

Abstract
Purpose:
Bcl-2 has been effectively targeted in lymphoid malignancies. However, resistance is inevitable, and novel approaches to target mitochondrial apoptosis are necessary. AZD5991, a selective BH3-mimetic in clinical trials, inhibits Mcl-1 with high potency.
Experimental Design:
We explored the pre-clinical activity of AZD5991 in diffuse large B-cell lymphoma (DLBCL) and ibrutinib-resistant mantle cell lymphoma (MCL) cell lines, MCL patient samples, and mice bearing DLBCL and MCL xenografts using flow cytometry, immunoblotting and Seahorse respirometry assay. Cas9 gene editing and ex vivo functional drug screen assays helped identify mechanisms of resistance to Mcl-1 inhibition.
Results:
Mcl-1 was expressed in DLBCL and MCL cell lines and primary tumors. Treatment with AZD5991 restricted growth of DLBCL cells independent of cell of origin and overcame ibrutinib resistance in MCL cells. Mcl-1 inhibition led to mitochondrial dysfunction as manifested by mitochondrial membrane depolarization, decreased mitochondrial mass and induction of mitophagy. This was accompanied by impairment of oxidative phosphorylation. TP53 and BAX were essential for sensitivity to Mcl-1, and oxidative phosphorylation was implicated in resistance to Mcl-1 inhibition. Induction of pro-survival proteins (e.g., Bcl-xL) in stromal conditions which mimic the tumor microenvironment rendered protection of primary MCL cells from Mcl-1 inhibition, while BH3-mimetics targeting Bcl-2/xL sensitized lymphoid cells to AZD5991. Treatment with AZD5991 reduced tumor growth in murine lymphoma models and prolonged survival of MCL PDX mice.
Conclusion:
Selective targeting Mcl-1 is a promising therapeutic approach in lymphoid malignancies. TP53 apoptotic network and metabolic reprogramming underlie susceptibility to Mcl-1 inhibition.
Keywords: Mcl-1, AZD5591, BH3-mimetics, mitochondria, diffuse large B-cell lymphoma, mantle cell lymphoma
Introduction
Intrinsic apoptosis is an evolutionarily conserved process which determines cell fate, and aberrations in apoptotic machinery are common in cancer. Bcl-2 family proteins govern mitochondrial outer membrane permeabilization (MOMP) and thereby control cytochrome C release which leads to irreversible stages of cell death. While Bcl-2 was the first identified endogenous inhibitor of cell death, the family is now known to comprise members with pro- and anti-apoptotic functions including: pro-apoptotic “activators” (Bim, Bid), “sensitizers” (e.g., BH3-only proteins Noxa, Puma), anti-apoptotic “guardians” (e.g., Bcl-2, Mcl-1, Bcl-xL, A1 and Bcl-w), and pro-apoptotic “effectors” (Bax and Bak) which form pores in the MOM, thus provoking molecular and ionic flux into the cytosol and mitochondrial depolarization (1).
Dysregulated homeostasis within the Bcl-2 family is integral to lymphomagenesis. Pro-survival proteins Bcl-2, Bcl-xL and Mcl-1 contribute to MYC-driven lymphomas in transgenic animal models (2–4). Overexpression of Bcl-2 as a result of chromosomal translocation t(14;18) is found in germinal-center neoplasms, including follicular lymphoma and diffuse large B-cell lymphoma (DLBCL). In addition, multiple other components of the Bcl-2 network are altered in human B-cell non-Hodgkin lymphomas (NHL) through genetic amplifications, deletions and chromosomal translocations as well as transcriptional and post-translational mechanisms. Upregulation of the anti-apoptotic Bcl-2 family members has been shown to contribute to drug resistance in NHL, including to Bruton tyrosine kinase (BTK) inhibitor ibrutinib (5,6).
Mcl-1 is an anti-apoptotic Bcl-2 family member that was discovered as an early response gene in ML-1 human myeloblastic leukemia cells (7). MCL1 gene amplifications have been identified as frequent chromosomal gains promoting survival in a study of more than 3000 samples representing 26 tumor types (8). MCL1 aberrations are prominent among the neoplasms of the hematopoietic system as well as solid tumors. MCL1 transgenic mice have been shown to develop B-cell lymphoma, predominantly of the aggressive subtypes (9). The critical role of Mcl-1 in lymphomagenesis is further supported by the demonstration of the dependence of MYC-driven lymphoma on its continued expression (10). Overexpression of Mcl-1 leads to enhanced survival and chemoresistance of human lymphoid tumors, including to Bcl-2 inhibitor venetoclax (11,12). Mcl-1 is expressed in aggressive B-cell lymphoma, including DLBCL and mantle cell lymphoma (MCL) (6,13,14). The latter is further characterized by loss of pro-apoptotic Bim (~20%) and downregulation of Noxa protein, both of which interact with Mcl-1 (15,16).
In the past 3 years, several BH3-mimetics which potently target MCL1 have entered drug development and demonstrated enhanced activity in hematologic compared with solid tumor cell lines. AZD5991, AMG-176, and S63845 are currently being studied in early phase clinical trials (17–20). AZD5991 is a highly selective, direct Mcl-1 inhibitor (Kd = 170 pM) (18). Preclinical evidence demonstrating the activity of AZD5991 in vitro (IC50<1 nM in Mcl-1-dependent cell lines) and in vivo led to an ongoing phase 1 clinical trial of this compound in hematologic malignancies (NCT03218683). However, data regarding pre-clinical efficacy of AZD5991 and its mechanistic effects in NHL are limited. Thus, we investigated pharmacologic inhibition of Mcl-1 using AZD5991 in aggressive NHL models (DLBCL and MCL).
Materials & Methods
Cell lines and primary cells
The following cell lines were obtained from the American Type Culture Collection (ATCC): MCL cells lines Mino and JeKo-1; DLBCL cell lines SU-DHL4, SU-DHL6, SU-DHL10 and mouse fibroblast (L) cell line. DLBCL cell lines OCI-LY3, U-2932, OCI-LY18, OCI-LY19 and VAL and the mouse fibroblast cell line engineered to express CD40L (L4.5) were obtained from DSMZ (Braunschweig, Germany). OCI-LY10 cell line is a kind gift from Dr. Jennifer Amengual (Columbia University, New York, USA). Ibrutinib resistance was developed in Mino and JeKo-1 cell lines (Mino_R and JeKo-1_R) via long-term exposure to increasing concentrations of ibrutinib (21). These lines were cultured in RPMI 1640 medium supplemented with fetal bovine serum (DLBCL - 10%, MCL - 20%), 100 U/mL penicillin and 100 μg/mL streptomycin (1% Pen-Strep). Stromal cell lines L and L4.5 were maintained in DMEM 1640 medium with 10% FBS and 1% Pen-Strep. B-cell activating factor (BAFF)-expressing Chinese hamster ovary cells described previously were cultured in MEM-α supplemented with 10% FBS and 1% Pen-Strep (22). Cell were used up to 10 passages and every 2 months underwent testing with MycoAlert Mycoplasma Detection Kit (Lonza, Walkersville, MD; latest testing in May 2021).
Following approval by the Institutional Review Board and provision of written consent, peripheral blood was obtained from patients with NHL treated at the City of Hope National Medical Center. Isolation of peripheral blood mononuclear cells (PBMCs) was performed using standard Ficoll-Hypaque technique (Amersham). Red blood cells were lysed using ACK lysing buffer (Life Technologies). Primary cells were cultured in RPMI-1640 medium supplemented with 20% FBS, 1% Pen-Strep, 1% L-glutamine, 1% HEPES and 1% non-essential amino acid solution.
Primary cells were cultured on stromal cells under standardized conditions (22). Briefly, stromal cells were seeded to achieve 60–70% confluence on the following day when primary cells were plated at a 50:1 ratio and incubated at 37°C in 5% CO2 for 24 hours before drug treatment. Prior to harvest, primary cells were gently washed from the stromal layer and transferred into a new well for 1 hour.
Cell viability testing and drugs
To measure cell proliferation, cells were plated in 96- or 384-well plates (3000 or 1250 cells/well, 3–6 wells/sample) with drugs and incubated for 48 hours at 37°C in 5% CO2. After incubation, relative numbers of viable cells were measured using a tetrazolium-based colorimetric assay (CellTiter Aqueous One Solution Cell Proliferation Assay, Promega). Inhibitor combination screening was carried out in pre-treated 384 well plates with inhibitors prepared in a 7-point concentration series ranging from 10 μM to 0.014 μM for each drug (23).
To measure cell apoptosis, cells (in duplicates) were resuspended in 100 μL of Annexin V binding buffer containing 1 μL of Annexin V-PE, 1 μL of 7-aminoactinomycin D (7-AAD) (Southern Biotech) and 1 μL of CD19-mAbs (BD Bioscience) followed by flow cytometry on FACSAria (BD Biosciences). Data analysis was performed using FlowJo software (Tree Star).
AZD5991 and navitoclax were purchased from Active Biochem; venetoclax and Q-VD-OPh were from MedChemExpress; AZD4320, IACS-010759 were from Selleck Chemicals. CCCP (carbonyl cyanide m-chlorophenyl hydrazine), NAC (N-Acetyl-L-cysteine) were from Sigma.
Reactive oxygen species (ROS)
Intracellular ROS were measured using the DCFDA/H2DCFDA Cellular ROS Assay kit (Abcam). Cells were incubated with DCFDA in PBS for 30 minutes, washed, resuspended at 1×105 cells/well in 96-well black-walled plates (Corning), and incubated with drugs for 24 hours. Florescence of the substrate was measured in SpectraMax M2 Microplates (Molecular devices).
