

Abstract
Immune sentinel cells initiate immune responses to pathogens and tissue injury and are capable of producing highly stimulus-specific responses. Insight into the mechanisms underlying such specificity has come from the identification of regulatory factors and biochemical pathways, as well as the definition of signaling circuits that enable combinatorial and temporal coding of information. Here we review the multi-layered molecular mechanisms that underlie stimulus-specific gene expression in macrophages. We categorize components of inflammatory and anti-pathogenic signaling pathways into five layers of regulatory control and discuss unifying mechanisms determining signaling characteristics at each layer. In this context, we review mechanisms that enable combinatorial and temporal encoding of information, identify recurring regulatory motifs and principles, and present strategies for integrating experimental and computational approaches towards the understanding of signaling specificity in innate immunity.
eTOC blurb
Immune sentinel cells are capable of highly specific responses to pathogens or tissue injury. Hoffmann and colleagues review the mechanisms that underlie stimulus-specific gene expression in macrophages and provide a framework for categorizing layers of regulatory control and understanding the mechanisms that enable temporal and combinatorial encoding of signal information in innate immunity.
Introduction
Immune sentinel cells such as macrophages and fibroblasts initiate and orchestrate immune responses to pathogen invaders or tissue injury. As first responders, they provide finely tuned immune activation to provide pathogen-appropriate protection, while avoiding excessive tissue damage and inflammation. Recent studies have revealed that immune sentinel cells produce gene expression responses that are remarkably more stimulus- and pathogen-specific (Amit et al., 2009; Cheng et al., 2017, 2019; Sheu et al., 2019) than previously thought (Nau et al., 2002). These support the theoretical consideration that because many immune response gene products are intrinsically toxic to the host they should only be expressed as-needed.
Immune sentinel cells respond to hundreds of ligands, including PAMPs present on or shed by pathogens, DAMPs from damaged host cells, and secreted cytokines, using dozens of pattern recognition receptors (PRRs) and numerous cytokine receptors. They reside in diverse cellular locations, such as the cell surface (e.g. Toll-like receptors (TLRs) and cytokine receptors such as TNFR, IL1R, and IFNAR), the endosome (TLRs), and the cytosol (e.g. the nucleic acid sensing cGAS and RIG-I like receptors, RLRs) (Boraschi et al., 2018; Dostert et al., 2019; Janeway and Medzhitov, 2002; Takeuchi and Akira, 2010; de Weerd and Nguyen, 2012). The optimal response to each threat involves a distinct mixture of dozens of functional responses that comprise cell-intrinsic pathogen defenses, cell death control, recruitment of diverse immune effector cells, tissue remodeling, and activation of systemic and adaptive immune mechanisms (Sheu et al., 2019).
The premise of this review is that integrating these mechanisms identified by molecular immunology with principles of systems biology will lead to substantial advances in our understanding of how immune sentinel responses are regulated. We specifically review the signaling mechanisms described by the innate immunology literature and relate these mechanisms to regulatory motifs commonly described by systems biology. This review therefore aims to build bridges to connect the two fields to advance our understanding of innate immune response regulation.
Connecting Molecular Immunology and Systems Biology
Molecular immunologists have generated a wealth of knowledge about the molecular signaling network which passes information from receptors to nuclear target genes, from identifying the components, to characterizing molecular interactions, to understanding the quantitative dose-response relationship. These have been summarized well in recent reviews (Ablasser and Hur, 2020; Barrat et al., 2019; Brignall et al., 2019; Caruso et al., 2014; Hu and Sun, 2016; Kawai and Akira, 2011; Newton and Dixit, 2012).
In parallel, systems biologists have posited that the observed stimulus specificity may be understood in terms of two main coding strategies by which information about the extra-cellular stimulus is conveyed to nuclear target genes (Figure 1A). The combinatorial code produces stimulus-specific gene expression when different stimuli activate distinct combinations of gene expression regulators that are differentially utilized by immune response genes (Buchler et al., 2003; Cheng et al., 2017; Doyle et al., 2002; Gottschalk et al., 2016; Hoffmann, 2016; Junkin et al., 2016; Scholes et al., 2017). The temporal code allows stimulus-specificity in gene activation through the action of a single effector, whose stimulus-specific temporal pattern of activity is decoded differentially by target genes (Behar and Hoffmann, 2010; Hoffmann, 2016; Purvis and Lahav, 2013; Sheu et al., 2019).
Figure 1. Principles of combinatorial and temporal coding for stimulus-specific gene expression and the layers of the stimulus-responsive network enabling it.
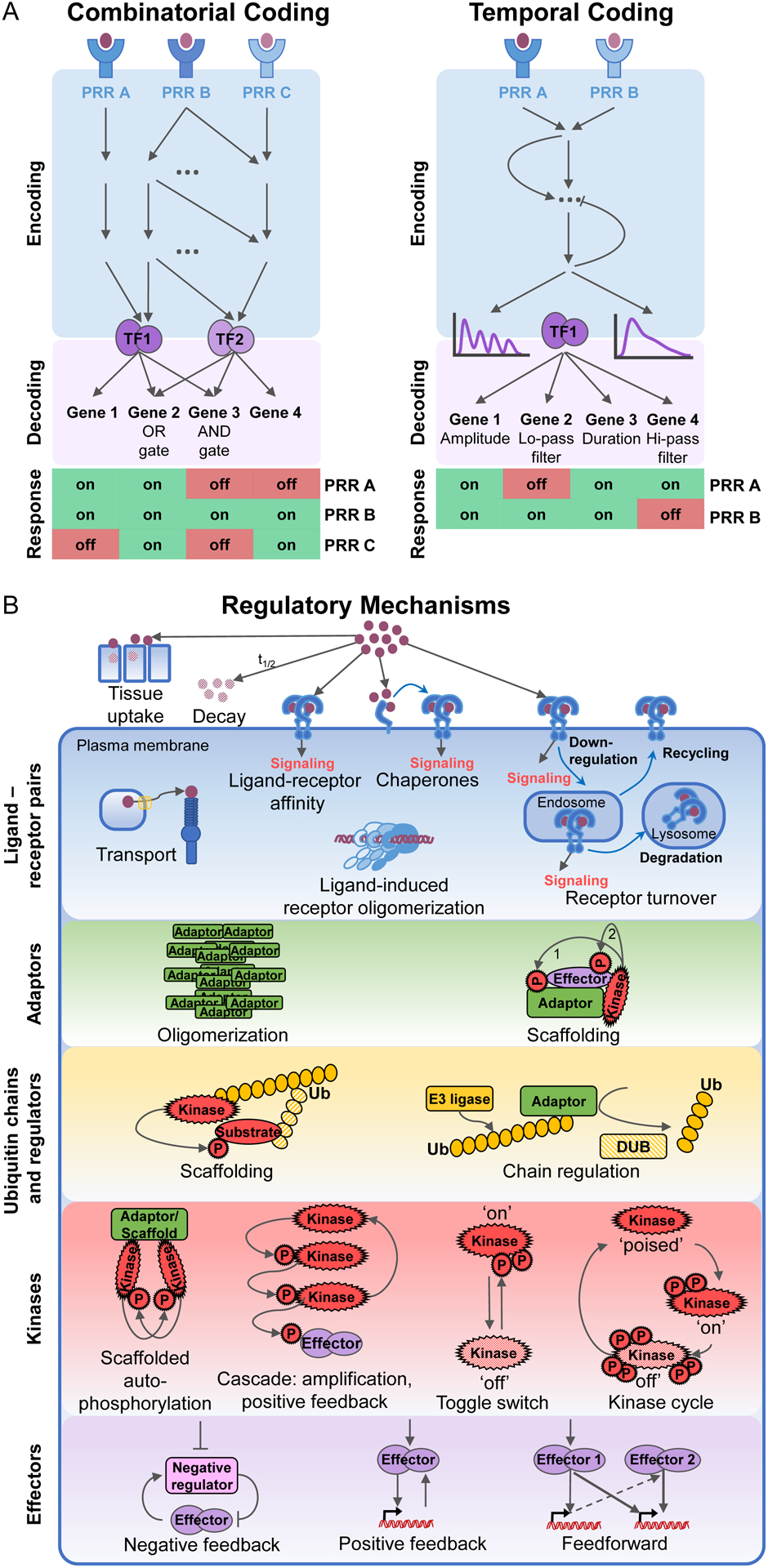
(A) In combinatorial encoding, receptor proximal mechanisms activate stimulus-specific effector combinations. Response genes decode this through Boolean logic gate-based regulation. Stimulus-specific activity profiles of single effector modules allow temporal encoding of information, which is decoded by gene-specific mechanisms. PRR: pattern recognition receptor; Lo-pass filter: low pass filter; Hi-pass filter: high pass filter.
(B) Regulatory strategies controlling immune sentinel cell responses in the signaling network. The stimulus-responsive network consists of multiple functionally distinguishable signaling layers: (1) ligand-receptor pairs, (2) receptor-associated adaptors, (3) ubiquitin signaling layer, (4) kinase hubs, (5) SDTF modules and other effectors. Biochemical and cell biological mechanisms are shared between the diverse components of each signaling layer that together enable stimulus-specific coding. Ligand-receptor interactions in different subcellular locations are regulated through ligand stability, diffusion, transport, binding chaperones, and receptor transport and turnover. Adaptor proteins scaffold the assembly of larger signaling complexes. The kinetics of adaptor complex oligomerization are a key determinant of downstream signaling. Ubiquitin chains of different linkages pass on, fine-tune, and eventually downregulate signaling. They are regulated by the complex interplay of E3 ligases and DUBs. Signaling converges on three key kinase families, which are activated through scaffolded autophosphorylation or kinase cascades with toggle switch or kinase cycle mechanisms. The effector molecules activated by kinases are transcription factors inducing gene expression or posttranscriptional regulators. They are regulated through negative and positive feedback and feedforward loops. DUBs: deubiquitinases, Ub:ubiquitin, P: phosphoryl group.
Yet, despite the significant advances made in both areas, a disconnect between the molecular immunology literature and the systems biology literature has slowed progress in understanding how immune sentinel cell responses. While early systems biology studies were focused on conceptual or proof-of-principle studies and therefore employed convenient experimental systems that do not always faithfully recapitulate complex biology, recent publications demonstrate that iteration between mathematical modeling and experimental studies can provide important biological insights when applied to primary cells, whether embryonic stem cells (Heemskerk et al., 2019), during gastrulation (Chhabra et al., 2019), epithelial cell oncogenesis (Aikin et al., 2019), cancer drug responses (Chopra et al., 2020), or epigenetic control of cell death (Vanaja et al., 2018).
However, in research of innate immunity, integrated experimental and mathematical modeling studies remain rare. Though several seminal studies have been carried out in related fields, including chemosensing (Manes et al., 2015; Meier-Schellersheim et al., 2006; Xu et al., 2010), TRAIL signaling to apoptosis (Albeck et al., 2008a, 2008b; Spencer et al., 2009), T cell receptor signaling (Chakraborty and Weiss, 2014; Ganti et al., 2020), and T cell signaling (Feinerman et al., 2008a, 2008b; Voisinne et al., 2015), the first signaling study employing primary macrophages has only just been reported (Adelaja et al., 2021). Still, recent advances in technology such as CRISPR-mediated gene insertion (Kundert et al., 2019), single molecule RNA in situ fluorescence (Eng et al., 2019; Foreman and Wollman, 2020; Xia et al., 2019), microfluidics (Junkin et al., 2016; Lin et al., 2019), or organoid culture (Zhang et al., 2019a) allow the generation of high-resolution, quantitative datasets that enable mathematical-model-aided systems biology studies of immune processes. These studies thereby illustrate how the wealth of molecular immunology knowledge may unravel how combinatorial and temporal codes control the stimulus-specificity of innate immune responses. We offer a supplementary note describing experimental methods perturbations used by molecular immunologists to dissect the components of signaling pathways and the regulatory motifs used by systems biologists to study the behavior of signaling pathways (Box1, Note S1).
Box 1. Experimental approaches and regulatory motifs to interrogate and understand signaling networks.
Immunologists can interrogate all layers of the signaling network with a variety of experimental methods.
In Supplemental Note S1, we describe a selection of commonly used methods to measure:
protein abundance and turnover kinetics
protein-protein interactions
protein localization
kinase activity
DNA-binding activity
chromatin state
RNA abundance • metabolic state
cytokine secretion
For each, we describe assay characteristics pertaining to quantitation, sensitivity, resolution, and multiplexing; we discuss the data generation and data interpretation challenges and provide references to protocols or prominent studies employing the method.
Perturbation studies are key to understanding a biological system. We describe a few commonly used approaches:
pharmacological perturbation
RNA interference
Genetic deletion by targeted recombination and CRISPR
ectopic gene expression by transfection/transduction
For each, we describe its purpose, advantages and caveats for interpretation, and provide references to protocols or prominent studies.
Regulatory motifs with characteristic dynamical behavior are found in the innate immune signaling network. In Supplemental Note S1, we describe some with the help of simple mathematical models:
Toggle switch vs. cycle switch
Multistep reaction cascades
Boolean logic gates • AND gates • OR gates
Coherent feedforward loop
Incoherent feedforward loop • pulse generator • fold change detector
Positive feedback loop • bistability • ultrasensitivity
Negative feedback loops • track input duration • improve responsiveness • pulse generator • oscillation generator
For each, we provide a general diagram of the reactions, the ordinary differential equations that model the system, a demonstration of key behaviors that the motif allows for, and brief examples of the molecular mechanisms the motifs apply to. This is discussed in greater detail in the main text.
Layers of Control in the Stimulus-Responsive Network
The signaling network that mediates inflammatory and anti-pathogenic gene expression consists of hundreds of proteins with diverse biochemical functions. We categorize them into five layers of regulatory control - receptors, signaling adaptors, ubiquitin chain regulators, signaling kinases, and effectors - and briefly describe the unifying molecular mechanisms determining signaling characteristics at each layer (Figure 1B).
