

Abstract
Identifying genes that interact to confer a biological function to an organism is one of the main goals of functional genomics. High‐throughput technologies for assessment and quantification of genome‐wide gene expression patterns have enabled systems‐level analyses to infer pathways or networks of genes involved in different functions under many different conditions. Here, we leveraged the publicly available, information‐rich RNA‐Seq datasets of the model plant Arabidopsis thaliana to construct a gene co‐expression network, which was partitioned into clusters or modules that harbor genes correlated by expression. Gene ontology and pathway enrichment analyses were performed to assess functional terms and pathways that were enriched within the different gene modules. By interrogating the co‐expression network for genes in different modules that associate with a gene of interest, diverse functional roles of the gene can be deciphered. By mapping genes differentially expressing under a certain condition in Arabidopsis onto the co‐expression network, we demonstrate the ability of the network to uncover novel genes that are likely transcriptionally active but prone to be missed by standard statistical approaches due to their falling outside of the confidence zone of detection. To our knowledge, this is the first A. thaliana co‐expression network constructed using the entire mRNA‐Seq datasets (>20,000) available at the NCBI SRA database. The developed network can serve as a useful resource for the Arabidopsis research community to interrogate specific genes of interest within the network, retrieve the respective interactomes, decipher gene modules that are transcriptionally altered under certain condition or stage, and gain understanding of gene functions.
One‐sentence summary
We present here an Arabidopsis gene co‐expression network constructed using RNA‐Seq datasets, which will serve as a useful resource for the Arabidopsis research community to gain insights into Arabidopsis gene interactions and functions.
1. INTRODUCTION
The exponentially growing availability of omics databases has spawned opportunities to leverage the power of computational models to interrogate different databases and mine new information that can illuminate complex molecular interactions underlying versatile phenotypes. One goal of this analysis is to integrate information from different types of omics data, for example, genomic, proteomic, and metabolomic, to build interactomes that can reveal complex interactions at a higher resolution. A combinatorial approach to understanding the biomolecular interactions is a key to deciphering new systems‐level information. Within the last decade, a massive influx of high‐throughput sequencing data has necessitated new strategies to unravel hidden information, and to this end, efforts have been made to derive biomolecular networks, for example, protein–protein interaction (PPI) networks, in various model organisms such as bacteria, yeast, fruit‐fly, and plants. The overarching goal of these efforts is to understand an organism at a systems level by illuminating multi‐scale interactions within cellular systems. Different biomolecular interaction networks have been constructed, including PPI networks, metabolic networks, gene transcriptional networks, and signal transduction networks. Additionally, theoretical advances in the field of network science have led to the elucidation of a number of features shared among networks emanating from many different disciplines, such as, small‐world property, network transitivity, network motif, and community structure, have enhanced our understanding of topological structure of the biological networks (Albert, 2005; Girvan & Newman, 2002; Joyce & Palsson, 2006; Kelley et al., 2003; Lee, 2004b; Wang et al., 2006; Zhang et al., 2007).
A biological network is characterized by nodes and edges; the former represents biomolecules such as genes, proteins, or metabolites, and the latter represents connections between nodes signifying interactions between biomolecules, such as physical interaction, metabolite flow, regulatory relationship, and/or co‐expression relationships. Biological networks are often modular; biomolecules belonging to the same module interact with each other to carry out a specific biological function. Deciphering modules and their associated functions is one of the primary goals in the studies of gene co‐expression networks. Here, we focus on the construction of a gene co‐expression network of the model plant, Arabidopsis thaliana. Arabidopsis gene expression networks have previously been primarily constructed and based mainly on microarray data. These networks have been extensively used for understanding biological pathways as well as their interactions in plants (Aoki et al., 2007; Bergmann et al., 2003; Carlson et al., 2006; Farahbod & Pavlidis, 2019; Freeman et al., 2007; Horvath & Dong, 2008; Jen et al., 2006; Jordan et al., 2005; Jordan et al., 2004; Lee, 2004a; Ma et al., 2007; Manfield et al., 2006; Mentzen, 2006; Obayashi et al., 2007; Rahme, 2003; Roszik & Woodman, 2014; Ruan & Zhang, 2006; Slonim & Yanai, 2009; Smith, 2018; Stuart, 2003; Wei et al., 2006). In addition, gene co‐expression in Arabidopsis has been investigated using RNA‐Seq data. For example, the ATTED‐II database provides an RNA‐seq‐based Arabidopsis co‐expression network that was derived using a mutual rank index approach (Obayashi et al., 2018). A recent study inferred Arabidopsis gene modules based on co‐expression derived from RNA‐Seq datasets; the published tool, EXPLICIT, infers genes regulated by various transcriptional factors in Arabidopsis (Geng et al., 2021).
In recent years, thousands of A. thaliana RNA‐Seq datasets representing many different conditions have been deposited into the NCBI GEO repository of expression data. The availability of these provides an information‐rich resource for an unbiased analysis that will advance plant functional genomics. With this goal in mind and to exploit the full extent of these datasets, we performed a gene co‐expression network analysis of A. thaliana by utilizing the RNA‐Seq data for this model plant.
We first constructed a co‐expression network based on expression correlation between genes and then decomposed the network into modules with genes cohesively linked within and sparsely between. The Arabidopsis gene co‐expression network constructed based on entire collection of Arabidopsis RNA‐Seq datasets at NCBI thus represents a multitude of genotypes and conditions for A. thaliana. Our investigation revealed a modular network comprised of distinct functional components representing a range of biological processes, including photosynthesis, stress, defense, and localization. As genes belonging to a module co‐expressed across thousands of diverse conditions, the network illuminated distinct functional entities in which the genes are strongly coupled by the same underlying co‐regulation mechanisms. The Arabidopsis gene co‐expression network developed provides a useful resource for the plant community, allowing researchers to interrogate the network with the genes of their interest, examine the gene modules to infer the functions of yet uncharacterized genes and uncover unknown pathways or networks of pathways, and map differentially expressing genes from an experiment onto the network to identify functional modules that are transcriptionally activated under certain condition, which could spur further investigations into novel regulatory pathways or yet unknown aspects of regulatory mechanisms in plants.
2. RESULTS
A gene co‐expression network of A. thaliana was constructed using weighted gene correlation network analysis (WGCNA) that allows examining the co‐expression patterns of genes from the entire collection of mRNA‐Seq datasets available at the NCBI Sequence Read Archive (SRA). Modules within the network comprised of genes that were found to be highly correlated in their expression under many different conditions and could thus be participating collectively in different biological processes. A gene module is considered to be equivalent of the retrieved subnetwork itself (Aoki et al., 2007; Ma et al., 2007; Manfield et al., 2006; Obayashi et al., 2007). Each module was subjected to functional enrichment analysis to determine enrichment of Gene Ontology (GO) terms in order to associate it with biological function(s). Additionally, we used the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and performed KEGG pathway enrichment analysis of the modules to investigate the roles of the constituent genes in different biological pathways. We describe below results from each step of our analysis and also emphasize the importance of network in gaining new insights into genes and their interactions.
2.1. Network construction
We used over 20,000 non‐redundant mRNA‐Seq Arabidopsis datasets from the NCBI SRA to construct a gene co‐expression network. The normalized gene expression values, quantified in terms of abundance of reads mapping onto a gene, were imported into WGCNA for network construction.
The Arabidopsis network is comprised of 21,332 nodes and 36,877,224 edges. Each node represents a gene, and each edge between two nodes represents a connection or association between the nodes (genes). The association is quantified based on the topological overlap value, ranging from 0 to 1, taking into consideration both the expression profile similarity, and the similarity of relationships each node has with all other nodes (Figure 1). A WGCNA network is fully interconnected, but each connection is weighted differently. Only genes that fall into a cluster annotated “zero” are disconnected from other genes in the network. Following a hierarchical clustering procedure, a specific cut height was used to clip the resultant clusters (modules). This resulted in the generation of a large WGCNA network with 54 gene modules. Unfortunately, Cytoscape (Shannon et al., 2003) was unable to import the entire network file and therefore, the visualization of the entire network could not be made. Additionally, annotation of the modules with the entire network loaded onto Cytoscape could not be accomplished. To circumvent this visualization challenge, we used an in‐house script to remove weak edges based on the network density. This is based on an approach used earlier for uncovering the modular structure of a network (Mao et al., 2009). Although a WGCNA derived network differs in that it is derived based on a soft‐thresholding approach (in contrast to hard thresholding by Pearson correlation coefficient; Mao et al., 2009), the network density approach may still aid in identifying weakly correlated edges that can be removed from the network only for the purpose of visualization with Cytoscape. In an attempt to choose an appropriate cutoff of topological overlap, in order to decide on edges to be included (greater than cutoff) or excluded (less than cutoff), we examined the changes in the number of nodes, and number of edges, as a function of the cutoff (varied from 0 to 1 at an increment of .01). We observed that as the cut off value increased, both the node number and the edge number decreased, and so did the network density, as expected (Figure S1). However, as the decreasing rate of edges became slower than that of the nodes, the network density increased beyond a certain cutoff. We observed that the network density reaches a minimum around .1 and increased thereafter. It would be appropriate to choose a cutoff value greater than .1 since that would enable selecting the edges that would densely connect a decreasing number of nodes. After attempting to maximize the number of retained nodes in the network, which can be imported and visualized in Cytoscape, we chose the cutoff value to be .12. Note that there are multiple inflection points (Figure S1), and we selected the first inflection point (of lowest value among all) to include as many edges as could be into the Cytoscape visualization. At this cutoff, only 3% of all possible edges were retained. If we allow a more relaxed threshold to include more edges, Cytoscape inevitably fails to load the network and prevents further processing. The resultant Arabidopsis network that could be visualized with Cytoscape consists of 11,158 nodes and 1,162,948 edges. Note that the aforementioned steps were performed to trim the network to enable visualization. The edge/node reduction was thus purely cosmetic, as the soft‐threshold nature of the WGCNA network does not lend well to traditional network visualization. All downstream analyses were performed with the complete (untrimmed) WGCNA network.
FIGURE 1.
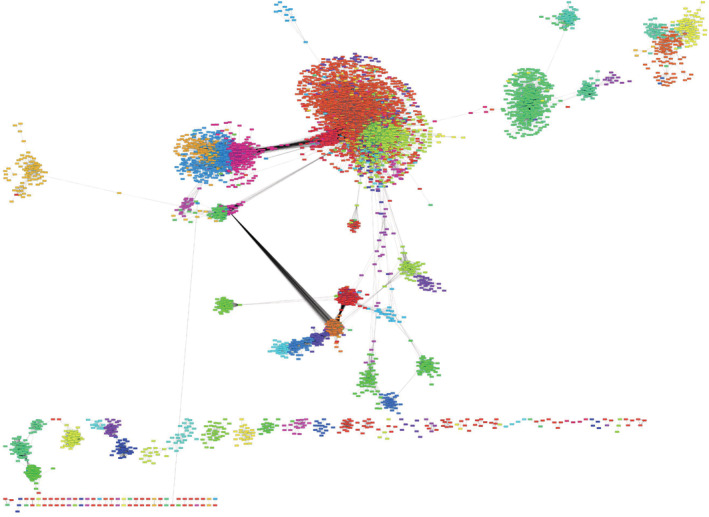
Arabidopsis weighted gene correlation network analysis (WGCNA) network. Nodes with correlation >.12 are shown. Each node (gene) is color‐coded to its respective modules. Each modules is designated to have a specific function based on gene enrichment analysis. The network topology is displayed using the Prefuse force directed layout algorithm in Cytoscape
2.2. Module annotation
Genes constituting a module co‐expressed under diverse conditions and it is therefore important to characterize the functions or functional pathways the modules represent. To this end, we performed GO term enrichment analysis using the TopGO analysis package (Alexa & Rahnenführer, 2009) to assess significantly enriched functional terms across all three aspects—Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). The enrichment analysis was performed on each of the 54 modules of the network. We shortlisted the enriched GO‐terms (FDR‐adjusted p value < .05) for all categories for each module. Based on the statistically significant BP, MF, and CC GO terms, and enrichment analysis of pathways (KEGG), we annotated each of the 54 modules as discussed below (Table 1). Refer to supporting information for complete data from GO analysis, tree maps and KEGG pathway enrichment analysis (Tables [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link] and Figures [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link]).
TABLE 1.
Enriched GO terms and KEGG pathways in modules 1–54
| Module number | Biological process | Molecular function | Cellular component | Enriched KEGG pathway | KEGG fold enrichment |
|---|---|---|---|---|---|
| 1 | RNA metabolic process | Nucleic acid binding | Nucleus | Basal transcription factors | 4.47 |
| 2 | Cell cycle | Microtubule binding | Cytoskeleton | DNA replication | 10.26 |
| 3 | Secondary metabolic process | Heme binding | Extracellular region | Flavonoid biosynthesis | 6.00 |
| 4 | Photosynthesis | Oxidoreductase activity | Chloroplast | Photosynthesis—antenna proteins | 15.19 |
| 5 | RNA modification | RNA binding | Mitochondrion/nucleolus | Ribosome biogenesis in eukaryotes | 8.52 |
| 6 | Vesicle−mediated transport | Transferase activity | Endomembrane system | Various types of N‐glycan biosynthesis | 9.71 |
| 7 | Plastid organization | Catalytic activity | Chloroplast/plastid | Porphyrin and chlorophyll metabolism | 9.11 |
| 8 | Translation | Structural constituent of ribosome | Ribosome | Ribosome | 10.37 |
| 9 | Catabolic process | Catalytic activity | Endomembrane system/Phagophore assembly | Autophagy—other | 10.71 |
| 10 | Ubiquitin‐dependent protein catabolic process | Peptidase activity | Proteosome complex | Proteasome | 21.85 |
| 11 | Cytoskeleton organization | Transferase activity | Golgi apparatus/cytoskeleton | One carbon pool by folate | 13.56 |
| 12 | Defense response | Kinase activity | Plasma membrane | Plant‐pathogen interaction | 11.48 |
| 13 | Localization | Phospholipase/lipase activity | Respiratory chain/membrane protein complex | Protein export | 12.65 |
| 14 | Purine metabolism | Metal ion/cation binding | Mitchondrial membrane/envelope | Citrate cycle (TCA cycle) | 12.67 |
| 15 | Protein modification | Ubiquitin protein ligase activity | Nucleus | — | — |
| 16 | Biotic stimulus | Kinase activity | Extracellular region | Plant‐pathogen interaction | 1.05 |
| 17 | Protein catabolic process | Ubiquitin conjugating enzyme activity | Ruffle membrane | SNARE interactions in vesicular transport | 14.07 |
| 18 | Cell wall organization or biogenesis | Oxidoreductase activity | Extracellular region | — | — |
| 19 | Pollen tube development | Structural constituent of cell wall | Pollen tube | Ether lipid metabolism | 65.71 |
| 20 | Response to stress | ADP binding | Plasma membrane/SMC loading complex | Alpha‐linolenic acid metabolism | 39.73 |
| 21 | Regulation of DNA replication | Sar guanyl‐nucleotide exchange factor | Telomere cap complex/CST complex | — | — |
| 22 | ATP metabolic process | NADH dehydrogenase activity | Mitochondrion/nucleolus | Oxidative phosphorylation | 23.87 |
| 23 | mRNA splicing | Nucleic acid binding | Nucleus | Spliceosome | 13.87 |
| 24 | Pollination/development | SNARE binding | Cell projection/Pollen tube | — | — |
| 25 | Response to water/chemical | Sucrose synthase activity | Monolayer‐surrounded lipid storage body | Glyoxylate and dicarboxylate metabolism | 16.43 |
| 26 | Chloropalst/plastid organization/embryo development | DNA supercoiling activity | Chloroplast/plastid | — | — |
| 27 | Phosphorylation | Transferase/kinase activity | Cell periphery | Cyanoamino acid metabolism | 16.27 |
| 28 | Protein phosphorylation | Catalytic activity/protein kinase activity/calmodulin binding | Plasma membrane/endosome | Plant‐pathogen interaction | 8.97 |
| 29 | Stomatal development | Transferase activity | Extracellular region | Fatty acid elongation | 37.96 |
| 30 | Response to hypoxia | Transcriptional regulation | Nucleus/CCR4‐NOT complex | Plant‐pathogen interaction | 11.17 |
| 31 | Response to biotic stimulus | Ligand‐gated ion channel | Extracellular region/Apoplast | Tryptophan metabolism | 20.34 |
| 32 | Protein amino acid modification | ADP binding | Extrinsic component of plasma membrane | — | — |
| 33 | Circadian rhythm/post‐embryonic development | Phosphorelay response regulator activity | Lipid droplet/vacuole | Circadian rhythm—plant | 41.07 |
| 34 | Electron transport chain | Chlorophyll binding/cofactor binding | Thylakoid | — | — |
| 35 | Carpel development | RNA polymerase II regulatory region sequence | Nucleus | Glycerolipid metabolism | 14.12 |
| 36 | Immune system response | Calmodulin binding | Plasma membrane | — | — |
| 37 | Lipid metabolic process | Hydrolase activity | Endomembrane system | — | — |
| 38 | Callose deposition/localization | Sucrose synthase activity | Anchored component of plasma membrane | Biotin metabolism | 53.39 |
| 39 | Fatty acid biosynthesis | Fatty acid synthesis | Chloroplast/plastid | Phosphatidylinositol signaling system | 66.56 |
| 40 | Protein phosphorylation | Protein kinase activity | Plasma membrane | Lysine biosynthesis | 53.39 |
| 41 | Amino acid biosynthetic process | Coenzyme binding | Chloroplast | — | — |
| 42 | Vascular/phloem transport | DNA binding transcription | Plasma membrane | Glucosinolate biosynthesis | 106.77 |
| 43 | Glucosinolate biosynthesis | Catalytic activity | Chloroplast | Protein export | 23.73 |
| 44 | Response to endoplasmic reticulum stress | Unfolded protein binding | Endoplasmic reticulum | — | — |
| 45 | Cellulose biosynthesis | Cellulose synthase | Trans‐Golgi network | — | — |
| 46 | Pollen tube development | Microfilament motor activity | Myosin complex | Endocytosis | 32.44 |
| 47 | Regulation of pollen developement | Protein kinase activity | Cell cortex | Thiamine metabolism | 48.81 |
| 48 | Translation/peptide biosynthesis | Structural constituent of ribosome | Plastid ribosome | Mismatch repair | 46.17 |
| 49 | Lactate catabolic process/monocarboxylic acid catabolic process/thiamine diphosphate biosynthetic process | Catalytic/transporter activity | Intracellular part | Starch and sucrose metabolism | 23.98 |
| 50 | DNA repair | Damaged DNA binding/DNA insertion or deletion | Anaphase‐promoting complex | Sulfur relay system | 43.80 |
| 51 | Starch catabolic process | Starch binding | Chloroplast | Inositol phosphate metabolism | 25.95 |
| 52 | RNA modification | Zinc ion binding | Mitochondrion | — | — |
| 53 | RNA splicing/gene expression | mRNA binding | Germ plasm | — | — |
| 54 | Embryonic meristem initiation/phosphorus metabolic process | Transferase/kinase activity | Plasma membrane | — | — |
Below, we describe some of these modules.
