

ABSTRACT
Mitochondrial dysfunction is involved in aging and multiple degenerative diseases, including intervertebral disc degeneration (IVDD) and osteoarthritis (OA). Thus, the maintenance of mitochondria homeostasis and function is important. Mitophagy, a process that selectively clears damaged or dysfunctional mitochondria through autophagic machinery, functions to maintain mitochondrial quality control and homeostasis. IVDD and OA are similar joint diseases involving the degradation of cartilaginous tissues that are mainly caused by oxidative stress, cell apoptosis and extracellular matrix (ECM) degradation. Over the past decade, accumulating evidence indicates the essential role of mitophagy in the pathogenesis of IVDD and OA. Importantly, strategies by the regulation of mitophagy exert beneficial effects in the pre-clinical experiments. Given the importance and novelty of mitophagy, we provide an overview of mitophagy pathways and discuss the roles of mitophagy in IVDD and OA. We also highlight the potential of targeting mitophagy for the treatment of degenerative joint diseases.
Abbreviations: AD: Alzheimer disease; AF: annulus fibrosus; ADORA2A/A2AR: adenosine A2a receptor; AMBRA1: autophagy and beclin 1 regulator 1; BMSCs: bone marrow mesenchymal stem cells; BNIP3: BCL2 interacting protein 3; BNIP3L/NIX: BCL2/adenovirus E1B interacting protein 3-like; CDH6: cadherin 6; CEP: cartilaginous endplates; circRNA: circular RNA; DNM1L/DRP1: dynamin 1-like; ECM: extracellular matrix; HIF1A: hypoxia inducible factor 1: alpha subunit; IL1B: interleukin 1 beta; IMM: inner mitochondrial membranes; IVDD: intervertebral disc degeneration; MAPK8/JNK: mitogen-activated protein kinase 8; MFN1: mitofusin 1; MFN2: mitofusin 2; MIA: monosodium iodoacetate; RHOT/MIRO: ras homolog family member T; MMP: mitochondrial transmembrane potential; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; NFE2L2: nuclear factor: erythroid 2 like 2; NP: nucleus pulposus; OA: osteoarthritis; OPA1: OPA1: mitochondrial dynamin like GTPase; OPTN: optineurin; PRKN: parkin RBR E3 ubiquitin protein ligase; PD: Parkinson disease; PGAM5: PGAM family member 5; PPARGC1A/PGC-1A: peroxisome proliferator activated receptor: gamma: coactivator 1 alpha; PHF23: PHD finger protein 23; PINK1: PTEN induced putative kinase 1; ROS: reactive oxygen species; SfMSCs: synovial fluid MSCs; SIRT1: sirtuin 1; SIRT2: sirtuin 2; SIRT3: sirtuin 3; SQSTM1/p62: sequestosome 1; TNF: tumor necrosis factor; Ub: ubiquitin; UBL: ubiquitin-like; VDAC: voltage-dependent anion channel.
KEYWORDS: Chondrocyte, intervertebral disc degeneration (IVDD), mitophagy, nucleus pulposus cells, osteoarthritis (OA), oxidative stress
Introduction
The intervertebral disc is a fibrocartilaginous tissue located between adjacent vertebrae, composed of three main parts, the inner nucleus pulposus (NP) containing chondrocyte-like cells, the outer annulus fibrosus (AF), and cartilaginous endplates (CEP) [1]. The resident NP cells serve for producing proteoglycan and type II collagen to control the ECM metabolism [2]. The highly specialized ECM within the IVD is essential for transmitting mechanical load and permitting movement of the spine [1]. Similarly, articular cartilage covers ends of long bones and allows joint movement, protecting bone tissue from mechanical damage. This cartilage consists of hyaluronic acid, type II collagen, and a rich proteoglycan matrix secreted by the only cartilage-resident cell, chondrocytes [3]. Like NP cells, chondrocytes govern the homeostasis of articular cartilage through controlling ECM metabolism [4]. Therefore, the dysfunction and loss of these two types of cell could lead to disorders in the ECM, which is greatly associated with the progression of IVDD and OA [5,6].
IVDD and OA are prevalent joint diseases, causing heavy socioeconomic effects on today’s society. IVDD is an important cause of chronic lower back pain while OA is the leading cause of disability among the elderly population, thereby both IVDD and OA seriously affect life quality of people [7,8]. Unfortunately, at present, there is a limited approach to retard and reverse these two diseases. Recent studies have focused on understanding the pathogenesis of IVDD and OA for developing effective therapies to alleviate their progression. Although the pathogenesis of IVDD and OA is complicated, the progressive degradation of cartilaginous tissue and the aberrance of NP cells and chondrocytes are considered as prominent factors involved in the development of both diseases. Hence, the protection of NP cells and chondrocytes is critical for maintenance of cartilage health and provide a therapeutic approach to IVDD and OA [9,10].
Mitochondria, the “powerhouse” of the cell, produce cellular energy in the form of ATP and involves in various vital cellular processes [11]. The dysfunction of mitochondria is correlated with cell damage and a wide range of age-related diseases, including degenerative joint disorders [12]. Therefore, it is quite important to maintain mitochondria homeostasis. Mitophagy, a specialized form of autophagy that functions in regulating the turnover of dysfunctional mitochondria, acts as a pivotal mechanism for mitochondrial homeostasis [13]. Impaired mitophagy is correlated with a series of physiological and pathological processes, such as differentiation, apoptosis, aging, neurodegenerative disorders [14–16]. Recently, this mechanism has been extensively studied in IVDD and OA and is tightly involved in their pathological processes. Mitophagy impairment gives rise to the progressive accumulation of defective mitochondria, leading to apoptosis of NP cells and ECM degradation, contributing to the degeneration of cartilage [17,18]. In this review, we will provide a synopsis of mitophagy, and summarize emerging literature to discuss the contributions of mitophagy in IVDD and OA pathogenesis, highlighting its therapeutic implications in degenerative joint diseases.
Overview of mitophagy pathway
Mitophagy is essential for the basal mitochondrial turnover and maintenance under physiological conditions while it could also be strongly provoked in response to various pathological stresses, such as mitochondrial transmembrane potential (MMP) collapse, hypoxia, oxidative stress, and iron starvation [19]. There are several mitophagy mechanisms that have been found and different stimulus induce mitophagy in distinct mechanisms. In general, mitophagy can be divided into PRKN (parkin RBR E3 ubiquitin protein ligase)-dependent and PRKN-independent pathways.
PRKN-dependent mitophagy
PRKN-dependent mitophagy is the best-studied pathway mediated by the PINK1 (PTEN induced putative kinase 1) and the E3-ubiquitin ligase PRKN [20]. This pathway controls ubiquitin-dependent mitophagy that affects a number of mitochondrial physiological processes, including mitochondrial biogenesis, dynamics, and autophagic machinery [21,22]. Under normal conditions, PINK1 is translocated into the inner mitochondrial membranes (IMM) through the translocase of the outer membrane and translocase of the inner mitochondrial membrane complexes in a membrane potential-dependent manner [23,24]. Subsequently, PINK1 is cleaved by a mitochondrial resident rhomboid serine protease, the presenilin-associated rhomboid-like protein [23], and the N-terminal truncated PINK1 is degraded by the (mitochondrial) proteasome, maintaining PINK1 at a low level [25]. Loss of MMP could disrupt PINK1 translocation into IMM and cause the accumulation of uncleaved PINK1 on outer mitochondrial membranes (OMM), owing to that pyruvate dehydrogenase kinase 2 phosphorylates presenilin-associated rhomboid-like protein, blocking the cleavage of PINK1 in response to MMP loss [26,27]. Afterward, the accumulated PINK1 promotes recruitment and activation of PRKN in the cytoplasm through directly phosphorylating the PRKN ubiquitin-like (UBL) domain. Additionally, PINK1 also phosphorylates ubiquitin (Ub) and poly-Ub chains that bind PRKN to facilitate its activation, and activated PRKN could further generate poly-Ub chains, thereby amplifying mitophagy signals [28–30]. The phosphorylated Ub and poly-Ub chains exhibit reduced hydrolysis by deubiquitinases. Deubiquitinating enzymes, like USP15, USP30 and USP35, function on eliminating PRKN-generated Ub chains from the mitochondrial surface, consequently resisting mitophagy [31–33]. Therefore, ubiquitination and deubiquitination events govern PRKN-dependent mitophagy, and a balance between them is critical for mitochondrial and cellular homeostasis [13].