Seahorse metabolism assay
Cells were treated with drugs and then were seeded in poly-lysine-coated (Sigma Aldrich) XFe96 Seahorse Cell Culture Microplates (Agilent) at 8×104 live cells /well. Plates were centrifuged at 300g for 5 minutes with no brake and incubated for 30 minutes with no CO2 at 37°C. Procedures were carried out per the Seahorse XFe96 Oxidative Stress Assay protocol. First, basal cellular respiration was recorded by simultaneously measuring oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) simultaneously. Next, 1.5 μM oligomycin (ATP synthase inhibitor) was injected to stop ATP-linked respiration, leading to a direct measurement of proton-leak, as well as non-mitochondrial oxygen consumption. Thereafter, 1.0 μM FCCP was injected to induce maximal respiration/uncoupled oxidative phosphorylation (OxPhos). Lastly, 0.5 μM rotenone and 0.5 μM antimycin A (Complex I and Complex III inhibitors, respectively) were injected, thereby inhibiting all OxPhos.
Immunohistochemistry (IHC)
Immunohistochemistry was performed on lymphoid tissues from 34 DLBCL patients. Cell of origin was determined by Hans IHC-based algorithm. Twenty-three germinal center B-cell like [GCB] and 11 non-GCB DLBCLs were analyzed. All staining was performed after antigen retrieval using intermittent heating for 4 cycles of 5 min each in a 625-W microwave oven to maintain the temperature of the buffer [(0.01 M citrate buffer, pH 6.0) at 95°C] and run in parallel with known positive and negative controls. After incubation with Mcl-1 or Bcl-xL antibodies (Cell Signaling Technology, 1:100) and relevant secondary antibodies, slides were stained using the Biotin-Streptavidin amplified system. Protein expression was considered positive when more than 30% of tumor cells demonstrated intense staining.
Ex vivo functional drug screen
Ex vivo functional drug screens were performed as previously described (23,24). Small-molecule inhibitors (a total of 189), purchased from LC Laboratories and Selleck Chemicals, were distributed into 384-well plates prepared with single agents in a seven-point concentration series (10–0.014 μM) in a seven-point equimolar ratio concentration series identical to those used for single agents. The final concentration of DMSO was ≤0.1% in all wells, and all plates were stored at −20 °C and thawed immediately prior to use. 1 μM AZD5991 or DMSO control were dispensed into wells at this stage. Cells were seeded into 384-well assay plates at 1250 cells/well. After 3 days of culture at 37 °C in 5% CO2, methanethiosulfonate reagent (CellTiter 96 AQueous One; Promega) was added to each well and absorbance was measured at 490 nm. IC50 values of different inhibitors with or without AZD5991 were calculated from cell viability values normalized to untreated cells and fitted to a curve using a probit regression.(24,25)
To validate findings of the screen assay, we performed matrix MTS assays (a 7-by-7 doses of each drug), and calculated synergy scores using Bliss algorithm with R_SynergyFinder.
Gene editing and overexpression
Ribonucleoprotein (RNP) electroporation genome editing experiments in DLBCL cells (OCI-LY3 and VAL) were performed using the Lonza Amaxa Nucleofector II System (Lonza, Basel, Switzerland), 4 μM (1:3, Cas9:sgRNA) Alt-R® (Integrated DNA Technologies, Inc) Cas9 RNP complex, and 4 μM Alt-R® Cas9 Electroporation Enhancer (Integrated DNA Technologies, Inc) as described (26). Chemically synthesized guide RNAs were purchased and prepared according to the manufacturer’s instructions (Synthego) and RNPs were formed by the addition of purified Cas9 protein (IDT). RNP complexes (5 μL) and 1.5 × 105 DLBCL cells (20 μL) were mixed and electroporated according to the manufacturer’s specifications using protocol V-001. Proteins were collected 6 days post electroporation. The targeting sequences were BAX-1: AGUUUCAUCCAGGAUCGAGC; BAX-2: AGUAGAAAAGGGCGACAACC; PMAIP1: CGAGUGUGCUACUCAACUC; TP53-1: GAUCCACUCACAGUUUCCAU; TP53-2: CCCCGGACGAUAUUGAACAA; NT: GAGATATCAATCCTCCCGC.
hBAX-C3-EGFP (Addgene #19741) was used for transient expression of Bax in OCI-LY3 cells. 1*106 cells were electroporated using the Lonza Nucleofector System, program T-020. Live-cell confocal imaging was performed after 24 h (Zeiss LSM 880), and cell proliferation was measured after 48 h in an MTS-based assay as described above (27).
In vivo models
All animal studies were carried out in accordance with institutional guidelines. Five-week old nonobese diabetic/severe combined immunodeficiency/null NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice were purchased from The Jackson Laboratory.
PDX MCL was obtained from ProXe (#96069). Mice were treated with 25 mg/kg busulfan via intraperitoneal injection. Next day, 3X106 MCL cells were inoculated intravenously. MCL cells were analyzed in the blood weekly by flow cytometry (CD45+CD5+CD19+) and once detected (approximately 2 weeks after inoculation) mice started treatment. Red blood cells were lysed using ACK lysis buffer (Life Technologies), washed with PBS, incubated on ice for 20 minutes in 100 μL PBS with 1 μL LIVE/DEAD Fixable Aqua (Invitrogen), 5 μL anti-CD19-FITC, 5 μL anti-CD5-PE, and 5 μL anti-CD45-APC (all from Becton Dickinson) and analyzed by flow cytometry. At the end of treatment, animals were euthanized and spleen tissue was collected.
3×106 OCI-LY3 cells were mixed with 50% Matrigel (BD Biosciences) and injected subcutaneously into the right flank. When tumors reached ~100 mm3, 10 to 14 days after implantation, mice started treatment. Tumor volume was assessed every other day. The data are expressed as average tumor diameter (in millimeters) per mouse as a function of time, normalized to pretreatment value. Animals were euthanized when tumor diameter exceeded 20 mm or after loss of >10% body weight. mice were euthanized and tissues were harvested immediately.
Mice were treated as follows: 60 mg/kg AZD5991, vehicle control, 50 mg/kg venetoclax, or a combination of AZD5991 and venetoclax. AZD5991 was administered every 10 days via tail vein, or intraperitoneally in case of tail vein damage. Venetoclax was administered Monday through Friday (5 days a week) through oral gavage except on days when AZD5991 was given.
Statistical Analysis
All experiments were conducted at least in triplicates. Statistical significance was calculated using the Student’s t-test on GraphPad Prism software. p<0.05 was considered to be statistically significant. *, p<0.05 and **, p<0.01 throughout the manuscript. Mean±SE refer to biological repeats. Drug combination effect and synergistic score was calculated for the Bliss independence dose-response surface model using R_SynergyFinder software.
Results
Selective targeting of Mcl-1 restricts growth of the neoplastic B cells
Treatment with AZD5591 diminished proliferation of DLBCL cells in a dose-dependent manner. Activated B-cell (ABC)-like cell lines OCI-LY10 and OCI-LY3 and germinal center B-cell (GCB)-like cell lines VAL, SU-DHL4 and SU-DHL6 were most sensitive to the drug (Fig. 1A). Meanwhile, other GCB (SU-DHL10, OCI-LY18) and ABC DLBCL cells (U-2932) showed resistance. Both parental and ibrutinib-resistant MCL cells were highly susceptible to Mcl-1 inhibition (IC50~0.3 μM; Fig. 1B). Correspondingly, we observed apoptosis following exposure of DLBCL and MCL cell lines to AZD5991 (Fig. 1C–D). Interestingly, ibrutinib-resistant JeKo-1 cells (JeKo-1_R) were more susceptible to Mcl-1 inhibition than parental cells, while the opposite was true for Mino_R cells (Fig. 1B, D). Treatment with AZD5591 displaced Bim from Mcl-1 (Fig. 1E). Consistent with earlier reports, Mcl-1 inhibition led to increased Mcl-1 protein levels but had no effect on Bcl-2/xL (Fig. 1F, Supplemental Fig. 1) (19).
Figure 1. Selective targeting Mcl-1 thwarts survival of the lymphoma cell lines.