2.3. Module 4—Photosynthesis
The most significantly over‐represented biological process GO terms detected in Module 4 are related to photosynthesis, as shown in Figure 2. We also observed that four of the five major GO terms are related to photosynthesis. Additionally, the classic Fisher and FDR adjusted p values were found to be very low for GO terms such as electron transport chain, chloroplast organization, chlorophyll metabolic process, and plastid membrane organization, highlighting the significance of this module in the photosynthesis process. The other significantly overrepresented processes such as cofactor metabolism, protein biosynthesis, and vitamin metabolism are also strongly associated with photosynthesis. Molecular function GO terms such as chlorophyll binding, heme binding, oxidoreductase activity, ADP binding, and ATPase binding were also over‐represented in this module. Additionally, the cellular component GO terms such as, chloroplast part, plastid part, thylakoid, and photosynthetic membrane were found to be over‐represented in this module. We also performed the KEGG pathway enrichment analysis. Among the enriched pathways, photosynthesis had the highest enrichment, closely followed by photosynthesis related pathways such as carotenoid biosynthesis, carbon fixation, Porphyrin and chlorophyll metabolism (Tables S13–S15). Additionally, pathways such as glycolysis/gluconeogenesis, starch and sucrose metabolism, pentose phosphate pathways, and thiamine metabolism were also significantly enriched (Table S16). The presence of all these related GO terms and the enrichment of associated KEGG pathways point to direct physiological relationship/association with photosynthesis and therefore we annotated Module 4 as the photosynthesis module.
FIGURE 2.
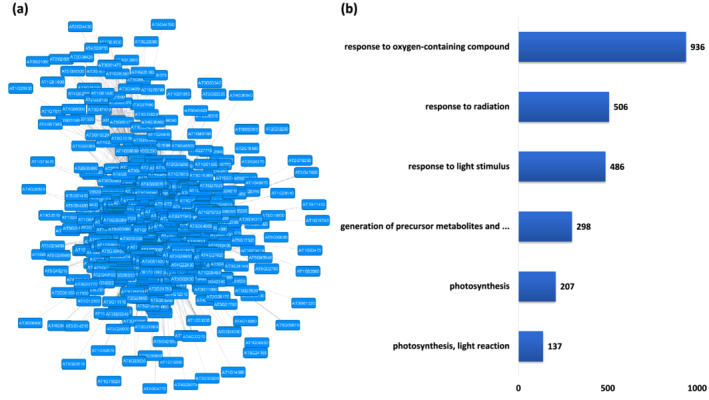
Functional analysis of Module 1. (a) Module 1 derived from the main Arabidopsis network showing genes associated with photosynthesis. (b) Five major biological process Gene Ontology (GO) terms derived from module 1. The number following each GO term refers to the number of genes that were found to be significant among the annotated to that category
2.4. Module 12—Defense response
In Module 12, the most significantly over‐represented biological process GO terms are related to defense response. Additionally, GO terms related to response to biotic stimulus, immune response, response to bacterium, innate immune response, response to drug, fungus, oomycetes, and so on were also found to be over‐represented (FDR‐adjusted p value < .05), as shown in Figure 3. The corresponding molecular function GO terms that were significantly enriched were of kinase activity and transferase activity. Overrepresented GO terms related to cellular component indicated enrichment of genes encoding proteins in the cell periphery, plasma membrane, extracellular region, Golgi transport complex, secretory vesicle/granules, and recycling endosome (Tables S45–S47). KEGG pathway enrichment analysis revealed plant‐pathogen interaction pathway as the significantly most enriched pathway (Table S48). Based on GO term and KEGG pathway enrichment, we annotated Module 12 as the defense response module.
FIGURE 3.
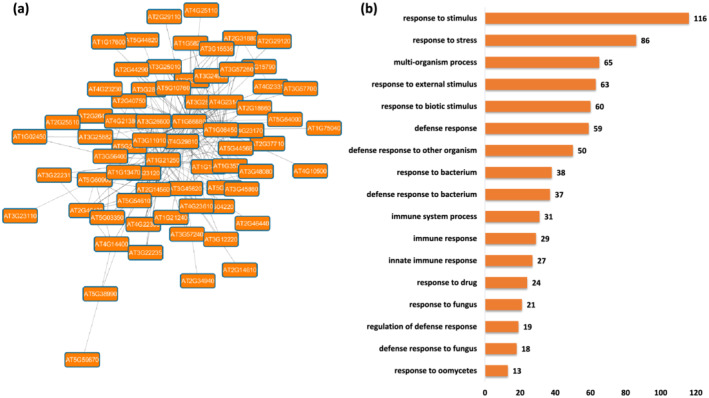
Functional analysis of Module 12. (a) Module 12 obtained from the main Arabidopsis network showing genes associated with defense. (b) Seventeen major biological process Gene Ontology (GO) terms derived from module 12. The number following each GO term refers to the number of genes that were found to be significant among the annotated to that category
2.5. Module 13—Localization
Module 13 is highly enriched with genes involved in localization and transport, as shown in Figure 4. The top overrepresented GO terms are related to intracellular transport, transport, and localization. Analysis of the GO terms related to molecular functions revealed significant over‐representation of genes encoding proteins that are involved in phosphorylation and phospholipase activation. Overrepresented GO terms associated with cellular component indicate enrichment of genes involved in the respiratory chain and thereby association with the mitochondrial complex (Tables S49–S51). On performing KEGG pathway enrichment analysis, the most enriched pathways were found to be protein export, oxidative phosphorylation, endocytosis, phagosome (Table S52). All the significant GO terms along with enriched KEGG pathways associated with this module indicate that the genes in this module are likely involved in localization and export, which led us to annotate Module 13 as localization module.
FIGURE 4.
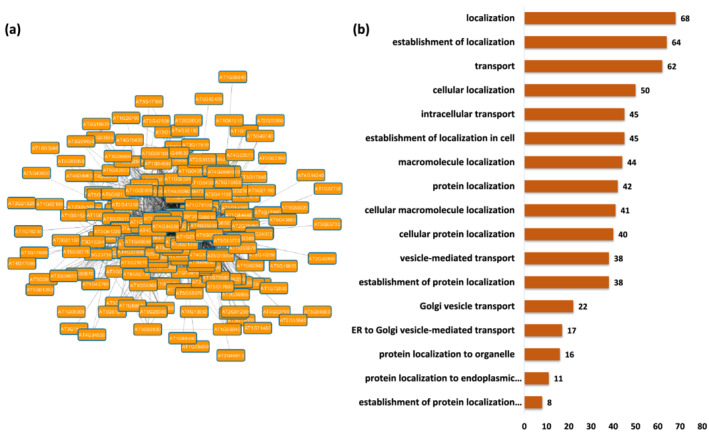
Functional analysis of Module 13. (a) Module 13 procured from the main Arabidopsis network showing genes associated with localization. (b) Seventeen major biological process Gene Ontology (GO) terms derived from module 13. The number following each GO term refers to the number of genes that were found to be significant among the annotated to that category
2.6. Module 16—Biotic stimulus
Module 16 is comprised of genes that are associated with GO terms related to response to biotic stimulus. GO terms such as response to fungus, oomycetes, bacteria, and antibiotic were found to be significantly over‐represented in this module. In addition, genes involved in defense response were over‐represented, as expected. We therefore noticed many of the GO terms are common among Module 12 (defense response) and Module 16. The associated molecular function GO terms that were significantly over‐represented include kinase activity, transferase activity, signal receptor activity, phosphotransferase activity, ion channel activity, and so on. The cellular component GO terms that are significantly enriched are or relate to extracellular region, plasma membrane, and cellular periphery (Figure 5 and Tables S61–S63). The KEGG pathway enrichment analysis revealed plant‐pathogen interaction as the only significantly enriched pathway in the module (Table S64). The presence of GO terms related to biotic stimulus and enrichment of plant‐pathogen interaction pathway led us to annotate this module as biotic stimulus module.
FIGURE 5.
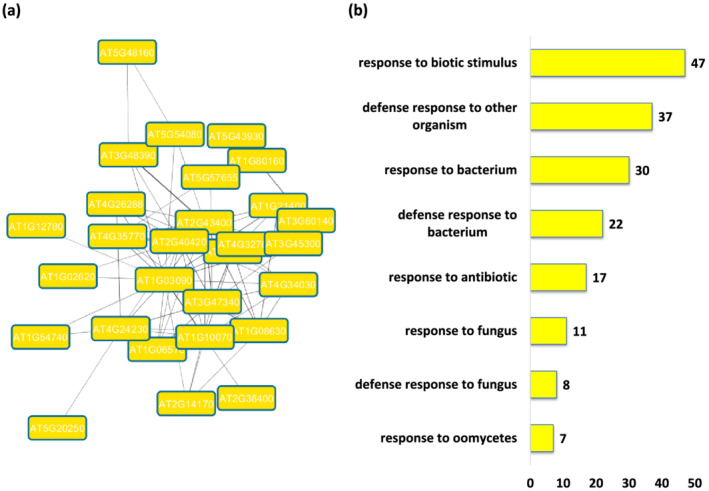
Functional analysis of Module 16. (a) Module 16 computed from the main Arabidopsis network showing genes associated with biotic stimulus. (b) Tree map representing the most statistically significantly overrepresented Biological Process (BP) GO terms. (c) Eight major biological process GO terms derived from module 16. The number following each GO term refers to the number of genes that were found to be significant among the annotated to that category
2.7. Module 20—Stress
The most overrepresented GO terms in Module 20 are related to response to stress. Most of the genes constituting this module are involved in response to either biotic (bacteria, chitin, other organisms) or abiotic stresses (oxygen, hypoxia, drug, and antibiotic). Several GO terms associated with regulation of defense to external stimulus as well as regulation of immune response were also found to be significantly enriched. The GO term related to molecular function ADP binding was found to be most over‐represented in the module. Considering the GO terms related to cellular component, plasma membrane raft and SMC loading complex were found most enriched (Tables S77–S79). On performing KEGG pathway analysis of genes comprising Module 20, alpha‐Linolenic acid metabolism and plant‐pathogen interaction pathways were found to be highly enriched (Table S80). Linolenic acid (Ln) released from chloroplast membrane galactolipids is a precursor of the phytohormone jasmonic acid (JA). The involvement of this hormone in different processes, such as responses to abiotic and biotic stress conditions, has been extensively studied (Wasternack, 2007). Seventy‐seven of the 88 genes in this module were found to be upregulated under high light stress (Figure 6). To further investigate this, we queried databases and literature and found a high proportion of these genes are responsive to many different abiotic stresses (e.g., cold, heat, excess light, salinity, ozone, wounding, and pathogen infection), ABA, externally applied ATP (eATP), methyl jasmonate, calcium, and singlet oxygen (Table 2) (Blanco et al., 2009; Choi et al., 2014; Consales et al., 2011; Davletova et al., 2005; Ding et al., 2014; Gadjev et al., 2006; Huang et al., 2008; Ikeuchi et al., 2017; Kleine et al., 2007; Larkindale & Vierling, 2007; Matsui et al., 2008; Nemhauser et al., 2006; Scarpeci et al., 2007; Tosti et al., 2006; Truman et al., 2006).
FIGURE 6.
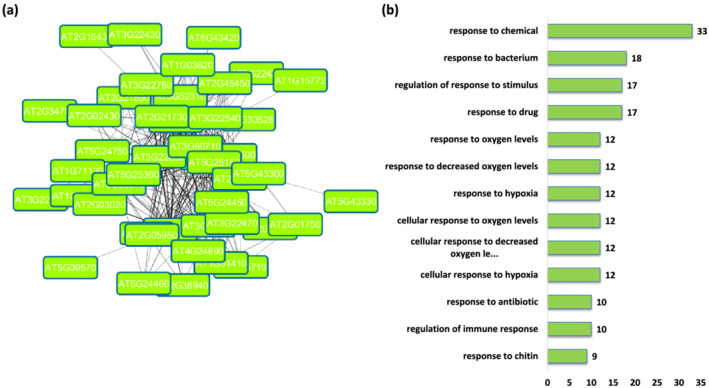
Functional analysis of Module 20. (a) Module 20 constructed from the main Arabidopsis network showing genes associated with stress. (b) Thirteen major biological process Gene Ontology (GO) terms derived from module 20. The number following each GO term refers to the number of genes that were found to be significant among the annotated to that category
TABLE 2.
Response of module 20 genes to different stresses, hormones, and stimuli
| Drought | Cold | Heat | High light | NaCl | Ozone | Wounding | Incompatible bacterial pathogen | eATP | Fe‐deficiency | Fe‐overload | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Abiotic stresses | 17 (19.31%) | 29 (32.95%) | 9 (10.22%) | 77 (87.5%) | 60 (68.18%) | 45 (51.14%) | 60 (68.18%) | 19 (21.6%) | 26 (29.54%) | 14 (15.9%) | 9 (10.22%) |
| ABA | ACC | Brassinolide | Cytokinin | Gibberellin | Indole‐3‐acetic acid | Methyl jasmonate | SA | H2O2 | O2 − | 1O2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Hormone/ROS | 5 (5.68%) | 1 (1.13%) | 1 (1.13%) | 0 (0%) | 1 (1.13%) | 1 (1.13%) | 10 (11.36%) | 11 (12.5%) | 34 (38.64%) | 22 (25%) | 2 (2.27%) |
Note: Top: Response of module 20 genes to different abiotic and biotic stresses. Bottom: Response of module 20 genes to different hormones, reactive oxygen species, and external ATP. (ABA, abscisic acid; ACC, 1‐aminocyclopropane‐1‐carboxylic acid; SA, salicylic acid; eATP, external ATP).
2.8. Gene‐specific networks
The Arabidopsis gene co‐expression network can also be interrogated for interactors of a gene of interest. We considered here genes encoding respiratory burst oxidase homolog D (RBOHD), A. thaliana NEET (AtNEET), and Heat shock transcription factor A1D (HSF1D) and extracted their direct neighbors from the Arabidopsis gene co‐expression network. We performed gene ontology and pathway analyses of each of these gene networks, and discuss below how this can be exploited to characterize proteins of yet unknown functions.
2.9. Respiratory burst oxidase homolog D (AT5G47910; RBOHD)
RBOHs are highly regulated membrane‐bound NADPH oxidases that help in catalyzing the formation of superoxide radical at the apoplast using the reducing power of NADPH at the cytosol (Lambeth, 2004; Sumimoto, 2008). They are part of a large protein family known as NOX and have been found to play a key signaling role in multiple developmental and stress response pathways via the regulated production of ROS (Lambeth, 2004; Sumimoto, 2008). In Arabidopsis, RBOHD (AT5G47910) has been shown to also be involved in mediating rapid systemic signaling (Miller et al., 2009). We interrogated the co‐expression network to decipher yet unknown genes involved in ROS signaling by identifying the interactors of RBOHD. In the co‐expression network, RBOHD is a part of module 36 and has direct connections to 583 genes. These connections are with genes that belong to 23 different modules (Table 1 and Figure 7), demonstrating the broad functions of RBOHD. GO analysis of the 583 gene set revealed that biological process terms related to stress, defense, hypoxia, signal transduction, and signaling were among the most significantly enriched terms. The GO cellular process terms related to the plasma membrane and cell periphery were the most enriched. The GO molecular function term protein kinase activity was found to be the most over‐represented (Figure 7). Furthermore, the KEGG pathway analysis revealed these genes to be enriched in plant‐pathogen interaction and MAPK signaling pathway. Among these 583 genes, 25 genes were found to be hypothetical protein genes (TAIR annotation) with functions yet not determined (Table S217). We further investigated the GO terms associated with these proteins and found that this set of genes has an enrichment in defense, stress, and oxygen‐related processes (Figure 7). To understand functional associations, we searched these hypothetical proteins in the STRING (Mering et al., 2003) database (along with RBOHD). Although the protein–protein association network did not show any high confidence link between these proteins and RBOHD, we isolated two subnetworks of connected proteins (Figure 7). In the smaller subnetwork (Figure 7), AT4G01090, a hypothetical protein (DUF3133) of unknown function, expressed at higher levels in the endodermis of the elongation zone of the root and the mature xylem (Winter et al., 2007), was found to interact with AT5G05190 (enhanced disease resistance 4; EDR4) (Mukhtar et al., 2011). Both of these proteins have also been found to modulate plant immunity by regulating clathrin heavy chain 2 (CHC2)‐mediated vesicle trafficking (Wu et al., 2015). Next, in the larger subnetwork (Figure 7), hypothetical protein AT1G32920 associate with proteins with enrichment of GO terms associated with response to wounding and stress (Figure 7). Additionally, both RBOHD (involved in oxidative stress) and AT1G32920 were found to be differentially regulated in shoots in the presence of whole soil microbial communities (Carvalhais et al., 2013). The overexpression of AT3G01470 (ATHB‐1) has been shown to mediate a pre‐adaption to hypoxic stress by reducing the endogenous level of nitric oxide (NO) in seeds (Thiel et al., 2011). Genes encoding transcription factors WRKY and AP2/EREBP, and genes related to hormone metabolism, namely, abscisic acid (ABA), salicylic acid (SA) and JA, have also been found to be upregulated in ATHB‐1 seeds (Thiel et al., 2011). Additionally, genes involved in signaling processes, such as those encoding MAPK kinases and receptor kinases, were also found to be strongly induced (Thiel et al., 2011), consistent with our KEGG pathway analysis. These genes with yet unknown functions may serve as a starting point for further analysis in deepening our understanding of RBOH proteins.
FIGURE 7.