The first layer of control for initiating stimulus-specific responses comprises the factors that govern ligand-receptor interactions. Ligands have distinct half-lives of bioactivity, differential diffusion, transport, and cell uptake characteristics. Receptors are expressed at distinct levels and sorted to distinct subcellular locations via intracellular trafficking (Bagnall et al., 2018; Chen et al., 2017) including endocytosis trafficking (Leifer and Medvedev, 2016). These processes may be modulated by signals from the tissue microenvironment and by prior immune threat exposure, providing context specificity to the first control layer (Roberts et al., 2017). A variety of proteins (e.g. TIRAP and TRAM) regulate receptor localization and availability to bind ligands (Kagan et al., 2008; Leifer and Medvedev, 2016). Ligand-receptor interaction affinity may not solely be determined by structures of ligand and receptors, but also by co-factors (e.g. CD14 for LPS-TLR4 binding) that facilitate ligand-receptor interaction or receptor oligomerization (Park and Lee, 2013; Zanoni et al., 2011). Ligand binding induces di- or multimerization of PRRs and cytokine receptors, often a prerequisite for adaptor recruitment and signal transduction. For example, TLRs dimerize upon ligand binding and RLRs multimerize into a filament on the RNA (Ablasser and Hur, 2020; Ohto and Shimizu, 2016). The cell-to-cell variability of receptor availability is an important determinant of phenotypic diversification. Systems biology modeling has proven very helpful in probing these relationships (Feinerman et al., 2008b; Gaudet et al., 2012; Martins et al., 2017).
Within the second control layer, signaling adaptors connect ligand-bound receptors with intra-cellular signaling cascades. Important adaptors in innate immune signaling include MyD88 for all TLRs except TLR3, TRIF for TLR3 and TLR4, MAVS for RLRs, and STING for cGAS. Among cytokine receptors, IL-1 signaling is also mediated by MyD88, but the large TNFR-superfamily has distinct adaptors (e.g. TRADD) (Ablasser and Hur, 2020; Boraschi et al., 2018; Chen and Jiang, 2013; Dostert et al., 2019; Luo et al., 2019). These adaptors generally lack enzymatic activity themselves but rather scaffold or nucleate the assembly of larger signaling complexes (e.g. the Myddosome). This often occurs through direct homotypic domain interactions with their receptors (e.g. CARD domains for RLR-MAVS interaction), but may also involve second messenger molecules (e.g. cGAMP in STING) (Ablasser and Hur, 2020; Cadena et al., 2019; Monie et al., 2009; Wu et al., 2013). Therefore, the precise biochemical characteristics and kinetic rate constants of receptor-adaptor interactions, adaptor oligomerization, and second messenger production, half-life and diffusion determine the signal transduction behavior of the second control layer. Interestingly, the biochemical characteristics may be further modulated by the subcellular location of the adaptor. MyD88 for example may function from both plasma membrane and endosomal compartments and may have distinct biochemical kinetic properties.
We define the third control layer by the complex network of E3 ubiquitin ligases and deubiquitinases (DUBs). These generate and degrade, respectively, K63-linked, K48-linked, and linear (M1-linked) ubiquitin chains. K63 and linear ubiquitin chains have scaffolding function to facilitate the interaction of downstream signaling kinases and substrates, while K48 ubiquitin chains induce proteasomal degradation of target proteins (Hu and Sun, 2016). Ubiquitin chains are attached either to the E3 ligases themselves (e.g. K63-linked ubiquitin on TRAF6 in MyD88 and MAVS signaling) or to upstream adaptors or bridge proteins (e.g. K63-linked chains on RIP1 kinase in TRIF and TRADD signaling) or may be unanchored (Hu and Sun, 2016; Xia et al., 2009). Regulatory specificity is achieved by the activation or recruitment of specific E3 ligases; the extent and duration of their activity may be modulated, and they also differ in the type of ubiquitin linkages that they generate (Hu and Sun, 2016; Parvatiyar et al., 2018). An interesting example is LUBAC, an E3 ligase that synthesizes linear ubiquitin chains which contribute to activation of the IκB kinase (IKK) in response to TNF signaling, but negatively affects RIG-I signaling (Hu and Sun, 2016; Tokunaga, 2013). Similarly, DUBs may have distinct roles in different pathways. CYLD, for example, attenuates RIG-I signaling by removing K63-linked chains, but promotes STING signaling by removing K48-linked chains (Friedman et al., 2008; Zhang et al., 2018). Interestingly, the anti-inflammatory protein TNFAIP3/A20 was reported to act as both a DUB and an E3 ligase to negatively regulate NFκB signaling by removing K63-linked chains and adding K48-linked chains to multiple targets (Catrysse et al., 2014; Wertz et al., 2004), though their physiological roles have been questioned (De et al., 2014; Skaug et al., 2011). Thus, the ubiquitin control layer regulates interactions, activity, and degradation of key signaling proteins, and provides many levers for fine-tuning of responses, cross-regulation and network memory.
The fourth control layer involves three central families of response kinases, namely MAPKs (JNK, p38, ERK), the IKK complex (consisting of IKKα, IKKβ, and IKKγ/NEMO), and TBK1 (Ablasser and Chen, 2019; Newton and Dixit, 2012). These kinases serve as the regulatory network hubs that directly or indirectly activate downstream effectors. Kinase activity in immune response pathways is generally initiated by trans-autophosphorylation when at least two kinase molecules are brought into proximity through oligomerized adaptors (e.g. activation of TBK1 downstream of STING) or ubiquitin chains (e.g. activation of TAK1 and IKK) (Emmerich et al., 2013; Häcker and Karin, 2006; Hu and Sun, 2016; Kensche et al., 2012; Ma et al., 2012; Polley et al., 2013; Shu et al., 2013; Xia et al., 2009; Zhang et al., 2019b). (In contrast, tyrosine kinase receptors are activated via ligand-induced conformational changes of receptor dimers (Livnah et al., 1998)). Kinase activity may then be amplified through kinase cascades, the most prominent being MAPK cascades (Kholodenko and Birtwistle, 2009; Witzel et al., 2012). Scaffolds may also connect kinases to their substrates. For example, for IKK, NEMO, which connects the catalytic components to ubiquitin chains, also contains an IκBα recruitment domain that ensures efficient activation of NFκB (Schröfelbauer et al., 2012). Kinase activity is then terminated via one of two mechanisms. In the classic toggle-switch model, inactivation occurs via dephosphorylation by phosphatases (Bardwell, 2008; Ferrell and Bhatt, 1997; Ferrell and Ha, 2014; Nolen et al., 2004; Zhao et al., 2012). (Phosphatases may also target signaling molecules further upstream, such as signaling adaptors STING, TRIF, MAVS (Ni et al., 2020; Xiang et al., 2016)). However, IKK is thought to be regulated via a multi-state reaction cycle, in which an autophosphorylated inactive state must be recycled to a phosphorylation-free poised state before re-activation can occur (Behar and Hoffmann, 2013). The two inactivation modalities impart fundamentally different dynamic control to the kinase activity, and phosphatases play distinct roles. Their potential for inducible expression provides another level of regulatory control.
The fifth control layer comprises the signaling pathway effectors that directly control gene expression. There are two main categories of effector proteins: transcription factors and proteins acting post-transcriptionally. Three prominent signal-dependent transcription factor (SDTF) families may be directly mapped to upstream kinases: MAPK-AP1/ATF, IKK-NFκB, and TBK1-IRF (Cargnello and Roux, 2011; Hayden and Ghosh, 2004; Honda et al., 2006). In addition, NFAT is activated in dendritic cells in a Ca2+-dependent manner (Fric et al., 2012; Zanoni and Granucci, 2012). Some kinases regulate post-transcriptional regulatory mechanisms. For example, IKK phosphorylates the mRNA decapping activator ECD4, affecting the levels of hundreds of mRNAs (Mikuda et al., 2018). Further, production of TNF involves not only NFκB-dependent transcription, but MAPK p38/ERK-dependent mRNA splicing, mRNA stabilization via tristetraprolin (TTP) (Mahtani et al., 2001), and pro-protein processing via TACE (Caldwell et al., 2014), explaining why TNF secretion by single cells does not correlate well with NFκB activity (Junkin et al., 2016). Effectors may be regulated in a simple toggle switch fashion with phosphatases terminating effector activity when kinase activity has diminished (e.g. IRFs and the MAPK substrates AP1 or TTP) (Gu et al., 2014; Long et al., 2014). Effectors may also be subject to regulatory feedback. NFκB is subject to complex negative feedback regulation via IκB proteins which inhibit NFκB through direct binding in the nucleus and escort NFκB back into the cytoplasm. This inhibition is reversible for some IκB isoforms (IκBα, IκBε), as their degradation is induced by IKK (Mitchell et al., 2016). In contrast, IRF induces the interferon-β (IFNβ) feedforward system to activate ISGF3-driven gene expression program in both primary response and bystander cells (Ourthiague et al., 2015). In addition, MAPK phosphatases such as dual-specificity phosphatases (DUSPs) are strongly upregulated by MAPK-activating PRRs, contributing to negative feedback on MAPK pathways (Brondello et al., 1999; Kuwano et al., 2008; Salojin et al., 2006). The strength and time delays in these feedback systems are key to imparting specific activation dynamics to the effectors. Further quantitative studies are needed to enable reliable mathematical modeling studies.
It is the stepwise transmission and modulation of the signal by these biochemical and cell biological mechanisms through the network layers that mediates combinatorial and temporal coding of response specificity. While numerous molecular mechanisms have been described for key TLR pathways, how they function together to generate stimulus-specific responses is generally less well understood. As will be discussed below, stimuli activate combinations of effectors through the molecular mechanisms within receptor, adaptor, ubiquitin, and kinase layers, while stimulus-specific dynamic control is mediated by molecular mechanisms in all five control layers.
Mechanisms that Enable Combinatorial Encoding
When stimuli activate different combinations of signaling pathways they convey stimulus-information by combinatorial coding (Figure 1A). Hence, branching of the signal into two (or more) signaling pathways is key to combinatorial coding. Here we first present the molecular mechanisms that allow for pathway branching and then describe how these may be deployed stimulus-specifically.
Pathway branching occurs at multiple control layers of the signaling network. Pathways usually branch when a regulator sequentially (not coincidently) engages two signaling complexes, often by moving from one sub-cellular compartment to another. A well-studied example of this in the receptor layer is how TLR4 engages two different adaptors at different subcellular locales, MyD88 at the cell surface (activating NFκB and MAPKs) and TRIF in the endosomes (additionally activating IRF3) (Figure 2A). This adaptor switch is mediated by a switch of the bridge proteins TIRAP to TRAM, which facilitate the association of the adaptors with TLR4 within the plasma or endosomal membranes, respectively (Balka and Nardo, 2019; Deguine and Barton, 2014). Thus, a key principle of combinatorial encoding by TLR4 is that adaptor engagement is sequential and may be regulated by regulating the transport of TLR4 from one compartment to the other (Leifer and Medvedev, 2016).
Figure 2. Molecular mechanisms for combinatorial encoding to achieve stimulus-specific responses.
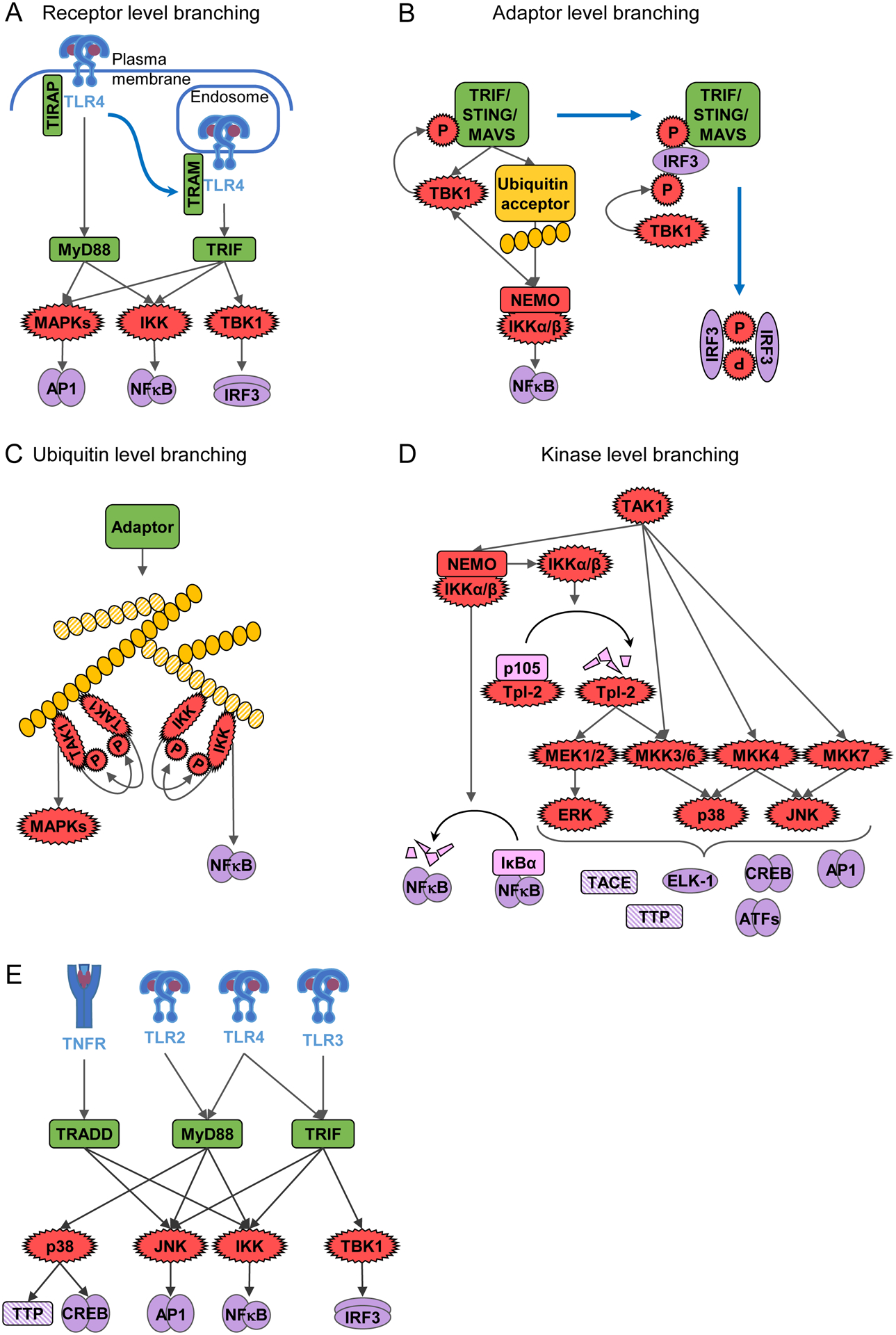
A-D. Pathway branching, in which one upstream protein activates multiple downstream signaling mediators, can occur at several signaling layers. Examples include branching (A) at the level of receptors with TLR4 activating the MyD88 and TRIF pathway, (B) at the level of adaptors with TRIF, MAVS, and STING being capable of IRF3 and NFκB activation, (C) at the level of ubiquitin chains, which scaffold activation of TAK1 for MAPK activation and of IKK for NFκB activation, and (D) at the level of kinase cascades which branch to activate IKK and the MAPKs p38, JNK, and ERK.