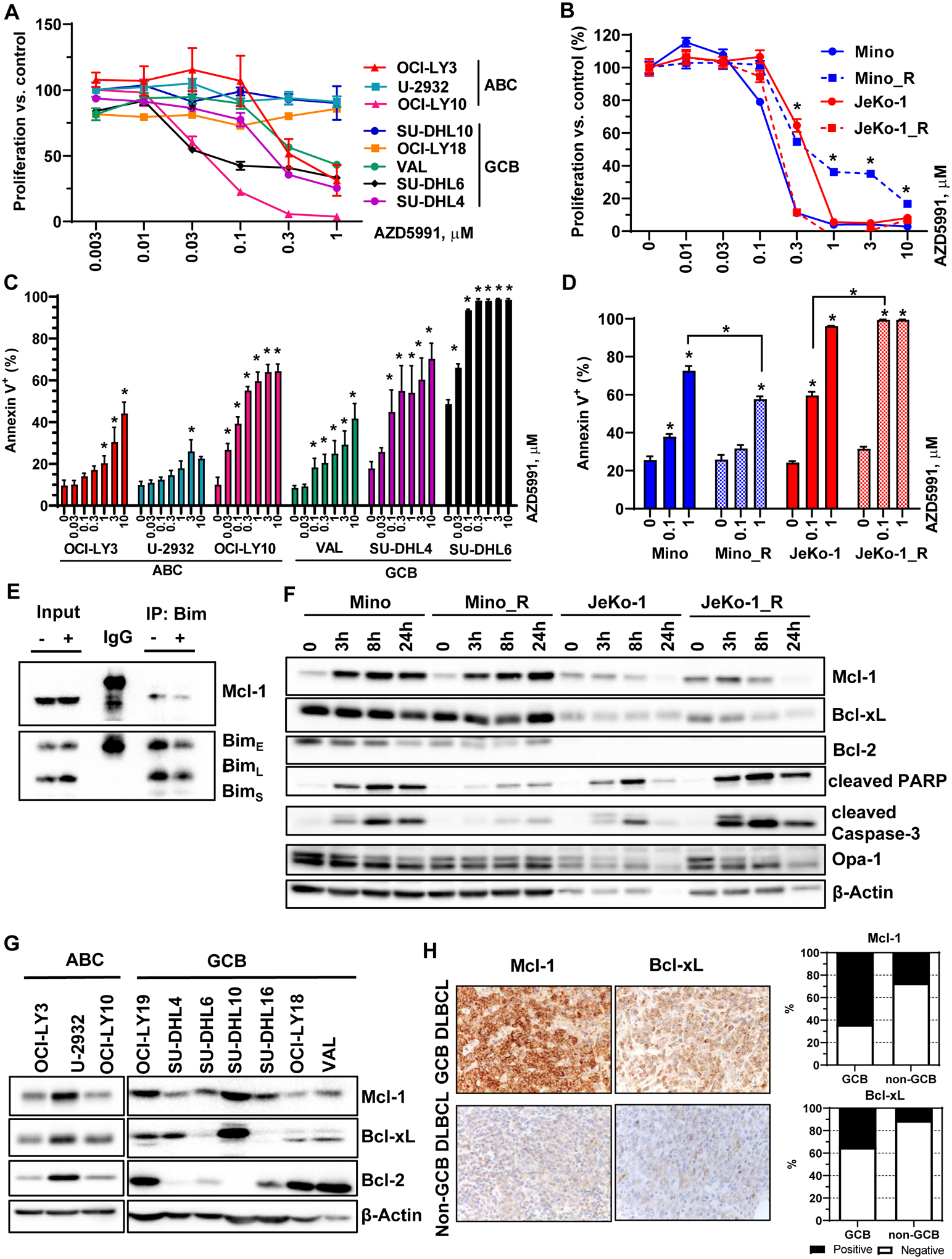
(A-B) DLBCL and MCL cell lines were treated with the indicated doses of AZD5991 for 48 hours. Cell proliferation was measured by colorimetric MTS assay. % viable cells, after normalization to untreated controls, was fit using non-linear regression analyses. *, p<0.05 Mino_R vs. Mino cells. (C-D) Cells were treated with indicated doses of AZD5991 for 24 hours. Apoptosis was determined by Annexin-V-FITC staining. Data are mean±SE. *, p<0.05 vs. untreated control. (E) SU-DHL6 cells were treated with 0.5 μM AZD5991 for 24 h. Protein lysates were subjected to immunoprecipitation experiments with Bim antibodies (in triplicates). (F) MCL cells were treated with 1 μM AZD5991 as shown. Cell lysates were subjected to immunoblotting (in triplicates here and subsequently). (G) Whole cell lysates of exponentially grown DLBCL cell lines were subjected to immunoblotting. (H) Expression of Mcl-1 and Bcl-xL was evaluated in 34 primary DLBCL lymph node tissues.
Mcl-1 was expressed at variable levels in all DLBCL cell lines irrespective of cell of origin (Fig. 1G). Anti-apoptotic proteins Bcl-xL and Bcl-2 were each overexpressed in four cell lines. AZD5591-sensitive cells (OCI-LY3/10, VAL, SU-DHL4/6) expressed relatively low Mcl-1 levels; of those, only VAL cells expressed abundant Bcl-2 (Fig. 1G). Similarly, Mino and JeKo-1 cells expressed low levels of Mcl-1. Bcl-xL was expressed in both, while Bcl-2 was only found in Mino cells (Fig. 1F). Expression of these proteins was similar in parental and ibrutinib-resistant cell lines. Thus, low Mcl-1 expression may in part underlie sensitivity to AZD5991.
Additionally, we analyzed a panel of primary lymph nodes and found that Mcl-1 and Bcl-xL were expressed (>30%) in 64% and 35% of GCB, and 27% and 11% of non-GCB DLBCL, respectively, reaffirming that Mcl-1 is a relevant target in NHL (Fig. 1H).
In sum, Mcl-1 inhibition restricted growth of DLBCL and MCL cells irrespective of cell of origin and ibrutinib resistance.
Mcl-1 inhibition induces mitochondrial stress
Treatment with AZD5991 has been shown to induce rapid dissociation of Bak/Mcl-1 complexes resulting in MOMP (18). Given the involvement of MOMP in regulation of mitochondrial bioenergetics, we studied the effects of AZD5991 on mitochondrial homeostasis. Treatment of NHL cells with AZD5591 resulted in mitochondrial depolarization, as visualized by a decrease in PE fluorescence (loss of JC-1 aggregates; Fig. 2A). We noted a decrease in mitochondrial mass following Mcl-1 inhibition, as evidenced by a left shift of the Mitotracker dye fluorescence peak (Fig. 2B). Both events were most prominent in OCI-LY10 and MCL cells and were not rescued by caspase inhibition (Supplemental Fig. 2A). Opa-1 regulates mitochondrial fusion and protects cells against apoptosis. Opa-1 was downmodulated in parental Mino and parental and ibrutinib-resistant Jeko-1 cells treated with AZD5991 (Fig. 1F), consistent with a disruption of the mitochondrial membrane potential and decreased mitochondrial mass.
Figure 2. Mcl-1 inhibition leads to mitochondrial dysfunction.
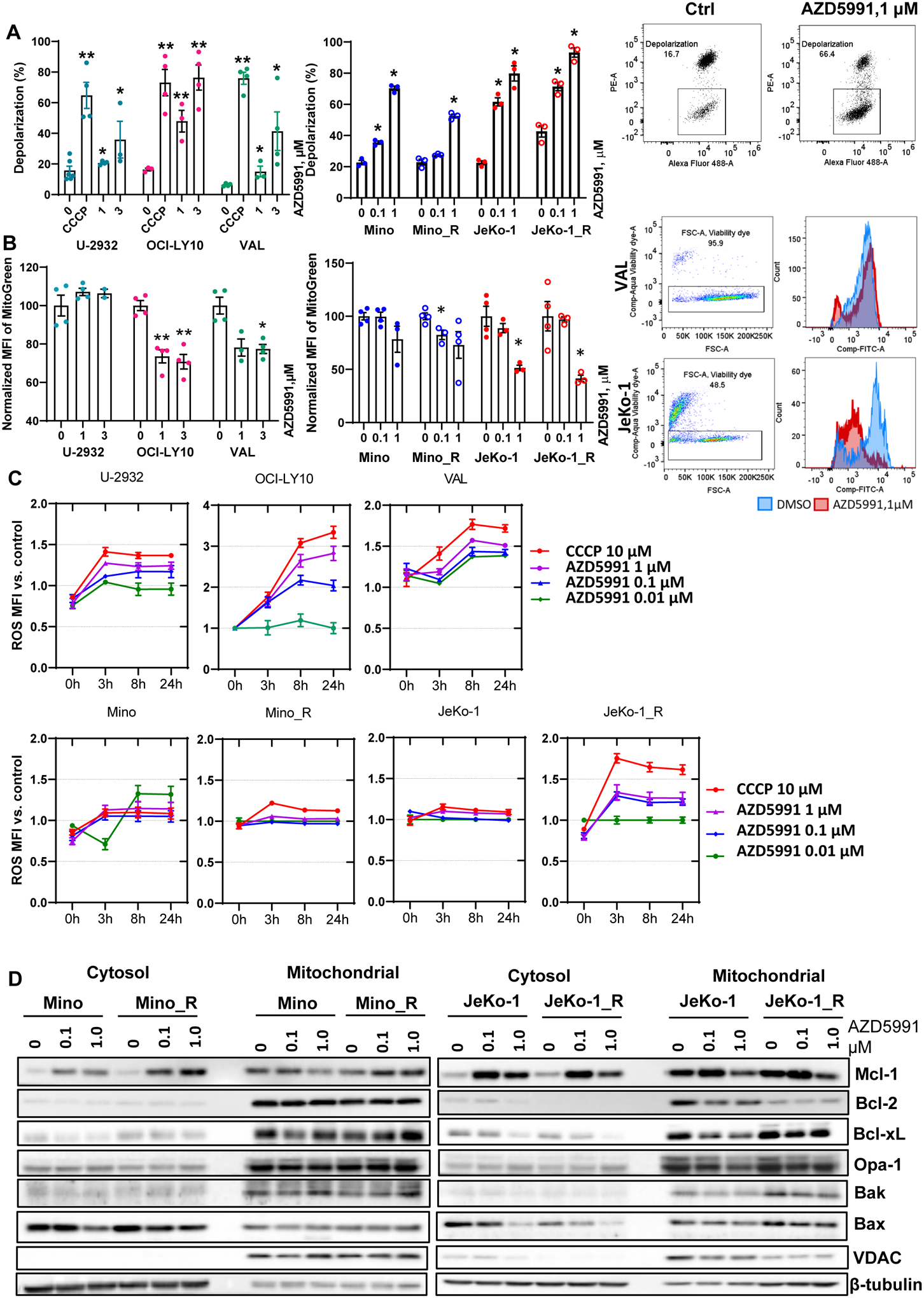
(A) Cells (in technical and biological triplicates) were treated with 10 μM CCCP, 0.1, 1 or 3 μM AZD5991 or vehicle control for 24 hours and analyzed for mitochondrial depolarization with JC-1 dye using flow cytometry. A representative dot plot image is shown (depolarized mitochondria is boxed). (B) Cell lines were treated with drugs for 24 hours as indicated and mitochondrial mass was assessed with Mitobright Green using flow cytometry. A representative histogram is shown. Data are mean±SE. *, p<0.05, **, p<0.01 vs. untreated control. (C) Cells were stained with DCFDA, treated with drugs for 24 h, followed by ROS MFI quantification at the indicated timepoints. MFI was normalized to untreated control. (D) Cells were treated with indicated doses of AZD5991 for 8 h followed by cellular fractionation and immunoblotting.
