

Abstract
Metabolic reprogramming in tumor‐immune interactions is emerging as a key factor affecting pro‐inflammatory carcinogenic effects and anticancer immune responses. Therefore, dysregulated metabolites and their regulators affect both cancer progression and therapeutic response. Here, we describe the molecular mechanisms through which microenvironmental, systemic, and microbial metabolites potentially influence the host immune response to mediate malignant progression and therapeutic intervention. We summarized the primary interplaying factors that constitute metabolism, immunological reactions, and cancer with a focus on mechanistic aspects. Finally, we discussed the possibility of metabolic interventions at multiple levels to enhance the efficacy of immunotherapeutic and conventional approaches for future anticancer treatments.
Keywords: aging, cancer, immune response, metabolism, microbes, obesity
In this review, we describe the molecular mechanisms through which systemic, microenvironmental and microbial metabolites potentially influence the host immune response to mediate malignant progression and therapeutic intervention. We summarize the primary interplaying factors that constitute metabolism, immunological reactions and cancer with a focus on mechanistic aspects. Finally, we discuss the possibility of metabolic interventions at multiple levels to enhance the efficacy of immunotherapeutic and conventional approaches for future antineoplastic treatments.
Abbreviations
- 2‐HG
2‐hydroxyglutarate
- α‐KG
α‐ketoglutaric acid
- ABCG1
ATP binding cassette transporter G1
- acetyl‐CoA
acetyl‐coenzyme A
- ACLY
ATP citrate lyase
- AHR
aryl hydrocarbon receptor
- AKT
protein kinase B
- AML
acute myeloid leukemia
- AMPK
adenosine 5‘‐monophosphate (AMP)‐activated protein kinase
- APCs
antigen presenting cells
- ARG1
arginase 1
- ASL
argininosuccinate lyase
- ASS1
argininosuccinate synthase
- ATP
adenosine triphosphate
- BAR
bile acid receptor
- BAs
bile acids
- BFs
bacterial biofilms
- BMDMs
bone‐marrow‐derived macrophages
- BPTES
bis‐2‐(5‐phenylacetamido‐1,2,4‐thiadiazol‐2‐yl)ethyl sulfide
- CAR‐T cell
chimeric antigen receptor T cell
- CDK4
cyclin‐dependent kinase 4
- CTLA‐4
cytotoxic T lymphocyte‐associated antigen‐4
- CR
caloric restriction
- CREB
cAMP response element‐binding protein
- CRMs
caloric restriction mimetics
- CTLs
cytotoxic T lymphocytes
- CXCL16
chemokine (C‐X‐C motif) ligand 16
- DCs
dendritic cells
- DI
dimethyl itaconate
- DMF
dimethyl fumarate
- FFAs
free fatty acids
- GLS1
glutaminase 1
- GPBAR1
G protein‐coupled BA receptor 1
- GPCRs
G protein‐coupled receptors
- GSH
glutathione
- H3K9Ac
histone H3 acetylation at the lysine 9 residue
- HDAC
histone deacetylase
- HCC
hepatocellular carcinoma
- HIF‐1α
hypoxia‐inducible factor‐1α
- IDH1
isocitrate dehydrogenase 1
- ICIs
immune checkpoint inhibitors
- IDO
indoleamine 2,3‐dioxygenase
- IFN‐γ
interferon γ
- IGF1
insulin‐like growth factor 1
- IRG1
immunoresponsive gene 1
- iTreg
induced Treg
- JNK1
c‐Jun N‐terminal protein kinase
- KA
kynurenic acid
- KEAP1
Kelch‐like ECH‐associated protein 1
- Kþ
potassium ions
- LDHA
lactate dehydrogenase A
- lncRNA
long noncoding RNA
- LPS
lipopolysaccharide
- MAP3K8
mitogen‐activated protein kinase kinase kinase 8
- MAPK
mitogen‐activated protein kinase
- MCTs
Monocarboxylate transporters
- MDSCs
myeloid‐derived suppressor cells
- MMF
monomethyl fumarate
- MS
multiple sclerosis
- mTOR
mammalian target of rapamycin
- mtROS
mitochondrial reactive oxygen species
- NAD
nicotinamide adenine dinucleotide
- NFAT
nuclear factor of activated T cells
- NF‐κB
nuclear factor kappa‐B
- NK
natural killer
- NKT
natural killer T
- NLRP3
NLR family pyrin domain containing 3
- NO
nitric oxide
- Nrf2
nuclear factor erythroid 2‐related factor 2
- OXPHOS
oxidative phosphorylation
- P2R
type‐2 purinergic receptors
- PD‐1
programmed death 1
- PD‐L1
programmed cell death‐ligand 1
- PKCθ
protein kinase C θ
- PPAR
peroxisome proliferator activated receptor
- PSA
polysaccharide A
- PUFA
polyunsaturated fatty acid
- RCC
renal cell carcinoma
- RORγt
retinoid‐related orphan receptor‐γt
- ROS
reactive oxygen species
- SASP
senescence‐associated secretory phenotype
- SCFAs
short‐chain fatty acids
- SERCA
sarco/ER Ca2+‐ATPase
- SIRT
sirtuin 3
- STAT
signal transducers and activators of transduction
- SUCNR1
succinate receptor 1
- TAMs
tumor‐associated macrophages
- TCA
tricarboxylic acid
- TCR
T cell receptor
- TDO
tryptophan 2,3‐dioxygenase
- Teff
effector T
- TET1
Ten eleven translocation 1
- TILs
tumor infiltrating lymphocytes
- TLR
Toll‐like receptor
- TME
tumor microenvironment
- TNF‐α
tumor necrosis factor‐α
- Treg
regulatory T
- VEGF
vascular endothelial growth factor
- Wnt
wingless and INT‐1
1. BACKGROUND
Metabolism is generally described as the process through which nutrients are decomposed into energy and small molecule metabolites by biochemical reaction networks, and these products are subsequently utilized by cells to generate energy and macromolecules, including proteins, lipids, and nucleic acids, needed for survival and the maintenance of physiological functions [1]. In addition, metabolic pathways are closely related to epigenetic networks and cell signalling because metabolic profiles provide a reflection of the cellular state [2]. Therefore, metabolism plays a crucial role in maintaining cellular homeostasis and facilitating adaptation to intracellular and extracellular stimuli.
Cancer progression is a consequence of the inability of the immune system to eliminate neoplastic cells. Tumor immune escape can be induced by many factors, including the loss of antigenicity, the loss of immunogenicity, and the immunosuppressive tumor microenvironment (TME), which are orchestrated by nutrient limitation and the build‐up of specific metabolites and signalling molecules [3, 4]. Tumor cells meet the needs for energy and macromolecules through significant metabolic reprogramming [5]. Moreover, the adaptability of tumor cells to a range of different hostile environments due to their metabolic flexibility further promotes their growth and metastasis [6]. Consequently, both metabolism disorders and failure of immunosurveillance to prevent malignancies are key drivers of cancer progression [6, 7].
Cellular metabolism serves a crucial function in regulating immune responses [8, 9]. Rapid and massive changes in immune cell function are involved in active immune responses, which are accompanied by substantial changes in cellular metabolism that are needed to support and, in many cases, force immune cell activation [8]. Cells can meet their particular requirements for energy and/or biosynthesis by regulating their metabolic pathways. Metabolic similarities have been observed between activated immune cells and tumor cells [10], and tumor cells are able to alter their metabolic profile to give them a survival advantage that allows them to maintain their optimal function by competing with and consuming essential nutrients or otherwise reducing the metabolic adaptability of tumor infiltrating lymphocytes (TILs). Metabolites in the TME, in turn, also affect the differentiation and effector function of immune cells [10].
Herein, we summarize various issues regarding the functions of microenvironmental and systemic metabolites in effective immune responses to tumors. The potential therapeutic opportunities arising from understanding these metabolic avenues could be exploited to enhance anticancer immunotherapies.
2. EFFECT OF LOCAL METABOLITES ON INFILTRATING LYMPHOCYTE FUNCTION
TILs are associated with good prognosis and responsiveness to therapy [11]. Similar to cancer cells, TILs need nutrients, including glucose, lipids, and amino acids found within the TME, and utilize these nutrients in metabolic pathways, primarily glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway, fatty acid oxidation, fatty acid synthesis, and amino acid metabolism, to support proliferation and differentiation [12]. On the one hand, cancer cells exhibit an enhanced ability to obtain nutrients, which increases the possibility of restricting nutrient consumption by TILs, thereby induces T‐cell exhaustion and immune escape [3]. On the other hand, immunosuppressive cells, such as regulatory T (Treg) cells, have a symbiotic metabolic relationship with cancer cells [13]. Taken together, these findings suggest that tumor cells can outcompete neighbouring immune cells for nutrients to sustain their proliferative programmes and to restrict the activation of anticancer immune cells by simultaneously recruiting and stimulating immunosuppressive cells via the release of oncometabolites.
2.1. Glucose metabolism
2.1.1. Glucose
High levels of glucose uptake and catabolism, which depend on nicotinamide adenine dinucleotide (NAD) produced by the conversion of pyruvate to lactate catalyzed by lactate dehydrogenase A (LDHA), are required for neoplastic cell growth [1]. Studies have shown that glucose metabolism in tumor cells are partly regulated by long noncoding RNAs (lncRNAs) in different ways, including directly regulating glycolytic enzymes (such as glucose transporters, hexokinase 2, LDHA, pyruvate kinase isoenzyme M2, and pyruvate dehydrogenase kinase 1) and indirectly modulating the signaling pathways [phosphatidylinositol‐3‐kinase/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathways and INT‐1 (Wnt)/Snail pathways] [14, 15]. The rapid consumption of glucose by tumors and the relative lack of vascular supply contribute to tumor interstitial glucose concentrations being less than one‐tenth that of normal organs [16], which might lead to local immunosuppression (Figure 1). Decreased glucose availability induces functional inhibition of effector T (Teff) cells.
FIGURE 1.
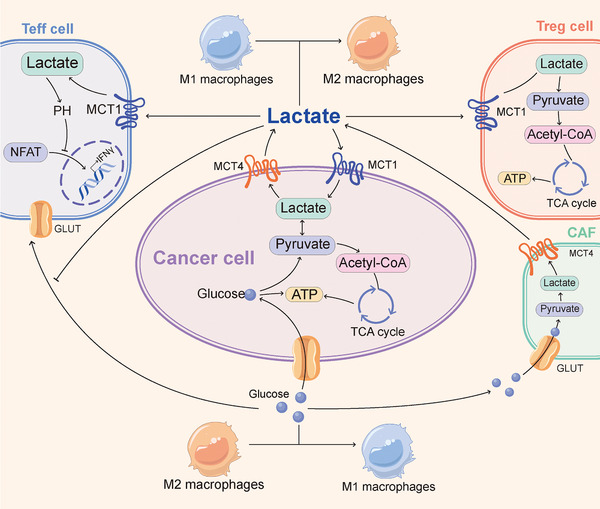
Effect of glucose and lactate metabolism on infiltrating lymphocyte function. In the TME, tumor cells consume large amounts of glucose and produce lactate. Meanwhile, tumor cells reprogram CAFs to undergo aerobic glycolysis and secrete lactate. Glucose promotes the function of Teff cells and stimulates the differentiation of macrophages into M1‐like phenotype. Relatively, lactate can be metabolized by cancer cells and Treg cells to maintain energy homeostasis, stimulates macrophages to differentiate into M2‐like phenotype, and inhibits T cell effector functions. Abbreviations: acetyl‐CoA: acetyl‐coenzyme A; ATP: adenosine triphosphate; CAFs: cancer‐associated fibroblasts; GLUT: glucose transporter; IFN‐γ: interferon γ; MCT: monocarboxylate transporters; NFAT: nuclear factor of activated T cells; pH: hydrogen ion concentration; TME: tumor microenvironment; TCA: tricarboxylic acid; Treg: regulatory T; Teff: effector T
Insufficient levels of the glycolysis metabolite phosphoenolpyruvate can increase sarco/ER Ca2+‐ATPase (SERCA)‐mediated Ca2+ re‐uptake, which results in defects in Ca2+‐nuclear factor of activated T cell (NFAT) signalling and T cell activation [16]. In addition, reduced glucose availability leads to downregulation of interferon‐γ (IFN‐γ) in T cells by enhancing the binding of the glycolytic enzyme glyceraldehyde‐3‐phosphate dehydrogenase to IFN‐γ transcripts and reducing histone acetylation [17], and this effect is accompanied by reduced CD4+ T cell differentiation into the Th1 phenotype [18]. Furthermore, limiting the glucose availability of Treg cells can support their growth and differentiation. The metabolic characteristics of Treg cells in the low glucose state include strengthened adenosine 5’‐monophosphate (AMP)‐activated protein kinase (AMPK) activity, increased fatty acid oxidation, and decreased glucose oxidation and support for energy homeostasis [19, 20]. Glucose availability is also involved in macrophage regulation. An important metabolic characteristic of activated macrophages is aerobic glycolysis [21]. The differentiation of macrophages into the M1 subtype is promoted by pyruvate dehydrogenase kinase, the key enzyme of aerobic glycolysis [22], and an enhanced glycolytic flux [23]. Thus, limiting glucose availability can inhibit macrophage activation [24]. Targeting the glucose supply in the TME yields mixed results. Reducing glycolytic metabolism by inhibiting glycolysis‐regulating enzymes or using the competitive glucose, analogue 2‐deoxy‐D‐glucose, can effectively reduce cancer cell proliferation [25] but can also inhibit the proliferation and function of Teff cells [26]. These data indicate the limitation of improving immunosuppression in the TME by targeting glycolysis, that is, the inhibition of tumor cells by targeting glycolysis might be accompanied by a decrease in TILs. Therefore, it is necessary to develop new targets that can differentially affect the glucose metabolism levels of Teff cells and tumor cells. For example, some studies have shown that acetate metabolism could enhance the function of CD8+ T cells under glucose‐restricted conditions [27, 28]. An increase in the levels of the enzyme phosphoenolpyruvate carboxykinase I in tumor‐specific CD4+ and CD8+ T cells increased the levels of the glycolytic intermediate phosphoenolpyruvate and thereby induced NFAT activity under glucose‐limited conditions [16].
2.1.2. Lactate
Lactate, a metabolic product of glycolysis, was identified as an important metabolic energy source for tumors and normal tissues in recent years [29]. High concentrations of lactate accumulate in the TME due to high glycolytic flux and decreased clearance attributed to the relative lack of a vascular supply, which increases the risk of metastasis and death [30]. Many recent studies have focused on the shuttling of lactate within tumors [31, 32, 33, 34, 35, 36, 37]. Monocarboxylate transporters (MCTs) are proteins that mediate the transport of lactate and monocarboxylate across membranes [38]. Among these transporters, MCT1 and MCT4 are closely related to tumors [33]. MCT1 is widely expressed in various tissues and promotes the uptake or efflux of lactate, depending on the cellular metabolic state and microenvironment [34]. In addition, MCT1 is usually expressed in tumor cells characterized by oxidative metabolism, particularly in cells that metabolize lactate [31]. MCT4 is only expressed in cancer or other cells, such as prostate cancer cells [35] and cancer‐associated adipocytes [36], with a high glycolytic rate and is induced by hypoxia to promote the efflux of lactate [31], which indicates the important role of MCT4 in lactate efflux in the presence of a high glycolytic flux. These findings indicate that tumor cells can flexibly choose to consume or secrete lactate depending on the extracellular or intracellular concentration of lactate. Moreover, metabolic tracer studies have further validated this possibility [37]. Notably, the accumulation of lactate in the TME cannot be attributed to cancer cells only. Recent studies have shown that cancer cells reprogramed stromal cells (mainly cancer‐associated fibroblasts) to undergo aerobic glycolysis and secrete lactate, and the secreted lactate feeds oxidative phosphorylation (OXPHOS) and mitochondrial biogenesis in cancer cells in less hypoxic regions of the TME [39, 40]. Lactate also exerts a regulatory effect on immune function. Studies showed that high concentrations of lactate induced the inhibition of glycolysis, which led to impaired function of Teff and natural killer (NK) cells [41, 42]. In contrast, high concentrations of lactate activated Treg cells because these cells could efficiently take up and metabolize lactate, particularly under low‐glucose conditions [19]. Mechanistically, lactate‐induced intracellular acidification prevents the upregulation of NFAT in T cells, which leads to diminished IFN‐γ production [41]. Of note, lactate is associated with not only the regulation of T cells but also the inhibition of both monocyte migration and the secretion of proinflammatory cytokines, the alteration of antigen presentation by monocyte‐derived dendritic cells (DCs), and promotion of the differentiation of macrophages into the M2 subtype, particularly under hypoxic conditions [43]. Based on the abovementioned information, both LDHs and MCTs are potential targets for cancer therapy. Similarly, although the inhibition of LDH might improve the lactate levels and tumor regression, it might also lead to the inhibition of glycolysis and decreases in the numbers of T cells and the production of IFN‐γ [44, 45]. Evidence showed that MCT inhibitors could effectively alleviate acidification in the TME and reduce the rate of glycolysis in cancer while enhancing the secretion of interferon‐2 (IL‐2) and IFN‐γ by T cells [45, 46]. Taken together, these findings suggest that lactate might act as a selective nutrient and paracrine signalling molecule to suppress immune activity.
2.1.3. TCA cycle
The TCA cycle, which serves as the hub for a variety of anabolic and catabolic functions, is an amphibolic pathway that occurs in the mitochondrial matrix. The TCA cycle is the primary oxidation pathway of acetyl‐coenzyme A (acetyl‐CoA) and the production pathway for the reducing equivalents nicotinamide adenine dinucleotide and flavine adenine dinucleotide. Importantly, several signalling molecules involved in the processes of immune cell activation and transformation are derived from metabolites of the TCA cycle [47]. Succinate, itaconate, fumarate, 2‐hydroxyglutarate (2‐HG), and acetyl‐CoA can affect immune regulation and/or tumor progression by inhibiting specific enzymes or driving covalent modifications of proteins. These processes result in remodelling of the epigenome and regulation of the expression of genes involved in immune cell effector functions and tumorigenesis (Figure 2).
FIGURE 2.