E. Combinatorial coding in the innate immune network is mediated by receptor-specific utilization of adapters and pathway branching at the adapter layer.
Signaling pathways may also branch at the adaptor layer (Figure 2B): TRIF-mediated activation of the TBK1-IRF3 axis and the IKK-NFκB axis is a prominent example (Chen and Jiang, 2013; Jiang et al., 2004), with MAVS and STING functioning similarly (Abe and Barber, 2014; Dunphy et al., 2018; Fang et al., 2017a, 2017b; Liu et al., 2013; de Oliveira Mann et al., 2019; Yoboua et al., 2010; Zhang et al., 2019b). On the one hand, TRIF oligomerization leads to the recruitment of RIP1 kinase, which is ubiquitinated by E3 ligases (Cusson-Hermance et al., 2005). Those ubiquitin chains lead to IKK recruitment and trans-autophosphorylation allowing it to activate NFκB (Hu and Sun, 2016; Kensche et al., 2012; Polley et al., 2013). On the other hand, oligomerized TRIF also recruits TBK1, though the exact mechanism is not fully elucidated (Funami et al., 2008). Although initial TBK1 activation may require a priming phosphorylation through IKK activity (Abe et al., 2020), trans-autophosphorylation of TBK1 leads to its full activation (Ma et al., 2012; Shu et al., 2013). Activated TBK1 phosphorylates the adaptor, allowing IRF3 binding to the adaptor, which subsequently serves as a scaffold for TBK1-mediated IRF3 phosphorylation (Abe et al., 2020; Liu et al., 2015b). Thus, activation of two pathways by a single adaptor appears to occur sequentially with the second pathway activation requiring a post-translational modification of the adaptor. Similarly, NFκB and IRF3 activation seem spatially regulated downstream of STING, with NFκB activation occurring from the ER, where STING is first activated, while IRF3 signaling requires STING trafficking to the Golgi (Ni et al., 2017; Stempel et al., 2019).
Ubiquitin chains constitute remarkably efficient signaling scaffolds that mediate pathway branching to the MAPK and NFκB pathways (Figure 2C). For example, ubiquitin chains attached to adaptor TRAF6 provide scaffolds for the trans-autoactivation of kinases, such as TAK1, a MAPK kinase kinase (MAPKKK) (Ishitani et al., 2003; Kanayama et al., 2004; Scholz et al., 2010; Schröfelbauer and Hoffmann, 2011; Xia et al., 2009). In addition, the IKK complex trans-autophosphorylates when clustered on (preferentially linear) ubiquitin chains through its ubiquitin-binding subunit NEMO (Ea et al., 2006; Haas et al., 2009; Kensche et al., 2012; Polley et al., 2013; Rahighi et al., 2009; Tokunaga, 2013; Xia et al., 2009). Thus, the ubiquitin layer provides numerous opportunities for signaling crosstalk and pathway branching.
Given the enzymatic activity of kinases, they may phosphorylate several substrates and thus mediate pathway branching (Figure 2D): TAK1 mediates pathway branching to the MKK4/7-JNK axis and the IKK-NFκB axis. TAK1 is the initiator of the MAPK cascade that leads to JNK activation (Cargnello and Roux, 2011). In addition, TAK1 may initiate IKK activation (Cohen and Strickson, 2017; Emmerich et al., 2013; Shim et al., 2005; Wang et al., 2001; Zhang et al., 2014), though this is stimulus-specific and may be cell type dependent. It is not clear what determines TAK1’s substrate specificity (Shim et al., 2005). Interestingly, IKK also engages in pathway branching by not only activating the IκB-NFκB axis, but also the p105-TPL2-ERK axis (Beinke et al., 2004; Gantke et al., 2012). IKK mediated p105 degradation is essential for Tpl2 activation, which is the MAPKKK for ERK (Beinke et al., 2004; Gantke et al., 2012; Waterfield et al., 2003). Indeed, IKK may further phosphorylate Tpl2 (Roget et al., 2012). IKK’s substrate specificity seems to be controlled by NEMO, which is essential for IKK’s activation by inflammatory stimuli: NEMO directs IKK’s substrate specificity to IκBα, but may then dissociate from the IKK dimer, broadening IKK’s specificity to include p105 (Schröfelbauer and Hoffmann, 2011; Schröfelbauer et al., 2012). Furthermore, in pDCs, IKK was reported to activate IRF7 downstream of MyD88 mediating TLR7 and TLR9 signals (Honda et al., 2004; Hoshino et al., 2006; Kawai et al., 2004; Pauls et al., 2012; Uematsu et al., 2005). Spatial segregation between distinct endolysosomal compartments may contribute to the switch from NFκB to IRF7 signaling (Honda et al., 2005; Sasai et al., 2010). This illustrates that pathway branching at the kinase control level also involves sequential mechanisms and may be cell-type specific.
To mediate stimulus-specific responses, pathway branching must be deployed stimulus-specifically. The combination of the IKK-NFκB and JNK-AP1 axes is activated by all adaptor proteins discussed here, i.e. MyD88, TRIF, STING, MAVS, and TRADD, ensured by pathway branching at the ubiquitin chain and the kinase layers, and thus generates a common core gene expression response (Amit et al., 2009; Cheng et al., 2017). However, originating at the receptor layer, stimulus-dose and exposure route modulate JNK and NFκB signaling to allow distinction of bacterial species, dose, and infection vs. bystander status (Lane et al., 2019). More obviously, the TBK1-IRF3 and the MAPKp38 axes are deployed stimulus-specifically by virtue of quantitative differences due to receptor-specific utilization of distinct adaptors, followed by pathway branching at the adaptor layer. Their stimulus-specific activation in conjunction with the common core pathways thus enables combinatorial coding (Figure 2E).
The TBK1-IRF3 axis mediates stimulus-specific induction of type I IFN (Kawai and Akira, 2011). In macrophages (and conventional dendritic cells), the IRF pathway is activated only in response to endosomal signaling by TLR4 and TLR3, and cytosolic signaling from cGAS and RLRs (Caruso et al., 2014; Kawai and Akira, 2011; Sparrer and Gack, 2015). This specificity is mediated by the ability of TLR3 and TLR4 to engage the TRIF adaptor in the endosome, while cGAS and RLRs engage STING and MAVS respectively. TRIF, STING and MAVS share an activation mechanism for the TBK1-IRF3 axis, while also being able to activate IKK-NFκB and TAK1-JNK axes (see Section 2). Thus, for example, early studies showed TLR4 activation by lipopolysaccharide (LPS) from gram-negative bacteria activates both NFκB and IRF3, while TLR2 activation by peptidoglycans from gram-positive bacteria lacks the IRF3 component (Doyle et al., 2002). As IRF3 induces expression of the cytokine IFNβ, which activates the ISGF3 TF complex in an autocrine and paracrine manner, the effect of stimulus-specific activation of IRF3 is dramatically amplified both within the first-responder cell and its neighbors.
A second, less well characterized example is the stimulus-specific activation of MAPKs p38 and ERK. Several lines of evidence indicate that functionally potent MAPKp38 activation is stimulus-specific, even if some level of activity can be detected in response to many stimuli, such as TLR ligands, cytosolic nucleic acids, and TNF. First, transcriptome profiling of fibroblasts and macrophages stimulated with a diverse set of PAMP and cytokine ligands revealed a set of about 80 genes that required both NFκB and MAPKp38 activity (Cheng et al., 2017). Second, MAPKs p38 and ERK have a key role in post-transcriptional processing, mRNA half-life, pro-protein processing and secretion of TNF, and cell stimulation with TNF failed to activate these processes (Caldwell et al., 2014). Finally, while stimulation with high concentrations of LPS led to TNF production, low concentrations activated NFκB but failed to produce TNF (Gottschalk et al., 2016), in line with the conclusion that single cell TNF secretion was better correlated with MAPK activation than NFκB (Junkin et al., 2016). However, the mechanism of stimulus-specific activation of the MAPKp38 axis remains poorly understood. One hypothesis is that the MyD88 oligomerization into the Myddosome complex results in a thresholded dose response (Cheng et al., 2015) such that p38 is only activated at high ligand concentrations. In addition, the MAPK cascade itself is thought to have a thresholded dose response curve (Huang and Ferrell, 1996), dependent on the level of phosphatase activity (Altan-Bonnet and Germain, 2005). Interestingly, MAPKp38 activity may be induced through two different pathways, namely MKK4 downstream of TAK1 MAPKKK activity and MKK3/6 activated by Tpl-2 (Cohen and Strickson, 2017; Pattison et al., 2016). This may suggest that while TNF induces MAPK p38 via MKK3/6, LPS engages both pathways to generate a more potent p38 activity (Pattison et al., 2016).
In sum, combinatorial effector activation may encode information about both the molecular identity and the dose of a stimulus. Quantitative differences in the dose response curves of co-activated pathways increase the capacity for producing stimulus-specific responses via combinatorial coding. Future studies should address not only the mechanisms of pathway branching (e.g. the role of IKK in TBK1-IRF activation and the mechanisms of NFκB and MAPK activation downstream of cytosolic nucleic acid sensing remain unclear), but also address dose-response behavior of each of the branched pathways, and whether they are modulated by the microenvironment and the cell’s exposure history. Importantly, during the infection process pathogens stimulate several PRRs and autocrine cytokines, which further activate related pathways, that in aggregate characterize pathogen-specific responses in vivo (Gottschalk et al., 2019). Given that in vitro studies with multiple ligands revealed instances of synergy or antagonism between pathways (Gottschalk et al., 2019; Gutschow et al., 2018; Lin et al., 2017; Pandey et al., 2020; Tan et al., 2014), further study of cross-regulatory connections between co-activated pathways is warranted.
Mechanisms that Enable Temporal Encoding
Signaling involves the transient perturbation of a homeostatic steady state. Hence, the activity of a signaling effector exhibits a temporal trajectory. The observation that two stimuli may trigger distinct temporal trajectories and thereby differential gene expression led to the hypothesis of a “temporal signaling code” (Figure 1A) (Hoffmann and Baltimore, 2006). Temporal trajectories may be quantitatively characterized by a set of features, such as peak amplitude, fold change, amplitude of the second signaling phase, total duration, or oscillations (Behar and Hoffmann, 2010; Levine et al., 2013; Li and Elowitz, 2019; Purvis and Lahav, 2013). Here we describe how such dynamic features may be generated, or encoded, by the successive modulation of signaling activity by multiple mechanisms as the signal passes through successive control layers.
Within the first control layer, the first determinant of signaling dynamics is necessarily ligand availability as determined by ligand diffusion, endocytosis, and decay. Secreted TNF, for example, has a short half-life, whereas the endotoxin LPS persists. In tissue environments, diffusion rates and rates of uptake by other cells may also be important (Bagnall et al., 2018). Next, receptor clustering upon ligand binding (Caré and Soula, 2011) may threshold the dose-response curve preventing spurious activation in the absence of ligand (Bray et al., 1998).
Following ligand binding, endocytosis of receptor-ligand complexes reduces the availability of receptors for further stimulation. The speed of receptor replenishment, through recycling or de novo synthesis, together with ligand half-life is a key determinant of the amplitude of second phase signaling (Becker et al., 2010) (Figure 3A).
Figure 3. Molecular mechanisms for temporal encoding to achieve stimulus-specific responses.
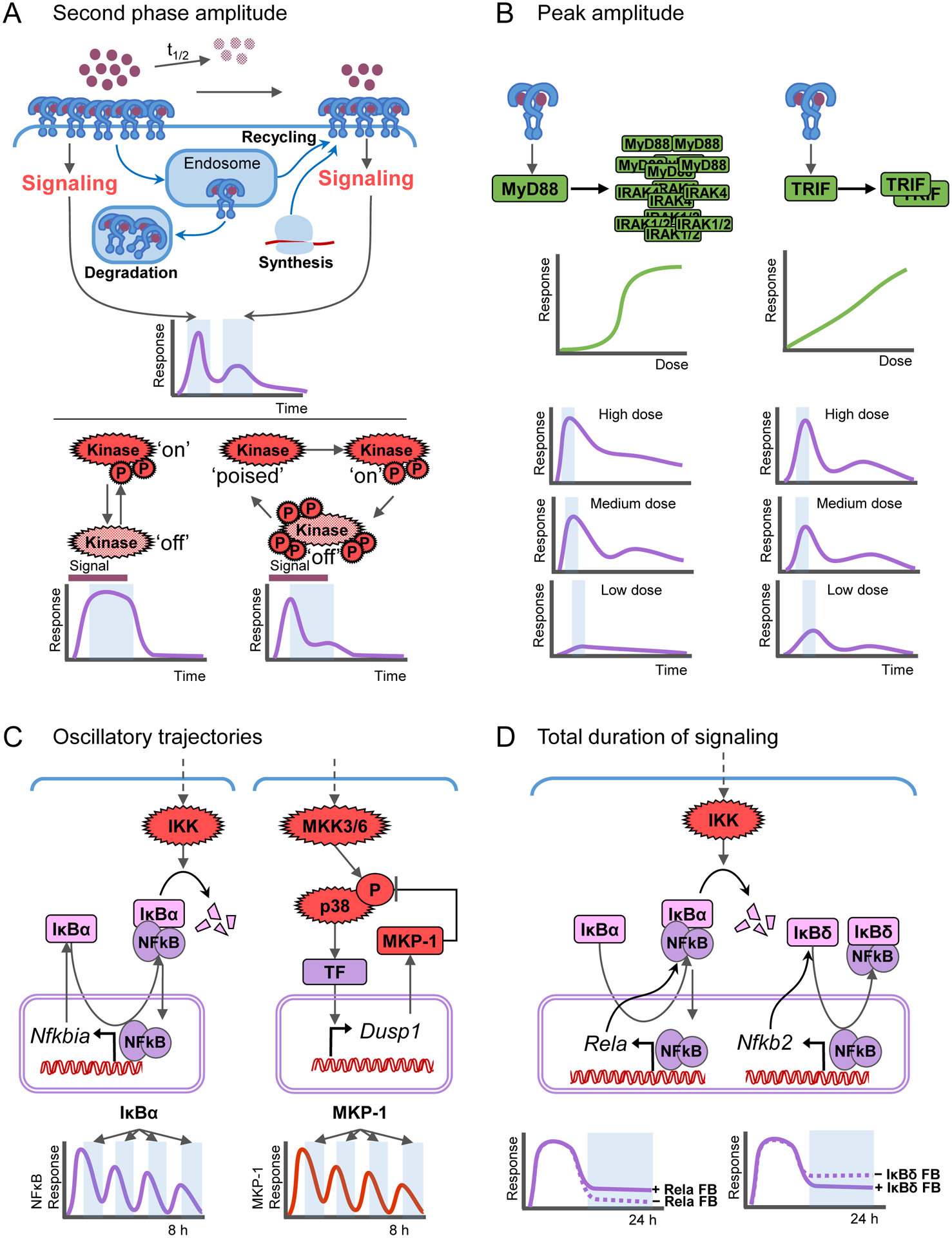
A-D. Mechanisms contributing to important features of temporal signaling dynamics: (A) The second phase amplitude is controlled by ligand stability, receptor turnover dynamics, and kinase activity kinetics. (B) Dose-to-peak-amplitude-encoding is dependent on adaptor oligomerization properties, which differ between MyD88 and TRIF. (C) Oscillations in effector activity trajectories of e.g. NFκB or MAPKp38 are introduced by negative feedback loops with delay. (D) Total duration of signaling is modulated by positive feedback or irreversible negative feedback, such as the induction of RelA and IκBδ expression, respectively.