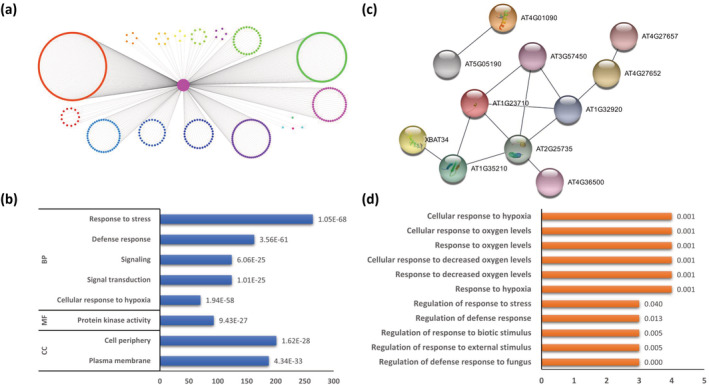
Gene specific network of respiratory burst oxidase homolog D (RBOHD). (a) Network represented using the first direct neighbors of RBOHD gene. Genes are color‐coded according to module assigned. (b) Enriched Gene Ontology (GO) terms in gene set constituting the first direct neighbors or RBOHD. The number following each GO term refers to the p‐value. (c) STRING‐DB network constructed out of hypothetical genes interacting with RBOHD. (d) GO terms found to be enriched in sub‐network of hypothetical genes
2.10. A. thaliana NEET (AT5G51720; AtNEET)
Iron–sulfur (Fe‐S) proteins play an integral role in various metabolic and regulatory pathways in plants (Balk & Pilon, 2011; Balk & Schaedler, 2014; Bernard et al., 2013; Hu et al., 2017; Lu, 2018; Przybyla‐Toscano et al., 2018). They likely originated under highly reducing environments during early evolution and are sensitive to damage by ROS (Andreini et al., 2017; Boyd et al., 2014; Lill, 2009; Sengupta et al., 2018). These proteins are also known to play an essential role as protein cofactors mediating diverse electron transfer reactions. Due to their inherent tendency to interact with oxygen to generate ROS that may inflict cellular damage, the biogenesis of their clusters is tightly regulated (Balk & Pilon, 2011; Balk & Schaedler, 2014; Bernard et al., 2013; Hu et al., 2017; Lu, 2018; Przybyla‐Toscano et al., 2018). In Arabidopsis, a single gene encoding a NEET protein (AT5G51720; AtNEET) has been previously proposed to play a significant role in maintaining iron and ROS homeostasis (Nechushtai et al., 2012). In the co‐expression network, AtNEET is a member of module 4 (the photosynthesis module) and has direct connections with 1023 genes from 9 different modules (Table 1 and Figure 8). On performing GO analysis on this set of genes, we found enrichment of terms related to photosynthesis, plastid, chloroplast, and vesicle organization (Figure 8). Most of these genes belong to module 4, followed by modules 6 and 13 that are associated with vesicle transport and localization respectively. Among the enriched cellular component GO terms were plastid, chloroplast, and thylakoid. AtNEET has previously been shown to be localized in mitochondria and chloroplasts (Khan et al., 2018; Su et al., 2013). Among the enriched molecular function GO terms were mRNA binding, oxidoreductase activity, and RNA binding. Several of these genes have already been associated with AtNEET, for example, AT1G44446 (Chlorophyllide a oxygenase), AT2G04700 (Ferredoxin thioredoxin reductase), AT2G24820 (TIC55), and AT3G05345 (Chaperon DnaJ) were found to have elevated expression when AtNEET was disrupted (Zandalinas et al., 2020).
FIGURE 8.
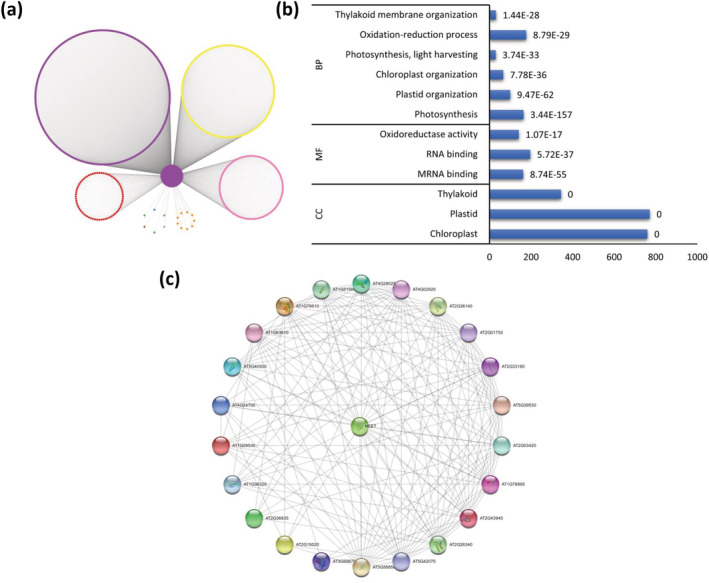
Gene specific network of AtNEET. (a) Network represented using the first direct neighbors of AtNEET. Genes are color‐coded according to module assigned. (b) Enriched Gene Ontology (GO) terms in gene set constituting the first direct neighbors or AtNEET. The number following each GO term refers to the p value. (c) STRING‐DB network constructed out of hypothetical genes interacting with AtNEET
Further investigation of these genes revealed 23 of the 1024 genes (Table S218) to be of unknown function. Ten of the 23 genes were found linked by STRING at a high confidence setting based on expression data across a large number of experiments (Figure 8). AT2G15020, a protein with unknown function, was found to be upregulated in SuperFifty (SF, an extract from the seaweed Ascophyllum nodosum) exposed Arabidopsis (Omidbakhshfard et al., 2020). It was shown that SF exposure largely prevents paraquat (PQ)‐induced oxidative stress in Arabidopsis (Omidbakhshfard et al., 2020). On lowering the confidence setting in STRING, all 23 genes got connected by means of co‐occurrence patterns across closely related genomes. As expected, due to lack of functional data for these genes, no GO terms were found to be enriched in this gene set. However, their association with AtNEET and strong connectivity among many of them as revealed by STRING points to their broader functional roles related to photosynthesis and stress.
2.11. Heat shock transcription factor A1D (AT1G32330; HSFA1D)
High temperature stress has a detrimental impact on many aspects of growth and development in plants (Lippmann et al., 2019). So far, histone sensors, unfolded protein response sensors in the endoplasmic reticulum (ER), plasma membrane channels, and phytochrome B are some of the well characterized heat sensors recorded in plants (Jung et al., 2016; Mittler et al., 2012; Vu et al., 2019). Recently, a class of Heat Shock Factor (HSF) family transcription factors (e.g., HSFA1s), was found to be involved in the cellular response to heat stress (Cortijo et al., 2017; Ohama et al., 2017). In our network, AT1G32330 or HSFA1D was found in module 6 and had direct connections to 8741 genes assigned to over 80% of the modules (Figure 9). GO analysis of these genes revealed enrichment of RNA metabolic process, nucleic acid metabolic process, cellular component organization, biogenesis, etc. (Figure 9). Furthermore, cellular component GO terms such as, protein‐containing complex, non‐membrane bound organelle, organelle lumen, and endomembrane system and molecular function GO terms such as, RNA binding, nucleic acid binding, small molecule binding, and mRNA binding, were found to be enriched (Figure 9). KEGG pathway analysis showed spliceosome, RNA transport, mRNA surveillance pathway, and protein processing in ER to be most enriched. These aforementioned enriched GO terms/KEGG pathways along with the high degree of connectivity (>8000 first direct neighbors) suggests the roles of HSFA1D in a plethora of system‐wide biological processes.
FIGURE 9.
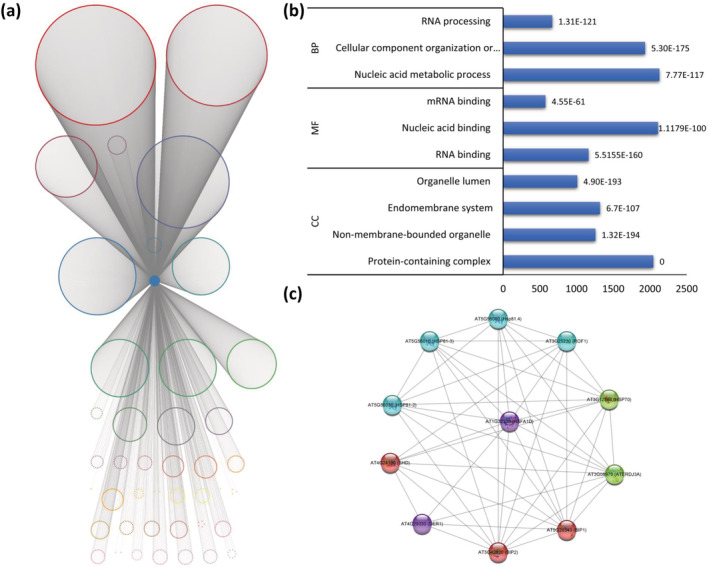
Gene specific network of HSFA1D. (a) Network represented using the first direct neighbors of HSFA1D. Genes are color‐coded according to module assigned. (b) Enriched Gene Ontology (GO) terms in gene set constituting the first direct neighbors or HSFA1D. The number following each GO term refers to the p value. (c) STRING‐DB network constructed out of hypothetical genes interacting with HSFA1D
Based on structural characteristics and phylogenetic analysis, Arabidopsis HSFs have been classified into three major classes (A, B, and C) and 14 groups as A1‐9, B1‐4, and C1 (Nover et al., 2001). Thus, the large number of HSFs and the complex modulation of their activities by hetero‐oligomerization render the attribution of specific functions very challenging. It has been shown previously that HSFA1D is involved in oxidative stress tolerance (Liu & Charng, 2013). The role of HSFA1D and HSFA1E in inducing HSFA2 expression under high light (HL) and heat stress (HS) has been established. Furthermore, HSFA1D and HSFA1E double knockout mutants showed impaired tolerance to HS stress. These findings suggest the pivotal role of HSFA1D and HSFA1E as a transcriptional regulator of HSFA2, and also as a key regulator for HSF signaling in response to environmental stress (Nishizawa‐Yokoi et al., 2011). The gene network of HSFA1D revealed 10 genes, namely, AT3G08970 (ATERDJ3A), AT5G28540 (BIP1), AT5G42020 (BIP2), AT4G29330 (DER1), AT3G12580 (HSP70), AT5G56030 (HSP81‐2), AT5G56010 (HSP81‐3), AT5G56000 (Hsp81.4), AT3G25230 (ROF1), and AT4G24190 (SHD) (Figure 9), which have been consistently shown to be involved in the response to stress and in protein folding (Bokszczanin et al., 2013; Dos Reis et al., 2012; Dossa et al., 2016; Guo et al., 2016; Jacob et al., 2017; Lu et al., 2016; Moumeni et al., 2011; Ohama et al., 2016; Shah et al., 2020; Swindell et al., 2007; Tiwari et al., 2020; Virdi et al., 2015; Wang et al., 2020, 2016; Wen et al., 2017; Yamada et al., 2007; Yamada & Nishimura, 2008; Zhang et al., 2015). Additionally, AT1G18080 (RACK1A), AT1G48630 (RACK1B), and AT3G18130 (RACK1C) have been shown to be involved in plant development (Chen et al., 2006) and abscisic acid (ABA) response, as well as in the interaction with eIF6, a key regulator of ribosome assembly (Guo et al., 2011). Along with the aforementioned genes, we also identified 174 novel genes (Table S219) with yet unknown functions (annotated as hypothetical proteins in the TAIR database). Analysis of these genes using the STRING database identified AT3G49490 as one of the downregulated stage 1 proteins that are involved in translation and protein folding during early leaf growth, along with AT2G04030 (HSP90.5) and AT1G18080 (RACK1A) (Baerenfaller et al., 2012). AT2G28330 was shown to bind with CYCB2;4 and CDKB1;1, which are associated with SMR1 and SMR2 proteins in the cell cycle interactome (Van Leene et al., 2010). Our analysis provides therefore new insights into the function of several different proteins with unknown function, associated with HSFA1D.
Together, these results demonstrate the ability of the network approach presented here to not just decipher functional modules but also derive gene‐specific networks with connections spanning multiple functional modules. Our analysis also uncovered many genes of unknown functions with connectivity to well characterized genes, thus helping understand their potential roles in different biological processes.
3. IDENTIFICATION OF MODULES ENRICHED IN DIFFERENTIALLY EXPRESSING GENES UNDER STRESS CONDITIONS
Balfagón et al., 2019 recently published a study investigating the response of Arabidopsis plants to a combination of high light (HL) and heat stress (HS). Combined HL + HS led to irreversible damage to photosystem II (PSII), decreased D1 (PsbA) protein levels, and an enhanced transcriptional response indicative of PSII repair activation. Several unique aspects of this stress combination were identified, including enhanced accumulation of JA and JA‐Ile, elevated expression of over 2200 different transcripts that are unique to the stress combination (included many that are JA‐associated), and distinctive structural changes to chloroplasts. Differentially expressing genes (DEGs) were determined based on statistical hypothetical testing by DESeq2. Here, we revisited this list and asked if our network approach can decipher genes that were likely altered in expression and therefore potentially biologically significant but were not deemed statistically significant by DESeq2. DEGs were identified by comparing plants exposed to HL, HS, and HL + HS with control (Balfagón et al., 2019). These DEGs were mapped onto the gene modules of the co‐expression network, for each of the HL, HS, and HL + HS experiments. Prior to mapping, genes with very low or no expression were removed to minimize potential artifacts. Gene modules enriched in DEGs were determined by performing Fisher's test (those with p value < .05 were deemed significantly enriched). For HL, we found 52 of the 54 modules had varying number of DEGs mapped onto them. Thirteen among these 52 modules harbored a large majority of genes (over 60%) that were DEGs or were upregulated or downregulated by two‐fold (equivalently, log2foldChange of 1) or more but not deemed significant in the Balfagón et al. (2019) study. Seven of these modules contained many genes that were two‐fold or more elevated or attenuated in expression level but were deemed insignificant by the (Balfagón et al., 2019) study. For HS and HL + HS, we found 6 and 10 such modules respectively (Table 3). Interrogating these modules of interest for each of the three stress conditions, we found three previously missed genes of yet unknown functions, that is, with expression fold‐change of two or more yet deemed insignificant. Since these genes lie within the DEG enriched modules, we posit that they are likely upregulated or downregulated and play significant roles in the plant response to stress. These genes are AT3G38630 (module 16; in HL, HS, HL + HS), AT3615518 (module 12; in HL), and AT2G07787 (module 22; in HS). These novel genes are harbored by modules associated with defense response (module 12) and biotic stimulus (module 16) and, therefore, are likely contributing to the response to stress in Arabidopsis.
TABLE 3.
Enrichment analysis of genes in network modules expressing differentially under high light (HL), heat (HS), and combination of high light and heat (HL + HS) stresses
| Stress | Module | DEG count | Up & down regulated non‐DEG count | Total DEG count | Module gene count | DEG percent in module | DEG Pval percent in module | Enrichment fold change | Enrichment fisher test |
|---|---|---|---|---|---|---|---|---|---|
| HL | 5 | 833 | 21 | 854 | 1,188 | 71.89 | 70.12 | 2.0 | 4.3E−237 |
| 8 | 455 | 1 | 456 | 584 | 78.08 | 77.91 | 2.3 | 2.4E−157 | |
| 12 | 149 | 9 | 158 | 242 | 65.29 | 61.57 | 1.8 | 3.4E−32 | |
| 16 | 71 | 14 | 85 | 124 | 68.55 | 57.26 | 1.7 | 8.6E−14 | |
| 17 | 77 | 0 | 77 | 116 | 66.38 | 66.38 | 1.9 | 3.5E−20 | |
| 20 | 36 | 19 | 55 | 89 | 61.80 | 40.45 | 1.2 | 2.2E−03 | |
| 26 | 61 | 0 | 61 | 69 | 88.41 | 88.41 | 2.6 | 6.3E−28 | |
| 27 | 56 | 2 | 58 | 68 | 85.29 | 82.35 | 2.4 | 1.6E−22 | |
| 32 | 40 | 0 | 40 | 57 | 70.18 | 70.18 | 2.0 | 2.3E−12 | |
| 41 | 35 | 0 | 35 | 41 | 85.37 | 85.37 | 2.5 | 1.6E−15 | |
| 52 | 18 | 5 | 23 | 29 | 79.31 | 62.07 | 1.8 | 3.8E−05 | |
| 53 | 16 | 0 | 16 | 25 | 64.00 | 64.00 | 1.9 | 6.0E−05 | |
| 54 | 18 | 0 | 18 | 25 | 72.00 | 72.00 | 2.1 | 1.6E−06 | |
| HS | 12 | 141 | 7 | 148 | 242 | 61.16 | 58.26 | 1.3 | 8.3E−18 |
| 15 | 97 | 0 | 97 | 144 | 67.36 | 67.36 | 1.5 | 8.7E−19 | |
| 16 | 101 | 7 | 108 | 124 | 87.10 | 81.45 | 1.8 | 1.4E−30 | |
| 20 | 46 | 15 | 61 | 89 | 68.54 | 51.69 | 1.1 | 7.9E−05 | |
| 22 | 40 | 8 | 48 | 79 | 60.76 | 50.63 | 1.1 | 3.8E−04 | |
| 23 | 53 | 0 | 53 | 78 | 67.95 | 67.95 | 1.5 | 4.2E−11 | |
| 27 | 49 | 3 | 52 | 68 | 76.47 | 72.06 | 1.6 | 6.6E−12 | |
| 32 | 37 | 0 | 37 | 57 | 64.91 | 64.91 | 1.4 | 2.1E−07 | |
| 40 | 26 | 0 | 26 | 42 | 61.90 | 61.90 | 1.4 | 7.1E−05 | |
| 44 | 30 | 0 | 30 | 35 | 85.71 | 85.71 | 1.9 | 4.2E−11 | |
| 51 | 19 | 1 | 20 | 29 | 68.97 | 65.52 | 1.4 | 1.8E−04 | |
| 53 | 15 | 0 | 15 | 25 | 60.00 | 60.00 | 1.3 | 3.9E−03 | |
| HL + HS | 4 | 886 | 12 | 898 | 1,417 | 63.37 | 62.53 | 1.2 | 1.0E−104 |
| 8 | 406 | 0 | 406 | 584 | 69.52 | 69.52 | 1.3 | 1.9E−66 | |
| 10 | 294 | 1 | 295 | 447 | 66.00 | 65.77 | 1.2 | 6.2E−41 | |
| 11 | 224 | 0 | 224 | 336 | 66.67 | 66.67 | 1.3 | 8.5E−33 | |
| 12 | 139 | 7 | 146 | 242 | 60.33 | 57.44 | 1.1 | 6.9E−13 | |
| 13 | 145 | 2 | 147 | 241 | 61.00 | 60.17 | 1.1 | 8.1E−16 | |
| 14 | 139 | 0 | 139 | 193 | 72.02 | 72.02 | 1.4 | 7.2E−26 | |
| 16 | 95 | 9 | 104 | 124 | 83.87 | 76.61 | 1.4 | 2.3E−21 | |
| 17 | 72 | 0 | 72 | 116 | 62.07 | 62.07 | 1.2 | 3.1E−09 | |
| 27 | 51 | 6 | 57 | 68 | 83.82 | 75.00 | 1.4 | 2.3E−11 | |
| 28 | 47 | 0 | 47 | 67 | 70.15 | 70.15 | 1.3 | 6.6E−09 | |
| 29 | 43 | 0 | 43 | 63 | 68.25 | 68.25 | 1.3 | 7.5E−08 | |
| 30 | 37 | 1 | 38 | 62 | 61.29 | 59.68 | 1.1 | 7.7E−05 | |
| 32 | 38 | 0 | 38 | 57 | 66.67 | 66.67 | 1.3 | 1.0E−06 | |
| 33 | 47 | 1 | 48 | 55 | 87.27 | 85.45 | 1.6 | 1.1E−14 | |
| 34 | 29 | 5 | 34 | 54 | 62.96 | 53.70 | 1.0 | 6.0E−03 | |
| 39 | 37 | 0 | 37 | 44 | 84.09 | 84.09 | 1.6 | 2.2E−11 | |
| 40 | 33 | 0 | 33 | 42 | 78.57 | 78.57 | 1.5 | 7.8E−09 | |
| 41 | 33 | 0 | 33 | 41 | 80.49 | 80.49 | 1.5 | 2.5E−09 | |
| 44 | 27 | 0 | 27 | 35 | 77.14 | 77.14 | 1.4 | 3.7E−07 | |
| 51 | 27 | 1 | 28 | 29 | 96.55 | 93.10 | 1.7 | 7.2E−11 | |
| 52 | 29 | 0 | 29 | 29 | 100.00 | 100.00 | 1.9 | 4.8E−14 | |
| 53 | 21 | 0 | 21 | 25 | 84.00 | 84.00 | 1.6 | 5.8E−07 |
Note: Differentially expressed genes (DEGs) from a previous study dataset (Balfagón et al., 2019) were mapped onto the gene modules generated using WGCNA. The gene modules that were enriched in genes expressing differentially during stress conditions were thus identified. Modules that are enriched with DEGs were determined by performing Fisher test; modules with p value ≤ .05 were deemed significantly enriched. Furthermore, modules with large majority of genes (over 60%) significantly differentially expressed and otherwise two‐fold or more upregulated or downregulated were identified. Stress: stress under consideration; Module: Modules with over 60% DEGs; DEG Count: Genes that are considered differentially expressing as per the study; Up & Down Regulated Non‐DEG Count: Genes that are 2‐fold or more up/down regulated but had p value > .05; Total DEG Count: DEG Count + Up & Down Regulated Non‐DEG Count; Module Gene Count: Number of genes in the module; DEG Percent In Module: Percentage of genes in Module that are considered as differentially expressing as per the study as well as genes that are 2‐fold or more up/down regulated but has p value > .05; DEG p value Percent In Module: Percentage of genes in Module that are considered as differentially expressing as per the study; Enrichment Fold Change: Fold enrichment of DEGs in a Module; Enrichment Fisher Test: p value generated using Fisher test to indicate the significance of DEG enrichment in a Module.