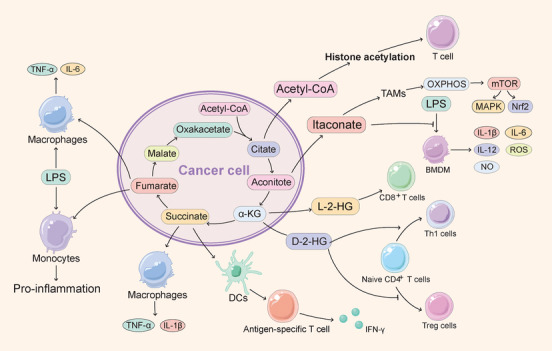
Effect of the TCA cycle on infiltrating lymphocyte function. Several signalling molecules involved in the process of immune cell activation and transformation are derived from TCA cycle metabolites, including succinate, itaconate, fumarate, 2‐HG, and acetyl‐CoA, which can affect immune regulation and/or tumor progression by inhibiting specific enzymes or driving covalent modification of proteins. Abbreviations: TCA: tricarboxylic acid; 2‐HG: 2‐hydroxyglutarate; acetyl‐CoA: acetyl‐coenzyme A; BMDM: bone‐marrow‐derived macrophage; DCs: dendritic cells; IFN‐γ: interferon γ; IL: interleukin; LPS: lipopolysaccharide; mtROS: mitochondrial reactive oxygen species; mTOR: mammalian target of rapamycin; Nrf2: nuclear factor erythroid 2‐related factor 2; NO: nitric oxide; OXPHOS: oxidative phosphorylation; MAPK: mitogen‐activated protein kinase; ROS: reactive oxygen species; TAMs: tumor‐associated macrophages; TNF‐α: tumor necrosis factor‐α; Treg: regulatory T; α‐KG: α‐ketoglutaric acid; Teff: effector T
Succinate accumulates in macrophages stimulated by lipopolysaccharide (LPS) and IFN‐γ and is a proinflammatory metabolite [48, 49]. The available evidence indicates that succinate plays its role as a proinflammatory effector through several pathways, including preventing hypoxia‐inducible factor‐1α (HIF‐1α) degradation [50] and binding to succinate receptor 1 (SUCNR1). SUCNR1 is widely expressed in different types of tissues, and high expression of SUCNR1 is found in DCs [51] and adipose tissue as well as in the liver, kidney, pancreas, and intestine [52]. The published evidence indicates that succinate, by acting as a chemokine, stimulates SUCNR1 to promote macrophage and DC migration [51]. In addition, extracellular succinate interacts with Toll‐like receptor (TLR) ligands to induce the expression of tumor necrosis factor‐α (TNF‐α) and IL‐1β, which activates the antigen presentation function of DCs, and this activation results in further activation of T cells and increases IFN‐γ production [51]. Both the autocrine and paracrine secretion of succinate induce IL‐1β secretion from macrophages [53]. LPS stimulation upregulates both extracellular SUCNR1 and intracellular succinate expression in macrophages, and succinate is then released from the cells, binds to SUCNR1 on the surface of surrounding cells, and exacerbates the production of IL‐1β, which in turn further induces the expression of SUCNR1 to generate a positive feedback loop [53]. Together these data demonstrate that succinate plays an antitumor role in M1 macrophages, DCs, and Teff cells. However, succinate is a known cancer promoter that stabilizes HIF‐1α [50] and inhibits 2‐oxo‐glutarate‐dependent histone and DNA demethylase [54]. Therefore, the current antitumor therapeutics targeting succinate mainly inhibit its accumulation [55].
Itaconate has emerged as an anti‐inflammatory metabolite [56, 57, 58]. The available evidence showed that bone marrow‐derived macrophages (BMDMs) pretreated with dimethyl itaconate (DI), a cell‐permeable methyl ester derivative of itaconate, exhibit potent inhibition of proinflammatory mediators, including reactive oxygen species (ROS), nitric oxide (NO), and the cytokines IL‐1β, IL‐12p70 and IL‐6 [56]. In addition, a research showed that itaconate played an anti‐inflammatory role by competitively inhibiting the succinate dehydrogenase‐mediated oxidation of succinate [56]. The knockout of immunoresponsive gene 1 (IRG1), which leads to itaconate synthesis inhibition, induces BMDMs to produce more NO, IL‐6, IL‐12, and HIF‐1α in response to LPS stimulation. Moreover, IL‐18 and IL‐1β expression in IRG1–/‐ BMDMs is increased under conditions of NLR family pyrin domain containing 3 (NLRP3) inflammasome activation [56]. Additional evidence also suggested that itaconate might suppress inflammation by alkylating the key redox‐sensing protein Kelch‐like ECH‐associated protein 1 (KEAP1) and thereby the anti‐inflammatory and antioxidant transcription factor nuclear factor erythroid 2 like 2 (Nrf2) [58]. Another potential mechanism of the itaconate‐induced activation of Nrf2 was revealed by a study of peritoneal tumors. The production of mitochondrial reactive oxygen species (mtROS) and OXPHOS in tumor‐infiltrating macrophages and the subsequent activation of mitogen‐activated protein kinase (MAPK) are enhanced by itaconate, which is the main metabolic feature of M2 macrophage polarization [58]. Moreover, ROS induced by itaconate participates in the activation of Nrf2 [59]. Accordingly, Nrf2 might play a key role in the anti‐inflammatory effect of itaconate. Given the anti‐inflammatory effect of itaconate, this molecule is an attractive target for cancer therapy, and the available evidence showed that the downregulation of IRG1 inhibited the progression of glioma [60] and peritoneal tumors [59]. Further research is needed to develop drugs that degrade or inhibit itaconate for tumor treatment.
Fumarate has been found to be an inflammatory signal in innate immune training [61, 62]. First, the available evidence showed that after restimulation with LPS, the accumulation of fumarate in β‐glucan‐treated monocytes upregulates the secretion of cytokines, which is important for trained immunity [62]. Second, elevated fumarate levels are one of the signs of glutamine anaplerosis, which means that the shunting of glutamine into the TCA cycle is increased. Published evidence showed that the glutaminase inhibitor bis‐2‐(5‐phenylacetamido‐1,2,4‐thiadiazol‐2‐yl)ethyl sulfide (BPTES) significantly reduced the concentrations of fumarate in the immune response induced by β‐glucan [63]. Third, fumarate is an epigenetic regulator that inhibits α‐ketoglutarate‐dependent dioxygenases [62]. Lysine demethylase 5 histone demethylase can be inhibited by cell‐permeable fumaric acid ester monomethyl fumarate (MMF) to promote histone methylation, which in turn increases the levels of the TNF and IL‐6 promoter H3K4me3. The evidence showed that the secretion of TNF‐α and IL‐6 from macrophages after β‐glucan training and LPS stimulation was further increased by MMF treatment [62]. These data establish a link between the TCA cycle and epigenetic regulation of the immune response [62]. In addition to trained immunity, the levels of fumarate are increased in LPS‐stimulated macrophages [49]. Induction of an inflammatory argininosuccinate shunt, which establishes a link between the TCA and urea cycles, has been revealed by an integrated high‐throughput transcriptional metabolic profiling and analysis pipeline (CoMBI‐T) [49]. The key to this pathway is the upregulation of argininosuccinate synthase 1 (ASS1), which catalyzes the synthesis of argininosuccinate from citrulline, aspartate, and adenosine triphosphate (ATP) [64]. Combined with the fact that argininosuccinate lyase (ASL) catalyzes the cleavage of fumarate and arginine from argininosuccinate, this pathway might explain the accumulation of fumarate in macrophages. Fumarate also contributes to the regulation of T cell function through the electrophilic properties of its derivatives. Although MMF cannot suppress the activation of T cells, dimethyl fumarate (DMF) is an effective potent electrophile for relapsing‐remitting multiple sclerosis (MS) and psoriasis [65]. Proteomics has revealed several proteins sensitive to the covalent modification of DMF that are involved in influencing T cell function, include protein kinase C θ (PKCθ) [65]. The mechanism underlying the PKCθ‐CD28 interaction is impaired by DMF, and this impairment inhibits T cell activation.
2‐HG is involved in tumor progression, as shown by many lines of evidence [66, 67, 68, 69]. Mutations in isocitrate dehydrogenase 1 (IDH1) and IDH2 have been widely observed in acute myeloid leukaemia (AML) and glioma, and most of these mutations have a common mechanism of pathogenesis [66, 67], which involves alteration of the enzymatic activity to convert α‐ketoglutaric acid (α‐KG) into 2‐HG by acting on several unique positions in the active site of the enzyme [70, 71]. 2‐HG has two enantiomers, namely, D‐2‐HG (also known as R‐2‐HG) and L‐2‐HG (also known as S‐2‐HG). D‐2‐HG is mainly produced by IDH1 and IDH2 and their mutants, whereas L‐2‐HG is mainly synthesized by LDHA or malate dehydrogenase under acidic or hypoxic conditions [72]. 2‐HG is involved in the regulation of T cells. Different metabolic remodelling processes occur during the activation of T cells to support proliferation, differentiation, and function [73]. For example, the conversion of OXPHOS to glycolysis, which is induced by HIF‐1α and retinoid‐related orphan receptor‐γt (RORγt), is necessary for the differentiation of naive CD4+ T cells into Th17 cells but is not needed for differentiation into induced Treg (iTreg) cells [74, 75]. Importantly, compared with iTreg cells, Th17 cells have increased D‐2‐HG levels and are associated with increased DNA methylation levels at the forkhead box protein P3 (FOXP3) locus, which inhibits iTreg differentiation [74]. In addition, exogenous d‐2‐HG or the silencing of ten‐eleven translocation 1 (TET1) and TET2, which regulate FOXP3 expression redundantly by demethylating the FOXP3 promoter and its intronic CpG island [76], induces the differentiation of naive CD4+ T cells into Th17 cells. Furthermore, TET1 and TET2 can be inhibited by 2‐HG [74, 76]. Mechanistically, aminooxyacetic acid can inhibit glutamic‐oxaloacetic transaminase 1, which can convert glutamic acid into α‐KG and promote the production of D‐2‐HG, and these effects reduce D‐2‐HG content and induce Th17 cell differentiation into iTreg cells [74], which indicates that D‐2‐HG acts as an epigenetic modifier to control T cell differentiation. In addition to D‐2‐HG, L‐2‐HG promotes the CD8+ T cell‐induced elimination of tumors [77]. Similarly, CD8+ T cells rely on HIF‐1α and glycolysis to function in hypoxic inflammatory or tumor environments [78]. The available evidence showed that the L‐2‐HG levels in CD8+ T cells increased through a HIF‐1α‐dependent pathway after T cell receptor (TCR) stimulation under physiological oxygen conditions [77]. Mechanistically, LDHA was induced by stabilized HIF‐1α and converts α‐KG into L‐2‐HG. L‐2‐HG further stabilized HIF‐1α and regulates the methylation of histones and DNA to alter CD8+ T cell differentiation [77]. Moreover, the proliferation, persistence, and antitumor capacity of adoptively transferred CD8+ T lymphocytes were all enhanced by L‐2‐HG treatment [77]. These data indicate that although 2‐HG is a protumor metabolite produced by tumor metabolic reprogramming, it can also promote the antitumor immune function of T cells through HIF‐1α and the epigenetic pathway in the TME.
Acetyl‐CoA is also a signalling molecule among metabolites involved in the TCA cycle. Acetyl‐CoA is critical for histone acetylation because it is the main provider of acetyl groups in histone acetylation, which is involved in tumor progression and immunity. Thus, acetyl‐CoA is involved in immune regulation. As mentioned above, glycolysis is needed for the function of Teff cells, and studies have further elaborated the mechanism and role of acetyl‐CoA in this required pathway [18]. In activated T cells, induced LDHA upregulated the acetyl‐CoA flux, which promoted histone acetylation at the IFN‐γ loci and thereby increased the expression of IFN‐γ. Consistently, the lack of LDHA in T cells limited the production of IFN‐γ and the progression of autoimmune diseases in mouse models, which ultimately prevents the death of the mice [18]. Mechanistically, LDHA deficiency in T cells leads to a decrease in the levels of acetyl‐coenzyme, and this effect is accompanied by a decrease in histone H3 acetylation at the lysine 9 residue (H3K9Ac) on the IFN‐γ promoter. Moreover, the expression of IFN‐γ is reduced by inhibiting ATP citrate lyase (ACLY), which converts citric acid into acetyl‐CoA, and then restores the raw material for the ACL‐independent synthesis pathway of acetyl‐CoA under conditions of acetate supplementation [18]. In addition, ACLY is also involved in T cell growth through histone acetylation [79]. A nuclear phosphoproteomic study demonstrated that IL‐2 enhanced histone acetylation and induced cell cycle regulation genes by phosphorylating ACLY. The genetic ablation or inhibition of ACLY also reduced the promotion of T cell growth by IL‐2 [79]. In addition, the outcome of chemotherapy depends on Treg depletion and can be improved by hydroxycitrate, an inhibitor of ACLY [80]. Taken together, the above‐described findings show that acetyl‐CoA promotes T cell activation by enhancing acetylation, but the mechanism and results of its actions in other immune cells need to be further characterized.
Although succinate, fumarate, 2‐HG, and acetyl‐CoA exert potential proinflammatory effects in the TME, it remains difficult to translate these effects to tumor treatment. Based on metabolic similarity, these metabolites also have the effect of promoting tumor progression [55, 66, 67, 81, 82], and further studies are needed to realize the full potential of these metabolites.
2.2. Lipid metabolism
Upregulated de novo fatty acid synthesis is a metabolic characteristic of neoplastic cells. This process utilizes the energy generated by glycolysis and other catabolism pathways to produce signalling molecules and plasma membrane phospholipids [83]. First, cancer‐associated adipocytes [38] and cancer‐associated fibroblasts [84] have been found to promote tumor progression and lipid accumulation in the TME. The available evidence showed that CD8+ TILs with high programmed death‐1 (PD‐1) expression levels presented both increased lipid intake and a higher lipid content and recognized tumor cells more efficiently [85]. Together, this evidence indicated the crucial role of lipid metabolism in the regulation of the tumor immune microenvironment and the feasibility of targeting lipid metabolism to regulate tumor immunity. To date, a variety of fatty acid and cholesterol metabolism inhibitors have been developed to suppress autoimmunity [86], and enhancing these metabolic pathways might also help improve antitumor immunity. The lipid composition of cell membranes is essential for TCR clustering and T cell synapses, which are involved in the activation of T cells [87]. Avasimibe, a sterol O‐acyltransferase 1 inhibitor, increases the proportion of cholesterol in CD8+ TIL plasma membranes by disrupting cholesterol esterification and thereby improves Teff cell function and promotes their proliferation. In addition, avasimibe significantly improves the efficacy of anti‐PD‐1 therapy in inhibiting tumor progression and prolonging survival [88]. Similarly, drugs that increase fatty acid oxidation by activating peroxisome proliferator activated receptor (PPAR) signalling also improve the efficacy of melanoma immunotherapy [89]. Moreover, the differentiation and proliferation of different subtypes of CD4+ T cells is promoted by different fatty acids. The differentiation of proinflammatory Th1 cells and Th17 cells is promoted by the long‐chain fatty acid lauric acid [90], while the development of Treg cells is supported by short‐chain fatty acids [91]. Therefore, both the content and species of lipids in the TME should be considered when targeting lipid metabolism to regulate T cells. In addition to T cells, tumor‐infiltrating myeloid cells, including tumor‐associated macrophages (TAMs) [92], DCs [93], and myeloid‐derived suppressor cells (MDSCs) [94], can take up exogenous lipids and exhibit intracellular lipid accumulation, leading to immune suppression or differentiation into anti‐inflammatory subtypes. A difference in lipid metabolism has been detected between M1 and M2 macrophages: fatty acid synthesis is enhanced in M1 macrophages, whereas M2 macrophages exhibit enhanced fatty acid oxidation [95]. The published evidence showed that the silencing of mitogen‐activated protein kinase kinase kinase 8 (MAP3K8) inhibited lipid catabolism and M2 macrophage polarization [96]. In addition, the antiviral immunity of macrophages was promoted by limiting the flux of cholesterol synthesis to induce a type I interferon response [97]. However, some lines of evidence also showed that macrophages could be promoted to switch from an M2 to M1 phenotype to enhance antitumor activity by inhibiting ATP binding cassette transporter G1 (ABCG1), which mediates cholesterol efflux [98]. Therefore, how cholesterol metabolism can be targeted to regulate antitumor activity in macrophages remains to be further studied.
2.3. Amino acid metabolism
2.3.1. Glutamine and glutamate
Increased glutamine intake and the decomposition of glutamine into glutamate, which depend on expression of the glutamine transporter alanine‐serine‐cysteine transporter 2 and glutaminase 1 (GLS1), respectively, are also metabolic characteristics of tumors [99]. Similarly, glutamine metabolism is upregulated in activated T cells and macrophages to support cell differentiation and function [100]. The removal of glutamine causes T cell proliferation arrest and prevents the secretion of IL‐2 and IFN‐γ from activated T cells [100]. Additional evidence showed that glutamine restriction inhibited Th1 cell differentiation and promotes Treg cell development [101]. However, some evidence also showed that CD8+ T cells cultured under glutamine restriction conditions were more effective in eliminating tumors in vivo [102], and GLS1 knockout reduced initial T cell activation and proliferation, impairing differentiation of Th17 cells but promoting differentiation and effector functions of CD4+ Th1 and CD8+ cytotoxic T lymphocytes (CTLs) [103]. Collectively, these lines of evidence indicate that the differential effects of glutamine metabolism on different states and subtypes of T cells need to be considered when targeting glutamine to regulate immune function in the TME. Currently, treatments targeting glutamine are being tested in a clinical trial. Specifically, CB‐839, an allosteric inhibitor of GLS1, can be used alone or in combination with PD‐1 inhibitors to treat solid [104, 105] or hematological malignancies [106, 107, 108]. Mechanistically, the synergistic effect of CB‐839 and PD‐1 inhibitors might be partially attributed to the enhancement of CTL‐mediated antitumor responses [103]. However, there are currently no data to indicate whether or how CB‐839 inhibits tumors by affecting the immune function of the TME in patients. In addition, the glutamate levels are also involved in the regulation of T cells. Glutamate receptors are upregulated in CD4+ and CD8+ T cells in response to TCR activation and promote IFN‐γ secretion [109]. However, excessive extracellular glutamate concentrations inhibit T cell activation, and this effect is partly attributed to the inhibition of potassium currents [110]. Accordingly, the role of glutamine and glutamate metabolism in regulating immune responses in the TME needs to be comprehensively considered (Figure 3).
FIGURE 3.
