

Abstract
Objective
To identify key determinants of the interactive pathophysiologic processes in subchondral bone and cartilage in osteoarthritis (OA).
Methods
We performed RNA sequencing on macroscopically preserved and lesional OA subchondral bone from patients in the Research Arthritis and Articular Cartilage study who underwent joint replacement surgery due to OA (n = 24 sample pairs: 6 hips and 18 knees). Unsupervised hierarchical clustering and differential expression analyses were conducted. Results were combined with data on previously identified differentially expressed genes in cartilage (partly overlapping samples) as well as data on recently identified OA risk genes.
Results
We identified 1,569 genes that were significantly differentially expressed between lesional and preserved subchondral bone, including CNTNAP2 (fold change [FC] 2.4, false discovery rate [FDR] 3.36 × 10−5) and STMN2 (FC 9.6, FDR 2.36 × 10−3). Among these 1,569 genes, 305 were also differentially expressed, and with the same direction of effect, in cartilage, including the recently recognized OA susceptibility genes IL11 and CHADL. Upon differential expression analysis with stratification for joint site, we identified 509 genes that were exclusively differentially expressed in subchondral bone of the knee, including KLF11 and WNT4. These genes that were differentially expressed exclusively in the knee were enriched for involvement in epigenetic processes, characterized by, e.g., HIST1H3J and HIST1H3H.
Conclusion
IL11 and CHADL were among the most consistently differentially expressed genes OA pathophysiology–related genes in both bone and cartilage. As these genes were recently also identified as robust OA risk genes, they classify as attractive therapeutic targets acting on 2 OA‐relevant tissues.
INTRODUCTION
Osteoarthritis (OA) represents multiple subtypes of degenerative joint diseases, characterized by progressive and irreversible degeneration of articular cartilage and structural changes in subchondral bone. Globally, OA is a highly prevalent and disabling disease that results in high social and economic burdens to society (1). Yet, there is no proven therapy to prevent OA or slow its progression. Development of OA is dependent on multiple factors, with both environmental and genetic components (2,3). To discover genes and underlying disease pathways, genetic investigations, such as large genome‐wide association studies, have been performed, identifying compelling OA risk single‐nucleotide polymorphisms (SNPs) (4, 5, 6). Functional follow‐up studies involve exploring the expression patterns in disease‐relevant tissues, behavior with pathophysiology, and/or expression quantitative trait locus (eQTL) or cis‐eQTL analysis. To date, major efforts have been made to characterize pathophysiologic processes of OA in articular cartilage. However, only a few studies have focused on OA pathophysiologic processes in the underlying bone (7,8).
In recent decades, there has been accumulating evidence that subchondral bone contributes to both onset and progression of OA (9, 10, 11, 12). In healthy bone there is a balanced process between bone resorption and bone deposition, as a consequence of dynamic adaptation to mechanical load. In OA this balance is disturbed, which results in changes in the architecture of the subchondral trabecular bone, increased thickness of the subchondral bone plate, formation of new bony structures, called osteophytes, at the joint margins, and development of subchondral bone cysts (2,13,14). In addition, studies have shown an association between bone mineral density and OA development, which suggests that subchondral bone is involved in the early stages of OA (13,15). This was also suggested in studies of subchondral bone marrow lesions, showing these to be very early markers of OA (8,16).
In contrast to cartilage and despite its relevance, only a limited number of studies have focused on the characterization of OA disease processes at the gene expression level in subchondral bone. Chou et al (7) performed whole‐genome expression profiling of non‐OA and OA subchondral bone using microarray analysis, which led to identification of genes involved in pathways such as lipid metabolism and mineral metabolism. Kuttapitiya et al (8) used microarray analysis to identify genes involved in bone remodeling, pain sensitization, and matrix turnover that were differentially expressed between OA bone marrow lesional tissue and control tissue. However, both of these studies included samples from the knee only.
In the present study, we explored RNA sequencing data on preserved and lesional OA subchondral bone to identify genes that change with progression of OA. The samples used were obtained from the joints of patients in the Research Arthritis and Articular Cartilage (RAAK) study who underwent total joint replacement surgery due to OA. In total, we compared paired subchondral bone samples (preserved and lesional) from 24 OA patients from whom preserved and lesional cartilage was also collected. The results presented here contribute to further understanding of the ongoing OA process in the subchondral bone and provide insight into the pathophysiology of the disease in bone relative to cartilage.
MATERIALS AND METHODS
Sample description
The current investigation included 26 patients from the RAAK study who underwent joint replacement surgery due to OA. Macroscopically preserved and lesional OA subchondral bone was collected from the joints of these patients. Of note, classification of OA subchondral bone as preserved or lesional was based on classification of its overlying cartilage as preserved or lesional, as described previously (17). The results reported here were compared to the results of our earlier study of macroscopically preserved and lesional OA articular cartilage from 35 patients from the RAAK study (18). Fourteen of these 35 patients were included in the present study, as samples of both preserved and lesional subchondral bone and preserved and lesional articular cartilage were available. The sample size for the current study was determined using the R package ssize.fdr, version 1.2 (19), with parameters based on our previous similar analysis of articular cartilage (18) and a desired power of 0.8 (see Supplementary Figure 1, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract). Since the parameters were based on cartilage, whereas bone is known to be more heterogeneous, we decided to include an excess of samples. The samples were either randomly selected or selected based on their overlap with the cartilage data. Informed consent was obtained from all participants in the RAAK study, and ethical approval for the RAAK study was granted by the medical ethics committee of Leiden University Medical Center (P08.239/P19.013).
RNA sequencing
RNA was isolated from subchondral bone using an RNeasy Mini kit (Qiagen). Paired‐end 2 × 100–bp RNA sequencing (Illumina TruSeq RNA Library Prep Kit, Illumina HiSeq2000, and Illumina HiSeq4000) was performed. Strand‐specific RNA sequencing libraries were generated, which yielded a mean of 20 million reads per sample. Data from both Illumina platforms were integrated and analyzed with the same in‐house pipeline. RNA sequencing reads were aligned using GSNAP (20) against GRCh38, with default parameters. Read abundance per sample was estimated using HTSeq count, version 0.11.1 (21). Only uniquely mapping reads were used for estimating expression. The quality of the raw reads for RNA sequencing was checked using MultiQC, version 1.7 (22). The adaptors were clipped using Cutadapt version 1.1 (23), applying default settings (minimum overlap length of 3). To identify outliers, principal components analysis and hierarchical clustering of the samples were applied, and 1 extreme outlier was identified. A sensitivity analysis was performed, which showed that the outlier had a large effect on the results in the overall data set. Based on this, the outlier was removed from the data set. There was 1 sample without paired data, which was also removed from the data set. After removal of these samples, only 24 participants were included for further analysis. The RNA sequencing data are deposited at the European Genome‐Phenome Archive (www.ega‐archive.org; accession no. EGAS00001004476).
