

Abstract
Intrauterine growth restriction (IUGR) increases the risk for perinatal complications and metabolic and cardiovascular disease later in life. The syncytiotrophoblast (ST) is the transporting epithelium of the human placenta and decreased expression of amino acid transporter isoforms in the ST plasma membranes is believed to contribute to IUGR. Placental mechanistic target of rapamycin Complex 2 (mTORC2) signaling is inhibited in IUGR and regulates the trafficking of key amino acid transporter (AAT) isoforms to the ST plasma membrane, however the molecular mechanisms are unknown. Cdc42 and Rac1 are Rho-GTPases that regulate actin-binding proteins, thereby modulating the structure and dynamics of the actin cytoskeleton. We hypothesized that inhibition of mTORC2 decreases AAT expression in the plasma membrane and amino acid uptake in primary human trophoblast (PHT) cells mediated by down-regulation of Cdc42 and Rac1. mTORC2, but not mTORC1, inhibition decreased the Cdc42 and Rac1 expression. Silencing of Cdc42 and Rac1 inhibited the activity of the System L and A transporters and markedly decreased the trafficking of LAT1 (System L isoform) and SNAT2 (System A isoform) to the plasma membrane. mTORC2 inhibition by silencing of rictor failed to decrease AAT following activation of Cdc42/Rac1. Placental Cdc42 and Rac1 protein expression was downregulated in human IUGR and was positively correlated with placental mTORC2 signaling. In conclusion, mTORC2 regulates AAT trafficking in PHT cells by modulating Cdc42 and Rac1. Placental mTORC2 inhibition in human IUGR may contribute to decreased placental amino acid transfer and reduced fetal growth mediated by down-regulation of Cdc42 and Rac1.
Keywords: Amino acid transport system L, amino acid transport system A, maternal-fetal exchange, placenta, fetal growth, pregnancy
Introduction
Intrauterine growth restriction (IUGR) increases the risk for perinatal complications (1, 2) and predisposes the infant for developing metabolic and cardiovascular disease later in life (3, 4, 5 ). A better understanding of the molecular mechanisms regulating fetal growth is critical for determining the causes of major pregnancy complications and developmental programming of adult disease. Fetal development and growth are critically dependent on an adequate nutrient supply, which is determined by placental nutrient transport (6-9). Altered fetal growth increases the risk for perinatal complications and predisposes the infant for the development of obesity, diabetes and cardiovascular disease later in life (10). The activity of key placental amino acid transporters is decreased in intrauterine growth restriction (IUGR) (6, 7, 11, 12) and up regulated in fetal overgrowth associated with maternal diabetes or obesity (8, 13), suggesting that changes in the activity of placental nutrient transporters may directly contribute to altered fetal growth. Studies in animal models indirectly support a cause-and-effect relationship between decreased placental amino acid transport and the development of IUGR (14-17) and between increased placental amino acid transport capacity and fetal overgrowth (13, 18, 19).
System A is a sodium-dependent transporter mediating the uptake of predominantly non-essential neutral amino acids into the cell (20). All three known isoforms of System A, SNAT1 (SLC38A1), SNAT2 (SLC38A2), and SNAT4 (SLC38A4) are expressed in the human placenta (21). Furthermore, reduced fetal amino acid concentrations (22) and reduced placental amino acid transport activity (12) in intrauterine growth restriction is related to a specific downregulation of SNAT2 expression (23). System L is a sodium-independent amino acid exchanger mediating cellular uptake of essential amino acids including leucine (24). The System L amino acid transporter is a heterodimer, consisting of a light chain, typically LAT1 (SLC7A5) or LAT2 (SLC7A8), and a heavy chain, 4F2hc/CD98 (SLC3A2). The molecular mechanisms regulating mammalian amino acid transporters are poorly understood, however recent studies in primary human trophoblast cells implicates mechanistic target of rapamycin (mTOR) signaling as a key regulator of System A and L amino acid transport (25) (26).
mTOR signaling controls cell growth in response to energy, nutrients, folate, oxygen, growth factors and other environmental cues (27, 28). mTOR forms two complexes with unique binding partners but distinct upstream regulators and downstream targets. mTOR complex 1 (mTORC1) is defined by its Raptor subunit which is replaced by Rictor in mTOR complex 2 (mTORC2) (29). mTORC1 phosphorylates S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E binding protein 1 (4E-BP1), which mediates many of the down-stream effects of mTORC1 (30). mTORC2 phosphorylates Akt, Protein Kinase C-α (PKC α) and Serum and Glucocorticoid-regulated Kinase (SGK) (31).
Both placental mTORC1 and mTORC2 activity is decreased in human IUGR (32-34) as well as in animal models of IUGR such as low protein diet in the rat (35). In addition, maternal nutrient restriction in the baboon inhibits placental mTOR signaling and nutrient transport and causes IUGR (17). These observations are consistent with an important role for placental mTOR signaling in regulating placental function and fetal growth and provide a possible mechanistic link between inhibition of placental mTOR signaling, down-regulation of placental nutrient transport and IUGR. mTORC2 has emerged as a key regulator of the actin cytoskeleton (36). Rho GTPases are a ubiquitous family of proteins that act as molecular switches, cycling between an active GTP-bound state and an inactive GDP-bound state. Activation of Rho-GTPases initiates the interaction with effector proteins and the activation of downstream signaling. In mammals, Rho, Rac and Cdc42 are among the most well characterized members of the Rho family of GTPases that regulate F-actin assembly and disassembly (37). Depletion of Rictor, a key component of mTORC2, in HeLa cells and fibroblasts disrupts stress fiber formation and reduces Rac activity (36, 38). In human neutrophils, inhibition of mTORC2 function by Rictor knockdown leads to cell polarity defects and uniform cortical F-actin accumulation (39). Furthermore, Rac and Cdc42 serve as downstream effectors of mTORC2 to regulate actin assembly and organization in neutrophils (40).
mTORC1 and mTORC2 independently regulate trophoblast System A and L amino acid transport activity by affecting the plasma membrane trafficking specifically of the SNAT2 (System A) and LAT1 (System L) isoforms (25). Furthermore, we have demonstrated that Nedd4-2, an E3 ubiquitin ligase, is required for the regulation of plasma membrane trafficking of amino acid transporter isoforms by mTORC1, but not mTORC2 (26). Therefore, the molecular mechanisms by which mTORC2 regulates the cell surface expression of trophoblast amino acid transporter isoforms remain unknown. We hypothesized that inhibition of mTORC2 decreases amino acid transporter expression in the plasma membrane and amino acid uptake in primary human trophoblast (PHT) cells mediated by down-regulation of Cdc42 and Rac1.
MATERIALS AND METHODS
Isolation and culture of primary human trophoblast (PHT) cells
Cytotrophoblast cells were isolated from uncomplicated term placentas and cultured in vitro. Tissue collection was approved by the Institutional Review Board of the University of Colorado Anchutz Medical Campus (COMIRB 14-1073). The research has been carried out in accordance with the World Medical Association Declaration of Helsinki, and that all subjects provided written informed consent. Cells were plated in either 60 mm culture dishes (~7.5×106 cells/dish for Western blot analysis) or six-well plates (for amino acid uptake experiments; ~3.75×106 cells/well for RNAi-mediated gene silencing) and cultured in 5% CO2, 95% atmosphere air at 37°C for 90 h. Cell culture medium (DMEM/Hams F-12, supplemented with L-glutamine, penicillin, streptomycin, gentamycin and 10% fetal bovine serum) was changed daily. We have previously reported that our cultured primary human trophoblast cells have a high expression of cytokeratin-7, a trophoblast specific marker, with no detectable expression of vimentin, a marker for mesenchyme-derived cells, confirming the high purity of our trophoblast cell population (25). Mononuclear cytotrophoblast cells undergo fusion and differentiate into multinucleated syncytiotrophoblast by 72 h in culture (25), as reflected by markedly increasing expression of syncytin, a marker of syncytialization (41). The cell viability and degree of syncytialization are maintained for 90 hours in culture. Studies at 90 h in cultured PHT cells ensure that maximum gene silencing has been achieved following siRNA transfection at 18 hours in culture, as in the current and previous reports (26, 41). The study design, including the time point of siRNA mediated silencing/activation of mTORC2 or Cdc42/Rac1 signaling and measurements of various parameters, is illustrated in Supplemental Figure 1.
Assessment of biochemical differentiation and viability
To confirm that trophoblast cells were undergoing biochemical differentiation, and to assess their viability with time in culture, the release of human chorion gonadotropin (hCG) by trophoblast cells into the culture medium 18, 42, 66 and 90 h after plating was measured using a commercial ELISA kit, which detects the β-subunit of hCG (Immuno Biological Labs).
RNA interference-mediated silencing
Dharmafect 2 transfection reagent (Thermo Scientific) and siRNAs (Sigma–Aldrich), targeting Raptor (100 nM; sense, 5′CAGUUCACCGCCAUCUACA′3), Rictor (100 nM; SASI_Hs02_00366683), Cdc42 (100 nM; SASI_Hs01_00113094) or Rac1 (100 nM; SASI_Hs01_00015565) were used. Control cells were transfected with a non-coding scrambled sequence (100 nM; sense: 5′GAUCA-UACGUGCGAUCAGATT). siRNAs were added to cultured PHT cells (~3.75×106 cells/well in six-well plate; ~7.5×106 cells in 60 mm dish) after 18 h in culture, incubated for 24 h and subsequently removed and replaced by fresh medium. At 90 h in culture, efficiency of target silencing was determined at the protein levels using Western blot.
