

Abstract
In many human cancers, deregulation of the Notch pathway has been shown to play a role in the initiation and maintenance of the neoplastic phenotype. Aberrant Notch activity also plays a central role in the maintenance and survival of cancer stem cells, which underlie metastasis and resistance to therapy. For these reasons, inhibition of Notch signaling has become an exceedingly attractive target for cancer therapeutic development. However, attempts to develop Notch pathway specific drugs have largely failed in the clinic, in part due to intestinal toxicity. Here we report the discovery of NADI-351, the first specific small molecule inhibitor of Notch1 transcriptional complexes. NADI-351 selectively disrupted Notch1 transcription complexes and reduced Notch1 recruitment to target genes. NADI-351 demonstrated robust anti-tumor activity without inducing intestinal toxicity in mouse models, and cancer stem cells were ablated by NADI-351 treatment. Our study demonstrates that NADI-351 is an orally available and potent inhibitor of Notch1-mediated transcription that inhibits tumor growth with low toxicity, providing a potential therapeutic approach for improved cancer treatment.
Introduction
Notch transcription is mediated by the formation of a multiprotein complex termed the Notch Ternary Complex (NTC) (1–4). Notch signaling is initiated at the cell membrane through contact between neighboring cells and results in the cleavage and release of the intracellular domain of the Notch receptor (NotchICD) (5). There are 4 Notch receptors expressed in humans (Notch1-4) (5). For all Notch paralogs, the NotchICD translocates to the nucleus and binds CSL, followed by the recruitment of Mastermind which binds an interface formed by Notch and CSL to form the NTC (4,6–8). The NTC subsequently recruits additional coactivators through the MAML C-terminal domain and drives transcription of target genes including HES, HEY and CCND1. (5–7). Therefore, approaches that disrupt NTC assembly will inhibit NotchICD-directed transcription, and since Notch signaling is uniquely stoichiometric, in contrast to other signal-amplifying cascades, and since transcription is extremely sensitive to even low levels of NotchICD, disruption of this critical node can significantly inhibit downstream effectors of Notch signaling (9–13).
Notch signaling is essential to drive the neoplastic progression of many cancers, including breast, prostate, esophageal adenocarcinoma (EAC), glioblastoma, T cell Acute Lymphoblastic Leukemia (T-ALL), and others (14–19). Because of this broad dependence, disruption of Notch-driven transcription has long been an attractive target in oncology. Inhibiting Notch signaling is challenging due to the paucity of druggable targets, as well as the important tissue-specific pleiotropic roles Notch signaling plays in the homeostasis of a number of adult tissues (20–25). Gamma secretase inhibitors (GSI), which prevent the cleavage of Notch receptors and release of NotchICD (16), have been extensively tested in the clinic for over a decade, originally against Alzheimer’s Disease and then against cancer (26–30). However, GSI’s are not selective for individual Notch paralogs, nor for Notch signaling in general as gamma secretase has >75 cleavage targets (31). Consequently, GSI’s have been clinically hampered by toxicities driven by the essential role of Notch signaling in the intestines, primarily orchestrated by Notch1 and Notch2, and most GSI’s are no longer under clinical development (32–36). Specifically, high-level inhibition of multiple Notch paralogs significantly disrupts the expression of the HES family of transcription factors, which cooperatively control cell fate determination in intestinal crypts, leading to differentiation of crypt precursor cells toward secretory lineages such as goblet cells (20–22, 37). This disruption unbalances the secretory/absorptive homeostasis of the intestines and is responsible for treatment emergent GI toxicity.
Approaches with greater Notch paralog selectivity have been shown to avoid severe GI toxicity in mice (38). Antibodies to the Notch1 and Notch2 receptors individually avoid intestinal toxicity over 12 days, but cotreatment results in goblet cell metaplasia, underlining the need for paralog selectivity. However, Notch1 mAbs failed in phase Ib clinical trials after proving intolerable in colorectal cancer patients cotreated with trifluridine/tipiracil (39). Thus, there exists a clear unmet need for selective Notch therapeutics which can maintain a clear therapeutic window between targeting individual paralogs required for oncogenic growth while avoiding toxicity in the intestines and other Notch-regulated tissues.
Deregulation of the Notch signaling pathway plays a role in the maintenance and survival of cancer stem cells (CSCs) which likely underlie resistance to chemotherapy, making it an exceedingly attractive target for anti-CSC therapies (40). Current approaches to inhibit the Notch signaling pathway include small molecules that target proteins upstream of the NTC. In spite of the critical role played by the NTC in cancer (40–41), there are no small molecule inhibitors in the clinic that successfully target the NTC and selectively inhibit stem-like Notch-driven tumors. Therefore, the full range of potential targets in the pathway have not been exploited.
We discovered the first potent, Notch1-selective inhibitor that dramatically attenuates Notch-dependent tumor cell growth in a variety of cancers, termed NADI-351. NADI-351 acts by selectively ablating the tumoral cancer stem cell (CSC) population by inhibiting Notch1-directed transcription.
Material and Methods
Cell lines
OE33 human esophageal adenocarcinoma cell line was obtained from the European Collection of Cell Culture. MDA-MB-231 (human triple negative breast cancer) was obtained from Dr. Caroline Briegel at the University of Miami, Miller School of Medicine (Miami, FL). Het1A (human immortalized esophageal epithelial cell line), MCF10A (human breast epithelial cells), PC-3 (human prostate cancer), MCF-7, and T47D (ER+ luminal breast cancer) were obtained from ATCC. All cell lines were tested monthly for mycoplasma contamination and propagated in growth media as specified by the provider (BI, 20-700-20). Cell lines, excluding OE19 were obtained between 2017 and 2019 and authenticated by ATCC (cell line authentication profiling utilizing short tandem repeat (STR) profiling). The OE19 cell line was obtained from the Leibniz Institute DSMZ in 2020, authenticated by STR profiling, and monitored monthly for mycoplasma contamination. Number of passages of cells between collection or thawing and use in the described experiments were between P2-P10.
Notch complex assembly assay (Alpha screen technology)
Recombinant proteins were expressed using baculovirus expression vectors in SF21 cells and purified as described previously (4). Notch complex assembly experiments were performed as described previously (40). Data analysis was performed using GraphPad Prism software (Version 8.4.3).
Real-time qPCR analysis
Cells (1 × 105) were seeded in cell culture dishes (100 mm) in RPMI 1640 or DMEM medium containing 10% FCS with antibiotics. After 24 h of incubation, cells were exposed to NADI-351 in a time-dependent manner. At the end of the treatment, total mRNA was isolated, and cDNA was synthesized according to the manufacturer’s protocol (4368814; Life Technologies). RT-qPCR analysis was performed using TaqMan probes according to manufacturer’s instructions (4324018; Applied Biosystems). Gene expression was normalized to HPRT or TBP gene. Data analysis was performed using GraphPad Prism software (Version 8.4.3).
Notch1-3 Reporter Assay
293A cells expressing 1) pCMV-Tet-On 3G, and 2) pCSL-RElement-Luc and one of the following: A) pLV[Tet]-Puro-TRE3G>Notch1ICD, or B) pLV[Tet]-Puro-TRE3G>Notch2ICD, or C) pLV[Tet]-Puro-TRE3G>Notch3ICD, respectively, were used. When doxycycline is added, the Tet-ON gene activates expression of hNotch1ICD, 2ICD, or 3ICD, which together with endogenous NTC components binds to CSL responsive elements (pCSL-RElement-Luc) and expresses luciferase. 1 × 104 cells are plated in 100 μL (96-well format). 24h later, compounds (10 mM stock in DMSO) are diluted in DMSO to 200X, then added (5 ul into 1 mL) to cell culture media containing 50 ng/mL Dox. This is then added 1:1 to cells. Final DMSO = 0.25%, Dox = 25 ng/mL. After 24h, media is discarded, and cells are lysed in passive lysis buffer (Promega). Cells are rocked at room temperature for 15 min and then lysate is divided for luciferase assay (Luciferase Assay System, Promega) and cell viability (CellTiter Glo 2.0, Promega). Raw luciferase is normalized to cell viability, and then scaled to DMSO wells. Results are analyzed and IC50’s are determined in GraphPad by nonlinear regression curve fitting (4 parameter) of dose response curves.