Cluster analysis
Prior to the cluster analysis, variance stabilizing transformation was performed on the data, and 1,000 genes were selected based on the highest coefficient of variation (24,25). To identify the optimal number of clusters in the unsupervised hierarchical clustering the silhouette width score approach was used, with a higher average silhouette width score indicating a more optimal number of clusters (26). Details on the cluster analyses and the stability of cluster solutions have been reported previously (25).
Differential expression analysis and pathway enrichment
Differential expression analysis was performed on paired lesional and preserved subchondral bone samples, using the DESeq2 R package, version 1.24.0 (27). A general linear model assuming a negative binomial distribution was applied, followed by a paired Wald test between lesional and preserved OA samples, with the preserved samples set as a reference. The Benjamini‐Hochberg method was used to correct for multiple testing, as indicated by the false discovery rate (FDR), with a significance cutoff value of 0.05. Gene enrichment was performed using the online functional annotation tool DAVID, selecting for the gene ontology (GO) terms Biological Processes (GOTERM_BP_DIRECT), Cellular Component (GOTERM_CC_DIRECT), and Molecular Function (GOTERM_MF_DIRECT) and for the Reactome Homo Sapiens (R‐HSA) and the KEGG pathways (28). Moreover, the protein–protein interactions were analyzed using the online tool STRING, version 11.0 (29). An analysis summary scheme is shown in Figure 1.
Figure 1.
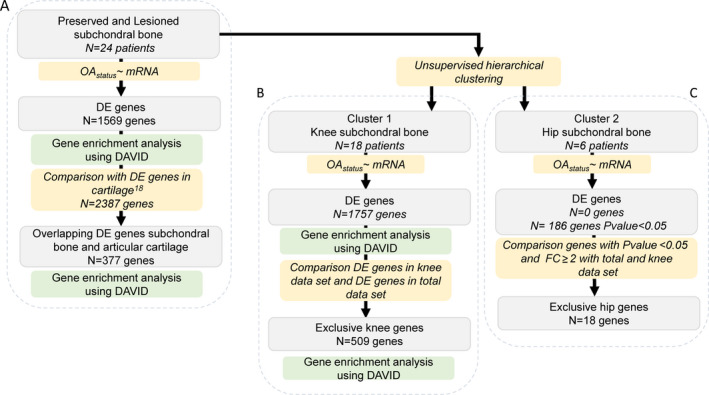
Overview of the study strategy. A, Determination of genes that were consistently differentially expressed (DE) between preserved and lesional osteoarthritis (OA) subchondral bone in the overall data set. B, Determination of genes that were differentially expressed in knee subchondral bone. C, Determination of genes that were differentially expressed in hip subchondral bone. Number of genes represents the significantly differentially expressed genes (according to the false discovery rate method), excluding those that were differentially expressed only in hip samples. FC = fold change.
Quantitative reverse transcriptase–polymerase chain reaction (qRT‐PCR) validation
Complementary DNA synthesis was performed with a Transcriptor First Strand cDNA Synthesis Kit (Roche), using 400 ng of RNA. We used qRT‐PCR to quantitatively determine gene expression of FRZB, CNTNAP2, STMN2, CHRDL2, POSTN, and ASPN. Relative gene expression was evaluated using −ΔCt values, with GAPDH and SDHA as internal controls. Generalized estimating equation analysis was performed to calculate the significance of differences between the lesional and preserved samples.
Comparison of subchondral bone and articular cartilage
The 1,569 genes that were significantly differentially expressed (by FDR) between preserved and lesional OA subchondral bone (24 paired samples) reported here were compared to the 2,387 genes that were significantly differentially expressed between preserved and lesional OA articular cartilage (35 paired samples) as determined in our earlier study (18). Genes that were significantly differentially expressed in both tissues were selected, and the directions of effect were explored.
RESULTS
Sample characteristics
To characterize the pathophysiologic process in subchondral bone with ongoing OA, we performed RNA sequencing on macroscopically preserved and lesional OA subchondral bone samples from patients in the RAAK study who underwent joint replacement surgery due to OA. The RNA sequencing was performed on 24 paired samples (6 from hips and 18 from knees (Supplementary Table 1, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract).
Prior to the differential expression analysis, we tested for possible contamination by cartilage tissue in the subchondral bone samples. We used RNA sequencing data on both tissue types from the same joint and evaluated the relative difference in expression levels of 3 cartilage‐specific genes (COL2A1, COMP, and CRTAC1) and 3 bone‐specific genes (COL1A1, SPP1, and BGLAP), as described previously (30). As shown in Supplementary Table 2 (http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract), we observed relatively low levels of cartilage‐specific genes and high levels of bone‐specific genes in the subchondral bone data set under study, suggesting no‐to‐minimal cross‐contamination. Next, we explored whether the expression pattern in subchondral bone was associated with any baseline characteristics of the patients (Supplementary Table 1), by performing unsupervised hierarchical clustering. To include the most informative genes in the cluster analysis, 1,000 genes were selected based on the highest coefficient of variation in the total data set (preserved and lesional; n = 24 pairs). As shown in Figure 2 and Supplementary Figure 2 (http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract), we identified 2 clusters. These appeared to be based on joint site, indicating an inherent difference between hip and knee subchondral bone.
Figure 2.
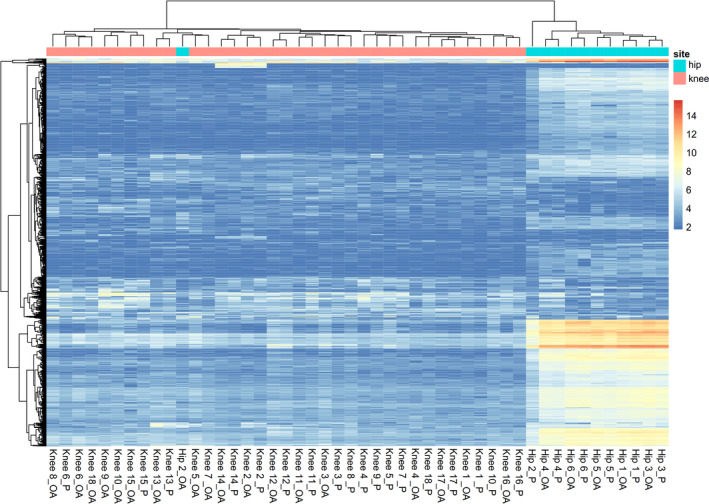
Cluster analysis using the 1,000 genes selected for their highest coefficient of variation. Two clusters based on joint site (knee or hip) were identified. OA = osteoarthritic; P = preserved.