System A and System L amino acid uptake assay
Amino acid uptake in PHT cells was determined at 90 h in culture. The activity of System A and System L amino acid transporters was assessed by measuring the Na+-dependent uptake of [14C] methyl-aminoisobutyric acid (MeAIB) and the 2-amino-2-norbornane-carboxylic acid (BCH)-inhibitable uptake of [3H] leucine, respectively, as described in detail previously (25).
Isolation of maternal facing microvillous plasma membranes from trophoblast cells
Microvillous plasma membranes (MVMs) were isolated from total homogenates of cultured PHT cells using differential centrifugation and Mg2+ precipitation (25). In brief, cells were lysed, scraped off the plate, homogenized and centrifuged. The pelleted crude membrane fraction was resuspended and 12 mM MgCl2 was added. The mixture was stirred slowly for 20 min on ice and then centrifuged. The supernatant containing MVM was centrifuged at 125,000 g for 30 min and the final pellet was resuspended. Protein concentration was determined using the Bradford assay. MVM enrichment was assessed using the MVM/total homogenate ratio of alkaline phosphatase protein expression as determined using Western blot after loading equal amounts of protein of MVM and cell lysates.
Isolation of fetal facing basal plasma membranes from trophoblast cells
Basal plasma membranes (BMs) were isolated from cultured PHT cells based on previously described method (42, 43) and described in supplemental methods.
Western blotting
For immunoblotting, cells were lysed in buffer containing phosphatase and protease inhibitors. Subsequently, cells were scraped, collected and sonicated. Proteins in cell lysates, MVM and BM were separated by electrophoresis. Western blotting was carried out as described (44). Protein expression of Cdc42, Rac1, Filamentous (F) actin and Globular (G) actin was determined in cell lysates using commercial antibodies (Cell Signaling Technology). MVM enrichment of alkaline phosphatase and insulin receptor expression were determined using commercial antibodies from Santa Cruz. Similarly, BM enrichment of VDAC1 expression was determined using VDAC1 antibody from Abcam, UK. Protein expression of the System A amino acid transporter isoform (SNAT2) and the System L amino acid transporter isoform (LAT1) was analyzed in MVM and BM preparations. A polyclonal SNAT2 antibody generated in rabbits was received as a generous gift from Dr. Prasad at University of Georgia, Augusta. The specificity of the SNAT2 antibody was validated by blocking peptide (25). An antibody targeting the LAT1 was produced in rabbits as described previously (45). LAT1 antibody specificity was validated using gene-silencing approaches (46). Anti-beta actin was from Sigma–Aldrich. The expression of actin or VDAC1 (BM) was used to control for any differences in loading and transfer. For each protein target the mean density of the control sample bands was assigned an arbitrary value of 1. All individual densitometry values were expressed relative to this mean.
Assay of G-actin and F-actin
PHT cells were homogenized in cold lysis buffer (10 mM K2HPO4, 100 mM NaF, 50 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.2 mM DTT, 0.5% Triton X-100, 1 mM sucrose, pH 7.0) supplemented with 1 mM ATP and protease inhibitor mixture for F-actin stabilization (Cytoskeleton). For preparation of G-actin and F-actin fractions from placenta homogenates, homogenates were mixed and resuspended with equal volume of cold lysis buffer and centrifuged at 1500 × g for 10 min. Further, G-actin and F-actin fractions were separated using the G-Actin/F-Actin In Vivo Assay Kit according to the manufacturer's instructions (Cytoskeleton Inc, Denver, CO; catalog #BK037). Protein concentrations were determined using the Bradford assay. Samples from the supernatant (G-actin) and pellet (F-actin) fractions were proportionally loaded and analyzed by western blot analysis using specific actin antibodies (Cell Signaling Technology, USA and Abcam, UK).
Immunoprecipitation
One mg of PHT cell lysates (1 μg/μl) was incubated with F-actin antibody overnight and antibodies were precipitated with protein G-Sepharose. Immunoprecipitates were denatured in sample buffer at 95°C, resolved by electrophoresis, and probed with SNAT2 or LAT1 antibody. The specificity of the F-actin antibody was validated by blocking peptide.
Proximity ligation assay (PLA) and confocal microscopy
Isolated cytotrophoblast cells were grown on chamber slides (Lab-Tek) for 90 hours. At 18 hours, cells were transfected with Rictor or scramble siRNA as described above. Next, at 90 hours the cells were fixed in ice-cold `methanol at −20C for 20 min and were blocked using 5% newborn calf serum (NCS) in PBS for 1 hour followed by incubation in anti-LAT1 (anti-rabbit) and anti-F actin (anti-mouse) for 3 hours. PLA probes anti-rabbit PLUS and anti-mouse MINUS were diluted in Duolink dilution buffer and incubated in a pre-heated humidity chamber for 1 hour. This was followed by ligation, amplification and detection according to the Duolink In Situ kit (Sigma-Aldrich) manufacturer’s protocol. Confocal microscopy was performed using Zeiss LSM 780 microscope at 63x magnification using oil immersion. Images were captured in the same laser settings with four Z-step of 0.4 um.
Placental mTORC2 signaling and Cdc42 and Rac1 expression in IUGR
Placentas from pregnancies complicated by intrauterine growth restriction (IUGR) and women delivering appropriate-for-gestational age (AGA) infants were collected within 15 minutes of delivery as described elsewhere (32). The decidua basalis and chorionic plate were removed, and villous tissue was dissected and rinsed in cold physiological saline. The villous tissue was transferred to cold buffer D (250 mM sucrose, 10 mM HEPES, pH 7.4) containing 1:100 dilution of protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO) and homogenized on ice with a Polytron (Kinematica, Luzern, Switzerland). The placental homogenates were frozen in liquid nitrogen and stored at −80°C until further processing. The expression of Rictor, Cdc42 and Rac1 were determined using Western blots as described for cells.
Data presentation and statistics
The number of experiments (n) represents the number of placentas studied. Data are presented as means ± S.E.M or + S.E.M. Differences between two independent groups were tested using unpaired Student's t test. Repeated measures analysis of variance (RMANOVA) with Tukey's post-hoc test was used to assess statistical differences between scramble siRNA, Raptor/Rictor siRNA, Raptor/ Rictor siRNA + Cdc42/Rac1 activator groups. A P value <0.05 was considered significant.
Results
mTORC2 but not mTORC1 inhibition decreases Cdc42 and Rac 1 expression
We determined whether mTORC2 regulates Rac1 and/or Cdc42 in PHT cells by determining the expression of Rac1 and Cdc42 in PHT cells following Raptor (mTORC1 inhibition) or Rictor siRNA silencing (mTORC2 inhibition). We have previously reported that raptor and rictor silencing in PHT is highly efficient, resulting in a 74 % (Raptor) and 70 % (Rictor) knockdown at the protein level (25). Silencing of Rictor, but not Raptor, decreased the expression of Cdc42 (−60 %, P=0.02, n=4) and Rac1 (−61 %, P=0.07, n=4) in PHT cells (Figure 1a-d).
Figure 1. Rictor silencing decreases the expression of Cdc42 and Rac1 and Cdc42 or Rac1 silencing inhibits PHT amino acid transport.
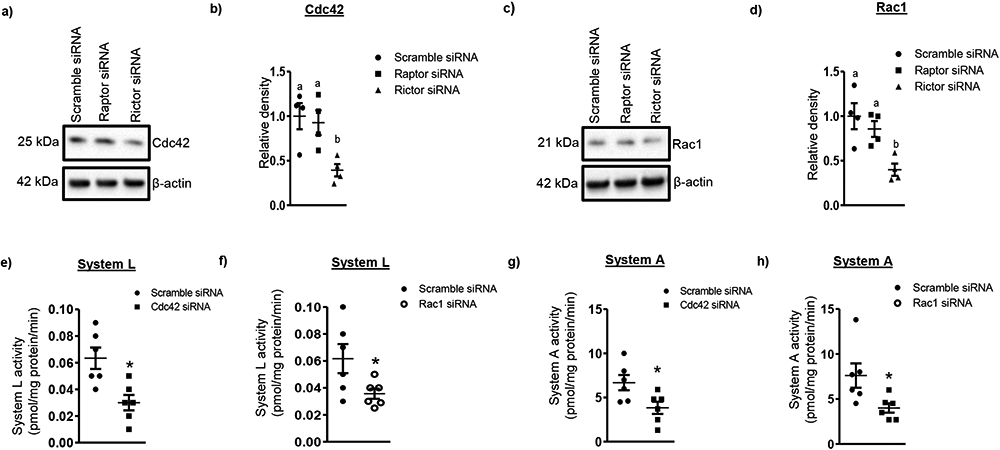
(a, c) Representative western blot of Cdc42 and Rac1 expression in cell lysates of scramble siRNA and Raptor/Rictor silenced cells. Equal loading was performed. (b, d) Summary of the western blot data. Values are given as means + S.E.M.; n=4/each group. Means without a common letter differ significantly (P<0.05) by RMANOVA with Tukey–Kramer multiple comparisons post hoc test. (e-f) Silencing of Cdc42 or Rac1 inhibits System L amino acid uptake in PHT cells. (g-h) Silencing of Cdc42 or Rac1 inhibits System A amino acid uptake in PHT cells. Values are means + S.E.M.; n=6 /each group for System L and System A. *P<0.05 compared with control; unpaired Student's t test.