Malignant cells often endure higher levels of oxidative stress than normal cells, partly due to increased metabolic activity generating ROS. Low ROS levels are required for cancer cell survival, but excessive oxidative stress can lead to cell death (28). MCL cell lines had higher basal ROS accumulation compared with DLBCL cell lines, and this was further increased in ibrutinib-resistant cells (Supplemental Fig. 3), suggesting that they may have higher level of metabolic distress. Treatment with AZD5991 led to a further increase in ROS production, likely indicative of damage-related reduction in mitochondrial antioxidant capacity (Fig. 2C). ROS scavengers did not rescue cells from Mcl-1 inhibition indicating that AZD5991-mediated cell death is not fully dependent on ROS (Supplemental Fig. 4).
Analysis of distribution between the cytosol and mitochondrial fractions demonstrated that while Mcl-1 accumulated in the cytosol following an 8-hour treatment with 1 μM AZD5991, mitochondrial Mcl-1 was diminished (Fig. 2D). By contrast, Bcl-2 and Bcl-xL were mostly confined to the mitochondrial fraction and their expression did not change significantly upon treatment with AZD5991. Somewhat unexpectedly, JeKo-1_R cells demonstrated a decrease in Bcl-2 in both fractions, compared with Jeko-1 parental cells. While localization of Bak remained unchanged, cytosolic sequestration of Bax diminished upon Mcl-1 inhibition. Opa-1 was also reduced in the mitochondrial fraction in JeKo-1 cells (Fig. 2D).
To probe for the effects of Mcl-1 inhibition on mitochondrial bioenergetics, we employed Seahorse respirometry assay. We measured the oxygen consumption rate (OCR) at baseline and over time in response to modulators of the mitochondrial electron transport chain (ETC) and OxPhos. Baseline rates of metabolic activity varied between cell lines, with VAL cells exhibiting higher OCR (130 pmol/min) compared with OCI-LY10 and U-2932 cells (~50 pmol/min; Fig. 3A). Ibrutinib-resistant JeKo-1_R cells exhibited increased metabolic activity versus parental cells (Fig. 3B). Treatment with AZD5991 inhibited both basal and uncoupled respiration in sensitive DLBCL and MCL cell lines, but not in resistant U-2932 cells (Fig. 3A–B). Reduction of maximal oxygen consumption capacity was particularly prominent in JeKo-1 and Mino cells, consistent with their enhanced susceptibility to Mcl-1 inhibition (Fig. 3B). Extracellular acidification rate (ECAR) was also impacted, indicating that cells failed to switch to glycolysis pathway despite a disruption in OxPhos (Fig. 3A–B). In these experiments, the impact of AZD5991 on OxPhos was independent of caspase activation-related apoptotic cell death, as it was not fully prevented with the caspase inhibitor QVD-OPh (Supplemental Fig. 2B).
Figure 3. Mcl-1 inhibition deregulates OxPhos and induces mitophagy.
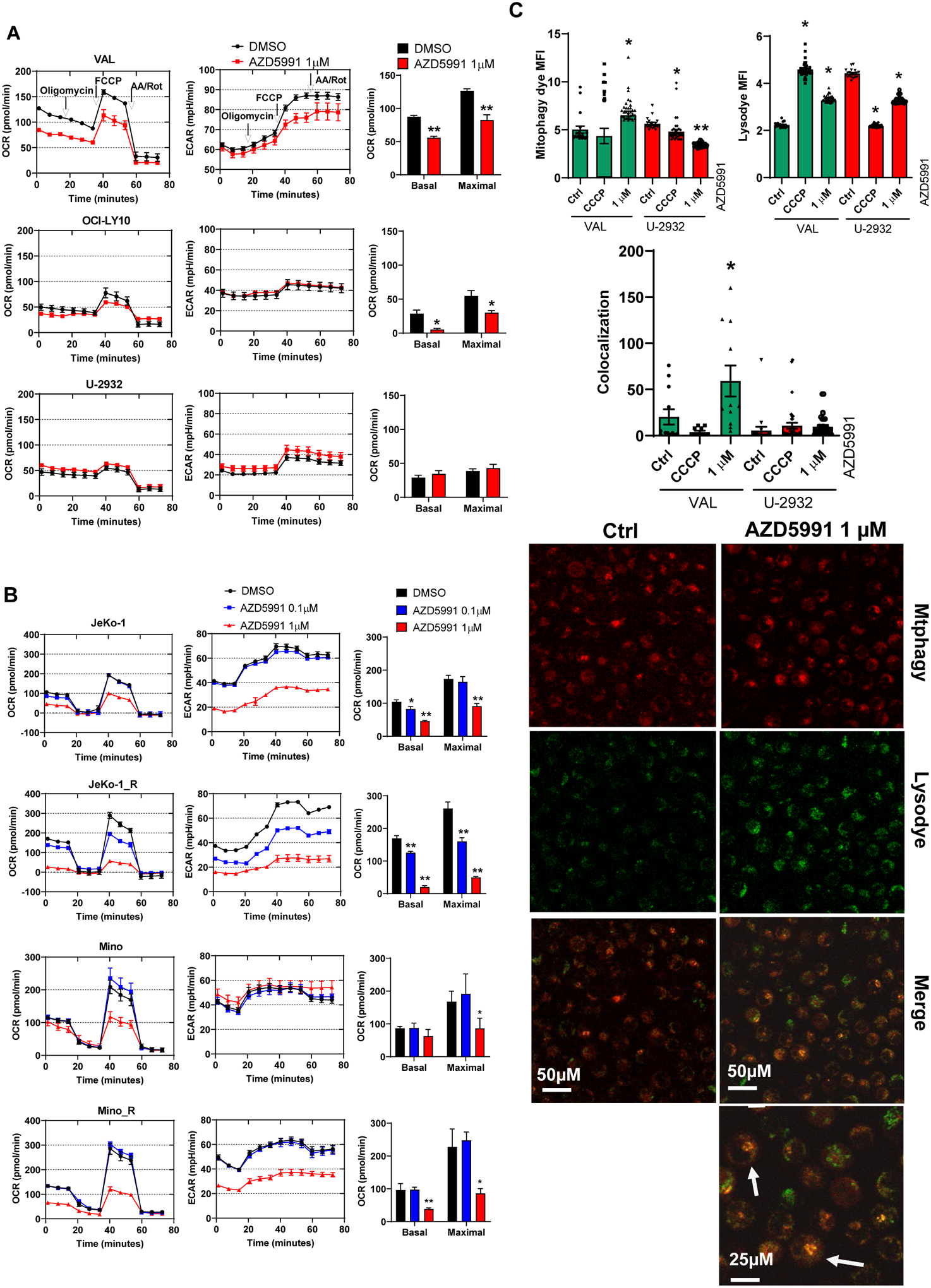
(A, B) Cells were treated with 1 μM AZD5991 or control for 24 h and subjected to Seahorse analysis. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) as a function of time, with exposure to inhibitors of the electron transport chain (ETC) and oxidative phosphorylation (OxPhos) to derive bioenergetics parameters of mitochondrial respiration are shown. (C) VAL (sensitive) and U-2932 (resistant) cells were treated with 1 μM AZD5991 or vehicle control for 24 h and stained with Mitophagy dye and Lysodye. Live cells were imaged with confocal microscopy. Arrows point to the mitophagy puncta. MFI was quantified, and co-localization was measured and calculated using Zen software. Data are mean±SE. A representative image (VAL cells) is shown. *, p<0.05 vs. untreated control.
Finally, we investigated the effect of Mcl-1 inhibition on mitophagy, a conserved process which eliminates dysfunctional mitochondria. Treatment of VAL cells with AZD5991 led to an increased number of damaged mitochondria and enhanced formation of lysosomes, accompanied by increased mitophagy as measured by dye colocalization (Fig. 3C). However, mitophagy was not observed in AZD5991-resistant cells.
Thus, Mcl-1 inhibition induced mitochondrial stress and disrupted cellular bioenergetics in NHL.
TP53 and BAX mediate resistance to Mcl-1 inhibition in DLBCL cells
Combination treatment approaches are poised to overcome intrinsic resistance to targeted agents. Our group has previously shown that inactivation of TP53, BAX, and PMAIP1 (Noxa) results in resistance to Bcl-2 inhibitor venetoclax in acute myeloid leukemia (29). Furthermore, Bax and Noxa are established Mcl-1 interactors and Bax was required for Mcl-1 inhibition-induced apoptosis in lymphoid cells (30,31). Hence, we investigated their role in resistance to AZD5991. Genetic knockout of TP53 or BAXresulted in loss of their expression in OCI-LY3 and VAL cells (Fig. 4A). Loss of either TP53 or BAX conferred resistance to AZD5591, indicating that the impact of AZD5991 is dependent on MOMP (Fig. 4B). Meanwhile, PMAIP1 was not essential to drug sensitivity in OCI-LY3 cells, and was not expressed in VAL cells. Loss of TP53 in OCI-LY3 cells resulted in downmodulation of Bax and Puma (Supplemental Fig. 5A). In these cells, we also observed stabilization of Bcl-xL and Bcl-2, while Bak levels remained unchanged. Overexpression of Bax in TP53-deficient cells led to re-sensitization to AZD5991 (Supplemental Fig. 5B), thus indicating that loss of TP53 mediates resistance to Mcl-1 inhibition at least in part via Bax (and Puma).