Differential expression analysis and pathway enrichment
We first determined the genes that were consistently differentially expressed between preserved and lesional OA subchondral bone in the overall data set, to explore the most consistent OA pathways (Figure 1A). Upon differential expression analysis in the 24 sample pairs, we identified 1,569 genes that were genome‐wide significantly differentially expressed between lesional and preserved OA subchondral bone tissue. Of these differentially expressed genes, 750 were up‐regulated and 819 were down‐regulated (Figure 3 and Supplementary Table 3, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract). The most significantly down‐regulated gene was FRZB (fold change [FC] 0.53, FDR 3.99 × 10−7), encoding Frizzled‐related protein, which is a well‐known OA gene showing consistently lower expression in lesional relative to preserved OA articular cartilage (17,18). The most significantly up‐regulated gene was CNTNAP2 (FC 2.42, FDR 3.36 × 10−5), encoding the contactin‐associated protein‐like 2 protein (CASPR2). Among the 1,569 differentially expressed genes, 53 had an absolute FC of ≥2 (35 up‐regulated and 18 down‐regulated). The most highly up‐regulated gene was STMN2 (FC 9.56, FDR 2.36 × 10−3), encoding stathmin 2, while the most down‐regulated gene was CHRDL2 (FC 0.14, FDR 1.20 × 10−4), encoding chordin‐like protein 2.
Figure 3.
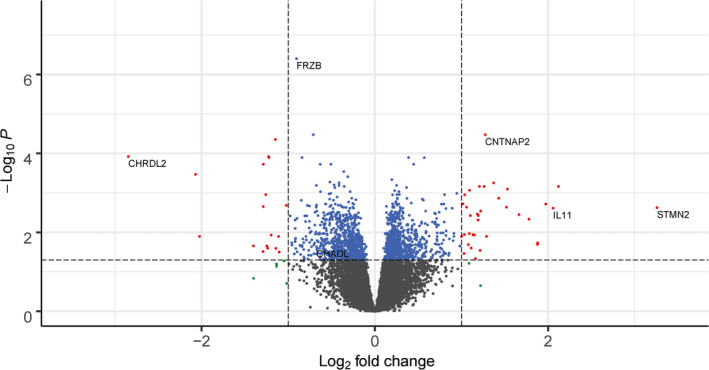
Volcano plot of differentially expressed genes in the subchondral bone. Blue dots represent genes that were significantly differentially expressed, red dots represent genes that were significantly differentially expressed and had an absolute fold change (FC) of ≥2, and green dots represent genes with an absolute FC of ≥2 that were not significantly differentially expressed.
Next, we explored whether the 1,569 significantly differentially expressed genes were enriched in relation to particular pathways or processes, using DAVID. The results demonstrated significantly enriched Gene Ontology (GO) terms regarding processes involved in translational and posttranslational processes, such as signal recognition particle–dependent cotranslational protein targeting to membrane (GO 0006614; 33 genes) (FDR 4.27 × 10−7) and translational initiation (GO 0006413; 36 genes) (FDR 1.95 × 10−4). These processes were both mainly characterized by ribosomal proteins such as RPS24, RPS4X, and RPS18 (Supplementary Table 4, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract). Gene enrichment analysis of the genes selected for the highest FC (FC ≥2; n = 53 genes) showed significant enrichment of processes regarding the extracellular matrix (GO 0005615; 16 genes) (FDR 1.19 × 10−5), characterized by up‐regulation of WNT16 (FC 4.35, FDR 6.88 × 10−4), CRLF1 (FC 2.32, FDR 2.86 × 10−2), and OGN (FC 3.43, FDR 4.62 × 10−3), and the proteinaceous extracellular matrix (GO 0005578; 7 genes) (FDR 4.50 × 10−2), characterized by up‐regulation of POSTN (FC 2.04, FDR 3.44 × 10−2), ASPN (FC 3.17, FDR 3.56 × 10−3), and CTHRC1 (FC 2.15, FDR 3.75 × 10−3) (Supplementary Table 5, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract). To explore interactions between proteins encoded by the 53 differentially expressed genes with an FC of ≥2, we used the online tool STRING. We identified significant enrichment for protein–protein interactions among 22 of 44 proteins (P = 3.20 × 10−9) (Figure 4).
Figure 4.
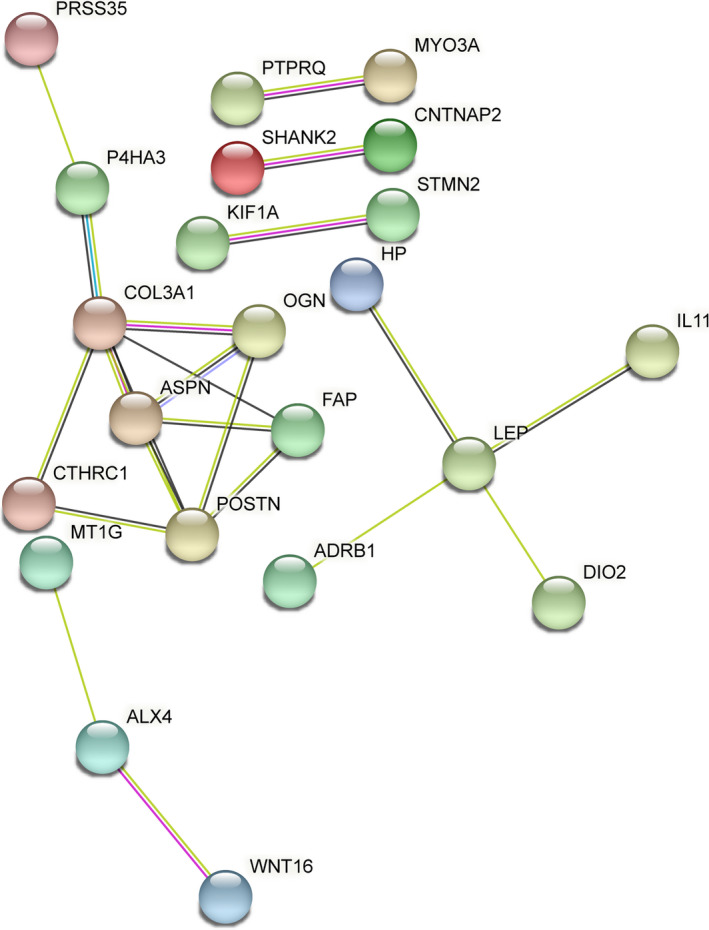
Protein–protein interaction network of proteins encoded by genes that showed an absolute fold change of ≥2 (n = 53 genes), created with the tool STRING.