Cdc42 or Rac1 silencing decreases System L and A amino acid transport
To determine the role of Cdc42 and Rac1 in regulating System A and L amino acid transport activity, we silenced Cdc42 or Rac1 in PHT cells and measured System A and L amino acid transport activity. Cdc42 siRNA decreased Cdc42 expression by 50% (Supplemental Figure 2a, c; n=4, P<0.05) as compared to scramble siRNA treated PHT cells. Similarly, Rac1 siRNA transfection in PHT cells significantly decreased Rac1 (−55%, n=4, P<0.05) expression as compared to control (Supplemental Figure 2b, d). As shown in Figure 1e-h, Cdc42 silencing decreased both System L and System A activity (System L: −50%, n=6, P=0.007; System A: −43 %, n=6, P=0.02). Likewise, Rac1 silencing (System L: −42%, n=6, P=0.04; System A: −47 %, n=6, P=0.03) also inhibited System L and System A amino acid transport activity compared to PHT cells transfected with scramble siRNA. Furthermore, hCG secretion profiles were similar in cells in which either Cdc42 or Rac1 had been silenced as compared cells transfected with scrambled siRNA (Supplemental Figure 3a). This finding supports that Rac1 or Cdc42 silencing did not affect differentiation/syncytialization and viability of cultured PHT cells and that the effects of Rac1/Cdc42 knockdown on amino acid uptake was not caused by unspecific effects.
Cdc42 or Rac1 silencing decreases the trafficking of LAT1 and SNAT2 to the microvillus plasma membrane (MVM) of PHT cells
We found that Cdc42 silencing caused a marked decrease in the expression of the System L amino acid transporter isoform LAT1 (−37 %, P=0.04, n=4, Figure 2a, b) and System A amino acid transporter isoform SNAT2 (−47%, P=0.02, n=3, Figure 2e, f) in the microvillus plasma membranes isolated from cultured PHT cells. Similarly, Rac1 silencing reduced the expression of LAT1 (−56 %, P=0.02, n=3, Figure 2c, d) and SNAT2 (−60 %, P=0.01, n=3, Figure 2g, h) in the microvillous plasma membrane fraction. Collectively, this data suggests that Cdc42 and Rac1 regulate System L and A amino acid transporter activity by modulating transporter trafficking of LAT1 and SNAT2 isoforms to the plasma membrane. The enrichment of alkaline phosphatase, a MVM marker (47), was determined by the ratio of alkaline phosphatase expression in MVM to total cell lysates. Alkaline phosphatase enrichment in MVM isolated from control cells was 17.4 ± 4.9, n=3, which was not significantly different from the alkaline phosphatase enrichment in MVM isolated from Cdc42 or Rac1 silenced cells. (Supplemental Figure 4a, b). As an additional enrichment marker, the expression of the insulin receptor, previously shown to be highly expressed in the MVM of human placenta (48), was determined using Western blot. MVM enrichment of the insulin receptor was not significantly different in the control (13.0 ± 3.5-fold, n=3) and Cdc42 silenced PHT cells (11.9 ± 4.5-fold, n=3) (Supplemental Figure 4c, d).
Figure 2. Cdc42 or Rac1 silencing decreases the trafficking of LAT1 and SNAT 2 to the maternal facing microvillus plasma membrane (MVM) of PHT cells.
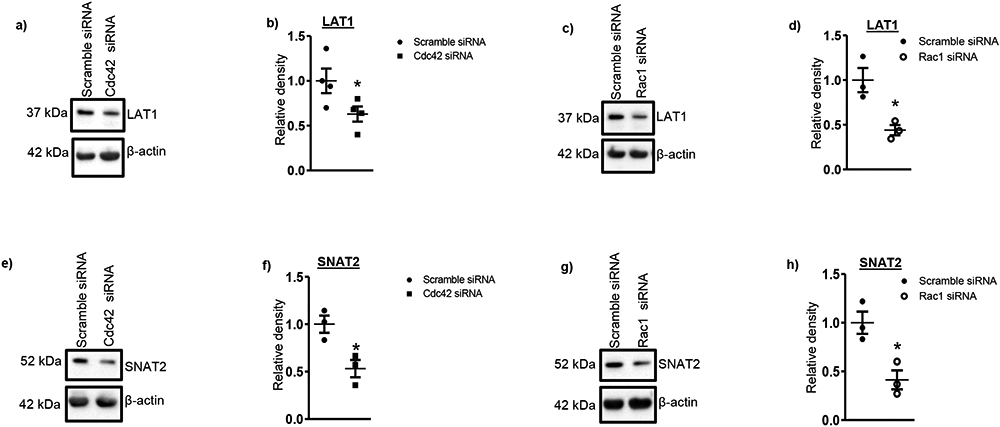
(a, c) Representative Western blots are shown for L-type amino acid transporter 1 (LAT1, 37 kDa) in MVMs of PHT cells transfected with scramble siRNA or siRNA targeting Cdc42 or Rac1. Equal loading was performed. Histograms (b, d) summarize the data. Expression in control cells for each isoform was arbitrarily assigned a value of one. (e, g) Protein expression of System A (SNAT2) amino acid transporter isoforms in MVMs isolated from scramble and Cdc42 or Rac1 siRNA silenced PHT cells. Equal loading was performed. Individual data point graphs (f, h) summarize the data. Expression in control cells for each isoform was arbitrarily assigned a value of one. Values are means + S.E.M.; n=3-4/each group. *P<0.05 compared with control; unpaired Student's t test
Cdc42 or Rac1 silencing decreases the trafficking of LAT1 to the basal plasma membrane (BM) of PHT cells
We determined the effect of Cdc42 or Rac1 silencing on expression of basal plasma membrane System L amino acid transporter isoforms in PHT cells. We found that Cdc42 silencing caused a significant decrease in the expression of the System L transporter isoform LAT1 (Supplemental Figure 5a, b; −52 %, P=0.04, n=4) in the BM fraction. Similarly, Rac1 silencing decreased the expression of System L amino acid transporter isoform LAT1 (Supplemental Figure 5c, d; −66 %, P=0.004, n=4) in the BM fraction. This data is the first to show that Cdc42 or Rac1 regulates the BM System L amino acid transporter isoform expression in any cell type. The enrichment of voltage-dependent anion channel (VDAC) (42), a BM marker, was determined by the ratio of VDAC expression in BM to total cell lysates. The VDAC enrichment in BM isolated from control cells was 36.3 ± 6.3, n=3, which was not different from BM isolated from Cdc42 or Rac1silenced cells (Supplemental Figure 6a, b). These data confirm significant enrichment of the basal plasma membrane from cultured PHT cells.
mTORC2 activation increases PHT System L and A amino acid transport activity and MVM isoform expression
In agreement with our previous report (25), inhibition of mTORC2 by silencing of Rictor in PHT cells decreased System L (Figure 3a; −52%, P=0.05; n=5) and System A (Figure 3b; −50%, P=0.001; n=5,) amino acid transporter activity as compared to cells transfected with scramble siRNA. In order to selectively activate mTORC2 in PHT, we combined Raptor siRNA to knock down mTORC1 with silencing of DEPTOR, an endogenous inhibitor of both mTORC1 and mTORC2, resulting in a selective activation of mTORC2. In Figure 3a, b, we demonstrate that specific activation of mTORC2 increased System L (+73 %, P=0.01) and System A (+ 70%, P=0.001; n=5) activity. As previously reported, we confirm that inhibition of mTORC2 alone with rictor siRNA caused a decreased expression of both the System L transporter isoform LAT1 (−56 %, P=0.05; n=4, Figure 3c, d) and System A transporter isoform SNAT2 (−44 %, P=0.05; n=4, Figure 3e, f) in the MVM fraction as compared to cells transfected with scramble siRNA. However, activation of mTORC2 accomplished by silencing raptor (mTORC1) + DEPTOR resulted in increased LAT1 (+60 %, P=0.001; n=4, Figure 3c, d) and SNAT 2 (+91 %, P=0.001; n=4, Figure 3e, f) expression in MVM. This data is consistent with the model that mTORC2 activation alone stimulates PHT amino acid uptake by increasing the cell surface expression of key amino acid transporter isoforms.
Figure 3. Activation of mTORC2 increases System L and System A amino acid uptake and LAT1 and SNAT2 MVM expression in PHT cells.
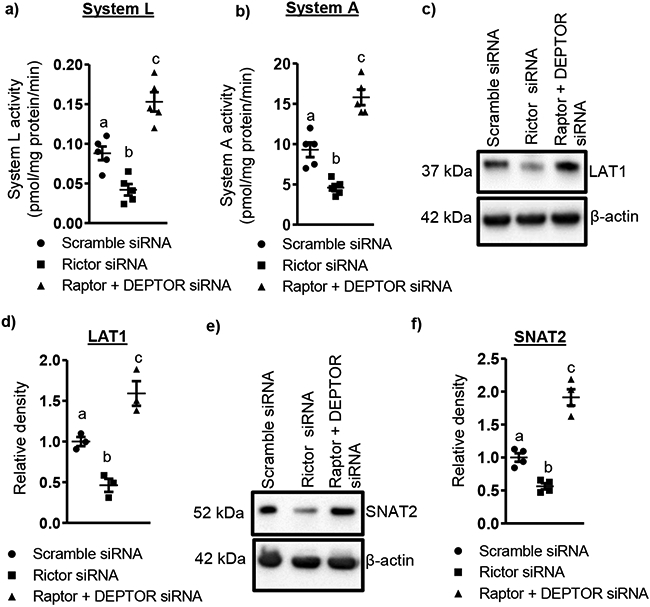
System L activity (a) was measured as the BCH-inhibitable uptake of [3H] leucine, and System A activity (b) was determined as the Na+-dependent uptake of [14C] MeAIB. Values are means ± S.E.M.; n= 5. Representative Western blots are shown for L-type amino acid transporter (LAT1, c) and sodium-coupled neutral amino acid transporter (SNAT2, e) in MVMs of scramble siRNA, Rictor siRNA and Raptor + DEPTOR silenced PHT cells. Equal loading was performed. Individual data point graphs (d, f) summarize the results. Expression in control cells for each isoform was arbitrarily assigned a value of one. Values are means ± S.E.M.; n=4. Means without a common letter differ significantly (P<0.05) by RMANOVA with Tukey–Kramer multiple comparisons post hoc test.
mTORC2, but not mTORC1 regulates the actin cytoskeleton in PHT cells
We explored the molecular mechanism by which mTORC2 regulates LAT1/SNAT2 transporter trafficking by first testing whether mTORC2 inhibition impairs actin dynamics in PHT cells. Actin exists in two states: as monomeric globular actin (G-actin), which polymerizes to form asymmetric two-stranded helical F-actin. mTORC2 inhibition resulted in a significant decrease (−43%, P=0.02; n=4) in F-actin levels compared with control (Figure 4a, b). This was accompanied by corresponding increase in G-actin levels (+59 %, P=0.03; n=4, Figure 4c, d), indicating that F-actin was depolymerizing to G-actin without any change in total actin concentration (Figure 4e, f). As a result, the ratio of F-actin to G-actin, which reflects the balance between actin polymerization and depolymerization, was reduced in mTORC2 inhibited PHT cells (−66 %, P=0.01; n=4, Figure 4g).