The activated PRKN will drive the ubiquitination of multiple substrates, such as MFN1 (mitofusin 1), MFN2 (mitofusin 2), VDAC (voltage-dependent anion channel), RHOT/MIRO (ras homolog family member T), and BAK proteins. The ubiquitination of mitochondrial fusion proteins MFN1 and MFN2 promotes mitochondria accessible for degradation and prevents fusion of damaged mitochondria [34]. In addition, Miro, located on the OMM, anchors mitochondria to the cytoskeleton, mediating mitochondrial motility and PRKN recruitment. PRKN can ubiquitinate Miro to promote its degradation and block mitochondrial transport [35,36]. VDAC1 also acts as mitochondrial docking sites to recruit PRKN from the cytosol to the damaged mitochondria [37]. BAK, an apoptotic protein identified as a PRKN target, establishes linkage between PRKN-dependent mitophagy and regulation of cellular apoptosis [38]. The PRKN-mediated ubiquitination events trigger the recruitment of autophagy receptors including CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2) [39], OPTN (optineurin) [40], and SQSTM1/p62 (sequestosome 1) [41] to the mitochondrial membrane. These autophagy receptors contain a LC3-interacting region (LIR) that selectively recognize autophagic cargo (mitochondria) and mediate its engulfment into autophagosomes by binding to the LC3 [42]. The cytosolic LC3-I is activated by the E1 enzyme ATG7, and transferred to the E2 enzyme ATG3, and finally is modified to LC3-II, by conjugating to the lipid phosphatidylethanolamine. Then, the ATG12–ATG5-ATG16L1 complex acts as an E3 enzyme for the conjugation reaction of LC3-II [43].
PRKN-independent mitophagy
PRKN-independent mitophagy pathways primarily depend on receptor proteins that directly interact with LC3 and GABARAP through their LIR motifs, conducing to the elimination of mitochondria. For instance, the LIR-containing proteins, BNIP3 (BCL2 interacting protein 3) and BNIP3L/NIX (BCL2/adenovirus E1B interacting protein 3-like) are capable of directly binding LC3-GABARAP and induce mitophagy without undergoing ubiquitination process. Both BNIP3 and BNIP3L/NIX are hypoxia-inducible genes and transcriptionally regulated by HIF1A (hypoxia inducible factor 1 subunit alpha) [44,45]. HIF1A could upregulate the expression of BNIP3 and BNIP3L to promote mitophagy under the hypoxia conditions [44]. Additionally, BNIP3 and BNIP3L are also under the transcriptional regulation of NFKB or FOXO3/FOXO3A, connecting the mitophagy with elementary signaling pathways [46,47]. It has been reported that BNIP3 and BNIP3L could interact with SPATA18/mitochondria-eating protein, to initial the removal of oxidized species [48] and with CDH6 (cadherin 6) to modulate mitophagy and DNM1L/DRP1 (dynamin 1-like)-mediated fission [49], maintaining homeostasis of mitochondrial. BNIP3L also plays a vital role in programmed mitophagy in cell differentiation and maturation [50]. BNIP3L expression is significantly increased, causing decreased MMP and the induction of mitophagy during erythrocyte differentiation and Bnip3l-deficient cells with accumulated mitochondria, results in the increase of apoptosis and developmental defects [51,52]. There exists an interaction between OPA1 (OPA1 mitochondrial dynamin like GTPase) and BNIP3 promoting mitochondrial fragmentation while phosphorylation of BNIP3 in its C-terminal domain will block this interaction, subsequently inducing mitophagy and reducing cell death [53].
Another mitophagy receptor, FUNDC1, is also an LIR-motif containing protein that regulates mitophagy, especially in hypoxic conditions. Under normoxic conditions, FUNDC1 LIR-motif is phosphorylated (at tyrosine 18 and serine 13) by the SRC and CSNK2/casein kinase 2 to suppress its activity. Under the circumstances of hypoxia or loss of MMP, FUNDC1 phosphorylation (of serine 17) by ULK1 and dephosphorylation (of tyrosine 18 and serine 13) mediated by the mitochondrial phosphatase PGAM5 (PGAM family member 5). FUNDC1 could promote its interaction with LC3 to enhance mitophagy [54,55]. In addition, mitochondrial dynamics also participate in FUNDC1-mediated mitophagy. FUNDC1 interacts with both key factors of mitochondrial fission and fusion, DNM1L and OPA1, and dephosphorylation of FUNDC1 stimulates the separation of the FUNDC1-OPA1 complex, in turn, increases the association of FUNDC1 with DNM1L, promoting mitochondrial fission and fostering mitophagy [53]. Similarly, the association of endoplasmic reticulum protein calnexin and FUNDC1 is reduced for liberating FUNDC1 to interact DNM1L, triggering mitochondrial fission in response to hypoxia [56].
AMBRA1 (autophagy and beclin 1 regulator 1) is also a mitophagy receptor that can directly bind to LC3 through an LIR motif. Of note, the emerging findings show that AMBRA1 is involved in both PRKN-dependent and PRKN-independent mitophagy [57]. Apart from the above receptors, some lipids also function on the interaction with LC3 to provoke mitophagy, such as ceramide and cardiolipin. Cardiolipin, a phospholipid normally located at the IMM, is able to externalize the outer membrane and acts as a mitophagy receptor, contributing to the removal of impaired mitochondria in neuronal cells [58]. For ceramide, it has been found as a possible receptor for anchoring LC3B-II autolysosome to mitochondrial membranes, thereby inducing lethal mitophagy [59] (Figure 1).
Figure 1.