Comparison of subchondral bone and articular cartilage
To investigate interacting OA pathophysiologic processes in subchondral bone and articular cartilage, we compared the differentially expressed genes identified in bone with our previously reported results on differentially expressed genes in articular cartilage (18) (Figure 1A) (24 sample pairs from bone and 35 from cartilage; 14 patients with available sample pairs from both bone and cartilage). This analysis revealed 337 genes that were differentially expressed in both subchondral bone and articular cartilage (Supplementary Figure 3, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract). Of these 337 overlapping genes, the majority (305 genes) showed similar directions of effect in cartilage and bone (Supplementary Table 6, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract), while 32 genes showed opposite directions of effect between the 2 tissue types (Supplementary Table 7, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract). ALX4¸ encoding aristaless‐like homeobox 4, was notable among the genes showing opposite directions of effect. ALX4 is known to be involved in osteogenesis and was one of the most highly up‐regulated genes in bone (Table 1). Among the 305 genes showing a similar direction of effect, 14 were among the top 25 genes with the highest FC in both tissues, such as WNT16, IL11, CRLF1, and FRZB (Table 1).
Table 1.
Genes classified as the top 25 genes based on the highest absolute FC in either bone or cartilage, 14 of which were among the top 25 with the highest FC in both tissues*
| Ensemble ID/gene name | Subchondral bone | Articular cartilage | Top 25 absolute FC | |||
|---|---|---|---|---|---|---|
| FC | FDR | FC | FDR | SB | AC | |
| WNT16 | 4.35 | 6.88 × 10−4 | 8.48 | 1.10 × 10−13 | x | x |
| IL11 | 4.16 | 2.44 × 10−3 | 22.8 | 1.53 × 10−20 | x | x |
| GDF6 | 3.67 | 2.02 × 10−2 | 1.58 | 3.19 × 10−2 | x | |
| OGN | 3.43 | 4.62 × 10−3 | 2.00 | 1.02 × 10−3 | x | |
| ASPN | 3.17 | 3.56 × 10−3 | 1.65 | 3.04 × 10−2 | x | |
| MYO3A | 2.44 | 1.27 × 10−2 | 2.25 | 1.16 × 10−4 | x | |
| CRLF1 | 2.32 | 2.86 × 10−2 | 3.04 | 2.96 × 10−10 | x | x |
| GPR158 | 2.31 | 6.88 × 10−4 | 2.73 | 3.63 × 10‐3 | x | x |
| PPP1R14C | 2.19 | 1.14 × 10−2 | 2.52 | 1.33 × 10−11 | x | |
| MT1G | 2.16 | 2.50 × 10−2 | 1.97 | 1.72 × 10−4 | x | |
| ALX4 | 2.08 | 2.30 × 10−3 | 0.55 | 2.75 × 10−2 | x | |
| P4HA3 | 2.05 | 1.12 × 10−3 | 1.84 | 1.49 × 10−5 | x | |
| FAP | 2.05 | 1.14 × 10−2 | 1.69 | 1.09 × 10−3 | x | |
| POSTN | 2.04 | 3.44 × 10−2 | 2.06 | 3.20 × 10−2 | x | |
| HOXB‐AS1 | 2.00 | 1.27 × 10−2 | 1.64 | 4.86 × 10−2 | x | |
| KIF20A | 1.97 | 2.22 × 10−2 | 1.59 | 4.44 × 10−2 | x | |
| TNFAIP | 1.93 | 1.03 × 10−3 | 3.58 | 2.48 × 10−8 | x | x |
| ERFE | 1.87 | 1.63 × 10−2 | 3.44 | 8.82 × 10−12 | x | x |
| PTGES | 1.64 | 1.63 × 10−2 | 3.06 | 3.61 × 10−12 | x | |
| TNFRSF12A | 1.50 | 2.31 × 10−2 | 2.68 | 1.14 × 10−8 | x | |
| WNT10B | 1.49 | 3.25 × 10−2 | 3.47 | 1.52 × 10−6 | x | |
| COCH | 1.46 | 4.21 × 10−2 | 3.30 | 1.01 × 10−8 | x | |
| CD55 | 1.46 | 2.48 × 10−2 | 2.96 | 1.05 × 10−14 | x | |
| P3H2 | 1.37 | 1.14 × 10−2 | 3.23 | 4.71 × 10−18 | x | |
| NGF | 1.36 | 3.26 × 10−2 | 4.91 | 2.53 × 10−14 | x | |
| SPP1 | 1.36 | 4.81 × 10−2 | 3.14 | 8.98 × 10−7 | x | |
| NTRK3 | 0.70 | 3.56 × 10−3 | 0.31 | 2.64 × 10−5 | x | |
| LMO3 | 0.58 | 3.82 × 10−3 | 0.28 | 1.67 × 10−5 | x | |
| FRZB | 0.53 | 3.99 × 10−7 | 0.27 | 1.87 × 10−9 | x | x |
| RELN | 0.53 | 2.56 × 10−2 | 0.22 | 7.37 × 10−12 | x | x |
| SLC14A1 | 0.53 | 1.71 × 10−2 | 0.51 | 7.05 × 10−6 | x | |
| CRISPLD1 | 0.51 | 1.84 × 10−2 | 0.36 | 9.29 × 10−6 | x | x |
| ZNF385C | 0.51 | 3.82 × 10−3 | 0.43 | 2.30 × 10−6 | x | |
| HIF3A | 0.49 | 2.07 × 10−3 | 0.58 | 2.72 × 10−2 | x | |
| AL845331.2 | 0.46 | 3.16 × 10−2 | 0.34 | 3.50 × 10−2 | x | x |
| GPC5 | 0.43 | 1.27 × 10−4 | 0.36 | 1.47 × 10−8 | x | x |
| AC005165.1 | 0.43 | 1.20 × 10−4 | 0.45 | 5.31 × 10−4 | x | |
| FGF14 | 0.41 | 1.89 × 10−4 | 0.58 | 2.01 × 10−4 | x | |
| AC084816.1 | 0.38 | 2.20 × 10−2 | 0.45 | 2.20 × 10−5 | x | |
| KIF1A | 0.25 | 1.27 × 10−2 | 0.37 | 8.64 × 10−8 | x | x |
| SPOCK3 | 0.24 | 3.41 × 10−4 | 0.22 | 1.56 × 10−9 | x | x |
| CHRDL2 | 0.14 | 1.20 × 10−4 | 0.13 | 7.07 × 10−9 | x | x |
FC = fold change; FDR = false discovery rate; SB = subchondral bone; AC = articular cartilage.