Among the modules with over 60% genes differentially expressing, five were identified across all three stress conditions—module 12 (defense response), module 16 (biotic stimulus), module 27 (phosphorylation), module 32 (amino acid modification), and module 53 (RNA splicing) (Table 1). Three of these contained genes with two‐fold or more expression change but deemed insignificant in the previous study (module 12, defense response; module 16, biotic stimulus; and module 27, phosphorylation). Our study therefore highlights the power of the co‐expression network in deciphering novel genes that are otherwise missed by the standard approach; indeed our approach identified many “insignificant” genes (Balfagón et al., 2019) study that are likely biologically significant and playing key roles in the stress response in Arabidopsis.
4. DISCUSSION
Although a number of methods have been developed for the construction of gene co‐expression network, there are computational challenges abound considering the vast amount of expression (RNA‐Seq) datasets to handle, particularly for model organisms such as Arabidopsis. Even before the application of a network building tool, this vast amount of data needs to be downloaded, quality checked, preprocessed, and then aligned against the reference genome. We utilized the services of High‐Performance Computing (HPC) of the University of North Texas to store the data in a custom local database. We could download terabytes of data from NCBI SRA using a parallelized version of the sra‐tools' fastq‐dump utility with the default prefetch utility. We were also apprehensive that the alignment may require extensive CPU and memory and therefore, we assessed computational efficiency of several alignment tools. We selected Salmon due to it being ultrafast with low memory requirement; Salmon could quickly align an RNA‐Seq dataset in less than 10 min on average with very low memory requirements, which, in turn, enabled us to parallelize the workflow over more CPUs than possible with popular aligners like STAR (Dobin et al., 2013). In addition, custom iteration scripts were written to iterate through the data for analysis rather than relying on packages like Numpy or Pandas that instead require loading the entirety of data at once. Although slower, it allowed us to use moderately powerful desktop workstations to process large data matrices. Similar pipelines for the analysis of RNA‐Seq data and subsequent generation of co‐expression network have previously been established, for example, LSTrAP (Large‐Scale Transcriptome Analysis Pipeline) that combines all essential tools to construct co‐expression networks based on RNA‐Seq data into a single, efficient workflow (Proost et al., 2017). We chose to develop our own workflow that has several components common to LSTrAP but also has distinct components such as Salmon for read alignment. We have chosen to utilize only those tools that have been extensively tested previously and have frequently been used for similar analysis.
Large amount of data collected from many different experiments does bring in additional challenges such as the batch effect. We attempted a few normalization methods to address the batch effect, such as TMM normalization (edgeR; Robinson et al., 2010) and variance‐stabilization (DESEq2; Love et al., 2014), but the lack of reliable annotation for many datasets made it difficult to apply batch correction properly. Our pre‐processing entailed removal of datasets with reads covering less than half of the TAIR10 transcriptome in the alignment and those that had more than 20% of their reads classified as unmapped, thereby providing us with only high‐quality datasets to render a robust network. Visualization of this large network using Cytoscape was another bottleneck, which we circumvented by using a density‐based metric to trim our network to a viewable form in Cytoscape. GO enrichment was exploited to optimize WGCNA's parametric setting or thresholds for our network, which was made possible by running a batch script over TopGO.
The Arabidopsis network revealed genes that were coupled by expression under many different conditions, and their organization into functional modules. Functional coupling, revealed in such networks, could be across tissues or could be specific to a tissue. Of course, tissue‐specific networks may reveal additional information (e.g., Burks & Azad, 2016), which can be investigated in follow‐up studies. The modules are an important feature of a co‐expression network and can be visualized at both coarse‐grained and fine‐grained resolutions. The module configurations can be retrieved from a wide‐range of inflation parameter in WGCNA. Note that the inflation parameter setting used in our analysis yielded fewer modules, with many of them large (coarse‐grained configuration), which could likely be representing many different pathways that are cross‐talking during a certain biological process. For this study, we annotated the modules based on only the most enriched GO BP term; however, it is plausible to have a module represented by multiple GO terms as genes harbored by the module might be participating in multiple processes or conferring multiple functions. In addition to this coarse‐grained configuration data, we have also made available an additional dataset in our project GitHub repository representing a fine‐grained module configuration (>250 clusters). This facilitates visualization of the modules at different resolutions.
In addition to extraction of gene modules from the network, gene‐specific networks were by derived by extracting direct neighbors of specific genes of interest, such as of RBOHD, AtNEET, and HSFA1D. We observed that, in addition to being well‐connected with other genes within their own modules, these genes were also directly associated with some genes from one or more other modules. This is not unexpected as due to functional relatedness or dependency, they might be connected to genes belonging to other modules as well. This could signify the relevance of a gene in many different processes; some genes could act as “hubs” in the network, they could not just have connections within their own modules but also many connections with genes from other modules, highlighting their functional significance in regulating many different processes or acting as mediator for enabling cross‐talks between different processes; on the other hand, some genes could have more specific roles and those could have connections only within their module or much fewer connections outside of their own module.
In addition to the innovations mentioned above that helped realize the network of this scale, we developed innovative approaches to interrogate the network to obtain biologically important information. For example, our approach to map differentially expressing genes from an experiment onto the network and then identify modules enriched in differentially expressing genes helped decipher putative transcriptionally altered genes by virtue of their association with differentially expressing genes within the enriched modules. These genes were two‐fold or more elevated or depressed in expression yet were deemed statistically insignificant in the original studies; network analysis provided support to these novel discoveries and close examination of known functions or associations of some of these genes rendered even more confidence over the novel predictions. Of course, more follow‐up analyses are needed to further validate these genes. Furthermore, the unbiased network approach has made possible identification of both known and unknown pathways or networks of genes that are regulated under certain conditions or stages, shining a light on molecular processes at a scale that is not possible to realize based on standard gene‐focused studies.
5. CONCLUSIONS
This study advances our knowledge of Arabidopsis functional genomics by constructing a new gene co‐expression network that leverages information from thousands of Arabidopsis RNA‐Seq datasets available for interrogation in public databases. The high amount of transcriptomics data rendered a robust network that can be a valuable resource for the Arabidopsis community to interrogate for genes, pathways or datasets of interest. The modules identified by our study represent pathways or networks of pathways that interact to confer certain biological functions. In addition to identifying differentially regulated genes by performing expression studies, researchers may use the network for an unbiased assessment of pathways or networks of pathways, both known and unknown, that are differentially regulated during different stages or conditions. In addition to providing the complete datasets as a supplement to this paper, we have made them available at https://doi.org/10.6084/m9.figshare.16752733.v4. All associated source codes are provided at the GitHub site: https://github.com/sohamsg90/WGCNA_Arabidopsis-main.
6. MATERIALS AND METHODS
6.1. Data collection and filtering
The entire collection of mRNA‐Seq A. thaliana datasets available at the NCBI SRA was retrieved and converted to FASTQ format using the prefetch and fastq‐dump utilities of SRA‐Tools v2.92 (Leinonen et al., 2010). Single and paired‐end read data representing a variety of experiments were processed and reads from each dataset were aligned to the TAIR10 transcriptome of the Ensembl Plants release 46 (Berardini et al., 2015; Bolser et al., 2016). Transcriptome indexing and alignment was performed using the v.12.0 release of the pseudoalignment program Salmon (Patro et al., 2017).
Datasets with reads covering less than half of the TAIR10 transcriptome in the alignment and those that had more than 20% of their reads classified as unmapped were removed from further analysis. For a gene with multiple isoforms (transcripts), the normalized expression values for transcripts of the gene were summed and log2 transformed. A 18,122 by 21,460 matrix representing the log transformed normalized expression values (log2 TPM), with rows corresponding to A. thaliana genes and columns to experiments, was imported into WGCNA for gene co‐expression network construction (Langfelder & Horvath, 2008).
6.2. Network construction
The aforementioned expression matrix was inputted into WGCNA with the blockwiseModules function to generate a signed network under a soft‐thresholding power of 12, minimum module size of 10, and merged cut height of .005. Gene associations, based on the underlying Pearson correlation and topological overlap of all gene to gene pairs, were generated directly from the expression matrix using the TOMsimilarityFromExpr function. Module assignments were exported using the exportNetworktoCytoscape function.
6.3. Enrichment analysis
Each cluster produced by WGCNA was assessed for gene ontology (GO) term enrichment using a batch script of the TopGO analysis package (Alexa & Rahnenführer, 2009). GO term enrichment was examined across all three categories, namely, Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). Unadjusted p‐values for GO terms were FDR‐adjusted using the p.adjust function of R. All enrichment tests within TopGO were based on “classic” algorithm with the statistics parameter set to “fisher”. GO annotations were imported from the org. At.tair.db package of the Bioconductor release v3.11 (Reimers & Carey, 2006). KEGG pathway enrichment was performed using the clusterProfiler R Bioconductor package (Yu et al., 2012).
6.4. Module annotation
The RNA‐Seq based Arabidopsis gene co‐expression network comprised of 54 gene modules. Genes within a module co‐express under diverse conditions, and therefore, functional coupling among the module members is expected. To annotate these modules, we performed enrichment analysis for BP, CC, and MF ontology terms in all of the 54 modules. We assigned labels to each module pertaining to the most significant GO terms for each module. This was performed primarily by construction of Tree Maps of GO terms using TopGO with the top 10 most significant terms (based on the p‐value and highest enrichment). The network provided a comprehensive insight into the relationships among genes in different functional modules.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
D.J.B. and R.K.A. conceived and designed the study. D.J.B., S.S., and R.D. performed the experiments. D.J.B., S.S., R.D., R.M., and R.K.A. analyzed and interpreted the results. S.S. and R.K.A wrote the manuscript, with inputs and contributions of D.J.B., R.D., and R.M. R.K.A. coordinated the project and agrees to serve as the author responsible for contact and ensures communication. All authors have read and approved the final manuscript.
Supporting information
Supplementary figure S1: Selection of cutoff values
Supplementary figure S2: Top 5 overrepresented BP terms of Module‐1
Supplementary figure S3: Top 5 overrepresented CC terms of Module‐1
Supplementary figure S4: Top 5 overrepresented MF terms of Module‐1
Supplementary figure S5: Top 5 overrepresented BP terms of Module‐2
Supplementary figure S6: Top 5 overrepresented CC terms of Module‐2
Supplementary figure S7: Top 5 overrepresented MF terms of Module‐2
Supplementary figure S8: Top 5 overrepresented BP terms of Module‐3
Supplementary figure S9: Top 5 overrepresented CC terms of Module‐3
Supplementary figure S10: Top 5 overrepresented MF terms of Module‐3
Supplementary figure S11: Top 5 overrepresented BP terms of Module‐4
Supplementary figure S12: Top 5 overrepresented CC terms of Module‐4
Supplementary figure S13: Top 5 overrepresented MF terms of Module‐4
Supplementary figure S14: Top 5 overrepresented BP terms of Module‐5
Supplementary figure S15: Top 5 overrepresented CC terms of Module‐5
Supplementary figure S16: Top 5 overrepresented MF terms of Module‐5
Supplementary figure S17: Top 5 overrepresented BP terms of Module‐6
Supplementary figure S18: Top 5 overrepresented CC terms of Module‐6
Supplementary figure S19: Top 5 overrepresented MF terms of Module‐6
Supplementary figure S20: Top 5 overrepresented BP terms of Module‐7
Supplementary figure S21: Top 5 overrepresented CC terms of Module‐7
Supplementary figure S22: Top 5 overrepresented MF terms of Module‐7
Supplementary figure S23: Top 5 overrepresented BP terms of Module‐8
Supplementary figure S24: Top 5 overrepresented CC terms of Module‐8
Supplementary figure S25: Top 5 overrepresented MF terms of Module‐8
Supplementary figure S26: Top 5 overrepresented BP terms of Module‐9
Supplementary figure S27: Top 5 overrepresented CC terms of Module‐9
Supplementary figure S28: Top 5 overrepresented MF terms of Module‐9
Supplementary figure S29: Top 5 overrepresented BP terms of Module‐10
Supplementary figure S30: Top 5 overrepresented CC terms of Module‐10
Supplementary figure S31: Top 5 overrepresented MF terms of Module‐10
Supplementary figure S32: Top 5 overrepresented BP terms of Module‐11
Supplementary figure S33: Top 5 overrepresented CC terms of Module‐11
Supplementary figure S34: Top 5 overrepresented MF terms of Module‐11
Supplementary figure S35: Top 5 overrepresented BP terms of Module‐12
Supplementary figure S36: Top 5 overrepresented CC terms of Module‐12
Supplementary figure S37: Top 5 overrepresented MF terms of Module‐12
Supplementary figure S38: Top 5 overrepresented BP terms of Module‐13
Supplementary figure S39: Top 5 overrepresented CC terms of Module‐13
Supplementary figure S40: Top 5 overrepresented MF terms of Module‐13
Supplementary figure S41: Top 5 overrepresented BP terms of Module‐14
Supplementary figure S42: Top 5 overrepresented CC terms of Module‐14
Supplementary figure S43: Top 5 overrepresented MF terms of Module‐14
Supplementary figure S44: Top 5 overrepresented BP terms of Module‐15
Supplementary figure S45: Top 5 overrepresented CC terms of Module‐15
Supplementary figure S46: Top 5 overrepresented MF terms of Module‐15
Supplementary figure S47: Top 5 overrepresented BP terms of Module‐16
Supplementary figure S48: Top 5 overrepresented CC terms of Module‐16
Supplementary figure S49: Top 5 overrepresented MF terms of Module‐16
Supplementary figure S50: Top 5 overrepresented BP terms of Module‐17
Supplementary figure S51: Top 5 overrepresented CC terms of Module‐17
Supplementary figure S52: Top 5 overrepresented MF terms of Module‐17
Supplementary figure S53: Top 5 overrepresented BP terms of Module‐18
Supplementary figure S54: Top 5 overrepresented CC terms of Module‐18
Supplementary figure S55: Top 5 overrepresented MF terms of Module‐18
Supplementary figure S56: Top 5 overrepresented BP terms of Module‐19
Supplementary figure S57: Top 5 overrepresented CC terms of Module‐19
Supplementary figure S58: Top 5 overrepresented MF terms of Module‐19
Supplementary figure S59: Top 5 overrepresented BP terms of Module‐20
Supplementary figure S60: Top 5 overrepresented CC terms of Module‐20
Supplementary figure S61: Top 5 overrepresented MF terms of Module‐20
Supplementary figure S62: Top 5 overrepresented BP terms of Module‐21
Supplementary figure S63: Top 5 overrepresented CC terms of Module‐21
Supplementary figure S64: Top 5 overrepresented MF terms of Module‐21
Supplementary figure S65: Top 5 overrepresented BP terms of Module‐22
Supplementary figure S66: Top 5 overrepresented CC terms of Module‐22
Supplementary figure S67: Top 5 overrepresented MF terms of Module‐22
Supplementary figure S68: Top 5 overrepresented BP terms of Module‐23
Supplementary figure S69: Top 5 overrepresented CC terms of Module‐23
Supplementary figure S70: Top 5 overrepresented MF terms of Module‐23
Supplementary figure S71: Top 5 overrepresented BP terms of Module‐24
Supplementary figure S72: Top 5 overrepresented CC terms of Module‐24
Supplementary figure S73: Top 5 overrepresented MF terms of Module‐24
Supplementary figure S74: Top 5 overrepresented BP terms of Module‐25
Supplementary figure S75: Top 5 overrepresented CC terms of Module‐25
Supplementary figure S76: Top 5 overrepresented MF terms of Module‐25
Supplementary figure S77: Top 5 overrepresented BP terms of Module‐26
Supplementary figure S78: Top 5 overrepresented CC terms of Module‐26
Supplementary figure S79: Top 5 overrepresented MF terms of Module‐26