CSL-DNA affinity pulldown assay (DAP)
DNA pulldown was performed as previously described (42). Streptavidin agarose beads (Pierce) were incubated with previously annealed 47 mer biotinylated dsDNA containing two high-affinity CSL binding sites facing forward (2× CSL binding DNA) or 2 mutated CSL binding sites (43).
Chromatin immunoprecipitation ChIP assay
MDA-MB-231 cells, OE33 cells and PC-3 cells were cross-linked with 1% formaldehyde for 10 min at 37°C and cross-linking was quenched by adding glycine to a final concentration of 0.125 M. Nuclear pellet was prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermofisher). Nuclear pellet was digested using Micrococcal Nuclease (NEB) to yield chromatin fragments of approximately 300 to 800 bp and lysed in ChIP buffer (1% Triton, 2 mM EDTA, 150 mM NaCl, and 20 mM Tris-HCl pH 8 and 0.01% SDS) containing a protease inhibitor cocktail (Roche). Lysates were immunoprecipitated with α-Notch1, α-Notch2 (Bethyl Laboratories), α-Notch3, α-Notch4 and α-Maml1 (CST) and α-IgG (Abcam) antibodies and were reverse cross-linked overnight at 65°C in 200 mM NaCl and proteinase K. DNA was purified using the PCR Purification Kit (Qiagen) and HES1 promoter were amplified by qPCR. Primer sequences:
Forward 5’: CGTGTCTCCTCCTCCCATT
Reverse 5’: GGGGGATTCCGCTGTTAT
Western blotting
Nuclear protein analysis was performed as previously described (44) using anti-Notch1val1744 (Cell Signaling Technology, 4147S), anti-Notch2 (Cell Signaling Technology, D76A6), anti-Notch3 (Cell Signaling Technology, D11B8), anti-Notch4 (Cell Signaling Technology, L5C5), anti-MAML1 (Cell Signaling Technology, D3K7B), anti-MAML2 (Cell Signaling Technology, D41E6), anti-MAML3 (Bethyl Laboratories, A300–6841), and anti-RBPSUH (CSL, Cell Signaling Technology, D104A).
Animal experiments
PDX cancer models and xenografts were established as described previously (45). We used the EAC74 PDX from our library of surgically resected patient tumors in order to evaluate the effect of NADI-351 on tumor growth. Six-week-old nude female mice were purchased from Charles River Laboratories. When the tumor size reached 200 mm3, the mice were treated with daily dose of 20 and 30 mg/kg for intraperitoneal injection (i.p.) and 40 mg/kg for oral gavage (p.o.), 6 mice per treatment/6 mice per negative control. Vehicle used for p.o.: 3.25% NMP, 50% PEG-400, 46.75% normal saline. Vehicle used for i.p.: DMSO. The stop/end point criteria are based on the days it takes for tumors to reach 2000 mm3 according to what is approved in our animal protocol. Tumor volume was measured by the formula: volume = (S × S × L)/2. Animal experiments were reviewed and approved by the University of Miami Institutional Animal Care and Use Committee (Miami, FL, USA)
Tissue sample preparation, PAS staining immunohistochemistry.
Immunohistochemical analysis of small intestine from C57BL/6 or Nude mice injected intraperitoneally with different concentrations of NADI-351 and DZB, daily for 5 or 30 days. Tissue sample preparation and PAS staining immunohistochemistry were performed as previously described (22). ALDH1A and ki67 stains were performed using anti-ALDH1A (Abcam, ab52492) and anti-ki67 (Leica biosystems, PA0118). ALDH1A scoring: negative (0), weak (1+), moderate (2+), strong (3+). Ki67 scoring: count % of positive cells in one 40x field with highest positive.
Colony Formation Assay and Cell Viability Assays
Cells were cultured at low density under treatment, and then colonies were stained with 0.01% crystal violet and counted. The cells were measured using the Cell Titer-Glo assay (G7572; Promega) for Cell Viability Assays.
Tumor Sphere Formation Assay
To obtain tumor spheres, cells were cultured in DMEM/F12 with 2% B-27 serum-free supplement (17504–044; Invitrogen), 20 ng/ml epidermal growth factor (EGF; PHG0311L; Invitrogen), and 20 ng/ml basic fibroblastic growth factor (FGF; PHG0266; Invitrogen) for 14 days to select for CSCs and early progenitor cells. Resulting tumor spheres were examined and counted under the microscope.
Flow Cytometric Analysis of Aldehyde Dehydrogenase (ALDH) and Notch1
Cells lines or single cells derived from tumors were stained using ALDEFLUOR kit (Stem Cell Tech) or a specific monoclonal BB515 anti-human Notch1 antibody (BD Horizon, 564781), following the manufacturer’s instructions and were analyzed by flow cytometry, as described previously (46). Singles cells were obtained from tumors following the manufacturer’s instructions (MACS, Tumor Dissociation kit, 130-095-929).
Nuclear DNA fragmentation
Tissue sample preparation and TUNEL staining immunofluorescence were performed following the manufacturer’s instructions (Promega, DeadEnd™ Fluorometric TUNEL System, G3250). TUNEL/DAPI staining was analyzed by Sp5 inverted confocal microscope (Leica Biosystems).
CEREP Safety44 panel
NADI-351 was assayed by Eurofins Discovery Services (CEREP) against the Safety44 panel (38 radioligand binding assays + 6 activity assays) at 10 μM, (n = 2). Results were compiled and analyzed in GraphPad Prism.
Wild type Kinase Assay
NADI-351 (10 μM) was assayed by Reaction Biology Corp. to determine changes in activity of 372 wild type human kinases, (n=2), [ATP] = 10 μM. Positive control compounds (staurosporine, or alternate kinase inhibitors, depending on kinase) were used in 10-concentration dose response assays to verify kinase activity. Results were analyzed in GraphPad Prism.
Computational methods
Modeling of the Notch1/CSL active site and small molecule docking experiments were performed using MOE (Molecular Operating Environment, 2019.01; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2020.
Statistics
P value was calculated using chi-square in contingency table. Data are presented as mean ± SD and were analyzed by 2-tailed Student’s t test. A P value of less than 0.05 was considered significant. In all other cases, statistical significance was determined by Student’s T test. P value < 0.05 was considered statistically significant.
Results
Discovery and characterization of a Notch1-selective small molecule inhibitor
Previously, we reported the first small molecule inhibitor of the NTC, IMR-1 (43). This small molecule binds to a pocket formed by NotchICD and CSL and prevents Mastermind from binding and forming a stable NTC. Although this demonstrated proof of principle that a small molecule could disrupt the formation of the NTC, IMR-1 is a pan-Notch inhibitor and lacked the necessary potency to advance. Therefore, a molecular modeling-focused SAR campaign was used to improve potency, Notch paralog specificity and drug-like properties and resulted in the identification of NADI-351 (Fig. S1A). Molecular modeling of NADI-351 in the context of the NTC suggests that NADI-351 interacts with both Notch1 and CSL residues (Fig. 1A–B and Fig. S1B). Activity of NADI-351 against an NTC AlphaScreen assay revealed that NADI-351 is 15 times more potent than IMR-1 (Fig 1C). NADI-130, an inactive analog of NADI-351 (Fig. S1C), displayed no activity and served as a negative control in the AlphaScreen. An inducible Notch1ICD-responsive luciferase reporter was employed to further analyze the effect of NADI-351 on Notch transcription in cells and, consistent with the AlphaScreen, there was a significant increase in potency (NADI-351 IC50 = 8.8 μM vs > 40 μM for IMR-1) (Fig. 1D).
Figure 1. NADI-351 displays increased potency against Notch1 over IMR-1.