Figure 4. mTORC2, but not mTORC1 regulates the actin cytoskeleton in PHT cells.
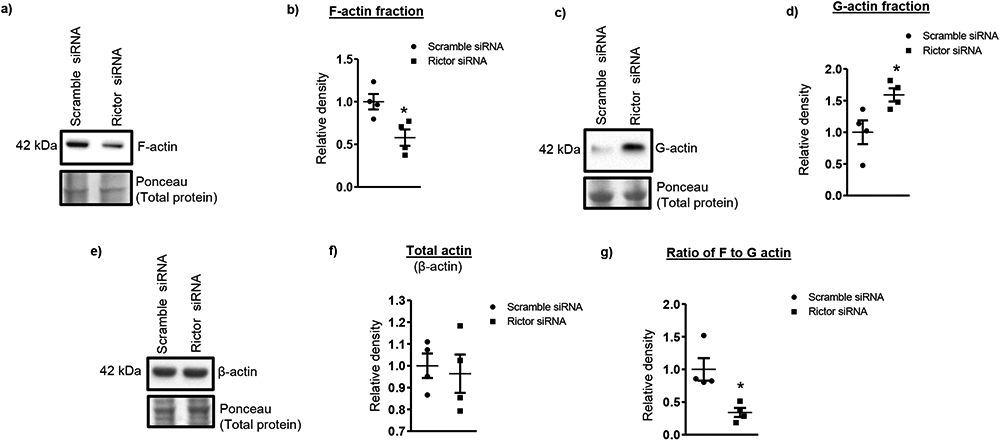
Enriched G-actin, F-actin and total actin fractions were isolated from Rictor and scramble siRNA silenced PHT cells. These fractions were resolved on SDS- PAGE gels and levels of F-actin, G-actin and total actin were analyzed by immunoblotting with anti-actin antibody. Densitometric analyses for actin levels were normalized to total lane proteins. Representative Western blots are shown for of F-actin (a), G-actin (c) and total actin (e) in actin enriched cell lysates of scramble siRNA, rictor siRNA PHT cells. Individual data point graphs (b, d, f, g) summarize the results. Values are given as means ± SEM.; n=4/each group, * P < 0.05 versus control; unpaired Student’s t test.
Changes in the ratio of F-actin to G-actin are regulated by different actin interacting molecules such as cofilin, paxillin, Cdc42, Rac1. To further investigate a role for mTOR complexes in the regulation of the actin cytoskeleton, we examined paxillin and cofilin phosphorylation in Rictor silenced PHT cells. Paxillin is a protein that when phosphorylated localizes and recruits other signaling molecules to focal adhesions (49). In control cells, paxillin was highly phosphorylated at Tyr 118 and Rictor silencing decreased paxillin Tyr 118 phosphorylation (−50 %, P<0.05, n=4, Figure 5a, b). In contrast, transfection of PHT cells with Raptor siRNA did not affect paxillin Tyr 118 phosphorylation (Figure 5a, b). Thus, inhibition of mTORC2, but not mTORC1, decreased paxillin phosphorylation and possibly inhibits assembly of focal adhesions.
Figure 5. Decreased phosphorylation of paxillin and cofilin in Rictor silenced PHT cells.
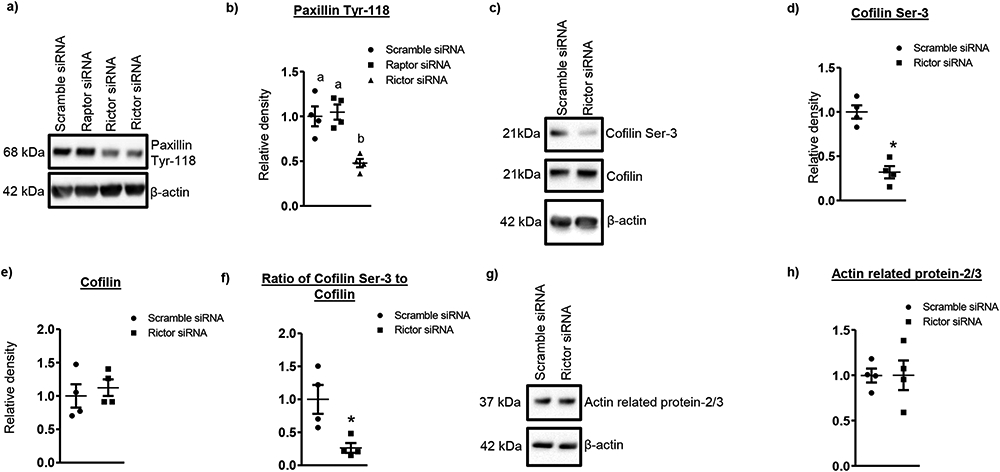
(a, c) Representative western blot of phosphorylated paxillin, and total and phosphorylated cofilin expression in cell lysates of scramble siRNA and rictor silenced cells. Equal loading was performed. (b, d, e, f) Summary of the western blot data. (g) Representative western blot of actin related protein 2/3 in cell lysates of scramble siRNA and Rictor silenced cells. Equal loading was performed. (h) Summary of the western blot data. Values are given as means ± SEM.; n=4/each group, * P < 0.05 versus control; unpaired Student’s t test.
Dephosphorylation activates cofilin, which binds to actin and promotes conversion of F-actin to G-actin. We observed significant decrease in cofilin phosphorylation in mTORC2 silenced cells (Figure 5c, d). The ratio of phosphorylated to total cofilin was also reduced (Figure 5f), which is expected to promote F-actin depolymerization as shown Figure 4b.
The actin-related protein-2/3 (ARP2/3) complex is a central player in cytoskeletal dynamics by controlling actin filament nucleation. However, protein expression of Arp2/3, which promotes actin polymerization, was unaffected in mTORC2 inhibited cells (Figure 5g, h), indicating that positive regulators of actin polymerization are not affected globally but rather selectively. Here, we have demonstrated that silencing of Rictor decreases paxillin and cofilin phosphorylation but not Arp2/3 in PHT cells.
mTORC2 inhibition decreases the association of F-actin with LAT1/SNAT2
Actin filaments are required for the trafficking of some ion channels and transporter proteins to the membrane and/or structurally anchoring the protein within the plasma membrane (50, 51). Some evidence suggests that F-actin directly binds to the epithelial sodium channel (ENaC) and translocate the transporter protein from cytosolic compartments to the plasma membrane (52). To study the molecular mechanisms linking mTORC2 signaling to system A and L amino acid transporter isoform trafficking, we measured system A (SNAT2) and system L (LAT1) transporter isoform association with F-actin in PHT cell lysates following mTORC2 inhibition. To this effect, we immunoprecipitated proteins in the cell lysate using an anti-F actin antibody. The proteins in the immunoprecipitate were then separated by SDS-PAGE and subsequently immunoblotted with anti-LAT1 or SNAT2 antibody. Rictor silencing (mTORC2 inhibition) resulted in decreased F-actin association with LAT1 (−62%, P=0.001; n=4, Figure 6a, b) and SNAT2 (−48 %, P=0.01; n=4, Figure 6c, d) as compared to control.
Figure 6: mTORC2 inhibition decreases the association of F-actin with SNAT2/LAT1.

(a, c) PHT cell lysates was used for IP with an anti-F actin antibody. Immunoprecipitated proteins were separated by SDS-PAGE and blotted (IB) with an anti- LAT1 or SNAT2 antibody. (b, d) Summary of the western blot data. Values are given as means ± SEM.; n=4/each group, * P < 0.05 versus control; unpaired Student’s t test. (e) Immunofluorescence confocal microscopy in combination with in situ proximity ligation assay (PLA), which detects protein–protein complexes, was used to explore interactions between LAT1 and F-actin following mTORC2 inhibition with Rictor siRNA. Trophoblast cells were transfected at 18 h in culture with scramble or rictor siRNA. At 90 h in culture, cells were fixed and F-actin LAT1 interaction (yellow) was visualized using PLA-confocal immunofluorescence Each detected F-actin-SNAT2 complex is represented by yellow dot. DNA was counterstained by (DAPI, 4′,6-diamidino-2-phenylindole) (blue). The data are from a representative experiment, and similar results were obtained from two other experiments.
To confirm that inhibition of mTORC2 decreases the interaction between F-actin and LAT1, we used proximity ligation assay and confocal microscopy, which allows the in-situ detection of interacting endogenous proteins. As shown in Figure 6e, LAT1/F-actin complexes were co-localized in PHT cells transfected with scramble siRNA. After Rictor siRNA the number of LAT-1/F-actin complexes decreased markedly, suggesting decreased F-actin interaction with LAT1 (Figure 6e).
