

Abstract
Heterogeneous nuclear ribonucleoprotein L‐like (HNRNPLL), a suppressor of colorectal cancer (CRC) metastasis, is transcriptionally downregulated when CRC cells undergo epithelial‐mesenchymal transition (EMT). Here we show that decrease of MYB mediates the downregulation of HNRNPLL during EMT. The promoter activity was attributed to a region from −273 to −10 base pairs upstream of the transcription start site identified by 5'‐RACE analysis, and the region contained potential binding sites for MYB and SP1. Luciferase reporter gene assays and knockdown or knockout experiments for genes encoding the MYB family proteins, MYB, MYBL1, and MYBL2, revealed that MYB was responsible for approximately half of the promoter activity. On the other hand, treatment with mithramycin A, an inhibitor for SP1 and SP3, suppressed the promoter activity and their additive contribution was confirmed by knockout experiments. The expression level of MYB was reduced on EMT while that of SP1 and SP3 was unchanged, suggesting that the downregulation of HNRNPLL during EMT was mediated by the decrease of MYB expression while SP1 and SP3 determine the basal transcription level of HNRNPLL. Histopathological analysis confirmed the accumulation of MYB‐downregulated cancer cells at the invasion front of clinical CRC tissues. These results provide an insight into the molecular mechanism underlying CRC progression.
Keywords: colorectal cancer, epithelial‐mesenchymal transition, HNRNP, MYB, SP1
Heterogeneous nuclear ribonucleoprotein L‐like (HNRNPLL), a suppressor of colorectal cancer (CRC) metastasis, is transcriptionally downregulated when CRC cells undergo epithelial‐mesenchymal transition (EMT). Here we show that decrease of MYB mediates the downregulation of HNRNPLL during EMT.

Abbreviations
- CRC
colorectal cancer
- EMT
epithelial‐mesenchymal transition
- HNRNPLL
heterogeneous nuclear ribonucleoprotein L‐like
- MET
mesenchymal‐epithelial transition
- MiA
mithramycin A
1. INTRODUCTION
Colorectal cancer (CRC) is one of the leading causes of cancer‐related deaths worldwide.1 Despite the recent advances in treatment of CRC patients, the prognosis remains unfavorable for those with distant metastasis; novel strategies for prevention and/or therapy of metastasis are awaited. We previously identified Hnrnpll (heterogeneous nuclear ribonucleoprotein L‐like) as a novel metastasis suppressor gene of CRC from an shRNA‐based functional in vivo screen using an orthotopic transplantation model of CRC metastasis.2 Hnrnpll encodes an RNA‐binding protein known as a regulator of the pre‐mRNA splicing in T and B lymphocytes.3, 4 Our study demonstrated that heterogeneous nuclear ribonucleoprotein L‐like (HNRNPLL) is downregulated at the transcript level in CRC cells during epithelial‐mesenchymal transition (EMT), a critical event in the early step of cancer metastasis,5, 6 and that the downregulation modulates the alternative splicing of CD44 variable exons leading to increased expression of the CD44v6 isoform, which enhances the invasion activity through an interaction with the hepatocyte growth factor receptor.2 In contrast, mesenchymal‐epithelial transition (MET) restores HNRNPLL, which then contributes to proliferation of CRC cells through increased expression of DNA replication factors, PCNA, RFC3, and FEN1, by binding to and stabilizing their pre‐mRNAs.7 This intriguing fluctuation of HNRNPLL expression during EMT and MET has prompted us to elucidate the underlying molecular mechanisms. Here, we demonstrate the role of MYB in transcriptional regulation of HNRNLLL in CRC cells undergoing EMT.
2. MATERIALS AND METHODS
2.1. Cell culture
Human colon cancer cell lines HT29 and DLD‐1 were obtained from the American Type Culture Collection (ATCC) and were maintained in DMEM (Nacalai Tesque) and RPMI (Nacalai Tesque) supplemented with 10% FBS (Thermo Fisher Scientific), respectively. EMT was induced as previously described using epidermal growth factor (Sigma‐Aldrich) at 20 ng/mL and basic fibroblast growth factor (Sigma‐Aldrich) at 10 ng/mL in the absence of FBS.8 In some experiments, mithramycin A (Sigma‐Aldrich) was added into the culture medium at a final concentration of 200 nmol/L 1 hour before transfection of reporter constructs in luciferase reporter assays or 24 hours before extraction of total RNA.
2.2. Rapid amplification of 5'‐complementary DNA ends
Total RNA prepared from HT29 cells was subjected to rapid amplification of 5'‐complementary DNA ends (5'‐RACE) using a SMARTer RACE 5', 3'‐Kit (Takara Bio) according to the manufacturer's protocol. Briefly, after first‐strand cDNA synthesis, PCR was performed using a Universal Primer Mix (UPM) and a gene specific primer for HNRNPLL with a sequence of 5'‐GATTACGCCAAGCTTCACCTGCCTCCGGCTGAGAGAAGCTCCGGC‐3'. The products were subjected to In‐Fusion Cloning using a pRACE vector and the subclones were processed with a BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) for DNA sequencing.