Figure 4. Loss of TP53 and BAX mediates resistance to Mcl-1 inhibition.
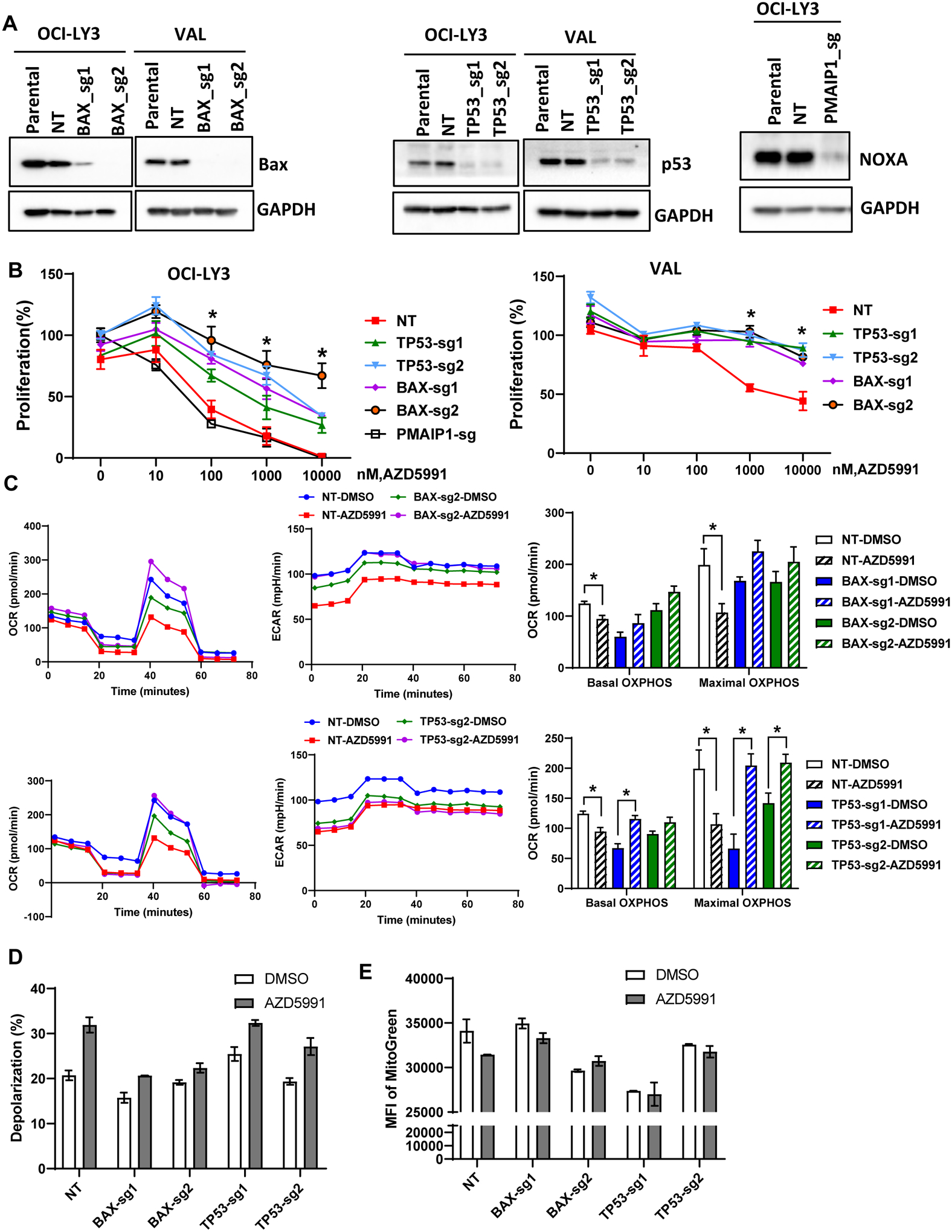
(A) OCI-LY3 and VAL cells were electroporated with RNP single sgRNA/Cas9 constructs targeting TP53, BAX, PMAIP1 or nontargeting sgRNA (NT). Whole protein lysates were subjected to immunoblotting. (B) Genetically manipulated cells were treated with the indicated concentrations of AZD5991 and proliferation was measured in the MTS assay. % viable cells, after normalization to untreated control, were fit using nonlinear regression analyses. Data are Mean±SE. * - p<0.05 vs. sgTP53/BAX vs. NT control (C-E) Genetically manipulated VAL cells were treated with 1 μM AZD5991 or vehicle control for 24 h and subjected to Seahorse analysis (C), analysis of mitochondrial depolarization (JC-1; D) and mitochondrial mass (Mitobright Green; E).
We next evaluated mitochondrial respiration in genetically manipulated VAL cells. Loss of BAX led to minimal reduction in maximal respiration, while TP53 had no effect (Fig. 4C). However, a decrease in maximal respiration following treatment with AZD5991 was fully reversed in cells lacking BAX or TP53. Loss of BAX (but not TP53) abrogated mitochondrial depolarization in AZD5991-treated VAL cells, consistent with its critical role in MOMP (Fig. 4D). Finally, BAX and TP53 were instrumental in loss of mitochondrial mass following Mcl-1 inhibition (Fig. 4E).
To further determine the role of the apoptotic network, we employed a functional drug screening assay using a panel of 189 small molecule inhibitors that target a variety of distinct signaling pathways activated in cancer. A number of agents demonstrated synergy with AZD5991 (Fig. 5A). Significant synergy was observed when AZD5991 was combined with Bcl-2/xL inhibitors(top two lanes; Fig. 5A), as well as ibrutinib. Our findings were validated using clinically available direct Bcl-2 inhibitor venetoclax and Bcl-2/xL inhibitors navitoclax and AZD4320 (Fig. 5B–D). Concurrent targeting Mcl-1 and Bcl-2/xL in OCI-LY3 cells was highly synergistic with a Bliss score of 35 (Fig. 5D). While ibrutinib-resistant MCL cells were less sensitive to Bcl-2 inhibitors than parental cell lines, their combination with AZD5991 also demonstrated significant synergy (Fig. 5E). Loss of either BAX of TP53 abrogated the combined effect of Mcl-1 and Bcl-2 inhibition in DLBCL cells (Fig. 5F).
Figure 5. Synergistic co-targeting Mcl-1 and Bcl-2/xL.
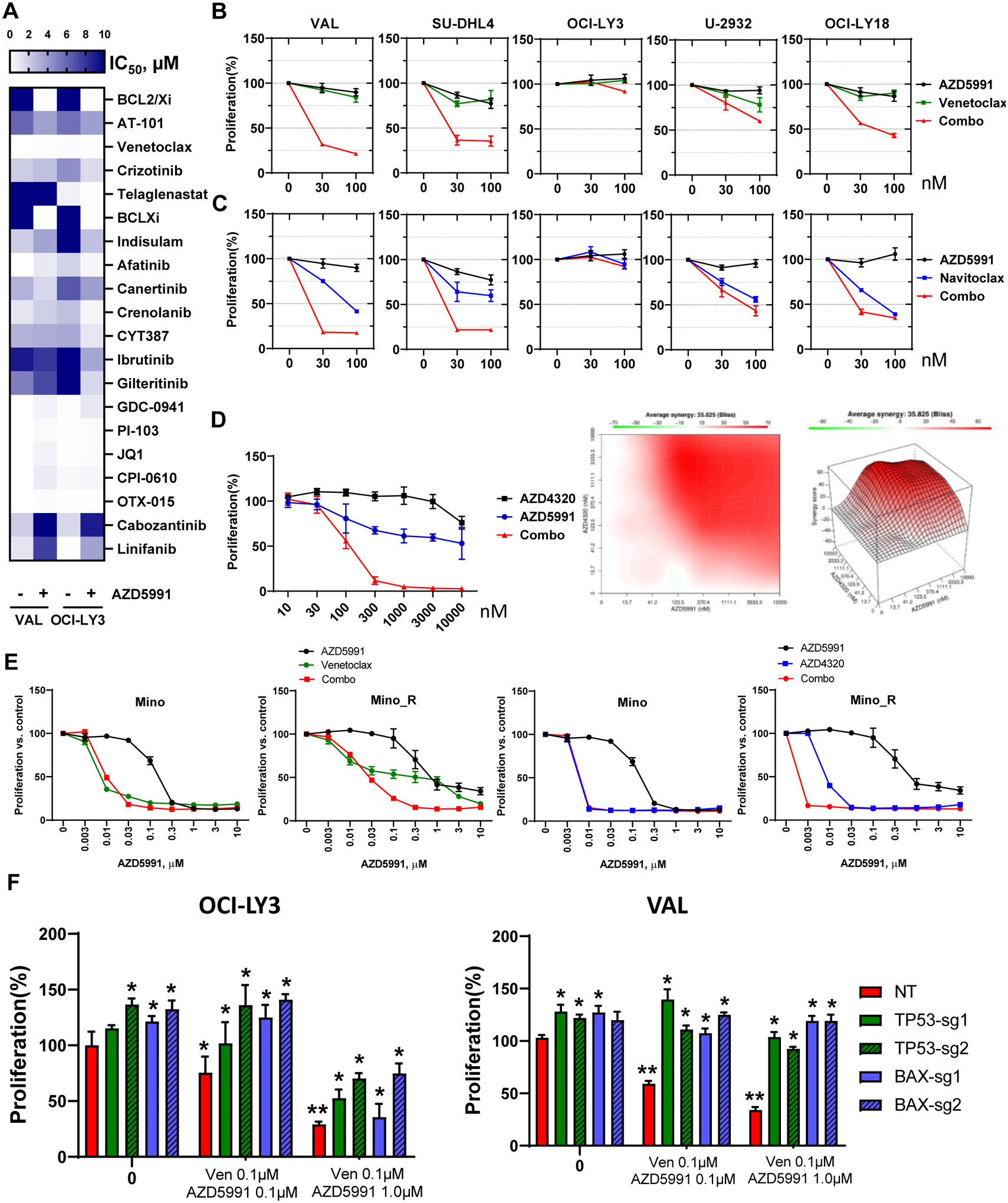
(A) VAL and OCI-LY3 cells were subjected to functional screening assay as described in the methods. IC50 values were calculated (represented in the color gradient). Drugs of interests that exhibit synergism or antagonism with AZD5991 are shown. (B, C) DLBCL cells were treated with drugs as indicated for 48 h. Cell proliferation was measured in an MTS assay; % viable cells were normalized to DMSO-treated control. (D) OCI-LY3 cells were treated with AZD5991 and AZD4320 for 48 h. Bliss analysis was conducted to determine the synergistic score. (E) MCL cells were treated with drugs as indicated for 48 h. Cell proliferation was measured in an MTS assay; % viable cells were normalized to DMSO-treated control. (F) Genetically manipulated DLBCL cells were treated with venetoclax and AZD5991 for 48 hours. Cell proliferation was measured in an MTS assay. % viable cells were normalized to DMSO-treated control. ** - p<0.05 vs. untreated control. * - p<0.05 vs. NT (non-targeting sgRNA).