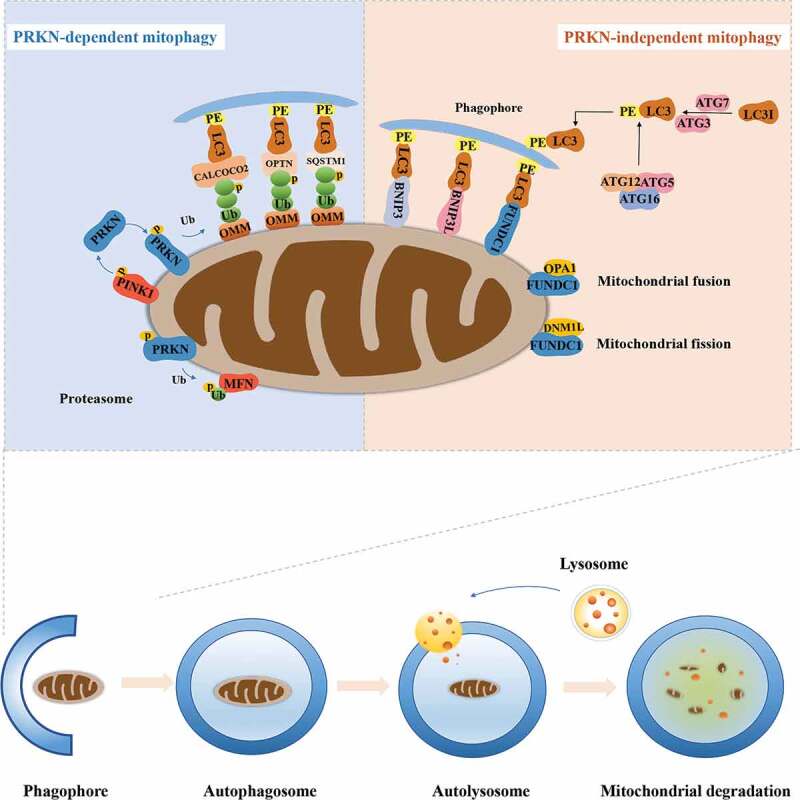
The overview of mitophagy pathway. PRKN-dependent mitophagy. The phosphorylated PINK1 accumulates on the OMM when mitochondria are depolarized under various cellular stress and subsequently recruits PRKN to mitochondria. The E3 ligase PRKN polyubiquitinates multiple OMM proteins, which will be recognized by LC3 receptors including CALCOCO2, OPTN, and SQSTM1 on the phagophore. PRKN-independent mitophagy. The PRKN-mediated ubiquitylation of MFN also promotes mitochondria accessible for degradation and prevents fusion of damaged mitochondria. Alternatively, FUNDC1, BNIP3 and BNIP3L could directly bind the LC3 molecules decorating the autophagosome. Dephosphorylation of FUNDC1 enhances mitochondrial fission by the disassembly of OPA1, and increasing the interaction with DNM1L on the mitochondrial surface. Impaired mitochondria are engulfed by phagophores to form mitophagosomes, followed by the fusion with lysosomes to form mitolysosomes that degrades damaged mitochondria
The roles of mitophagy in IVDD
Recently, a large-scale genome-wide association study has identified that a variant in the PRKN/PARK2 gene is significantly associated with IVDD and differential methylation at one CpG island of the PRKN/PARK2 promoter is observed in a small subset of subjects [60]. Shortly afterward, Zhang and his colleagues found that PRKN is markedly upregulated in degenerative NP tissues and in NP cells under the stimulation of TNF (tumor necrosis factor), a commonly elevated cytokine in degenerated disc tissues [17]. Besides, the expression of PRKN is dependent on the degree of disc degeneration, suggesting an association between PRKN and IVDD progression. They further found that knockdown of Prkn promotes TNF-stimulated mitochondria-induced ROS generation and aggravated apoptosis of NP cells. Whereas, salidroside, considered as an activator of PRKN and autophagy, is able to enhance PRKN expression and the translocation of cytosolic PRKN to the surface of defective mitochondria, repressing ROS generation and NP cells apoptosis. In addition, salidroside is also beneficial for IVDD in rats through the role of PRKN [17]. Their data indicate the role of PRKN in the eliminating of damaged mitochondria and NP cells survival.
Reactive oxygen species (ROS), caused by external stimulus including pro-inflammatory cytokines, nutrition deprivation, and mechanical loading, is another crucial intermediator of the occurrence and progression of IVDD [61]. It has been reported that oxidative stress induces excessive mitophagy and further cause cell death in human and rat NP cells [62,63]. In another study conducted by Wang et al., oxidative stress induces mitochondria dysfunction and mitophagy in NP cells while depletion of PINK1 by short hairpin RNA further impairs mitophagy, and aggravates NP cells senescence under oxidative stress [63], suggesting that PINK1-mediated mitophagy is indispensable in the protection of oxidative stress-related mitochondrial damage and cell senescence. However, the role of activated PINK1 in the pathological condition remains unclear. In line with that, mitophagy and mitochondrial dynamics in endplate chondrocytes are also stimulated by oxidative stress. Both PRKN and NFE2L2 (nuclear factor, erythroid 2 like 2) deficiency accelerate mitochondrial dysfunction and apoptosis in endplate chondrocytes under oxidative stress, whereas polydatin-mediated upregulation of PRKN and NFE2L2 could inhibit oxidative stress-induced cell damage and alleviate cartilaginous endplate and disc degeneration in puncture-induced rat models [64]. MFN2, as mentioned above, is an OMM protein that motivates mitochondrial fusion and maintains the homeostasis of mitochondrial dynamics [65]. The results of Chen and his collogues showed that MFN2 is indispensable for autophagic flux and mitochondrial function while overexpression of Mfn2 can promote PINK1-PRKN-mediated mitophagy in rat NP cells in a ROS-dependent and attenuate cell damage and the development of IVDD in rats [66]. The finding of this study indicates loss of MFN2 is closely involved in IVDD pathogenesis owing to its impairment on mitophagy and mitochondrial function.
Circular RNA (circRNA), a large endogenous class of non-coding RNA with a closed-loop structure, could act as miRNA sponges and provide binding sites for miRNAs to regulate the expression of the target gene [67]. Increasing evidence demonstrates that some circRNAs could serve as miRNA inhibitors involved in the development of IVDD, among which circERCC2 is observed to be downregulated in IVDD and contributes to the disease [68]. Mir182-5p, identified as a direct target of circErcc2, downregulates SIRT1 (sirtuin 1) expression, thereby promoting apoptosis and repressing mitophagy. Therefore, overexpression of circErcc2 could significantly attenuate apoptosis, ECM degradation and enhance mitophagy via targeting Mir182-5p-SIRT1 in response to oxidative stress [68]. This study provides a crucial regulatory mechanism of mitophagy referring circErcc2, Mir182-5p and SIRT1, which helps develop potential therapeutic approaches for IVDD.
Melatonin, an endogenous molecule, has been widely applied in health care [69] and current studies have highlighted its correlation with mitophagy in several tissues [70,71]. Chen et al. have found that melatonin could induce the activation of mitophagy in a PRKN-dependent manner in NP cells, which counteracts oxidative stress‐caused ECM degradation and cell apoptosis while suppression of mitophagy by an inhibitor (CsA) abrogates these effects. Moreover, melatonin also ameliorates IVDD in vivo. These findings suggest that mitophagy activation is necessary for Melatonin to protect NP cells against oxidative stress‐caused cell apoptosis and ECM degeneration [72]. Like melatonin, a metabolite of ellagitannins and ellagic acid [73], urolithin A, also protects against mitochondrial dysfunction and cell apoptosis by promoting mitophagy. Interestingly, the AMPK signaling pathway mediates the initiation of mitophagy under urolithin A treatment and protective roles of urolithin A in vivo and in vitro [74]. In another study, Honokiol, a small molecule natural compound agonist of SIRT3 (sirtuin 3) [75], was reported not only to upregulate the expression of two mitophagic markers, BNIP3 and BNIP3L, but also increase lipidation of the marker of autophagic vacuoles, LC3. Moreover, more colocalization of LC3 and BNIP3L and more autophagic vesicles containing mitochondria are observed in Honokiol-treated NP cells compared with untreated NP cells. Whereas, SIRT3 knockdown abolishes these mitophagy outcomes caused by Honokiol, suggesting that Honokiol is conducive to initial mitophagy via SIRT3 [76]. Although Wang et al. demonstrated the beneficial effects of Honokiol in the improvement of anti-oxidation, mitochondrial dynamics, mitophagy and cell survivals in NP cells and in IVDD rats, a direct association between Honokiol-mediated mitophagy and these protective effects is unknown.
Excessive mechanical stress is considered as a critical contributor to the development of IVDD. In the study of Huang et al., compression significantly promotes PINK1- PRKN-mediated mitophagy, senescence, and mitochondrial impairment of NP cells in a time-dependent manner. Also, enhanced expression of PINK1-PRKN was found in rat-degenerated discs and in human-herniated NP tissues. Their data show a positive relationship between mitophagy in IVDD tissues and IDD [77]. Impaired mitochondria are engulfed by autophagosomes to form mitophagosomes, followed by the fusion with lysosomes to form mitolysosomes that degrades damaged mitochondria. During this process, mitophagic flux outcome is prone to be interrupted by abnormal lysosomal function or disturbed mitophagosome-lysosome fusion [78]. Kang et al. have reported that 1.0 MPa compression could initiate mitophagy, but blocks mitophagic flux mainly attributed by impairments in mitophagosome‐lysosome fusion and lysosomal degradation in human NP cells. Fortunately, they found that mitochondria-targeted antioxidant, MitoQ promotes PINK1-PRKN-mediated mitophagy, restores the mitochondrial dynamics balance and further maintains mitophagic flux, which rescues mitochondrial dysfunction and NP cell apoptosis under the compression [79].