Supplementary figure S80: Top 5 overrepresented BP terms of Module‐27
Supplementary figure S81: Top 5 overrepresented CC terms of Module‐27
Supplementary figure S82: Top 5 overrepresented MF terms of Module‐27
Supplementary figure S83: Top 5 overrepresented BP terms of Module‐28
Supplementary figure S84: Top 5 overrepresented CC terms of Module‐28
Supplementary figure S85: Top 5 overrepresented MF terms of Module‐28
Supplementary figure S86: Top 5 overrepresented BP terms of Module‐29
Supplementary figure S87: Top 5 overrepresented CC terms of Module‐29
Supplementary figure S88: Top 5 overrepresented MF terms of Module‐29
Supplementary figure S89: Top 5 overrepresented BP terms of Module‐30
Supplementary figure S90: Top 5 overrepresented CC terms of Module‐30
Supplementary figure S91: Top 5 overrepresented MF terms of Module‐30
Supplementary figure S92: Top 5 overrepresented BP terms of Module‐31
Supplementary figure S93: Top 5 overrepresented CC terms of Module‐31
Supplementary figure S94: Top 5 overrepresented MF terms of Module‐31
Supplementary figure S95: Top 5 overrepresented BP terms of Module‐32
Supplementary figure S96: Top 5 overrepresented CC terms of Module‐32
Supplementary figure S97: Top 5 overrepresented MF terms of Module‐32
Supplementary figure S98: Top 5 overrepresented BP terms of Module‐33
Supplementary figure S99: Top 5 overrepresented CC terms of Module‐33
Supplementary figure S100: Top 5 overrepresented MF terms of Module‐33
Supplementary figure S101: Top 5 overrepresented BP terms of Module‐34
Supplementary figure S102: Top 5 overrepresented CC terms of Module‐34
Supplementary figure S103: Top 5 overrepresented MF terms of Module‐34
Supplementary figure S104: Top 5 overrepresented BP terms of Module‐35
Supplementary figure S105: Top 5 overrepresented CC terms of Module‐35
Supplementary figure S106: Top 5 overrepresented MF terms of Module‐35
Supplementary figure S107: Top 5 overrepresented BP terms of Module‐36
Supplementary figure S108: Top 5 overrepresented CC terms of Module‐36
Supplementary figure S109: Top 5 overrepresented MF terms of Module‐36
Supplementary figure S110: Top 5 overrepresented BP terms of Module‐37
Supplementary figure S111: Top 5 overrepresented CC terms of Module‐37
Supplementary figure S112: Top 5 overrepresented MF terms of Module‐37
Supplementary figure S113: Top 5 overrepresented BP terms of Module‐38
Supplementary figure S114: Top 5 overrepresented CC terms of Module‐38
Supplementary figure S115: Top 5 overrepresented MF terms of Module‐38
Supplementary figure S116: Top 5 overrepresented BP terms of Module‐39
Supplementary figure S117: Top 5 overrepresented CC terms of Module‐39
Supplementary figure S118: Top 5 overrepresented MF terms of Module‐39
Supplementary figure S119: Top 5 overrepresented BP terms of Module‐40
Supplementary figure S120: Top 5 overrepresented CC terms of Module‐40
Supplementary figure S121: Top 5 overrepresented MF terms of Module‐40
Supplementary figure S122: Top 5 overrepresented BP terms of Module‐41
Supplementary figure S123: Top 5 overrepresented CC terms of Module‐41
Supplementary figure S124: Top 5 overrepresented MF terms of Module‐41
Supplementary figure S125: Top 5 overrepresented BP terms of Module‐42
Supplementary figure S126: Top 5 overrepresented CC terms of Module‐42
Supplementary figure S127: Top 5 overrepresented MF terms of Module‐42
Supplementary figure S128: Top 5 overrepresented BP terms of Module‐43
Supplementary figure S129: Top 5 overrepresented CC terms of Module‐43
Supplementary figure S130: Top 5 overrepresented MF terms of Module‐43
Supplementary figure S131: Top 5 overrepresented BP terms of Module‐44
Supplementary figure S132: Top 5 overrepresented CC terms of Module‐44
Supplementary figure S133: Top 5 overrepresented MF terms of Module‐44
Supplementary figure S134: Top 5 overrepresented BP terms of Module‐45
Supplementary figure S135: Top 5 overrepresented CC terms of Module‐45
Supplementary figure S136: Top 5 overrepresented MF terms of Module‐45
Supplementary figure S137: Top 5 overrepresented BP terms of Module‐46
Supplementary figure S138: Top 5 overrepresented CC terms of Module‐46
Supplementary figure S139: Top 5 overrepresented MF terms of Module‐46
Supplementary figure S140: Top 5 overrepresented BP terms of Module‐47
Supplementary figure S141: Top 5 overrepresented CC terms of Module‐47
Supplementary figure S142: Top 5 overrepresented MF terms of Module‐47
Supplementary figure S143: Top 5 overrepresented BP terms of Module‐48
Supplementary figure S144: Top 5 overrepresented CC terms of Module‐48
Supplementary figure S145: Top 5 overrepresented MF terms of Module‐48
Supplementary figure S146: Top 5 overrepresented BP terms of Module‐49
Supplementary figure S147: Top 5 overrepresented CC terms of Module‐49
Supplementary figure S148: Top 5 overrepresented MF terms of Module‐49
Supplementary figure S149: Top 5 overrepresented BP terms of Module‐50
Supplementary figure S150: Top 5 overrepresented CC terms of Module‐50
Supplementary figure S151: Top 5 overrepresented MF terms of Module‐50
Supplementary figure S152: Top 5 overrepresented BP terms of Module‐51
Supplementary figure S153: Top 5 overrepresented CC terms of Module‐51
Supplementary figure S154: Top 5 overrepresented MF terms of Module‐51
Supplementary figure S155: Top 5 overrepresented BP terms of Module‐52
Supplementary figure S156: Top 5 overrepresented CC terms of Module‐52
Supplementary figure S157: Top 5 overrepresented MF terms of Module‐52
Supplementary figure S158: Top 5 overrepresented BP terms of Module‐53
Supplementary figure S159: Top 5 overrepresented CC terms of Module‐53
Supplementary figure S160: Top 5 overrepresented MF terms of Module‐53
Supplementary figure S161: Top 5 overrepresented BP terms of Module‐54
Supplementary figure S162: Top 5 overrepresented CC terms of Module‐54
Supplementary figure S163: Top 5 overrepresented MF terms of Module‐54
Supplementary table S1: BP terms enriched in Module‐1
Supplementary table S2: CC terms enriched in Module‐1
Supplementary table S3: MF terms enriched in Module‐1
Supplementary table S4: Pathways enriched in Module‐1
Supplementary table S5: BP terms enriched in Module‐2
Supplementary table S6: CC terms enriched in Module‐2
Supplementary table S7: MF terms enriched in Module‐2
Supplementary table S8: Pathways enriched in Module‐2
Supplementary table S9: BP terms enriched in Module‐3
Supplementary table S10: CC terms enriched in Module‐3
Supplementary table S11: MF terms enriched in Module‐3
Supplementary table S12: Pathways enriched in Module‐3
Supplementary table S13: BP terms enriched in Module‐4
Supplementary table S14: CC terms enriched in Module‐4
Supplementary table S15: MF terms enriched in Module‐4
Supplementary table S16: Pathways enriched in Module‐4
Supplementary table S17: BP terms enriched in Module‐5
Supplementary table S18: CC terms enriched in Module‐5
Supplementary table S19: MF terms enriched in Module‐5
Supplementary table S20: Pathways enriched in Module‐5
Supplementary table S21: BP terms enriched in Module‐6
Supplementary table S22: CC terms enriched in Module‐6
Supplementary table S23: MF terms enriched in Module‐6
Supplementary table S24: Pathways enriched in Module‐6
Supplementary table S25: BP terms enriched in Module‐7
Supplementary table S26: CC terms enriched in Module‐7
Supplementary table S27: MF terms enriched in Module‐7
Supplementary table S28: Pathways enriched in Module‐7
Supplementary table S29: BP terms enriched in Module‐8
Supplementary table S30: CC terms enriched in Module‐8
Supplementary table S31: MF terms enriched in Module‐8
Supplementary table S32: Pathways enriched in Module‐8
Supplementary table S33: BP terms enriched in Module‐9
Supplementary table S34: CC terms enriched in Module‐9
Supplementary table S35: MF terms enriched in Module‐9
Supplementary table S36: Pathways enriched in Module‐9
Supplementary table S37: BP terms enriched in Module‐10
Supplementary table S38: CC terms enriched in Module‐10
Supplementary table S39: MF terms enriched in Module‐10
Supplementary table S40: Pathways enriched in Module‐10
Supplementary table S41: BP terms enriched in Module‐11
Supplementary table S42: CC terms enriched in Module‐11
Supplementary table S43: MF terms enriched in Module‐11
Supplementary table S44: Pathways enriched in Module‐11
Supplementary table S45: BP terms enriched in Module‐12
Supplementary table S46: CC terms enriched in Module‐12
Supplementary table S47: MF terms enriched in Module‐12
Supplementary table S48: Pathways enriched in Module‐12
Supplementary table S49: BP terms enriched in Module‐13
Supplementary table S50: CC terms enriched in Module‐13
Supplementary table S51: MF terms enriched in Module‐13
Supplementary table S52: Pathways enriched in Module‐13
Supplementary table S53: BP terms enriched in Module‐14
Supplementary table S54: CC terms enriched in Module‐14
Supplementary table S55: MF terms enriched in Module‐14
Supplementary table S56: Pathways enriched in Module‐14
Supplementary table S57: BP terms enriched in Module‐15
Supplementary table S58: CC terms enriched in Module‐15
Supplementary table S59: MF terms enriched in Module‐15
Supplementary table S60: Pathways enriched in Module‐15
Supplementary table S61: BP terms enriched in Module‐16
Supplementary table S62: CC terms enriched in Module‐16
Supplementary table S63: MF terms enriched in Module‐16
Supplementary table S64: Pathways enriched in Module‐16
Supplementary table S65: BP terms enriched in Module‐17
Supplementary table S66: CC terms enriched in Module‐17
Supplementary table S67: MF terms enriched in Module‐17
Supplementary table S68: Pathways enriched in Module‐17
Supplementary table S69: BP terms enriched in Module‐18
Supplementary table S70: CC terms enriched in Module‐18
Supplementary table S71: MF terms enriched in Module‐18
Supplementary table S72: Pathways enriched in Module‐18
Supplementary table S73: BP terms enriched in Module‐19
Supplementary table S74: CC terms enriched in Module‐19
Supplementary table S75: MF terms enriched in Module‐19
Supplementary table S76: Pathways enriched in Module‐19
Supplementary table S77: BP terms enriched in Module‐20
Supplementary table S78: CC terms enriched in Module‐20
Supplementary table S79: MF terms enriched in Module‐20
Supplementary table S80: Pathways enriched in Module‐20
Supplementary table S81: BP terms enriched in Module‐21
Supplementary table S82: CC terms enriched in Module‐21
Supplementary table S83: MF terms enriched in Module‐21
Supplementary table S84: Pathways enriched in Module‐21
Supplementary table S85: BP terms enriched in Module‐22
Supplementary table S86: CC terms enriched in Module‐22
Supplementary table S87: MF terms enriched in Module‐22
Supplementary table S88: Pathways enriched in Module‐22
Supplementary table S89: BP terms enriched in Module‐23
Supplementary table S90: CC terms enriched in Module‐23
Supplementary table S91: MF terms enriched in Module‐23
Supplementary table S92: Pathways enriched in Module‐23
Supplementary table S93: BP terms enriched in Module‐24
Supplementary table S94: CC terms enriched in Module‐24
Supplementary table S95: MF terms enriched in Module‐24
Supplementary table S96: Pathways enriched in Module‐24
Supplementary table S97: BP terms enriched in Module‐25
Supplementary table S98: CC terms enriched in Module‐25
Supplementary table S99: MF terms enriched in Module‐25
Supplementary table S100: Pathways enriched in Module‐25
Supplementary table S101: BP terms enriched in Module‐26
Supplementary table S102: CC terms enriched in Module‐26
Supplementary table S103: MF terms enriched in Module‐26
Supplementary table S104: Pathways enriched in Module‐26
Supplementary table S105: BP terms enriched in Module‐27
Supplementary table S106: CC terms enriched in Module‐27
Supplementary table S107: MF terms enriched in Module‐27
Supplementary table S108: Pathways enriched in Module‐27
Supplementary table S109: BP terms enriched in Module‐28
Supplementary table S110: CC terms enriched in Module‐28
Supplementary table S111: MF terms enriched in Module‐28
Supplementary table S112: Pathways enriched in Module‐28
Supplementary table S113: BP terms enriched in Module‐29
Supplementary table S114: CC terms enriched in Module‐29
Supplementary table S115: MF terms enriched in Module‐29
Supplementary table S116: Pathways enriched in Module‐29
Supplementary table S117: BP terms enriched in Module‐30
Supplementary table S118: CC terms enriched in Module‐30
Supplementary table S119: MF terms enriched in Module‐30
Supplementary table S120: Pathways enriched in Module‐30
Supplementary table S121: BP terms enriched in Module‐31
Supplementary table S122: CC terms enriched in Module‐31
Supplementary table S123: MF terms enriched in Module‐31
Supplementary table S124: Pathways enriched in Module‐31
Supplementary table S125: BP terms enriched in Module‐32
Supplementary table S126: CC terms enriched in Module‐32
Supplementary table S127: MF terms enriched in Module‐32
Supplementary table S128: Pathways enriched in Module‐32
Supplementary table S129: BP terms enriched in Module‐33
Supplementary table S130: CC terms enriched in Module‐33
Supplementary table S131: MF terms enriched in Module‐33
Supplementary table S132: Pathways enriched in Module‐33
Supplementary table S133: BP terms enriched in Module‐34
Supplementary table S134: CC terms enriched in Module‐34
Supplementary table S135: MF terms enriched in Module‐34
Supplementary table S136: Pathways enriched in Module‐34
Supplementary table S137: BP terms enriched in Module‐35
Supplementary table S138: CC terms enriched in Module‐35
Supplementary table S139: MF terms enriched in Module‐35
Supplementary table S140: Pathways enriched in Module‐35
Supplementary table S141: BP terms enriched in Module‐36
Supplementary table S142: CC terms enriched in Module‐36
Supplementary table S143: MF terms enriched in Module‐36
Supplementary table S144: Pathways enriched in Module‐36
Supplementary table S145: BP terms enriched in Module‐37
Supplementary table S146: CC terms enriched in Module‐37
Supplementary table S147: MF terms enriched in Module‐37
Supplementary table S148: Pathways enriched in Module‐37
Supplementary table S149: BP terms enriched in Module‐38
Supplementary table S150: CC terms enriched in Module‐38
Supplementary table S151: MF terms enriched in Module‐38
Supplementary table S152: Pathways enriched in Module‐38
Supplementary table S153: BP terms enriched in Module‐39
Supplementary table S154: CC terms enriched in Module‐39
Supplementary table S155: MF terms enriched in Module‐39
Supplementary table S156: Pathways enriched in Module‐39
Supplementary table S157: BP terms enriched in Module‐40
Supplementary table S158: CC terms enriched in Module‐40
Supplementary table S159: MF terms enriched in Module‐40
Supplementary table S160: Pathways enriched in Module‐40
Supplementary table S161: BP terms enriched in Module‐41
Supplementary table S162: CC terms enriched in Module‐41
Supplementary table S163: MF terms enriched in Module‐41
Supplementary table S164: Pathways enriched in Module‐41
Supplementary table S165: BP terms enriched in Module‐42
Supplementary table S166: CC terms enriched in Module‐42
Supplementary table S167: MF terms enriched in Module‐42
Supplementary table S168: Pathways enriched in Module‐42
Supplementary table S169: BP terms enriched in Module‐43
Supplementary table S170: CC terms enriched in Module‐43
Supplementary table S171: MF terms enriched in Module‐43
Supplementary table S172: Pathways enriched in Module‐43
Supplementary table S173: BP terms enriched in Module‐44
Supplementary table S174: CC terms enriched in Module‐44
Supplementary table S175: MF terms enriched in Module‐44
Supplementary table S176: Pathways enriched in Module‐44
Supplementary table S177: BP terms enriched in Module‐45
Supplementary table S178: CC terms enriched in Module‐45
Supplementary table S179: MF terms enriched in Module‐45
Supplementary table S180: Pathways enriched in Module‐45
Supplementary table S181: BP terms enriched in Module‐46
Supplementary table S182: CC terms enriched in Module‐46
Supplementary table S183: MF terms enriched in Module‐46
Supplementary table S184: Pathways enriched in Module‐46
Supplementary table S185: BP terms enriched in Module‐47
Supplementary table S186: CC terms enriched in Module‐47
Supplementary table S187: MF terms enriched in Module‐47
Supplementary table S188: Pathways enriched in Module‐47
Supplementary table S189: BP terms enriched in Module‐48
Supplementary table S190: CC terms enriched in Module‐48
Supplementary table S191: MF terms enriched in Module‐48
Supplementary table S192: Pathways enriched in Module‐48
Supplementary table S193: BP terms enriched in Module‐49
Supplementary table S194: CC terms enriched in Module‐49
Supplementary table S195: MF terms enriched in Module‐49
Supplementary table S196: Pathways enriched in Module‐49
Supplementary table S197: BP terms enriched in Module‐50
Supplementary table S198: CC terms enriched in Module‐50
Supplementary table S199: MF terms enriched in Module‐50
Supplementary table S200: Pathways enriched in Module‐50
Supplementary table S201: BP terms enriched in Module‐51
Supplementary table S202: CC terms enriched in Module‐51
Supplementary table S203: MF terms enriched in Module‐51
Supplementary table S204: Pathways enriched in Module‐51
Supplementary table S205: BP terms enriched in Module‐52
Supplementary table S206: CC terms enriched in Module‐52
Supplementary table S207: MF terms enriched in Module‐52
Supplementary table S208: Pathways enriched in Module‐52
Supplementary table S209: BP terms enriched in Module‐53
Supplementary table S210: CC terms enriched in Module‐53
Supplementary table S211: MF terms enriched in Module‐53
Supplementary table S212: Pathways enriched in Module‐53
Supplementary table S213: BP terms enriched in Module‐54
Supplementary table S214: CC terms enriched in Module‐54
Supplementary table S215: MF terms enriched in Module‐54
Supplementary table S216: Pathways enriched in Module‐54
Supplementary table S217: List of novel genes identified as the first neighbors of RBOHD.
Supplementary table S218: List of novel genes identified as the first neighbors of AtNEET.