Together, our data suggest that TP53 and Bcl-2 apoptotic network mediates resistance to Mcl-1 inhibition.
MCL1 inhibition induces apoptosis in primary MCL cells
We then evaluated the effect of Mcl-1 inhibition in primary MCL cells. Since primary cells rapidly undergo spontaneous apoptosis in vitro, stromal co-culture conditions were used. To partially mimic the lymph node microenvironment ex vivo, we employed CD40L- and BAFF (B-cell activation factor)-expressing models which abrogate spontaneous and ibrutinib-induced apoptosis (22,32,33). Consistent with our previous data, CD40L-stimulated primary MCL cells robustly upregulated Bcl-xL, while BAFF-stimulated cells induced Mcl-1 (Fig. 6A). Treatment of primary MCL cells with AZD5991 induced apoptosis (Fig. 6A–B). While BAFF-expressing stroma conferred partial resistance to venetoclax, cells remained sensitive to AZD5991. By contrast, CD40L stroma conferred resistance to both drugs, presumably due to protection afforded by Bcl-xL. Combined treatment demonstrated enhanced cytotoxicity in stromal conditions (Fig. 6B).
Figure 6. Mcl-1 inhibition overcomes stromal rescue of the primary MCL cells and demonstrates efficacy in vivo.
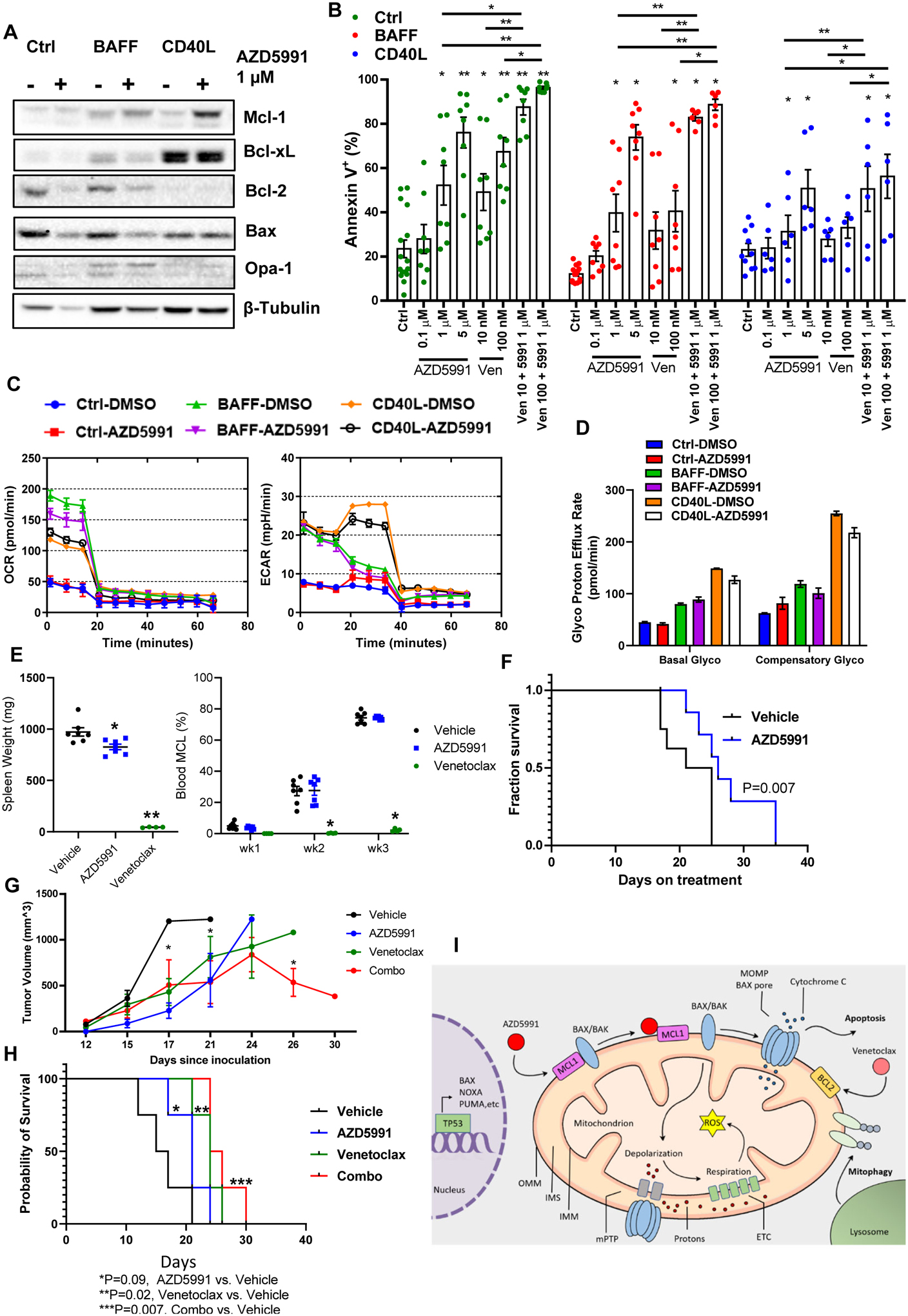
(A-B) Primary MCL cells (4_individual patient samples, in duplicates) were co-cultured with BAFF-, CD40L-expressing or control stroma for 24 h and then treated with the indicated drugs for additional 24 h. Cells were assayed for apoptosis and protein lysates were analyzed by immunoblotting. (C, D) Primary MCL cells were cultured on stroma as indicated, treated with 1 μM AZD5991 for 24 hours and subjected to Seahorse respiration assay. (E-F) MCL PDX mice received treatment with AZD5991 (N=8), venetoclax (N=4) or vehicle control (N=8) as described in the methods. Treatment was begun upon detection of the circulating MCL cells in the peripheral blood. Spleen weight was measured at the time of sacrifice. % MCL involvement and survival are shown since start of treatment. (G-H) Mice xenografted with OCI-LY3 cells were treated with AZD5991, venetoclax, the combination or vehicle control as discussed in the methods (N=4 each). Tumor volume and survival are shown. X axis represents days since tumor inoculation. * - p<0.05 each treated versus untreated tumor. (I) Pharmacologic targeting Mcl-1 induces mitochondrial dysfunction and apoptosis in B-cell lymphoma cells in TP53- and BAX-dependent manner.
Primary MCL cells sustained OxPhos when cultured in stromal conditions. Both BAFF and CD40L stimulation dramatically enhanced basal OxPhos and glycolysis pathways as compared with control stroma, and this was partially reversed by Mcl-1 inhibition (Fig. 6C). However, when basal OxPhos was inhibited, CD40L-stimulated MCL cells demonstrated the capability to rapidly switch to glycolytic pathway (compensatory glycolysis), and this was not fully overcome by treatment with AZD5991 (Fig. 6D).
Encouraged by our data, we conducted a preliminary investigation of AZD5991 in a PDX MCL mouse model. While Mcl-1 inhibition (see Methods) did not block tumor expansion in the peripheral blood, AZD5991-treated mice demonstrated a reduced spleen weight compared with vehicle-control treated mice (Fig. 6E). Treatment with AZD5991 prolonged survival of MCL PDX mice (Fig. 6F). This model was very sensitive to venetoclax. While AZD5991 is not expected to target murine Mcl-1, no significant treatment toxicities were observed (Supplemental Fig. 6). Mice xenografted with OCI-LY3 cells demonstrated a significant delay in tumor growth when treated with AZD5991 alone (Fig. 6G), as well as a trend toward prolonged survival (Fig. 6H). Treatment with venetoclax had a similar effect, whereas a combination of the two drugs was particularly effective (treatment vs, control, p=0.007, Fig. 6H).