During aging and degeneration of intervertebral discs, the nutrition supply including glucose, oxygen, and serum, significantly reduced and even disappeared, driving NP microenvironment more acidic and anoxic, which has been considered as a factor involved in the disc degeneration [80]. To mimic this pathological condition, Liu and his colleagues cultured the NP cells in a nutrition deprivation condition and observed that BNIP3 is upregulated and translocated to mitochondria with decreased MMP and increased cell apoptosis during oxygen-glucose-serum deprivation, implying a possible correlation between BNIP3 and cell apoptosis. Afterward, the results from Liu et al. and He et al. demonstrated that nutrient deprivation-induced cell apoptosis in NP cells and cartilage endplate stem cells indeed rely on the activation of the BNIP3 pathway [81,82]. These results are accordance with other reports that BNIP3 is a pro-apoptotic protein, mediating apoptotic process of multiple type of cells [83]. Interestingly, it has been found that BNIP3-mediated mitophagy also contributes to mitochondrial apoptosis as increased mitophagy overtly consumes mitochondrial mass causing the mitochondrial metabolism disorder [84]. Nevertheless, there is no direct evidence to show the relationship of BNIP3-mediated mitophagy and apoptosis in degenerative NP cells. FOXO3 is a critical key regulator of cellular quality control and regulates mitophagy [85]. In NP cells, FOXO3 inhibits mitochondrial dysfunction, apoptosis and ECM degradation under nutrient deficiency through activating autophagy, especially for PINK1-mediated autophagy [86], suggesting that FOXO3-mediated mitophagy might be a promising therapeutic target to retard disc degeneration. Of note, BNIP3 and BNIP3L are also under the transcriptional regulation of FOXO3. How FOXO3-BNIP3- BNIP3L pathway controls mitophagy and affects cellular processes in NP cells or chondrocytes deserves to be considered.
Although mitophagy is activated under multiple pathological stimulus and with the development of IVDD, there exists some controversy about the role of mitophagy in IVDD pathogenesis. In Huang et al. study, inhibition of PINK1-PRKN-mediated mitophagy by CsA and PINK1 knockdown strongly rescues the compression-induced senescence of NPCs. They explained that continuous excessive compression stimulates aberrant PINK1-PRKN‐mediated mitophagy, causing excessive removal of mitochondria which might accelerate the senescence of NP cells. In addition, Xu et al. suppressed mitophagy with the mitophagy inhibitor CsA and observed that NP cells are protected against oxidative stress-induced apoptosis. They further found that the upregulation of mitochondrial NDUFA4L2 facilitates the survival of NP cells by repressing excessive mitophagy, ultimately alleviating IVDD [62]. Their another study displayed that apart from NP cells, there is a similar association between mitophagy and oxidative stress in AF cells. Importantly, PPARGC1A/PGC-1A (peroxisome proliferator activated receptor, gamma, coactivator 1 alpha) could act as a target of SIRT2 (sirtuin 2) to protect AF cells against apoptosis under oxidative stress by restraining mitophagy [87], which provides a bridge between PPARGC1A and mitophagy in AF cells and probably in various cell types with mitochondrial quality control. All these findings show a differential role of mitophagy in IVDD. According to the existing evidence, it could be speculated that a proper level of mitophagy serves to facilitate cell survival owing to selective elimination of damaged mitochondria to counteract mitochondrial dysfunction, oxidative stress, and apoptosis in response to multiple moderate stimuli, while excessive mitophagy promotes undue clearance of mitochondria, causing the mitochondrial metabolism disorder and eventually cell death. Besides, the differential role of mitophagy might depend on the types of cells and stimulus that affect different mechanisms. In fact, the autophagy including mitophagy is not fully understood, and it might function in a stage-dependent pattern during the development of IVDD. However, given the essential role of mitophagy in IVDD pathogenesis, the detailed regulatory mechanism and agents of mitophagy need further investigation.
The roles of mitophagy in OA
Like the roles of mitophagy in IVDD, accumulating evidence also demonstrates the critical roles of mitophagy in the progression of OA. It has been found that autophagy and mitophagy-related proteins including LC3B, SQSTM1, and PINK1 are highly expressed in cartilage from OA patients and monosodium iodoacetate (MIA)-induced rodent model of OA [88], which shows a strong link between mitophagy and OA. IL1B (interleukin 1 beta), a key mediator implicated in OA, is commonly used to mimic in vitro pathological conditions [89]. In Haqqi et al. study, IL1B-stimulated OA chondrocytes show overproduction of ROS, MMP collapse, accumulation of damaged mitochondria and elevated cell apoptosis with activated PRKN-mediated mitophagy. Furthermore, depletion of PRKN in chondrocytes impairs mitophagy and give rise to increased mitochondrial dysfunction and oxidative stress under the IL1B stimulation while PRKN overexpression reduces mitochondrial ROS and apoptosis of chondrocyte through the clearance of dysfunctional mitochondria [18]. Additionally, metformin also serves as an activator of the SIRT3-PINK1-PRKN signaling pathway to counteract IL1B-provoked oxidative stress and imbalance of anabolism and catabolism in chondrocytes [90]. These lines of evidence confirm the essential role of PRKN for mitophagy and maintenance of chondrocyte homeostasis under pathological conditions, and suggests that metformin could regulate mitophagy process, which provides a promising choice for degenerative diseases treatment. However, this promising agent needs to be further confirmed using in vivo models. Recently, some autophagy regulators have been reported to affect mitophagy. For example, PHF23 (PHD finger protein 23), newly identified as an autophagy inhibitor, its deficiency in IL1B-induced chondrocytes shows the promotion of autophagy and mitophagy as well as mitigation in ECM degradation, demonstrating the therapeutic potential of PHF23 inhibition in OA and a correlation between PHF23 and mitophagy [91]. Another autophagy regulator, spermidine, has been reported to exert anti-oxidative and anti-inflammatory effects in oxidative stress-disturbed chondrocytes through activating autophagy [92]. This study also revealed the property of spermidine in inducing mitophagy [92]. Unfortunately, the exact role of mitophagy induced by PHF23 and spermidine in the protection of OA chondrocytes remains unclear. Trehalose is a natural disaccharide and also a novel autophagy activator in multiple cells [93]. This disaccharide could restore oxidative stress-induced interrupted autophagic flux and mitophagy, thereby attenuating chondrocyte apoptosis in vitro by activating BNIP3 and PGAM5 while BNIP3 knockdown partially reverses the anti-apoptotic effects of trehalose but does not alter autophagy. Trehalose also retards OA in a destabilized medial meniscus mouse model probably via promoting autophagic flux [94]. These data indicate that these effects of trehalose might be an association with the activity of BNIP3-mediated mitophagy.
Adenosine, a metabolite of ATP could regulate chondrocyte function by binding to its ADORA2A/A2AR (adenosine A2a receptor) [95]. Mice lacking A2AR will develop spontaneous OA [95,96]. Furthermore, there is mitochondrial dysfunction with increased ROS burden and diminished mitophagy in A2AR−/- chondrocytes. Importantly, Castro et al. found that A2AR agonism improves mitochondrial dynamics and function in IL1B-stimulated human chondrocytes and in the obesity-induced OA mouse model, and their preliminary data showed A2AR ligation also modulates mitophagy and autophagy [96], suggesting A2AR ligation is mitoprotective in OA. Nevertheless, the investigation about the direct relationship between mitophagy and A2AR-mediated protection in OA is expected.
In contrast, the data from Shin et al. revealed that PINK1-mediated mitophagy results in mitochondrial fragmentation and cell death in human chondrocytes and rats following MIA treatment, while there are reduced cartilage damage and pain behaviors in pink1-knockout mice with MIA-induced OA compared with the control [88]. This data, along with the above results exhibits two-side effects of mitophagy on cell survival and function that is possibly attributed to the mitophagy level under the pathological conditions (Table 1).