Supplementary table S219: List of novel genes identified as the first neighbors of HSFA1D.
ACKNOWLEDGMENTS
This work was supported by a funding from the National Science Foundation (award number: IOS‐1932639).
Burks, D. J. , Sengupta, S. , De, R. , Mittler, R. , & Azad, R. K. (2022). The Arabidopsis gene co‐expression network. Plant Direct, 6(4), e396. 10.1002/pld3.396
David J. Burks and Soham Sengupta are joint first‐authors.
DATA AVAILABILITY STATEMENT
All data have been made publicly available at https://doi.org/10.6084/m9.figshare.16752733.v4, and the associated source codes have been provided at the project's GitHub site: https://github.com/sohamsg90/WGCNA_Arabidopsis-main.
REFERENCES
- Albert, R. (2005). Scale‐free networks in cell biology. Journal of Cell Science, 118, 4947–4957. 10.1242/jcs.02714 [DOI] [PubMed] [Google Scholar]
- Alexa, A. , & Rahnenführer, J. (2009). Gene set enrichment analysis with topGO. Bioconductor Improv, 27, 1–26. [Google Scholar]
- Andreini, C. , Rosato, A. , & Banci, L. (2017). The relationship between environmental dioxygen and Iron‐sulfur proteins explored at the genome level. PLoS ONE, 12, e0171279. 10.1371/journal.pone.0171279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki, K. , Ogata, Y. , & Shibata, D. (2007). Approaches for extracting practical information from gene co‐expression networks in plant biology. Plant and Cell Physiology, 48, 381–390. 10.1093/pcp/pcm013 [DOI] [PubMed] [Google Scholar]
- Baerenfaller, K. , Massonnet, C. , Walsh, S. , Baginsky, S. , Bühlmann, P. , Hennig, L. , Hirsch‐Hoffmann, M. , Howell, K. A. , Kahlau, S. , & Radziejwoski, A. (2012). Systems‐based analysis of Arabidopsis leaf growth reveals adaptation to water deficit. Molecular Systems Biology, 8(1), 606. 10.1038/msb.2012.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balfagón, D. , Sengupta, S. , Gómez‐Cadenas, A. , Fritschi, F. B. , Azad, R. K. , Mittler, R. , & Zandalinas, S. I. (2019). Jasmonic acid is required for plant acclimation to a combination of high light and heat stress. Plant Physiology, 181(4), 1668–1682. 10.1104/pp.19.00956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balk, J. , & Pilon, M. (2011). Ancient and essential: The assembly of iron–sulfur clusters in plants. Trends in Plant Science, 16(4), 218–226. 10.1016/j.tplants.2010.12.006 [DOI] [PubMed] [Google Scholar]
- Balk, J. , & Schaedler, T. A. (2014). Iron cofactor assembly in plants. Annual Review of Plant Biology, 65, 125–153. 10.1146/annurev-arplant-050213-035759 [DOI] [PubMed] [Google Scholar]
- Berardini, T. Z. , Reiser, L. , Li, D. , Mezheritsky, Y. , Muller, R. , Strait, E. , & Huala, E. (2015). The arabidopsis information resource: Making and mining the. “Gold Standard” Annotated Reference Plant Genome. Genesis, 53(8), 474–485. 10.1002/dvg.22877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann, S. , Ihmels, J. , & Barkai, N. 2003. Similarities and differences in genome‐wide expression data of six organisms. PLoS Biology, 2, e9. 1 10.1371/journal.pbio.0020009, E9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard, D. G. , Netz, D. J. A. , Lagny, T. J. , Pierik, A. J. , & Balk, J. (2013). Requirements of the cytosolic iron–sulfur cluster assembly pathway in Arabidopsis. Philosophical Transactions of the Royal Society B: Biological Sciences, 368(1622), 20120259. 10.1098/rstb.2012.0259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco, F. , Salinas, P. , Cecchini, N. M. , Jordana, X. , Van Hummelen, P. , Alvarez, M. E. , & Holuigue, L. (2009). Early genomic responses to salicylic acid in Arabidopsis. Plant Molecular Biology, 70(1–2), 79–102. 10.1007/s11103-009-9458-1 [DOI] [PubMed] [Google Scholar]
- Bokszczanin, K. L. , Solanaceae Pollen Thermotolerance Initial Training Network, C. , & Fragkostefanakis, S. (2013). Perspectives on deciphering mechanisms underlying plant heat stress response and thermotolerance. Frontiers in Plant Science, 4, 315. 10.3389/fpls.2013.00315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolser, D. , Staines, D. M. , Pritchard, E. , & Kersey, P. (2016). Ensembl plants: Integrating tools for visualizing, mining, and analyzing plant genomics data. In Plant Bioinformatics. Springer. 10.1007/978-1-4939-3167-5_6 [DOI] [PubMed] [Google Scholar]
- Boyd, E. S. , Thomas, K. M. , Dai, Y. , Boyd, J. M. , & Outten, F. W. (2014). Interplay between oxygen and Fe‐S cluster biogenesis: Insights from the Suf pathway. Biochemistry, 53(37), 5834–5847. 10.1021/bi500488r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burks, D. J. , & Azad, R. K. (2016). Identification and network‐enabled characterization of auxin response factor genes in Medicago truncatula. Frontiers in Plant Science, 7, 1857. 10.3389/fpls.2016.01857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, M. R. J. , Zhang, B. , Fang, Z. , Mischel, P. S. , Horvath, S. , & Nelson, S. F. (2006). Gene connectivity, function, and sequence conservation: Predictions from modular yeast co‐expression networks. BMC Genomics, 7(1). 10.1186/1471-2164-7-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalhais, L. C. , Muzzi, F. , Tan, C.‐H. , Choo, J. H. , & Schenk, P. M. (2013). Plant growth in Arabidopsis is assisted by compost soil‐derived microbial communities. Frontiers in Plant Science, 4, 235. 10.3389/fpls.2013.00235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J.‐G. , Ullah, H. , Temple, B. , Liang, J. , Guo, J. , Alonso, J. M. , Ecker, J. R. , & Jones, A. M. (2006). RACK1 mediates multiple hormone responsiveness and developmental processes in Arabidopsis. Journal of Experimental Botany, 57(11), 2697–2708. 10.1093/jxb/erl035 [DOI] [PubMed] [Google Scholar]
- Choi, J. , Tanaka, K. , Liang, Y. , Cao, Y. , Lee, S. y. , & Stacey, G. (2014). Extracellular ATP, a danger signal, is recognized by DORN1 in Arabidopsis. Biochemical Journal, 463(3), 429–437. 10.1042/BJ20140666 [DOI] [PubMed] [Google Scholar]
- Consales, F. , Schweizer, F. , Erb, M. , Gouhier‐Darimont, C. , Bodenhausen, N. , Bruessow, F. , Sobhy, I. , & Reymond, P. (2011). Insect oral secretions suppress wound‐induced responses in Arabidopsis. Journal of Experimental Botany, 63, 727–737. 10.1093/jxb/err308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortijo, S. , Charoensawan, V. , Brestovitsky, A. , Buning, R. , Ravarani, C. , Rhodes, D. , Van Noort, J. , Jaeger, K. E. , & Wigge, P. A. (2017). Transcriptional regulation of the ambient temperature response by H2A.Z nucleosomes and HSF1 transcription factors in Arabidopsis. Molecular Plant, 10, 1258–1273. 10.1016/j.molp.2017.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davletova, S. , Schlauch, K. , Coutu, J. , & Mittler, R. (2005). The zinc‐finger protein Zat12 plays a central role in reactive oxygen and abiotic stress signaling in Arabidopsis. Plant Physiology, 139, 847–856. 10.1104/pp.105.068254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, F. , Cui, P. , Wang, Z. , Zhang, S. , Ali, S. , & Xiong, L. (2014). Genome‐wide analysis of alternative splicing of pre‐mRNA under salt stress in Arabidopsis. BMC Genomics, 15, 431. 10.1186/1471-2164-15-431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C. A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , Batut, P. , Chaisson, M. , & Gingeras, T. R. (2013). STAR: ultrafast universal RNA‐seq aligner. Bioinformatics, 29, 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Reis, S. P. , Lima, A. M. , & De Souza, C. R. B. (2012). Recent molecular advances on downstream plant responses to abiotic stress. International Journal of Molecular Sciences, 13(7), 8628–8647. 10.3390/ijms13078628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossa, K. , Diouf, D. , & Cissé, N. (2016). Genome‐wide investigation of Hsf genes in sesame reveals their segmental duplication expansion and their active role in drought stress response. Frontiers in Plant Science, 7, 1522. 10.3389/fpls.2016.01522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farahbod, M. , & Pavlidis, P. (2019). Untangling the effects of cellular composition on coexpression analysis. Cold Spring Harbor Laboratory. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman, T. C. , Goldovsky, L. , Brosch, M. , Van Dongen, S. , Mazière, P. , Grocock, R. J. , Freilich, S. , Thornton, J. , & Enright, A. J. 2007. Construction, visualisation, and clustering of transcription networks from microarray expression data. PLoS Computational Biology, 3, e206. 10 10.1371/journal.pcbi.0030206, 2032, 2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadjev, I. , Vanderauwera, S. , Gechev, T. S. , Laloi, C. , Minkov, I. N. , Shulaev, V. , Apel, K. , Inzé, D. , Mittler, R. , & Van Breusegem, F. (2006). Transcriptomic footprints disclose specificity of reactive oxygen species signaling in Arabidopsis. Plant Physiology, 141, 436–445. 10.1104/pp.106.078717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng, H. , Wang, M. , Gong, J. , Xu, Y. , & Ma, S. (2021). An Arabidopsis expression predictor enables inference of transcriptional regulators for gene modules. The Plant Journal, 107, 597–612. 10.1111/tpj.15315 [DOI] [PubMed] [Google Scholar]
- Girvan, M. , & Newman, M. E. (2002). Community structure in social and biological networks. Proceedings of the National Academy of Sciences, 99(12), 7821–7826. 10.1073/pnas.122653799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, J. , Wang, S. , Valerius, O. , Hall, H. , Zeng, Q. , Li, J.‐F. , Weston, D. J. , Ellis, B. E. , & Chen, J.‐G. (2011). Involvement of Arabidopsis RACK1 in protein translation and its regulation by abscisic acid. Plant Physiology, 155, 370–383. 10.1104/pp.110.160663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, M. , Liu, J.‐H. , Ma, X. , Luo, D.‐X. , Gong, Z.‐H. , & Lu, M.‐H. (2016). The plant heat stress transcription factors (HSFs): Structure, regulation, and function in response to abiotic stresses. Frontiers in Plant Science, 7, 114. 10.3389/fpls.2016.00114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, S. , & Dong, J. (2008). Geometric interpretation of gene Coexpression Network analysis. PLoS Computational Biology, 4(8), e1000117. 10.1371/journal.pcbi.1000117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, X. , Kato, Y. , Sumida, A. , Tanaka, A. , & Tanaka, R. (2017). The SUFBC2D complex is required for the biogenesis of all major classes of plastid Fe‐S proteins. The Plant Journal, 90, 235–248. 10.1111/tpj.13483 [DOI] [PubMed] [Google Scholar]
- Huang, D. , Wu, W. , Abrams, S. R. , & Cutler, A. J. (2008). The relationship of drought‐related gene expression in Arabidopsis thaliana to hormonal and environmental factors. Journal of Experimental Botany, 59(11), 2991–3007. 10.1093/jxb/ern155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi, M. , Iwase, A. , Rymen, B. , Lambolez, A. , Kojima, M. , Takebayashi, Y. , Heyman, J. , Watanabe, S. , Seo, M. , De Veylder, L. , Sakakibara, H. , & Sugimoto, K. (2017). Wounding triggers callus formation via dynamic hormonal and transcriptional changes. Plant Physiology, 175(3), 1158–1174. 10.1104/pp.17.01035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob, P. , Hirt, H. , & Bendahmane, A. (2017). The heat‐shock protein/chaperone network and multiple stress resistance. Plant Biotechnology Journal, 15, 405–414. 10.1111/pbi.12659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen, C.‐H. , Manfield, I. W. , Michalopoulos, I. , Pinney, J. W. , Willats, W. G. T. , Gilmartin, P. M. , & Westhead, D. R. (2006). The Arabidopsis co‐expression tool (act): A WWW‐based tool and database for microarray‐based gene expression analysis. The Plant Journal, 46, 336–348. 10.1111/j.1365-313X.2006.02681.x [DOI] [PubMed] [Google Scholar]
- Jordan, I. K. , MariñO‐RamíRez, L. , Wolf, Y. I. , & Koonin, E. V. (2004). Conservation and coevolution in the scale‐free human gene Coexpression Network. Molecular Biology and Evolution, 21(11), 2058–2070. 10.1093/molbev/msh222 [DOI] [PubMed] [Google Scholar]
- Jordan, I. K. , Mariño‐Ramírez, L. , & Koonin, E. V. (2005). Evolutionary significance of gene expression divergence. Gene, 345, 119–126. 10.1016/j.gene.2004.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce, A. R. , & Palsson, B. Ø. (2006). The model organism as a system: Integrating 'omics' data sets. Nature Reviews Molecular Cell Biology, 7(3), 198–210. 10.1038/nrm1857 [DOI] [PubMed] [Google Scholar]
- Jung, J.‐H. , Domijan, M. , Klose, C. , Biswas, S. , Ezer, D. , Gao, M. , Khattak, A. K. , Box, M. S. , Charoensawan, V. , Cortijo, S. , Kumar, M. , Grant, A. , Locke, J. C. W. , Schäfer, E. , Jaeger, K. E. , & Wigge, P. A. (2016). Phytochromes function as thermosensors inArabidopsis. Science, 354(6314), 886–889. 10.1126/science.aaf6005 [DOI] [PubMed] [Google Scholar]
- Kelley, B. P. , Sharan, R. , Karp, R. M. , Sittler, T. , Root, D. E. , Stockwell, B. R. , & Ideker, T. (2003). Conserved pathways within bacteria and yeast as revealed by global protein network alignment. Proceedings of the National Academy of Sciences, 100, 11394–11399. 10.1073/pnas.1534710100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M. A. , Castro‐Guerrero, N. A. , Mcinturf, S. A. , Nguyen, N. T. , Dame, A. N. , Wang, J. , Bindbeutel, R. K. , Joshi, T. , Jurisson, S. S. , Nusinow, D. A. , & Mendoza‐Cozatl, D. G. (2018). Changes in iron availability in Arabidopsis are rapidly sensed in the leaf vasculature and impaired sensing leads to opposite transcriptional programs in leaves and roots. Plant, Cell & Environment, 41(10), 2263–2276. 10.1111/pce.13192 [DOI] [PubMed] [Google Scholar]
- Kleine, T. , Kindgren, P. , Benedict, C. , Hendrickson, L. , & Strand, A. S. (2007). Genome‐wide gene expression analysis reveals a critical role for CRYPTOCHROME1 in the response of Arabidopsis to high irradiance. Plant Physiology, 144(3), 1391–1406. 10.1104/pp.107.098293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth, J. D. (2004). NOX enzymes and the biology of reactive oxygen. Nature Reviews Immunology, 4(3), 181–189. 10.1038/nri1312 [DOI] [PubMed] [Google Scholar]
- Langfelder, P. , & Horvath, S. (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics, 9(1), 1–13. 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkindale, J. , & Vierling, E. (2007). Core genome responses involved in acclimation to high temperature. Plant Physiology, 146, 323–324. 10.1104/pp.107.112060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. K. (2004a). Coexpression analysis of human genes across many microarray data sets. Genome Research, 14(6), 1085–1094. 10.1101/gr.1910904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, I. (2004b). A probabilistic functional Network of yeast genes. Science, 306(5701), 1555–1558. 10.1126/science.1099511 [DOI] [PubMed] [Google Scholar]
- Leinonen, R. , Sugawara, H. , & Shumway, M. (2010). The sequence read archive. Nucleic Acids Research, 39(Database), D19–D21. 10.1093/nar/gkq1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill, R. (2009). Function and biogenesis of iron–Sulphur proteins. Nature, 460(7257), 831–838. 10.1038/nature08301 [DOI] [PubMed] [Google Scholar]
- Lippmann, R. , Babben, S. , Menger, A. , Delker, C. , & Quint, M. (2019). Development of wild and cultivated plants under global warming conditions. Current Biology, 29(24), R1326–R1338. 10.1016/j.cub.2019.10.016 [DOI] [PubMed] [Google Scholar]
- Liu, H.‐C. , & Charng, Y.‐Y. (2013). Common and distinct functions of Arabidopsis class A1 and A2 heat shock factors in diverse abiotic stress responses and development. Plant Physiology, 163, 276–290. 10.1104/pp.113.221168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15(12), 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Y. (2018). Assembly and transfer of Iron–sulfur clusters in the plastid. Frontiers in Plant Science, 9. 10.3389/fpls.2018.00336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, K. , Xiao, Z. , Jian, H. , Peng, L. , Qu, C. , Fu, M. , He, B. , Tie, L. , Liang, Y. , Xu, X. , & Li, J. (2016). A combination of genome‐wide association and transcriptome analysis reveals candidate genes controlling harvest index‐related traits in Brassica napus. Scientific Reports, 6(1), 36452. 10.1038/srep36452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, S. , Gong, Q. , & Bohnert, H. J. (2007). An Arabidopsis gene network based on the graphical Gaussian model. Genome Research, 17(11), 1614–1625. 10.1101/gr.6911207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfield, I. W. , Jen, C. H. , Pinney, J. W. , Michalopoulos, I. , Bradford, J. R. , Gilmartin, P. M. , & Westhead, D. R. (2006). Arabidopsis co‐expression tool (ACT): Web server tools for microarray‐based gene expression analysis. Nucleic Acids Research, 34, W504–W509. 10.1093/nar/gkl204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, L. , Van Hemert, J. L. , Dash, S. , & Dickerson, J. A. (2009). Arabidopsis gene co‐expression network and its functional modules. BMC Bioinformatics, 10, 1–24. 10.1186/1471-2105-10-346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui, A. , Ishida, J. , Morosawa, T. , Mochizuki, Y. , Kaminuma, E. , Endo, T. A. , Okamoto, M. , Nambara, E. , Nakajima, M. , Kawashima, M. , Satou, M. , Kim, J. M. , Kobayashi, N. , Toyoda, T. , Shinozaki, K. , & Seki, M. (2008). Arabidopsis transcriptome analysis under drought, cold, high‐salinity and ABA treatment conditions using a tiling Array. Plant and Cell Physiology, 49(8), 1135–1149. 10.1093/pcp/pcn101 [DOI] [PubMed] [Google Scholar]
- Mentzen, W. I. (2006). From pathway to regulon in Arabidopsis. Iowa State University. https://www.proquest.com/docview/305316049?accountid=7113 [Google Scholar]