Discussion
DLBCL is the most prevalent NHL subtype worldwide, with >25,000 cases diagnosed in in the United States annually (34). While 50–60% of patients are cured with standard chemo-immunotherapy, those who exhibit chemo-refractoriness succumb to their disease. Novel targeted agents including BTK inhibitors (e.g., ibrutinib) and Bcl-2 inhibitor venetoclax have limited efficacy in DLBCL. The search for novel therapeutic approaches is complicated by the genomic heterogeneity of DLBCL, as illustrated by recent molecular analyses and novel classification systems (35,36). Here we demonstrate that direct targeting Mcl-1 using a selective BH3-mimetic AZD5991 restricts cell proliferation and induces apoptosis in DLBCL cells independent of cell of origin, a finding not entirely surprising considering that Mcl-1 is expressed in both ABC- and GCB DLBCL subsets, as evidenced by this and other studies (14). Interestingly, we found that B-cell receptor (BCR)-dependent cell lines SU-DHL4, SU-DHL6 and OCI-LY10 (“cluster 3” per the Chapuy et al.) are susceptible to Mcl-1 inhibition (35,37,38). These cell lines expressed low levels of Mcl-1 and Bcl-2, and have been previously shown to be susceptible to phosphoinositide-3 kinase (PI3K) inhibition in part via loss of Mcl-1 (38). Our data align with a recent report demonstrating pre-clinical efficacy of direct Mcl-1 inhibitors S63845 and A1210477 in DLBCL (31). We also observed that low Mcl-1 expression may be associated with enhanced sensitivity to Mcl-1 inhibition, as previously seen in other models (19). Complete Mcl-1 abrogation in low-expressing cells may be one possible explanation, yet this phenomenon will require further study. Future efforts to elucidate the genetic subsets of DLBCL most vulnerable to Mcl-1 inhibition will advance personalized approaches to DLBCL therapy in the clinic.
By contrast, inhibitors of BTK and Bcl-2 are efficacious in MCL, an aggressive lymphoma subtype characterized by recurrent translocation of CCND1. However, patients who progress on these agents have poor outcomes. In particular, resistance to BTK inhibition in MCL is associated with survival of ~3 months and thus represents an unmet medical need, justifying a search for novel therapeutic modalities (39). Our work builds on earlier findings where MCL1 gene knockout with an inducible CRISPR/Cas9 system or treatment with S63845 triggered spontaneous apoptosis of MCL cell lines, including those which show primary resistance to ibrutinib (6). Here we demonstrate that MCL cell lines with acquired resistance to ibrutinib remain sensitive to AZD5991. Furthermore, primary MCL cells were sensitive to AZD5991 in BAFF-expressing stromal co-cultures which confer partial resistance to ibrutinib and venetoclax (22). Taken together, the above data ascertain Mcl-1 as a tractable target in MCL resistant to novel agents.
Metabolic reprogramming is a hallmark of cancer. Elevated glycolysis in cancer cells, even in the presence of sufficient oxygen, termed Warburg effect, had been postulated to result from defective mitochondrial respiration. However, mitochondrial integrity and consumption of oxidizable substrates have been increasingly appreciated as critical contributors to cancer growth (40,41). In some models, high mitochondrial output contributes to tumor progression, metastasis and chemoresistance (42). Inhibition of OxPhos can slow tumor growth and act as a chemo-sensitizer (43,44). DLBCL tumors significantly enriched in genes involved in mitochondrial respiration comprise the OxPhos-DLBCL subgroup and are characterized by enhanced mitochondrial energy transduction and relative independence of BCR signaling (45). Relevant to our work, metabolic reprogramming towards OxPhos contributed to resistance to ibrutinib in MCL and venetoclax resistance in chronic lymphocytic leukemia (12,46). Therefore, OxPhos represents an important drug target in NHL. In addition to their well-established roles in regulating intrinsic apoptosis programs, the pro-survival Bcl-2 family proteins have been recognized as critical regulators of mitochondrial respiration (29). We found that Mcl-1 inhibition led to mitochondrial stress, as manifested by mitochondrial depolarization, reduction of mitochondrial mass and induction of mitophagy in drug-sensitive DLBCL and MCL cells. Mitochondrial bioenergetics is governed by a complex network of metabolic pathways ultimately leading to generation of the electrochemical proton gradient and ATP synthesis. Mitochondrial dysfunction induced by AZD5991 was accompanied by a compromise in mitochondrial respiration, a finding which was particularly prominent in MCL cells.
How exactly Mcl-1 regulates metabolism remains understudied. Mammalian mitochondria are dynamic organelles that can grow, fuse and undergo lysis depending on the energetic needs of the cell. While there are many factors that can impact mitochondrial mass, a decrease in mitochondrial mass can indicate a compromise in mitochondrial quality. Mcl-1 has been shown to facilitate mitochondrial fusion and ATP production when immersed in the mitochondrial matrix (47), which serves as a likely explanation of metabolic effects of Mcl-1 inhibition seen in our studies. Furthermore, we observed loss of Opa-1, a mitochondrial fusion protein, which, together with a decrease in mitochondrial mass and mitophagy, could further perturbate the mitochondrial dynamics.
Substrates that promote OxPhos through TCA cycle include glucose/pyruvates, glutamine, and fatty acids and relative importance of each may vary between cancer types. For example, triple-negative breast cancer cells exhibited dependence on fatty acid oxidation (48). We show that unlike parental MCL cells which have unbiased nutrient dependence, ibrutinib-resistant cells become addicted to glutamine oxidation that may be suppressed by Mcl-1 inhibition. Here we pave way for future studies which could elucidate metabolic addiction pattern in cancer cells and thus inform therapy.
Earlier studies suggested that sensitivity of DLBCL cells to Mcl-1 inhibitors correlated positively with Mcl-1:Bim expression ratio, and negatively – with expression of Bcl-xL, but not Mcl-1 (17,31). Other studies have found that sensitivity of primary chronic lymphocytic leukemia cells to direct Mcl-1 inhibitor AMG-176 significantly correlated with expression of Mcl-1, but not Bcl-2/xL (19,20). Mechanistically, BH3-mimetics lead to reduced binding of pro-apoptotic Bcl-2 proteins to their targets (31). Mcl-1 may complex with several BH3-only proteins (Bim, Noxa) and pro-apoptotic effectors (Bak, Bax). Since the balance between these proteins likely depends on multiple cell-intrinsic and extrinsic factors and shifts over time, targeting multiple pro-survival Bcl-2 family members may be necessary to skew towards apoptosis. Here we demonstrate that loss of BAX or TP53 confers resistance to Mcl-1 inhibition, and this is accompanied by restoration of OxPhos. This aligns with our earlier work which implicated these genes in resistance to venetoclax in acute myeloid leukemia (29). TP53 likely mediates response to Mcl-1 inhibition via multiple pathways, however our data suggest that it in part relies no functional Bax and possibly Puma.. Thus, a mechanism involving TP53 apoptotic network underlies resistance to distinct BH3-mimetics across a spectrum of hematologic malignancies (Fig. 6I).
We also show that concurrent targeting Mcl-1 and Bcl-2/xL is synergistic in both DLBCL and MCL cells. This result is similar to those obtained previously by us and others using either direct or indirect Mcl-1 inhibitors (6,14,49). However, whether such an approach may be safely translated to the bedside remains to be seen. In preclinical studies, direct Mcl-1 inhibition led to a decrease in monocytes, B cells, and neutrophils, suggesting that immunosuppression may become the dose-limiting factor (20). Furthermore, Mcl-1 inhibition has been shown to suppress hematopoiesis (50). Thus, it is likely that combinations of BH3-mimetics will need to be carefully titrated in the clinic
In sum, direct targeting Mcl-1 interferes with cell survival and metabolic function and is therefore a promising therapeutic approach in DLBCL and MCL. Sensitivity to Mcl-1 inhibition in NHL is mediated via TP53 and BAX apoptotic network.
Supplementary Material
Translational Relevance.
BH3-mimetics targeting the anti-apoptotic protein Bcl-2 have entered clinical practice in lymphoid malignancies. Venetoclax has been approved in therapy of chronic lymphocytic leukemia and demonstrated efficacy in other lymphoid tumors. However, primary and acquired resistance to venetoclax is inevitable and thus novel approaches to target mitochondrial apoptosis are necessary. Mcl-1 is a pro-survival Bcl-2 family member implicated in resistance to venetoclax and other therapeutics. Here we investigated the pre-clinical efficacy of AZD5991, a selective Mcl-1 inhibitor, in lymphoid tumor models in vitro and in vivo. We demonstrate that Mcl-1 inhibition leads to cell metabolic reprogramming and apoptosis. Meanwhile, a dysfunctional TP53 apoptotic network ultimately account for drug resistance, necessitating combination approaches. Our results provide an impetus further to develop Mcl-1 as a target in lymphoid malignancies and highlights potential combination strategies to overcome drug resistance.
Acknowledgements:
A.V.D. is a Leukemia and Lymphoma Society Clinical Scholar (#2319-19). This was work supported by the Leukemia & Lymphoma Society TRP award (#6542-18), American Society of Hematology Bridge Award, and NCI 1R01CA244576-01A1 (to AVD).
Conflict of Interest:
A.V.D. has received consulting fees from Abbvie, AstraZeneca, Bayer Oncology, BeiGene, Genentech, GenMab, Karyopharm, Nurix, Pharmacyclics and TG Therapeutics and has ongoing research funding from AstraZeneca, Bayer Oncology, Bristol Meyers Squibb, Genentech, MEI Pharma, SecuraBio and Takeda Oncology.
J.W.T. received research support from Agios, Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Petra, Seattle Genetics, Syros, Tolero, and Takeda.