To explore common underlying pathways in subchondral bone and articular cartilage, we performed gene enrichment analysis with the 305 genes that showed similar directions of effect in cartilage and bone. We found significant enrichment for the GO terms extracellular region (GO 0005576; 36 genes) (FDR 4.56 × 10−3), characterized by the expression of, for example, COL6A3, FGF14, and GDF6, proteinaceous extracellular matrix (GO 0005578; 17 genes) (FDR 7.98 × 10−3), characterized by the expression of, for example, CHADL, ADAMTS17, and SPOCK3, and extracellular space (GO 0005615; 37 genes) (FDR 4.42 × 10−3), characterized by the expression of, for example, CD63, SPP1, and RELN (Supplementary Table 8, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract).
Differential expression analysis stratified by joint site
Since hip and knee samples showed different gene expression profiles in the cluster analysis (Figure 2), we repeated the differential expression analysis with stratification by joint site to explore whether we could identify exclusive OA pathways that occur in subchondral bone of knees only or hips only. Differential expression analysis of the 18 knee sample pairs revealed 1,757 genes that were significantly differentially expressed (Figure 1B), of which 902 genes were up‐regulated and 855 were down‐regulated in lesional compared to preserved OA subchondral bone (Supplementary Table 9, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract). Moreover, we identified 509 genes that were differentially expressed exclusively in the knee (Supplementary Table 10, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract), i.e., these genes were not differentially expressed in an analysis of the total data set (Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract) or the hip data set (Supplementary Table 11, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract). Enrichment analysis of these genes that were differentially expressed exclusively in the knee showed significant enrichment for processes involved in epigenetic regulation, such as nucleosome (GO 0000786; 20 genes) (FDR 1.81 × 10−9), DNA methylation (R‐HSA 5334118; 15 genes) (FDR 2.48 × 10−6), and regulation of gene silencing (GO 0060968; 6 genes) (FDR 1.90 × 10−2), all characterized by members of the histone H3 family, such as HIST1H3J and HIST1H3H (Supplementary Table 12, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract).
Differential expression analysis using only the hip samples (6 pairs) did not reveal any genes that were significantly differentially expressed by the FDR method when comparing preserved and lesional subchondral bone. However, among the genes with a P value of <0.05 and an absolute FC of ≥2 (Supplementary Table 11), 18 genes appeared to be differentially expressed exclusively in the hip (Figure 1C); i.e., not differentially expressed in an analysis of the total data set (Supplementary Table 3) or the knee data set (Supplementary Table 9). Included among these genes with differential expression exclusively in the hip were CALCR, LGR5, and COL2A1 (Supplementary Table 13, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract).
Validation of differentially expressed genes
To validate and replicate the findings of the differential expression analysis performed using RNA sequencing, we used a set of 20 samples to conduct both technical replication (10 samples) and biologic replication (10 samples) by qRT‐PCR. Validation analysis of 6 genes, FRZB, CNTNAP2, STMN2, CHRDL2, POSTN, and ASPN, showed significant differences between preserved and lesional subchondral bone, with directions of effect similar to those found by RNA sequencing. Replication analysis also showed significant differences, with the same directions of effects as shown by RNA sequencing (Supplementary Table 14, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract).
Differential expression of previously identified risk genes
In recent genome‐wide association studies of hip and knee OA (5,6), 27 loci conferring risk to OA were identified (Table 2). To assess whether those OA susceptibility genes are also involved in OA pathophysiology in articular cartilage, subchondral bone, or both, we explored their expression levels and differential expression between lesional and preserved tissue in our data sets. As shown in Table 2, we identified 2 risk genes, IL11 and CHADL, that were differentially expressed in both subchondral bone and articular cartilage. In addition, IL11 showed both significant differential expression in knee subchondral bone (FC 4.07, FDR 7.00 × 10−3) and a high FC (FC 4.77, P = 4.43 × 10−2) in hip subchondral bone. This indicates that, based on our data set, IL11 has an effect in both tissues and at both joint sites, albeit not significant according to FDR in hip subchondral bone.
Table 2.