Cdc42 and Rac1 is required for mTORC2 but not mTORC1 regulation of System L and System A amino acid transport activity
To mechanistically link mTORC2, Cdc42 or Rac1 and cellular amino acid uptake, PHT cells were transfected with raptor or rictor siRNA then Rac and Cdc42 were activated. At 89.5 h of culture PHT cells were treated with Rac + Cdc42 Activator (Cytoskeleton, Inc, Denver, CO), which efficiently activates Rac1, Rac2, Rac3 and Cdc42 in a variety of cultured cells (53). Subsequently, System L and System A amino acid transport activity was measured at 90 h (Supplemental Figure 7). As shown in Figure 7, silencing of Raptor or Rictor markedly inhibited basal levels of System L and System A amino acid transporter activity in cultured primary human trophoblast cells as compared with scramble siRNA transfected cells. These data confirm our previous observations that mTORC1 and mTORC2 independently regulate trophoblast amino acid transport (25). Rac and Cdc42 activation prevented the inhibitory effect of mTORC2 silencing on System L and System A amino acid transport activity (Figure 7a, b). In contrast, Rac and Cdc42 activation failed to reverse the inhibition in amino acid uptake as a result of raptor silencing (Figure 7c, d). These data support that mTORC1 and 2 both regulate amino acid transport activity but by different mechanisms.
Figure 7. Cdc42 and Rac1 is required for mTORC2 but not mTORC1 regulation of System L and System A amino acid transport activity.
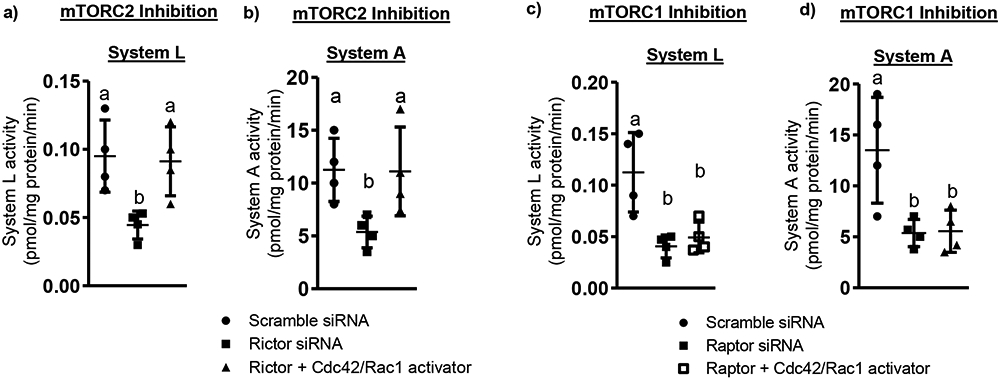
Scramble, Raptor and Rictor silenced PHT cells were preincubated at 88.5 hours of culture with Cdc42/Rac1 activator and System L/A amino acid transporter activity was measured at 90 hours of culture. Values are means ± S.E.M.; n=4 for System L and System A activity. Means without a common letter differ significantly (P<0.05) by RMANOVA with Tukey–Kramer multiple comparisons post hoc test.
Placental mTORC2 signaling is associated with protein expression of Cdc42/Rac1 in human IUGR
To explore the clinical relevance of our findings, we examined the relationship between placental mTORC2 signaling and the protein expression of Cdc42, Rac1 in placentas collected from normal healthy appropriately grown for gestational age (AGA) and IUGR pregnancies (Table 1). The protein expression of Cdc42 (−35 %, P=0.001) and Rac1 (−26 %, P=0.02) were significantly reduced in IUGR placentas as compared to AGA (Figure 8a-c). Rictor expression in placental homogenates of AGA and IUGR pregnancies were both positively correlated with expression of placental Cdc42 and Rac1expression (Figure 8d, e). Furthermore, to determine whether the loss of F-actin observed in mTORC2 inhibited PHT cells also occurs in the IUGR placenta, we determined F-actin and G-actin expression. As expected, F-actin expression was decreased (−36%, P=0.002; n=4), G-actin expression increased (+63 %, P=0.002, n=4) with no change in total actin in the IUGR placentas (Supplemental Figure 8a-c), consistent with a shift from actin polymerization to depolymerization. We further examined the F-actin interaction with SNAT2 in IUGR placentas using co-immunoprecipitation and found that F-actin-SNAT2 interaction was reduced in IUGR placentas as compared to AGA (−60 %, P=0.005; n=4, Supplemental Figure 9a-b). These findings are consistent with the model of mTOR regulating amino acid transport in human placenta and that inhibition of placental mTORC2, decreased Rac1/Cdc42 expression and lower F actin levels are mechanistically linked to the decreased SNAT 2 expression and lower System A amino acid transport capacity that we previously reported in syncytiotrophoblast plasma membranes isolated from human IUGR placentas (32).
Table 1.
Selected clinical data for AGA and IUGR pregnancies
| AGA (n=19) | IUGR (n=25) | |
|---|---|---|
| Maternal age (years) | 25.9±1.29 | 28.7±1.23 |
| BMI (kg/m2) * | 28.3±2.6 | 26.8±2.0 |
| Gestational age (weeks) | 33.9±0.95 | 35.7±0.61 |
| Birth weight (g) | 2493±236 | 1804±110† |
| Birth weight percentile‡ | 55.9±4.6 | 2.4±0.3§ |
| Placental weight (g) | 566±42.0 | 394±18.4∥ |
| Fetal sex (M/F) | 7/12 | 8/17 |
| Mode of delivery (C/V) | 6/13 | 15/10 |
Data are presented as means ± S.E.M. Abbreviations: AGA, appropriate for gestational age; IUGR, intrauterine growth restriction; F, female; M, male; C, caesarean section; V, vaginal delivery.
data from n=10 AGA and 18 IUGR
by corresponding gestational age
P<0.05
P<0.01
P<0.0001.
None of the mothers had any concurrent disease and apart from some preterm deliveries (both groups) and IUGR (IUGR group) no other pregnancy complication, such as preeclampsia or gestational diabetes, was present.
Figure 8. Placental mTORC2 signaling is associated with the protein expression of Cdc42/Rac1 expression in human IUGR pregnancy.
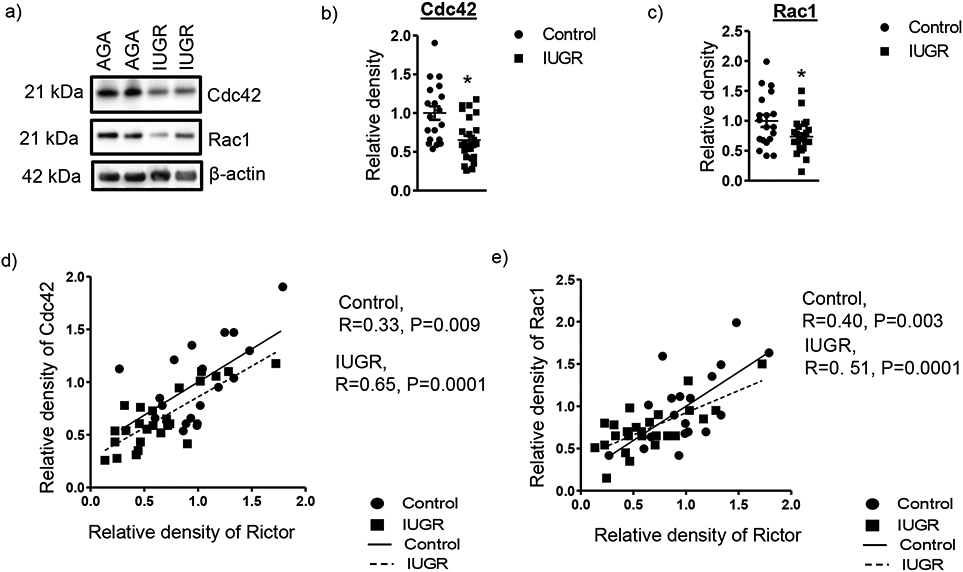
(a) Representative western blot of Cdc42 and Rac1 expression in placental homogenates of AGA and IUGR group. (b-c) Relative expression of Cdc42 and Rac1 in placental homogenates of AGA and IUGR. After normalization to β-actin, the mean density of C samples was assigned an arbitrary value of 1. Subsequently, individual IUGR density values were expressed relative to this mean. Control, n=19; IUGR, n= 25. Values are given as means ± S.E.M.; *P < 0.05 vs. AGA; unpaired Student’s t test. (d, e) Correlation between placental Rictor and Cdc42 or Rac1 expression in AGA and IUGR group. r = Pearson correlation coefficient, n = Control, 19; IUGR, 25.
Discussion
We show for the first time that mTORC2 signaling modulates plasma membrane trafficking of specific amino acid transporter isoforms in human primary cells mediated by Rac1 and Cdc42 regulation of actin polymerization. This new information will increase our understanding of the molecular mechanisms regulating key mammalian amino acid transporters. For example, our findings may have an implication for the cancer field given the efforts to develop interventions specifically targeting System L amino acid transporter isoforms in this disease (54, 55). In addition, these findings provide new insight into the link between changes in placental function and abnormal fetal growth in human pregnancy. Notably, we found that inhibition of mTORC2 signaling disrupts actin polymerization, actin regulatory signaling and selectively suppresses LAT1/SNAT2 transporter trafficking to the plasma membrane. We then identified Cdc42/Rac1 signaling as a critical link between mTORC2 and LAT1/SNAT2 plasma membrane trafficking. In general support of our findings, Huang and coworkers reported that mTORC2 mediated stabilization of the actin cytoskeleton in neurons promotes the trafficking and insertion of AMPA receptors clustered at the synapse (56).