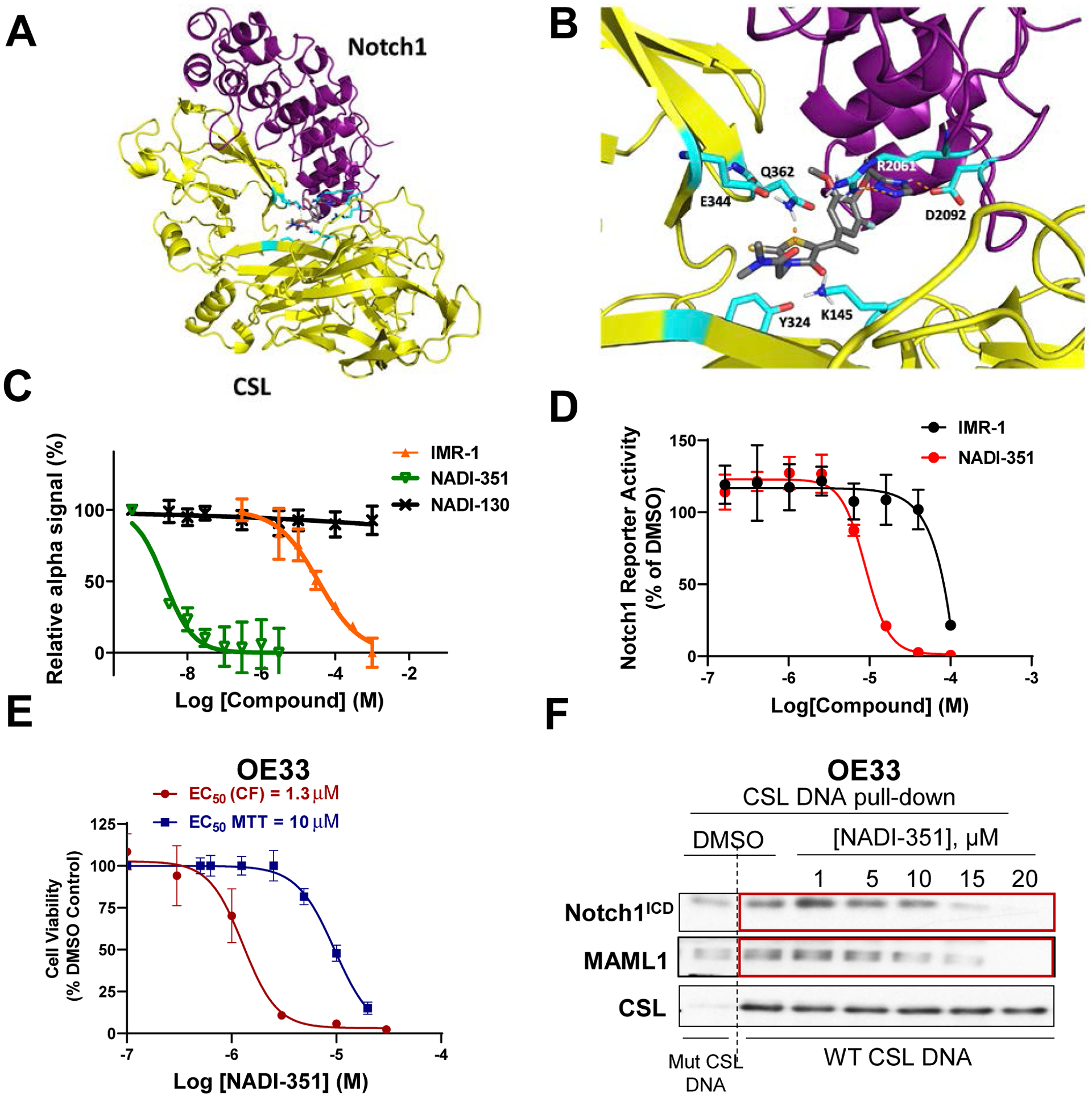
(A) The crystal structure of the ternary complex (Notch1/CSL/MAML1/DNA – PDB code 2F8X) was edited to remove DNA and MAML1 followed by structure preparation. Potential small molecule binding sites were mapped, and docking was performed with NADI-351. (B) Binding site residues are highlighted in cyan including the salt bridge between Arg2061 and Gln362. NADI-351 binds in the Arg-rich cavity between Notch1 and CSL and forms hydrogen-bonding interactions with Arg2061, Asp2092, and Gln362. (C) Inhibition of NTC assembly and IC50 determination for compounds IMR-1, NADI-351, and NADI-130 by NTC AlphaScreen assay. NADI-351 is 15 times more potent than IMR-1. (D) Effect of IMR-1 and NADI-351 against an inducible Notch1ICD-driven luciferase reporter (NADI-351 IC50 = 8.8 μM vs > 40 μM for IMR-1). (E) Dose-response curves of cell viability from OE33 colonies treated with NADI-351 every 48h for 2 weeks vs. single dose MTT assay on OE33 cells treated for 72 h. Cell viability assays were normalized to the control (DMSO). (F) CSL/Notch1ICD pulldown on DNA using OE33 nuclear extract after 48 h of NADI-351 treatment (10 μM).
Our previous work demonstrated the dependence of esophageal adenocarcinoma (EAC) on Notch signaling (14). NADI-351 inhibited OE33 cell growth in single dose MTT assays (EC50= 10 μM) and, more potently, in multi-dose colony formation assays (EC50 = 1.3 μM) (Fig. 1E). Similar results were observed in other reported Notch-dependent cell lines, including MDA-MB-231 cells (human triple-negative breast cancer) and PC-3 cells (human prostate cancer) (Fig. S2A–B). As previously reported, DAPT (a GSI) and IMR-1 inhibited OE33 colony formation and other Notch-dependent cell lines, with EC50 values between 10–15 μM (43), underscoring the higher comparative potency of NADI-351 against Notch-dependent cell growth. No significant effect was observed in Notch-independent cell lines MCF-7 and T47D (ER+ Luminal breast cancer) or on normal esophageal cells (Het1A) or breast epithelial cells (MCF10A) (Fig. S2C–F). To demonstrate NADI-351 can disrupt the DNA-bound NTC in cells, we utilized a CSL-DNA affinity pulldown assay (DAP). When OE33 cells are treated with NADI-351, we observed a dose-dependent concomitant inhibition of Notch1ICD and Maml1 binding to CSL on DNA, without a loss of CSL binding. This result demonstrates NADI-351 inhibits Notch1-driven transcription by disruption of the NTC in cells (Fig. 1F).
NADI-351 selectively disrupts Notch1 transcription complexes and prevents binding to the HES1 promoter.
Since OE33 cells only express Notch 1–3, we next sought to confirm the selectivity of NTC inhibition by NADI-351. In order to fully validate the proposed mechanism of action, we employed the triple negative breast cancer (TNBC) cell line MDA-MB-231, which are Notch-dependent and express Notch1-4. MDA-MB-231 cells were treated with the determined single-dose MTT EC50 value (10 μM, Fig. S2A) and were harvested at several time-points for NTC DAP assays. NADI-351 selectively inhibited Notch1 and MAML1 binding to DNA-bound CSL, whereas no effect was observed in Notch 2–4 or CSL binding to DNA (Fig. 2A and Fig. S3A). No reduction in binding of either MAML2 or MAML3 was observed, and no other NTC proteins displayed a similar kinetic effect over time or magnitude of inhibition as displayed by Notch1 and MAML1 (Fig. 2A). RT-qPCR analysis of MDA-MB-231 cells indicated that NADI-351 treatment significantly decreased transcription of Notch target genes HES1 and HES5 (Fig. 2B). Furthermore, chromatin immunoprecipitation (ChIP) in MDA-MB-231 cells indicated that NADI-351 selectively inhibits the recruitment of Notch1 (Fig. 2C) and MAML1 (Fig. 2D) to the HES1 promoter. We also observed that only Notch1 was inhibited from binding at the HES1 promoter with NADI-351 treatment, while Notch2-4 had no significant reduction in binding (Fig. 2E), thereby confirming DAP results. We then evaluated the selectivity index for Notch1 NTCs using higher concentrations of NADI-351 (1, 2, and 3X the EC50) in NTC DAP assays. This analysis revealed that although cellular complexes comprised of Notch1ICD and MAML1 were dramatically disrupted (Fig. 2F and Fig. S3B–C), Notch 2–4 complexes remained largely intact, therefore demonstrating exquisite selectivity for Notch1 NTCs. Similar results were observed in PC-3 and OE33 cell lines (Fig. S3D–E and Fig. S4A–F). Consistent with DAP assays, selectivity for Notch1 over Notch2 and Notch3 was observed using inducible Notch1, 2, or 3-driven luciferase reporter assays (Fig. 2G).
Figure 2. NADI-351 inhibits recruitment of Notch1 to the NTC and to the HES1 promoter.
