalysisAssign: a flexible platform for integrated NMR analysis. J Biomol NMR 66: 111–124. 10.1007/s10858-016-0060-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, Allis CD, Lucchesi JC. 2000. The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol Cell Biol 20: 312–318. 10.1128/MCB.20.1.312-318.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soruco MM, Chery J, Bishop EP, Siggers T, Tolstorukov MY, Leydon AR, Sugden AU, Goebel K, Feng J, Xia P, et al. 2013. The CLAMP protein links the MSL complex to the X chromosome during Drosophila dosage compensation. Genes Dev 27: 1551–1556. 10.1101/gad.214585.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Neumann MF, Prestel M, Kremmer E, Kaether C, Haass C, Becker PB. 2005. Stable chromosomal association of MSL2 defines a dosage-compensated nuclear compartment. Chromosoma 114: 352–364. 10.1007/s00412-005-0020-x [DOI] [PubMed] [Google Scholar]
- Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB. 2008. The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet 4: e1000302. 10.1371/journal.pgen.1000302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Zabel A, Gilfillan GD, Feller C, Becker PB. 2013. Different chromatin interfaces of the Drosophila dosage compensation complex revealed by high-shear ChIP-seq. Genome Res 23: 473–485. 10.1101/gr.146407.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sural TH, Peng S, Li B, Workman JL, Park PJ, Kuroda MI. 2008. The MSL3 chromodomain directs a key targeting step for dosage compensation of the Drosophila melanogaster X chromosome. Nat Struct Mol Biol 15: 1318–1325. 10.1038/nsmb.1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova E, Fedotova A, Bonchuk A, Mogila V, Larschan EN, Georgiev P, Maksimenko O. 2019. The simultaneous interaction of MSL2 with CLAMP and DNA provides redundancy in the initiation of dosage compensation in Drosophila males. Development 146: dev179663. 10.1242/dev.179663 [DOI] [PubMed] [Google Scholar]
- Valsecchi CIK, Basilicata MF, Georgiev P, Gaub A, Seyfferth J, Kulkarni T, Panhale A, Semplicio G, Manjunath V, Holz H, et al. 2021. RNA nucleation by MSL2 induces selective X chromosome compartmentalization. Nature 589: 137–142. 10.1038/s41586-020-2935-z [DOI] [PubMed] [Google Scholar]
- Villa R, Forné I, Müller M, Imhof A, Straub T, Becker PB. 2012. MSL2 combines sensor and effector functions in homeostatic control of the Drosophila dosage compensation machinery. Mol Cell 48: 647–654. 10.1016/j.molcel.2012.09.012 [DOI] [PubMed] [Google Scholar]
- Villa R, Schauer T, Smialowski P, Straub T, Becker PB. 2016. PionX sites mark the X chromosome for dosage compensation. Nature 537: 244–248. 10.1038/nature19338 [DOI] [PubMed] [Google Scholar]
- Yang S, Yalamanchili HK, Li X, Yao KM, Sham PC, Zhang MQ, Wang J. 2011. Correlated evolution of transcription factors and their binding sites. Bioinformatics 27: 2972–2978. 10.1093/bioinformatics/btr503 [DOI] [PubMed] [Google Scholar]
- Zheng S, Villa R, Wang J, Feng Y, Wang J, Becker PB, Ye K. 2014. Structural basis of X chromosome DNA recognition by the MSL2 CXC domain during Drosophila dosage compensation. Genes Dev 28: 2652–2662. 10.1101/gad.250936.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Yang Y, Scott MJ, Pannuti A, Fehr KC, Eisen A, Koonin EV, Fouts DL, Wrightsman R, Manning JE. 1995. Male-specific lethal 2, a dosage compensation gene of Drosophila, undergoes sex-specific regulation and encodes a protein with a RING finger and a metallothionein-like cysteine cluster. Embo J 14: 2884–2895. 10.1002/j.1460-2075.1995.tb07288.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Żylicz JJ, Heard E. 2020. Molecular mechanisms of facultative heterochromatin formation: an X-chromosome perspective. Annu Rev Biochem 89: 255–282. 10.1146/annurev-biochem-062917-012655 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NMR chemical shift assignments of the MSL2 CXC domain from D. virilis were deposited to the BMRB under accession number 50724. The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD023747. Sequencing data were deposited to GEO with accession number GSE165833.