F-actin is the major cytoskeletal protein and actin status (i.e., dynamics of F-actin/G-actin ratio) is critical for GLUT4 vesicle trafficking (57). Several reports have shown that mTORC2 modulates the organization of the actin cytoskeleton. Disruption of mTORC2 signaling by Rictor knockdown in fibroblasts prevented actin polymerization and cell spreading (36). mTORC1 and mTORC2 dual inhibitor KU0063794 specifically caused abnormal accumulation of F-actin and disordered distribution of microtubules, thereby markedly impairing endothelial cell elongation and tube formation (58). Here, we demonstrate that mTORC2 inhibition decreases the F-actin to G-actin ratio due to depolymerization because total actin levels were unchanged in PHT cells. In the IUGR placenta F-actin to G-actin ratio was reduced, which was associated with decreased mTORC2-Cdc42/Rac1 signaling and MVM expression of SNAT2. These findings suggest that inhibition of placental mTORC2 in IUGR (32) causes dysregulation of the actin skeleton contributing to the decreased MVM System A activity (17, 32) and SNAT2 protein expression (32) reported in this pregnancy complication (Figure 9).
Figure 9. A proposed model linking mTORC2 inhibition to decreased placental amino acid transport in human IUGR.
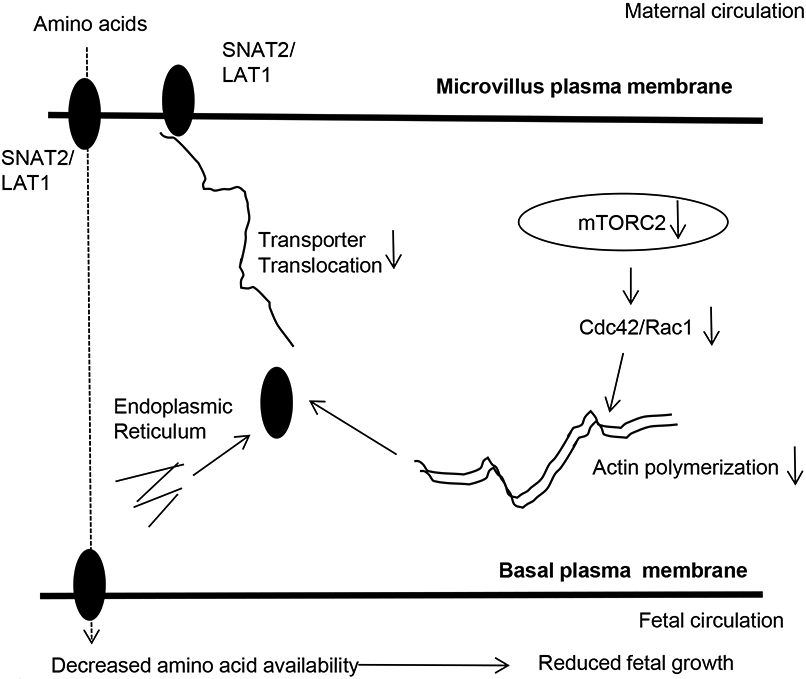
The findings of the present study are consistent with the model that inhibition of placental mTORC2 in human IUGR reduces the Rac1 and Cdc42 expression, resulting in inhibition of actin signaling mediated transporter trafficking of amino acid transporter isoforms, which contributes to a decreased fetal amino acid supply and restricted fetal growth.
Our study provides insight into the signaling pathways linking mTORC2 to the actin skeleton and SNAT2/LAT1 isoform trafficking in human cells. The Rho family of small GTPases, including Cdc42, Rac and Rho, are molecular switches that regulate actin cytoskeleton organization and are also implicated in intracellular trafficking (59). Rac-dependent actin remodeling was shown to be crucial for insulin-stimulated plasma membrane translocation of GLUT4, a glucose transporter highly expressed in muscle and adipocytes (60). Both Rac2 and Cdc42 have been shown to promote actin polymerization in different cell types. For example, Rac2 deletion in mouse neutrophils (61) and inhibition of Cdc42 in smooth muscle prevents actin polymerization (62). mTORC2 appears to regulate the actin assembly through a signaling pathway involving Rac1 and Cdc42 (56). In our experiments, mTORC2 but not mTORC1 inhibition significantly reduced the Rac1 and Cdc42 expression in PHT cells. Silencing of Cdc42 or Rac1 in PHT cells inhibited cellular amino acid uptake mediated by System A and L amino acid transporters and decreased MVM LAT1/SNAT2 isoform expression, which strongly suggest an essential role of Cdc42 and Rac1 in trafficking of LAT1 and SNAT2 isoforms to the PHT cell plasma membrane. Collectively, our findings in the current study, together with these literature reports, suggest that mTORC2 regulates amino acid transporter trafficking mediated by the effects of Rho GTPases Cdc42 and Rac1 on the actin cytoskeleton.
This present study is first to demonstrate that mTORC2-Cdc42/Rac1 signaling regulate fetal facing basal plasma membrane trafficking of LAT1 in PHT cells. System L transporters mediates the efflux of essential amino acids from the syncytiotrophoblast across the BM into the fetal circulation (63, 64). In vivo stable isotope tracer studies have demonstrated reduced transfer of leucine from mother to fetus in human IUGR (9). We have reported a decreased BM System L amino acid transport activity in human IUGR (6). Here we demonstrate that placental Cdc42 and Rac1 expression is decreased in human IUGR. Collectively, these observations are consistent with the model that inhibition of mTORC2 (17) and decreased Cdc42/Rac1 expression are linked to decreased transporter trafficking of LAT1 and lower BM System L transporter activity in IUGR placentas (6). Given the importance of fetal amino acid availability for fetal growth (14, 35), our findings have implications for our understanding of the development of IUGR.
We recently demonstrated that both mTORC1 and mTORC2 signaling regulates System A and L mediated amino acid uptake by governing the cell surface expression of SNAT2 and LAT1 isoforms (25). Specifically, mTORC1 regulation of cellular amino acid uptake was dependent on changes in Nedd4-2-mediated ubiquitination of SNAT2 and LAT1 whereas mTORC2 modulated transporter plasma membrane trafficking was independent of Nedd4-2 (44). Similarly, in the current study we show that Cdc42 and Rac1 is required for mTORC2 but not mTORC1 regulation of System A and System L amino acid transport activity. Thus, the mechanisms by which mTORC1 and mTORC2 regulate cellular amino acid uptake are distinct possibly representing a regulatory redundancy reflecting the critical importance of amino acid uptake for cellular protein synthesis, growth and survival.
Normal fetal growth and development is critically dependent on a continuous supply of nutrients and altered fetal nutrient availability impairs fetal development and causes abnormal fetal growth (65-67). Indeed, both under-nutrition and over-nutrition in fetal life are associated with a range of pregnancy complications and program the infant for future metabolic and cardiovascular disease (3, 4, 67, 68). Placental amino acid transport capacity has been consistently shown to be reduced in IUGR, both in women (69-75) and in a range of animal models (14, 15, 17, 35, 76-79). We have reported that down-regulation of key placental amino acid transport systems precedes the development of IUGR in rodents (15, 35) and non-human primates (14, 17, 80), supporting the concept that down-regulation of placental nutrient transport is a primary event, which directly contributes to IUGR. Thus, understanding the molecular mechanism regulating placental amino acid transport can not only help us better understand the underpinnings of important human diseases but may also provide the foundation for development of novel intervention strategies. The data generated in the current study using cultured primary human trophoblast cells and placental tissue from human IUGR pregnancies implicate a mechanistic link between placental mTORC2 inhibition, decreased Cdc42 and Rac1 expression, depolymerization of actin filaments, reduced plasma membrane trafficking of specific amino acid transporter isoforms, which results in decreased placental amino acid transfer and subsequent limitations in fetal amino acid availability, directly contributing to the restricted fetal growth.
An incomplete trophoblast invasion resulting in suboptimal spiral artery remodeling, which restricts the normal increase in utero-placental blood flow is believed to be the most common etiology of IUGR in developed countries. However, placental insufficiency is much more than just a matter of reduced blood flow because IUGR is associated with complex, coordinated and highly regulated changes in placental signaling and nutrient transport capacity that directly may contribute to poor outcomes (81). Thus, approaches targeting the trophoblast to improve placental function may improve fetal growth and alleviate adverse outcomes in IUGR. Given that mTORC1 and mTORC2 signaling regulates a numerous key placental functions including amino acid and folate transport, mitochondrial biogenesis and protein synthesis and that IUGR is associated with inhibition of placental mTOR signaling, specific approaches to activate trophoblast mTOR signaling in IUGR hold promise for improving fetal growth and preventing poor outcomes in this important pregnancy complication.
Supplementary Material
Summary statement.
We demonstrate that mTOR Complex 2 signaling modulates plasma membrane trafficking of specific amino acid transporter isoforms in human primary cells mediated by the regulation of actin polymerization by Rac1 and Cdc42, which may constitute a molecular link between changes in placental function and abnormal fetal growth in human pregnancy.
Clinical perspectives.
Alterations in placental amino acid transport directly contribute to altered fetal growth, which increases the risk for perinatal complications and predisposes for the development of obesity, diabetes and cardiovascular disease later in life.
We demonstrate that mTOR Complex 2 signaling modulates plasma membrane trafficking of specific amino acid transporter isoforms in human primary cells mediated by the regulation of actin polymerization by Rac1 and Cdc42.