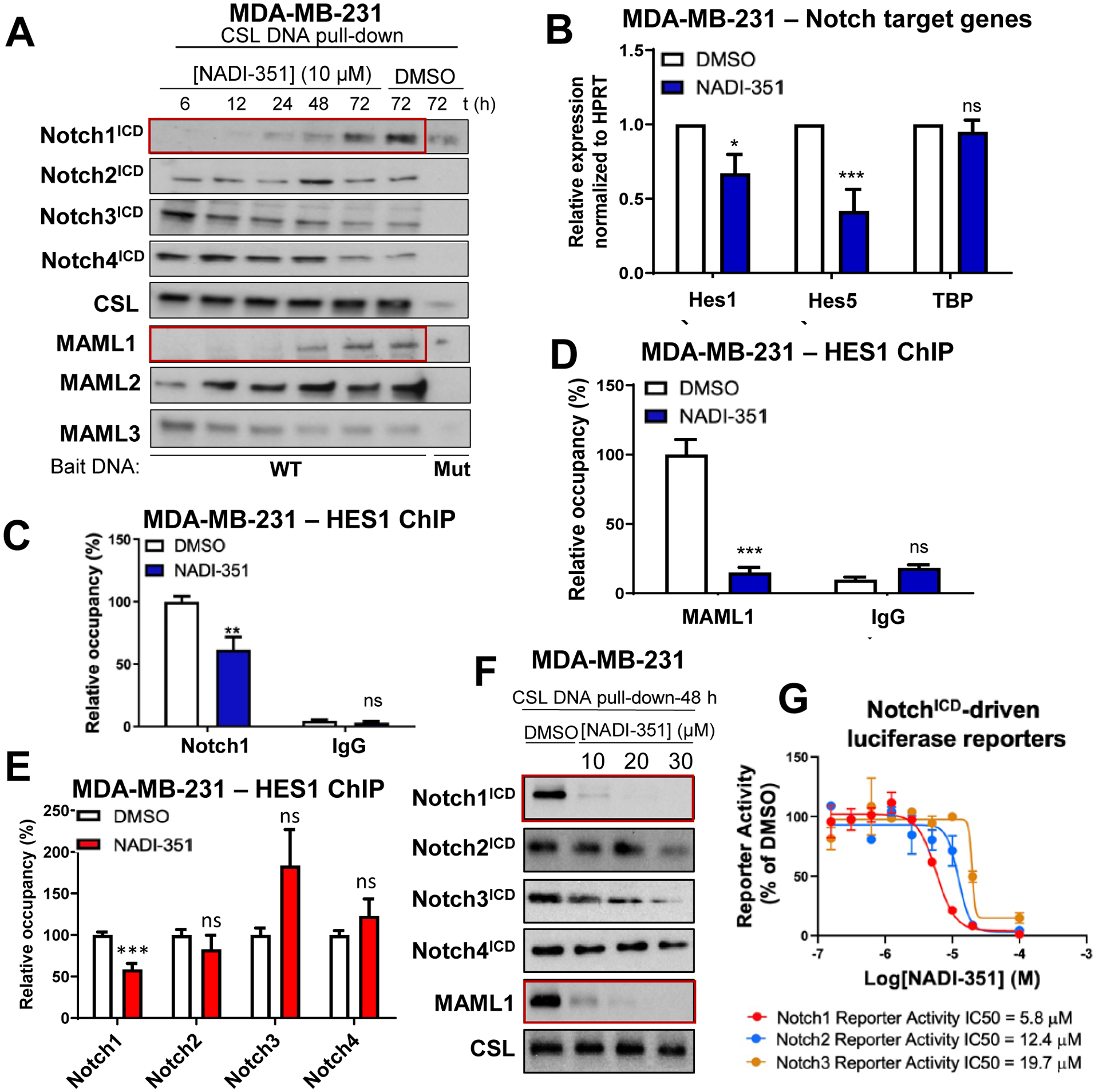
(A) CSL-dependent affinity pulldown indicates NADI-351 (10 μM) selectively inhibits Notch1ICD and MAML1 NTC assembly in MDA-MB-231 in a time-dependent manner (red boxes). (B) NADI-351 (10 μM) inhibits Notch target genes (HES1, HES5 and HEY1) expression in MDA-MB-231 cells through RT-qPCR at 6 h of treatment (n = 3). NADI-351 (10 μM) selectively inhibits recruitment of Notch1ICD (C,E) and MAML1 (D) to the NTC and to the HES1 promoter in MDA-MB-231 cells by ChIP at 6 h of treatment (n = 6). (F) CSL-dependent affinity pulldown indicates that high concentrations of NADI-351 selectively inhibits the recruitment of Notch1ICD and MAML1 to DNA-bound CSL at 48h in MDA-MB-231. (G) Effect of NADI-351 against individual inducible NotchICD-driven luciferase reporters (Notch1-3). Error bars are representative of at least three independent experiments, and values indicate mean ± SD [(B), (C), (D) and (E)]. p values ≤ 0.05 are considered statistically significant and indicated by an asterisk. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ns (no significant difference).
NADI-351 inhibits Notch-dependent tumor growth without induction of goblet cell metaplasia, consistent with selective Notch1 inhibition.
To evaluate the effect of NADI-351 on tumor growth we employed several Notch-dependent cell lines and patient-derived xenograft models. MDA-MB-231, PC-3, and OE19 cell line-derived tumors were formed and grown in nude mice to approximately 200 mm3 prior to treatment. Both oral (p.o) or intraperitoneal (i.p.) treatment routes resulted in significant inhibition of tumor growth in all xenograft models and had no significant effect on mouse weight and no observable effects on overall appearance (Fig. 3A & S5A–I). We further investigated effects on the intestines; other Notch inhibitors (such as GSIs) have demonstrated toxicity (goblet cell metaplasia) due to inhibition of transcription driven by multiple NotchICD’s (20–22). In contrast to GSI treatment (DBZ, 10 mg/kg), we found no evidence of GI toxicity after 5 daily treatments with NADI-351, even at the highest dose tested (40 mg/kg) (Fig 3B). NADI-351 treated mice displayed intact crypts and normal intestinal architecture with no evidence of goblet cell metaplasia (PAS staining), comparable to the vehicle group (DMSO). To assess the activity of NADI-351 on a more clinically relevant model, we utilized an EAC patient-derived xenograft model (EAC47 PDX) (14). NADI-351 significantly inhibited tumor growth in the EAC model (Fig. 3C and S5J) with no effect on weight, compared to vehicle (Fig. S5K). We examined tumors from NADI-351-treated EAC47 PDX mice and found that apoptosis was increased, while the CSC marker ALDH1A and Ki67 were lower in comparison to vehicle-treated controls (Fig. 3D & S6A–D). Examination of small intestines from these animals further demonstrated that 30 days of treatment (30 mg/kg) with NADI-351 produced no effect on villi architecture or proliferation and did not drive goblet cell metaplasia (Fig. 3E). To further assess potential off-target interactions, NADI-351 was assayed against a panel of 44 common targets responsible for toxicity in human trials (Fig. S7A). No targets were bound or inhibited to a significant degree by NADI-351 (10 μM). Profiling of 372 wild type human kinases similarly showed no significant off-target activity by NADI-351 (10 μM) (Fig. S7B). These results indicate a clear therapeutic window for the use of NADI-351 as a Notch inhibitor without the dose-limiting toxicity of previous approaches and hint at the mechanisms underlying in vivo activity.
Figure 3. NADI-351 inhibits tumor growth in Notch-dependent cancers and does not exert gastrointestinal toxicity associated with goblet cell metaplasia.