- Mering, C. V. , Huynen, M. , Jaeggi, D. , Schmidt, S. , Bork, P. , & Snel, B. (2003). STRING: A database of predicted functional associations between proteins. Nucleic Acids Research, 31(1), 258–261. 10.1093/nar/gkg034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, G. , Schlauch, K. , Tam, R. , Cortes, D. , Torres, M. A. , Shulaev, V. , Dangl, J. L. , & Mittler, R. 2009. The plant NADPH oxidase RBOHD mediates rapid systemic signaling in response to diverse stimuli. Science Signaling, 2(84), ra45. 10.1126/scisignal.2000448 [DOI] [PubMed] [Google Scholar]
- Mittler, R. , Finka, A. , & Goloubinoff, P. (2012). How do plants feel the heat? Trends in Biochemical Sciences, 37, 118–125. 10.1016/j.tibs.2011.11.007 [DOI] [PubMed] [Google Scholar]
- Moumeni, A. , Satoh, K. , Kondoh, H. , Asano, T. , Hosaka, A. , Venuprasad, R. , Serraj, R. , Kumar, A. , Leung, H. , & Kikuchi, S. (2011). Comparative analysis of root transcriptome profiles of two pairs of drought‐tolerant and susceptible rice near‐isogenic lines under different drought stress. BMC Plant Biology, 11, 174. 10.1186/1471-2229-11-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtar, M. S. , Carvunis, A.‐R. , Dreze, M. , Epple, P. , Steinbrenner, J. , Moore, J. , Tasan, M. , Galli, M. , Hao, T. , & Nishimura, M. T. (2011). Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science, 333(6042), 596–601. 10.1126/science.1203659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtai, R. , Conlan, A. R. , Harir, Y. , Song, L. , Yogev, O. , Eisenberg‐Domovich, Y. , Livnah, O. , Michaeli, D. , Rosen, R. , Ma, V. , Luo, Y. , Zuris, J. A. , Paddock, M. L. , Cabantchik, Z. I. , Jennings, P. A. , & Mittler, R. (2012). Characterization of Arabidopsis NEET reveals an ancient role for NEET proteins in Iron metabolism. The Plant Cell, 24(5), 2139–2154. 10.1105/tpc.112.097634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemhauser, J. L. , Hong, F. , & Chory, J. (2006). Different plant hormones regulate similar processes through largely nonoverlapping transcriptional responses. Cell, 126(3), 467–475. 10.1016/j.cell.2006.05.050 [DOI] [PubMed] [Google Scholar]
- Nishizawa‐Yokoi, A. , Nosaka, R. , Hayashi, H. , Tainaka, H. , Maruta, T. , Tamoi, M. , Ikeda, M. , Ohme‐Takagi, M. , Yoshimura, K. , & Yabuta, Y. (2011). HsfA1d and HsfA1e involved in the transcriptional regulation of HsfA2 function as key regulators for the Hsf signaling network in response to environmental stress. Plant and Cell Physiology, 52, 933–945. 10.1093/pcp/pcr045 [DOI] [PubMed] [Google Scholar]
- Nover, L. , Bharti, K. , Döring, P. , Mishra, S. K. , Ganguli, A. , & Scharf, K.‐D. (2001). Arabidopsis and the heat stress transcription factor world: How many heat stress transcription factors do we need? Cell Stress & Chaperones, 6(3), 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi, T. , Kinoshita, K. , Nakai, K. , Shibaoka, M. , Hayashi, S. , Saeki, M. , Shibata, D. , Saito, K. , & Ohta, H. (2007). ATTED‐II: A database of co‐expressed genes and cis elements for identifying co‐regulated gene groups in Arabidopsis. Nucleic Acids Research, 35, D863–D869. 10.1093/nar/gkl783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi, T. , Aoki, Y. , Tadaka, S. , Kagaya, Y. , & Kinoshita, K. (2018). ATTED‐II in 2018: A plant coexpression database based on investigation of the statistical property of the mutual rank index. Plant and Cell Physiology, 59(1), e3. 10.1093/pcp/pcx191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohama, N. , Kusakabe, K. , Mizoi, J. , Zhao, H. , Kidokoro, S. , Koizumi, S. , Takahashi, F. , Ishida, T. , Yanagisawa, S. , Shinozaki, K. , & Yamaguchi‐Shinozaki, K. (2016). The transcriptional Cascade in the heat stress response of Arabidopsis is strictly regulated at the level of transcription factor expression. The Plant Cell, 28, 181–201. 10.1105/tpc.15.00435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohama, N. , Sato, H. , Shinozaki, K. , & Yamaguchi‐Shinozaki, K. (2017). Transcriptional regulatory Network of plant heat stress response. Trends in Plant Science, 22(1), 53–65. 10.1016/j.tplants.2016.08.015 [DOI] [PubMed] [Google Scholar]
- Omidbakhshfard, M. A. , Sujeeth, N. , Gupta, S. , Omranian, N. , Guinan, K. J. , Brotman, Y. , Nikoloski, Z. , Fernie, A. R. , Mueller‐Roeber, B. , & Gechev, T. S. (2020). A biostimulant obtained from the seaweed Ascophyllum nodosum protects Arabidopsis thaliana from severe oxidative stress. International Journal of Molecular Sciences, 21, 474. 10.3390/ijms21020474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro, R. , Duggal, G. , Love, M. I. , Irizarry, R. A. , & Kingsford, C. (2017). Salmon provides fast and bias‐aware quantification of transcript expression. Nature Methods, 14, 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proost, S. , Krawczyk, A. , & Mutwil, M. (2017). LSTrAP: Efficiently combining RNA sequencing data into co‐expression networks. BMC Bioinformatics, 18, 1–9. 10.1186/s12859-017-1861-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyla‐Toscano, J. , Roland, M. , Gaymard, F. , Couturier, J. , & Rouhier, N. (2018). Roles and maturation of iron–sulfur proteins in plastids. JBIC Journal of Biological Inorganic Chemistry, 23(4), 545–566. 10.1007/s00775-018-1532-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahme, L. (2003). Faculty opinions recommendation of a gene‐coexpression network for global discovery of conserved genetic modules. In Faculty opinions—Post‐publication peer review of the biomedical literature. Faculty Opinions Ltd. [Google Scholar]
- Reimers, M. , & Carey, V. J. (2006). [8] Bioconductor: An open source framework for bioinformatics and computational biology. In Methods in enzymology. Elsevier. 10.1016/S0076-6879(06)11008-3 [DOI] [PubMed] [Google Scholar]
- Robinson, M. D. , Mccarthy, D. J. , & Smyth, G. K. (2010). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszik, J. , & Woodman, S. E. (2014). HotSpotter: Efficient visualization of driver mutations. BMC Genomics, 15, 1044. 10.1186/1471-2164-15-1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan, J. , & Zhang, W. (2006). Identification and evaluation of functional modules in gene co‐expression networks. In Lecture notes in computer science. Springer. [Google Scholar]
- Scarpeci, T. E. , Zanor, M. I. , Carrillo, N. , Mueller‐Roeber, B. , & Valle, E. M. (2007). Generation of superoxide anion in chloroplasts of Arabidopsis thaliana during active photosynthesis: A focus on rapidly induced genes. Plant Molecular Biology, 66(4), 361–378. 10.1007/s11103-007-9274-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta, S. , Nechushtai, R. , Jennings, P. A. , Onuchic, J. N. , Padilla, P. A. , Azad, R. K. , & Mittler, R. (2018). Phylogenetic analysis of the CDGSH iron‐sulfur binding domain reveals its ancient origin. Scientific Reports, 8(1), 4840. 10.1038/s41598-018-23305-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, Z. , Shah, S. H. , Ali, G. S. , Munir, I. , Khan, R. S. , Iqbal, A. , Ahmed, N. , & Jan, A. (2020). Introduction of Arabidopsis's heat shock factor HsfA1d mitigates adverse effects of heat stress on potato (Solanum tuberosum L.) plant. Cell Stress & Chaperones, 25(1), 57–63. 10.1007/s12192-019-01043-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N. S. , Wang, J. T. , Ramage, D. , Amin, N. , Schwikowski, B. , & Ideker, T. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slonim, D. K. , & Yanai, I. (2009). Getting started in gene expression microarray analysis. PLoS Computational Biology, 5(10), e1000543. 10.1371/journal.pcbi.1000543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, L. M. (2018). Identification of woodland strawberry gene Coexpression networks. Plant Physiology, 178, 7–8. 10.1104/pp.18.00880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart, J. M. (2003). A gene‐Coexpression Network for global discovery of conserved genetic modules. Science, 302(5643), 249–255. 10.1126/science.1087447 [DOI] [PubMed] [Google Scholar]
- Su, L.‐W. , Chang, S. H. , Li, M.‐Y. , Huang, H.‐Y. , Jane, W.‐N. , & Yang, J.‐Y. (2013). Purification and biochemical characterization of Arabidopsis at‐NEET, an ancient iron‐sulfur protein, reveals a conserved cleavage motif for subcellular localization. Plant Science, 213, 46–54. 10.1016/j.plantsci.2013.09.001 [DOI] [PubMed] [Google Scholar]
- Sumimoto, H. (2008). Structure, regulation and evolution of Nox‐family NADPH oxidases that produce reactive oxygen species. FEBS Journal, 275(13), 3249–3277. 10.1111/j.1742-4658.2008.06488.x [DOI] [PubMed] [Google Scholar]
- Swindell, W. R. , Huebner, M. , & Weber, A. P. (2007). Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non‐heat stress response pathways. BMC Genomics, 8, 125. 10.1186/1471-2164-8-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel, J. , Rolletschek, H. , Friedel, S. , Lunn, J. E. , Nguyen, T. H. , Feil, R. , Tschiersch, H. , Müller, M. , & Borisjuk, L. (2011). Seed‐specific elevation of non‐symbiotic hemoglobin AtHb1: Beneficial effects and underlying molecular networks in Arabidopsis thaliana. BMC Plant Biology, 11, 1–18. 10.1186/1471-2229-11-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari, L. D. , Khungar, L. , & Grover, A. (2020). AtHsc70‐1 negatively regulates the basal heat tolerance in Arabidopsis thaliana through affecting the activity of HsfAs and Hsp101. The Plant Journal, 103(6), 2069–2083. 10.1111/tpj.14883 [DOI] [PubMed] [Google Scholar]
- Tosti, N. , Pasqualini, S. , Borgogni, A. , Ederli, L. , Falistocco, E. , Crispi, S. , & Paolocci, F. (2006). Gene expression profiles of O3‐treated Arabidopsis plants. Plant, Cell and Environment, 29(9), 1686–1702. 10.1111/j.1365-3040.2006.01542.x [DOI] [PubMed] [Google Scholar]
- Truman, W. , Zabala, M. T. , & Grant, M. (2006). Type III effectors orchestrate a complex interplay between transcriptional networks to modify basal defence responses during pathogenesis and resistance. The Plant Journal, 46, 14–33. 10.1111/j.1365-313X.2006.02672.x [DOI] [PubMed] [Google Scholar]
- Van Leene, J. , Hollunder, J. , Eeckhout, D. , Persiau, G. , Van De Slijke, E. , Stals, H. , Van Isterdael, G. , Verkest, A. , Neirynck, S. , & Buffel, Y. (2010). Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Molecular Systems Biology, 6(1), 397. 10.1038/msb.2010.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virdi, A. S. , Singh, S. , & Singh, P. (2015). Abiotic stress responses in plants: Roles of calmodulin‐regulated proteins. Frontiers in Plant Science, 6, 809. 10.3389/fpls.2015.00809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu, L. D. , Gevaert, K. , & De Smet, I. (2019). Feeling the heat: Searching for plant Thermosensors. Trends in Plant Science, 24(3), 210–219. 10.1016/j.tplants.2018.11.004 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Joshi, T. , Zhang, X.‐S. , Xu, D. , & Chen, L. (2006). Inferring gene regulatory networks from multiple microarray datasets. Bioinformatics, 22(19), 2413–2420. 10.1093/bioinformatics/btl396 [DOI] [PubMed] [Google Scholar]
- Wang, X. , Huang, W. , Yang, Z. , Liu, J. , & Huang, B. (2016). Transcriptional regulation of heat shock proteins and ascorbate peroxidase by CtHsfA2b from African bermudagrass conferring heat tolerance in Arabidopsis. Scientific Reports, 6(1), 28021. 10.1038/srep28021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Ma, K.‐B. , Lu, Z.‐G. , Ren, S.‐X. , Jiang, H.‐R. , Cui, J.‐W. , Chen, G. , Teng, N.‐J. , Lam, H.‐M. , & Jin, B. (2020). Differential physiological, transcriptomic and metabolomic responses of Arabidopsis leaves under prolonged warming and heat shock. BMC Plant Biology, 20(1), 86. 10.1186/s12870-020-2292-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasternack, C. (2007). Jasmonates: An update on biosynthesis, signal transduction and action in plant stress response, growth and development. Annals of Botany, 100(4), 681–697. 10.1093/aob/mcm079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, H. , Persson, S. , Mehta, T. , Srinivasasainagendra, V. , Chen, L. , Page, G. P. , Somerville, C. , & Loraine, A. (2006). Transcriptional coordination of the metabolic Network in Arabidopsis. Plant Physiology, 142, 762–774. 10.1104/pp.106.080358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, F. , Wu, X. , Li, T. , Jia, M. , Liu, X. , Li, P. , Zhou, X. , Ji, X. , & Yue, X. (2017). Genome‐wide survey of heat shock factors and heat shock protein 70s and their regulatory network under abiotic stresses in Brachypodium distachyon. PLoS ONE, 12, e0180352. 10.1371/journal.pone.0180352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter, D. , Vinegar, B. , Nahal, H. , Ammar, R. , Wilson, G. V. , & Provart, N. J. (2007). An “electronic fluorescent pictograph” browser for exploring and analyzing large‐scale biological data sets. PLoS ONE, 2(8), e718. 10.1371/journal.pone.0000718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G. , Liu, S. , Zhao, Y. , Wang, W. , Kong, Z. , & Tang, D. (2015). ENHANCED DISEASE RESISTANCE4 associates with CLATHRIN HEAVY CHAIN2 and modulates plant immunity by regulating relocation of EDR1 in Arabidopsis. The Plant Cell, 27(3), 857–873. 10.1105/tpc.114.134668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, K. , & Nishimura, M. (2008). Cytosolic heat shock protein 90 regulates heat shock transcription factor in Arabidopsis thaliana. Plant Signaling & Behavior, 3, 660–662. 10.4161/psb.3.9.5775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, K. , Fukao, Y. , Hayashi, M. , Fukazawa, M. , Suzuki, I. , & Nishimura, M. (2007). Cytosolic HSP90 regulates the heat shock response that is responsible for heat acclimation in Arabidopsis thaliana. Journal of Biological Chemistry, 282(52), 37794–37804. 10.1074/jbc.M707168200 [DOI] [PubMed] [Google Scholar]
- Yu, G. , Wang, L.‐G. , Han, Y. , & He, Q.‐Y. (2012). clusterProfiler: An R package for comparing biological themes among gene clusters. Omics: A Journal of Integrative Biology, 16(5), 284–287. 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandalinas, S. I. , Song, L. , Sengupta, S. , Mcinturf, S. A. , Grant, D. G. , Marjault, H. B. , Castro‐Guerrero, N. A. , Burks, D. , Azad, R. K. , & Mendoza‐Cozatl, D. G. (2020). Expression of a dominant‐negative AtNEET‐H89C protein disrupts iron–sulfur metabolism and iron homeostasis in Arabidopsis. The Plant Journal, 101(5), 1152–1169. 10.1111/tpj.14581 [DOI] [PubMed] [Google Scholar]
- Zhang, S. , Jin, G. , Zhang, X. S. , & Chen, L. (2007). Discovering functions and revealing mechanisms at molecular level from biological networks. Proteomics, 7(16), 2856–2869. 10.1002/pmic.200700095 [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Liu, B. , Li, J. , Zhang, L. , Wang, Y. , Zheng, H. , Lu, M. , & Chen, J. (2015). Hsf and Hsp gene families in Populus: Genome‐wide identification, organization and correlated expression during development and in stress responses. BMC Genomics, 16, 181. 10.1186/s12864-015-1398-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure S1: Selection of cutoff values
Supplementary figure S2: Top 5 overrepresented BP terms of Module‐1
Supplementary figure S3: Top 5 overrepresented CC terms of Module‐1
Supplementary figure S4: Top 5 overrepresented MF terms of Module‐1
Supplementary figure S5: Top 5 overrepresented BP terms of Module‐2
Supplementary figure S6: Top 5 overrepresented CC terms of Module‐2
Supplementary figure S7: Top 5 overrepresented MF terms of Module‐2
Supplementary figure S8: Top 5 overrepresented BP terms of Module‐3
Supplementary figure S9: Top 5 overrepresented CC terms of Module‐3
Supplementary figure S10: Top 5 overrepresented MF terms of Module‐3
Supplementary figure S11: Top 5 overrepresented BP terms of Module‐4
Supplementary figure S12: Top 5 overrepresented CC terms of Module‐4
Supplementary figure S13: Top 5 overrepresented MF terms of Module‐4
Supplementary figure S14: Top 5 overrepresented BP terms of Module‐5
Supplementary figure S15: Top 5 overrepresented CC terms of Module‐5
Supplementary figure S16: Top 5 overrepresented MF terms of Module‐5
Supplementary figure S17: Top 5 overrepresented BP terms of Module‐6
Supplementary figure S18: Top 5 overrepresented CC terms of Module‐6
Supplementary figure S19: Top 5 overrepresented MF terms of Module‐6
Supplementary figure S20: Top 5 overrepresented BP terms of Module‐7
Supplementary figure S21: Top 5 overrepresented CC terms of Module‐7
Supplementary figure S22: Top 5 overrepresented MF terms of Module‐7
Supplementary figure S23: Top 5 overrepresented BP terms of Module‐8
Supplementary figure S24: Top 5 overrepresented CC terms of Module‐8
Supplementary figure S25: Top 5 overrepresented MF terms of Module‐8
Supplementary figure S26: Top 5 overrepresented BP terms of Module‐9
Supplementary figure S27: Top 5 overrepresented CC terms of Module‐9
Supplementary figure S28: Top 5 overrepresented MF terms of Module‐9
Supplementary figure S29: Top 5 overrepresented BP terms of Module‐10
Supplementary figure S30: Top 5 overrepresented CC terms of Module‐10
Supplementary figure S31: Top 5 overrepresented MF terms of Module‐10
Supplementary figure S32: Top 5 overrepresented BP terms of Module‐11
Supplementary figure S33: Top 5 overrepresented CC terms of Module‐11
Supplementary figure S34: Top 5 overrepresented MF terms of Module‐11
Supplementary figure S35: Top 5 overrepresented BP terms of Module‐12
Supplementary figure S36: Top 5 overrepresented CC terms of Module‐12
Supplementary figure S37: Top 5 overrepresented MF terms of Module‐12