Placental mTORC2 is inhibited in fetal growth restriction and activated in fetal overgrowth. We propose that placental mTORC2 inhibition, decreased Cdc42 and Rac1 expression, depolymerization of actin filaments, reduced plasma membrane trafficking of specific amino acid transporter isoforms which results in decreased placental amino acid transfer and subsequent limitations in fetal amino acid availability, directly contributing to the restricted fetal growth. This data advances our mechanistic understanding of common pregnancy complications and may provide a foundation for the development of intervention strategies for fetal growth restriction and overgrowth.
Acknowledgements
The authors are grateful to Anita Kramer, Kathryn Erickson and Kelsey Barner for technical assistance and the Perinatal CTRC for collection of placental tissue.
Funding information
This work was supported by NIH grant HD068370 and the NIH/NCATS Colorado CTSA Grant Number UL1 TR002535. Imaging experiments were performed in the University of Colorado Anschutz Medical Campus Advance Light Microscopy Core supported in part by NIH/NCATS Colorado CTSI Grant Number UL1 TR001082.
List of abbreviations
- AATs
amino acid transporters
- BCH-2
amino-2-norbornane-carboxylic acid
- BM
basal plasma membrane
- Cdc42
cell division control protein 42
- DEPTOR
DEP domain-containing mTOR-interacting protein
- G-actin
globular actin
- IUGR
intrauterine growth restriction
- IGF-I
insulin-like growth factor I
- LAT
large neutral amino acid transporter
- MeAIB
methyl-aminoisobutyric acid
- mTOR
mechanistic target of rapamycin
- mTORC1/2
mechanistic target of rapamycin complex 1/2
- MVM
microvillous plasma membrane
- NCS
newborn calf serum
- Nedd4-2
neuronal precursor cell-expressed, developmentally downregulated gene 4 isoform 2
- PHT
primary human trophoblast
- PIKK
phosphoinositide 3-kinase (PI3K)-related protein kinase
- PKCα
protein kinase C-α
- PLA
proximity ligation assay
- Raptor
raptor regulatory associated protein of mTOR
- Rictor
rapamycin-insensitive companion of mTOR
- S6K1
p70 S6 kinase
- SGK
serum and glucocorticoid-regulated kinase
- siRNA
small interfering RNA
- SNAT
sodium-dependent neutral amino acid transporter
- VDAC
voltage-dependent anion channel
- 4F2hc
4F2 cell-surface antigen heavy chain
- 4EBP-1
4E-eukaryotic initiation factor binding protein-1
Footnotes
Declarations of interest
The authors have nothing to disclose.
References:
- 1.Dall'Asta A, Brunelli V, Prefumo F, Frusca T, Lees CC. Early onset fetal growth restriction. Matern Health Neonatol Perinatol. 2017;3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Araujo Junior E, Peixoto AB, Zamarian AC, Elito Junior J, Tonni G. Macrosomia. Best Pract Res Clin Obstet Gynaecol. 2017;38:83–96. [DOI] [PubMed] [Google Scholar]
- 3.Gluckman PD, Hanson MA. Living with the past: Evolution, development, and patterns of disease. Science. 2004;305:1733–6. [DOI] [PubMed] [Google Scholar]
- 4.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barker DJP, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993;341:938–41. [DOI] [PubMed] [Google Scholar]
- 6.Jansson T, Scholtbach V, Powell TL. Placental transport of leucine and lysine is reduced in intrauterine growth restriction. Pediatr Res. 1998;44(4):532–7. [DOI] [PubMed] [Google Scholar]
- 7.Norberg S, Powell TL, Jansson T. Intrauterine growth restriction is associated with a reduced activity of placental taurine transporters. Pediatr Res. 1998;44(2):233–8. [DOI] [PubMed] [Google Scholar]
- 8.Jansson T, Ekstrand Y, Bjorn C, Wennergren M, Powell TL. Alterations in the activity of placental amino acid transporters in pregnancies complicated by diabetes. Diabetes. 2002;51(7):2214–9. [DOI] [PubMed] [Google Scholar]
- 9.Paolini CL, Marconi AM, Ronzoni S, Di Noio M, Fennessey PV, Pardi G, et al. Placental transport of leucine, phenylalanine, glycine, and proline in intrauterine growth-restricted pregnancies. J Clin Endocrinol Metab. 2001;86(11):5427–32. [DOI] [PubMed] [Google Scholar]
- 10.Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305(5691):1733–6. [DOI] [PubMed] [Google Scholar]
- 11.Mahendran D, Donnai P, Glazier JD, D'Souza SW, Boyd RD, Sibley CP. Amino acid (system A) transporter activity in microvillous membrane vesicles from the placentas of appropriate and small for gestational age babies. Pediatr Res. 1993;34(5):661–5. [DOI] [PubMed] [Google Scholar]
- 12.Glazier JD, Cetin I, Perugino G, Ronzoni S, Grey AM, Mahendran D, et al. Association between the activity of the system A amino acid transporter in the microvillous plasma membrane of the human placenta and severity of fetal compromise in intrauterine growth restriction. Pediatr Res. 1997;42(4):514–9. [DOI] [PubMed] [Google Scholar]
- 13.Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, et al. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab. 2013;98(1):105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pantham P, Rosario FJ, Nijland M, Cheung A, Nathanielsz PW, Powell TL, et al. Reduced placental amino acid transport in response to maternal nutrient restriction in the baboon. Am J Physiol Regul Integr Comp Physiol. 2015;309(7):R740–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jansson N, Pettersson J, Haafiz A, Ericsson A, Palmberg I, Tranberg M, et al. Down-regulation of placental transport of amino acids precedes the development of intrauterine growth restriction in rats fed a low protein diet. J Physiol. 2006;576(Pt 3):935–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malandro MS, Beveridge MJ, Kilberg MS, Novak DA. Effect of low-protein diet-induced intrauterine growth retardation on rat placental amino acid transport. Am J Physiol. 1996;271(1 Pt 1):C295–303. [DOI] [PubMed] [Google Scholar]
- 17.Kavitha JV, Rosario FJ, Nijland MJ, McDonald TJ, Wu G, Kanai Y, et al. Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. Faseb J. 2014;28(3):1294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aye IL, Rosario FJ, Powell TL, Jansson T. Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc Natl Acad Sci U S A. 2015;112(41):12858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosario FJ, Kanai Y, Powell TL, Jansson T. Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obesity (Silver Spring). 2015;23(8):1663–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mackenzie B, Erickson JD. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch. 2004;447(5):784–95. [DOI] [PubMed] [Google Scholar]
- 21.Desforges M, Lacey HA, Glazier JD, Greenwood SL, Mynett KJ, Speake PF, et al. SNAT4 isoform of system A amino acid transporter is expressed in human placenta. Am J Physiol Cell Physiol. 2006;290(1):C305–12. [DOI] [PubMed] [Google Scholar]
- 22.Cetin I, Ronzoni S, Marconi AM, Perugino G, Corbetta C, Battaglia FC, et al. Maternal concentrations and fetal-maternal concentration differences of plasma amino acids in normal and intrauterine growth-restricted pregnancies. Am J Obstet Gynecol. 1996;174(5):1575–83. [DOI] [PubMed] [Google Scholar]
- 23.Mando C, Tabano S, Pileri P, Colapietro P, Marino MA, Avagliano L, et al. SNAT2 expression and regulation in human growth-restricted placentas. Pediatr Res. 2013;74(2):104–10. [DOI] [PubMed] [Google Scholar]
- 24.Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch. 2004;447(5):532–42. [DOI] [PubMed] [Google Scholar]
- 25.Rosario FJ, Kanai Y, Powell TL, Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol. 2013;591:609–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosario FJ, Dimasuay KG, Kanai Y, Powell TL, Jansson T. Regulation of Amino Acid Transporter Trafficking by mTORC1 in Primary Human Trophoblast cells is Mediated by the Ubiquitin Ligase Nedd4-2. Clin Sci (Lond). 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silva E, Rosario FJ, Powell TL, Jansson T. Mechanistic Target of Rapamycin Is a Novel Molecular Mechanism Linking Folate Availability and Cell Function. J Nutr. 2017;147(7):1237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med. 2012;18(9):524–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015;517(7534):302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alessi DR, Pearce LR, Garcia-Matinez JM. New insights into mTOR signaling: mTORC2 and beyond. Sci Signaling 2009;2:1–4. [DOI] [PubMed] [Google Scholar]
- 32.Chen YY, Rosario FJ, Abu Shehab M, Powell TL, Gupta MB, Jansson T. Increased ubiquitination and reduced plasma membrane trafficking of placental amino acid transporter SNAT-2 in human IUGR. Clin Sci (Lond). 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roos S, Jansson N, Palmberg I, Saljo K, Powell TL, Jansson T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J Physiol. 2007;582(Pt 1):449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yung HW, Calabrese S, Hynx D, Hemmings BA, Cetin I, Charnock-Jones DS, et al. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am J Pathol. 2008;173(2):451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosario FJ, Jansson N, Kanai Y, Prasad PD, Powell TL, Jansson T. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology. 2011;152(3):1119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–8. [DOI] [PubMed] [Google Scholar]
- 37.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–35. [DOI] [PubMed] [Google Scholar]
- 38.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–302. [DOI] [PubMed] [Google Scholar]