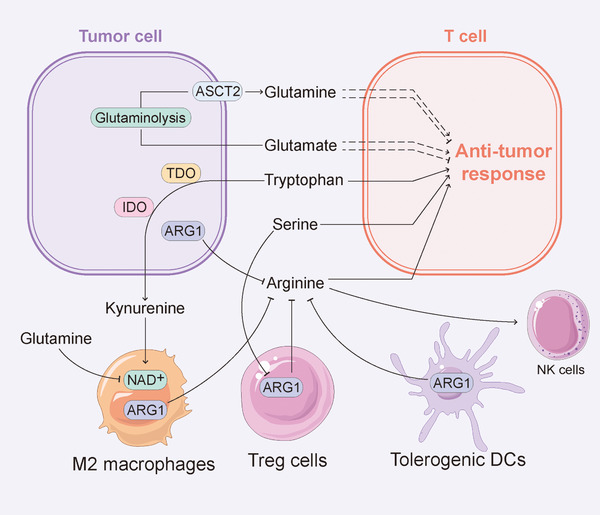
Effect of amino acid metabolism on infiltrating lymphocyte function. Amino acid metabolism plays a critical role in the regulation of immune response. Glutamine and glutamate metabolisms have different effects on the antitumor response of T cells under different states and of different subtypes. The removal of glutamine causes T cell proliferation arrest, while CD8+ T cells cultured under glutamine restriction conditions are more effective in eliminating tumor cells. Arginine is necessary for the activation of Teff cells. Thus, immune regulatory cells and tumor cells can degrade arginine by expressing ARG1 to limit the availability of arginine to T cells, thereby inhibiting antitumor immune activity. Tryptophan deprivation impairs the cell cycle and differentiation of T cells, and TDO and IDO, the catabolic enzymes of tryptophan, are expressed in tumor cells and various stromal cells in the TME to promote tumor progression. Serine is crucial for Teff cell response and reduces the immunomodulatory capacity of Treg cells. Abbreviations: ARG1: arginase 1; ASCT2: alanine‐serine‐cysteine transporter 2; DC: dendritic cell; IDO: indoleamine 2,3‐dioxygenase; NAD: nicotinamide adenine dinucleotide; Teff: effector T; Treg: regulatory T; TDO: tryptophan 2,3‐dioxygenase; TME: tumor microenvironment
2.3.2. Arginine
Arginine metabolism is involved in immune responses. Activated arginine catabolism through arginase 1 (ARG1) is one of the metabolic markers of the immune regulation response [111]. Immune regulatory cells, including Treg cells, tolerogenic DCs, and M2 macrophages, can degrade arginine by expressing ARG1 to limit the availability of arginine to T cells, which results in the inhibition of antitumor immune activity [112]. Arginine restriction can arrest T cells at the G0‐G1 phase by inhibiting the expression of cyclin‐dependent kinase 4 (CDK4) and cyclin D3 [113]. Similarly, L‐arginine depletion contributes to downregulation of the TCR z chain, a critical signalling element of the TCR, and thereby inhibits T cell activation [114]. It has been revealed that adaptive immunity could be suppressed by increased arginine breakdown, which was associated with enhanced Treg cell function and restricted Teff cell function [115]. Specifically, MDSCs promoted tumor immune escape by expressing ARG1 to stabilize Treg cells and inhibit CD8+ Teff cells [116, 117]. Consistently, the published evidence showed that supplementation with arginine promoted the production of cytokines in NK cells and T cells and synergized with anti‐programmed cell death‐ligand 1 (PD‐L1) antibodies to enhance antiimmune cell and antitumor activity [118]. In addition, the differentiation of T cells into central memory‐like T cells, a type of T cell with long‐term memory and stronger antitumor activity, was promoted by arginine supplementation [119], and this promotion maintained the concentration of arginine in the TME by supplementing or preventing degradation, which might represent an effective strategy to activate antitumor immunity. Specifically, INCB001158, an ARG1 inhibitor, and ADI‐PEG 20, a pegylated arginine deiminase, have been applied in clinical trials in combination with pembrolizumab, an immune checkpoint inhibitor (ICI) (Table 1), and the evidence showed that the tumor infiltration and cytokine secretion of CD8+ Teff cells and NK cells was promoted by INCB001158 in vivo [120] and that ADI‐PEG 20 inhibited arginine auxotrophic tumors, including small‐cell lung cancer [121] and breast cancer [122] by reducing the arginine levels. Notably, the evidence showed that ADI‐PEG 20 treatment activated T cells, induced T cell infiltration into tumors, modulated T cell exhaustion, and reduced Treg cell accumulation [123]. Thus, the specific mechanisms of treatments that target arginine metabolism need to be further explored prior to its application.
TABLE 1.
Clinical trials of INCB001158 and ADI‐PEG 20 in cancer therapy
| ClinicalTrials.gov Identifier | Intervention/treatment | Condition or disease | Phase |
|---|---|---|---|
| NCT03837509 |
INCB001158 (ARG1 inhibitor) Daratumumab SC (CD38 inhibitor) |
Relapsed or Refractory Multiple Myeloma | I/II |
| NCT03910530 |
Retifanlimab (PD‐1 inhibitor) INCB001158 (ARG1 inhibitor) |
Advanced Solid Tumors Metastatic Solid Tumors |
I |
| NCT03361228 |
INCB001158 (ARG1 inhibitor) Epacadostat (IDO1 inhibitor) Pembrolizumab (PD‐1 inhibitor) |
Solid Tumors | I/II |
| NCT02903914 |
INCB001158 (ARG1 inhibitor) Pembrolizumab (PD‐1 inhibitor) |
Metastatic Cancer Solid Tumors Colorectal Cancer Gastric Cancer Head and Neck Cancer Lung Cancer Renal Cell Carcinoma Bladder Cancer Urothelial Cancer Mesothelioma |
I/II |
| NCT03498222 |
Atezolizumab (PD‐L1 inhibitor) Pemetrexed Carboplatin ADI PEG20 (pegylated arginine deiminase) |
Carcinoma Non‐Small‐Cell Lung Cancer |
I |
| NCT03922880 |
ADI PEG20 (pegylated arginine deiminase) Nivolumab (PD‐1 inhibitor) Ipilimumab (CTLA‐4 inhibitor) |
Uveal Melanoma | I |
Abbreviations: ARG1: arginase 1; CTLA‐4: cytotoxic T lymphocyte‐associated antigen‐4; PD‐1: programmed death 1; PD‐L1: programmed cell death‐ligand 1.
2.3.3. Tryptophan
Tryptophan is a common hallmark of the immunological TME at the clinical presentation of cancer. First, tryptophan deprivation impairs the cell cycle and the differentiation of T cells. Recent evidence showed that tryptophan deprivation induced the arrest of T cells at the mid‐G1 phase and makes these cells more prone to the induction of apoptosis through Fas‐induced and other signalling pathways [124]. Additionally, tryptophan 2,3‐dioxygenase (TDO) and indoleamine 2,3‐dioxygenase (IDO), the catabolic enzymes of tryptophan, are expressed in tumor cells and various stromal cells in the TME [125]. These enzymes were reported to promote tumor progression and are associated with shorter survival [111]. TDO and IDO reduce the levels of tryptophan in the TME to inhibit the activity of CTLs [126], and the catabolite kynurenine further promotes differentiation of Treg cells and inhibits Teff cells [127], which results in the induction of tumor immune escape. Moreover, kynurenine and its downstream catabolite kynurenic acid (KA) are natural aryl hydrocarbon receptor (AHR) agonists. AHR signalling regulates immunity and will be detailed in the following paragraphs. Furthermore, kynurenine production promotes NAD synthesis and mitochondrial OXPHOS in macrophages to activate phagocytosis and anti‐inflammatory effects. Furthermore, the inhibition of IDO suppresses the function of M2 macrophages [128]. In addition, a series of lines of evidence have revealed the inhibitory effect of IDO on antitumor immunity in vivo. Compared with treatment consisting of antigen‐loaded DCs without IDO silencing, the introduction of antigen‐loaded DCs with IDO silencing into breast tumor‐bearing mice enhanced tumor antigen‐specific T cell proliferation and CTL activity, which reduced the number of Treg cells [129]. IDO inhibitors suppressed metastatic liver tumors and bladder tumors in mouse models by promoting tumor infiltration of neutrophils and T cells as well as the secretion of IL‐12 and IFN‐γ [130]. IDO inhibitors inhibited the recruitment of MDSCs by reducing the number of Treg cells and diminishing their immunomodulatory effects in mouse models and humans with melanoma [131]. A series of IDO inhibitors and TDO inhibitors have been developed and used in different clinical trials (Table 2), and these include indoximod (IDO1 and IDO2 inhibitor), navoximod (IDO1 inhibitor), HTI‐1090 and DN1406131 (IDO1/TDO dual inhibitor). Specifically, indoximod has been combined with the anti‐PD‐1 antibody nivolumab (NCT02073123) or pembrolizumab (NCT03301636) or the anti‐cytotoxic T lymphocyte‐associated antigen‐4 (CTLA‐4) antibody ipilimumab (NCT02073123) in clinical studies to treat melanoma. The results of a phase Ib study (NCT02073123) showed that indoximod combined with ipilimumab exhibits no dose‐limiting toxicity [132]. In addition, indoximod has been combined with therapeutic anticancer vaccines across several tumor types (NCT02460367, NCT01302821, NCT01042535 and NCT01560923). Navoximod (NCT02471846) and HTI‐1090 (NCT03491631) have been combined with the anti‐PD‐L1 antibodies atezolizumab and camrelizumab, respectively. Furthermore, phase I–III clinical trials of epacadostat (an IDO1 selective inhibitor) in combination with ICIs are currently in progress [133]. However, compared with pembrolizumab plus placebo, epacadostat combined with pembrolizumab did not improve progression‐free survival and is unlikely to improve overall survival in patients with unresectable or metastatic melanoma in the phase III ECHO‐301/KEYNOTE‐252 trial [134]. Because upregulation of IDO and TDO expression is induced by chronic inflammation in the TME and is involved in immunosuppression, the immune status in the TME might partially determine the therapeutic effect of IDO or TDO inhibitors combined with ICIs. Therefore, evaluating the expression of IDO and TDO in tumors and the inflammatory state of the TME before treatment might serve as a strategy for improving the efficacy of targeted tryptophan metabolism.
TABLE 2.
Clinical trials testing IDO inhibitors and TDO inhibitors in cancer therapy
| ClinicalTrials.gov Identifier | Intervention/treatment | Condition or disease | Phase |
|---|---|---|---|
| NCT02073123 |
Indoximod (IDO inhibitor) Ipilimumab (CTLA‐4 inhibitor) Nivolumab (PD‐1 inhibitor) Pembrolizumab (PD‐1 inhibitor) |
Metastatic Melanoma Stage III Melanoma Stage IV Melanoma | I/II |
| NCT03301636 |
Pembrolizumab (PD‐1 inhibitor) Nivolumab (PD‐1 inhibitor) Indoximod (IDO inhibitor) |
Melanoma | II/III |
| NCT02460367 |
Docetaxel Tergenpumatucel‐L (Lung cancer vaccine) Indoximod (IDO inhibitor) |
Non‐small Cell Lung Cancer Progression of Non‐small Cell Lung Cancer Recurrent Non‐small Cell Lung Cancer |
I |
| NCT01302821 |
Adenovirus‐p53 transduced dendritic cell vaccine 1‐Methyl‐D‐tryptophan (IDO inhibitor) Carbon C 11 alpha‐methyltryptophan |
Breast Cancer | Not applicable |
| NCT01042535 |
Adenovirus‐p53 transduced dendritic cell vaccine 1‐Methyl‐D‐tryptophan (IDO inhibitor) Laboratory biomarker analysis |
Male Breast Cancer Recurrent Breast Cancer Stage IV Breast Cancer Unspecified Adult Solid Tumor, Protocol Specific |
I/II |
| NCT01560923 |
Indoximod (IDO inhibitor) Sipuleucel‐T (Prostate cancer vaccine) |
Metastatic Prostate Cancer | II |
| NCT02471846 |
Atezolizumab (PD‐L1 inhibitor) GDC‐0919 (IDO inhibitor) |
Solid Tumor | I |
| NCT03491631 |
SHR9146 (IDO/TDO inhibitor) SHR‐1210 (PD‐1 inhibitor) Apatinib (tyrosine kinase inhibitor) |
Solid Tumor, Metastatic Cancer, Malignant Neoplasm |
I |
| NCT02752074 |
Pembrolizumab (PD‐1 inhibitor) Epacadostat (IDO inhibitor) |
Melanoma | III |
Abbreviations: CTLA‐4: cytotoxic T lymphocyte‐associated antigen‐4; IDO: indoleamine 2,3‐dioxygenase; TDO: tryptophan 2,3‐dioxygenase; PD‐1: programmed death 1; PD‐L1: programmed cell death‐ligand 1.
2.3.4. Serine
Serine, a crucial non‐essential amino acid for cell growth and survival [135], participates in the synthesis of cell membranes, muscle, and nerve sheaths. As a precursor of many essential biomacromolecules such as phospholipids, ATPs, and nucleic acids, serine is reportedly needed for rapidly propagating cells [136, 137]. Furthermore, it has been found that the pathways of serine uptake and synthesis are usually upregulated in cancers, which indicates the dependence of tumor growth on serine. Similar to tumor cells, proliferating immune cells in vitro and in vivo are highly dependent on the serine supply [138]. Some key regulators that can mediate serine synthesis and metabolism also affect the growth, proliferation, and differentiation of immune cells [138, 139, 140]. Thus, both tumor cells and immune cells need an abundant supply of serine, which implies that immune cells compete with tumor cells for limited serine resources during the antitumor immune response. Rapidly proliferating Teff cells need a serine supply both in vitro and in vivo [138]. In response to stimulation by tumor‐specific antigens, primordial T cells transform into an active state characterized by rapid proliferation to generate a Teff cell pool that can mediate antitumor immunity. The activation of Teff cells requires the heavy consumption of various nutrient factors, including serine, to support their proliferative demands and exert their effector functions. Similarly, dietary serine restriction can limit T cell proliferation in vivo [138]. In addition, glutathione (GSH) is composed of cysteine, glutamine, and glycine and is synthesized by glutathione synthase and glutamate cysteine ligase (GCL). It has been found that GSH synthesis could be activated by serine, and the synthesized GSH then entered the one‐carbon metabolic network, which is crucial for the Teff cell response [141]. GCL consists of two subunits, Gclc and Gclm. Gclc deficiency in Treg cells increased the levels of serine metabolism in Treg cells with FOXP3 downregulation, which led to activated autoimmunity and antitumor immunity in mice, and the restriction of cellular serine in Gclc‐deficient Tregs restored FOXP3 expression and was crucial for maintaining the immunomodulatory capacity of Tregs both in vitro and in vivo [142]. This finding suggests that the uptake and synthesis of serine are affected by the disruption of GSH synthesis, and this feedback loop is crucial to the immunomodulatory capacity of Treg cells. In addition to T cells, serine availability is also vital to macrophages. Both NADPH produced by the pentose phosphate pathway [23] and the maintenance of intracellular reduced glutathione pools [143] are needed by M1 macrophages to neutralize ROS generated during respiratory bursts. Because serine metabolism is involved in the maintenance of NADPH and glutathione, tumor cells can induce excessive accumulation of ROS in M1 macrophages in the TME to toxic levels through the excessive consumption of serine during the chronic antitumor response [143]. Therefore, for tumor cells and activated immune cells, it is necessary to mobilize pathways of serine acquisition to meet their demand for serine, which is the theoretical basis for intervening in antitumor immunity from the perspective of serine metabolism.
2.4. ATP and adenosine signalling
ATP, the energy currency of cells, and its derivatives serve as biochemical constituents involved in the function of immunomodulation in the TME [144, 145]. Intracellular nucleotides are released into the extracellular space to activate immune responses when cells undergo apoptosis or stimulation, including chemical stress, mechanical damage, hypoxia, and cytotoxic agents [146]. ATP is released through a variety of methods, including pannexin channels [147], connexin hemichannels [148], exocytosis [149], and transmembrane transport via ATP‐transporters specific for the ATP‐binding cassette family, such as the cystic fibrosis transmembrane conductance regulator [150]. Furthermore, the available evidence showed that compared with autophagy‐competent tumor cells, autophagy‐deficient tumor cells released less ATP after treatment with antitumor chemotherapy, which suggested that autophagy was crucial for antitumor chemotherapy‐induced ATP release [151].
ATP binds to type‐2 purinergic receptor (P2R) family receptors, including ionotropic P2XR and metabotropic P2YR purinergic receptors [152]. ATP that originates from dying cells promotes chemotaxis and the recruitment of antigen‐presenting cells (APCs) by ligating purinergic receptors of the P2RY2 and P2RX7 types [152], whereas ATP that originates from immunogenic cell death released via the autophagy‐dependent pathway interacts with P2RX7 in synergy with its metabolites to recruit and activate DCs [153, 154]. The activation of purinergic signals further activates the NLRP3 inflammasome, resulting in the proteolytic activation of caspase‐1 and promoting the processing and immunostimulatory release of IL‐1β, which leads to the activation of IL‐17‐producing γδ T cells [155].
In the extracellular space, ATP is gradually decomposed by the nucleotidases CD39 and CD73 into adenosine, which exerts immunosuppressive effects [156]. Adenosine activates the protein kinase A signalling cascade by ligating four G protein‐coupled purinergic type 1 receptors, namely A1R, A2AR, A2BR, and A3R, to enhance the activity of adenylyl cyclase‐mediated production of cAMP. The levels of extracellular nucleotides can be regulated by hypoxia. Specifically, the expression of CD39 and CD73 can be elevated by hypoxia through a HIF‐1‐dependent pathway [157]. Hypoxia also inhibits the expression of equilibrative nucleoside transporters (ENTs), which reduces the uptake of adenosine and thereby increases the level of extracellular adenosine [158]. High levels of CD39 and CD73 are associated with poor prognosis in many human malignancies [159, 160], whereas the ecto‐nucleoside triphosphate diphosphohydrolase inhibitor polyoxometalate‐1 inhibits tumor growth [161]. In addition to tumor cells, regulatory immune Treg cells [162] and M2 [163] macrophages express CD39 and CD73. Treg cells and M2 macrophages degrade ATP and produce adenosine via CD39 and CD73 to limit the proinflammatory effects of ATP and further lead to immunosuppression through the adenosine‐A2AR or adenosine‐A2BR signalling axis [162, 163]. In addition, published evidence showed that Treg cells inhibited the antitumor activity of NK cells to promote the metastasis of melanoma in a CD39‐dependent pathway in mice [161]. Notably, the activities of A2AR and A2BR effectively promote immunosuppression in inflammatory diseases [164]. It has been found that the activation of the adenosine‐A2AR signalling axis promoted the expression of PD‐1 [165] and inhibited the expression of the IL‐2 receptor and proliferation after TCR stimulation of tumor‐infiltrating T cells [166], which thereby inhibited antitumor immune function. Moreover, activation of the adenosine‐A2AR signalling axis promoted the recruitment of MDSCs and the expression of vascular endothelial growth factor (VEGF) in tumors of mouse models [167], and these effects promoted tolerogenic DCs to express PD‐L2 and IL‐10 to inactivate Teff cells [168]. Consistently, PSB1115, an inhibitor of A2BR, inhibits tumor angiogenesis and promotes the accumulation of T cells in the TME [167].
Therefore, targeting ATP/adenosine metabolism is an effective strategy to relieve immunosuppression. Specifically, CD39 deletion has been found to lead to defects in angiogenesis and melanoma cell migration, which inhibits tumor progression in a mouse model [169]. Adenosine 5'‐(α,β‐methylene)diphosphate, a specific CD73 inhibitor, prevented breast cancer cell migration by affecting adenosine production[170]. Moreover, some lines of evidence indicated that both CD73 inhibitors [165] and A2AR antagonists [171] synergized with ICIs to inhibit tumor progression in mouse models. Drugs targeting the adenosine signalling pathway combined with ICIs in cancer patients are currently in clinical trials. CPI‐144, an A2AR inhibitor, has been demonstrated to be effective in controlling refractory renal cell carcinoma (RCC; disease control rate of 60%) and can further significantly improve the control effect when combined with atezolizumab (anti‐PD‐L1 antibody; disease control rate of 100%) in a phase I/Ib trial (NCT02655822) [172]. A list of clinical trials testing CD39 and CD73 inhibitors are shown in Table 3.