2.3. Reporter constructs and luciferase assays
Genomic DNA prepared from HT29 cells using ISOGEN reagent (Nippon Gene) was subjected to PCR with oligonucleotide primers including synthetic BglII or HindIII sites and PrimeSTAR Max DNA Polymerase (Takara Bio). Primer sequences used in this study are listed in Table S1. After digestion with BglII and HindIII (New England Biolabs), the PCR products were subcloned into a pGL4.10[luc2] vector (Promega). Mutant reporter constructs were prepared by PCR using primers with mutated DNA sequences followed by DpnI‐treatment and subcloning. For luciferase assays, the reporter constructs (1 μg) together with a phRL‐TK vector for normalization (Promega, 50 ng) were transfected into cells with Lipofectamine 3000 (Thermo Fisher Scientific). After 24 hours of cultivation, luciferase activity was measured using Dual‐Luciferase Reporter Assay System (Promega) according to the manufacturer's instruction.
2.4. Western blot
Cells were lysed using RIPA buffer containing blends of protease inhibitors (Nacalai Tesque), and the lysate was subjected to SDS‐PAGE followed by transfer onto PVDF membranes (Bio‐Rad). After blocking with Blocking‐One reagent (Nacalai Tesque) at room temperature for 60 minutes, the membranes were blotted with primary antibodies overnight at 4°C and with HRP‐conjugated secondary antibody (Southern Biotech) for 60 minutes at room temperature. The signals were visualized using Immobilon Western Chemiluminescent HRP Substrate (Merck Millipore). The antibodies used in this study are listed in Table S2.
2.5. Quantitative RT‐PCR
First‐strand cDNA was prepared from ISOGEN‐extracted total RNA using a High‐Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) with oligo (dT) primers. Quantitative RT‐PCR was performed with FAM‐labeled TaqMan Gene Expression Assays (listed in Table S3) using 7500 Fast Real‐Time PCR System (Thermo Fisher Scientific) according to the manufacturer's protocol. The results were obtained by the comparative CT method, by which a relative expression level of the gene of interest to that of GAPDH was obtained as 2‐ΔΔCT.
2.6. Constructs and lentiviral transduction
Single‐guide RNAs (sgRNAs) for the CRISPR‐Cas9 system were designed using GPP sgRNA Designer (https://portals.broadinstitute.org/gpp/public/analysis‐tools/sgrna‐design). The synthetic target oligonucleotides (Table S4) and its complementary oligonucleotides were hybridized and subcloned into a lentiCRISPR v2 vector (Addgene) and the sequences were confirmed by DNA sequencing. Expression vectors were prepared by subcloning PCR‐amplified cDNA into the lentiviral vector pLEX‐MCS (GE Healthcare). Lentivirus was prepared by introducing the vectors together with packaging vectors into HEK293T cells using Lipofectamine 3000. The supernatants were subjected to viral transduction with polybrene (Sigma‐Aldrich) at a final concentration of 8 μg/mL.
2.7. ChIP assay
Chromatin samples were prepared using a ChIP‐IT Express Enzymatic kit (Active Motif) according to the manufacturer's instructions. Enzymatic shearing was performed for 12 minutes at 37°C. The samples were incubated with anti‐FLAG (clone M2, mouse IgG1) or isotype control antibody overnight at 4°C. Primers used for PCR analysis are listed in Table S1.
2.8. Immunostaining
Paraffin sections of surgically resected human colon cancer tissues were obtained from Aichi Cancer Center Hospital under informed consent. The samples were subjected to deparaffinization, antigen retrieval in 95°C‐heated citrate buffer, blocking with PBS (‐) containing 5% normal goat serum and 0.3% Triton X‐100 for 60 minutes, incubation with primary antibodies overnight at 4°C, and incubation with Alexa‐Fluor‐conjugated secondary antibodies for 90 minutes at room temperature. Nuclear staining was performed using Hoechst 33342 (Dojindo) for 5 minutes before observation with the confocal microscope LSM800 (Carl Zeiss).
3. RESULTS
3.1. Identification of the 5'‐regulatory region of HNRNPLL in HT29 cells
To elucidate the mechanism of the transcriptional downregulation of HNRNPLL in CRC cells during EMT, we first performed a 5'‐RACE analysis to identify the transcription start site of HNRNPLL in HT29 cells, a human CRC cell line. Because the RefSeq database (https://www.ncbi.nlm.nih.gov/refseq/) lists two curated mRNA sequences for HNRNPLL (NM_138394 and NM_001142650), of which the latter lacks a segment of 256 bp found in the exon 1 of the former (Figure 1A), we designed a gene‐specific primer in the exon 1 region shared between the two mRNA sequences (Figure 1A). Electrophoresis of the RACE PCR product showed a major band around 550 bp and a minor band around 200 bp, corresponding to NM_138394 and NM_001142650, respectively (Figure 1B). These PCR products were subcloned and the transcription start site of HNRNPLL was determined by DNA sequencing of the subcloned vectors. To crudely identify the region including the promoter, we generated a luciferase reporter construct covering the 1650 bp upstream from the transcription start site (designated as R1, Figure 1C) and four additional deletion constructs (R2‐R5, Figure 1C). Luciferase activity of R2, R3, and R4 was statistically equivalent to that of R1, while R5 showed significantly decreased activity, suggesting that the region from −273 to −10 contains the promoter of HNRNPLL (Figure 1D). We then looked for potential transcription factor‐binding sites in the region using the public online program MATCH (http://gene‐regulation.com/cgi‐bin/pub/programs/match/bin/match.cgi) (Figure 1E). The result showed a number of candidate transcription factors, among which MYB and SP1 drew our attention. MYB was reported to be downregulated during EMT in CRC cells9 as well as in breast cancer cells10 and SP1 is known as a key transcriptional regulator of a variety of TATA‐less genes,11 a feature we found in the HNRNPLL gene (Figure 1E). We therefore decided to determine whether MYB and/or SP1 are involved in the transcriptional regulation of HNRNPLL.