Expression levels and differential expression of OA risk genes identified in recent genome‐wide association studies*
| Gene | Bone, total data set | Cartilage, total data set | ||||
|---|---|---|---|---|---|---|
| Expression† | FC, preserved vs. OA | FDR | Expression† | FC, preserved vs. OA | FDR | |
| COL11a1 | 1 | 1.19 | 6.21 × 10−1 | 1 | 1.07 | 7.59 × 10−1 |
| HDAC9 | 2 | 0.97 | 6.75 × 10−1 | 1 | 0.59 | 9.10 × 10−6 |
| SMO | 2 | 1.00 | 9.91 × 10−1 | 1 | 0.69 | 7.85 × 10−5 |
| TNC | 1 | 1.18 | 2.58 × 10−1 | 1 | 1.41 | 1.09 × 10−2 |
| LMX1B | NE | NA | NA | 3 | 0.99 | 9.80 × 10−1 |
| LTBP3 | 1 | 0.87 | 3.08 × 10−1 | 1 | 1.08 | 6.95 × 10−1 |
| FAM101A (RFLNA) | 4 | 0.99 | 9.77 × 10−1 | 2 | 0.49 | 6.48 × 10−5 |
| IL11 | 3 | 4.16 | 2.44 × 10−3 | 1 | 22.80 | 1.53 × 10−20 |
| ITIH1 | NE | NA | NA | NE | NA | NA |
| FILIP1 | 2 | 0.84 | 7.07 × 10−2 | 3 | 1.23 | 2.38 × 10−1 |
| RUNX2 | 1 | 1.07 | 4.75 × 10−1 | 2 | 0.93 | 7.79 × 10−1 |
| ASTN2 | 4 | 0.87 | 2.42 × 10−1 | 4 | 0.82 | 2.43 × 10−1 |
| SMAD3 | 1 | 0.93 | 3.74 × 10−1 | 1 | 0.84 | 2.83 × 10−2 |
| HFE | 3 | 1.01 | 9.07 × 10−1 | 2 | 0.88 | 1.32 × 10−1 |
| CHADL | 4 | 0.63 | 2.33 × 10−2 | 1 | 0.63 | 1.29 × 10−1 |
| LTBP1 | 1 | 0.97 | 6.91 × 10−1 | 1 | 1.15 | 1.70 × 10−1 |
| SBNO1 | 1 | 0.98 | 6.79 × 10−1 | 1 | 1.10 | 3.94 × 10−1 |
| WWP2 | 1 | 0.82 | 2.47 × 10−1 | 1 | 0.79 | 3.43 × 10−2 |
| GDF5 | 4 | 0.92 | 8.08 × 10−1 | 1 | 1.23 | 3.09 × 10−1 |
| TGFB1 | NE | NA | NA | NE | NA | NA |
| TNFSF15 | 4 | 1.23 | 2.42 × 10−1 | 3 | 1.00 | 9.91 × 10−1 |
| FGF18 | NE | NA | NA | 2 | 1.58 | 9.51 × 10−4 |
| CTSK | 1 | 1.41 | 3.23 × 10−1 | 1 | 1.03 | 8.91 × 10−1 |
| DPEP1 (MBD1) | 1 | 0.95 | 2.83 × 10−1 | 1 | 0.96 | 6.20 × 10−1 |
| DIABLO | 4 | 0.95 | 7.37 × 10−1 | 3 | 1.08 | 6.46 × 10−1 |
| CRHR1 | NE | NA | NA | 4 | 0.62 | 5.10 × 10−1 |
| MAPT | 3 | 0.61 | 3.47 × 10−2 | 4 | 0.70 | 1.56 × 10−1 |
OA = osteoarthritis; FC = fold change; FDR = false discovery rate; NE = not expressed; NA = not applicable.
In quartiles, with 1 being the highest expressed quartile and 4 being the lowest expressed quartile.
DISCUSSION
Differential expression analysis of gene expression levels in preserved and lesional OA subchondral bone (n = 24 paired samples) revealed 1,569 genes that were significantly differentially expressed, including CNTNAP2 and STMN2. Upon comparing these 1,569 differentially expressed genes with the 2,387 genes with OA pathophysiology previously shown to be differentially expressed in cartilage, we found an overlap of 305 genes that had the same direction of effect. These 305 overlapping genes were enriched for processes related to the extracellular matrix, characterized by the expression of, among others, COL6A3, GDF6, and SPP1. Moreover, among the 305 overlapping genes were IL11 and CHADL (Table 2 and Supplementary Table 6, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract), which were previously identified as being OA risk genes (5, 6). By applying hierarchical clustering on the overall RNA sequencing data set from subchondral bone, we observed 2 clusters based on joint site (knee and hip). When stratifying the analysis for joint site, we identified 1,757 genes that were differentially expressed between preserved and lesional knee OA bone, 509 of which were differentially expressed in the knee exclusively, including genes such as WNT4 and KLF11. These OA genes that were differentially expressed exclusively in the knee were enriched for regulation of gene silencing by epigenetic processes such as DNA methylation and histone modification, characterized by genes such as HIST1H3J and HIST1H3H, as well as being enriched for other processes.
Among the 1,569 genes that were significantly differentially expressed between lesional and preserved OA subchondral bone using the FDR method in the complete data set, we identified CNTNAP2 (FC 2.42, FDR 3.36 × 10−5) and STMN2 (FC 9.56, FDR 2.36 × 10−3) as the most significantly up‐regulated gene and the gene with the highest FC, respectively. CNTNAP2, encoding CASPR2, is known for its effect on cell–cell interactions in the nervous system, synapse development, neural migration, and neural connectivity (31,32). Neither CNTNAP2 nor its encoded protein were previously identified as being related to OA. STMN2 also plays a role in the control of neuronal differentiation. Moreover, STMN2 is expressed during osteogenesis, and it was previously shown to be highly up‐regulated in OA bone marrow lesions as compared to control bone samples (8,33). In addition, we found other neural markers to be up‐regulated in lesional compared to preserved OA subchondral bone, such as NGF and THBS3 (Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.41600/abstract). Based on these findings, we hypothesize that the formation of new neuronal structures in bone is increased with ongoing OA, which might suggest that OA‐related pain originates from bone (8). However, functional follow‐up research is needed to confirm this hypothesis.
The hierarchical clustering was done on the top 1,000 genes that showed the highest coefficient of variation between samples; hence, the clusters reflect particularly large differences. Based on the results observed here, it could thus be concluded that these highly variable genes reflect consistent differences between subchondral bone in the knee and subchondral bone in the hip, which was not previously seen in similar analyses of cartilage (25). Consequently, the fact that neither preserved and lesional samples from the same individual nor preserved samples or lesional samples as a group cluster together indicated that the 1,000 genes with the highest coefficient of variation are marking differences between knees and hips only. This does not rule out the relevance of the highly consistently differentially expressed genes reflecting OA subchondral bone pathology described here.
Upon differential expression analysis with stratification by joint site, we discovered 509 genes that were unique to the knee compared to the complete data set, which were significantly enriched for epigenetic processes such as DNA methylation, reflected by the expression of, among others, HIST1H3J and HIST1H3H. The significant enrichment of these epigenetic processes among the knee‐exclusive genes indicates a change in epigenetics with ongoing knee OA, which is not seen with ongoing hip OA. This was also previously demonstrated in articular cartilage, where hip and knee methylation profiles clustered apart irrespective of OA status. However, this was characterized by the expression of different genes, such as the homeobox genes (34,35). We did not find FDR‐significant genes when selecting the hip samples, which is likely due to the small sample size (n = 6 sample pairs). Nonetheless, we identified 18 genes that were exclusively differentially expressed in the hip based on the nominal P value and an absolute FC of ≥2, including genes such as CALCR, LGR5, and COL2A1. However, replication is needed to confirm our findings regarding these genes differentially expressed exclusively in the hip.