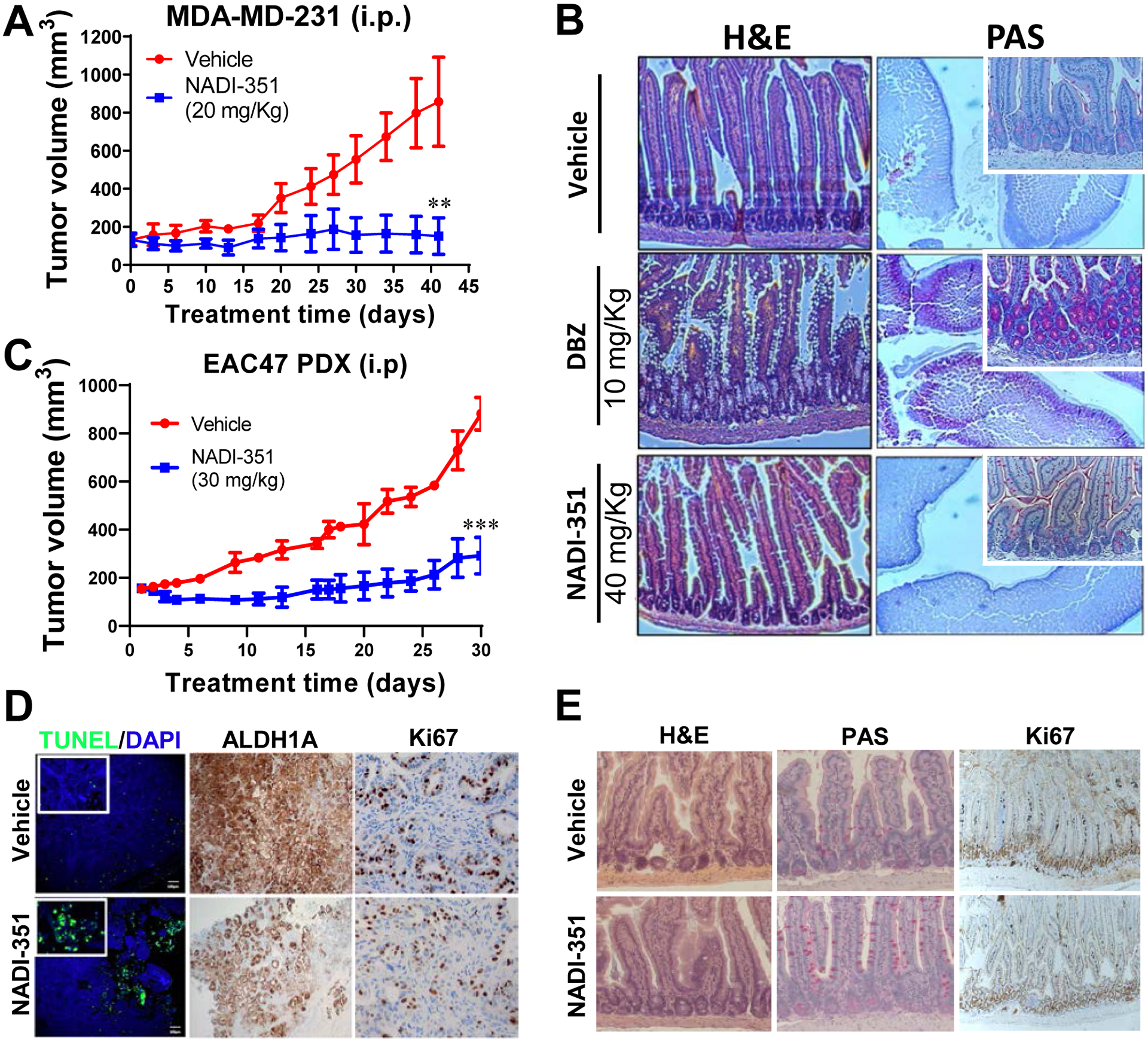
(A) Tumor growth of MDA-MB-231 cell line-derived xenografts are inhibited upon daily treatment with NADI-351 (20 mg/kg intraperitoneal injection i.p. routes, respectively) in nude mice (n=6). (B) Immunohistochemical analysis of small intestines from C57BL/6 mice injected intraperitoneally with NADI-351, daily for 5 days. NADI-351 does not exert gastrointestinal toxicity, as evident by intact crypts (PAS staining - larger picture is 2X, insert is 20X magnification). The gamma secretase DBZ (10 mg/kg) was used as a positive control for induction of goblet cell metaplasia (n=3). (C) Patient derived EAC xenografts (EAC47) are similarly sensitive to NADI-351 treatment, demonstrating significant abrogation of tumor growth, (30 mg/kg) in nude mice (n=6). (D) NADI-351 treated EAC47 tumors (C) displayed increased apoptosis, decreased ALDH1A expression, and decreased proliferation. (E) NADI-351 does not exert gastrointestinal toxicity or change intestinal cell proliferation after daily treatment in EAC47-PDX (C). Error bars are representative of independent experiments, and values indicate mean ± SD [(A-D) and (F-G)]. p values ≤ 0.05 are considered statistically significant and indicated by an asterisk. **P ≤ 0.01; ***P ≤ 0.001.
Cancer stem-like populations exhibit a profound sensitivity to NADI-351.
It is thought that cancer stem cell (CSC) populations drive tumor formation and are responsible for much of the neoplastic phenotype (40–41). In addition, many studies have attributed progression, metastasis and resistance to therapy to CSCs (47). Since NADI-351 had significantly increased potency against EAC and TNBC clonogenic growth compared to general proliferation assays and reduced ALDH1A in EAC47 tumors, we reasoned that NADI-351 might exert its effects primarily through ablation of CSC populations. In many tumor cells types, certain cell populations have been identified as CSCs (43). In the TNBC cell line MDA-MB-231, the primary CSC population is marked by CD44+/CD24+/low while the CD44+/CD24− does not possess CSC properties (48). Sorting (Fig. S8A–B) and treatment of these populations revealed that NADI-351 more potently inhibited CD44+/CD24+/low populations with an EC50 = 2.3 μM vs 10 μM for CD44+/CD24− (Fig. S8C). Enriched CD44+/CD24+/low cells had higher Notch activity than CD44+/CD24− (24) and we found that NADI-351 inhibited the formation of primary secondary spheres derived from enriched MDA-MB-231 CD44+/CD24+/low in a dose-dependent manner unlike CD44+/CD24− spheres (Fig. S8D–E).
EAC cell lines are Notch-dependent and display characteristics of CSC according to their expression of ALDH1A (14), making them a good model to study the effect of NADI-351 on CSCs. We first examined the effect of NADI-351 on colony formation of OE33 cells sorted by ALDH expression (Fig. 4A, S9A–B). NADI-351 more potently inhibited ALDH+ OE33 colony (EC50 = 1 μM) compared with ALDH− colonies (EC50 = 3.4 μM, Fig. S9C). Further RT-qPCR analysis demonstrated that NADI-351 selectively inhibited Notch target gene expression in OE33 ALDH+ cells only (Fig. 4B/C). Since cell proliferation is well correlated to the regulation of cell cycle progression, we evaluated the effect of NADI-351 on the cell cycle of ALDH+ OE33 cells. We observed that NADI-351 modulates an S-phase arrest in ALDH+ cells after 24 h of treatment (Fig. S9D). We also observed that ALDH+ populations experienced a significant, dose-dependent increase in apoptosis following NADI-351 treatment (Fig. S9E). Together, these data indicate that cell cycle arrest and activation of apoptosis are a direct consequence of the inhibition of Notch-dependent transcriptional regulation by NADI-351 in EAC CSC populations.
Figure 4. NADI-351 selectively inhibits stem-like EAC cell populations.