References
- 1.Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 2018;25(1):65–80 doi 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strasser A, Elefanty AG, Harris AW, Cory S. Progenitor tumours from Emu-bcl-2-myc transgenic mice have lymphomyeloid differentiation potential and reveal developmental differences in cell survival. EMBO J 1996;15(15):3823–34. [PMC free article] [PubMed] [Google Scholar]
- 3.Kelly PN, Grabow S, Delbridge AR, Strasser A, Adams JM. Endogenous Bcl-xL is essential for Myc-driven lymphomagenesis in mice. Blood 2011;118(24):6380–6 doi 10.1182/blood-2011-07-367672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T, et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev 2014;28(1):58–70 doi 10.1101/gad.232009.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agarwal R, Chan YC, Tam CS, Hunter T, Vassiliadis D, Teh CE, et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med 2019;25(1):119–29 doi 10.1038/s41591-018-0243-z. [DOI] [PubMed] [Google Scholar]
- 6.Dengler MA, Teh CE, Thijssen R, Gangoda L, Lan P, Herold MJ, et al. Potent efficacy of MCL-1 inhibitor-based therapies in preclinical models of mantle cell lymphoma. Oncogene 2020;39(9):2009–23 doi 10.1038/s41388-019-1122-x. [DOI] [PubMed] [Google Scholar]
- 7.Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A 1993;90(8):3516–20 doi 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463(7283):899–905 doi 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou P, Levy NB, Xie H, Qian L, Lee CY, Gascoyne RD, et al. MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood 2001;97(12):3902–9 doi 10.1182/blood.v97.12.3902. [DOI] [PubMed] [Google Scholar]
- 10.Grabow S, Kelly GL, Delbridge AR, Kelly PN, Bouillet P, Adams JM, et al. Critical B-lymphoid cell intrinsic role of endogenous MCL-1 in c-MYC-induced lymphomagenesis. Cell Death Dis 2016;7:e2132 doi 10.1038/cddis.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Awan FT, Kay NE, Davis ME, Wu W, Geyer SM, Leung N, et al. Mcl-1 expression predicts progression-free survival in chronic lymphocytic leukemia patients treated with pentostatin, cyclophosphamide, and rituximab. Blood 2009;113(3):535–7 doi 10.1182/blood-2008-08-173450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guieze R, Liu VM, Rosebrock D, Jourdain AA, Hernandez-Sanchez M, Martinez Zurita A, et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019;36(4):369–84 e13 doi 10.1016/j.ccell.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khoury JD, Medeiros LJ, Rassidakis GZ, McDonnell TJ, Abruzzo LV, Lai R. Expression of Mcl-1 in mantle cell lymphoma is associated with high-grade morphology, a high proliferative state, and p53 overexpression. J Pathol 2003;199(1):90–7 doi 10.1002/path.1254. [DOI] [PubMed] [Google Scholar]
- 14.Klanova M, Andera L, Brazina J, Svadlenka J, Benesova S, Soukup J, et al. Targeting of BCL2 Family Proteins with ABT-199 and Homoharringtonine Reveals BCL2- and MCL1-Dependent Subgroups of Diffuse Large B-Cell Lymphoma. Clin Cancer Res 2016;22(5):1138–49 doi 10.1158/1078-0432.CCR-15-1191. [DOI] [PubMed] [Google Scholar]
- 15.Dengler MA, Weilbacher A, Gutekunst M, Staiger AM, Vohringer MC, Horn H, et al. Discrepant NOXA (PMAIP1) transcript and NOXA protein levels: a potential Achilles’ heel in mantle cell lymphoma. Cell Death Dis 2014;5:e1013 doi 10.1038/cddis.2013.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, et al. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 2005;24(8):1348–58 doi 10.1038/sj.onc.1208300. [DOI] [PubMed] [Google Scholar]
- 17.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016;538(7626):477–82 doi 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 18.Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun 2018;9(1):5341 doi 10.1038/s41467-018-07551-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yi X, Sarkar A, Kismali G, Aslan B, Ayres M, Iles LR, et al. AMG-176, an Mcl-1 Antagonist, Shows Preclinical Efficacy in Chronic Lymphocytic Leukemia. Clin Cancer Res 2020;26(14):3856–67 doi 10.1158/1078-0432.CCR-19-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov 2018;8(12):1582–97 doi 10.1158/2159-8290.CD-18-0387. [DOI] [PubMed] [Google Scholar]
- 21.Best S, Liu T, Bruss N, Kittai A, Berger A, Danilov AV. Pharmacologic inhibition of the ubiquitin-activating enzyme induces ER stress and apoptosis in chronic lymphocytic leukemia and ibrutinib-resistant mantle cell lymphoma cells. Leuk Lymphoma 2019;60(12):2946–50 doi 10.1080/10428194.2019.1616190. [DOI] [PubMed] [Google Scholar]
- 22.Paiva C, Rowland TA, Sreekantham B, Godbersen C, Best SR, Kaur P, et al. SYK inhibition thwarts the BAFF - B-cell receptor crosstalk and thereby antagonizes Mcl-1 in chronic lymphocytic leukemia. Haematologica 2017;102(11):1890–900 doi 10.3324/haematol.2017.170571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eide CA, Kurtz SE, Kaempf A, Long N, Agarwal A, Tognon CE, et al. Simultaneous kinase inhibition with ibrutinib and BCL2 inhibition with venetoclax offers a therapeutic strategy for acute myeloid leukemia. Leukemia 2020;34(9):2342–53 doi 10.1038/s41375-020-0764-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurtz SE, Eide CA, Kaempf A, Khanna V, Savage SL, Rofelty A, et al. Molecularly targeted drug combinations demonstrate selective effectiveness for myeloid- and lymphoid-derived hematologic malignancies. Proc Natl Acad Sci U S A 2017;114(36):E7554–E63 doi 10.1073/pnas.1703094114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018;562(7728):526–31 doi 10.1038/s41586-018-0623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med 2018;24(8):1216–24 doi 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ader NR, Hoffmann PC, Ganeva I, Borgeaud AC, Wang C, Youle RJ, et al. Molecular and topological reorganizations in mitochondrial architecture interplay during Bax-mediated steps of apoptosis. Elife 2019;8 doi 10.7554/eLife.40712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 2016;1863(12):2977–92 doi 10.1016/j.bbamcr.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 29.Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D’Alessandro A, et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov 2019;9(7):910–25 doi 10.1158/2159-8290.CD-19-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rooswinkel RW, van de Kooij B, de Vries E, Paauwe M, Braster R, Verheij M, et al. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood 2014;123(18):2806–15 doi 10.1182/blood-2013-08-519470. [DOI] [PubMed] [Google Scholar]
- 31.Smith VM, Dietz A, Henz K, Bruecher D, Jackson R, Kowald L, et al. Specific interactions of BCL-2 family proteins mediate sensitivity to BH3-mimetics in diffuse large B-cell lymphoma. Haematologica 2020;105(8):2150–63 doi 10.3324/haematol.2019.220525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rauert-Wunderlich H, Rudelius M, Berberich I, Rosenwald A. CD40L mediated alternative NFkappaB-signaling induces resistance to BCR-inhibitors in patients with mantle cell lymphoma. Cell Death Dis 2018;9(2):86 doi 10.1038/s41419-017-0157-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, et al. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res 2014;20(6):1576–89 doi 10.1158/1078-0432.CCR-13-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66(6):443–59 doi 10.3322/caac.21357. [DOI] [PubMed] [Google Scholar]
- 35.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018;24(5):679–90 doi 10.1038/s41591-018-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 2018;378(15):1396–407 doi 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci U S A 2015;112(44):13447–54 doi 10.1073/pnas.1514944112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bojarczuk K, Wienand K, Ryan JA, Chen L, Villalobos-Ortiz M, Mandato E, et al. Targeted inhibition of PI3Kalpha/delta is synergistic with BCL-2 blockade in genetically defined subtypes of DLBCL. Blood 2019;133(1):70–80 doi 10.1182/blood-2018-08-872465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin P, Maddocks K, Leonard JP, Ruan J, Goy A, Wagner-Johnston N, et al. Postibrutinib outcomes in patients with mantle cell lymphoma. Blood 2016;127(12):1559–63 doi 10.1182/blood-2015-10-673145. [DOI] [PubMed] [Google Scholar]
- 40.Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochim Biophys Acta 2011;1807(6):552–61 doi 10.1016/j.bbabio.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 41.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 2012;15(1):110–21 doi 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Heredia JM, Carnero A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020;9(7) doi 10.3390/cells9071693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013;23(3):287–301 doi 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 2010;107(19):8788–93 doi 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012;22(4):547–60 doi 10.1016/j.ccr.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med 2019;11(491) doi 10.1126/scitranslmed.aau1167. [DOI] [PubMed] [Google Scholar]
- 47.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 2012;14(6):575–83 doi 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab 2017;26(4):633–47 e7 doi 10.1016/j.cmet.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paiva C, Godbersen JC, Soderquist RS, Rowland T, Kilmarx S, Spurgeon SE, et al. Cyclin-Dependent Kinase Inhibitor P1446A Induces Apoptosis in a JNK/p38 MAPK-Dependent Manner in Chronic Lymphocytic Leukemia B-Cells. PLoS One 2015;10(11):e0143685 doi 10.1371/journal.pone.0143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohler S, Afreen S, Fernandez-Orth J, Demmerath EM, Molnar C, Wu Y, et al. Inhibition of the anti-apoptotic protein MCL-1 severely suppresses human hematopoiesis. Haematologica 2020;Online ahead of print doi 10.3324/haematol.2020.252130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.