Given the accumulating awareness of cross‐talk between articular cartilage and subchondral bone in OA (10,36), we compared RNA sequencing data from subchondral bone and from articular cartilage (24 sample pairs, and 35 sample pairs, respectively, with an overlap of 14 patients). Compared to the number of genes identified as being significantly differentially expressed between preserved and lesional OA articular cartilage based on FDR (n = 2,387 genes), we found fewer genes that were significantly differentially expressed by FDR between preserved and lesional OA subchondral bone (n = 1,569 genes). This difference might be due to the difference in sample size. However, it could also reflect the fact that bone as multicellular tissue is more heterogeneous. The relatively small overlap in genes that were differentially expressed in the same direction in both subchondral bone and cartilage (305 of 3,619; 8.43%) subchondral bone and cartilage suggests that there is a difference in OA pathophysiology between the 2 tissues.
To find genes that are most likely causal in OA, we explored 27 previously published genes with SNPs that were identified as being genome‐wide significantly associated with OA (Table 2), suggesting that those genes have a more causal relationship to OA and making them attractive potential treatment targets (5,6). To examine whether the previously identified OA risk genes are involved in the OA pathophysiologic process in both cartilage and subchondral bone, we compared the expression levels and the differential expression between preserved and lesional samples (Table 2). We found that the OA risk genes IL11 and CHADL were differentially expressed in both cartilage and subchondral bone and with the same direction of effect, thus making them attractive potential therapeutic targets with effects in both tissue types. CHADL, encoding chondroadherin‐like protein, is involved in collagen binding and is a negative modulator of chondrocyte differentiation. The OA susceptibility allele rs117018441‐T, located in an intron of CHADL, marks higher expression of CHADL compared to rs117018441‐G in skeletal muscle and adipose tissue according to the Genotype‐Tissue Expression Project (5,37). This may indicate that increased expression of CHADL has a negative regulatory role in both bone and cartilage and that inhibition of this gene could be a therapeutic strategy. However, when stratifying by joint site, we found CHADL to be differentially expressed specifically in the knee subchondral bone, suggesting that it is a treatment target for knee OA exclusively.
IL11, encoding interleukin‐11 (IL‐11), is known for its role in bone remodeling, and lack of IL‐11 function is associated with impaired bone formation (38). Notably, IL11 was recently proposed as a potential therapeutic target for OA in cartilage (6), since the OA risk allele rs4252548‐T, a missense variant p.Arg112His, acts via reduced function of the IL‐11 protein. As such, increasing IL‐11 protein levels was proposed as a strategy for treatment of OA. In this study we have again shown that IL11 is highly up‐regulated in lesional versus preserved OA tissue in both subchondral bone and articular cartilage (FC 4.16 and 22.8, respectively). Taken together, these data indicate that reduced function of IL‐11 predisposes to OA onset and that the up‐regulation of IL11 with OA pathophysiology could be considered an attempt of chondrocytes to enhance extracellular matrix integrity. Nonetheless, the consistent and considerable up‐regulation of IL11 in both subchondral bone and articular cartilage may not necessarily reflect a lack of potency to produce IL‐11, unless translation of the protein is hampered. This requires further functional investigation, preferably in an in vitro model of OA. CHADL and IL11 could both be highly suitable treatment targets with effects in both bone and cartilage. However, further functional research is needed to confirm the effects of these genes on bone and cartilage metabolism.
The classification of OA subchondral bone as preserved or lesional is derived from its overlying cartilage. We acknowledge that this ascertainment strategy is bound to introduce heterogeneity between samples. Nonetheless, we identified FDR‐significant, and hence very consistent, differentially expressed genes. In other words, despite the fact that there may be heterogeneity in the preserved cartilage, we found consistent markers of the OA pathophysiologic process in subchondral bone.
To our knowledge, this is the first reported study of large‐scale differential gene expression patterns in OA subchondral bone, performed using RNA sequencing in both hip and knee samples. We identified distinct differences in expression patterns between hips and knees. Moreover, we identified multiple genes that were previously demonstrated in OA articular cartilage, in addition to genes that were subchondral bone specific. These results will contribute to a better understanding of the pathophysiologic processes underlying the development of OA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Ms Tuerlings had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Tuerlings, Ramos, Coutinho de Almeida, Meulenbelt.
Acquisition of data
Tuerlings, van Hoolwerff, Houtman, Suchiman, Lakenberg, Mei, van der Linden, Nelissen, Ramos, Coutinho de Almeida, Meulenbelt.
Analysis and interpretation of data
Tuerlings, Ramos, Coutinho de Almeida, Meulenbelt.
Supporting information
Fig S1
Fig S2
Fig S3
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Table S8
Table S9
Table S10
Table S11
Table S12
Table S13
Table S14
Supplementary Material
Acknowledgments
We thank all of the participants of the RAAK study. We also thank Demien Broekhuis, Robert van der Wal, Peter van Schie, Shaho Hasan, Maartje Meijer, Daisy Latijnhouwers, and Geert Spierenburg for collecting the study samples. We thank the personnel of the Sequence Analysis Support Core of Leiden University Medical Center for their support. Data were generated within the scope of the Medical Delta Regenerative Medicine 4D programs Generating Complex Tissues with Stem Cells and Printing Technology and Improving Mobility with Technology.
Supported by the Dutch Scientific Research Council (NOW/ZonMW Vici grant 91816631/528), the European Union Seventh Framework Programme (project TREAT‐OA; 200800), and the Dutch Arthritis Society (grant DAA_10_1‐402). The RAAK study is supported by Leiden University Medical Center.
No potential conflicts of interest relevant to this article were reported.