Table 1.
Regulatory molecules of mitophagy in IVDD and OA
| Regulators | Target cell/tissue | Mitophagy pathway | Main findings | Reference |
|---|---|---|---|---|
| IVDD↓ PRKN |
Rat NP cells | PRKN-mediated mitophagy↓ | Knockdown of Prkn promotes TNF-stimulated mitochondria-induced ROS generation and aggravates apoptosis of NP cells. | [17] |
| PINK1↓ | Human NP cells | PRKN-mediated mitophagy↓ | Aggravates NP cells senescence under oxidative stress | [63] |
| PRKN↓ | Human endplate chondrocytes |
PRKN-mediated mitophagy↓ | Accelerates mitochondrial dysfunction and apoptosis in endplate chondrocytes under oxidative stress | [64] |
| MFN2↑ | Rat NP cells | PRKN-mediated mitophagy↑ | Overexpression of Mfn2 attenuates cell damage and the development of IVDD in rats | [66] |
| circErcc2↑ | Rat NP cells |
Mir182-5p-SIRT1-PRKN -mediated mitophagy↑ |
Attenuates apoptosis, ECM degradation and enhances mitophagy in response to oxidative stress and retards IVDD in rats. | [68] |
| FOXO3↑ | Rat NP cells | PRKN-mediated mitophagy↑ | Inhibits mitochondrial dysfunction, cell apoptosis and ECM degradation under nutrient deficiency | [86 |
| NDUFA4L2↑ | Rat NP cells | PRKN-mediated mitophagy↓ | Facilitates the survival of NP cells by repressing excessive mitophagy, and alleviates IVDD in rats | [62] |
| PPARGC1A↑ | Rat AF cells | SIRT2-PPARGC1A-PRKN -mediated mitophagy↓ |
Protect AF cells against apoptosis under oxidative stress | [87] |
| OA PRKN↓ |
Human chondrocytes | PRKN-mediated mitophagy↓ | Depletion of PRKN increases mitochondrial dysfunction and oxidative stress under the IL1B stimulation | [18] |
| PRKN↑ | Human chondrocytes | PRKN-mediated mitophagy↑ | PRKN overexpression reduces mitochondrial ROS and apoptosis of chondrocyte through the clearance of dysfunctional mitochondria | [18] |
| PHF23↓ | C28/I2 chondrocytes | PRKN-mediated mitophagy↑ | PHF23 deficiency reduces in IL1B-induced ECM degradation. | [91] |
| A2AR↓ | Murine chondrocytes | Mitophagy↓ | Promotes mitochondrial swelling and dysfunction with increased ROS burden and diminished mitophagy | [96] |
| PINK1↓ | Mice cartilage | PINK1-mediated mitophagy↓ | Reduces cartilage damage and pain behaviors in pink1-knockout mice with MIA-induced OA | [88] |
Conclusion and future prospects
Both OA and IVDD are degenerative diseases that strongly correlate with age and the breakdown of cartilaginous tissue. There is a similar pathological process between IVDD and OA in term of mitochondria dysfunction, senescence, cell apoptosis and ECM degradation [1,97] (Figure 2). Particularly, the maintenance of mitochondria homeostasis governs fate and characteristics of functional cells including NP cells, chondrocytes and other cells with rich mitochondria. Therefore, selective autophagy toward impaired mitochondria, mitophagy, is tightly associated with IVDD and OA and could be a critical regulatory mechanism to develop therapeutic approaches for these degenerative diseases. Based on the current evidence, we summarized mechanisms and regulators of mitophagy involved in IVDD and OA, among which PINK1, PRKN, BNIP3, and MFN2 exert their roles in mitophagy and pathological conditions of IVDD or OA. PINK1-PRKN is the most-studied mitophagy pathway and play a prominent role in the activation of mitophagy in NP cells and chondrocytes. As for other pathways of mitophagy, such as FUNDC1, BNIP3L or lipid-mediated mitophagy, these mechanisms have not been reported in IVDD and OA. However, these topics are essential for a comprehensive understanding of mitophagy in degenerative joint diseases and deserve to be investigated. Additionally, some molecules, such as AMPK, SIRT1, SIRT2, SIRT3, FOXO3, NDUFA4L2, A2AR, PHF23, and PPARGC1A, serving as potential upstream regulators of mitophagy, also develop and expand the mechanism of mitophagy and may represent new therapeutic alternatives in IVDD and OA (Figure 3). To better develop pharmaceuticals by targeting these proteins, more functional experiments under various pathological conditions and in vivo experiments should be considered. Fortunately, a number of compounds, such as polydatin, Melatonin, urolithin A and Honokiol have been found to contribute to the regulation of mitophagy, directly or indirectly benefiting the attenuation of IVDD or OA (Table 2). Of note, urolithin A, a natural compound with a great mitophagy-inducing ability, has been demonstrated to extend lifespan and attenuate aging [98]. It could significantly protect against age-related IVDD and sarcopenia [74,98]. Besides, melatonin and metformin, widely used as medicines in clinical practice, are more likely to be applied in the treatment of IVDD or OA. These findings highlight the health benefits of mitophagy enhancers in preservation of degenerative changes with aging. However, the clinical trials of feasibility and efficacy of these medicines are warranted. Moreover, MSCs is considered as a potential approach for cartilage repair and regeneration [99]. However, aging can seriously compromise MSCs’ characteristics and ability, particularly in chondrogenic differentiation [100]. The data from Mazzotti et al. exhibited a reduced efficiency in the chondrogenic differentiation of aged equine synovial fluid MSCs (SfMSCs) compared to young SfMSCs. Interestingly, mitophagy was distinctly observed only in aged SfMSCs, implying activation of mitophagy in aged MSCs [100]. Oxidative stress is closely involved in the fate of MSCs and the pathogenesis of age-related diseases. It has been demonstrated that oxidative stress rapidly improved mitophagy via MAPK8/JNK (mitogen-activated protein kinase 8) signaling at an early stage while limited mitophagy and facilitated apoptosis at a late stage in bone marrow mesenchymal stem cells (BMSCs) [101]. In agreement with previous studies, inhibition of mitophagy by CsA obviously aggravated oxidative stress-caused cell apoptosis [101]. These findings inspire us that targeting the mitophagy pathway in MSCs may improve their capacity of survival and chondrogenic differentiation for cell-based therapies in OA and IVDD.
Figure 2.
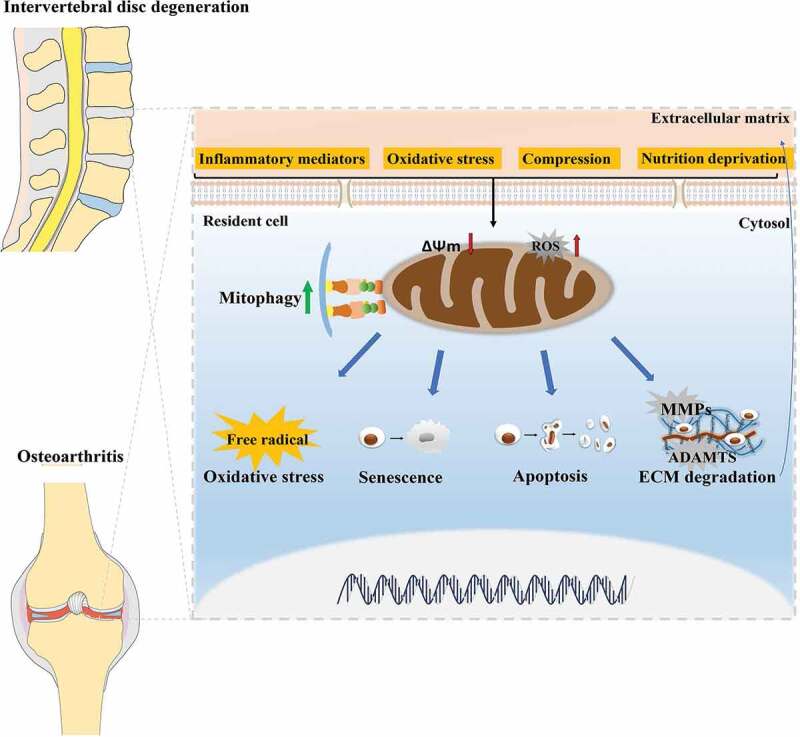
The similar pathological processes between IVDD and OA, and the role of mitophagy in the pathogenesis of IVDD and OA. There exist similar pathological processes between IVDD and OA in term of mitochondria dysfunction, oxidative stress, senescence, cell apoptosis, and ECM degradation of cartilage-resident cells including NP cells and chondrocytes. During the development of IVDD and OA, mitophagy is activated to clear dysfunctional mitochondria and involved in these pathological processes
Figure 3.