In the next layer, the kinetic characteristics of adaptors are important determinants of signaling dynamics. For example, stepwise assembly of the Myddosome imparts the MyD88 pathway a thresholded, close-to-digital dose response curve, whereas TRIF dimerization leads to a more linear dose-response curve and translates ligand concentration into effector amplitude more faithfully (Cheng et al., 2015) (Figure 3B). Interestingly, Myddosome-mediated signaling is transient and limited to a few hours, but how it is turned off remains unclear. In the case of IL1 signaling, kinase mediator IRAK4 is proteolyzed after signaling (Ferrao et al., 2014), which may limit signal duration, but how Myd88 signaling is temporally limited demands further study. In the case of TRIF-signaling, the progressive acidification of the endosome appears to be a determinant of its duration, allowing for long-lasting and variable signal trajectories (Cheng et al., 2015; Husebye et al., 2006; McGettrick and O’Neill, 2010).
Within the ubiquitin layer, complex chain regulatory dynamics and their interplay with ubiquitin binding proteins likely also control signaling; however, a quantitative, time-resolved understanding requires further study. Anti-inflammatory A20, one prominent regulator of ubiquitin chain dynamics, blunts the dose-response of TNF signaling and therefore limits the weaker signals of later time points. Both induced A20 feedback expression and constitutive A20 expression at certain levels can fulfill this function (Werner et al., 2008).
The kinase layer provides for interesting temporal modulation of signaling activity. The two-state toggle switch kinase transmits the incoming signal, but there is an inherent trade-off between faithful turn-off with cessation of the signal (requiring high phosphatase activity) and reduction of the peak amplitude dynamic range. This is resolved by kinase cascades (e.g. in the MAPK pathway), which amplify the signal’s dynamic range without losing too much temporal control (Heinrich et al., 2002; Kholodenko and Birtwistle, 2009). An alternative topology is the kinase cycle (e.g. canonical IKK). This allows for a strong peak of activation which triggers the kinase’s transition to an inactive state (Delhase et al., 1999). From this, the kinase has to be recycled (via a phosphatase) to a poised state before reactivation. This results in a two-phased temporal trajectory with the duration of the first phase determined by the inactivation reaction and the amplitude of the second phase determined by the recycling reaction (Behar and Hoffmann, 2013). Thus, the distinction between first vs. second signaling phases is enforced by kinase cycles or diminished by toggle kinase cascades (Behar and Hoffmann, 2013) (Figure 3A). When positive feedback is included in the kinase layer, an excitable dose-response curve may produce digital all-or-none responses (Shinohara et al., 2014).
Within the effector layer, feedback mechanisms are prominent, and they may reach back to upstream layers. The functional effects of feedback are diverse, determined by the speed, reach, and molecular mechanism of the regulator. Proximal negative feedback that is rapid and mediated by a directly binding but reversible inhibitor (e.g. IκBα) or a highly active inhibitory enzyme (e.g. MAPK phosphatase) can mediate oscillatory effector activity if the input signal to the feedback loop persists (Hoffmann et al., 2002; Tomida et al., 2015) (Figure 3C). The IKK-IκBα-NFκB system generates particularly robust oscillations due to the rapid NFκB-responsive IκBα synthesis (including an appropriate delay to the negative feedback), rapid nuclear import of IκBα, efficient removal NFκB from the DNA through IκBα binding, rapid nuclear export of the complex, and responsiveness to IKK, which allows for efficient IκBα degradation after each cycle of re-synthesis (Mitchell et al., 2016). A delay in the negative feedback (e.g. IκBε) can diminish oscillatory propensity (Kearns et al., 2006). Irreversible negative feedback, such as mediated by A20 or by IκBδ, which, unlike other IκB isoforms, is not targeted for degradation by canonical IKK, can lead to a shutdown or attenuation of signaling that may impact both the amplitude of the second signaling phase, the total signal duration, and the response to a secondary signal (Shih et al., 2009; Werner et al., 2008) (Figure 3D). Hence, these proteins also mediate signaling crosstalk (Werner et al., 2008), as do the SOCS proteins regulating IRF/ISGF3 responses (Krebs and Hilton, 2001; Liu et al., 2015a).
Positive feedback and feedforward mechanisms mediated by a) expression of signaling substrates or b) induction of cytokines may extend the duration of effector activity. NFκB-responsive expression of the main NFκB dimer component, RelA, sustains long-term NFκB activity (Sung et al., 2014) (Figure 3D), while ISGF3-inducible expression of its constituents (STAT1/2/IRF9) renders cells more responsive to subsequent IFN stimulation (Ivashkiv and Donlin, 2014). Importantly, induced cytokines not only amplify or prolong signaling in primary response cell, but also trigger signaling in previously un-exposed or un-responsive neighboring cells. For example, TNF was found to have an autocrine role in prolonging NFκB signaling in response to synthetic TLR9 ligand CpG, but primarily paracrine functions in response to LPS (Caldwell et al., 2014). Similarly, IRF3-dependent IFNβ induces amplification of type I IFN signaling in the primary response cell, but also initiates antiviral responses in neighboring cells. These cytokines are primarily controlled by a feedforward mechanism rather than positive feedback, as potent paracrine signaling coupled to positive feedback can lead to runaway inflammation (Ourthiague et al., 2015): In the case of TNF, MAPK-p38 is critical for its expression, but is not strongly activated by it (Caldwell et al., 2014). In the case of IFNβ, IRF3 is critical for its expression, but is not activated by it (Honda et al., 2006). That regulatory feature also allows these cytokines to extend the duration of signaling, but not indefinitely.
In order to mediate stimulus-specific responses, temporal trajectories must be stimulus-specific. Thus far this has been most extensively described for NFκB, where the temporal trajectory may encode stimulus duration, dose, and molecular identity. Early studies examined how the duration of the TNF stimulus is encoded in the temporal profile of NFκB. Persistent TNF stimulation leads to characteristic oscillatory NFκB activity, while short TNF pulses lead to only a single peak whose duration of approximately 45 minute is independent of stimulus pulse duration (as short as 1 minute) (Hoffmann et al., 2002; Werner et al., 2008). Two temporal amplification steps explain this. First, the receptor-ligand complex and mechanisms in the ubiquitin layer provide for at least 10 minutes of IKK activity, and, second, the IκBα feedback loop ensures 45 minutes of activity. When TNF pulses exceed 60 minutes, subsequent cycles of oscillations are activated allowing for faithful stimulus-duration to response duration encoding (Hoffmann et al., 2002).
Stimulus dose can also be encoded in the temporal profile of the effector. For TNF, dose is encoded in amplitude and duration (Cheong et al., 2006, 2011; Tay et al., 2010; Turner et al., 2010). Amplitude provides only partial dose information as single cell studies observed some degree of ultrasensitivity (Tay et al., 2010) and amplitude measurements at 30 min for doses over 3 orders of magnitude contained less than 1 bit of information (Cheong et al., 2011). Instead, the short half-life of TNF mediates dose-dependent control of NFκB duration (Adelaja et al., 2021). For PAMPs, the MyD88 and TRIF pathways encode dose information differently (Cheng et al., 2015). MyD88-dependent PAMPs show an all-or-none response, with the threshold determined by receptor and MyD88 abundance. PAMPs engaging the TRIF pathway show a more graded dose-response with dose-dependent amplitudes. However, in both pathways the speed of response increases with dose.
The important question of whether molecular identity is represented in the temporal pattern of NFκB activity had until recently received confusing answers. Early population level biochemical studies in primary fibroblasts showed that, in contrast to the oscillatory activity induced by TNF, the NFκB response to LPS-TLR4 signaling is persistent and non-oscillatory (Covert et al., 2005; Werner et al., 2005). However, studies with immortalized cell lines did not provide confirmation at the single cell level as NFκB responses to several ligands showed noisy, largely oscillatory trajectories (Hughey et al., 2015). A recent study with primary macrophages has provided clearer answers and identified six features within NFκB temporal trajectories correlated with the molecular identity of the ligand (Adelaja et al., 2021). For example, whereas TNF, TLR2/1 ligand Pam3CSK4, and LPS activate NFκB rapidly, the response to CpG and TLR3 ligand poly(I:C) was slower, due to a delay in endosomal ligand availability. Remarkably, oscillatory content was substantially higher in response to cytokine TNF than to PAMPs, largely due to the distinct amplitude of the second phase of IKK activity that either overrides IκBα negative feedback (in case of PAMPs) or allows it to generate oscillations (in case of TNF). Each ligand generated a unique combination of these six features of the temporal NFκB code that may allow target genes to be expressed stimulus-specifically.
Thus, the dynamic features in the temporal profile of an effector, modulated by biochemical and cell biological mechanisms at every signaling layer, can encode information about the stimulus such as molecular identity, dose, and duration. While two decades of research into temporal encoding in the NFκB pathway have provided some mechanistic explanations for a handful of ligand receptor combinations, the temporal encoding of other effectors, such as the IRF and MAPK p38, remains underexplored. Indeed, how sequentially administered pulses of stimulation or multiple simultaneously administered stimuli are registered by the temporal activity profiles of effectors can provide insights about the intrinsic dynamic regulation of the signaling pathway (Ashall et al., 2009; Gutschow et al., 2018; Kellogg and Tay, 2015).
Decoding Mechanisms that Enable Stimulus-Specific Gene Expression
For immune sentinels to generate stimulus-specific gene expression, the gene regulatory network must interpret the combinatorial and temporal features of effector proteins. The gene regulatory network associated with each immune response gene involves DNA-associated mechanisms that move nucleosomes, establish enhancers, catalyze chromatin looping, or recruit components of the pre-initiation complex; RNA-associated mechanisms involving transcription initiation and RNA polymerase promoter clearance, transcription elongation, mRNA processing, splicing and export, or cytoplasmic decay; and translation-associated mechanisms such as pre-protein processing or activation.
Combinatorial decoding mediates stimulus-specific gene expression when immune response genes are differentially responsive to the various signaling effectors. Transcriptome profiling revealed that genes may contain AP1 binding sites, κB elements or ISRE sites, or combinations of these (Amit et al., 2009; Cheng et al., 2017). However, two binding sites being present does not indicate whether the gene is responsive to either pathway, or only when both pathways are active. The promiscuity of the former and specificity of the latter are aptly described by the conceptual framework of Boolean OR and AND logic gates, respectively (Buchler et al., 2003; Morris et al., 2010). Beyond combinatorial activation of signaling pathways, the number of combinatorial possibilities increases drastically when differential TF-promoter binding affinities and activation strengths are also considered (Brignall et al., 2019).
Within the pathogen-responsive transcriptome, a cluster of about 100 genes were identified as being activated by either NFκB or ISGF3, their regulatory regions tend to contain both κB sites and ISREs, and therefore classified as OR gate genes (Cheng et al., 2017). However, NFκB and ISGF3 do not have redundant roles as they show distinct temporal control: only their combined action ensures that such genes are both rapidly induced (first by NFκB) and expressed for extended times (then by ISGF3) (Figure 4A). Indeed, when such genes encode mRNAs with a long half-life, transient TF activity may be insufficient for full gene expression (Hao and Baltimore, 2009; Sen et al., 2020). In this regard, the OR gate may be thought of as providing an additional mechanism for temporal coding, as it allows for the decoding of the combined temporal trajectory of both NFκB and ISGF3.
Figure 4. Mechanisms that decode combinatorial and temporal codes of pathway activity to achieve stimulus-specific responses.