TABLE 3.
Clinical trials testing CD39 inhibitors and CD73 inhibitors in cancer therapy
| ClinicalTrials.gov Identifier | Intervention/treatment | Condition or disease | Phase |
|---|---|---|---|
| NCT04262375 |
Durvalumab (PD‐L1 inhibitor) Oleclumab (CD73 inhibitor) |
Non‐small Cell Lung Cancer Renal Cell Carcinoma |
II |
| NCT03773666 |
Durvalumab (PD‐L1 inhibitor) Oleclumab (CD73 inhibitor) |
Muscle Invasive Bladder Cancer | I |
| NCT03616886 |
Paclitaxel Carboplatin MEDI4736 (PD‐L1 inhibitor) MEDI9447 (CD73 inhibitor) |
Triple Negative Breast Cancer | I/II |
| NCT03822351 |
Durvalumab (PD‐L1 inhibitor) Oleclumab (CD73 inhibitor) Monalizumab (NKG2A inhibitor) |
Stage III Non‐small Cell Lung Cancer Unresectable |
II |
| NCT03884556 |
TTX‐030 (CD39 inhibitor) Pembrolizumab (PD‐1 inhibitor) Docetaxel Gemcitabine Nab‐paclitaxel |
Solid Tumor Lymphoma |
I |
| NCT03381274 |
MEDI9447 (CD73 inhibitor) Osimertinib (EGFR inhibitor) AZD4635 (A2AR inhibitor) |
Non‐small Cell Lung Cancer | I/II |
| NCT04089553 |
AZD4635 (A2AR inhibitor) Oleclumab (CD73 inhibitor) Durvalumab (PD‐L1 inhibitor) |
Prostate Cancer Metastatic Castration‐Resistant Prostate Cancer |
II |
| NCT04261075 |
IPH5201 (CD39 inhibitor) Durvalumab (PD‐L1 inhibitor) Oleclumab (CD73 inhibitor) |
Advanced Solid Tumors | I |
Abbreviations: A2AR: A2a receptor; EGFR: epidermal growth factor receptor; NKG2A: NK group 2 member A; PD‐1: programmed death 1; PD‐L1: programmed cell death‐ligand 1.
2.5. Hypoxia
Hypoxia is a common feature of the TME caused by rapid cancer cell proliferation and an abnormal tumor vasculature [173]. Hypoxia is sensed by the O2/prolyl hydroxylase/Von Hippel Lindau axis, which stabilizes and activates HIF‐1 and contributes to the immunosuppressive microenvironment [173]. HIF‐1 shifts metabolism from OXPHOS to aerobic glycolysis by activating the expression of glucose transporter, which is necessary for cells to increase their glucose uptake and glycolysis, and the expression of genes encoding glycolytic enzymes and lactate dehydrogenase [174]. Recent evidence showed that lncRNA calcium‐dependent kinase activation activated the nuclear factor kappa‐B (NF‐κB) pathway and induced tumor microenvironment remodeling under hypoxic tumor conditions [175, 176]. In addition, HIF‐1 increases extracellular adenosine production and accumulation by inducing CD39 and CD73 [157] and inhibiting ENT [158]. HIF‐1 also promotes tumor progression via other pathways, including cell immortalization, stem cell maintenance, genetic instability, and chemotherapy resistance, by directly or indirectly regulating the transcription of multiple target genes [177]. Although the stability of HIF‐1α enhances the function of cytotoxic T cells, T cell activation is inhibited under hypoxic conditions, and this effect is enhanced under glucose‐limited conditions [178]. Increasing lines of evidence show that glycolytic tumor mitochondrial metabolism plays a role in immunosuppression [179]. The available evidence showed that TILs in the TME derived from cancer cells with stronger OXPHOS exhibited reduced IFN‐γ secretion and cytolytic activity, whereas OXPHOS‐deficient tumors showed less hypoxia and more functional TIL activity. In addition, OXPHOS‐deficient tumors were more responsive to anti‐PD‐1 therapy, which highlighted the effect of OXPHOS‐mediated hypoxia on TIL function and ICI efficacy [180]. Therefore, hypoxia/HIF‐1 is a promising therapeutic target. The exposure of a mouse model to respiratory hyperoxia resulted in reduced hypoxia, increased T cell infiltration, and a stronger antitumor response in the tumor, which indicated that hypoxia played a crucial role in suppressing the antitumor immune response. Several HIF inhibitors have been found to have antitumor activities. The first class of drugs is cardiac glycosides, which inhibit HIF‐1α synthesis [181]. The second class of drugs is anthracycline chemotherapy drugs, including daunorubicin and doxorubicin, and these drugs impair the DNA‐binding activity of HIF‐1 [182]. The third class of drugs includes acriflavine, which blocks HIF‐dependent gene transcription by binding to the PAS‐B subregions of HIF‐1α and HIF‐2α [183]. However, the results of clinical trials with HIF‐1 inhibitors are not ideal [184] due in part to the complex signal network and overlapping mechanisms mediated by HIF‐1 in tumor cells and immune cells in the TME.
In addition, HIF‐1 and TME acidification drive the expression of VEGF to activate VEGF‐R, which triggers angiogenesis and vascular aberrations. Vascular aberrations in the TME lead to nutritional deficiency and damage the infiltration of lymphocytes into tumors, which leads to tumor immune escape [185]. In addition to inducing vascular malformations, VEGF is also directly involved in inhibiting antitumor immune responses [186]. VEGF inhibits the maturation of DCs, which leads to the inactivation of CTLs [187]. Some evidence showed that VEGF significantly reduced the cytotoxic activity of T cells extracted from peripheral blood samples, and anti‐VEGFR2 reversed the suppression of T cells caused by VEGF [188]. VEGF also promotes the immunosuppressive TME by inducing Tregs, TAMs, and MDSCs [189]. Consistently, the published evidence demonstrated that vessel normalization contributed to the efficacy of immunotherapy by promoting the delivery of ICIs and improving lymphocyte infiltration. This approach might provide an effective strategy for the treatment of tumors. Several clinical trials have been conducted for multiple tumor types and with different environments to evaluate the combination of anti‐PD‐L1, anti‐PD‐1, or anti‐CTLA‐4 and anti‐VEGF therapies (Table 4).
TABLE 4.
Clinical trials testing VEGF inhibitors in cancer therapy
| ClinicalTrials.gov Identifier | Intervention/treatment | Condition or disease | Phase |
|---|---|---|---|
| NCT00790010 |
Bevacizumab (VEGF inhibitor) Ipilimumab (CTLA‐4 inhibitor) |
Melanoma | I |
| NCT01633970 |
5‐FU Atezolizumab (PD‐L1 inhibitor) Bevacizumab (VEGF inhibitor) Carboplatin Leucovorin Nab‐paclitaxel Oxaliplatin Paclitaxel Pemetrexed |
Advanced Solid Tumors | I |
| NCT01984242 |
Atezolizumab (PD‐L1 inhibitor) Bevacizumab (VEGF inhibitor) Sunitinib (VEGFR inhibitor) |
Renal Cell Carcinoma | II |
| NCT02039674 |
Pembrolizumab (PD‐1 inhibitor) Paclitaxel Carboplatin Bevacizumab (VEGF inhibitor) Pemetrexed Ipilimumab (CTLA‐4 inhibitor) Erlotinib (EGFR inhibitor) Gefitinib (EGFR inhibitor) |
Non‐small Cell Lung Carcinoma | I/II |
| NCT02366143 |
Atezolizumab (PD‐L1 inhibitor) Bevacizumab (VEGF inhibitor) Carboplatin Paclitaxel |
Carcinoma Non‐Small‐Cell Lung Cancer |
III |
| NCT02420821 |
Atezolizumab (PD‐L1 inhibitor) Bevacizumab (VEGF inhibitor) Sunitinib (VEGFR inhibitor) |
Renal Cell Carcinoma | III |
| NCT03434379 |
Atezolizumab (PD‐L1 inhibitor) Bevacizumab (VEGF inhibitor) Sorafenib (VEGFR inhibitor) |
Carcinoma Hepatocellular |
III |
| NCT03596281 |
Pembrolizumab (PD‐1 inhibitor) Bevacizumab (VEGF inhibitor) Pegylated liposomal doxorubicin |
Ovarian Cancer | I |
| NCT03847428 |
Durvalumab (PD‐L1 inhibitor) Bevacizumab (VEGF inhibitor) |
Hepatocellular Carcinoma | III |
| NCT04091217 |
Atezolizumab (PD‐L1 inhibitor) Bevacizumab (VEGF inhibitor) |
Melanoma | II |
Abbreviations: CTLA‐4: cytotoxic T lymphocyte‐associated antigen‐4; EGFR: epidermal growth factor receptor; PD‐1: programmed death 1; PD‐L1: programmed cell death‐ligand 1; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor.
2.6. Ionic environment
Extracellular ions in the TME determine the immune response. Tumors often contain necrotic regions due to nutritional deficiencies. A recent study found a markedly higher concentration of potassium ions (K+) in tumor interstitial fluid compared with that in serum, which could inhibit CD8+ T effector function through suppression of AKT‐mTOR signalling. CD8+ T effector function could be restored by decreasing intracellular [K+] in CD8+ T cells, either pharmacologically or through the overexpression of a K+ efflux channel [190]. Abnormal accumulation of K+ in the TME contributes to impaired T cell effector function without inhibiting T cell proliferative potential. Mechanistically, a high concentration of extracellular K+ limits nutrient absorption by Teff cells and induces a starvation response, which is characterized by increased autophagy, upregulated mitochondrial metabolic activity, and decreased histone acetylation, and these effects lead to the inhibition of effector functions [191]. Intriguingly, although the activity of T cells is inhibited at high potassium concentrations, adoptively transferred CD8+ T cells activated in the presence of high potassium levels exhibit improvements in their durability, antitumor activity, and pluripotency due to metabolic reprogramming caused by the starvation response [191]. Thus, potassium induces T cell metabolic reprograming in the TME, which further results in improved antitumor immune responses.
3. ROLE OF MICROBIAL METABOLITES IN CANCER IMMUNITY
Because the intestinal flora is a key component of digestion, food residues and the compounds produced by the host and microorganisms are fermented by the intestinal flora to generate abundant metabolites. Intestinal microbes and their metabolites further interact with host cells through intestinal epithelial monolayer cells, involving host immune activities and cancer progression. In addition, a link has been established between the microbiota composition and the immune checkpoint blockade efficacy in patients with melanoma [192, 193] or epithelial tumors [192, 193]. Moreover, the available data show that nearly half of the sources of plasma metabolites are closely related to bacteria, which indicates the crucial role of the microbiota in human metabolism [194]. Furthermore, bacterial metabolites, including short‐chain fatty acids (SCFAs), AHR ligands, secondary bile acids (BAs), polyamines, and polysaccharides, might be involved in cancer progression and the efficacy of anticancer therapies by influencing the host immune system (Figure 4).
FIGURE 4.
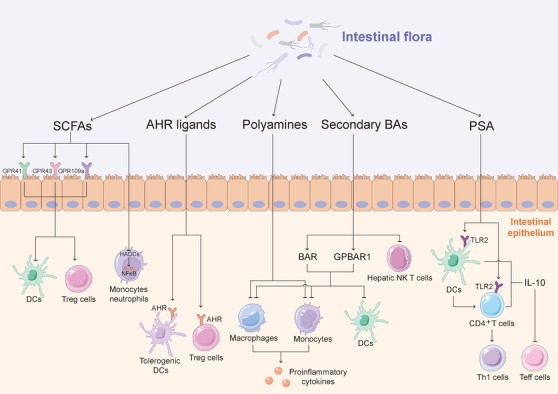
Role of microbial metabolites in cancer immunity. SCFAs, AHR ligands, secondary BAs, polyamines and PSA, the bacterial metabolites, are involved in cancer progression and the efficacy of anticancer therapies by influencing the host immune system. SCFAs inactivate DCs and promote proliferation and immune regulation of Treg cells through GPR41, GPR43 and GPR109A, and inhibit the activity of NF‐κB and the secretion of TNF in peripheral blood mononuclear cells and neutrophils by inhibiting HDAC. AHR ligands serve a function in regulating adaptive immunity through binding to AHR. BAs activate BAR and GPBAR1 to inactivate inflammation. In addition, secondary BAs, the products of primary BAs metabolized by bacteria, reduce hepatic NKT cells. PSA promotes CD4+ T cells to express IL‐10, induces naive CD4+ T cell differentiation toward the Th1 subtype via a DC‐dependent manner, and activates the expression of TLR2 to induce the secretion of IL‐10, thereby inactivating Teff cells. Abbreviations: AHR: aryl hydrocarbon receptor; BAR: bile acids receptor; BAs: bile acids; DC: dendritic cell; GPBAR1: G protein‐coupled BA receptor 1; GPR: G protein‐coupled receptor; HDAC: histone deacetylase; IL: interleukin; NF‐κB: nuclear factor kappa‐B; NKT: natural killer T; PSA: polysaccharide A; Teff: effector T; TLR 2: Toll‐like receptor 2; TNF: tumor necrosis factor; Treg: regulatory T; SCFA: short‐chain fatty acid
3.1. SCFAs
SCFAs, primarily acetate, butyrate, and propionate, are products of the metabolism of polysaccharides and dietary fibre by Clostridium clusters IV and XIVa in the Firmicutes phylum, including species in the genera Eubacterium, Roseburia, Faecalibacterium, and Coprococcus, in the colon. Evidence showed that SCFAs, particularly propionate and butyrate, are involved in host immune activities. SCFAs activate the proliferation and function of immune cells by inhibiting histone deacetylase (HADC) and activating G protein‐coupled receptors (GPCRs). Because SCFAs are inhibitors of HDAC, SCFAs inhibit the activity of NF‐κB and the secretion of TNF in peripheral blood mononuclear cells [195] and neutrophils [196]. Furthermore, the activities of macrophages and DCs are also inhibited by SCFAs [197, 198]. Thus, SCFAs are crucial for the regulation of innate immunity. In addition to innate immunity, the HDAC inhibitory function of SCFAs acts on Treg cells to induce FOXP3 expression and thereby maintain intestinal homeostasis [199, 200]. In addition, animal models have revealed the immunoregulatory effect of HDAC inhibitors via Treg cells dependent on inflammatory diseases [201]. In addition to inhibiting HDAC, SCFAs regulate host immunity through GPCRs, including GPR41, GPR43, and GPR109A, which are expressed in a variety of cells, including intestinal epithelial cells and immune cells. The SCFA‐induced activation of GPR41 inactivates DCs and improves the severity of allergic inflammation in a mouse model [198]. GPR43 stimulation induces chemotaxis of neutrophils and promotes the proliferation and immune regulation of Treg cells [196, 202]. In a dextran sulphate sodium‐induced and T cell transfer inflammatory bowel disease mouse model, SCFAs improve the severity of inflammation by recruiting and activating Treg cells in a GPR43‐dependent manner [202, 203]. In addition, SCFAs promote the differentiation of Treg cells and IL‐10‐producing T cells, which results in the suppression of colitis and colon cancer in a GPR109a‐dependent manner [204]. Taken together, the above‐described findings demonstrate that SCFAs, the metabolites of microbes, are crucial to the regulation of inflammation and the prevention of tumourigenesis.
3.2. AHR ligands
AHR ligands are a type of metabolite that binds to AHR to exert their function. AHR is a ligand‐dependent transcription factor involved in the metabolism of exogenous compounds. Recent evidence has revealed the existence of endogenous AHR ligands [205]. As mentioned above, kynurenine, a catabolite of tryptophan, is an endogenous ligand of AHR, and evidence showed that kynurenine is induced by TDO and inhibits antitumor immune activity through the activation of AHR in both autocrine and paracrine forms [206]. Studies have suggested that AHR serves a function in regulating adaptive immunity and the differentiation of T cells and APCs [207, 208]. AHR participates in promoting the suppressive capacity of Treg cells by modulating DCs and also promotes the direct transactivation and induction of epigenetic modifications that control FOXP3 transcription [209]. The available evidence showed that AHR activation led to increased retinoic acid levels in DCs and deactivates signal transducer and activator of transcription 1 in T cells, which promoted the differentiation of Treg cells and suppressed Teff cells [209, 210]. Notably, the composition of the intestinal flora is an important factor in the activation of AHR. Specific bacterial subgroups, primarily Lactobacillus, can catabolize tryptophan to generate AHR ligands that activate AHR in innate lymphoid cells, which induces a balanced mucosal response that prevents colonization of the fungus Candida albicans [211]. Therefore, endogenous microbe‐derived AHR ligands assist the host in preventing pathogen invasion and inflammation. Intriguingly, indole and 3‐methyl indole, which are AHR ligands, exhibit species specificity for AHR activation, which indicates that ligands common to the microbiota and host have the potential for coevolution [212]. In summary, microbe‐derived AHR ligands might play an important role in immunoregulation and immunopathology.
3.3. Polyamines
Polyamines, the primary examples of which are putrescine, spermidine, and spermine, exist in all forms of organisms [213]. The synthesis of polyamines in mammals is gradually catalysed by ARG1, ornithine decarboxylase (ODC), and sequential enzymes that interconvert putrescine, spermidine, and spermine. Among these enzymes, ODC is the primary rate‐limiting enzyme [213], and polyamines are produced by the intestinal flora via constitutive or inducible forms of amino acid decarboxylase, supporting the survival and toxicity of a variety of pathogens [214]. Polyamines are involved in key physiological processes that are crucial to host cells, such as gene transcription and translation, cell growth and death. Thus, the downregulation of ODC and polyamines leads to the suppression of cell growth, whereas the upregulation of polyamines promote tumorigenesis and tumor progression [213]. Polyamines also serve the function of regulating immunity. First, polyamines affect the function of innate immune cells. A high availability of polyamines inhibits the secretion of proinflammatory cytokines in macrophages and monocytes in response to LPS‐stimulation. Spermine inactivates M1 macrophages by suppressing the expression of ODC and the production of proinflammatory cytokines without influencing the secretion of IL‐10 or transforming growth factor‐β [215]. Consistently, treatment with polyamines improves the severity of localized and systemic inflammation in mice [216], and arginine in combination with probiotic Bifidobacteria LKM512 increases the level of polyamines, which in turn suppresses inflammation by decreasing TNF and IL‐6 [217]. Accordingly, the colonic metabolic state might be improved by altering the dietary structure and through the appropriate intake of probiotics. Second, polyamines are involved in the modulation of adaptive immunity. Evidence showed that breast milk polyamines promoted the maturation of T cells, B cells, and NK cells in rats [218]. The role of polyamine metabolism in tumor progression was revealed in recent years. Similarly, a high availability of polyamines is needed to meet the fuel demands of tumor cells [213]. Samples from patients with intestinal cancer showed increased levels of polyamine metabolites, particularly N1‐ and N12‐diacetylspermine, which are also associated with the mutual promotion of cancer cells and bacterial biofilms (BFs). Furthermore, antibiotic treatment could remove BFs and decrease the N1‐ and N12‐diacetylspermine levels [219]. Thus, N1‐ and N12‐diacetylspermine might represent BF‐associated cancer biomarkers. In addition, evidence from a mouse model showed that polyamines were involved in suppression of antitumor immunity, and the depletion of polyamines activates Teff cells to limit tumor progression, which indicated that reducing polyamines might represent a strategy for relieving immunosuppression in the TME [220]. In contrast, spermidine, one of the CR mimetics, induces autophagy and enhances the inhibitory effect of chemotherapy on tumor growth, and this effect depends on the depletion of Treg cells in the TME [80]. Similarly, a high spermidine intake is associated with an increased survival time in patients with cancer [221]. In addition to its metabolic function, spermidine act as a competitive inhibitor of acetyltransferase P300 to reduce the levels of acetyl‐CoA and thereby induce autophagy [222, 223]. Notably, spermidine‐induced autophagy is beneficial for anticancer immune surveillance [153]. Altogether, the abovementioned findings show that the regulatory mechanism of polyamines and the complex role of polyamines between microorganisms and hosts require further exploration.