References
- 1. Woolf AD, Erwin J, March L. The need to address the burden of musculoskeletal conditions. Best Pract Res Clin Rheumatol 2012;26:183–224. [DOI] [PubMed] [Google Scholar]
- 2. Goldring MB, Goldring SR. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann N Y Acad Sci 2010;1192:230–7. [DOI] [PubMed] [Google Scholar]
- 3. Vina ER, Kwoh CK. Epidemiology of osteoarthritis: literature update [review]. Curr Opin Rheumatol 2018;30:160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zeggini E, Panoutsopoulou K, Southam L, Rayner NW, Day‐Williams AG, Lopes MC, et al, for the arcOGEN Consortium and the arcOGEN Collaborators . Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome‐wide association study. Lancet 2012;380:815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Styrkarsdottir U, Lund SH, Thorleifsson G, Zink F, Stefansson OA, Sigurdsson JK, et al. Meta‐analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nat Genet 2018;50:1681–7. [DOI] [PubMed] [Google Scholar]
- 6. Tachmazidou I, Hatzikotoulas K, Southam L, Esparaza‐Gordillo J, Haberland V, Zheng J, et al. Identification of new therapeutic targets for osteoarthritis through genome‐wide analyses of UK Biobank data. Nat Genet 2019;51:230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chou CH, Wu CC, Song IW, Chuang HP, Lu SL, Chang JH, et al. Genome‐wide expression profiles of subchondral bone in osteoarthritis. Arthritis Res Ther 2013;15:R190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuttapitiya A, Assi L, Laing K, Hing C, Mitchell P, Whitley G, et al. Microarray analysis of bone marrow lesions in osteoarthritis demonstrates up‐regulation of genes implicated in osteochondral turnover, neurogenesis and inflammation. Ann Rheum Dis 2017;76:1764–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fellows CR, Matta C, Mobasheri A. Applying proteomics to study crosstalk at the cartilage‐subchondral bone interface in osteoarthritis: current status and future directions. EBioMedicine 2016;11:2–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goldring SR, Goldring MB. Changes in the osteochondral unit during osteoarthritis: structure, function and cartilage‐bone crosstalk [review]. Nat Rev Rheumatol 2016;12:632–44. [DOI] [PubMed] [Google Scholar]
- 11. Funck‐Brentano T, Cohen‐Solal M. Crosstalk between cartilage and bone: when bone cytokines matter. Cytokine Growth Factor Rev 2011;22:91–7. [DOI] [PubMed] [Google Scholar]
- 12. Pan J, Wang B, Li W, Zhou X, Scherr T, Yang Y, et al. Elevated cross‐talk between subchondral bone and cartilage in osteoarthritic joints. Bone 2012;51:212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Funck‐Brentano T, Cohen‐Solal M. Subchondral bone and osteoarthritis. Curr Opin Rheumatol 2015;27:420–6. [DOI] [PubMed] [Google Scholar]
- 14. Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ [review]. Arthritis Rheum 2012;64:1697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barbour KE, Murphy LB, Helmick CG, Hootman JM, Renner JB, Jordan JM. Bone mineral density and the risk of hip and knee osteoarthritis: the Johnston County Osteoarthritis Project. Arthritis Care Res (Hoboken) 2017;69:1863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roemer FW, Guermazi A, Javaid MK, Lynch JA, Niu J, Zhang Y, et al. Change in MRI‐detected subchondral bone marrow lesions is associated with cartilage loss: the MOST Study. A longitudinal multicentre study of knee osteoarthritis. Ann Rheum Dis 2009;68:1461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramos YF, den Hollander W, Bovée JV, Bomer N, van der Breggen R, Lekenberg N, et al. Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage: the RAAK study. PLoS One 2014;9:e103056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coutinho de Almeida R, Ramos YF, Mahfouz A, den Hollander W, Lakenberg N, Houtman E, et al. RNA sequencing data integration reveals an miRNA interactome of osteoarthritis cartilage. Ann Rheum Dis 2019;78:270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu P, Hwang JT. Quick calculation for sample size while controlling false discovery rate with application to microarray analysis. Bioinformatics 2007;23:739–46. [DOI] [PubMed] [Google Scholar]
- 20. Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005;21:1859–75. [DOI] [PubMed] [Google Scholar]
- 21. Anders S, Pyl PT, Huber W. HTSeq: a Python framework to work with high‐throughput sequencing data. Bioinformatics 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32:3047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin M. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J 2011;17:10–2. [Google Scholar]
- 24. Li W, Fan M, Xiong M. SamCluster: an integrated scheme for automatic discovery of sample classes using gene expression profile. Bioinformatics 2003;19:811–7. [DOI] [PubMed] [Google Scholar]
- 25. Coutinho de Almeida R, Mahfouz A, Mei H, Houtman E, den Hollander W, Soul J, et al. Identification and characterization of two consistent osteoarthritis subtypes by transcriptome and clinical data integration. Rheumatology (Oxford) 2021;60:1166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Charrad M, Ghazzali N, Boiteau V, Niknafs A. NbClust: an R package for determining the relevant number of clusters in a data set. J Stat Softw 2014;61:1–36. [Google Scholar]
- 27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 29. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta‐Cepas J, et al. STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res 2019;47:D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chou CH, Lee CH, Lu LS, Song IW, Chuang HP, Kuo SY, et al. Direct assessment of articular cartilage and underlying subchondral bone reveals a progressive gene expression change in human osteoarthritic knees. Osteoarthritis Cartilage 2013;21:450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu Z, Reddy MV, Liu J, Kalichava A, Liu J, Zhang L, et al. Molecular architecture of contactin‐associated protein‐like 2 (CNTNAP2) and its interaction with contactin 2 (CNTN2). J Biol Chem 2016;291:24133–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rodenas‐Cuadrado P, Ho J, Vernes SC. Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet 2014;22:171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chiellini C, Grenningloh G, Cochet O, Scheideler M, Trajanoski Z, Ailhaud G, et al. Stathmin‐like 2, a developmentally‐associated neuronal marker, is expressed and modulated during osteogenesis of human mesenchymal stem cells. Biochem Biophys Res Commun 2008;374:64–8. [DOI] [PubMed] [Google Scholar]
- 34. Reynard LN. Analysis of genetics and DNA methylation in osteoarthritis: what have we learnt about the disease? [review]. Semin Cell Dev Biol 2017;62:57–66. [DOI] [PubMed] [Google Scholar]
- 35. Den Hollander W, Ramos YF, Bos SD, Bomer N, van der Breggen R, Lakenberg N, et al. Knee and hip articular cartilage have distinct epigenomic landscapes: implications for future cartilage regeneration approaches. Ann Rheum Dis 2014;73:2208–12. [DOI] [PubMed] [Google Scholar]
- 36. Tat SK, Lajeunesse D, Pelletier JP, Martel‐Pelletier J. Targeting subchondral bone for treating osteoarthritis: what is the evidence? Best Pract Res Clin Rheumatol 2010;24:51–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Styrkarsdottir U, Helgason H, Sigurdsson A, Norddahl GL, Agustsdottir AB, Reynard LN, et al. Whole‐genome sequencing identifies rare genotypes in COMP and CHADL associated with high risk of hip osteoarthritis. Nat Genet 2017;49:801–5. [DOI] [PubMed] [Google Scholar]
- 38. Lokau J, Göttert S, Arnold P, Düsterhöft S, López DM, Grötzinger J, et al. The SNP rs4252548 (R112H) which is associated with reduced human height compromises the stability of IL‐11. Biochim Biophys Acta Mol Cell Res 2018;1865:496–506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Table S8
Table S9
Table S10
Table S11
Table S12
Table S13
Table S14
Supplementary Material