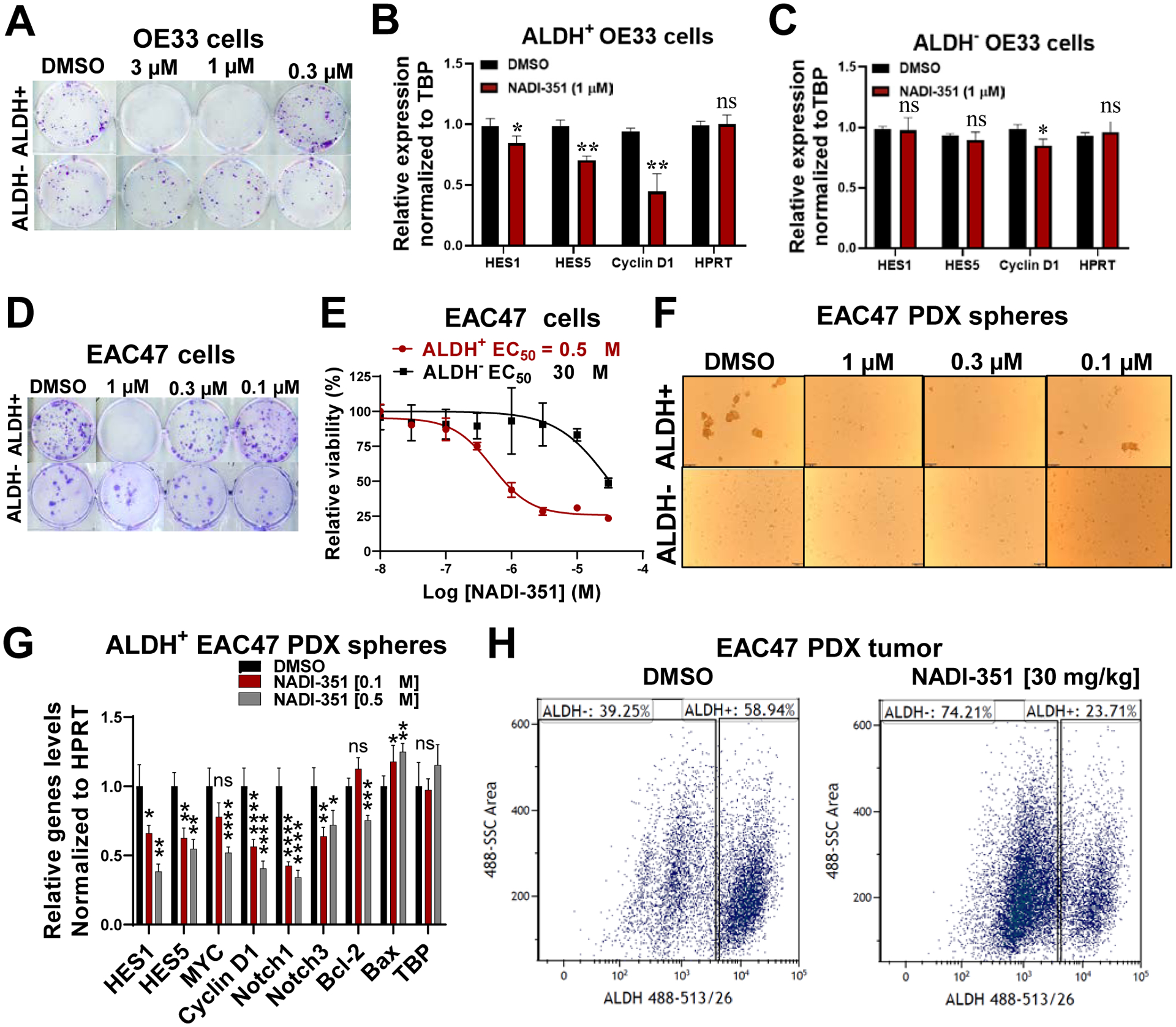
(A) Colony formation assays for NADI-351 were performed using sorted OE33 ALDH+ and ALDH− cells (n=3). (B & C) Relative Notch target gene expression in sorted OE33 ALDH+ and ALDH− cells exposed to 1 μM of NADI-351 for 24 h (RT-qPCR) (n=3). mRNA levels were normalized to HPRT expression. (D-F) EAC47 PDX tumors (n=3) were FACS-sorted to isolate ALDH+ and ALDH− cells. The effect of NADI-351 on colony formation (D), cell viability (E, MTT assay), and tumor spheres (F) were determined for each cell population. (G) Relative expression of Notch and apoptosis related genes was determined in sorted ALDH+ tumor-spheres from EAC47 tumors after treatment with 0.1 and 0.5 μM of NADI-351 for 24 h (RT-qPCR) (n=3). (H) FACS analysis of treated EAC47 PDX tumor cells (30 mg/kg NADI-351, 14 days) stained with ALDEFLUOR kit (n=3). Error bars are representative of three independent experiments and indicate mean ± SD. p values ≤ 0.05 are considered statistically significant and indicated by asterisk(s). *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Consistent with colony formation results in ALDH-sorted OE33 populations, NADI-351 more potently inhibited EAC47 PDX ALDH+ colony formation and cell viability compared with ALDH− colonies (Fig. 4D–E and Fig. S10A–B). NADI-351 selectivity against ALDH-sorted EAC47 subpopulations was further examined in tumorspheres. Intriguingly, ALDH− EAC47 cells did not form tumorspheres suggesting CSC activity may be crucial for tumorsphere formation. In contrast, ALDH+ EAC47 cells readily formed tumorspheres which were potently inhibited by NADI-351 (EC50 = 110 nM, Fig. 4F, S10C). RT-qPCR analysis of ALDH+ EAC47 cells revealed NADI-351 treatment significantly decreased Notch target genes and the anti-apoptosis regulator Bcl-2 using sub-micromolar concentrations (Fig. 4G).
To determine whether CSC ablation occurs in vivo in response to NADI-351 treatment, mice bearing EAC47 PDX tumors were treated with vehicle (DMSO) or 30 mg/kg NADI-351 for 14 days after which tumors were excised and isolated tumor cells were analyzed for ALDH expression by FACS analysis. Indeed, in contrast to vehicle, NADI-351 ablated ALDH expressing cells in vivo in EAC47 PDX tumors (Fig. 4H). Together, these results confirm NADI-351 inhibits Notch transcription and target CSCs in Notch-driven EAC tumors.
Notch1-driven tumor cell population is the target of NADI-351 and underlies the dramatic anti-tumor effects of NADI-351.
As Notch plays a critical role in CSC populations (14), we sought to further determine the specificity of NADI-351 inhibition in cells sorted on the basis on Notch1 expression. OE33 cells were sorted by Notch1 expression (Notch1+ and Notch1−) (Fig. 5A–B), using a specific monoclonal anti-Notch1 antibody. We observed that treatment with NADI-351 significantly inhibited colony formation in the Notch1+ population in contrast to Notch1− colonies, which were not inhibited. (Fig. 5C & S10D). Furthermore, NADI-351 selectively inhibited the transcription of Notch target genes in a dose-dependent manner only in Notch1+ cells and caused no significant changes in the transcription of target genes in Notch1− cells (Fig. 5D). These results demonstrate that Notch1 is crucial to the mechanism and activity of NADI-351. To confirm that NADI-351 targets Notch1 in tumors, we treated nude mice bearing EAC47 PDX tumors for 14 days with NADI-351 after which tumors were harvested and cells were analyzed for Notch1 expression by FACS analysis. Vehicle treated tumors have high numbers of Notch1 expressing cells, but in contrast, NADI-351 treatment dramatically reduces Notch1-expressing cells in tumors (Fig. 5E). Further RT-qPCR analysis of these ex vivo tumor cells demonstrated NADI-351 treatment inhibited Notch target gene expression and induced a pro-apoptotic transcriptional program (Fig. 5F) consisted with results obtained from ALDH+ EAC47 PDX spheres (Fig. 4G) These results confirm NADI-351’s selective anti-Notch1 activity in tumors and further underscore the Notch-dependent, anti-CSC mechanism through which NADI-351 acts in vivo.
Figure 5. NADI-351 selectively targets Notch1-driven cells and tumors.