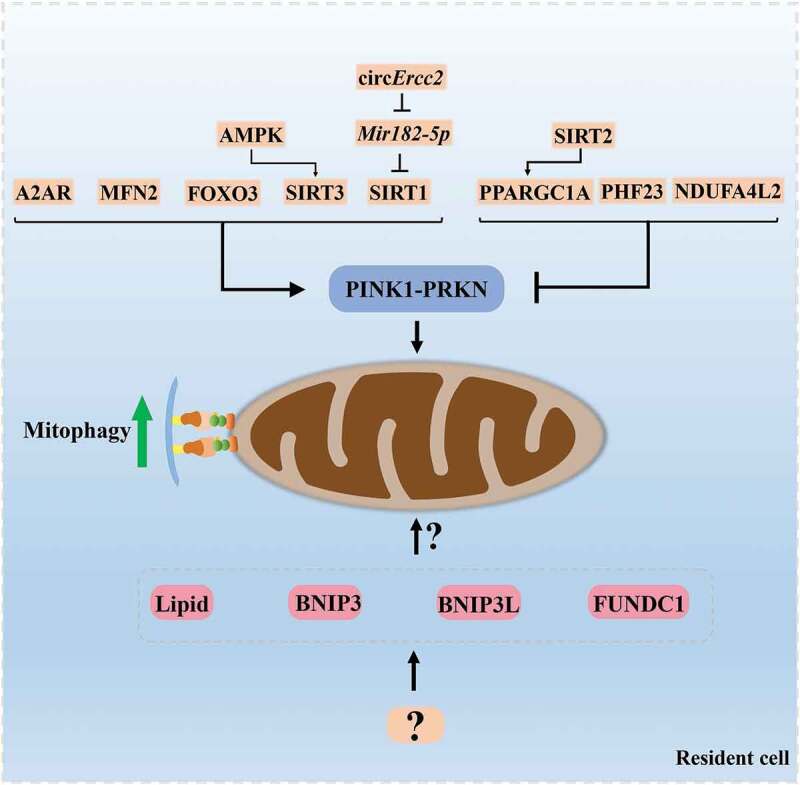
The regulatory molecules of mitophagy in the resident cell of intervertebral disc and articular cartilage. The molecules described in this review including MFN2, AMPK, SIRT1, SIRT2, SIRT3, FOXO3, NDUFA4L2, A2AR, PHF23, PPARGC1A, circErcc2, and Mir182-5p act as potential upstream regulators of PINK1-PRKN-mediated mitophagy and may represent new therapeutic targets in IVDD and OA. Other mechanisms of mitophagy, such as BNIP3, FUNDC1, BNIP3L or lipid-mediated mitophagy, have not been well studied in IVDD and OA. These topics are essential for a comprehensive understanding of mitophagy in degenerative joint diseases and deserve to be investigated
Table 2.
Regulatory compounds of mitophagy in IVDD and OA
| Compounds | Target cell/tissue | Mitophagy pathway↑ | Main findings | Reference |
|---|---|---|---|---|
| Salidroside | Rat NP cells | PRKN-mediated mitophagy↑ | Represses ROS generation and NP cells apoptosis, and protects against IVDD in rats through PRKN. | [17] |
| Polydatin | Human endplate chondrocytes |
PRKN-mediated mitophagy↑ | Inhibits oxidative stress-induced cell damage and alleviate cartilaginous endplate and disc degeneration in puncture-induced rat models. | [64] |
| Melatonin | Rat NP cells | PRKN-mediated mitophagy↑ | Counteracts oxidative stress‐caused ECM degradation and cell apoptosis | [72] |
| Urolithin A | Rat NP cells | AMPK-mediated mitophagy↑ | Protects against mitochondrial dysfunction and cell apoptosis, and alleviates IVDD in rats. | [74] |
| Honokiol | Rat NP cells | AMPK-PPARGC1A-SIRT3-BNIP3 -mediated mitophagy↑ |
Improves anti-oxidation, mitochondrial dynamics, mitophagy and cell survivals in NP cells under oxidative and ameliorated IVDD in rats. | [76] |
| MitoQ | Human NP cells | PRKN-mediated mitophagy↑ | Restores the mitochondrial dynamics balance and maintains mitophagic flux, which rescues mitochondrial dysfunction and NP cell apoptosis under the compression | [79] |
| Metformin | Murine chondrocytes | SIRT3-PINK1-PRKN -mediated mitophagy↑ |
Counteracts IL1B-provoked oxidative stress and imbalance of anabolism and catabolism in chondrocyte. | [90] |
| Spermidine | Human chondrocytes | PRKN-mediated mitophagy↑ | Induces mitophagy, and inhibits oxidative stress-induced cell death through activating autophagy. | [92] |
| Trehalose | Human chondrocytes | BNIP3-mediated mitophagy↑ | Attenuates oxidative stress-induced chondrocyte apoptosis and retard OA in mice. | [94] |
Mitophagy is a common and essential mechanism in physiological and pathological conditions. The regulation and roles of mitophagy exert many similarities in OA, IVDD and other age-related diseases. Thus, those vital regulators and compounds targeting mitophagy are available and promising in the research and clinical practice of degenerative joint diseases and a series of age-related diseases, such as Parkinson disease (PD) and Alzheimer disease (AD). As expected, some compounds mentioned in our review, including urolithin A and spermidine, exert significate protection against degenerative changes in models of AD through the induction of mitophagy [102,103]. Also, it is not surprise to find that the mitophagy-inducing drugs reported in AD or PD including actinonin [102] and resveratrol [104] could have a similar effect and a therapeutic potential in IVDD and OA as well as other age-related diseases. However, how to regulate mitophagy in a proper level is also warranted to be addressed in these fields. For those inherited diseases linked to mitophagy defect, the mutations or deletions of mitophagy-related genes are prone to cause complex multisystem pathologies and neurodegeneration particularly [105]. For example, early onset recessive familial PD could be caused by mutations in the genes PINK1, PRKN OR PARK7 [106]. The deficiency of each of them results in increased sensitivity toward oxidative stress along with mitochondrial and energetic dysfunction [106]. Intriguingly, overexpression of Pink1 and Prkn rescues Park7 deficiency-induced phenotype, suggesting partial redundancies in the mitophagic apparatus [107]. An alternative pathway of mitophagy might be a target to compensate the function of mitophagy. In addition, it could be speculated that the pathologies with mitophagy defects may result from the mild oxidative stress by mitochondrial ROS or oxidized protein aggregates [108]. Therefore, therapies that targeting alterative approach of mitophagy or targeting mitochondrial ROS might bring new hope for treatments of these diseases. Moreover, the association between mitophagy defects and joint degenerative diseases remains unclear and deserves to be explored.
Taken together, mitophagy is closely involved in IVDD and OA. Thus, targeting mitophagy is a promising therapeutic approach. Despite these, there is a critical need to understand how mitophagy develops during the progression of these diseases, and to ascertain the time-window of pharmacological intervention. The development of studies in this area will bring a bright future to the treatment of OA, IVDD, and other age-related diseases.