FIGURE 1.
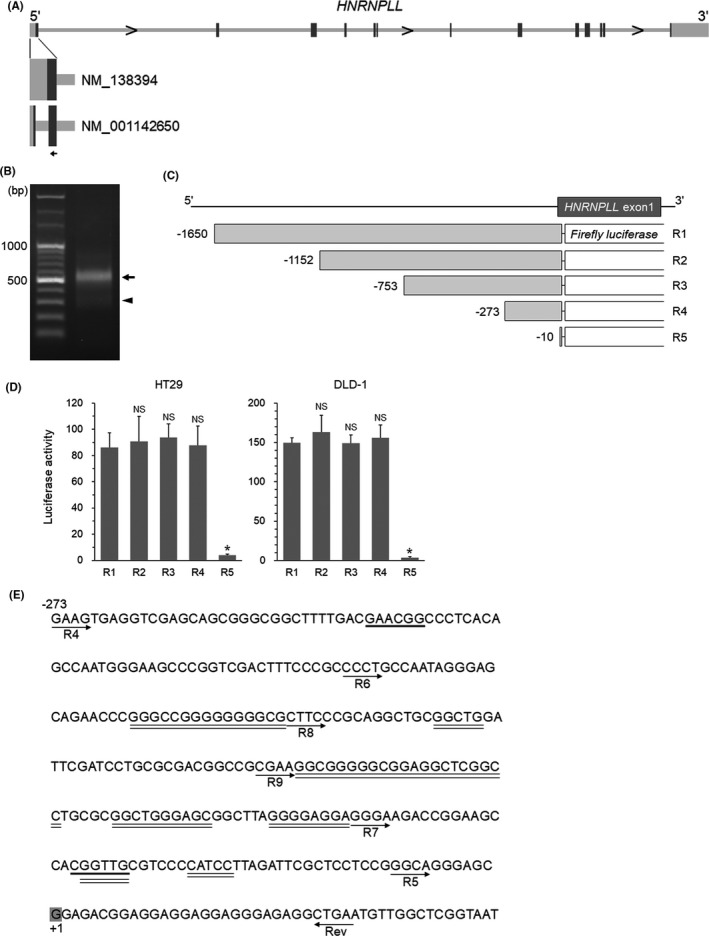
Identification of the 5'‐regulatory region of HNRNPLL. A, The gene structure of HNRNPLL. Boxes: exons; dark boxes, coding region; light boxes, non‐coding region; arrow, gene‐specific primer site for 5'‐RACE analysis. Two variants for the first exon are shown with RefSeq accession numbers. B, Electrophoresis of the 5'‐RACE PCR product. Arrow and arrowhead show bands corresponding to NM_138394 and NM_001142650, respectively. C, Schema of the reporter constructs. D, Luciferase activity of the R1‐5 reporters measured in HT29 and DLD‐1 cells. *P < .001; NS, not significant (P > .05). E, Nucleic acid sequence including the 5'‐regulatory region of HNRNPLL (−273 to +46). Locations of the individual forward primers used for the reporter constructs and their common reverse primer (Rev) are indicated with arrows. (The R6‐9 reporter constructs are used in Figures 2 and 3) The transcription start site is indicated with shaded "G". Potential binding sites for MYB and SP1 are shown with bold and double underlines, respectively
3.2. MYB positively regulates the transcription of HNRNPLL
To address the possible role of MYB, we first tested whether MYB is actually downregulated during EMT in CRC cells. We induced EMT in HT29 and DLD‐1 cells as we previously reported2, 7, 8 and confirmed EMT‐specific phenotypic alterations, that is, fibroblastic morphology (Figure S1A), increased SNAI1 (SNAIL) protein expression (Figure S1B), and decreased CDH1 (E‐cadherin) mRNA expression (Figure S1C). We were able to confirm that EMT induction in these cells resulted in decreased expression of MYB at the mRNA level (Figure S2A), in agreement with the previous report.9 As MYB has two closely related family member genes, MYBL1 and MYBL2,12 we also examined the expression levels of these genes and found that they were both also downregulated during EMT (Figure S2A). Western blot analysis indicated that the MYB protein level was drastically reduced upon EMT induction, whereas MYBL1 was hardly detectable and the MYBL2 level was very low and not affected by EMT (Figure 2A). We next prepared reporter constructs with mutations in the two potential MYB‐binding sites suggested by the MATCH program (Figure 2B). The results of luciferase assays indicated that the mutation at the distal MYB site in R4 (R4‐M1) significantly reduced the reporter activity to the level comparable to that of R6 (Figure 2C). Similarly, the mutation at the proximal MYB site in R7 (R7‐M2) significantly decreased the reporter activity to the level comparable to that of R5 (Figure 2C). To determine relative contribution of MYB and MYBL2 in regulating transcription of HNRNPLL at these sites, we prepared CRC cells lacking MYB and/or MYBL2 (Figure S2B) using the CRISPR‐Cas9 technique. Quantitative RT‐PCR analysis indicated that MYB depletion significantly suppressed HNRNPLL mRNA expression, whereas knockout of MYBL2 did not affect HNRNPLL mRNA levels (Figure 2D). The luciferase activity was comparable between R4 and R4‐M1 in MYB‐knockout cells, indicating the essential role of MYB in the distal MYB binding site. Unexpectedly, however, the luciferase activity of R7 was higher than that of R7‐M2 in MYB‐knockout cells (Figure 2E), suggesting that the proximal MYB binding site is regulated by transcription factors other than MYB. ChIP analysis of FLAG‐MYB‐introduced cells using a primer set bracketing the distal MYB binding site (Figure S2C) detected a PCR product both in HT29 and DLD‐1 