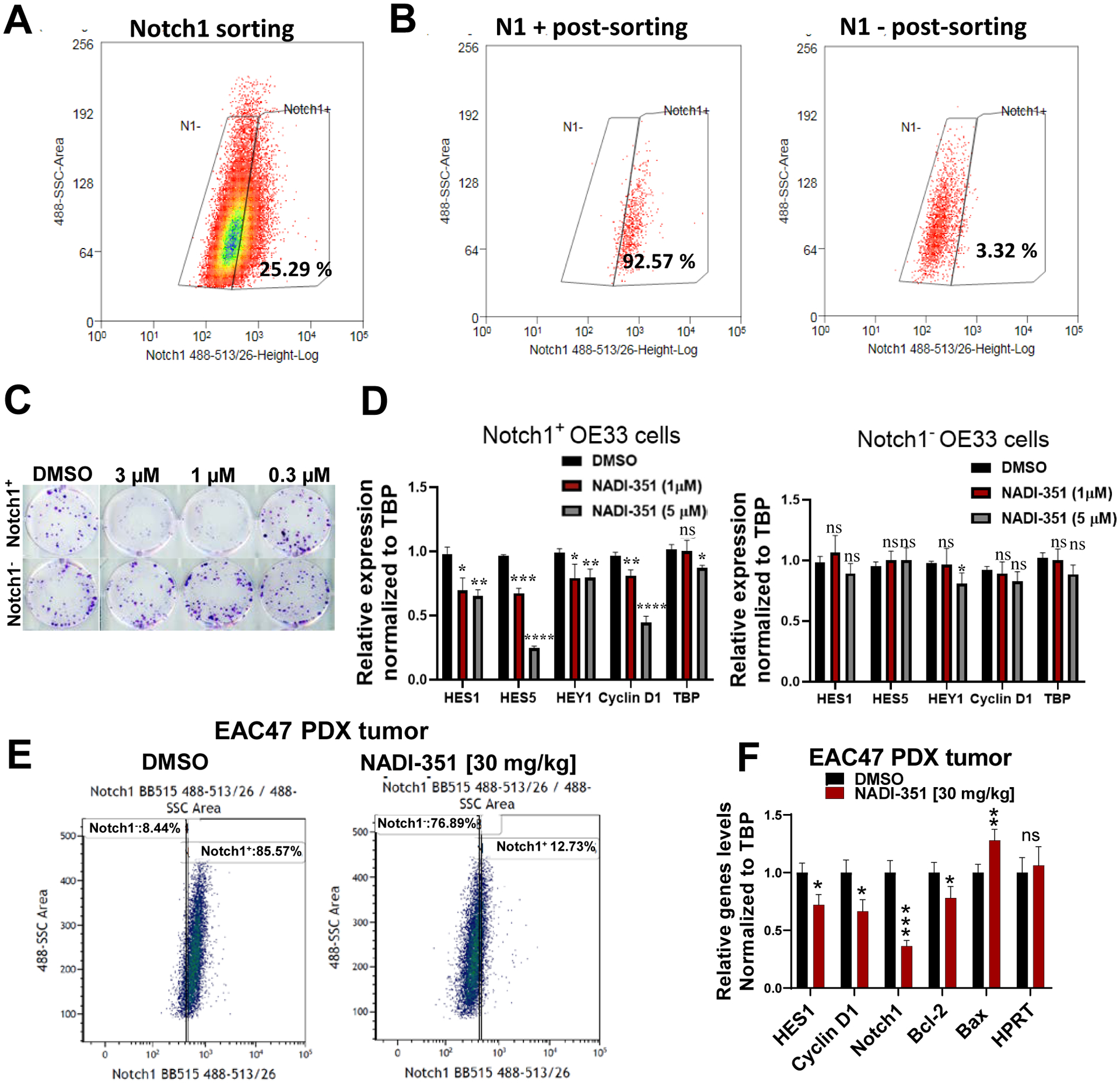
(A) FACS sorting of OE33 cells stained with Notch1 Ab. (B) Post-sort analysis of cells gated in (A). (C) Colony formation assays using NADI-351 were performed using each sorted OE33 Notch1 population. Cells were dosed every 48h for 2 weeks with the indicated concentration of NADI-351. (D) Relative Notch target gene expression in Notch1+ and Notch1− cells treated with NADI-351 for 24h (RT-qPCR). (E) FACS analysis of cells from EAC47 PDX tumor stained with Notch1 Ab and treated with 30 mg/kg of NADI-351 daily for 14 days. (F) EAC47 PDX tumors treated with vehicle (DMSO) or 30 mg/kg NADI-351, i.p. daily for 14 days were excised and examined by RT-qPCR for changes in Notch and apoptosis target genes (n=3). Error bars are representative of three independent experiments and indicate mean ± SD. p values ≤ 0.05 are considered statistically significant and indicated by asterisk(s). *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Discussion
Specific inhibition of Notch signaling has been of interest in oncology drug development for more than 2 decades. To date however, all approaches have largely failed in the clinic, most commonly due to dose-limiting GI toxicity caused by non-selective Notch inhibition, which has precluded attainment of a therapeutic dose (34–36). Even more selective approaches, including Notch1 mAbs, have failed due to limited efficacy or toxicity when combined with standard of care therapies, perhaps due to chronic (weeks-long) Notch inhibition by long systemic exposure inherent to mAb modalities (49).
Herein we describe the first Notch1-selective small molecule inhibitor of the NTC, NADI-351. NADI-351 is an orally available and potent inhibitor of Notch 1-mediated transcription that dramatically attenuates tumor cell growth. NADI-351 acts by selectively driving cell cycle arrest and apoptosis of the CSC population by inhibiting Notch-directed transcription. Unlike other attempts to target Notch signaling in cancer, NADI-351 does not induce goblet cell metaplasia, the dose limiting toxicity of pan-Notch inhibitors. Therefore, with its potent activity and lack of apparent toxicity, NADI-351 provides the necessary selectivity and therapeutic window required for a cancer therapeutic targeting the Notch pathway. Moreover, NADI-351 is the first compound with demonstrable selectivity in the inhibition of the CSC population of tumors.
Notch signaling is mediated by the formation of specific transcriptional activation complexes. These complexes are formed from Notch- and MAML-specific NTC’s bound to DNA via CSL (3–4). Although it is known that the 4 Notch proteins can form NTC with various MAML proteins (MAML1–3), it is not known what the contextual or functional differences are in Notch transcriptional signaling (50–51). Many cells, including tumor cells, express multiple Notch proteins. But the specific contributions of Notch1-4 NTC in normal or pathological physiology is not well understood. For the first time, we have been able to specifically inhibit Notch1ICD/MAML1 transcriptional complexes in tumor cells, which has provided a clearer understanding of the functional consequences of selective Notch inhibition. These data further underscore the importance of selectively targeting individual Notch paralogs and that drugging the Notch1 NTC specifically is essential for high efficacy with low toxicity.
Finally, our extensive characterization demonstrated that NADI-351 activity is derived through downstream targeted ablation of CSCs, likely as a consequence of Notch1-specific inhibition. Developmental pathways, specifically Notch, Hedgehog and Wnt, have long been desirable targets due to their role in early development and influence in governing stem cell biology (40). Various studies have implicated a role for these pathways in CSC, which has generated hope that targeted therapies could lead to more durable responses and improved patient outcomes. However, well-controlled studies on CSC-specific effects have been limited using currently approved inhibitors and acquired drug resistance indicates that these agents do not meaningfully impact CSCs clinically, therefore there has not been a clear validation of this hypothesis (40–41,44,52). We demonstrate that NADI-351 selectively ablates multiple independent Notch-dependent CSC populations from breast and EAC cell lines and from both in vivo and ex vivo EAC47 PDX cells and tumor spheres by selectively inhibiting Notch-mediated transcription in the Notch positive CSC, resulting in cell cycle arrest and induced apoptosis, thereby attenuating viability and tumor growth. Our studies provide compelling evidence that selective inhibition of Notch1 transcriptional complexes can profoundly arrest tumor growth while avoiding GI toxicity through specific depletion of the CSC population and suggests that NADI-351 has promise in translation to the clinic for Notch-dependent cancers.
Supplementary Material
Statement of Significance.
This study showcases the first Notch1-selective inhibitor that suppresses tumor growth with limited toxicity by selectively ablating cancer stem cells.
Acknowledgements
Financial support:
This work was supported by the Bankhead Coley Cancer Research from Florida Department of health (6BC02 to A.J. Capobianco, Lead optimization and preclinical evaluation of small molecule inhibitors of Notch transcriptional activation). This project was also generously supported by funding from the Dewitt Daughtry Family Department of Surgery and the Sylvester Comprehensive Cancer Center to A.J. Capobianco.
We thank Dr. Caroline Briegel (The DeWitt Daughtry Family Department of Surgery, Miller School of Medicine, University of Miami) for the MDA-MB-231 cell line. We thank MS. Teresa De Tony (Diabetes Research Institute, Miller School of Medicine, University of Miami) for the assistance with the confocal microscope and images. We want to thank Dr. Daniel Bilbao (Director of cancer modelling shared resource from the Sylvester Comprehensive Cancer Center) and members of his lab for technical assistance in animal experiments.
Footnotes
Conflict of interest: D.J.R. and A.J.C. are co-founders in StemSynergy Therapeutics, a company commercializing small-molecule cell signaling inhibitors. W.G. and D.O. are employees of StemSynergy Therapeutics.