Funding Statement
This study was supported by the National Natural Science Foundation of China [no. 81874020].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Vergroesen PPA, Kingma I, Emanuel KS, et al. Mechanics and biology in intervertebral disc degeneration: a vicious circle. Osteoarthr Cartilage. 2015;23:1057–1070. [DOI] [PubMed] [Google Scholar]
- [2].Risbud MV, Schoepflin ZR, Mwale F, et al. Defining the phenotype of young healthy nucleus pulposus cells: recommendations of the Spine Research Interest Group at the 2014 annual ORS meeting. J Orthop Res. 2015;33:283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sophia Fox AJ, Bedi A, Rodeo SA.. The basic science of articular cartilage: structure, composition, and function. Sports Health. 2009;1:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Deng ZH, Li YS, Gao X, et al. Bone morphogenetic proteins for articular cartilage regeneration. Osteoarthr Cartilage. 2018;26:1153–1161. [DOI] [PubMed] [Google Scholar]
- [5].Adams MA, Roughley PJ.. What is Intervertebral Disc Degeneration, and What Causes It? Spine (Phila Pa 1976). 2006;31(18):2151–2161. [DOI] [PubMed] [Google Scholar]
- [6].Rahmati M, Nalesso G, Mobasheri A, et al. Aging and osteoarthritis: central role of the extracellular matrix. 2017;20–30. [DOI] [PubMed] [Google Scholar]
- [7].Maher C, Underwood M, Buchbinder R. Non-specific low back pain. Lancet. 2017;389:736–747. [DOI] [PubMed] [Google Scholar]
- [8].Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393:1745–1759. [DOI] [PubMed] [Google Scholar]
- [9].Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12:412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wu PH, Kim HS, Jang I. Intervertebral disc diseases PART 2: a review of the current diagnostic and treatment strategies for intervertebral disc disease. 202021(6):2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wallace DC. A mitochondrial bioenergetic etiology of disease. J Clin Invest. 2013;123:1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Giorgi C, Marchi S, Simoes ICM, et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int Rev Cell Mol Biol. 2018;340:209–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20:1013–1022. [DOI] [PubMed] [Google Scholar]
- [14].Esteban-Martínez L, Boya P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. AUTOPHAGY. 2018;14:915–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Peña-Blanco A, Haschka MD, Jenner A, et al. Drp1 modulates mitochondrial stress responses to mitotic arrest. Cell Death Differ. 2020;27(9):2620–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shaltouki A, Hsieh C, Kim MJ, et al. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. ACTA NEUROPATHOL. 2018;136:607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang Z, Xu T, Chen J, et al. Parkin-mediated mitophagy as a potential therapeutic target for intervertebral disc degeneration. Cell Death Dis. 2018;9:980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ansari MY, Khan NM, Ahmad I, et al. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthr Cartilage. 2018;26(8):1087–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen G, Kroemer G, Kepp O. Mitophagy: an emerging role in aging and age-associated diseases. Front Cell Dev Biol. 2020;8:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Clark IE, Dodson MW, Jiang C, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. NATURE. 2006;441:1162–1166. [DOI] [PubMed] [Google Scholar]
- [21].Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Harper JW, Ordureau A, Heo J. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Bio. 2018;19:93–108. [DOI] [PubMed] [Google Scholar]
- [23].Jin SM, Lazarou M, Wang C, et al. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Meer C, Lorenz H, Weihofen A, et al. The mitochonrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem. 2011;117:856–867. [DOI] [PubMed] [Google Scholar]
- [25].Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. AUTOPHAGY. 2013;9:1758–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kondapalli C, Kazlauskaite A, Zhang N, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shiba-Fukushima K, Imai Y, Yoshida S, et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep-uk. 2012;2:1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ordureau A, Sarraf SA, Duda DM, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56:360–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. [DOI] [PubMed] [Google Scholar]
- [30].Aguirre JD, Dunkerley KM, Mercier P, et al. Structure of phosphorylated UBL domain and insights into PINK1-orchestrated parkin activation. Proc Natl Acad Sci U S A. 2017;114:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bingol B, Tea JS, Phu L, et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. NATURE. 2014;510:370–375. [DOI] [PubMed] [Google Scholar]
- [32].Wang Y, Serricchio M, Jauregui M, et al. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. AUTOPHAGY. 2015;11:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cornelissen T, Haddad D, Wauters F, et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet. 2014;23:5227–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tanaka A, Cleland MM, Xu S, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Safiulina D, Kuum M, Choubey V, et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. Embo J. 2019;38:e99384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shlevkov E, Kramer T, Schapansky J, et al. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc Natl Acad Sci U S A. 2016;113:E6097–E6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sun Y, Vashisht AA, Tchieu J, et al. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem. 2012;287:40652–40660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bernardini JP, Brouwer JM, Tan IK, et al. Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. Embo J. 2019;38:e99916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. NATURE. 2015;524:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wong YC, Holzbaur ELF. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111:E4439–E4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. [DOI] [PubMed] [Google Scholar]
- [42].Narendra D, Tanaka A, Suen D, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [45].Sowter HM, Ratcliffe PJ, Watson P, et al. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669. [PubMed] [Google Scholar]
- [46].Dhingra R, Gang H, Wang Y, et al. Bidirectional regulation of nuclear factor-κB and mammalian target of rapamycin signaling functionally links bnip3 gene repression and cell survival of ventricular myocytes. Circulation. 2013;6:335–343. [DOI] [PubMed] [Google Scholar]
- [47].Chaanine AH, Kohlbrenner E, Gamb SI, et al. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am J Physiol Heart Circ Physiol. 2016;311:H1540–H1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nakamura Y, Kitamura N, Shinogi D, et al. BNIP3 and NIX mediate Mieap-induced accumulation of lysosomal proteins within mitochondria. Plos One. 2012;7:e30767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gugnoni M, Sancisi V, Gandolfi G, et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. ONCOGENE. 2017;36:667–677. [DOI] [PubMed] [Google Scholar]
- [50].Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. Embo Rep. 2010;11:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Aerbajinai W, Giattina M, Lee YT, et al. The proapoptotic factor Nix is coexpressed with Bcl-xL during terminal erythroid differentiation. BLOOD. 2003;102:712–717. [DOI] [PubMed] [Google Scholar]
- [52].Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–19505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen M, Chen Z, Wang Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. AUTOPHAGY. 2016;12:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wu W, Tian W, Hu Z, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. Embo Rep. 2014;15:566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chen G, Han Z, Feng D, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54:362–377. [DOI] [PubMed] [Google Scholar]
- [56].Wu W, Lin C, Wu K, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. Embo J. 2016;35:1368–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Strappazzon F, Nazio F, Corrado M, et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015;22:419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Williams FMK, Bansal AT, van Meurs JB, et al. Novel genetic variants associated with lumbar disc degeneration in northern Europeans: a meta-analysis of 4600 subjects. Ann Rheum Dis. 2013;72:1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Feng C, Yang M, Lan M, et al. ROS: crucial intermediators in the pathogenesis of intervertebral disc degeneration. Oxid Med Cell Longev. 2017;2017:5601593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Xu W, Zheng H, Yang R, et al. Mitochondrial NDUFA4L2 attenuates the apoptosis of nucleus pulposus cells induced by oxidative stress via the inhibition of mitophagy. Exp Mol Med. 2019;51:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wang Y, Shen J, Chen Y, et al. PINK1 protects against oxidative stress induced senescence of human nucleus pulposus cells via regulating mitophagy. Biochem Biophys Res Commun. 2018;504:406–414. [DOI] [PubMed] [Google Scholar]