3.4. Bile acids
The synthesis of BAs occurs in the liver. Initially, cholesterol is oxidized by cytochrome P450 to generate primary BAs, which are conjugated to glycine or taurine to form bile salts before secretion. After being secreted into the intestinal tract, bile salts are further modified by intestinal bacteria into secondary BAs. Finally, approximately 95% of BAs are reabsorbed from the intestine and circulate to the liver via the portal vein [224]. Accordingly, the intestinal flora is crucial to the enterohepatic circulation of BAs. In addition to digestion, BAs have the function of regulating immune activities. First, BAs inhibit the secretion of proinflammatory cytokines from DCs, monocytes, macrophages, and Kupffer cells (resident macrophages in the liver) [225] via the bile acid receptor (BAR) and G protein‐coupled BA receptor 1 (GPBAR1). In addition, both primary and secondary BAs can activate the two receptors [226]. BAR, a nuclear receptor, is a ligand‐activated transcription factor that forms heterodimers with the retinoid X receptor. The activation of BAR in LPS‐activated macrophages induces the transcription of BAR‐dependent genes and inhibits the transcription of NF‐κB‐dependent genes through sumoylation‐dependent stabilization of the nuclear receptor corepressor protein on the promoter, which results in the inhibition of proinflammatory cytokine expression [227]. Consistently, the inflammatory damage of colitis was alleviated in response to the administration of a BAR synthetic agonist [227, 228], but aggravated in BAR‐deficient mice [227]. In addition to macrophages, hepatocytes and natural killer T (NKT) cells are also involved in the BAR deficiency‐mediated activation of inflammation [229]. The activation of GPBAR1 by BAs contributes to increased intracellular cAMP levels, which activate extracellular signal‐regulated kinases [230]. Similarly, synthetic agonists of GPBAR1 suppress the secretion of LPS‐activated proinflammatory cytokines in macrophages and Kupffer cells by interfering with NF‐κB‐dependent transcription, and this effect is associated with direct inactivation of NF‐κB and transcriptional competition between NF‐κB and the cAMP‐activated transcription factor cAMP response element‐binding protein (CREB) [231]. Furthermore, the gut microbiota alter immune status through the metabolism of BAs. The expression of chemokine (C‐X‐C motif) ligand 16 (CXCL16), which inhibits tumor liver metastasis by inducing NKT cells, is increased by primary BAs but is decreased by secondary BAs in liver sinusoidal endothelial cells [232]. Moreover, vancomycin monotherapy leads to a reduction in Gram‐positive Clostridium species, which play a central role in the production of secondary BAs, and thereby increase hepatic NKT cells. In addition, treatment with secondary BAs or colonization by Clostridium scindens leads to decreased hepatic NKT cells and increased liver metastases in vancomycin‐treated mice [232]. Finally, 3‐oxoLCA and isoalloLCA, metabolites of lithocholic acid, are involved in the regulation of T cells. The differentiation of Th17 cells is inhibited by the direct binding of 3‐oxoLCA to the key transcription factor RORγt, and the differentiation of Treg cells is promoted by isoalloLCA through the production of mitochondrial ROS [233]. Taken together, these data indicate that targeting the intestinal flora to regulate the metabolism of BAs might represent a promising strategy for cancer treatment through the regulation of immune cells.
3.5. Polysaccharide
Zwitterionic polysaccharide A (PSA), which is produced by the common gut bacterium Bacteroides fragilis, is another microbial metabolite that serves as an immune modulator [234]. PSA promotes CD4+ T cells to express IL‐10 and induces naive CD4+ T cell differentiation towards the Th1 subtype in a DC‐dependent manner. Mechanistically, DCs stimulate the activation and clonal expansion of T cells by internalizing PSA and then presenting the antigen to CD4+ T cells [235]. In addition, TLR2 expression on the surface of DCs [236] and Treg cells [237] is activated by PSA to induce the secretion of IL‐10, and PSA‐induced IL‐10 inactivates Teff cells, particularly Th17 cells, and thereby alleviates the symptoms of colitis in a mouse model [236]. Intriguingly, the immunosuppressive properties of PSA are essential for the intestinal colonization of Bacteroides fragilis, which indicates the mutualistic nature of the interactions between PSA and the immune system [237]. Moreover, the expression of CD39 on Treg cells is increased by PSA. Thus, PSA might be involved in modulating ATP and adenosine signalling with systemic consequences [238].
3.6. Intestinal flora and immunotherapy
Emerging knowledge on the ability of the gut microbiota to modulate the response to immunotherapy offers new possibilities to improve its efficacy by targeting the microbiota [192, 193, 239, 240]. CTLA‐4 antagonists alter the composition of the intestinal flora and induce T cell‐mediated mucosal damage in the ileum and colon. In general, treatment with CTLA‐4 antagonists decreases both Bacteroidales and Burkholderiales (for example, Bacteroides thetaiotaomicron, Bacteroides uniformis and Burkholderia cepacia) while increasing Clostridiales in a tumor‐bearing mouse model [239]. The oral administration of either B. thetaiotaomicron or B. fragilis to microbiota‐depleted mice restores the therapeutic response to anti‐CTLA‐4 by promoting the maturation of intratumoral DCs and inducing a Th1 response in the tumor‐draining lymph nodes [239], which indicates that anti‐CTLA‐4 treatment might alter the composition of the gut microbiota in a direction that favours its antitumor activity in some patients. Similarly, the composition of the intestinal flora is also altered by PD‐1/PD‐L1 blockers to influence antitumor immunity. Moreover, the alpha diversity and relative abundance of bacteria in the Ruminococcaceae family are significantly higher in faecal microbiome samples from patients responding to checkpoint blockade immunotherapy. The results from an immune profile analysis showed that patients with a favourable gut microbiome, as well as germ‐free mice receiving faecal transplants from responding patients, exhibited enhanced systemic and antitumor immunity [192]. In addition, a metagenomics analysis of patient stool samples obtained at diagnosis revealed that the abundance of Akkermansia muciniphila was most significantly associated with the clinical responses to ICIs. The oral administration of Akkermansia muciniphila after faecal microbiota transplantation with nonresponder faeces restores the efficacy of the PD‐1 blockade by increasing the recruitment of CCR9+CXCR3+CD4+ T lymphocytes into mouse tumor beds [193]. Roberti et al. [241] also discovered that B. fragilis and Erysipelotrichaceae near ileal crypts induced DCs to activate the immune response of follicular helper T cells, which contributed to immunosurveillance against colon cancer and thus promotes the antitumor effects of chemotherapy and immunotherapy. Similarly, the administration of Enterococcus hirae containing a bacteriophage activates T cell responses after treatment with chemotherapy or immunotherapy [242]. The mechanism by which changes in the gut microbiota alters systemic immunity is under investigation, but a recently published study by Tanoue et al. [243] showed that colonization of mice with a consortium of 11 bacterial strains from healthy human donors could improve CD8 T‐cell–mediated immunity in several mouse models, and they demonstrated that the 11‐strain consortium was associated with cecal and systemic elevation in metabolites, such as mevalonate and dimethylglycine, which might potentially augment immune function.
Based on the interaction between the microbiota and the host, the effect of using antibiotics to regulate the microbiome during cancer immunotherapy is complex. The oral administration of vancomycin improves the outcomes of cancer immunotherapy that targets CTLA‐4 by depleting gram‐positive Clostridium species [232]. Similarly, thiostrepton, an antibiotic produced by Streptomyces azureus and Streptomyces laurentii, synergizes with chemotherapy by enhancing anticancer immunogenicity [244]. However, antibiotics inhibit the effect of ICIs in advanced cancer [193], which indicates that the antibiotic‐mediated effects might be context‐dependent. Thus, natural antibiotics, which have narrow activity ranges, might be suitable for eliminating specific harmful microbial species. Evidence showed that the use of antianaerobic drugs, including carbapenem, cefmetazole, flomoxef, cefpirome, cefepime, clindamycin, and pazufloxacin, was associated with poor prognosis [245]. Accordingly, the targeted use of specific antibiotics and/or probiotics instead of the short‐term or repeated use of broad‐spectrum antibiotics to adjust the composition of the microbiome might be beneficial to tumor treatment efficacy.
4. INFLUENCE OF SYSTEMIC METABOLISM ON REGULATION OF THE ANTITUMOR IMMUNE RESPONSE
The concept of “immunonutrition” has been developed due to awareness of the status of systemic nutritional influences on immune responses observed in epidemiological studies and clinical trials [246]. Nutritive variations, particularly obesity and fasting, exert immunomodulatory effects in the whole body. In addition, aging is characterized by generalized degeneration in a progressive manner, which leads to metabolic alterations and immunosuppression that drive tumorigenesis [247, 248] (Figure 5).
FIGURE 5.
Influence of systemic metabolism on regulation of the antitumor immune response. (A) Obesity (left) is correlated with increased glucose, FFAs, leptin, insulin, and IGF1, along with decreased adiponectin, and induces the conversion of adipose‐tissue macrophages from the M2 subtype to the M1 subtype, and is associated with aging‐related immune dysfunctions. Relatively, fasting (right) induces histone acetylation, enhances autophagy, inactivates NLRP3 inflammasome, and decreases the bioavailability of IGF1. B. In response to various stimuli, cells undergo senescence and product SASP, which is involved in eliminating incipient tumor cells during cancer initiation, while leading to immunosuppression after cancer formation. Abbreviations: IGF1: insulin‐like growth factor 1; IGFBP1: insulin‐like growth factor‐binding protein‐1; IL: interleukin; TNF‐α: tumor necrosis factor‐α; SIRT: sirtuin 3; NK cell: NK: natural killer cell.; NLRP3: NLR family pyrin domain containing 3; SASP: senescence‐associated secretory phenotype; SOD2: Superoxide dismutase 2; TLR 2: Toll‐like receptor 2; FFAs: free fatty acids
4.1. Obesity
Obesity affects the occurrence and progression of a variety of cancers and is one of the most important avoidable causes of cancer [249]. A related link between obesity concurrent with metabolic syndrome and cancer has been established. Metabolically, obesity is correlated with increased glucose, FFAs, leptin, insulin and insulin‐like growth factor 1 (IGF1), decreased adiponectin, and an imbalance in the intestinal flora with a reduced barrier function, which contributes to endotoxinemia and the overgrowth of bacterial species that produce carcinogenic metabolites [250, 251]. FFA induces TLR4 signalling in a fetuin‐A‐dependent manner, which culminates in increased activation of proinflammatory cytokines such as NF‐κB and c‐Jun N‐terminal protein kinase 1 (JNK1) [252]. In addition, the conversion of adipose‐tissue macrophages from the M2 subtype to the M1 subtype can be induced by obesity, and this conversion promotes the secretion of various proinflammatory cytokines, such as TNF, IL‐6 and IL‐1β [95, 253]. In addition, consistent with the theory that obesity is an inducer of aging [247], a variety of aging‐related immune dysfunctions have been observed in individuals with obesity [254]. Specifically, the number and function of cells are inhibited, and the numbers of circulating Vγ9+Vδ2+ T cells are reduced in these individuals [255]. These effects are accompanied by decreased levels of cytotoxic CD8+ T cells [256], which are properly induced by thymic atrophy [257] and the functional decline of DCs [258]. Evidence showed that the function of CD8+ T cells is impaired by high‐fat diet‐induced obesity through increases in the fat uptake of MC38 colorectal adenocarcinoma cells [259]. In addition, mammary adipose tissue macrophages enhance tumorigenesis of breast cancer by inducing stem‐like properties in obese mice [181]. Obesity‐related leptin reduction leads to increased PD‐1 expression [260]. Consistently, some tumor immunotherapies are ineffective and even toxic in obese mice but show effectiveness in lean mice [261, 262]. The immunotherapy combination of αCD40 and IL‐2 induces a rapidly lethal cytokine storm in obese or aging mice caused by heightened production of TNF released from increased proinflammatory M1 macrophages [262]. These findings emphasize that obesity serves a function of promoting resistance to tumor immunotherapy.
Alleviating obesity is thought to be an effective strategy for preventing and treating cancer based on all the evidence relating obesity to a pro‐tumor state both within the microenvironment and in peripheral circulation. Adjusting the diet and/or exercise to reduce weight restores multiple pathways that are dysfunctional under obese conditions and contributes to the prevention of tumor occurrence and progression. Several clinical studies have shown that significant calorie restriction and ketogenic or specific low‐nutrient diets resulted in improvements in the subjective well‐being, which indicates a reduction in side effects [263] and in inflammatory gene expression signatures within subcutaneous adipose tissues [264]. Patients with cancer who are overweight and obese could, in theory, benefit from medications that stimulate weight loss. Finally, several medications, including metformin, everolimus and aspirin, could be beneficial in alleviating local and systemic inflammatory symptoms [265]. However, although preclinical data support the notion that metformin could enhance the effect of anti‐PD‐1 blockade [266], clinical trial data show that metformin can exert a synergistic effect to improve the curative effect of ICIs, but the effect was not statistically significant [267, 268, 269]. Although the website ClinicalTrials.gov provides information on multiple clinical trials that are either ongoing or completed, outcome information is generally lacking. It is worth investigating to see whether any of these studies have observed or will document a clinical benefit linked to a specific nutritional intervention.
In contrast, some recent data suggest an inverse relationship between body mass index and mortality in patients with cancer who receive immunotherapy [270, 271, 272]. The phenomenon might be explained by several reasons. Excess adipose tissue can serve as a potential energy reserve for cancer treatment, whereas malnutrition and cachexia might impair immune function [273]. In addition, the proinflammatory microenvironment in obesity might also be a reason. As mentioned above, although the inflammatory microenvironment caused by obesity is a recognized risk factor for tumorigenesis, this microenvironment also makes the level of immune cell infiltration in the TME significantly higher than that of patients with a normal weight. The immune cells in the TME are activated after immunotherapy, which results in a better prognosis in patients with obesity [270]. However, these concepts have not been verified experimentally, and no clinical trial has verified whether weight gain serves as an adjuvant treatment for malignant tumors. Taken together, the above‐described findings indicate that obesity‐overweight is a clear risk factor for tumorigenesis, but its association with cancer outcomes remains unclear. The pathophysiological mechanism underlying this process still needs to be further explored.
4.2. Fasting
Fasting plays important roles in the metabolic response and immune function. Evidence showed that histone acetylation is reduced and autophagy is enhanced in leukocytes from both starved mice and human volunteers [274]. Similarly, caloric restriction exerts considerable anti‐inflammatory effects and improves the function of immune cells [275]. Furthermore, a 24‐hour fast can induce resistance to NLRP3 inflammasome activation by activating sirtuin 3 biology, and thereby reduces IL‐1β secretion in human subjects [276]. In contrast, the coeffect of bacterial products and glucose induces increases in the expression of macrophage‐derived IL‐1β in lean mice after feeding, which promotes the secretion of postprandial insulin [277]. Furthermore, some scholars have proposed that sickness‐associated anorexia is an evolutionarily conserved reaction that regulates the immune response through the systemic upregulation of autophagic processes [278]. Moreover, caloric restriction mimetics (CRMs), a class of drugs or dietary supplements that reproduce the effects of CR, are reputed to promote health and even prolong the lifespan [279]. Evidence showed that starvation and multiple CRMs decrease the bioavailability of IGF1 by upregulating insulin‐like growth factor‐binding protein‐1, which leads to large convergent metabolomic perturbations [80]. In addition, hydroxycitrate, a type of CRM, improves the effects of chemotherapy in a T cell‐dependent manner [80]. Taken together, the findings indicate that CRMs appear to substantially facilitate immunosurveillance through systemic metabolic remodelling.
4.3. Aging
Initially, cellular senescence was considered an irreversible exit from the cell cycle. However, senescence has since been identified as a stress response induced in response to various stimuli [280]. A number of studies have focused on aging‐related metabolic characteristics [247] and further explored age‐related biological changes through metabolomic characteristics [281]. The metabolic age score calculated by urine metabolomics can be used to evaluate the state of aging and predict survival and is independent of the chronological age and other risk factors [281]. Similarly, age‐related characteristics and early pathological features of age‐related diseases are reflected by serum metabolomics [282]. Recently, the relationship between longevity and the metabolic characteristics of different organs has been established by transspecies comparisons [283]. Moreover, the aging‐induced accumulation of metabolites has been demonstrated to promote tumor progression and invasion [284].
Evidence indicates that longevity is positively correlated with sphingomyelin levels but negatively correlated with triacylglycerols containing polyunsaturated fatty acid (PUFA) side chains and by‐products of inflammatory processes. PUFAs containing triacylglycerols have been consistently associated with shorter lifespans in human females [282]. In addition, naked mole rats, which have a life expectancy greater than 10 times that of rodents of the same size, have more polyamines and alpha‐tocopherol (also known as vitamin E) in their plasma metabolites than do mice, and these metabolites have been shown to contribute to health and longevity in mice [285]. Longevity is negatively correlated with the hepatic levels of enzymatic cofactors that are involved in amino acid metabolism and the hepatic concentrations of tryptophan degradation products in several mammals. Therefore, the lifespan of animal models might be prolonged by reducing their intake of amino acids (particularly tryptophan and methionine), and daily weight‐based caloric consumption might be one of the metabolic characteristics of long‐lived mammals.