References:
- 1.Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, Honjo T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H). Curr Biol 5, 1416–1423 (1995). [DOI] [PubMed] [Google Scholar]
- 2.Aster JC, Robertson ES, Hasserjian RP, Turner JR, Kieff E, Sklar J. Oncogenic forms of NOTCH1 lacking either the primary binding site for RBP-Jkappa or nuclear localization sequences retain the ability to associate with RBP-Jkappa and activate transcription. J Biol Chem 272, 11336–11343 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat. Rev. Cancer 11, 338–351 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Jeffries S, Robbins DJ, Capobianco AJ. Characterization of a high-molecular-weight Notch complex in the nucleus of Notchic-transformed RKE cells and in a human T-cell leukemia cell line. J. Mol. Cell Biol 22, 3927–3941 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science 284, 770–776 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Ranganathan P, Vasquez-Del Carpio R, Kaplan FM, Wang H, Gupta A, VanWye JD, et al. Hierarchical phosphorylation within the ankyrin repeat domain defines a phosphoregulatory loop that regulates Notch transcriptional activity. J Biol Chem 286, 28844–28857 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nam Y, Sliz P, Pear WS, Aster JC, Blacklow SC. Cooperative assembly of higher-order Notch complexes functions as a switch to induce transcription. PNAS 104, 2103–2108 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu L, Sun T, Kobayashi K, Gao P, Griffin JD. Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Molecular and cellular biology 22, 7688–7700 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bray SJ. Notch signaling: A simple pathway becomes complex. Nat Rev Mol Cell Biol, 7, 678–689 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Lecourtois M, Schweisguth F. Indirect evidence for Delta-dependent intracellular processing of Notch in Drosophila embryos. Curr Biol, 8, 771–775 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature, 393, 382–386 (1998). [DOI] [PubMed] [Google Scholar]
- 12.Struhl G, Adachi A. Nuclear access and action of Notch in vivo. Cell, 93, 649–660 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Falo-Sanjuan J, Bray SJ. Decoding the Notch signal. Develop Growth Differ. 00, 1–11. (2019). [DOI] [PubMed] [Google Scholar]
- 14.Wang Z, Da Silva TG, Jin K, Han X, Ranganathan P, Zhu X, et al. Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer research 74, 6364–6374 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farah E, Li C, Cheng L, Kong Y, Lanman NA, Pascuzzi P, Lorenz GR, et al. NOTCH signaling is activated in and contributes to resistance in enzalutamide-resistant prostate cancer cells. Journal of Biological Chemistry 294, 8543–8554 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Xie ZY, Guo XT, Xiao XH, Xiong LX. Notch and breast cancer metastasis: Current knowledge, new sights and targeted therapy. Oncol. Lett 18, 2743–2755 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stockhausen MT, Kristoffersen K, Poulsen HS. The functional role of Notch signaling in human gliomas. Neuro-oncology 12, 199–211 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aster JC, Blacklow SC, Pear WS. Notch signalling in T‐cell lymphoblastic leukaemia/lymphoma and other haematological malignancies. The Journal of pathology 223, 263–274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donnem T, Andersen S, Al-Shibli K, Al-Saad S, Busund LT, Bremnes RM “Prognostic impact of Notch ligands and receptors in non-small cell lung cancer. Cancer 116, 5676–5685 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, et al. Modulation of Notch Processing by γ-Secretase Inhibitors Causes Intestinal Goblet Cell Metaplasia and Induction of Genes Known to Specify Gut Secretory Lineage Differentiation. Toxicological Sciences 82, 341–358 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 435, 964–968 (2005). [DOI] [PubMed] [Google Scholar]
- 22.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, et al. Notch/γ-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435, 959–963 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Moriyama M, Durham AD, Moriyama H, Hasegawa K, Nishikawa S, Radtke F, Osawa M. Multiple Roles of Notch Signaling in the Regulation of Epidermal Development. Developmental Cell 14, 594–604 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Zhou M, Cui ZL, Guo XJ, Ren LP, Yang M, Fan ZW, et al. Blockade of Notch Signaling by γ-Secretase Inhibitor in Lung T Cells of Asthmatic Mice Affects T Cell Differentiation and Pulmonary Inflammation. Inflammation 38, 1281–1288 (2015). [DOI] [PubMed] [Google Scholar]
- 25.O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. J Exp Med 204, 1813–1824 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piha-Paul SA, Munster PN, Hollebecque A, Argilés G, Dajani O, Cheng JD, et al. Results of a phase 1 trial combining ridaforolimus and MK-0752 in patients with advanced solid tumours. European Journal of Cancer 51, 1865–1873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee SM et al. “Phase 2 study of RO4929097, a gamma-secretase inhibitor in metastatic melanoma: SWOG 0933.” Cancer 121, 432–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha Y, Othus M, et al. A phase II study of single-agent RO4929097, a gamma-secretase inhibitor of Notch signaling, in patients with recurrent platinum-resistant epithelial ovarian cancer: A study of the Princess Margaret, Chicago and California phase II consortia. Gynecol Oncol 137, 216–222 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer 48, 997–1003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kummar S, O’Sullivan Coyne G, Do KT, Turkbey B, Meltzer PS, et al. Clinical activity of the g-secretase inhibitor PF-03084014 in adults with desmoid tumors (aggressive fibromatosis). J Clin Oncol 35, 1561–1569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haapasalo A, Kovacs DM. The many substrates of presenilin/γ-secretase. J Alzheimers Dis. 25, 3–28, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR, et al. Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional gamma-secretase inhibitor. J Biol Chem 278, 46107–46116, (2003). [DOI] [PubMed] [Google Scholar]
- 33.Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem 279, 12876–12882, (2004). [DOI] [PubMed] [Google Scholar]
- 34.Takebe N, Nguyen D, Yang SX, et al. Targeting notch signaling pathway in cancer: clinical development advances and challenges. Pharmacol Ther 141, 140–149, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. New England Journal of Medicine 369, 341–350 (2013). [DOI] [PubMed] [Google Scholar]
- 36.De Strooper B. Lessons from a Failed γ-Secretase Alzheimer Trial. Cell 159, 721–726 (2014). [DOI] [PubMed] [Google Scholar]
- 37.VanDussen KL, Carulli AJ, Keeley TM, Patel SR, Puthoff BJ, Magness ST, et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 139, 488–497 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature 464,1052–1057 (2010). [DOI] [PubMed] [Google Scholar]
- 39. https://www.globenewswire.com/news-release/2017/04/17/961251/0/en/OncoMed-s-Phase-2-Trial-of-Tarextumab-in-Small-Cell-Lung-Cancer-Does-Not-Meet-Endpoints.html.
- 40.Takebe N, Harris PJ, Warren RQ, Ivy SP, et al. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nature reviews Clinical oncology 8, 97–106 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell 24, 25–40 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Wu KK. “Analysis of protein-DNA binding by streptavidin-agarose pulldown.” Gene Mapping, Discovery, and Expression. Humana Press, 281–290 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Astudillo L, Da Silva TG, Wang Z, Han X, Jin K, VanWye J, et al. The small molecule IMR-1 inhibits the notch transcriptional activation complex to suppress tumorigenesis. Cancer research 76, 3593–3603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weaver KL, Alves-Guerra MC, Jin K, Wang Z, Han X, Ranganathan P, et al. NACK is an integral component of the Notch transcriptional activation complex and is critical for development and tumorigenesis. Cancer research 74, 4741–4751 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, et al. Establishment of patient-derived non–small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clinical cancer research 14, 6456–6468 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Liu P, Brown S, Goktug T, Channathodiyil P, Kannappan V, Hugnot JP, et al. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. British journal of cancer 107, 1488–1497 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal transduction and targeted therapy 5, 1–35 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Azzam DJ, Zhao D, Sun J, Minn AJ, Ranganathan P, Drews-Elger K, et al. Triple negative breast cancer initiating cell subsets differ in functional and molecular characteristics and in γ‐secretase inhibitor drug responses. EMBO molecular medicine 5, 1502–1522 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferrarotto R, Eckhardt G, Patnaik A, LoRusso P, Faoro L, Heymach JV, et al. A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol 29 1561–1568 (2018) [DOI] [PubMed] [Google Scholar]
- 50.Wang H, Zang C, Liu XS, Aster JC, et al. The role of Notch receptors in transcriptional regulation. J. Cell. Physiol 230, 982–988 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vasquez-Del Carpio R, Kaplan FM, Weaver KL, VanWye JD, Alves-Guerra MC, Robbins DJ, et al. Assembly of a Notch transcriptional activation complex requires multimerization. Mol Cell Biol 31, 1396–1408 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z, Li Y, Ahmad A, Azmi AS, Banerjee S, Kong D, Sarkar FH, et al. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. BBA 1806, 258–267 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.