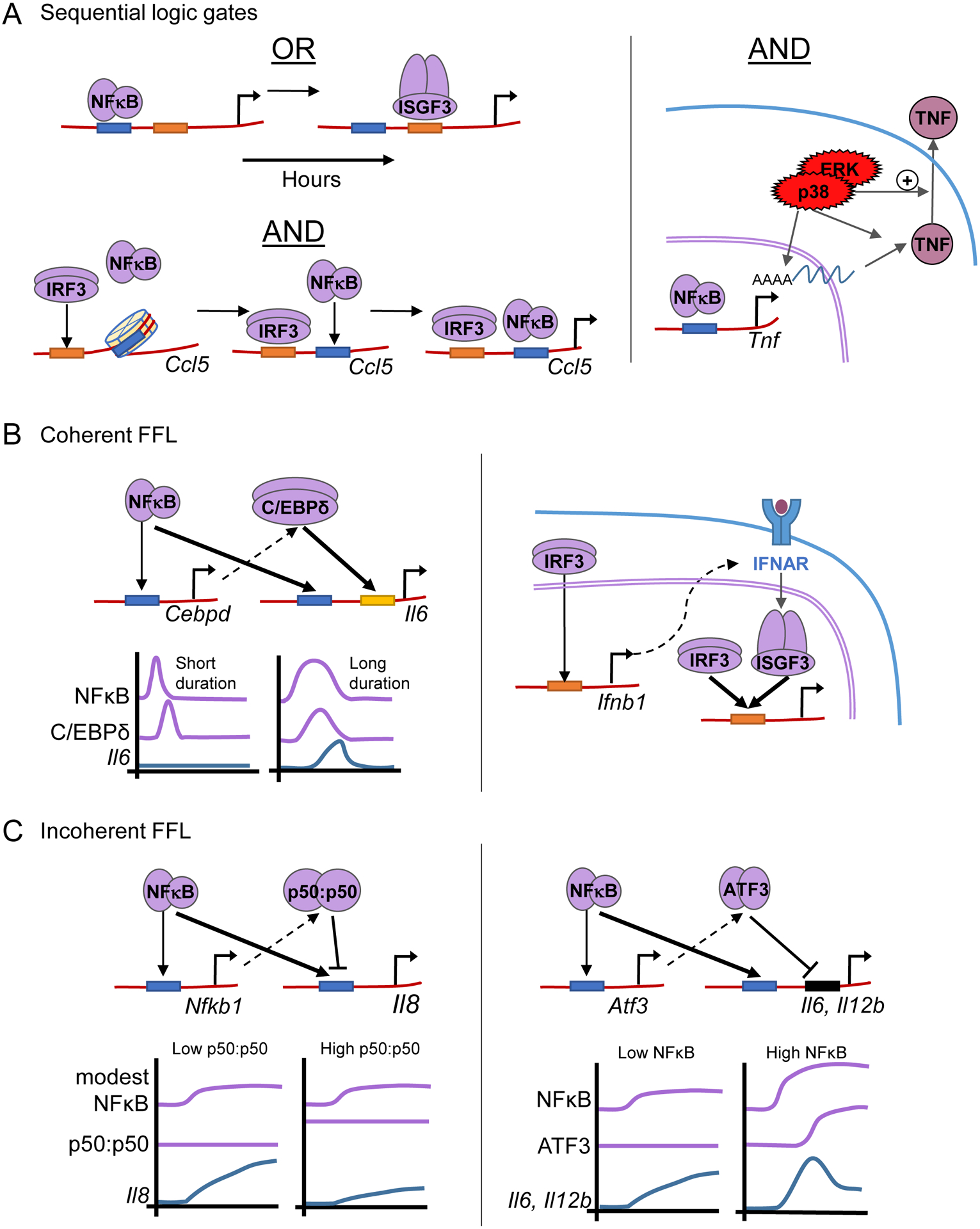
A. Decoding of pathway-specific effector combinations occurs through sequential logic gates that involve pre-transcriptional mechanisms, such as (left top) NFκB and ISGF3 forming a sequential OR gate, allowing sustained responses, (left bottom) IRF3-mediated nucleosome movement for Ccl5, allowing NFκB binding and forming a sequential AND gate, and (right) posttranscriptional mechanisms combining NFκB-induced transcription AND MAPKp38 processing and stabilization of Tnf mRNA.
B. Coherent feedforward loops, where one regulator positively controls a target and another positive regulator. Left: NFκB induces transcription of Cebpd, which activates a second wave of response genes controlled by both NFκB and CEPBδ. Here, coherent FFLs combined with AND gates allow duration decoding. Right: Similarly, IRF3-induced Ifnb1 generates a coherent feedforward loop through activation of IFNAR and subsequently ISGF3, which binds similar gene promoters as IRF3.
C. Incoherent feedforward loops, where one regulator positively controls a target as well as a negative regulator. Together with combinatorial decoding principles they allow for fold change detection and mediate biphasic responses. Left: NFκB-induced expression of the competing p50:p50 homodimer provides memory of previous NFκB abundances for fold-change detection on NFκB target genes such as Il8. Right: NFκB-induced transcription of Atf3, which in turn represses NFκB target genes. This results in the diminishing of the second phase of response gene expression when NFκB activity is high.
Corner arrow indicates active transcription.
In the established conceptual framework of Boolean logic gates, AND gate logic provides for high stimulus-selectivity, as both signaling pathway effectors must be active. It was thought that synergy between two TFs is achieved either by cooperative binding of DNA or by the cooperative recruitment of co-activators such as CBP/p300, which contains multiple TF interaction domains (Merika et al., 1998). However, in the pathogen-responsive transcriptome, there is little evidence for TFs needing to present at the same time, or “coincident AND gates” at gene promoters. Rather, the sequential actions of signaling pathway effectors are required (Cheng et al., 2017). The discovery of sequential Boolean gates emphasizes the importance of integrating combinatorial and temporal signaling concepts for a full understanding of decoding.
One example of sequential AND gates are the small number of genes whose transcriptional initiation is regulated by both IRF3 and NFκB (Freaney et al., 2013; Tong et al., 2016). While Ifnb1 is the most well-known example, the molecular mechanism enabling NFκB and IRF3’s synergy is not clearly understood (Panne et al., 2007). In the case of Ccl5, recent studies suggest that IRF3 may facilitate a chromatin opening step that is required for the recruitment of NFκB, which then activates transcription (Figure 4A) (Ramirez-Carrozzi et al., 2009; Tong et al., 2016). A second example of sequential AND gates pertains to a number of important cytokine genes such as Tnf, Il6, and Il1b, that are dependent on both NFκB and MAPKp38 activation downstream of the MyD88 adaptor (Cheng et al., 2017). While NFκB is sufficient for transcriptional initiation, it appears that several post-transcriptional control steps, particularly mRNA stabilization, are p38-dependent (Figure 4A). In the case of TNF, the combined action of MAPKp38 and ERK enhance pre-mRNA splicing, cytoplasmic mRNA half-life, translation, and pro-protein processing (Caldwell et al., 2014). Thus, the sequential action of diverse biochemical processes in several different cellular compartments form a sequential AND gate in the production of secreted TNF. AND gate function may also control epigenomic reprogramming: a study on IFNγ-induced gene repression showed that dissociation of active enhancers was a result of an AND gate at MAF-bound enhancers, by IFNγ-induced repression of MAF transcription and coordinate dissociation of PU.1 and C/EBP (Kang et al., 2017).
While combinatorial decoding interprets the activities of multiple TF pathways, temporal decoding distinguishes between distinct temporal trajectories of a single signaling effector and requires mechanisms that are kinetically regulated. For example, a kinetically slow mechanistic step not only transduces the instantaneous activity but also stores information about past activity (Behar and Hoffmann, 2010; Purvis and Lahav, 2013). Three molecular mechanisms have been identified: mechanisms controlling mRNA half-life, nucleosome displacement, and enhancer formation.
Long mRNA half-life has been shown by both mathematical modelling and experimental data to decode the duration of signaling effector activity. As short-half life transcripts achieve their half-maximal induction levels faster than long-half life transcripts, only long duration signals may allow full induction of long-half life mRNAs (Behar and Hoffmann, 2010; Hao and Baltimore, 2009). In the pathogen-response, the long duration of IKK/NFκB activity induced by LPS allows for induction genes that are poorly induced by transient TNF stimulation (Werner et al., 2005). A recent study quantified the contribution of long mRNA half-life to decoding differential duration of NFκB activity for several dozen genes, but also showed that chromatin-associated mechanisms play important roles as well (Sen et al., 2020).
Nucleosome displacement also constitutes a kinetically ‘slow’ step such that signal-dependent transcription factors (SDTF) binding to promoters or enhancers (Kaikkonen et al., 2013; Tong et al., 2016), co-activator recruitment, and pre-initiation complex (PIC) assembly and polymerase recruitment (Bhaumik, 2011) may in principle decode temporal dynamics of signaling. For example, LPS-induced NFκB target genes fall into two categories, those with open chromatin immediately available for NFκB binding and ready transcriptional induction, and those requiring chromatin remodeling before NFκB can bind (Kayama et al., 2008; Natoli, 2009; Ramirez-Carrozzi et al., 2009; Saccani et al., 2001). One recent study quantified the contribution of chromatin-associated mechanisms for differentiating between LPS- versus TNF-induced (2–3 versus 1 hour) NFκB activity, for 81 immune response genes (Sen et al., 2020). Interestingly, the chromatin-associated mechanisms provided a higher degree of specificity than the aforementioned long mRNA half-life mechanism and pertained particularly to immune effectors such as Mmp3, Pfr1, Pilra, and also Ccl5.
The formation of de novo enhancers is a third kinetically slow step that can decode temporal dynamics, but it is less reversible than the two previous examples. While poised target gene expression did not markedly differ in cells with oscillatory versus non-oscillatory NFκB activity (Barken et al., 2005), a recent study demonstrated that the absence of oscillations in NFκB activity determines whether stimulus-responsive de novo enhancers are established (Cheng et al., 2021). (While the presence vs absence of oscillations in NFκB dynamics is stimulus-specific, their frequency is not (Longo et al., 2013)). The capacity to distinguish oscillatory and non-oscillatory NFκB is based on the multi-step unwrapping and rewrapping dynamics of nucleosomal DNA-histone interactions, displacing the nucleosome and allowing the deposition of histone marks. A negative feedback mutant that converted normally oscillatory TNF-induced NFκB activity to non-oscillatory activity allowed for more efficient nucleosome eviction and increased chromatin accessibility, and the subsequent formation of de novo enhancers enabled novel gene expression programs upon a secondary stimulation. SDTF dynamics may thus also be a critical determinant for epigenomic reprogramming that may allow immune sentinel cells to function in an exposure-history-dependent manner, potentially conferring innate immune memory (Netea et al., 2016).
A gene regulatory network where an effector positively controls a target both directly and indirectly via another positive regulator forms a coherent feedforward loop. Coherent feedforward loops, which comprise an AND logic gate, may decode differential TF durations (Figure 4B). For example, IL6 expression was found to depend not only on NFκB but also the secondary TF C/EBPδ (Litvak et al., 2009). Because NFκB activity induces the expression of C/EBPδ, they form a coherent feedforward loop to activate IL6. However, a short exposure to LPS will not provide long-lasting NFκB that can synergize with de novo C/EBPδ, thus preventing induction of secondary IL6-mediated inflammation. The regulatory relationship of IRF3 and ISGF3 may also constitute a coherent feedforward loop on genes that require both IRF3 and ISGF3 for full activation. If for example IRF3 triggers a key chromatin remodeling step that then allows ISGF3 to bind and activate transcription, such target genes would primarily be induced in the infected (pathogen-exposed) cell, rather than the bystander cells. A transcriptomic analysis yielded just a few candidates (e.g. Cxcl10, Ifit3) for such a regulatory mechanism (Ourthiague et al., 2015) and whether they differentiate IRF3-ISGF3 temporal features was not explored.
Another type of regulatory network motif, in which a signaling effector positively controls a target as well as a negative regulator of the same target, is referred to as an incoherent feedforward loop. Incoherent feedforward loops provide decoding mechanism for differences in fold changes (Figure 4C). For example, the decoding of fold changes of NFκB activity on a set of target genes was reported to be governed by an incoherent feedforward loop involving the activation-domain lacking p50:p50 homodimer (Lee et al., 2014). A subset of TNF-induced genes were found to correlate particularly well with NFκB fold changes in single cells. Interestingly, p50:p50 homodimers were also shown to bind a subset of ISREs and thus tune the responsiveness of ISGF3 target genes (Cheng et al., 2011). In an earlier example, an incoherent feedforward loop was shown to prevent run-away expression of inflammatory cytokines (Gilchrist et al., 2006). ATF3 was found to repress the NFκB-induced expression of IL6 and IL-12B. As ATF3 itself is induced by NFκB, its repressive role on IL6 and IL-12B becomes relevant when NFκB activity is high and sustained, thereby preventing over-production of these potent inflammatory cytokines.
In further illustration of the precise temporal regulation of gene expression, incoherent feed forward loops may also involve post-transcriptional mechanisms. MicroRNAs known as inflammamIRs, mir155 and mir146, are NFκB inducible and target immune response mRNAs (Rothchild et al., 2016; Testa et al., 2017). Mir146 targets regulators of the TLR signaling pathway and this repressive function serves to fine tune the innate immune response, whereas mir155 acts in an opposing and dominant function to further potentiate NFκB response (Mann et al., 2017). Similarly, the RNA editing protein ADAR1 provides incoherent feedforward control, as it is induced by IFN and then acts to repress RLR signaling, dampening the IFN response and preventing autoimmunity (Lamers et al., 2019; Mannion et al., 2014). While these regulatory circuits have the potential to decode dynamic features of the pathogen-responsive temporal code, their true functional impact will need to be addressed in future studies.
Decoding mechanisms are intricately tied to immune sentinel functions of context-dependence and memory. Beyond generating the stimulus-specificity of immediate gene expression responses, decoding mechanisms that invoke chromatin remodeling are particularly important to the establishment of stimulus-specific innate immune memory. The eviction of nucleosomes and the deposition of new histone marks are a downstream consequence of stimulus-specific chromatin remodeling, though the functional and physiological consequences of this reprogramming, especially in disease contexts, remains to be explored.
Concluding Remarks
Recent work has begun to document the capacity of immune sentinel cells to mount pathogen-specific responses, as well as provide a set of mechanistic studies to support the notions of combinatorial and temporal coding. These insights may serve as a basis for addressing key questions through future studies.
First, while the mechanisms underlying temporal coding in the NFκB signaling module are relatively well understood, the integration of combinatorial and temporal signaling via coordinated activities of IRF, JNK, and MAPK p38 pathways is a next big challenge in the immune signaling field. Temporal dynamics and pathway branching of signaling initiated by cytosolic PRRs such as cGAS, RLRs, and NLRs is also understudied.
Second, understanding how combinatorial and temporal signaling codes are interpreted to produce gene expression requires substantial effort, in part because every gene is controlled by a seemingly unique gene regulatory network. To study the connection between encoding and decoding of signaling activity it will be necessary to develop efficient workflows that directly connect temporal effector activity profiles, ideally for multiple effectors, with gene expression or cytokine secretion measurements in the same cells (Gutschow et al., 2018; Lane et al., 2017).