- [64].Kang L, Liu S, Li J, et al. Parkin and Nrf2 prevent oxidative stress-induced apoptosis in intervertebral endplate chondrocytes via inducing mitophagy and anti-oxidant defenses. Life Sci. 2020;243:117244. [DOI] [PubMed] [Google Scholar]
- [65].Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chen Y, Lin J, Chen J, et al. Mfn2 is involved in intervertebral disc degeneration through autophagy modulation. Osteoarthr Cartilage. 2020;28:363–374. [DOI] [PubMed] [Google Scholar]
- [67].Kristensen LS, Andersen MS, Stagsted LVW, et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20:675–691. [DOI] [PubMed] [Google Scholar]
- [68].Xie L, Huang W, Fang Z, et al. CircERCC2 ameliorated intervertebral disc degeneration by regulating mitophagy and apoptosis through miR-182-5p/SIRT1 axis. Cell Death Dis. 2019;10:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Cipolla-Neto J, Amaral FGD. Melatonin as a Hormone: new Physiological and Clinical Insights. Endocr Rev. 2018;39:990–1028. [DOI] [PubMed] [Google Scholar]
- [70].Kang J, Hong J, Lee S. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J Pineal Res. 2016;60:383–393. [DOI] [PubMed] [Google Scholar]
- [71].Lee JH, Yoon YM, Song K, et al. Melatonin suppresses senescence-derived mitochondrial dysfunction in mesenchymal stem cells via the HSPA1L-mitophagy pathway. Aging Cell. 2020;19:e13111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chen Y, Wu Y, Shi H, et al. Melatonin ameliorates intervertebral disc degeneration via the potential mechanisms of mitophagy induction and apoptosis inhibition. J Cell Mol Med. 2019;23:2136–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cerdá B, Periago P, Espín JC, et al. Identification of Urolithin A as a Metabolite Produced by Human Colon Microflora from Ellagic Acid and Related Compounds. J Agr Food Chem. 2005;53:5571–5576. [DOI] [PubMed] [Google Scholar]
- [74].Lin J, Zhuge J, Zheng X, et al. Urolithin A-induced mitophagy suppresses apoptosis and attenuates intervertebral disc degeneration via the AMPK signaling pathway. Free Radical Bio Med. 2020;150:109–119. [DOI] [PubMed] [Google Scholar]
- [75].Pillai VB, Samant S, Sundaresan NR, et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun. 2015;6:6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang J, Nisar M, Huang C, et al. Small molecule natural compound agonist of SIRT3 as a therapeutic target for the treatment of intervertebral disc degeneration. Exp Mol Med. 2018;50:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Huang D, Peng Y, Li Z, et al. Compression-induced senescence of nucleus pulposus cells by promoting mitophagy activation via the PINK1/PARKIN pathway. J Cell Mol Med. 2020;24:5850–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ni H, Williams JA, Ding W. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kang L, Liu S, Li J, et al. The mitochondria-targeted anti-oxidant MitoQ protects against intervertebral disc degeneration by ameliorating mitochondrial dysfunction and redox imbalance. Cell Proliferat. 2020;53:e12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Liu J, Wang J, Zhou Y. Upregulation of BNIP3 and translocation to mitochondria in nutrition deprivation induced apoptosis in nucleus pulposus cells. Joint Bone Spine. 2012;79:186–191. [DOI] [PubMed] [Google Scholar]
- [81].Liu J, Yuan C, Pu L, et al. Nutrient deprivation induces apoptosis of nucleus pulposus cells via activation of the BNIP3/AIF signalling pathway. Mol Med Rep. 2017;16:7253–7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].He Z, Pu L, Yuan C, et al. Nutrition deficiency promotes apoptosis of cartilage endplate stem cells in a caspase-independent manner partially through upregulating BNIP3. Acta Bioch Bioph Sin. 2017;49:25–32. [DOI] [PubMed] [Google Scholar]
- [83].Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. ONCOGENE. 2008;27(Suppl 1):S2–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jin Q, Li R, Hu N, et al. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018;14:576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39:159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wang Y, Yang Y, Zuo R, et al. FOXO3 protects nucleus pulposus cells against apoptosis under nutrient deficiency via autophagy. Biochem Biophys Res Commun. 2020;524:756–763. [DOI] [PubMed] [Google Scholar]
- [87].Xu W, Yang R, Zheng H, et al. PGC-1α acts as an mediator of Sirtuin2 to protect annulus fibrosus from apoptosis induced by oxidative stress through restraining mitophagy. Int J Biol Macromol. 2019;136:1007–1017. [DOI] [PubMed] [Google Scholar]
- [88].Shin HJ, Park H, Shin N, et al. Pink1-Mediated Chondrocytic Mitophagy Contributes to Cartilage Degeneration in Osteoarthritis. J Clin Med. 2019;8:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kapoor M, Martel-Pelletier J, Lajeunesse D, et al. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2010;7:33. [DOI] [PubMed] [Google Scholar]
- [90].Wang C, Yang Y, Zhang Y, et al. Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci Trends. 2018;12:605–612. [DOI] [PubMed] [Google Scholar]
- [91].Maimaitijuma T, Yu J, Ren Y, et al. PHF23 negatively regulates the autophagy of chondrocytes in osteoarthritis. Life Sci. 2020;253:117750. [DOI] [PubMed] [Google Scholar]
- [92].D’Adamo S, Cetrullo S, Guidotti S, et al. Spermidine rescues the deregulated autophagic response to oxidative stress of osteoarthritic chondrocytes. Free Radical Bio Med. 2020;153:159–172. [DOI] [PubMed] [Google Scholar]
- [93].Hosseinpour-Moghaddam K, Caraglia M, Sahebkar A. Autophagy induction by trehalose: molecular mechanisms and therapeutic impacts. J Cell Physiol. 2018;233:6524–6543. [DOI] [PubMed] [Google Scholar]
- [94].Tang Q, Zheng G, Feng Z, et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis. 2017;8:e3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Corciulo C, Lendhey M, Wilder T, et al. Endogenous adenosine maintains cartilage homeostasis and exogenous adenosine inhibits osteoarthritis progression. Nat Commun. 2017;8:15019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Castro CM, Corciulo C, Solesio ME, et al. Adenosine A2A receptor (A2AR) stimulation enhances mitochondrial metabolism and mitigates reactive oxygen species-mediated mitochondrial injury. FASEB J. 2020;34:5027–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Rustenburg CME, Emanuel KS, Peeters M, et al. Osteoarthritis and intervertebral disc degeneration: quite different, quite similar. JOR spine. 2018;1:e1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Ryu D, Mouchiroud L, Andreux PA, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016;22:879–888. [DOI] [PubMed] [Google Scholar]
- [99].Fitzsimmons REB, Mazurek MS, Soos A, et al. Mesenchymal stromal/stem cells in regenerative medicine and tissue engineering. Stem Cells Int. 2018;2018:8031718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Mazzotti E, Teti G, Falconi M, et al. Age-Related Alterations Affecting the Chondrogenic Differentiation of Synovial Fluid Mesenchymal Stromal Cells in an Equine Model. CELLS-BASEL. 2019;8:1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Fan P, Yu X, Xie X, et al. Mitophagy is a protective response against oxidative damage in bone marrow mesenchymal stem cells. Life Sci. 2019;229:36–45. [DOI] [PubMed] [Google Scholar]
- [102].Fang EF, Hou Y, Palikaras K, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22:401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Madeo F, Eisenberg T, Pietrocola F, et al. Spermidine in health and disease. SCIENCE. 2018;359:n2788. [DOI] [PubMed] [Google Scholar]
- [104].Wang H, Jiang T, Li W, et al. Resveratrol attenuates oxidative damage through activating mitophagy in an in vitro model of Alzheimer’s disease. Toxicol Lett. 2018;282:100–108. [DOI] [PubMed] [Google Scholar]
- [105].Bakula D, Scheibye-Knudsen M. MitophAging: mitophagy in Aging and Disease. Front Cell Dev Biol. 2020;8:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Dodson MW, Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson’s disease. Curr Opin Neurobiol. 2007;17:331–337. [DOI] [PubMed] [Google Scholar]
- [107].Irrcher I, Aleyasin H, Seifert EL, et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet. 2010;19:3734–3746. [DOI] [PubMed] [Google Scholar]
- [108].Bravo-San PJM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. 2017;120:1812–1824. [DOI] [PubMed] [Google Scholar]