cells, indicating a binding of MYB to the region (Figure 2F). We next examined whether downregulation of HNRNPLL during EMT could be rescued by restoring MYB. Overexpression of MYB in CRC cells undergoing EMT significantly increased the expression level of HNRNPLL to the level comparable to that in non‐EMT cells (Figures S2D and 2G). Taken together, these results demonstrate that MYB positively regulates transcription of HNRNPLL through binding to the distal MYB binding site in the HNRNPLL promoter and that the downregulation of HNRNPLL during EMT is mediated by decrease of MYB.
FIGURE 2.
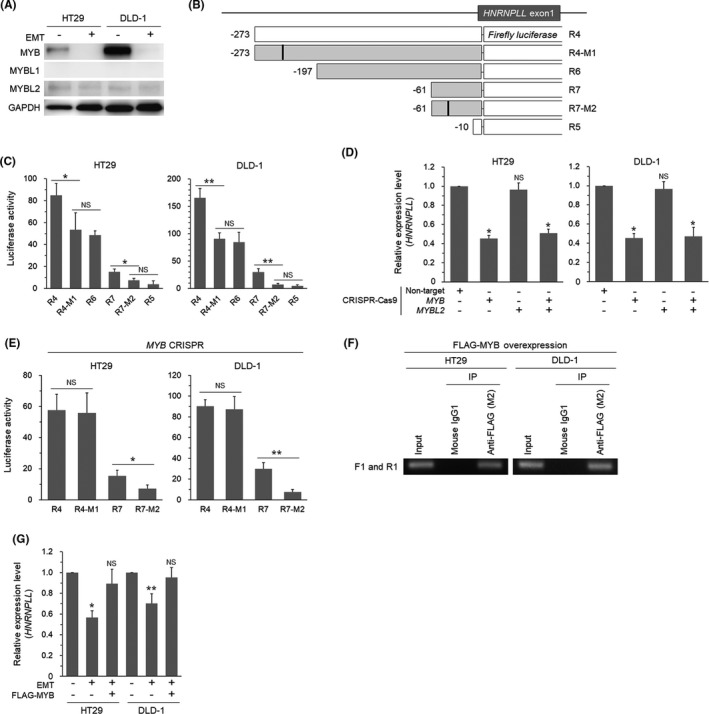
The MYB family proteins are involved in the transcriptional regulation of HNRNPLL. A, Western blot analysis for expression of MYB, MYBL1, and MYBL2 in HT29 and DLD‐1 cells with or without EMT‐induction. B, Reporter constructs for addressing the two potential MYB‐binding sites. Filled squares indicate additional reporters prepared for experiments in Figure 2. R4‐M1 and R7‐M2 are mutant constructs with synthetic base substitution in the distal and proximal MYB sites, respectively. C, Luciferase activity of the reporter constructs in HT29 and DLD‐1 cells. *P < .05; **P < .01; NS, not significant (P > .05). D, Quantitative RT‐PCR analysis for expression of HNRNPLL in HT29 and DLD‐1 cells transduced with CRISPR‐Cas9 for MYB and/or MYBL2. *P < .001; NS, not significant (P > .05). E, Luciferase activity of the reporter constructs in MYB‐knocked out cells. *P < .05; **P < .01; NS, not significant (P > .05). F, Agarose gel electrophoresis of the PCR products amplified from chromatins immunoprecipitated from FLAG‐MYB‐introduced cells using the ChIP primers F1 and R1. G, Quantitative RT‐PCR analysis for expression levels of HNRNPLL in EMT‐induced cells with or without MYB‐overexpression. *P < .0005; **P < .01; NS, not significant (P > .05)
3.3. SP1 and SP3 determine the basal transcription level of HNRNPLL
Since the results above showed that MYB regulates approximately a half of the promoter activity of HNRNPLL in CRC cells, we next tried to identify the transcription factors responsible for the rest of the activity, as indicated by the R6 construct (Figure 2C). The lack of a TATA box and the existence of many potential SP1‐binding sites in the HNRNPLL promoter region led us to focus on SP1. As the potential SP1‐binding sites were overlapping with each other, it was difficult to address an involvement of each site individually. We therefore performed luciferase reporter assays using two additional deletion constructs (Figure 3A) and found that the reporter activity decreased gradually with stepwise deletion from R6 to R5 (Figure 3B). Treatment with mithramycin A (MiA), an inhibitor for SP1 and SP3, suppressed the activity of the R6 reporter to the level comparable to that of the R5 reporter (Figure 3B), suggesting that the activity of the region −197 to −10 bp is regulated by SP1 and/or SP3. We next generated CRC cells lacking SP1 and/or SP3 (Figure S3A). The quantitative RT‐PCR analysis showed that knockout of SP1 and SP3 additively reduced the expression level of HNRNPLL (Figure 3C). ChIP assays using FLAG‐SP1/SP3‐introduced cells with two sets of PCR primers (Figure S3B) indicated the binding of SP1 and SP3 to the region (Figure 3D). Since the expression levels of SP1 and SP3 were not altered on EMT induction (Figure 3E) in HT29 and DLD‐1 cells, these transcription factors are likely to determine the basal expression level of HNRNPLL in CRC cells.