Third, and related, at the single cell level, both stimulus-responsive signaling activity and gene expression are highly heterogeneous, which may be essential for the breadth, robustness, and adaptability of the immune response, but may also contribute to pathology (Satija and Shalek, 2014). While live cell imaging has been successful in characterizing the cell-to-cell variability of signaling activity, the stochasticity of decoding mechanisms at the gene promoter likely plays an especially important role in determining gene expression responses of a specific cell. This requires innovative integration of live cell imaging and necessarily destructive single cell technologies such as scRNAseq, smFISH, scATACseq, or scChIPseq to probe expression of native chromatin and endogenous genes (rather than ectopic reporters), via data driven and mechanistic modeling approaches.
Fourth, with such workflows in place, future work will be able to explore how encoding and decoding mechanisms are a function of microenvironmental context and the cell’s exposure history. Cytokines in the microenvironment, which guide the functional specialization of immune sentinel cells, may alter the encoding of combinatorial and temporal signaling profiles, while exposure history that changes the cell’s baseline epigenome affects how signals are decoded (Cheng et al., 2019; Kang et al., 2017; Park et al., 2017).
Detailed and predictive knowledge of how immune sentinel responses are regulated will undoubtedly impact our understanding of a range of immune-related disease, whether immunopathological responses to infectious disease (e.g. as seen during SARS-CoV2 infections), or autoinflammatory and autoimmune disorders (Funes et al., 2018; Gustine and Jones, 2021). As pathogenesis may be triggered by small subsets of outlier cells, understanding the basis for the heterogeneity of responses at the single cell level is key. By characterizing macrophages derived from a Sjögren’s syndrome mouse model, a recent study revealed that a minority of cells show stimulus-confused NFκB dynamics and gene expression (Adelaja et al., 2021). We therefore suggest that a quantitative and mechanistic understanding of immune sentinel responses will enable a better characterization and diagnosis of auto-inflammatory and auto-immune diseases and support the development of therapeutic strategies aimed at correcting disease-associated miscoding.
Supplementary Material
Acknowledgments
SL is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 419234150. KMS is supported by the UCLA Medical Scientist Training Program (NIH NIGMS T32 GM008042). Research in the Hoffmann lab on described topics is funded by NIH R01AI127867, R01AI132835, R01AI127864, R01GM117134.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe T, and Barber GN (2014). Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol 88, 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe H, Satoh J, Shirasaka Y, Kogure A, Kato H, Ito S, and Fujita T (2020). Priming Phosphorylation of TANK-Binding Kinase 1 by IκB Kinase β Is Essential in Toll-Like Receptor 3/4 Signaling. Mol. Cell. Biol 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, and Chen ZJ (2019). cGAS in action: Expanding roles in immunity and inflammation. Science 363, eaat8657. [DOI] [PubMed] [Google Scholar]
- Ablasser A, and Hur S (2020). Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat Immunol 21, 17–29. [DOI] [PubMed] [Google Scholar]
- Adelaja A, Taylor B, Sheu KM, Liu Y, Luecke S, and Hoffmann A (2021). Six distinct NFκB signaling codons convey discrete information to distinguish stimuli and enable appropriate macrophage responses. Immunity 54, 916–930.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikin TJ, Peterson AF, Pokrass MJ, Clark HR, and Regot S (2019). Collective MAPK Signaling Dynamics Coordinates Epithelial Homeostasis. BioRxiv 826917. [Google Scholar]
- Akira S, Uematsu S, and Takeuchi O (2006). Pathogen Recognition and Innate Immunity. Cell 124, 783–801. [DOI] [PubMed] [Google Scholar]
- Albeck JG, Burke JM, Spencer SL, Lauffenburger DA, and Sorger PK (2008a). Modeling a Snap-Action, Variable-Delay Switch Controlling Extrinsic Cell Death. PLOS Biology 6, e299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albeck JG, Burke JM, Aldridge BB, Zhang M, Lauffenburger DA, and Sorger PK (2008b). Quantitative Analysis of Pathways Controlling Extrinsic Apoptosis in Single Cells. Molecular Cell 30, 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altan-Bonnet G, and Germain RN (2005). Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol 3, e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, et al. (2009). Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science (New York, N.Y.) 326, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashall L, Horton CA, Nelson DE, Paszek P, Harper CV, Sillitoe K, Ryan S, Spiller DG, Unitt JF, Broomhead DS, et al. (2009). Pulsatile stimulation determines timing and specificity of NF-kappaB-dependent transcription. Science 324, 242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnall J, Boddington C, England H, Brignall R, Downton P, Alsoufi Z, Boyd J, Rowe W, Bennett A, Walker C, et al. (2018). Quantitative analysis of competitive cytokine signaling predicts tissue thresholds for the propagation of macrophage activation. Sci. Signal 11, eaaf3998. [DOI] [PubMed] [Google Scholar]
- Balka KR, and Nardo DD (2019). Understanding early TLR signaling through the Myddosome. Journal of Leukocyte Biology 105, 339–351. [DOI] [PubMed] [Google Scholar]
- Bardwell L (2008). Signal Transduction: Turning a Switch into a Rheostat. Current Biology 18, R910–R912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barken D, Wang CJ, Kearns J, Cheong R, Hoffmann A, and Levchenko A (2005). Comment on “Oscillations in NF-κB Signaling Control the Dynamics of Gene Expression”. Science 308, 52–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat FJ, Crow MK, and Ivashkiv LB (2019). Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol 20, 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker V, Schilling M, Bachmann J, Baumann U, Raue A, Maiwald T, Timmer J, and Klingmüller U (2010). Covering a broad dynamic range: information processing at the erythropoietin receptor. Science 328, 1404–1408. [DOI] [PubMed] [Google Scholar]
- Behar M, and Hoffmann A (2010). Understanding the temporal codes of intra-cellular signals. Current Opinion in Genetics & Development 20, 684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar M, and Hoffmann A (2013). Tunable Signal Processing through a Kinase Control Cycle: the IKK Signaling Node. Biophysical Journal 105, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinke S, Robinson MJ, Hugunin M, and Ley SC (2004). Lipopolysaccharide activation of the TPL-2/MEK/extracellular signal-regulated kinase mitogen-activated protein kinase cascade is regulated by IkappaB kinase-induced proteolysis of NF-kappaB1 p105. Mol. Cell. Biol 24, 9658–9667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler BA (2009). TLRs and innate immunity. Blood 113, 1399–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik SR (2011). Distinct regulatory mechanisms of eukaryotic transcriptional activation by SAGA and TFIID. Biochim Biophys Acta 1809, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boraschi D, Italiani P, Weil S, and Martin MU (2018). The family of the interleukin-1 receptors. Immunol. Rev 281, 197–232. [DOI] [PubMed] [Google Scholar]
- Botos I, Segal DM, and Davies DR (2011). The Structural Biology of Toll-like Receptors. Structure 19, 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray D, Levin MD, and Morton-Firth CJ (1998). Receptor clustering as a cellular mechanism to control sensitivity. Nature 393, 85–88. [DOI] [PubMed] [Google Scholar]
- Brignall R, Moody AT, Mathew S, and Gaudet S (2019). Considering Abundance, Affinity, and Binding Site Availability in the NF-κB Target Selection Puzzle. Front. Immunol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brondello J-M, Pouysségur J, and McKenzie FR (1999). Reduced MAP Kinase Phosphatase-1 Degradation After p42/p44MAPK-Dependent Phosphorylation. Science 286, 2514–2517. [DOI] [PubMed] [Google Scholar]
- Buchler NE, Gerland U, and Hwa T (2003). On schemes of combinatorial transcription logic. Proc Natl Acad Sci USA 100, 5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena C, Ahmad S, Xavier A, Willemsen J, Park S, Park JW, Oh S-W, Fujita T, Hou F, Binder M, et al. (2019). Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 177, 1187–1200.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell AB, Cheng Z, Vargas JD, Birnbaum HA, and Hoffmann A (2014). Network dynamics determine the autocrine and paracrine signaling functions of TNF. Genes & Development 28, 2120–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caré BR, and Soula HA (2011). Impact of receptor clustering on ligand binding. BMC Syst Biol 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargnello M, and Roux PP (2011). Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol Mol Biol Rev 75, 50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso R, Warner N, Inohara N, and Núñez G (2014). NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity 41, 898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catrysse L, Vereecke L, Beyaert R, and van Loo G (2014). A20 in inflammation and autoimmunity. Trends in Immunology 35, 22–31. [DOI] [PubMed] [Google Scholar]
- Chakraborty AK, and Weiss A (2014). Insights into the initiation of TCR signaling. Nature Immunology 15, 798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, and Jiang Z (2013). The essential adaptors of innate immune signaling. Protein Cell 4, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Almo SC, and Wu Y (2017). General principles of binding between cell surface receptors and multi-specific ligands: A computational study. PLOS Computational Biology 13, e1005805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CS, Feldman KE, Lee J, Verma S, Huang DB, Huynh K, Chang M, Ponomarenko JV, Sun SC, Benedict CA, et al. (2011). The specificity of innate immune responses is enforced by repression of interferon response elements by NF-kappaB p50. Science Signaling 4, ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CS, Behar MS, Suryawanshi GW, Feldman KE, Spreafico R, and Hoffmann A (2017). Iterative Modeling Reveals Evidence of Sequential Transcriptional Control Mechanisms. Cell Systems 4, 330–343.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Behzadi F, Sen S, Ohta S, Spreafico R, Teles R, Modlin RL, and Hoffmann A (2019). Sequential conditioning-stimulation reveals distinct gene- and stimulus-specific effects of Type I and II IFN on human macrophage functions. Scientific Reports 9, 5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng QJ, Ohta S, Sheu KM, Spreafico R, Adelaja A, Taylor B, and Hoffmann A (2021). NF-κB dynamics determine the stimulus specificity of epigenomic reprogramming in macrophages. Science 372, 1349–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Taylor B, Ourthiague DR, and Hoffmann A (2015). Distinct single-cell signaling characteristics are conferred by the MyD88 and TRIF pathways during TLR4 activation. Science Signaling 8, ra69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong R, Bergmann A, Werner SL, Regal J, Hoffmann A, and Levchenko A (2006). Transient IkappaB kinase activity mediates temporal NF-kappaB dynamics in response to a wide range of tumor necrosis factor-alpha doses. The Journal of Biological Chemistry 281, 2945–2950. [DOI] [PubMed] [Google Scholar]
- Cheong R, Rhee A, Wang CJ, Nemenman I, and Levchenko A (2011). Information transduction capacity of noisy biochemical signaling networks. Science (New York, N.Y.) 334, 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra S, Liu L, Goh R, Kong X, and Warmflash A (2019). Dissecting the dynamics of signaling events in the BMP, WNT, and NODAL cascade during self-organized fate patterning in human gastruloids. PLoS Biol 17, e3000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra SS, Jenney A, Palmer A, Niepel M, Chung M, Mills C, Sivakumaren SC, Liu Q, Chen J-Y, Yapp C, et al. (2020). Torin2 Exploits Replication and Checkpoint Vulnerabilities to Cause Death of PI3K-Activated Triple-Negative Breast Cancer Cells. Cell Systems 10, 66–81.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, and Strickson S (2017). The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death & Differentiation 24, 1153–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert MW, Leung TH, Gaston JE, and Baltimore D (2005). Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science (New York, N.Y.) 309, 1854–1857. [DOI] [PubMed] [Google Scholar]
- Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, and Kelliher MA (2005). Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-kappaB activation but does not contribute to interferon regulatory factor 3 activation. J. Biol. Chem 280, 36560–36566. [DOI] [PubMed] [Google Scholar]
- De A, Dainichi T, Rathinam CV, and Ghosh S (2014). The deubiquitinase activity of A20 is dispensable for NF-κB signaling. EMBO Rep. 15, 775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguine J, and Barton GM (2014). MyD88: a central player in innate immune signaling. F1000Prime Rep 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhase M, Hayakawa M, Chen Y, and Karin M (1999). Positive and Negative Regulation of IκB Kinase Activity Through IKKβ Subunit Phosphorylation. Science 284, 309–313. [DOI] [PubMed] [Google Scholar]
- Dostert C, Grusdat M, Letellier E, and Brenner D (2019). The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev 99, 115–160. [DOI] [PubMed] [Google Scholar]
- Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, et al. (2002). IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17, 251–263. [DOI] [PubMed] [Google Scholar]
- Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, Jakobsen MR, Nevels MM, Bowie AG, and Unterholzner L (2018). Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell 71, 745–760.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea C-K, Deng L, Xia Z-P, Pineda G, and Chen ZJ (2006). Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 22, 245–257. [DOI] [PubMed] [Google Scholar]
- Emmerich CH, Ordureau A, Strickson S, Arthur JSC, Pedrioli PGA, Komander D, and Cohen P (2013). Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. U.S.A 110, 15247–15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng C-HL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan G-C, et al. (2019). Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Jiang Q, Zhou X, Wang C, Guan Y, Tao J, Xi J, Feng J-M, and Jiang Z (2017a). MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 13, e1006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Wang C, Jiang Q, Lv M, Gao P, Yu X, Mu P, Zhang R, Bi S, Feng JM, et al. (2017b). NEMO-IKKbeta Are Essential for IRF3 and NF-kappaB Activation in the cGAS-STING Pathway. Journal of Immunology (Baltimore, Md. : 1950) 199, 3222–3233. [DOI] [PubMed] [Google Scholar]
- Feinerman O, Germain RN, and Altan-Bonnet G (2008a). Quantitative challenges in understanding ligand discrimination by αβ T cells. Molecular Immunology 45, 619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinerman O, Veiga J, Dorfman JR, Germain RN, and Altan-Bonnet G (2008b). Variability and Robustness in T Cell Activation from Regulated Heterogeneity in Protein Levels. Science 321, 1081–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrao R, Zhou H, Shan Y, Liu Q, Li Q, Shaw DE, Li X, and Wu H (2014). IRAK4 Dimerization and trans-Autophosphorylation Are Induced by Myddosome Assembly. Molecular Cell 55, 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE, and Bhatt RR (1997). Mechanistic Studies of the Dual Phosphorylation of Mitogen-activated Protein Kinase. J. Biol. Chem 272, 19008–19016. [DOI] [PubMed] [Google Scholar]