- 39.Liu L, Das S, Losert W, Parent CA. mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev Cell. 2010;19(6):845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He Y, Li D, Cook SL, Yoon MS, Kapoor A, Rao CV, et al. Mammalian target of rapamycin and Rictor control neutrophil chemotaxis by regulating Rac/Cdc42 activity and the actin cytoskeleton. Mol Biol Cell. 2013;24(21):3369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen YY, Powell TL, Jansson T. 1,25-Dihydroxy vitamin D3 stimulates system A amino acid transport in primary human trophoblast cells. Mol Cell Endocrinol. 2017;442:90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oh SY, Hwang JR, Lee Y, Choi SJ, Kim JS, Kim JH, et al. Isolation of basal membrane proteins from BeWo cells and their expression in placentas from fetal growth-restricted pregnancies. Placenta. 2016;39:24–32. [DOI] [PubMed] [Google Scholar]
- 43.Goode RJ, Simpson RJ. Purification of basolateral integral membrane proteins by cationic colloidal silica-based apical membrane subtraction. Methods Mol Biol. 2009;528:177–87. [DOI] [PubMed] [Google Scholar]
- 44.Rosario FJ, Dimasuay KG, Kanai Y, Powell TL, Jansson T. Regulation of Amino Acid Transporter Trafficking by mTORC1 in Primary Human Trophoblast cells is Mediated by the Ubiquitin Ligase Nedd4-2. Clin Sci (Lond). 2016;130(7):499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park SY, Kim JK, Kim IJ, Choi BK, Jung KY, Lee S, et al. Reabsorption of neutral amino acids mediated by amino acid transporter LAT2 and TAT1 in the basolateral membrane of proximal tubule. Arch Pharm Res. 2005;28(4):421–32. [DOI] [PubMed] [Google Scholar]
- 46.Gaccioli F, Aye IL, Roos S, Lager S, Ramirez VI, Kanai Y, et al. Expression and functional characterisation of System L amino acid transporters in the human term placenta. Reprod Biol Endocrinol. 2015;13:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johansson M, Karlsson L, Wennergren M, Jansson T, Powell TL. Activity and protein expression of Na+/K+ ATPase are reduced in microvillous syncytiotrophoblast plasma membranes isolated from pregnancies complicated by intrauterine growth restriction. J Clin Endocrinol Metab. 2003;88(6):2831–7. [DOI] [PubMed] [Google Scholar]
- 48.Tavare JM, Holmes CH. Differential expression of the receptors for epidermal growth factor and insulin in the developing human placenta. Cell Signal. 1989;1(1):55–64. [DOI] [PubMed] [Google Scholar]
- 49.Schaller MD. Paxillin: a focal adhesion-associated adaptor protein. Oncogene. 2001;20(44):6459–72. [DOI] [PubMed] [Google Scholar]
- 50.Condrescu M, Reeves JP. Actin-dependent regulation of the cardiac Na(+)/Ca(2+) exchanger. Am J Physiol Cell Physiol. 2006;290(3):C691–701. [DOI] [PubMed] [Google Scholar]
- 51.Ikari A, Harada H, Takagi K. Role of actin in the cAMP-dependent activation of sodium/glucose cotransporter in renal epithelial cells. Biochim Biophys Acta. 2005;1711(1):20–4. [DOI] [PubMed] [Google Scholar]
- 52.Mazzochi C, Benos DJ, Smith PR. Interaction of epithelial ion channels with the actin-based cytoskeleton. Am J Physiol Renal Physiol. 2006;291(6):F1113–22. [DOI] [PubMed] [Google Scholar]
- 53.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70(3):401–10. [DOI] [PubMed] [Google Scholar]
- 54.Kim CS, Cho SH, Chun HS, Lee SY, Endou H, Kanai Y, et al. BCH, an inhibitor of system L amino acid transporters, induces apoptosis in cancer cells. Biol Pharm Bull. 2008;31(6):1096–100. [DOI] [PubMed] [Google Scholar]
- 55.Wang Q, Holst J. L-type amino acid transport and cancer: targeting the mTORC1 pathway to inhibit neoplasia. Am J Cancer Res. 2015;5(4):1281–94. [PMC free article] [PubMed] [Google Scholar]
- 56.Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, et al. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat Neurosci. 2013;16(4):441–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tunduguru R, Thurmond DC. Promoting Glucose Transporter-4 Vesicle Trafficking along Cytoskeletal Tracks: PAK-Ing Them Out. Front Endocrinol (Lausanne). 2017;8:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsuji-Tamura K, Ogawa M. Dual inhibition of mTORC1 and mTORC2 perturbs cytoskeletal organization and impairs endothelial cell elongation. Biochem Biophys Res Commun. 2018;497(1):326–31. [DOI] [PubMed] [Google Scholar]
- 59.Chi X, Wang S, Huang Y, Stamnes M, Chen JL. Roles of rho GTPases in intracellular transport and cellular transformation. Int J Mol Sci. 2013;14(4):7089–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.JeBailey L, Wanono O, Niu W, Roessler J, Rudich A, Klip A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes. 2007;56(2):394–403. [DOI] [PubMed] [Google Scholar]
- 61.Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, et al. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170(11):5652–7. [DOI] [PubMed] [Google Scholar]
- 62.Tang DD, Gunst SJ. The small GTPase Cdc42 regulates actin polymerization and tension development during contractile stimulation of smooth muscle. J Biol Chem. 2004;279(50):51722–8. [DOI] [PubMed] [Google Scholar]
- 63.Cleal JK, Glazier JD, Ntani G, Crozier SR, Day PE, Harvey NC, et al. Facilitated transporters mediate net efflux of amino acids to the fetus across the basal membrane of the placental syncytiotrophoblast. J Physiol. 2011;589(Pt 4):987–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Furesz TC, Smith CH. Identification of two leucine-sensitive lysine transport activities in human placental basal membrane. Placenta. 1997;18(8):649–55. [DOI] [PubMed] [Google Scholar]
- 65.Gaccioli F, Lager S, Powell TL, Jansson T. Placental transport in response to altered maternal nutrition. J DOHaD 2013;4:101–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jansson T, Powell TL. IFPA 2005 Award in Placentology Lecture. Human Placental Transport in Altered Fetal Growth: Does the Placenta Function as a Nutrient Sensor? - A Review. Placenta. 2006;27 SupplA S91–7. [DOI] [PubMed] [Google Scholar]
- 67.Jansson T, Powell TL. Role of placental nutrient sensing in developmental programming. Clin Obstet Gynecol. 2013;56(3):591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gluckman PD, Hanson MA. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab 2004;15:183–7. [DOI] [PubMed] [Google Scholar]
- 69.Norberg S, Powell TL, Jansson T. Intrauterine growth restriction is associated with a reduced activity of placental taurine transporters. Pediatr Res. 1998;44:233–8. [DOI] [PubMed] [Google Scholar]
- 70.Jansson T, Scholtbach V, Powell TL. Placental transport of leucine and lysine is reduced in intrauterine growth restriction. Pediatr Res. 1998;44:532–7. [DOI] [PubMed] [Google Scholar]
- 71.Chen YY, Rosario FJ, Shehab MA, Powell TL, Gupta MB, Jansson T. Increased ubiquitination and reduced plasma membrane trafficking of placental amino acid transporter SNAT-2 in human IUGR. Clin Sci (Lond). 2015;129(12):1131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mahendran D, Donnai P, Glazier JD, D'Souza SW, Boyd RDH, Sibley CP. Amino acid (System A) transporter activity in microvillous membrane vesicles from the placentas of appropriate and small for gestational age babies. Pediatr Res. 1993;34:661–5. [DOI] [PubMed] [Google Scholar]
- 73.Glazier JD, Cetin I, Perugino G, Ronzoni S, Grey AM, Mahendran D, et al. Association between the activity of the system A amino acid transporter in the microvillous plasma membrane of the human placenta and severity of fetal compromise in intrauterine growth restriction. Pediatr Res. 1997;42:514–9. [DOI] [PubMed] [Google Scholar]
- 74.Marconi AM, Paolini CL, Stramare L, Cetin I, Fennessey PV, Pardi G, et al. Steady state maternal-fetal leucine enrichments in normal and intrauterine growth-restricted pregnancies. Pediatr Res. 1999;46(1):114–9. [DOI] [PubMed] [Google Scholar]
- 75.Paolini CL, Marconi AM, Ronzoni S, Di Noio M, Fennessey PV, Pardi G, et al. Placental transport of leucine, phenylalanine, glycine, and proline in intrauterine growth-restricted pregnancies. J Clin Endocrinol Metab. 2001;86:5427–32. [DOI] [PubMed] [Google Scholar]
- 76.Ross JC, Fennessey PV, Wilkening RB, Battaglia FC, Meschia G. Placental transport and fetal utilization of leucine in a model of fetal growth retardation. Am J Physiol. 1996;270:E491–E503. [DOI] [PubMed] [Google Scholar]
- 77.Anderson AH, Fennessey PV, Meschia G, Wilkening RB, Battaglia FC. Placental transport of threonine and its utilization in the normal and growth-restricted fetus. Am J Physiol. 1997;272(5 Pt 1):E892–900. [DOI] [PubMed] [Google Scholar]
- 78.Thureen PJ, Trembler KA, Meschia G, Makowski EL, Wilkening RB. Placental glucose transport in heat-induced fetal growth retardation. Am J Physiol. 1992;263(3 Pt 2):R578–85. [DOI] [PubMed] [Google Scholar]
- 79.de Vrijer B, Regnault TR, Wilkening RB, Meschia G, Battaglia FC. Placental uptake and trnsport of ACP, a nutral nonmetabolizable amino acid, in an ovine model of fetal growth restriction. Am J Physiol. 2004;287:E1114–E24. [DOI] [PubMed] [Google Scholar]
- 80.Pantham P, Rosario FJ, Weintraub S, Nathanielsz PW, Powell TL, Li C, et al. Down-regulation of Placental Transport of Amino Acids Precedes the Development of Intrauterine Growth Restriction in Maternal Nutrient Restricted Baboons. Biol Reprod. 2016;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chassen S, Jansson T. Complex, coordinated and highly regulated changes in placental signaling and nutrient transport capacity in IUGR. Biochim Biophys Acta Mol Basis Dis. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.