FIGURE 3.
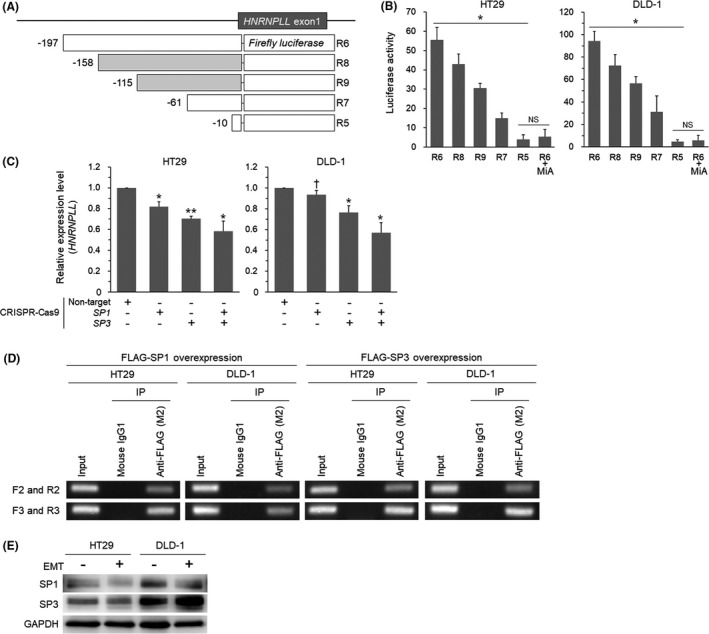
SP1 and SP3 are involved in the transcriptional regulation of HNRNPLL. A, Reporter constructs for addressing the potential SP1‐binding sites. Filled squares indicate reporters additionally prepared for experiments in Figure 3. B, Luciferase activity of the reporter constructs in HT29 and DLD‐1 cells. The effect of the SP1/3 inhibitor mithramycin A (MiA) was also tested for R6. *P < .001; NS, not significant (P > .05). C, Quantitative RT‐PCR analysis for expression levels of HNRNPLL in HT29 and DLD‐1 cells transduced with CRISPR‐Cas9 for SP1 and/or SP3. *P < .01; **P < .001; † P < .05. D, Agarose gel electrophoresis of the PCR products amplified from chromatins immunoprecipitated from FLAG‐SP1‐ or FLAG‐SP3‐overexpressed cells. E, Western blot analysis for expression levels of SP1 and SP3 in HT29 and DLD‐1 cells with or without EMT induction
3.4. MYB and HNRNPLL are downregulated at the invasion front of CRC tissues
We next assessed the clinical relevance of the downregulation of MYB and HNRNPLL in CRC cells undergoing EMT by immunostaining of the paraffin‐embedded sections obtained from surgically resected CRC tissues. Because we could not find a good combination of anti‐MYB and anti‐HNRNPLL antibodies suitable for double staining, MYB and HNRNPLL were individually stained. We first stained clinical samples with anti‐MYB and anti‐CDH1 antibodies and found that MYB was highly expressed in the nucleus of most of the cancer cells with high cell‐surface CDH1 expression composing glandular structure at the tumor center, whereas its nuclear staining was weak in a portion of cancer cells scattered around the invasion front with low cell‐surface CDH1 expression, a feature of EMT (Figures 4A and S4A). In line with this observation, HNRNPLL was downregulated in a portion of migrating CRC cells at the invasion front, as we previously reported (Figures 4B and S4B).2 In contrast, the levels of SP1 and SP3 in CRC cells were similar between the tumor center and the invasion front (Figures S5 and S6).
FIGURE 4.