- Ferrell JE, and Ha SH (2014). Ultrasensitivity part III: cascades, bistable switches, and oscillators. Trends Biochem Sci 39, 612–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman R, and Wollman R (2020). Mammalian gene expression variability is explained by underlying cell state. Mol. Syst. Biol 16, e9146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freaney JE, Kim R, Mandhana R, and Horvath CM (2013). Extensive Cooperation of Immune Master Regulators IRF3 and NFκB in RNA Pol II Recruitment and Pause Release in Human Innate Antiviral Transcription. Cell Reports 4, 959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fric J, Zelante T, Wong AYW, Mertes A, Yu H-B, and Ricciardi-Castagnoli P (2012). NFAT control of innate immunity. Blood 120, 1380–1389. [DOI] [PubMed] [Google Scholar]
- Friedman CS, O’Donnell MA, Legarda-Addison D, Ng A, Cárdenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, et al. (2008). The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9, 930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funami K, Sasai M, Oshiumi H, Seya T, and Matsumoto M (2008). Homo-oligomerization is essential for Toll/interleukin-1 receptor domain-containing adaptor molecule-1-mediated NF-kappaB and interferon regulatory factor-3 activation. J. Biol. Chem 283, 18283–18291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funes SC, Rios M, Escobar-Vera J, and Kalergis AM (2018). Implications of macrophage polarization in autoimmunity. Immunology 154, 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganti RS, Lo W-L, McAffee DB, Groves JT, Weiss A, and Chakraborty AK (2020). How the T cell signaling network processes information to discriminate between self and agonist ligands. PNAS 117, 26020–26030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantke T, Sriskantharajah S, Sadowski M, and Ley SC (2012). IκB kinase regulation of the TPL-2/ERK MAPK pathway. Immunol. Rev 246, 168–182. [DOI] [PubMed] [Google Scholar]
- Gaudet S, Spencer SL, Chen WW, and Sorger PK (2012). Exploring the contextual sensitivity of factors that determine cell-to-cell variability in receptor-mediated apoptosis. PLoS Comput Biol 8, e1002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Kennedy K, Hai T, Bolouri H, and Aderem A (2006). Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173. [DOI] [PubMed] [Google Scholar]
- Gottschalk RA, Martins AJ, Angermann BR, Dutta B, Ng CE, Uderhardt S, Tsang JS, Fraser ID, Meier-Schellersheim M, and Germain RN (2016). Distinct NF-kappaB and MAPK Activation Thresholds Uncouple Steady-State Microbe Sensing from Anti-pathogen Inflammatory Responses. Cell Systems 2, 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk RA, Dorrington MG, Dutta B, Krauss KS, Martins AJ, Uderhardt S, Chan W, Tsang JS, Torabi-Parizi P, Fraser ID, et al. (2019). IFN-mediated negative feedback supports bacteria class-specific macrophage inflammatory responses. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Zhang T, lin W, Liu Z, Lai R, Xia D, Huang H, and Wang X (2014). Protein phosphatase PP1 negatively regulates the Toll-like receptor- and RIG-I-like receptor-triggered production of type I interferon by inhibiting IRF3 phosphorylation at serines 396 and 385 in macrophage. Cellular Signalling 26, 2930–2939. [DOI] [PubMed] [Google Scholar]
- Gustine JN, and Jones D (2021). Immunopathology of Hyperinflammation in COVID-19. Am J Pathol 191, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutschow MV, Mason JC, Lane KM, Maayan I, Hughey JJ, Bajar BT, Amatya DN, Valle SD, and Covert MW (2018). Combinatorial processing of bacterial and host-derived innate immune stimuli at the single-cell level. MBoC 30, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, et al. (2009). Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36, 831–844. [DOI] [PubMed] [Google Scholar]
- Häcker H, and Karin M (2006). Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13. [DOI] [PubMed] [Google Scholar]
- Hao S, and Baltimore D (2009). The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol 10, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, and Ghosh S (2004). Signaling to NF-kappaB. Genes Dev. 18, 2195–2224. [DOI] [PubMed] [Google Scholar]
- Heemskerk I, Burt K, Miller M, Chhabra S, Guerra MC, Liu L, and Warmflash A (2019). Rapid changes in morphogen concentration control self-organized patterning in human embryonic stem cells. ELife 8, e40526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich R, Neel BG, and Rapoport TA (2002). Mathematical models of protein kinase signal transduction. Mol. Cell 9, 957–970. [DOI] [PubMed] [Google Scholar]
- Hoffmann A (2016). Immune Response Signaling: Combinatorial and Dynamic Control. Trends in Immunology 37, 570–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, and Baltimore D (2006). Circuitry of nuclear factor kappaB signaling. Immunological Reviews 210, 171–186. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, and Baltimore D (2002). The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science (New York, N.Y.) 298, 1241–1245. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh W-C, and Taniguchi T (2004). Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. PNAS 101, 15416–15421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, and Taniguchi T (2005). Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434, 1035–1040. [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, and Taniguchi T (2006). Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 25, 349–360. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M, Hemmi H, Ohara O, Akira S, and Kaisho T (2006). IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 440, 949–953. [DOI] [PubMed] [Google Scholar]
- Hu H, and Sun S-C (2016). Ubiquitin signaling in immune responses. Cell Research 26, 457–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, and Ferrell JE (1996). Ultrasensitivity in the mitogen-activated protein kinase cascade. PNAS 93, 10078–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughey JJ, Gutschow MV, Bajar BT, and Covert MW (2015). Single-cell variation leads to population invariance in NF-kappaB signaling dynamics. Molecular Biology of the Cell 26, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husebye H, Halaas Ø, Stenmark H, Tunheim G, Sandanger Ø, Bogen B, Brech A, Latz E, and Espevik T (2006). Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 25, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishitani T, Takaesu G, Ninomiya-Tsuji J, Shibuya H, Gaynor RB, and Matsumoto K (2003). Role of the TAB2-related protein TAB3 in IL-1 and TNF signaling. EMBO J 22, 6277–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB, and Donlin LT (2014). Regulation of type I interferon responses. Nature Reviews Immunology 14, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA, and Medzhitov R (2002). Innate Immune Recognition. Annu. Rev. Immunol 20, 197–216. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Mak TW, Sen G, and Li X (2004). Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc. Natl. Acad. Sci. U.S.A 101, 3533–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junkin M, Kaestli AJ, Cheng Z, Jordi C, Albayrak C, Hoffmann A, and Tay S (2016). High-Content Quantification of Single-Cell Immune Dynamics. Cell Reports 15, 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Su T, Horng T, Chow A, Akira S, and Medzhitov R (2008). TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat Immunol 9, 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, et al. (2013). Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 51, 310–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanayama A, Seth RB, Sun L, Ea C-K, Hong M, Shaito A, Chiu Y-H, Deng L, and Chen ZJ (2004). TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol. Cell 15, 535–548. [DOI] [PubMed] [Google Scholar]
- Kang K, Park SH, Chen J, Qiao Y, Giannopoulou E, Berg K, Hanidu A, Li J, Nabozny G, Kang K, et al. (2017). Interferon-γ Represses M2 Gene Expression in Human Macrophages by Disassembling Enhancers Bound by the Transcription Factor MAF. Immunity 47, 235–250.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650. [DOI] [PubMed] [Google Scholar]
- Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, et al. (2004). Interferon-α induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 5, 1061–1068. [DOI] [PubMed] [Google Scholar]
- Kayama H, Ramirez-Carrozzi VR, Yamamoto M, Mizutani T, Kuwata H, Iba H, Matsumoto M, Honda K, Smale ST, and Takeda K (2008). Class-specific Regulation of Pro-inflammatory Genes by MyD88 Pathways and IκBζ. J. Biol. Chem 283, 12468–12477. [DOI] [PubMed] [Google Scholar]
- Kearns JD, Basak S, Werner SL, Huang CS, and Hoffmann A (2006). IkappaBepsilon provides negative feedback to control NF-kappaB oscillations, signaling dynamics, and inflammatory gene expression. J. Cell Biol 173, 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg RA, and Tay S (2015). Noise Facilitates Transcriptional Control under Dynamic Inputs. Cell 160, 381–392. [DOI] [PubMed] [Google Scholar]
- Kensche T, Tokunaga F, Ikeda F, Goto E, Iwai K, and Dikic I (2012). Analysis of nuclear factor-κB (NF-κB) essential modulator (NEMO) binding to linear and lysine-linked ubiquitin chains and its role in the activation of NF-κB. J. Biol. Chem 287, 23626–23634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodenko BN, and Birtwistle MR (2009). Four-dimensional dynamics of MAPK information-processing systems. WIREs Systems Biology and Medicine 1, 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs DL, and Hilton DJ (2001). SOCS proteins: negative regulators of cytokine signaling. Stem Cells 19, 378–387. [DOI] [PubMed] [Google Scholar]
- Kundert K, Lucas JE, Watters KE, Fellmann C, Ng AH, Heineike BM, Fitzsimmons CM, Oakes BL, Qu J, Prasad N, et al. (2019). Controlling CRISPR-Cas9 with ligand-activated and ligand-deactivated sgRNAs. Nat Commun 10, 2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwano Y, Kim HH, Abdelmohsen K, Pullmann R, Martindale JL, Yang X, and Gorospe M (2008). MKP-1 mRNA stabilization and translational control by RNA-binding proteins HuR and NF90. Mol Cell Biol 28, 4562–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers MM, van den Hoogen BG, and Haagmans BL (2019). ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front Immunol 10, 1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane K, Van Valen D, DeFelice MM, Macklin DN, Kudo T, Jaimovich A, Carr A, Meyer T, Pe’er D, Boutet SC, et al. (2017). Measuring Signaling and RNA-Seq in the Same Cell Links Gene Expression to Dynamic Patterns of NF-kappaB Activation. Cell Systems 4, 458–469.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane K, Andres-Terre M, Kudo T, Monack DM, and Covert MW (2019). Escalating Threat Levels of Bacterial Infection Can Be Discriminated by Distinct MAPK and NF-kappaB Signaling Dynamics in Single Host Cells. Cell Systems 8, 183–196.e4. [DOI] [PubMed] [Google Scholar]
- Lee REC, Walker SR, Savery K, Frank DA, and Gaudet S (2014). Fold change of nuclear NF-κB determines TNF-induced transcription in single cells. Mol. Cell 53, 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifer CA, and Medvedev AE (2016). Molecular mechanisms of regulation of Toll-like receptor signaling. J. Leukoc. Biol 100, 927–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Lin Y, and Elowitz MB (2013). Functional Roles of Pulsing in Genetic Circuits. Science 342, 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, and Elowitz MB (2019). Communication codes in developmental signaling pathways. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Dutta B, and Fraser IDC (2017). Systematic Investigation of Multi-TLR Sensing Identifies Regulators of Sustained Gene Activation in Macrophages. Cell Syst 5, 25–37.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Jordi C, Son M, Van Phan H, Drayman N, Abasiyanik MF, Vistain L, Tu H-L, and Tay S (2019). Ultra-sensitive digital quantification of proteins and mRNA in single cells. Nat Commun 10, 3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvak V, Ramsey SA, Rust AG, Zak DE, Kennedy KA, Lampano AE, Nykter M, Shmulevich I, and Aderem A (2009). Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat. Immunol 10, 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Sheng C, Gao S, Yao C, Li J, Jiang W, Chen H, Wu J, Pan C, Chen S, et al. (2015a). SOCS3 Drives Proteasomal Degradation of TBK1 and Negatively Regulates Antiviral Innate Immunity. Molecular and Cellular Biology 35, 2400–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chen J, Cai X, Wu J, Chen X, Wu Y-T, Sun L, and Chen ZJ (2013). MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2, e00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, et al. (2015b). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (New York, N.Y.) 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- Livnah O, Johnson DL, Stura EA, Farrell FX, Barbone FP, You Y, Liu KD, Goldsmith MA, He W, Krause CD, et al. (1998). An antagonist peptide-EPO receptor complex suggests that receptor dimerization is not sufficient for activation. Nat Struct Biol 5, 993–1004. [DOI] [PubMed] [Google Scholar]
- Long L, Deng Y, Yao F, Guan D, Feng Y, Jiang H, Li X, Hu P, Lu X, Wang H, et al. (2014). Recruitment of phosphatase PP2A by RACK1 adaptor protein deactivates transcription factor IRF3 and limits type I interferon signaling. Immunity 40, 515–529. [DOI] [PubMed] [Google Scholar]
- Longo DM, Selimkhanov J, Kearns JD, Hasty J, Hoffmann A, and Tsimring LS (2013). Dual Delayed Feedback Provides Sensitivity and Robustness to the NF-κB Signaling Module. PLOS Computational Biology 9, e1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Lucas RM, Liu L, and Stow JL (2019). Signalling, sorting and scaffolding adaptors for Toll-like receptors. J. Cell. Sci 133. [DOI] [PubMed] [Google Scholar]
- Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, and Dueber EC (2012). Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. U.S.A 109, 9378–9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, and Clark AR (2001). Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol. Cell. Biol 21, 6461–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes NP, Angermann BR, Koppenol-Raab M, An E, Sjoelund VH, Sun J, Ishii M, Germain RN, Meier-Schellersheim M, and Nita-Lazar A (2015). Targeted Proteomics-Driven Computational Modeling of Macrophage S1P Chemosensing. Mol Cell Proteomics 14, 2661–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M, Mehta A, Zhao JL, Lee K, Marinov GK, Garcia-Flores Y, Lu L-F, Rudensky AY, and Baltimore D (2017). An NF-κB-microRNA regulatory network tunes macrophage inflammatory responses. Nat Commun 8, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. (2014). The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9, 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins AJ, Narayanan M, Prüstel T, Fixsen B, Park K, Gottschalk RA, Lu Y, Andrews-Pfannkoch C, Lau WW, Wendelsdorf KV, et al. (2017). Environment Tunes Propagation of Cell-to-Cell Variation in the Human Macrophage Gene Network. Cell Syst 4, 379–392.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGettrick AF, and O’Neill LAJ (2010). Localisation and trafficking of Toll-like receptors: an important mode of regulation. Curr. Opin. Immunol 22, 20–27. [DOI] [PubMed] [Google Scholar]
- Meier-Schellersheim M, Xu X, Angermann B, Kunkel EJ, Jin T, and Germain RN (2006). Key Role of Local Regulation in Chemosensing Revealed by a New Molecular Interaction-Based Modeling Method. PLOS Computational Biology 2, e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merika M, Williams AJ, Chen G, Collins T, and Thanos D (1998). Recruitment of CBP/p300 by the IFNβ Enhanceosome Is Required for Synergistic Activation of Transcription. Molecular Cell 1, 277–287. [DOI] [PubMed] [Google Scholar]