Importantly, the available evidence suggests that senescent cells can affect the TME. Ageing can cause immune dysfunction, which contributes to alterations in the TME during aging [286]. The senescence‐associated secretory phenotype (SASP), the general term given to the combination of cytokines, chemokines, extracellular matrix proteases, growth factors, and other signalling molecules, is produced by senescent cells [287]. There are complex reciprocal interactions between the immune response and the SASP. The SASP is involved in recruiting immune cells to clear aging cells, which allows the SASP to recruit Th1 cells, NK cells, and macrophages to eliminate incipient tumor cells [288]. However, the SASP might also lead to immunosuppression. The coexistence of hepatocellular carcinoma (HCC) cells and premalignant senescent hepatocytes leads to the recruitment of immature myeloid cells by the SASP, which contributes to the impairment of NK cell function and thus promotes HCC [289]. Accordingly, there are multiple interactions among immune cells, senescent cells, and cancer cells.
5. CONCLUSIONS AND PERSPECTIVES
In recent years, the field of cancer immunometabolism has attracted much attention [290, 291]. Studies on the metabolism of the TME have revealed the remarkable plasticity of cancer cells to obtain nutrients for growth and survival under the harshest conditions. In contrast, non‐malignant cells are sensitive to nutrient consumption and metabolic waste accumulation. Specifically, both tumor cells and antitumor immune cells consume high amounts of glucose and glutamine, but tumor cells are more able to tolerate or even utilize lactate and hypoxia, which are harmful to antitumor immune cells and beneficial to immune regulatory cells. These differences provide opportunities for targeted metabolism. However, we need to be mindful of potential treatment toxicities because both tumor cells and immune cells often utilize the same metabolic pathways for proliferation. Some strategies might be applied to overcome this limitation. (A) One of these strategies involve developing new drug delivery methods that selectively modify specific immune cell subgroups or phenotypic metabolism. It is important to consider that tumor cells and lymphocytes might differentially express amino acids and nutrient transporters, especially given the plethora of carriers involved in metabolism. The identification of metabolic targets that are selectively utilized in tumor cells might greatly improve antitumor immunity by avoiding detrimental effects on immune cells, and identifying these differences might allow the selective inhibition of metabolic pathways in cancer cells. (B) The second strategy combines metabolic intervention with chimeric antigen receptor T (CAR‐T) cell therapy based on which manipulation of metabolic pathways can be precisely defined by genetic means. In addition, the metabolic characteristics of tumors that are responsive or nonresponsive to ICIs are different; thus, the relative utilization of glucose, amino acids, oxygen, or other nutrients might also be different. Therefore, studies aiming to determine the metabolic characteristics of ICI‐resistant tumors might help identify targetable metabolic pathways in cancer cells that influence ICIs.
Caloric restriction promotes some anticancer therapies, and a high‐fat diet might exacerbate malignant tumors. Similarly, calorie restriction can improve the incidence and severity of certain cancers. However, the operation of the diet will eventually regulate the intestinal microbiota, which, in turn, affects tumor and immune metabolism and even exerts a significant regulatory effect on various cancer types and immunotherapies. Although interference with the intestinal flora (directly through probiotics and/or faecal transplantation or indirectly through dietary changes) might be a promising strategy for individualized immunotherapy, caution should be taken because the impact of the diverse intestinal flora on the human body remains unclear.
At present, many topics in tumor immunonutrition remain to be improved, and these include the relationship between carcinogenic mutations and metabolic changes, which and how metabolic changes define the composition of the immunosuppressive TME, and whether the different metabolic characteristics of tumors of different types, stages and metastatic sites will affect multiple immune responses. Clarifying these issues will contribute to the development of personalised medicine for tumors. Further studies should include critically assessing the systemic changes in local metabolic phenotypes, whole body metabolism, and microbial communities.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS' CONTRIBUTIONS
QW and SS performed and conceived the review. QW and XY wrote the manuscript. JL prepared all the figures. YT helped improved the quality of language and helped supply financial support. All the authors read and approved the final manuscript.
ACKNOWLEDGEMENTS
We thank a professional English editor (American Journal Experts) for assistance in improving the quality of language.
This work was partially supported by a National Natural Science Foundation of China (NSFC) grant (Grant NO: 81471781) and a National Major Scientific Instruments and Equipment Development Projects grant (Grant NO: 2012YQ160203) to Dr. Shengrong Sun, and a Natural Science Foundation of Hubei Province (Grant NO: 2020CFA026) to Dr. Yi Tu.
Wu Qi, Yu X, Li J, Sun S, Tu Yi. Metabolic regulation in the immune response to cancer. Cancer Commun. 2021;41:661–694. 10.1002/cac2.12182
Contributor Information
Shengrong Sun, Email: rm000752@whu.edu.cn.
Yi Tu, Email: rm000750@whu.edu.cn.
REFERENCES
- 1.Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–9. 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell SL, Wellen KE. Metabolic signaling to the nucleus in cancer. Mol Cell. 2018;71(3):398–408. 10.1016/j.molcel.2018.07.015. [DOI] [PubMed] [Google Scholar]
- 3.Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–41. 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wellenstein MD, de Visser KE. Cancer‐cell‐intrinsic mechanisms shaping the tumor immune landscape. Immunity. 2018;48(3):399–416. 10.1016/j.immuni.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Tang Z, Xu Z, Zhu X, Zhang J. New insights into molecules and pathways of cancer metabolism and therapeutic implications. Cancer Commun (London, England). 2021;41(1):16–36. 10.1002/cac2.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. 2017;168(4):657–69. 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47. 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Sullivan D, Sanin DE, Pearce EJ, Pearce EL. Metabolic interventions in the immune response to cancer. Nat Rev Immunol. 2019;19(5):324–35. 10.1038/s41577-019-0140-9. [DOI] [PubMed] [Google Scholar]
- 9.Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. 2017;169(4):570–86. 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrejeva G, Rathmell JC. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 2017;26(1):49–70. 10.1016/j.cmet.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fridman WH, Galon J, Pages F, Tartour E, Sautes‐Fridman C, Kroemer G. Prognostic and predictive impact of intra‐ and peritumoral immune infiltrates. Cancer Res. 2011;71(17):5601–5. 10.1158/0008-5472.CAN-11-1316. [DOI] [PubMed] [Google Scholar]
- 12.O'Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–65. 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang YA, Li XL, Mo YZ, Fan CM, Tang L, Xiong F, et al. Effects of tumor metabolic microenvironment on regulatory T cells. Mol Cancer. 2018;17(1):168. 10.1186/s12943-018-0913-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin YH. Crosstalk of lncRNA and cellular metabolism and their regulatory mechanism in cancer. Int J Mol Sci. 2020;21(8). 10.3390/ijms21082947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan YT, Lin JF, Li T, Li JJ, Xu RH, Ju HQ. LncRNA‐mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Communications (London, England). 2021;41(2):109–20. 10.1002/cac2.12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti‐tumor T cell responses. Cell. 2015;162(6):1217–28. 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang CH, Curtis JD, MaggiLB, Jr., Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153(6):1239–51. 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, MO Li. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354(6311):481–4. 10.1126/science.aaf6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angelin A, Gil‐de‐Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low‐glucose, high‐lactate environments. Cell Metab. 2017;25(6):1282–93 e7. 10.1016/j.cmet.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gualdoni GA, Mayer KA, Goschl L, Boucheron N, Ellmeier W, Zlabinger GJ. The AMP analog AICAR modulates the Treg/Th17 axis through enhancement of fatty acid oxidation. FASEB J. 2016;30(11):3800–9. 10.1096/fj.201600522R. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez‐Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin‐Sanz P, et al. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J Immunol. 2010;185(1):605–14. 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 22.Tan Z, Xie N, Cui H, Moellering DR, Abraham E, Thannickal VJ, et al. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J Immunol. 2015;194(12):6082–9. 10.4049/jimmunol.1402469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)‐mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. 2014;289(11):7884–96. 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Venter G, Oerlemans FT, Wijers M, Willemse M, Fransen JA, Wieringa B. Glucose controls morphodynamics of LPS‐stimulated macrophages. PLoS One. 2014;9(5):e96786. 10.1371/journal.pone.0096786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang D, Li J, Wang F, Hu J, Wang S, Sun Y. 2‐Deoxy‐D‐glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014;355(2):176–83. 10.1016/j.canlet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 26.Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479–88. 10.1172/jci69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu J, Villa M, Sanin DE, Buck MD, O'Sullivan D, Ching R, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep. 2019;27(7):2063–74.e5. 10.1016/j.celrep.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366(6468):1013–21. 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551(7678):115–8. 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000;60(4):916–21. [PubMed] [Google Scholar]
- 31.Wu Q, Li J, Li Z, Sun S, Zhu S, Wang L, et al. Exosomes from the tumour‐adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J Exp Clin Cancer Res. 2019;38(1):223. 10.1186/s13046-019-1210-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Pérez‐Escuredo J, Dadhich RK, Dhup S, Cacace A, Van Hée VF, De Saedeleer CJ, et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle. 2016;15(1):72–83. 10.1080/15384101.2015.1120930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pucino V, Cucchi D, Mauro C. Lactate transporters as therapeutic targets in cancer and inflammatory diseases. Expert Opin Ther Targets. 2018;22(9):735–43. 10.1080/14728222.2018.1511706. [DOI] [PubMed] [Google Scholar]
- 34.Halestrap AP, Wilson MC. The monocarboxylate transporter family–role and regulation. IUBMB Life. 2012;64(2):109–19. 10.1002/iub.572. [DOI] [PubMed] [Google Scholar]
- 35.Kelsey R. Prostate cancer: MCT4 is a novel target for prostate cancer. Nat Rev Urol. 2016;13(3):123. 10.1038/nrurol.2016.14. [DOI] [PubMed] [Google Scholar]
- 36.Li Z, Wu Q, Sun S, Wu J, Li J, Zhang Y, et al. Monocarboxylate transporters in breast cancer and adipose tissue are novel biomarkers and potential therapeutic targets. Biochem Biophys Res Commun. 2018;501(4):962–7. 10.1016/j.bbrc.2018.05.091. [DOI] [PubMed] [Google Scholar]
- 37.Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate metabolism in human lung tumors. Cell. 2017;171(2):358–71 e9. 10.1016/j.cell.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Q, Li B, Li Z, Li J, Sun S, Sun S. Cancer‐associated adipocytes: Key players in breast cancer progression. J Hematol Oncol. 2019;12(1):95. 10.1186/s13045-019-0778-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duda P, Janczara J, McCubrey JA, Gizak A, Rakus D. The reverse Warburg effect is associated with Fbp2‐dependent Hif1α regulation in cancer cells stimulated by fibroblasts. Cells. 2020;9(1). 10.3390/cells9010205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilde L, Roche M, Domingo‐Vidal M, Tanson K, Philp N, Curry J, et al. Metabolic coupling and the reverse Warburg effect in cancer: Implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44(3):198–203. 10.1053/j.seminoncol.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA‐associated lactic acid production blunts tumor immunosurveillance by T and NK Cells. Cell Metab. 2016;24(5):657–71. 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 42.Haas R, Smith J, Rocher‐Ros V, Nadkarni S, Montero‐Melendez T, D'Acquisto F, et al. Lactate regulates metabolic and pro‐inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 2015;13(7):e1002202. 10.1371/journal.pbio.1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour‐associated macrophages by tumour‐derived lactic acid. Nature. 2014;513(7519):559–63. 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kouidhi S, Ben Ayed F, Benammar Elgaaied A. Targeting tumor metabolism: A new challenge to improve immunotherapy. Front Immunol. 2018;9:353. 10.3389/fimmu.2018.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de la Cruz‐López KG, Castro‐Muñoz LJ, DO Reyes‐Hernández, García‐Carrancá A, Manzo‐Merino J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol. 2019;9:1143. 10.3389/fonc.2019.01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chirasani SR, Leukel P, Gottfried E, Hochrein J, Stadler K, Neumann B, et al. Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model. Int J Cancer. 2013;132(4):843–53. 10.1002/ijc.27712. [DOI] [PubMed] [Google Scholar]
- 47.Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O'Neill LA, et al. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nature Metabolism. 2019;1(1):16–33. 10.1038/s42255-018-0014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tannahill GM, Curtis AM, Adamik J, Palsson‐McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL‐1beta through HIF‐1alpha. Nature. 2013;496(7444):238–42. 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42(3):419–30. 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 50.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF‐alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 51.Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido‐Perrig N, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol. 2008;9(11):1261–9. 10.1038/ni.1657. [DOI] [PubMed] [Google Scholar]
- 52.He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, et al. Citric acid cycle intermediates as ligands for orphan G‐protein‐coupled receptors. Nature. 2004;429(6988):188–93. 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 53.Littlewood‐Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med. 2016;213(9):1655–62. 10.1084/jem.20160061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α‐KG‐dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–38. 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dalla Pozza E, Dando I, Pacchiana R, Liboi E, Scupoli MT, Donadelli M, et al. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin Cell Dev Biol. 2020;98:4–14. 10.1016/j.semcdb.2019.04.013. [DOI] [PubMed] [Google Scholar]
- 56.Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016;24(1):158–66. 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, et al. Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta‐ATF3 inflammatory axis. Nature. 2018;556(7702):501–4. 10.1038/s41586-018-0052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti‐inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556(7699):113–7. 10.1038/nature25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weiss JM, Davies LC, Karwan M, Ileva L, Ozaki MK, Cheng RY, et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J Clin Invest. 2018;128(9):3794–805. 10.1172/JCI99169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi HZ, Wang D, Sun XN, Sheng L. MicroRNA‐378 acts as a prognosis marker and inhibits cell migration, invasion and epithelial‐mesenchymal transition in human glioma by targeting IRG1. Eur Rev Med Pharmacol Sci. 2018;22(12):3837–46. 10.26355/eurrev_201806_15268. [DOI] [PubMed] [Google Scholar]
- 61.Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR‐ and HIF‐1alpha‐mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1250684. 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24(6):807–19. 10.1016/j.cmet.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30(4):406–10. 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 64.Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, et al. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer. 2010;126(12):2762–72. 10.1002/ijc.25202. [DOI] [PubMed] [Google Scholar]
- 65.Blewett MM, Xie J, Zaro BW, Backus KM, Altman A, Teijaro JR, et al. Chemical proteomic map of dimethyl fumarate‐sensitive cysteines in primary human T cells. Sci Signal. 2016;9(445):rs10. 10.1126/scisignal.aaf7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cancer Genome Atlas Research N, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, et al. Comprehensive, integrative genomic analysis of diffuse lower‐grade gliomas. N Engl J Med. 2015;372(26):2481–98. 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ye D, Guan KL, Xiong Y. Metabolism, activity, and targeting of D‐ and L‐2‐hydroxyglutarates. Trends Cancer. 2018;4(2):151–65. 10.1016/j.trecan.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prensner JR, Chinnaiyan AM. Metabolism unhinged: IDH mutations in cancer. Nat Med. 2011;17(3):291–3. 10.1038/nm0311-291. [DOI] [PubMed] [Google Scholar]
- 69.Waitkus MS, Diplas BH, Yan H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell. 2018;34(2):186–95. 10.1016/j.ccell.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature. 2009;462(7274):739–44. 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ward PS, Patel J, Wise DR, Abdel‐Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia‐associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha‐ketoglutarate to 2‐hydroxyglutarate. Cancer Cell. 2010;17(3):225–34. 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ježek P. 2‐Hydroxyglutarate in cancer cells. Antioxid Redox Signaling. 2020;33(13):903–26. 10.1089/ars.2019.7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–83. 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature. 2017;548(7666):228–33. 10.1038/nature23475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia‐inducible factor 1. Cell. 2011;146(5):772–84. 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)‐2‐hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339(6127):1621–5. 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S‐2‐hydroxyglutarate regulates CD8(+) T‐lymphocyte fate. Nature. 2016;540(7632):236–41. 10.1038/nature20165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia‐inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. 2013;14(11):1173–82. 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Osinalde N, Mitxelena J, Sanchez‐Quiles V, Akimov V, Aloria K, Arizmendi JM, et al. Nuclear phosphoproteomic screen uncovers ACLY as mediator of IL‐2‐induced proliferation of CD4+ T lymphocytes. Mol Cell Proteomics. 2016;15(6):2076–92. 10.1074/mcp.M115.057158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30(1):147–60. 10.1016/j.ccell.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schmidt C, Sciacovelli M, Frezza C. Fumarate hydratase in cancer: A multifaceted tumour suppressor. Semin Cell Dev Biol. 2020;98:15–25. 10.1016/j.semcdb.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martínez‐Reyes I, Chandel NS. Acetyl‐CoA‐directed gene transcription in cancer cells. Genes Dev. 2018;32(7‐8):463–5. 10.1101/gad.315168.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Currie E, Schulze A, Zechner R, Walther TC, FareseRV, Jr.Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18(2):153–61. 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martinez‐Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer‐associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. 2014;25:47–60. 10.1016/j.semcancer.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 85.Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD‐1(+) CD8(+) T cell pool with predictive potential in non‐small‐cell lung cancer treated with PD‐1 blockade. Nat Med. 2018;24(7):994–1004. 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McDonald G, Deepak S, Miguel L, Hall CJ, Isenberg DA, Magee AI, et al. Normalizing glycosphingolipids restores function in CD4+ T cells from lupus patients. J Clin Invest. 2014;124(2):712–24. 10.1172/JCI69571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zech T, Ejsing CS, Gaus K, de Wet B, Shevchenko A, Simons K, et al. Accumulation of raft lipids in T‐cell plasma membrane domains engaged in TCR signalling. EMBO J. 2009;28(5):466–76. 10.1038/emboj.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. 2016;531(7596):651–5. 10.1038/nature17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32(3):377–91 e9. 10.1016/j.ccell.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haghikia A, Jorg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. 2015;43(4):817–29. 10.1016/j.immuni.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 91.Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, et al. Short‐chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR‐S6K pathway. Mucosal Immunology. 2015;8(1):80–93. 10.1038/mi.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Niu Z, Shi Q, Zhang W, Shu Y, Yang N, Chen B, et al. Caspase‐1 cleaves PPARgamma for potentiating the pro‐tumor action of TAMs. Nat Commun. 2017;8(1):766. 10.1038/s41467-017-00523-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cubillos‐Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales‐Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti‐tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527–38. 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Al‐Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor‐associated myeloid‐derived suppressor cells. Oncoimmunology. 2017;6(10):e1344804. 10.1080/2162402X.2017.1344804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu Q, Li B, Sun S, Sun S. Macrophage plasticity in adipose tissue: implication for obesity‐related cancer. Under Review.