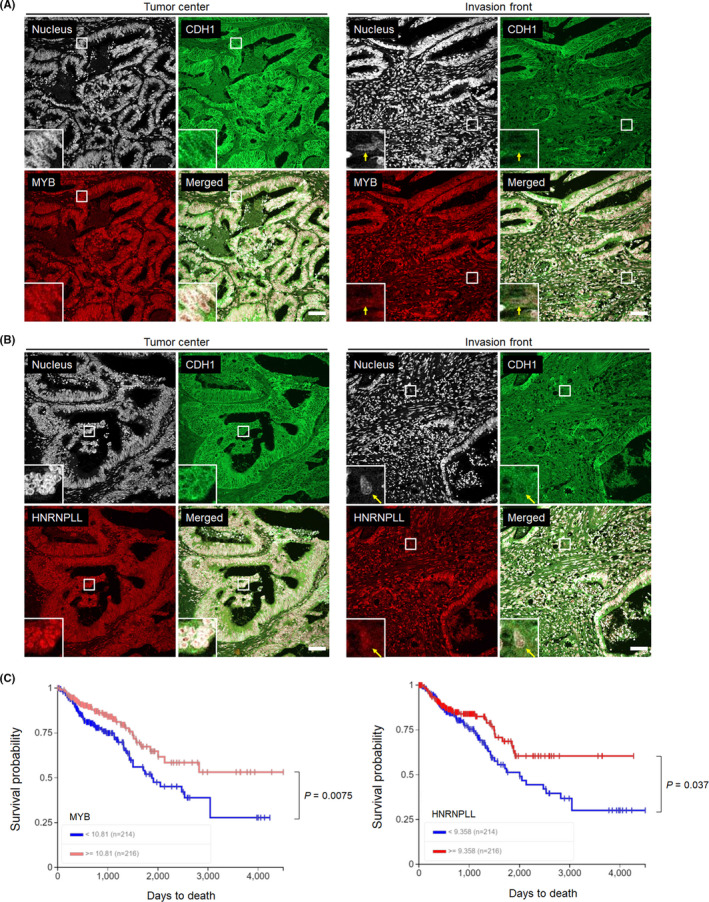
Assessment of the clinical relevance of MYB and HNRNPLL to CRC progression. A, Immunohistochemical analysis for MYB (green) and E‐cadherin (CDH1, red) in paraffin‐embedded sections obtained from surgically resected primary colon cancer tissues. Bars, 20 μm. A magnified view of the squared area is shown in the left‐bottom corner of each panel. Arrows indicate cancer cells showing EMT‐phenotypes, namely, low cell‐surface CDH1 expression and isolation from glandular structures. B, Sections from the same patient were stained for HNRNPLL (green) and CDH1 (red). Bars, 20 μm. C, Kaplan‐Meier analysis of the survival probability (days to death) of CRC patients according to high or low expression levels of MYB (left) or HNRNPLL (right) in the TCGA database. Statistical analysis was performed using the log‐rank chi‐square test
We finally addressed whether expression levels of MYB and HNRNPLL are associated with the prognosis of CRC patients. Kaplan‐Meier curves obtained from the Cancer Genome Atlas (TCGA) Research Network (https://www.cancer.gov/tcga) indicated that CRC patients with low MYB or HNRNPLL expression levels showed significantly poorer prognosis compared with those with high expression levels (Figure 4C). These results strongly suggest the clinical relevance of the EMT‐MYB‐HNRNPLL axis indicated from our in vitro experiments.
4. DISCUSSION
In this study, we identified MYB, SP1, and SP3 as transcriptional factors positively regulating the expression of HNRNPLL in CRC cells. Although our search by the MATCH program gave other potential transcription factors, results of luciferase assays using mutant reporter constructs (Figures 2C and 3B), MYB knockout cells (Figure 2E), and mithramycin A‐treated cells (Figure 3B) indicated that MYB, SP1, and SP3 are responsible for regulating the activity of the promoter region that we identified in this study. Nevertheless, it is possible that other transcription factors may also regulate the transcription of HNRNPLL through distant enhancer elements that remain to be identified.
The MYB protein is encoded by c‐Myb, a homologue of v‐Myb avian myeloblastosis viral oncogene, and has been implicated in not only in hematopoietic malignancies but also some solid tumors in humans, particularly CRC and breast cancer.13, 14, 15, 16, 17 In general, overexpression of MYB is believed to promote the initiation and maintenance of these malignancies. Many studies have demonstrated the contribution of MYB to the homeostasis of colonic tissues. For example, tissue‐specific depletion of MYB resulted in a major defect in the epithelial organization of the colon,18 and overexpression of MYB was shown to drive cell proliferation at least partly by enhancing CCNE1 (Cyclin E1) expression in cooperation with E2F1 and TCF4.19 We previously demonstrated that HNRNPLL promotes cell cycle progression through upregulating the expression of three DNA synthesis regulator genes, PCNA, RFC3, and FEN1, by binding with and stabilizing their pre‐messenger and messenger RNAs.7 Therefore, the positive link between MYB and HNRNPLL shown in this study suggests that MYB may drive cell proliferation by enhancing DNA synthesis as well. On the other hand, we previously identified HNRNPLL as a metastasis suppressor of CRC and proposed that its transient downregulation during EMT contributes to invasion and metastasis of CRC cells.2 This study has uncovered the molecular mechanism underlying this downregulation of HNRNPLL during EMT, and is, to our knowledge, the first to demonstrate the mechanism by which MYB downregulation during EMT can contribute to the enhanced invasion activity of CRC cells.