- Mikuda N, Kolesnichenko M, Beaudette P, Popp O, Uyar B, Sun W, Tufan AB, Perder B, Akalin A, Chen W, et al. (2018). The IκB kinase complex is a regulator of mRNA stability. The EMBO Journal 37, e98658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell S, Vargas J, and Hoffmann A (2016). Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med 8, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monie TP, Moncrieffe MC, and Gay NJ (2009). Structure and regulation of cytoplasmic adapter proteins involved in innate immune signaling. Immunol. Rev 227, 161–175. [DOI] [PubMed] [Google Scholar]
- Morris MK, Saez-Rodriguez J, Sorger PK, and Lauffenburger DA (2010). Logic-Based Models for the Analysis of Cell Signaling Networks. Biochemistry 49, 3216–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli G (2009). Control of NF-kappaB-dependent transcriptional responses by chromatin organization. Cold Spring Harb Perspect Biol 1, a000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau GJ, Richmond JFL, Schlesinger A, Jennings EG, Lander ES, and Young RA (2002). Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. U.S.A 99, 1503–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, et al. (2004). Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science (New York, N.Y.) 306, 704–708. [DOI] [PubMed] [Google Scholar]
- Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, ONeill LAJ, and Xavier RJ (2016). Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098–aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, and Dixit VM (2012). Signaling in Innate Immunity and Inflammation. Cold Spring Harbor Perspectives in Biology 4, a006049–a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni G, Konno H, and Barber GN (2017). Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni G, Ma Z, Wong JP, Zhang Z, Cousins E, Major MB, and Damania B (2020). PPP6C Negatively Regulates STING-Dependent Innate Immune Responses. MBio 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen B, Taylor S, and Ghosh G (2004). Regulation of Protein Kinases. Molecular Cell 15, 661–675. [DOI] [PubMed] [Google Scholar]
- Ohto U, and Shimizu T (2016). Structural aspects of nucleic acid-sensing Toll-like receptors. Biophys Rev 8, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Mann CC, Orzalli MH, King DS, Kagan JC, Lee ASY, and Kranzusch PJ (2019). Modular Architecture of the STING C-Terminal Tail Allows Interferon and NF-κB Signaling Adaptation. Cell Reports 27, 1165–1175.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ourthiague DR, Birnbaum H, Ortenlof N, Vargas JD, Wollman R, and Hoffmann A (2015). Limited specificity of IRF3 and ISGF3 in the transcriptional innate-immune response to double-stranded RNA. Journal of Leukocyte Biology 98, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Gruenbaum A, Kanashova T, Mertins P, Cluzel P, and Chevrier N (2020). Pairwise Stimulations of Pathogen-Sensing Pathways Predict Immune Responses to Multi-adjuvant Combinations. Cell Syst 11, 495–508.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panne D, Maniatis T, and Harrison SC (2007). An Atomic Model of the Interferon-β Enhanceosome. Cell 129, 1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BS, and Lee J-O (2013). Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med 45, e66–e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Kang K, Giannopoulou E, Qiao Y, Kang K, Kim G, Park-Min K-H, and Ivashkiv LB (2017). Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat Immunol 18, 1104–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvatiyar K, Pindado J, Dev A, Aliyari SR, Zaver SA, Gerami H, Chapon M, Ghaffari AA, Dhingra A, and Cheng G (2018). A TRAF3-NIK module differentially regulates DNA vs RNA pathways in innate immune signaling. Nat Commun 9, 2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison MJ, Mitchell O, Flynn HR, Chen C-S, Yang H-T, Ben-Addi H, Boeing S, Snijders AP, and Ley SC (2016). TLR and TNF-R1 activation of the MKK3/MKK6-p38α axis in macrophages is mediated by TPL-2 kinase. Biochem. J 473, 2845–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauls E, Shpiro N, Peggie M, Young ER, Sorcek RJ, Tan L, Choi HG, and Cohen P (2012). Essential Role for IKKβ in Production of Type 1 Interferons by Plasmacytoid Dendritic Cells. J. Biol. Chem 287, 19216–19228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polley S, Huang D-B, Hauenstein AV, Fusco AJ, Zhong X, Vu D, Schröfelbauer B, Kim Y, Hoffmann A, Verma IM, et al. (2013). A structural basis for IκB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation. PLoS Biol. 11, e1001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, and Lahav G (2013). Encoding and decoding cellular information through signaling dynamics. Cell 152, 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, et al. (2009). Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 136, 1098–1109. [DOI] [PubMed] [Google Scholar]
- Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, and Smale ST (2009). A Unifying Model for the Selective Regulation of Inducible Transcription by CpG Islands and Nucleosome Remodeling. Cell 138, 114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, and Barton GM (2017). Tissue-Resident Macrophages Are Locally Programmed for Silent Clearance of Apoptotic Cells. Immunity 47, 913–927.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roget K, Ben-Addi A, Mambole-Dema A, Gantke T, Yang H-T, Janzen J, Morrice N, Abbott D, and Ley SC (2012). IκB kinase 2 regulates TPL-2 activation of extracellular signal-regulated kinases 1 and 2 by direct phosphorylation of TPL-2 serine 400. Mol. Cell. Biol 32, 4684–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothchild AC, Sissons JR, Shafiani S, Plaisier C, Min D, Mai D, Gilchrist M, Peschon J, Larson RP, Bergthaler A, et al. (2016). MiR-155–regulated molecular network orchestrates cell fate in the innate and adaptive immune response to Mycobacterium tuberculosis. PNAS 113, E6172–E6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccani S, Pantano S, and Natoli G (2001). Two waves of nuclear factor kappaB recruitment to target promoters. J. Exp. Med 193, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, and Oravecz T (2006). Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol 176, 1899–1907. [DOI] [PubMed] [Google Scholar]
- Sasai M, Linehan MM, and Iwasaki A (2010). Bifurcation of Toll-Like Receptor 9 Signaling by Adaptor Protein 3. Science 329, 1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, and Shalek AK (2014). Heterogeneity in immune responses: from populations to single cells. Trends Immunol. 35, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholes C, DePace AH, and Sánchez Á (2017). Combinatorial Gene Regulation through Kinetic Control of the Transcription Cycle. Cell Systems 4, 97–108.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz R, Sidler CL, Thali RF, Winssinger N, Cheung PCF, and Neumann D (2010). Autoactivation of Transforming Growth Factor β-activated Kinase 1 Is a Sequential Bimolecular Process. J. Biol. Chem 285, 25753–25766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröfelbauer B, and Hoffmann A (2011). How do pleiotropic kinase hubs mediate specific signaling by TNFR superfamily members? Immunol. Rev 244, 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröfelbauer B, Polley S, Behar M, Ghosh G, and Hoffmann A (2012). NEMO ensures signaling specificity of the pleiotropic IKKbeta by directing its kinase activity toward IkappaBalpha. Mol. Cell 47, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Cheng Z, Sheu KM, Chen YH, and Hoffmann A (2020). Gene Regulatory Strategies that Decode the Duration of NFκB Dynamics Contribute to LPS- versus TNF-Specific Gene Expression. Cell Syst 10, 169–182.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu KM, Luecke S, and Hoffmann A (2019). Stimulus-specificity in the responses of immune sentinel cells. Current Opinion in Systems Biology 18, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih VF-S, Kearns JD, Basak S, Savinova OV, Ghosh G, and Hoffmann A (2009). Kinetic control of negative feedback regulators of NF-kappaB/RelA determines their pathogen- and cytokine-receptor signaling specificity. Proc Natl Acad Sci U S A 106, 9619–9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J-H, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee K-Y, Bussey C, Steckel M, Tanaka N, et al. (2005). TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19, 2668–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara H, Behar M, Inoue K, Hiroshima M, Yasuda T, Nagashima T, Kimura S, Sanjo H, Maeda S, Yumoto N, et al. (2014). Positive Feedback Within a Kinase Signaling Complex Functions as a Switch Mechanism for NF- B Activation. Science 344, 760–764. [DOI] [PubMed] [Google Scholar]
- Shu C, Sankaran B, Chaton CT, Herr AB, Mishra A, Peng J, and Li P (2013). Structural insights into the functions of TBK1 in innate antimicrobial immunity. Structure 21, 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaug B, Chen J, Du F, He J, Ma A, and Chen ZJ (2011). Direct, noncatalytic mechanism of IKK inhibition by A20. Mol. Cell 44, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrer KM, and Gack MU (2015). Intracellular detection of viral nucleic acids. Current Opinion in Microbiology 26, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SL, Gaudet S, Albeck JG, Burke JM, and Sorger PK (2009). Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stempel M, Chan B, Juranić Lisnić V, Krmpotić A, Hartung J, Paludan SR, Füllbrunn N, Lemmermann NA, and Brinkmann MM (2019). The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-κB signaling and STING’s dual role during MCMV infection. EMBO J. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung M-H, Li N, Lao Q, Gottschalk RA, Hager GL, and Fraser IDC (2014). Switching of the Relative Dominance Between Feedback Mechanisms in Lipopolysaccharide-Induced NF-κB Signaling. Sci. Signal 7, ra6–ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, and Akira S (2010). Pattern Recognition Receptors and Inflammation. Cell 140, 805–820. [DOI] [PubMed] [Google Scholar]
- Tan RST, Ho B, Leung BP, and Ding JL (2014). TLR cross-talk confers specificity to innate immunity. Int Rev Immunol 33, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, and Covert MW (2010). Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature 466, 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa U, Pelosi E, Castelli G, and Labbaye C (2017). miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development. Noncoding RNA 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F (2013). Linear ubiquitination-mediated NF-κB regulation and its related disorders. J. Biochem 154, 313–323. [DOI] [PubMed] [Google Scholar]
- Tomida T, Takekawa M, and Saito H (2015). Oscillation of p38 activity controls efficient pro-inflammatory gene expression. Nat Commun 6, 8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, Allred AL, Barish GD, and Smale ST (2016). A Stringent Systems Approach Uncovers Gene-Specific Mechanisms Regulating Inflammation. Cell 165, 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DA, Paszek P, Woodcock DJ, Nelson DE, Horton CA, Wang Y, Spiller DG, Rand DA, White MRH, and Harper CV (2010). Physiological levels of TNFalpha stimulation induce stochastic dynamics of NF-kappaB responses in single living cells. J. Cell. Sci 123, 2834–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, et al. (2005). Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-α induction. J Exp Med 201, 915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanaja KG, Timp W, Feinberg AP, and Levchenko A (2018). A Loss of Epigenetic Control Can Promote Cell Death through Reversing the Balance of Pathways in a Signaling Network. Mol. Cell 72, 60–70.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisinne G, Nixon BG, Melbinger A, Gasteiger G, Vergassola M, and Altan-Bonnet G (2015). T Cells Integrate Local and Global Cues to Discriminate between Structurally Similar Antigens. Cell Reports 11, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, and Chen ZJ (2001). TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351. [DOI] [PubMed] [Google Scholar]
- Waterfield MR, Zhang M, Norman LP, and Sun SC (2003). NF-kappaB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the Tpl2 kinase. Mol. Cell 11, 685–694. [DOI] [PubMed] [Google Scholar]
- de Weerd NA, and Nguyen T (2012). The interferons and their receptors--distribution and regulation. Immunol. Cell Biol 90, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner SL, Barken D, and Hoffmann A (2005). Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science (New York, N.Y.) 309, 1857–1861. [DOI] [PubMed] [Google Scholar]
- Werner SL, Kearns JD, Zadorozhnaya V, Lynch C, O’Dea E, Boldin MP, Ma A, Baltimore D, and Hoffmann A (2008). Encoding NF-kappaB temporal control in response to TNF: distinct roles for the negative regulators IkappaBalpha and A20. Genes & Development 22, 2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. (2004). De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430, 694. [DOI] [PubMed] [Google Scholar]
- Witzel F, Maddison L, and Blüthgen N (2012). How scaffolds shape MAPK signaling: what we know and opportunities for systems approaches. Frontiers in Physiology 3, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (New York, N.Y.) 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C, Fan J, Emanuel G, Hao J, and Zhuang X (2019). Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc Natl Acad Sci USA 116, 19490–19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z-P, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, and Chen ZJ (2009). Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 461, 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang W, Zhang Q, Lin X, Wu S, Zhou Y, Meng F, Fan Y, Shen T, Xiao M, Xia Z, et al. (2016). PPM1A silences cytosolic RNA sensing and antiviral defense through direct dephosphorylation of MAVS and TBK1. Sci Adv 2, e1501889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Meckel T, Brzostowski JA, Yan J, Meier-Schellersheim M, and Jin T (2010). Coupling Mechanism of a GPCR and a Heterotrimeric G Protein During Chemoattractant Gradient Sensing in Dictyostelium. Sci. Signal 3, ra71–ra71. [DOI] [PubMed] [Google Scholar]
- Yoboua F, Martel A, Duval A, Mukawera E, and Grandvaux N (2010). Respiratory syncytial virus-mediated NF-kappa B p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKK beta. J. Virol 84, 7267–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, and Granucci F (2012). Regulation and dysregulation of innate immunity by NFAT signaling downstream of pattern recognition receptors (PRRs). European Journal of Immunology 42, 1924–1931. [DOI] [PubMed] [Google Scholar]
- Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, and Kagan JC (2011). CD14 controls the LPS-induced endocytosis of Toll-like Receptor 4. Cell 147, 868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Tu H-L, Jia G, Mukhtar T, Taylor V, Rzhetsky A, and Tay S (2019a). Ultra-multiplexed analysis of single-cell dynamics reveals logic rules in differentiation. Sci. Adv 5, eaav7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Shang G, Gui X, Zhang X, Bai X-C, and Chen ZJ (2019b). Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Clark K, Lawrence T, Peggie MW, and Cohen P (2014). An unexpected twist to the activation of IKKβ: TAK1 primes IKKβ for activation by autophosphorylation. Biochem. J 461, 531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wei N, Cui Y, Hong Z, Liu X, Wang Q, Li S, Liu H, Yu H, Cai Y, et al. (2018). The deubiquitinase CYLD is a specific checkpoint of the STING antiviral signaling pathway. PLOS Pathogens 14, e1007435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Liang L, Fan Y, Sun S, An L, Shi Z, Cheng J, Jia W, Sun W, Mori-Akiyama Y, et al. (2012). PPM1B negatively regulates antiviral response via dephosphorylating TBK1. Cell Signal 24, 2197–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.