- 96.Kannan Y, Perez‐Lloret J, Li Y, Entwistle LJ, Khoury H, Papoutsopoulou S, et al. TPL‐2 regulates macrophage lipid metabolism and M2 differentiation to control TH2‐mediated immunopathology. PLoS Pathog. 2016;12(8):e1005783. 10.1371/journal.ppat.1005783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell. 2015;163(7):1716–29. 10.1016/j.cell.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sag D, Cekic C, Wu R, Linden J, Hedrick CC. The cholesterol transporter ABCG1 links cholesterol homeostasis and tumour immunity. Nat Commun. 2015;6:6354. 10.1038/ncomms7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li T, Le A. Glutamine metabolism in cancer. Adv Exp Med Biol. 2018;1063:13–32. 10.1007/978-3-319-77736-8_2. [DOI] [PubMed] [Google Scholar]
- 100.Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. 2010;185(2):1037–44. 10.4049/jimmunol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine‐dependent alpha‐ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal. 2015;8(396):ra97. 10.1126/scisignal.aab2610. [DOI] [PubMed] [Google Scholar]
- 102.Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, et al. Reinforce the antitumor activity of CD8(+) T cells via glutamine restriction. Cancer Sci. 2018;109(12):3737–50. 10.1111/cas.13827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.MO Johnson, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase‐dependent metabolism. Cell. 2018;175(7):1780–95 e19. 10.1016/j.cell.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gross MI, Demo SD, Dennison JB, Chen L, Chernov‐Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB‐839 in triple‐negative breast cancer. Mol Cancer Ther. 2014;13(4):890–901. 10.1158/1535-7163.MCT-13-0870. [DOI] [PubMed] [Google Scholar]
- 105.Tannir NM, Motzer RJ, Agarwal N, Liu P‐Y, Whiting SH, O'Keeffe B, et al. CANTATA: A randomized phase 2 study of CB‐839 in combination with cabozantinib vs. placebo with cabozantinib in patients with advanced/metastatic renal cell carcinoma. J Clin Oncol. 2018;36(15_suppl):TPS4601‐TPS. 10.1200/JCO.2018.36.15_suppl.TPS4601. [DOI] [Google Scholar]
- 106.Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL‐2 inhibition. Blood. 2015;126(11):1346–56. 10.1182/blood-2015-01-621870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parlati F, Bromley‐Dulfano S, Demo S, Janes J, Gross M, Lewis E, et al. Antitumor activity of the glutaminase inhibitor CB‐839 in hematological malignances. Blood. 2013;122(21):4226–. . [DOI] [Google Scholar]
- 108.Vogl DT, Younes A, Stewart K, Orford KW, Bennett M, Siegel D, et al. Phase 1 study of CB‐839, a first‐in‐class, glutaminase inhibitor in patients with multiple myeloma and lymphoma. Blood. 2015;126(23):3059–. . [DOI] [Google Scholar]
- 109.Shanker A, de Aquino MTP, Hodo T, Uzhachenko R. Glutamate receptors provide costimulatory signals to improve T cell immune response. J Immunol. 2018;200(1 Supplement):47.24–47.24. [Google Scholar]
- 110.Poulopoulou C, Markakis I, Davaki P, Nikolaou C, Poulopoulos A, Raptis E, et al. Modulation of voltage‐gated potassium channels in human T lymphocytes by extracellular glutamate. Mol Pharmacol. 2005;67(3):856–67. 10.1124/mol.67.3. [DOI] [PubMed] [Google Scholar]
- 111.Lemos H, Huang L, Prendergast GC, Mellor AL. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat Rev Cancer. 2019;19(3):162–75. 10.1038/s41568-019-0106-z. [DOI] [PubMed] [Google Scholar]
- 112.Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. 2016;16(10):599–611. 10.1038/nri.2016.80. [DOI] [PubMed] [Google Scholar]
- 113.Rodriguez PC, Quiceno DG, Ochoa AC. L‐arginine availability regulates T‐lymphocyte cell‐cycle progression. Blood. 2007;109(4):1568–73. 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by L‐arginine. J Biol Chem. 2002;277(24):21123–9. 10.1074/jbc.M110675200. [DOI] [PubMed] [Google Scholar]
- 115.Mondanelli G, Ugel S, Grohmann U, Bronte V. The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr Opin Pharmacol. 2017;35:30–9. 10.1016/j.coph.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 116.Shipp C, Speigl L, Janssen N, Martens A, Pawelec G. A clinical and biological perspective of human myeloid‐derived suppressor cells in cancer. Cellular and Molecular Life Sciences: CMLS. 2016;73(21):4043–61. 10.1007/s00018-016-2278-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bronte V, Zanovello P. Regulation of immune responses by L‐arginine metabolism. Nat Rev Immunol. 2005;5(8):641–54. 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 118.He X, Lin H, Yuan L, Li B. Combination therapy with L‐arginine and alpha‐PD‐L1 antibody boosts immune response against osteosarcoma in immunocompetent mice. Cancer Biol Ther. 2017;18(2):94–100. 10.1080/15384047.2016.1276136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L‐arginine modulates T cell metabolism and enhances survival and anti‐tumor activity. Cell. 2016;167(3):829–42 e13. 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB‐1158 blocks myeloid cell‐mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5(1):101. 10.1186/s40425-017-0308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kelly MP, Jungbluth AA, Wu BW, Bomalaski J, Old LJ, Ritter G. Arginine deiminase PEG20 inhibits growth of small cell lung cancers lacking expression of argininosuccinate synthetase. Br J Cancer. 2012;106(2):324–32. 10.1038/bjc.2011.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Qiu F, Chen YR, Liu X, Chu CY, Shen LJ, Xu J, et al. Arginine starvation impairs mitochondrial respiratory function in ASS1‐deficient breast cancer cells. Sci Signal. 2014;7(319):ra31. 10.1126/scisignal.2004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brin E, Wu K, Lu HT, He Y, Dai Z, He W. PEGylated arginine deiminase can modulate tumor immune microenvironment by affecting immune checkpoint expression, decreasing regulatory T cell accumulation and inducing tumor T cell infiltration. Oncotarget. 2017;8(35):58948–63. 10.18632/oncotarget.19564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee GK, Park HJ, Macleod M, Chandler P, Munn DH, Mellor AL. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology. 2002;107(4):452–60. 10.1046/j.1365-2567.2002.01526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Amobi A, Qian F, Lugade AA, Odunsi K. Tryptophan catabolism and cancer immunotherapy targeting IDO mediated immune suppression. Adv Exp Med Biol. 2017;1036:129–44. 10.1007/978-3-319-67577-0_9. [DOI] [PubMed] [Google Scholar]
- 126.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3‐dioxygenase. Immunity. 2005;22(5):633–42. 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 127.Cronin SJF, Seehus C, Weidinger A, Talbot S, Reissig S, Seifert M, et al. The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature. 2018;563(7732):564–8. 10.1038/s41586-018-0701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat Immunol. 2019;20(1):50–63. 10.1038/s41590-018-0255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zheng X, Koropatnick J, Chen D, Velenosi T, Ling H, Zhang X, et al. Silencing IDO in dendritic cells: A novel approach to enhance cancer immunotherapy in a murine breast cancer model. Int J Cancer. 2013;132(4):967–77. 10.1002/ijc.27710. [DOI] [PubMed] [Google Scholar]
- 130.Yen MC, Lin CC, Chen YL, Huang SS, Yang HJ, Chang CP, et al. A novel cancer therapy by skin delivery of indoleamine 2,3‐dioxygenase siRNA. Clin Cancer Res. 2009;15(2):641–9. 10.1158/1078-0432.CCR-08-1988. [DOI] [PubMed] [Google Scholar]
- 131.Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor‐expressed IDO recruits and activates MDSCs in a Treg‐dependent manner. Cell Rep. 2015;13(2):412–24. 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zakharia Y, Drabick J, Khleif S, Munn D, Link C, Vahanian N, et al. Results of phase 1b trial of the indoleamine 2,3‐dioxygenase (IDO) pathway inhibitor indoximod plus ipilimumab for the treatment of unresectable stage III or IV melanoma. Eur J Cancer. 2015;51:S108. 10.1016/S0959-8049(16)30315-X. [DOI] [Google Scholar]
- 133.Yue EW, Sparks R, Polam P, Modi D, Douty B, Wayland B, et al. INCB24360 (epacadostat), a highly potent and selective indoleamine‐2,3‐dioxygenase 1 (IDO1) inhibitor for immuno‐oncology. ACS Med Chem Lett. 2017;8(5):486–91. 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO‐301/KEYNOTE‐252): A phase 3, randomised, double‐blind study. Lancet Oncol. 2019;20(8):1083–97. 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- 135.Qi Wu XC, Li Juanjuan, Sun Shengrong. Serine and metabolism regulation: A novel mechanism in antitumor immunity and senescence. Aging and Disease. 2020;0–. 10.14336/ad.2020.0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang M, Vousden KH. Serine and one‐carbon metabolism in cancer. Nat Rev Cancer. 2016;16(10):650–62. 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- 137.Ouyang Y, Wu Q, Li J, Sun S, S‐adenosylmethionine Sun S.: A metabolite critical to the regulation of autophagy. Cell Prolif. 2020:e12891. 10.1111/cpr.12891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 2017;25(2):345–57. 10.1016/j.cmet.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 139.Jones RG, Pearce EJ. MenTORing immunity: mTOR signaling in the development and function of tissue‐resident immune cells. Immunity. 2017;46(5):730–42. 10.1016/j.immuni.2017.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Casey SC, Baylot V, Felsher DW. MYC: Master regulator of immune privilege. Trends Immunol. 2017;38(4):298–305. 10.1016/j.it.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30(1‐2):42–59. 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kurniawan H, Franchina DG, Guerra L, Bonetti L, Baguet LS, Grusdat M, et al. Glutathione restricts serine metabolism to preserve regulatory T cell function. Cell Metab. 2020. 10.1016/j.cmet.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nabeyama A, Kurita A, Asano K, Miyake Y, Yasuda T, Miura I, et al. xCT deficiency accelerates chemically induced tumorigenesis. PNAS. 2010;107(14):6436–41. 10.1073/pnas.0912827107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Khalafalla FG, Greene S, Khan H, Ilves K, Monsanto MM, AlvarezR, Jr., et al. P2Y2 nucleotide receptor prompts human cardiac progenitor cell activation by modulating hippo signaling. Circ Res. 2017;121(11):1224–36. 10.1161/CIRCRESAHA.117.310812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pitt JM, Kroemer G, Zitvogel L. Immunogenic and non‐immunogenic cell death in the tumor microenvironment. Adv Exp Med Biol. 2017;1036:65–79. 10.1007/978-3-319-67577-0_5. [DOI] [PubMed] [Google Scholar]
- 146.Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16(3):177–92. 10.1038/nri.2016.4. [DOI] [PubMed] [Google Scholar]
- 147.Bao L, Locovei S, Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004;572(1‐3):65–8. 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 148.Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277(12):10482–8. 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 149.Sakaki H, Tsukimoto M, Harada H, Moriyama Y, Kojima S. Autocrine regulation of macrophage activation via exocytosis of ATP and activation of P2Y11 receptor. PLoS One. 2013;8(4):e59778. 10.1371/journal.pone.0059778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lazarowski ER, Sesma JI, Seminario‐Vidal L, Kreda SM. Molecular mechanisms of purine and pyrimidine nucleotide release. Adv Pharmacol. 2011;61:221–61. 10.1016/B978-0-12-385526-8.00008-4. [DOI] [PubMed] [Google Scholar]
- 151.Qi W, Hanpu Z, Si S, Lijun W, Shengrong S. Extracellular vesicles and immunogenic stress in cancer. Under review.
- 152.Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. 2018. 10.1038/s41568-018-0037-0. [DOI] [PubMed] [Google Scholar]
- 153.Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy‐induced intratumoral recruitment and differentiation of antigen‐presenting cells. Immunity. 2013;38(4):729–41. 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 154.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy‐dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334(6062):1573–7. 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 155.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL‐1beta‐dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–8. 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 156.Kepp O, Loos F, Liu P, Kroemer G. Extracellular nucleosides and nucleotides as immunomodulators. Immunol Rev. 2017;280(1):83–92. 10.1111/imr.12571. [DOI] [PubMed] [Google Scholar]
- 157.Steingold JM, Hatfield SM. Targeting hypoxia‐A2A adenosinergic immunosuppression of antitumor T cells during cancer immunotherapy. Front Immunol. 2020;11:570041. 10.3389/fimmu.2020.570041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Krys D, Hamann I, Wuest M, Wuest F. Effect of hypoxia on human equilibrative nucleoside transporters hENT1 and hENT2 in breast cancer. FASEB J. 2019;33(12):13837–51. 10.1096/fj.201900870RR. [DOI] [PubMed] [Google Scholar]
- 159.Turcotte M, Spring K, Pommey S, Chouinard G, Cousineau I, George J, et al. CD73 is associated with poor prognosis in high‐grade serous ovarian cancer. Cancer Res. 2015;75(21):4494–503. 10.1158/0008-5472.CAN-14-3569. [DOI] [PubMed] [Google Scholar]
- 160.Inoue Y, Yoshimura K, Kurabe N, Kahyo T, Kawase A, Tanahashi M, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non‐small‐cell lung cancer. Oncotarget. 2017;8(5):8738–51. 10.18632/oncotarget.14434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. 2010;139(3):1030–40. 10.1053/j.gastro.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD‐L1‐blockade resistance in tumor. Nat Immunol. 2017;18(12):1332–41. 10.1038/ni.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ohradanova‐Repic A, Machacek C, Charvet C, Lager F, Le Roux D, Platzer R, et al. Extracellular purine metabolism is the switchboard of immunosuppressive macrophages and a novel target to treat diseases with macrophage imbalances. Front Immunol. 2018;9:852. 10.3389/fimmu.2018.00852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Boison D, Yegutkin GG. Adenosine metabolism: Emerging concepts for cancer therapy. Cancer Cell. 2019;36(6):582–96. 10.1016/j.ccell.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti‐PD‐1 and anti‐CTLA‐4 mAbs. Clin Cancer Res. 2013;19(20):5626–35. 10.1158/1078-0432.CCR-13-0545. [DOI] [PubMed] [Google Scholar]
- 166.Butler JJ, Mader JS, Watson CL, Zhang H, Blay J, Hoskin DW. Adenosine inhibits activation‐induced T cell expression of CD2 and CD28 co‐stimulatory molecules: Role of interleukin‐2 and cyclic AMP signaling pathways. J Cell Biochem. 2003;89(5):975–91. 10.1002/jcb.10562. [DOI] [PubMed] [Google Scholar]
- 167.Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Myeloid‐derived suppressor cells contribute to A2B adenosine receptor‐induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget. 2015;6(29):27478–89. 10.18632/oncotarget.4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li L, Huang L, Ye H, Song SP, Bajwa A, Lee SJ, et al. Dendritic cells tolerized with adenosine A(2)AR agonist attenuate acute kidney injury. J Clin Invest. 2012;122(11):3931–42. 10.1172/JCI63170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jackson SW, Hoshi T, Wu Y, Sun X, Enjyoji K, Cszimadia E, et al. Disordered purinergic signaling inhibits pathological angiogenesis in cd39/Entpd1‐null mice. Am J Pathol. 2007;171(4):1395–404. 10.2353/ajpath.2007.070190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang L, Zhou X, Zhou T, Ma D, Chen S, Zhi X, et al. Ecto‐5'‐nucleotidase promotes invasion, migration and adhesion of human breast cancer cells. J Cancer Res Clin Oncol. 2008;134(3):365–72. 10.1007/s00432-007-0292-z. [DOI] [PubMed] [Google Scholar]
- 171.Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine receptor 2A blockade increases the efficacy of anti‐PD‐1 through enhanced antitumor T‐cell responses. Cancer Immunol Res. 2015;3(5):506–17. 10.1158/2326-6066.CIR-14-0211. [DOI] [PubMed] [Google Scholar]
- 172.McCaffery I, Laport G, Hotson A, Willingham S, Patnaik A, Beeram M, et al. Biomarker and clinical activity of CPI‐444, a novel small molecule inhibitor of A2A receptor (A2AR), in a Ph1b study in advanced cancers. Ann Oncol. 2016;27:vi124. 10.1093/annonc/mdw368.32. [DOI] [Google Scholar]
- 173.Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18(1):157. 10.1186/s12943-019-1089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM, Karagiannis TC. Cancer metabolism and the Warburg effect: The role of HIF‐1 and PI3K. Mol Biol Rep. 2015;42(4):841–51. 10.1007/s11033-015-3858-x. [DOI] [PubMed] [Google Scholar]
- 175.Luo J, Langer LF, Liu J. A novel role of LncRNA in regulating tumor metabolism and angiogenesis under hypoxia. Cancer Communications (London, England). 2019;39(1):2. 10.1186/s40880-019-0348-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sang LJ, Ju HQ, Liu GP, Tian T, Ma GL, Lu YX, et al. LncRNA CamK‐A regulates Ca(2+)‐signaling‐mediated tumor microenvironment remodeling. Mol Cell. 2018;72(1):71–83.e7. 10.1016/j.molcel.2018.08.014. [DOI] [PubMed] [Google Scholar]
- 177.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8(9):705–13. 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 178.Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32(3):377–91.e9. 10.1016/j.ccell.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 180.Najjar YG, Menk AV, Sander C, Rao U, Karunamurthy A, Bhatia R, et al. Tumor cell oxidative metabolism as a barrier to PD‐1 blockade immunotherapy in melanoma. JCI Insight. 2019;4(5). 10.1172/jci.insight.124989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tiwari P, Blank A, Cui C, Schoenfelt KQ, Zhou G, Xu Y, et al. Metabolically activated adipose tissue macrophages link obesity to triple‐negative breast cancer. J Exp Med. 2019;216(6):1345–58. 10.1084/jem.20181616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL. Anthracycline chemotherapy inhibits HIF‐1 transcriptional activity and tumor‐induced mobilization of circulating angiogenic cells. PNAS. 2009;106(7):2353–8. 10.1073/pnas.0812801106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 183.Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF‐1 dimerization, tumor growth, and vascularization. PNAS. 2009;106(42):17910–5. 10.1073/pnas.0909353106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 184.Yu T, Tang B, Sun X. Development of inhibitors targeting hypoxia‐inducible factor 1 and 2 for cancer therapy. Yonsei Med J. 2017;58(3):489–96. 10.3349/ymj.2017.58.3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Viallard C, Larrivée B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis. 2017;20(4):409–26. 10.1007/s10456-017-9562-9. [DOI] [PubMed] [Google Scholar]
- 186.Kim JM, Chen DS. Immune escape to PD‐L1/PD‐1 blockade: Seven steps to success (or failure). Annals of Oncology: Official Journal of the European Society for Medical Oncology. 2016;27(8):1492–504. 10.1093/annonc/mdw217. [DOI] [PubMed] [Google Scholar]
- 187.Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096–103. 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 188.Ziogas AC, Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Terpos E, et al. VEGF directly suppresses activation of T cells from ovarian cancer patients and healthy individuals via VEGF receptor type 2. Int J Cancer. 2012;130(4):857–64. 10.1002/ijc.26094. [DOI] [PubMed] [Google Scholar]
- 189.Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: Myeloid‐derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138(2):105–15. 10.1111/imm.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 2016;537(7621):539–43. 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH, et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 2019;363(6434). 10.1126/science.aau0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti‐PD‐1 immunotherapy in melanoma patients. Science. 2017. 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD‐1‐based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–7. 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 194.Martin FP, Dumas ME, Wang Y, Legido‐Quigley C, Yap IK, Tang H, et al. A top‐down systems biology view of microbiome‐mammalian metabolic interactions in a mouse model. Mol Syst Biol. 2007;3:112. 10.1038/msb4100153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Usami M, Kishimoto K, Ohata A, Miyoshi M, Aoyama M, Fueda Y, et al. Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr Res. 2008;28(5):321–8. 10.1016/j.nutres.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 196.Vinolo MA, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R. Suppressive effect of short‐chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem. 2011;22(9):849–55. 10.1016/j.jnutbio.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 197.Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. PNAS. 2014;111(6):2247–52. 10.1073/pnas.1322269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom‐Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20(2):159–66. 10.1038/nm.3444. [DOI] [PubMed] [Google Scholar]