In this study, we also assessed the involvement of other MYB family members, MYBL1 and MYBL2, in the transcriptional regulation of HNRNPLL. MYBL1 is known to show highly restricted distribution, with major expression in the developing mammary gland and the spermatogenic tissue, and with minor expression in the central nervous system and T and B lymphocytes.20, 21, 22, 23 Accordingly, MYBL1 expression was undetectable in HT29 and DLD‐1 cells. Although unneglectable MYBL2 expression was detected in HT29 and DLD‐1 cells, MYBL2 was not downregulated during EMT (Figure 2A) and knockout of MYBL2 failed to enhance the suppression of HNRNPLL expression caused by MYB knockout (Figure 2D). Despite their nearly identical DNA‐binding domains, the MYB family proteins are known to exert distinct biological functions. For example, Rushton et al.24 demonstrated individual roles of MYB, MYBL1, and MYBL2 in mammary cells. Our data also indicate a functional segregation of MYB and MYBL2 in CRC cells.
We demonstrated that the basal transcription level of HNRNPLL is regulated by SP1 and SP3, members of the Specificity protein/Krüppel‐like factor (SP/KLF) transcription factor family. These factors are similar in the amino acid sequence of the DNA binding domains, and both of their common and distinct functions have been reported.25 In this study, the expression level of HNRNPLL mRNA was suppressed significantly by knockout of either SP1 or SP3, and to a greater extent by double‐knockout (Figure 3C), indicating that these factors additively enhance the transcription of HNRNPLL. The similar cooperative action of SP1 and SP3 has been reported for regulation of a number of genes, such as PKM, CHRNB4, and MET.26, 27, 28
Our data mining analysis using the TCGA database confirmed the negative correlation between MYB expression and the overall survival of CRC patients. In line with this result, there is a report that downregulation of MYB is associated with poor prognosis of CRC patients.29, 30 However, the opposite result has been reported, in which the MYB expression level was negatively correlated with the prognosis of CRC patients.31 We speculate that this discrepancy may be caused by the dual role of MYB in CRC progression through regulation of HNRNPLL; HNRNPLL enhances cancer cell proliferation while it suppresses cancer cell invasion. It will therefore be difficult to consider MYB or HNRNPLL as a target for monotherapy of CRC. Future studies are expected to identify novel downstream targets of HNRNPLL that positively regulate invasion and metastasis of CRC.
DISCLOSURE
The authors have no conflict of interest to declare.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Table S1‐S4
ACKNOWLEDGMENTS
This work was supported in part by Grant‐in‐Aid for Scientific Research C (16K07138 and 20K07581) to KS and Grant‐in‐Aid for Scientific Research B (18H02686) to MA from the Japan Society for the Promotion of Science, grants from the Takeda Science Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, the Nagono Medical Foundation, the Daiko Foundation, and the Princess Takamatsu Cancer Research Fund, Japan.
Sakuma K, Sasaki E, Hosoda W, et al. MYB mediates downregulation of the colorectal cancer metastasis suppressor heterogeneous nuclear ribonucleoprotein L‐like during epithelial‐mesenchymal transition. Cancer Sci. 2021;112:3846–3855. 10.1111/cas.15069
Contributor Information
Keiichiro Sakuma, Email: ksakuma@aichi-cc.jp.
Masahiro Aoki, Email: msaoki@aichi-cc.jp.