- 199.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T‐cell generation. Nature. 2013;504(7480):451–5. 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe‐derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–50. 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 201.Wang L, de Zoeten EF, Greene MI, Hancock WW. Immunomodulatory effects of deacetylase inhibitors: Therapeutic targeting of FOXP3+ regulatory T cells. Nat Rev Drug Discovery. 2009;8(12):969–81. 10.1038/nrd3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short‐chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341(6145):569–73. 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–6. 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40(1):128–39. 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gutierrez‐Vazquez C, Quintana FJ. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity. 2018;48(1):19–33. 10.1016/j.immuni.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour‐promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203. 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 207.Singh NP, Singh UP, Rouse M, Zhang J, Chatterjee S, Nagarkatti PS, et al. Dietary indoles suppress delayed‐type hypersensitivity by inducing a switch from proinflammatory Th17 cells to anti‐inflammatory regulatory T cells through regulation of MicroRNA. J Immunol. 2016;196(3):1108–22. 10.4049/jimmunol.1501727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S, et al. AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep. 2016;17(5):1318–29. 10.1016/j.celrep.2016.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. PNAS. 2010;107(48):20768–73. 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Pino‐Lagos K, Guo Y, Brown C, Alexander MP, Elgueta R, Bennett KA, et al. A retinoic acid‐dependent checkpoint in the development of CD4+ T cell‐mediated immunity. J Exp Med. 2011;208(9):1767–75. 10.1084/jem.20102358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin‐22. Immunity. 2013;39(2):372–85. 10.1016/j.immuni.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 212.Hubbard TD, Murray IA, Bisson WH, Lahoti TS, Gowda K, Amin SG, et al. Adaptation of the human aryl hydrocarbon receptor to sense microbiota‐derived indoles. Sci Rep. 2015;5:12689. 10.1038/srep12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.CaseroRA, Jr., Murray Stewart T, Pegg AE. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat Rev Cancer. 2018. 10.1038/s41568-018-0050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Di Martino ML, Campilongo R, Casalino M, Micheli G, Colonna B, Prosseda G. Polyamines: Emerging players in bacteria‐host interactions. International Journal of Medical Microbiology: IJMM. 2013;303(8):484–91. 10.1016/j.ijmm.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 215.Zhang M, Wang H, Tracey KJ. Regulation of macrophage activation and inflammation by spermine: A new chapter in an old story. Crit Care Med. 2000;28(4 Suppl):N60–6. 10.1097/00003246-200004001-00007. [DOI] [PubMed] [Google Scholar]
- 216.Zhu S, Ashok M, Li J, Li W, Yang H, Wang P, et al. Spermine protects mice against lethal sepsis partly by attenuating surrogate inflammatory markers. Mol Med. 2009;15(7‐8):275–82. 10.2119/molmed.2009.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kibe R, Kurihara S, Sakai Y, Suzuki H, Ooga T, Sawaki E, et al. Upregulation of colonic luminal polyamines produced by intestinal microbiota delays senescence in mice. Sci Rep. 2014;4:4548. 10.1038/srep04548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Perez‐Cano FJ, Gonzalez‐Castro A, Castellote C, Franch A, Castell M. Influence of breast milk polyamines on suckling rat immune system maturation. Dev Comp Immunol. 2010;34(2):210–8. 10.1016/j.dci.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 219.Johnson CH, Dejea CM, Edler D, Hoang LT, Santidrian AF, Felding BH, et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 2015;21(6):891–7. 10.1016/j.cmet.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hayes CS, Shicora AC, Keough MP, Snook AE, Burns MR, Gilmour SK. Polyamine‐blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol Res. 2014;2(3):274–85. 10.1158/2326-6066.CIR-13-0120-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pietrocola F, Castoldi F, Kepp O, Carmona‐Gutierrez D, Madeo F, Kroemer G. Spermidine reduces cancer‐related mortality in humans. Autophagy. 2018:1–4. 10.1080/15548627.2018.1539592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Madeo F, Eisenberg T, Pietrocola F, Kroemer G. Spermidine in health and disease. Science. 2018;359(6374). 10.1126/science.aan2788. [DOI] [PubMed] [Google Scholar]
- 223.Pietrocola F, Lachkar S, Enot DP, Niso‐Santano M, Bravo‐San Pedro JM, Sica V, et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015;22(3):509–16. 10.1038/cdd.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7(1):22–39. 10.1080/19490976.2015.1127483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol. 2013;14(7):676–84. 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Martinot E, Sedes L, Baptissart M, Lobaccaro JM, Caira F, Beaudoin C, et al. Bile acids and their receptors. Mol Aspects Med. 2017;56:2–9. 10.1016/j.mam.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 227.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol. 2009;183(10):6251–61. 10.4049/jimmunol.0803978. [DOI] [PubMed] [Google Scholar]
- 228.Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60(4):463–72. 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 229.Mencarelli A, Renga B, Migliorati M, Cipriani S, Distrutti E, Santucci L, et al. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. J Immunol. 2009;183(10):6657–66. 10.4049/jimmunol.0901347. [DOI] [PubMed] [Google Scholar]
- 230.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein‐coupled receptor responsive to bile acids. J Biol Chem. 2003;278(11):9435–40. 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 231.Guo C, Qi H, Yu Y, Zhang Q, Su J, Yu D, et al. The G‐protein‐coupled bile acid receptor Gpbar1 (TGR5) inhibits gastric inflammation through antagonizing NF‐kappaB signaling pathway. Frontiers in Pharmacology. 2015;6:287. 10.3389/fphar.2015.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome‐mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360(6391). 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature. 2019;576(7785):143–8. 10.1038/s41586-019-1785-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Sharma S, Erickson KM, Troutman JM. Complete tetrasaccharide repeat unit biosynthesis of the immunomodulatory bacteroides fragilis capsular polysaccharide A. ACS Chem Biol. 2017;12(1):92–101. 10.1021/acschembio.6b00931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Johnson JL, Jones MB, Cobb BA. Polysaccharide A from the capsule of bacteroides fragilis induces clonal CD4+ T cell expansion. J Biol Chem. 2015;290(8):5007–14. 10.1074/jbc.M114.621771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Dasgupta S, Erturk‐Hasdemir D, Ochoa‐Reparaz J, Reinecker HC, Kasper DL. Plasmacytoid dendritic cells mediate anti‐inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host & Microbe. 2014;15(4):413–23. 10.1016/j.chom.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The toll‐like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332(6032):974–7. 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Telesford KM, Yan W, Ochoa‐Reparaz J, Pant A, Kircher C, Christy MA, et al. A commensal symbiotic factor derived from bacteroides fragilis promotes human CD39(+)Foxp3(+) T cells and Treg function. Gut Microbes. 2015;6(4):234–42. 10.1080/19490976.2015.1056973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA‐4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–84. 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sivan A, Corrales L, Hubert N, Williams JB, Aquino‐Michaels K, Earley ZM, et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti‐PD‐L1 efficacy. Science. 2015;350(6264):1084–9. 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Roberti MP, Yonekura S, Duong CPM, Picard M, Ferrere G, Tidjani Alou M, et al. Chemotherapy‐induced ileal crypt apoptosis and the ileal microbiome shape immunosurveillance and prognosis of proximal colon cancer. Nat Med. 2020;26(6):919–31. 10.1038/s41591-020-0882-8. [DOI] [PubMed] [Google Scholar]
- 242.Fluckiger A, Daillère R, Sassi M, Sixt BS, Liu P, Loos F, et al. Cross‐reactivity between tumor MHC class I–restricted antigens and an enterococcal bacteriophage. Science. 2020;369(6506):936. 10.1126/science.aax0701. [DOI] [PubMed] [Google Scholar]
- 243.Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A defined commensal consortium elicits CD8 T cells and anti‐cancer immunity. Nature. 2019;565(7741):600–5. 10.1038/s41586-019-0878-z. [DOI] [PubMed] [Google Scholar]
- 244.Wang Y, Xie W, Humeau J, Chen G, Liu P, Pol J, et al. Autophagy induction by thiostrepton improves the efficacy of immunogenic chemotherapy. J Immunother Cancer. 2020;8(1). 10.1136/jitc-2019-000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Iida N, Mizukoshi E, Yamashita T, Terashima T, Arai K, Seishima J, et al. Overuse of anti‐anaerobic drug is associated with poor post‐chemotherapy prognosis of patients with hepatocellular carcinoma. Int J Cancer. 2019. 10.1002/ijc.32339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Prieto I, Montemuino S, Luna J, de Torres MV, Amaya E. The role of immunonutritional support in cancer treatment: Current evidence. Clin Nutr. 2017;36(6):1457–64. 10.1016/j.clnu.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 247.Lopez‐Otin C, Galluzzi L, Freije JMP, Madeo F, Kroemer G. Metabolic control of longevity. Cell. 2016;166(4):802–21. 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 248.Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762. 10.1038/ncomms11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA. Obesity and cancer mechanisms: Tumor microenvironment and inflammation. J Clin Oncol. 2016;34(35):4270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Font‐Burgada J, Sun B, Karin M. Obesity and cancer: The oil that feeds the flame. Cell Metab. 2016;23(1):48–62. 10.1016/j.cmet.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 251.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity‐induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499(7456):97–101. 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 252.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin‐A acts as an endogenous ligand of TLR4 to promote lipid‐induced insulin resistance. Nat Med. 2012;18(8):1279–85. 10.1038/nm.2851. [DOI] [PubMed] [Google Scholar]
- 253.Engin AB, Engin A, Gonul, II. The effect of adipocyte‐macrophage crosstalk in obesity‐related breast cancer. J Mol Endocrinol. 2019;62(3):R201–R22. 10.1530/JME-18-0252. [DOI] [PubMed] [Google Scholar]
- 254.Gruneberg RN. In vitro studies with combinations of sulfamoxole and trimethoprim (co‐trifamole). Curr Med Res Opin. 1982;8(2):128–33. 10.1185/03007998209109768. [DOI] [PubMed] [Google Scholar]
- 255.Conroy MJ, Dunne MR, Donohoe CL, Reynolds JV. Obesity‐associated cancer: An immunological perspective. Proc Nutr Soc. 2016;75(2):125–38. 10.1017/S0029665115004176. [DOI] [PubMed] [Google Scholar]
- 256.Lamas O, Marti A, Martinez JA. Obesity and immunocompetence. Eur J Clin Nutr. 2002;56(Suppl 3):S42–5. 10.1038/sj.ejcn.1601484. [DOI] [PubMed] [Google Scholar]
- 257.Yang H, Youm YH, Vandanmagsar B, Rood J, Kumar KG, Butler AA, et al. Obesity accelerates thymic aging. Blood. 2009;114(18):3803–12. 10.1182/blood-2009-03-213595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Macia L, Delacre M, Abboud G, Ouk TS, Delanoye A, Verwaerde C, et al. Impairment of dendritic cell functionality and steady‐state number in obese mice. J Immunol. 2006;177(9):5997–6006. 10.4049/jimmunol.177.9.5997. [DOI] [PubMed] [Google Scholar]
- 259.Ringel AE, Drijvers JM, Baker GJ, Catozzi A, García‐Cañaveras JC, Gassaway BM, et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti‐tumor immunity. Cell. 2020;183(7):1848–66.e26. 10.1016/j.cell.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD‐1 checkpoint blockade. Nat Med. 2019;25(1):141–51. 10.1038/s41591-018-0221-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.James BR, Tomanek‐Chalkley A, Askeland EJ, Kucaba T, Griffith TS, Norian LA. Diet‐induced obesity alters dendritic cell function in the presence and absence of tumor growth. J Immunol. 2012;189(3):1311–21. 10.4049/jimmunol.1100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Mirsoian A, Bouchlaka MN, Sckisel GD, Chen M, Pai CC, Maverakis E, et al. Adiposity induces lethal cytokine storm after systemic administration of stimulatory immunotherapy regimens in aged mice. J Exp Med. 2014;211(12):2373–83. 10.1084/jem.20140116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Levesque S, Pol JG, Ferrere G, Galluzzi L, Zitvogel L, Kroemer G. Trial watch: Dietary interventions for cancer therapy. Oncoimmunology. 2019;8(7):1591878. 10.1080/2162402X.2019.1591878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Clement K, Viguerie N, Poitou C, Carette C, Pelloux V, Curat CA, et al. Weight loss regulates inflammation‐related genes in white adipose tissue of obese subjects. FASEB J. 2004;18(14):1657–69. 10.1096/fj.04-2204com. [DOI] [PubMed] [Google Scholar]
- 265.O'Flanagan CH, Smith LA, McDonell SB, Hursting SD. When less may be more: Calorie restriction and response to cancer therapy. BMC Medicine. 2017;15(1):106. 10.1186/s12916-017-0873-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD‐1 blockade is potentiated by metformin‐induced reduction of tumor hypoxia. Cancer Immunol Res. 2017;5(1):9–16. 10.1158/2326-6066.Cir-16-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Afzal MZ, Dragnev K, Sarwar T, Shirai K. Clinical outcomes in non‐small‐cell lung cancer patients receiving concurrent metformin and immune checkpoint inhibitors. Lung Cancer Management. 2019;8(2):Lmt11. 10.2217/lmt-2018-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Gandhi S, Pandey M, Ammannagari N, Wang C, Bucsek MJ, Hamad L, et al. Impact of concomitant medication use and immune‐related adverse events on response to immune checkpoint inhibitors. Immunotherapy. 2020;12(2):141–9. 10.2217/imt-2019-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Afzal MZ, Mercado RR, Shirai K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti‐PD‐1/anti‐CTLA‐4) in metastatic malignant melanoma. J Immunother Cancer. 2018;6(1):64. 10.1186/s40425-018-0375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Sanchez A, Furberg H, Kuo F, Vuong L, Ged Y, Patil S, et al. Transcriptomic signatures related to the obesity paradox in patients with clear cell renal cell carcinoma: A cohort study. Lancet Oncol. 2020;21(2):283–93. 10.1016/S1470-2045(19)30797-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.McQuade JL, Daniel CR, Hess KR, Mak C, Wang DY, Rai RR, et al. Association of body‐mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: A retrospective, multicohort analysis. Lancet Oncol. 2018;19(3):310–22. 10.1016/s1470-2045(18)30078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Richtig G, Hoeller C, Wolf M, Wolf I, Rainer BM, Schulter G, et al. Body mass index may predict the response to ipilimumab in metastatic melanoma: An observational multi‐centre study. PLoS One. 2018;13(10):e0204729. 10.1371/journal.pone.0204729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Hollander D, Kampman E, van Herpen CM. Pretreatment body mass index and head and neck cancer outcome: A review of the literature. Crit Rev Oncol Hematol. 2015;96(2):328–38. 10.1016/j.critrevonc.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 274.Pietrocola F, Demont Y, Castoldi F, Enot D, Durand S, Semeraro M, et al. Metabolic effects of fasting on human and mouse blood in vivo. Autophagy. 2017;13(3):567–78. 10.1080/15548627.2016.1271513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Madeo F, Carmona‐Gutierrez D, Hofer SJ, Kroemer G. Caloric restriction mimetics against age‐associated disease: Targets, mechanisms, and therapeutic potential. Cell Metab. 2019;29(3):592–610. 10.1016/j.cmet.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 276.Traba J, Kwarteng‐Siaw M, Okoli TC, Li J, Huffstutler RD, Bray A, et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J Clin Invest. 2015;125(12):4592–600. 10.1172/JCI83260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Dror E, Dalmas E, Meier DT, Wueest S, Thevenet J, Thienel C, et al. Postprandial macrophage‐derived IL‐1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol. 2017;18(3):283–92. 10.1038/ni.3659. [DOI] [PubMed] [Google Scholar]
- 278.van Niekerk G, Loos B, Nell T, Engelbrecht AM. Autophagy–A free meal in sickness‐associated anorexia. Autophagy. 2016;12(4):727–34. 10.1080/15548627.2016.1147672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Madeo F, Pietrocola F, Eisenberg T, Kroemer G. Caloric restriction mimetics: Towards a molecular definition. Nat Rev Drug Discovery. 2014;13(10):727–40. 10.1038/nrd4391. [DOI] [PubMed] [Google Scholar]
- 280.Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. 2018;128(4):1238–46. 10.1172/JCI95148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Hertel J, Friedrich N, Wittfeld K, Pietzner M, Budde K, Van der Auwera S, et al. Measuring biological age via metabonomics: The metabolic age score. J Proteome Res. 2016;15(2):400–10. 10.1021/acs.jproteome.5b00561. [DOI] [PubMed] [Google Scholar]
- 282.Gonzalez‐Covarrubias V, Beekman M, Uh HW, Dane A, Troost J, Paliukhovich I, et al. Lipidomics of familial longevity. Aging Cell. 2013;12(3):426–34. 10.1111/acel.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Ma S, Lee SG, Kim EB, Park TJ, Seluanov A, Gorbunova V, et al. Organization of the mammalian ionome according to organ origin, lineage specialization, and longevity. Cell Reports. 2015;13(7):1319–26. 10.1016/j.celrep.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Gomes AP, Ilter D, Low V, Endress JE, Fernández‐García J, Rosenzweig A, et al. Age‐induced accumulation of methylmalonic acid promotes tumour progression. Nature. 2020. 10.1038/s41586-020-2630-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Viltard M, Durand S, Perez‐Lanzon M, Aprahamian F, Lefevre D, Leroy C, et al. The metabolomic signature of extreme longevity: Naked mole rats versus mice. Aging. 2019;11(14):4783–800. 10.18632/aging.102116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Li B, Geng R, Wu Q, Yang Q, Sun S, Zhu S, et al. Alterations in immune‐related genes as potential marker of prognosis in breast cancer. Front Oncol. 2020;10:333. 10.3389/fonc.2020.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence‐associated secretory phenotype: The dark side of tumor suppression. Annual Review of Pathology. 2010;5:99–118. 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–51. 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 289.Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence‐associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30(4):533–47. 10.1016/j.ccell.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer. 2020;20(9):516–31. 10.1038/s41568-020-0273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Giannone G, Ghisoni E, Genta S, Scotto G, Tuninetti V, Turinetto M, et al. Immuno‐metabolism and microenvironment in cancer: Key players for immunotherapy. Int J Mol Sci. 2020;21(12). 10.3390/ijms21124414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.