REFERENCES
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: Cancer J Clin. 2011;61:69‐90. [DOI] [PubMed] [Google Scholar]
- 2.Sakuma K, Sasaki E, Kimura K, et al. HNRNPLL, a newly identified colorectal cancer metastasis suppressor, modulates alternative splicing of CD44 during epithelial‐mesenchymal transition. Gut. 2018;67:1103‐1111. [DOI] [PubMed] [Google Scholar]
- 3.Benson MJ, Aijo T, Chang X, et al. Heterogeneous nuclear ribonucleoprotein L‐like (hnRNPLL) and elongation factor, RNA polymerase II, 2 (ELL2) are regulators of mRNA processing in plasma cells. Proc Natl Acad Sci USA. 2012;109:16252‐16257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oberdoerffer S, Moita LF, Neems D, Freitas RP, Hacohen N, Rao A. Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL. Science. 2008;321:686‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalluri R, Weinberg RA. The basics of epithelial‐mesenchymal transition. J Clin Invest. 2009;119:1420‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21‐45. [DOI] [PubMed] [Google Scholar]
- 7.Sakuma K, Sasaki E, Kimura K, et al. HNRNPLL stabilizes mRNA for DNA replication proteins and promotes cell cycle progression in colorectal cancer cells. Cancer Sci. 2018;109:2458‐2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakuma K, Aoki M, Kannagi R. Transcription factors c‐Myc and CDX2 mediate E‐selectin ligand expression in colon cancer cells undergoing EGF/bFGF‐induced epithelial‐mesenchymal transition. Proc Natl Acad Sci USA. 2012;109:7776‐7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beyes S, Andrieux G, Schrempp M, et al. Genome‐wide mapping of DNA‐binding sites identifies stemness‐related genes as directly repressed targets of SNAIL1 in colorectal cancer cells. Oncogene. 2019;38:6647‐6661. [DOI] [PubMed] [Google Scholar]
- 10.Hugo HJ, Pereira L, Suryadinata R, et al. Direct repression of MYB by ZEB1 suppresses proliferation and epithelial gene expression during epithelial‐to‐mesenchymal transition of breast cancer cells. Breast Cancer Res. 2013;15:R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wierstra I. Sp1: emerging roles–beyond constitutive activation of TATA‐less housekeeping genes. Biochem Biophys Res Commun. 2008;372:1‐13. [DOI] [PubMed] [Google Scholar]
- 12.Oh IH, Reddy EP. The myb gene family in cell growth, differentiation and apoptosis. Oncogene. 1999;18:3017‐3033. [DOI] [PubMed] [Google Scholar]
- 13.Mitra P. Transcription regulation of MYB: a potential and novel therapeutic target in cancer. Ann Transl Med. 2018;6:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer. 2008;8:523‐534. [DOI] [PubMed] [Google Scholar]
- 15.Pattabiraman DR, Gonda TJ. Role and potential for therapeutic targeting of MYB in leukemia. Leukemia. 2013;27:269‐277. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Xu Y, Han L, Yi Y. Reassessing the potential of MYB‐targeted anti‐cancer therapy. J Cancer. 2018;9:1259‐1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciciro Y, Sala A. MYB oncoproteins: emerging players and potential therapeutic targets in human cancer. Oncogenesis. 2021;10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malaterre J, Carpinelli M, Ernst M, et al. c‐Myb is required for progenitor cell homeostasis in colonic crypts. Proc Natl Acad Sci USA. 2007;104:3829‐3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheasley D, Pereira L, Sampurno S, et al. Defective Myb function ablates cyclin E1 expression and perturbs intestinal carcinogenesis. Mol Cancer Res. 2015;13:1185‐1196. [DOI] [PubMed] [Google Scholar]
- 20.Golay J, Broccoli V, Lamorte G, et al. The A‐Myb transcription factor is a marker of centroblasts in vivo. J Immunol. 1998;160:2786‐2793. [PubMed] [Google Scholar]
- 21.Trauth K, Mutschler B, Jenkins NA, Gilbert DJ, Copeland NG, Klempnauer KH. Mouse A‐myb encodes a trans‐activator and is expressed in mitotically active cells of the developing central nervous system, adult testis and B lymphocytes. EMBO J. 1994;13:5994‐6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toscani A, Mettus RV, Coupland R, et al. Arrest of spermatogenesis and defective breast development in mice lacking A‐myb. Nature. 1997;386:713‐717. [DOI] [PubMed] [Google Scholar]
- 23.Mettus RV, Litvin J, Wali A, et al. Murine A‐myb: evidence for differential splicing and tissue‐specific expression. Oncogene. 1994;9:3077‐3086. [PubMed] [Google Scholar]
- 24.Rushton JJ, Davis LM, Lei W, Mo X, Leutz A, Ness SA. Distinct changes in gene expression induced by A‐Myb, B‐Myb and c‐Myb proteins. Oncogene. 2003;22:308‐313. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Davie JR. The role of Sp1 and Sp3 in normal and cancer cell biology. Ann Anat. 2010;192:275‐283. [DOI] [PubMed] [Google Scholar]
- 26.Netzker R, Weigert C, Brand K. Role of the stimulatory proteins Sp1 and Sp3 in the regulation of transcription of the rat pyruvate kinase M gene. Eur J Biochem. 1997;245:174‐181. [DOI] [PubMed] [Google Scholar]
- 27.Bigger CB, Melnikova IN, Gardner PD. Sp1 and Sp3 regulate expression of the neuronal nicotinic acetylcholine receptor beta4 subunit gene. J Biol Chem. 1997;272:25976‐25982. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Li Y, Dai C, Yang J, Mundel P, Liu Y. Sp1 and Sp3 transcription factors synergistically regulate HGF receptor gene expression in kidney. Am J Physiol Renal Physiol. 2003;284:F82‐F94. [DOI] [PubMed] [Google Scholar]
- 29.Tichý M, Knopfová L, Jarkovský J, et al. High c‐Myb expression associates with good prognosis in colorectal carcinoma. J Cancer. 2019;101393‐1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tichy M, Knopfova L, Jarkovsky J, et al. Overexpression of c‐Myb is associated with suppression of distant metastases in colorectal carcinoma. Tumour Biol. 2016;37:10723‐10729. [DOI] [PubMed] [Google Scholar]
- 31.Biroccio A, Benassi B, D'Agnano I, et al. c‐Myb and Bcl‐x overexpression predicts poor prognosis in colorectal cancer: clinical and experimental findings. Am J Pathol. 2001;158:1289‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Table S1‐S4