Supplementary figure S38: Top 5 overrepresented BP terms of Module‐13
Supplementary figure S39: Top 5 overrepresented CC terms of Module‐13
Supplementary figure S40: Top 5 overrepresented MF terms of Module‐13
Supplementary figure S41: Top 5 overrepresented BP terms of Module‐14
Supplementary figure S42: Top 5 overrepresented CC terms of Module‐14
Supplementary figure S43: Top 5 overrepresented MF terms of Module‐14
Supplementary figure S44: Top 5 overrepresented BP terms of Module‐15
Supplementary figure S45: Top 5 overrepresented CC terms of Module‐15
Supplementary figure S46: Top 5 overrepresented MF terms of Module‐15
Supplementary figure S47: Top 5 overrepresented BP terms of Module‐16
Supplementary figure S48: Top 5 overrepresented CC terms of Module‐16
Supplementary figure S49: Top 5 overrepresented MF terms of Module‐16
Supplementary figure S50: Top 5 overrepresented BP terms of Module‐17
Supplementary figure S51: Top 5 overrepresented CC terms of Module‐17
Supplementary figure S52: Top 5 overrepresented MF terms of Module‐17
Supplementary figure S53: Top 5 overrepresented BP terms of Module‐18
Supplementary figure S54: Top 5 overrepresented CC terms of Module‐18
Supplementary figure S55: Top 5 overrepresented MF terms of Module‐18
Supplementary figure S56: Top 5 overrepresented BP terms of Module‐19
Supplementary figure S57: Top 5 overrepresented CC terms of Module‐19
Supplementary figure S58: Top 5 overrepresented MF terms of Module‐19
Supplementary figure S59: Top 5 overrepresented BP terms of Module‐20
Supplementary figure S60: Top 5 overrepresented CC terms of Module‐20
Supplementary figure S61: Top 5 overrepresented MF terms of Module‐20
Supplementary figure S62: Top 5 overrepresented BP terms of Module‐21
Supplementary figure S63: Top 5 overrepresented CC terms of Module‐21
Supplementary figure S64: Top 5 overrepresented MF terms of Module‐21
Supplementary figure S65: Top 5 overrepresented BP terms of Module‐22
Supplementary figure S66: Top 5 overrepresented CC terms of Module‐22
Supplementary figure S67: Top 5 overrepresented MF terms of Module‐22
Supplementary figure S68: Top 5 overrepresented BP terms of Module‐23
Supplementary figure S69: Top 5 overrepresented CC terms of Module‐23
Supplementary figure S70: Top 5 overrepresented MF terms of Module‐23
Supplementary figure S71: Top 5 overrepresented BP terms of Module‐24
Supplementary figure S72: Top 5 overrepresented CC terms of Module‐24
Supplementary figure S73: Top 5 overrepresented MF terms of Module‐24
Supplementary figure S74: Top 5 overrepresented BP terms of Module‐25
Supplementary figure S75: Top 5 overrepresented CC terms of Module‐25
Supplementary figure S76: Top 5 overrepresented MF terms of Module‐25
Supplementary figure S77: Top 5 overrepresented BP terms of Module‐26
Supplementary figure S78: Top 5 overrepresented CC terms of Module‐26
Supplementary figure S79: Top 5 overrepresented MF terms of Module‐26
Supplementary figure S80: Top 5 overrepresented BP terms of Module‐27
Supplementary figure S81: Top 5 overrepresented CC terms of Module‐27
Supplementary figure S82: Top 5 overrepresented MF terms of Module‐27
Supplementary figure S83: Top 5 overrepresented BP terms of Module‐28
Supplementary figure S84: Top 5 overrepresented CC terms of Module‐28
Supplementary figure S85: Top 5 overrepresented MF terms of Module‐28
Supplementary figure S86: Top 5 overrepresented BP terms of Module‐29
Supplementary figure S87: Top 5 overrepresented CC terms of Module‐29
Supplementary figure S88: Top 5 overrepresented MF terms of Module‐29
Supplementary figure S89: Top 5 overrepresented BP terms of Module‐30
Supplementary figure S90: Top 5 overrepresented CC terms of Module‐30
Supplementary figure S91: Top 5 overrepresented MF terms of Module‐30
Supplementary figure S92: Top 5 overrepresented BP terms of Module‐31
Supplementary figure S93: Top 5 overrepresented CC terms of Module‐31
Supplementary figure S94: Top 5 overrepresented MF terms of Module‐31
Supplementary figure S95: Top 5 overrepresented BP terms of Module‐32
Supplementary figure S96: Top 5 overrepresented CC terms of Module‐32
Supplementary figure S97: Top 5 overrepresented MF terms of Module‐32
Supplementary figure S98: Top 5 overrepresented BP terms of Module‐33
Supplementary figure S99: Top 5 overrepresented CC terms of Module‐33
Supplementary figure S100: Top 5 overrepresented MF terms of Module‐33
Supplementary figure S101: Top 5 overrepresented BP terms of Module‐34
Supplementary figure S102: Top 5 overrepresented CC terms of Module‐34
Supplementary figure S103: Top 5 overrepresented MF terms of Module‐34
Supplementary figure S104: Top 5 overrepresented BP terms of Module‐35
Supplementary figure S105: Top 5 overrepresented CC terms of Module‐35
Supplementary figure S106: Top 5 overrepresented MF terms of Module‐35
Supplementary figure S107: Top 5 overrepresented BP terms of Module‐36
Supplementary figure S108: Top 5 overrepresented CC terms of Module‐36
Supplementary figure S109: Top 5 overrepresented MF terms of Module‐36
Supplementary figure S110: Top 5 overrepresented BP terms of Module‐37
Supplementary figure S111: Top 5 overrepresented CC terms of Module‐37
Supplementary figure S112: Top 5 overrepresented MF terms of Module‐37
Supplementary figure S113: Top 5 overrepresented BP terms of Module‐38
Supplementary figure S114: Top 5 overrepresented CC terms of Module‐38
Supplementary figure S115: Top 5 overrepresented MF terms of Module‐38
Supplementary figure S116: Top 5 overrepresented BP terms of Module‐39
Supplementary figure S117: Top 5 overrepresented CC terms of Module‐39
Supplementary figure S118: Top 5 overrepresented MF terms of Module‐39
Supplementary figure S119: Top 5 overrepresented BP terms of Module‐40
Supplementary figure S120: Top 5 overrepresented CC terms of Module‐40
Supplementary figure S121: Top 5 overrepresented MF terms of Module‐40
Supplementary figure S122: Top 5 overrepresented BP terms of Module‐41
Supplementary figure S123: Top 5 overrepresented CC terms of Module‐41
Supplementary figure S124: Top 5 overrepresented MF terms of Module‐41
Supplementary figure S125: Top 5 overrepresented BP terms of Module‐42
Supplementary figure S126: Top 5 overrepresented CC terms of Module‐42
Supplementary figure S127: Top 5 overrepresented MF terms of Module‐42
Supplementary figure S128: Top 5 overrepresented BP terms of Module‐43
Supplementary figure S129: Top 5 overrepresented CC terms of Module‐43
Supplementary figure S130: Top 5 overrepresented MF terms of Module‐43
Supplementary figure S131: Top 5 overrepresented BP terms of Module‐44
Supplementary figure S132: Top 5 overrepresented CC terms of Module‐44
Supplementary figure S133: Top 5 overrepresented MF terms of Module‐44
Supplementary figure S134: Top 5 overrepresented BP terms of Module‐45
Supplementary figure S135: Top 5 overrepresented CC terms of Module‐45
Supplementary figure S136: Top 5 overrepresented MF terms of Module‐45
Supplementary figure S137: Top 5 overrepresented BP terms of Module‐46
Supplementary figure S138: Top 5 overrepresented CC terms of Module‐46
Supplementary figure S139: Top 5 overrepresented MF terms of Module‐46
Supplementary figure S140: Top 5 overrepresented BP terms of Module‐47
Supplementary figure S141: Top 5 overrepresented CC terms of Module‐47
Supplementary figure S142: Top 5 overrepresented MF terms of Module‐47
Supplementary figure S143: Top 5 overrepresented BP terms of Module‐48
Supplementary figure S144: Top 5 overrepresented CC terms of Module‐48
Supplementary figure S145: Top 5 overrepresented MF terms of Module‐48
Supplementary figure S146: Top 5 overrepresented BP terms of Module‐49
Supplementary figure S147: Top 5 overrepresented CC terms of Module‐49
Supplementary figure S148: Top 5 overrepresented MF terms of Module‐49
Supplementary figure S149: Top 5 overrepresented BP terms of Module‐50
Supplementary figure S150: Top 5 overrepresented CC terms of Module‐50
Supplementary figure S151: Top 5 overrepresented MF terms of Module‐50
Supplementary figure S152: Top 5 overrepresented BP terms of Module‐51
Supplementary figure S153: Top 5 overrepresented CC terms of Module‐51
Supplementary figure S154: Top 5 overrepresented MF terms of Module‐51
Supplementary figure S155: Top 5 overrepresented BP terms of Module‐52
Supplementary figure S156: Top 5 overrepresented CC terms of Module‐52
Supplementary figure S157: Top 5 overrepresented MF terms of Module‐52
Supplementary figure S158: Top 5 overrepresented BP terms of Module‐53
Supplementary figure S159: Top 5 overrepresented CC terms of Module‐53
Supplementary figure S160: Top 5 overrepresented MF terms of Module‐53
Supplementary figure S161: Top 5 overrepresented BP terms of Module‐54
Supplementary figure S162: Top 5 overrepresented CC terms of Module‐54
Supplementary figure S163: Top 5 overrepresented MF terms of Module‐54
Supplementary table S1: BP terms enriched in Module‐1
Supplementary table S2: CC terms enriched in Module‐1
Supplementary table S3: MF terms enriched in Module‐1
Supplementary table S4: Pathways enriched in Module‐1
Supplementary table S5: BP terms enriched in Module‐2
Supplementary table S6: CC terms enriched in Module‐2
Supplementary table S7: MF terms enriched in Module‐2
Supplementary table S8: Pathways enriched in Module‐2
Supplementary table S9: BP terms enriched in Module‐3
Supplementary table S10: CC terms enriched in Module‐3
Supplementary table S11: MF terms enriched in Module‐3
Supplementary table S12: Pathways enriched in Module‐3
Supplementary table S13: BP terms enriched in Module‐4
Supplementary table S14: CC terms enriched in Module‐4
Supplementary table S15: MF terms enriched in Module‐4
Supplementary table S16: Pathways enriched in Module‐4
Supplementary table S17: BP terms enriched in Module‐5
Supplementary table S18: CC terms enriched in Module‐5
Supplementary table S19: MF terms enriched in Module‐5
Supplementary table S20: Pathways enriched in Module‐5
Supplementary table S21: BP terms enriched in Module‐6
Supplementary table S22: CC terms enriched in Module‐6
Supplementary table S23: MF terms enriched in Module‐6
Supplementary table S24: Pathways enriched in Module‐6
Supplementary table S25: BP terms enriched in Module‐7
Supplementary table S26: CC terms enriched in Module‐7
Supplementary table S27: MF terms enriched in Module‐7
Supplementary table S28: Pathways enriched in Module‐7
Supplementary table S29: BP terms enriched in Module‐8
Supplementary table S30: CC terms enriched in Module‐8
Supplementary table S31: MF terms enriched in Module‐8
Supplementary table S32: Pathways enriched in Module‐8
Supplementary table S33: BP terms enriched in Module‐9
Supplementary table S34: CC terms enriched in Module‐9
Supplementary table S35: MF terms enriched in Module‐9
Supplementary table S36: Pathways enriched in Module‐9
Supplementary table S37: BP terms enriched in Module‐10
Supplementary table S38: CC terms enriched in Module‐10
Supplementary table S39: MF terms enriched in Module‐10
Supplementary table S40: Pathways enriched in Module‐10
Supplementary table S41: BP terms enriched in Module‐11
Supplementary table S42: CC terms enriched in Module‐11
Supplementary table S43: MF terms enriched in Module‐11
Supplementary table S44: Pathways enriched in Module‐11
Supplementary table S45: BP terms enriched in Module‐12
Supplementary table S46: CC terms enriched in Module‐12
Supplementary table S47: MF terms enriched in Module‐12
Supplementary table S48: Pathways enriched in Module‐12
Supplementary table S49: BP terms enriched in Module‐13
Supplementary table S50: CC terms enriched in Module‐13
Supplementary table S51: MF terms enriched in Module‐13
Supplementary table S52: Pathways enriched in Module‐13
Supplementary table S53: BP terms enriched in Module‐14
Supplementary table S54: CC terms enriched in Module‐14
Supplementary table S55: MF terms enriched in Module‐14
Supplementary table S56: Pathways enriched in Module‐14
Supplementary table S57: BP terms enriched in Module‐15
Supplementary table S58: CC terms enriched in Module‐15
Supplementary table S59: MF terms enriched in Module‐15
Supplementary table S60: Pathways enriched in Module‐15
Supplementary table S61: BP terms enriched in Module‐16
Supplementary table S62: CC terms enriched in Module‐16
Supplementary table S63: MF terms enriched in Module‐16
Supplementary table S64: Pathways enriched in Module‐16
Supplementary table S65: BP terms enriched in Module‐17
Supplementary table S66: CC terms enriched in Module‐17
Supplementary table S67: MF terms enriched in Module‐17
Supplementary table S68: Pathways enriched in Module‐17
Supplementary table S69: BP terms enriched in Module‐18
Supplementary table S70: CC terms enriched in Module‐18
Supplementary table S71: MF terms enriched in Module‐18
Supplementary table S72: Pathways enriched in Module‐18
Supplementary table S73: BP terms enriched in Module‐19
Supplementary table S74: CC terms enriched in Module‐19
Supplementary table S75: MF terms enriched in Module‐19
Supplementary table S76: Pathways enriched in Module‐19
Supplementary table S77: BP terms enriched in Module‐20
Supplementary table S78: CC terms enriched in Module‐20
Supplementary table S79: MF terms enriched in Module‐20
Supplementary table S80: Pathways enriched in Module‐20
Supplementary table S81: BP terms enriched in Module‐21
Supplementary table S82: CC terms enriched in Module‐21
Supplementary table S83: MF terms enriched in Module‐21
Supplementary table S84: Pathways enriched in Module‐21
Supplementary table S85: BP terms enriched in Module‐22
Supplementary table S86: CC terms enriched in Module‐22
Supplementary table S87: MF terms enriched in Module‐22
Supplementary table S88: Pathways enriched in Module‐22
Supplementary table S89: BP terms enriched in Module‐23
Supplementary table S90: CC terms enriched in Module‐23
Supplementary table S91: MF terms enriched in Module‐23
Supplementary table S92: Pathways enriched in Module‐23
Supplementary table S93: BP terms enriched in Module‐24
Supplementary table S94: CC terms enriched in Module‐24
Supplementary table S95: MF terms enriched in Module‐24
Supplementary table S96: Pathways enriched in Module‐24
Supplementary table S97: BP terms enriched in Module‐25
Supplementary table S98: CC terms enriched in Module‐25
Supplementary table S99: MF terms enriched in Module‐25
Supplementary table S100: Pathways enriched in Module‐25
Supplementary table S101: BP terms enriched in Module‐26
Supplementary table S102: CC terms enriched in Module‐26
Supplementary table S103: MF terms enriched in Module‐26
Supplementary table S104: Pathways enriched in Module‐26
Supplementary table S105: BP terms enriched in Module‐27
Supplementary table S106: CC terms enriched in Module‐27
Supplementary table S107: MF terms enriched in Module‐27
Supplementary table S108: Pathways enriched in Module‐27
Supplementary table S109: BP terms enriched in Module‐28
Supplementary table S110: CC terms enriched in Module‐28
Supplementary table S111: MF terms enriched in Module‐28
Supplementary table S112: Pathways enriched in Module‐28
Supplementary table S113: BP terms enriched in Module‐29
Supplementary table S114: CC terms enriched in Module‐29
Supplementary table S115: MF terms enriched in Module‐29
Supplementary table S116: Pathways enriched in Module‐29
Supplementary table S117: BP terms enriched in Module‐30
Supplementary table S118: CC terms enriched in Module‐30
Supplementary table S119: MF terms enriched in Module‐30
Supplementary table S120: Pathways enriched in Module‐30
Supplementary table S121: BP terms enriched in Module‐31
Supplementary table S122: CC terms enriched in Module‐31
Supplementary table S123: MF terms enriched in Module‐31
Supplementary table S124: Pathways enriched in Module‐31
Supplementary table S125: BP terms enriched in Module‐32
Supplementary table S126: CC terms enriched in Module‐32
Supplementary table S127: MF terms enriched in Module‐32
Supplementary table S128: Pathways enriched in Module‐32
Supplementary table S129: BP terms enriched in Module‐33
Supplementary table S130: CC terms enriched in Module‐33
Supplementary table S131: MF terms enriched in Module‐33
Supplementary table S132: Pathways enriched in Module‐33
Supplementary table S133: BP terms enriched in Module‐34
Supplementary table S134: CC terms enriched in Module‐34
Supplementary table S135: MF terms enriched in Module‐34
Supplementary table S136: Pathways enriched in Module‐34
Supplementary table S137: BP terms enriched in Module‐35
Supplementary table S138: CC terms enriched in Module‐35
Supplementary table S139: MF terms enriched in Module‐35
Supplementary table S140: Pathways enriched in Module‐35
Supplementary table S141: BP terms enriched in Module‐36
Supplementary table S142: CC terms enriched in Module‐36
Supplementary table S143: MF terms enriched in Module‐36
Supplementary table S144: Pathways enriched in Module‐36
Supplementary table S145: BP terms enriched in Module‐37
Supplementary table S146: CC terms enriched in Module‐37
Supplementary table S147: MF terms enriched in Module‐37
Supplementary table S148: Pathways enriched in Module‐37
Supplementary table S149: BP terms enriched in Module‐38
Supplementary table S150: CC terms enriched in Module‐38
Supplementary table S151: MF terms enriched in Module‐38
Supplementary table S152: Pathways enriched in Module‐38
Supplementary table S153: BP terms enriched in Module‐39
Supplementary table S154: CC terms enriched in Module‐39
Supplementary table S155: MF terms enriched in Module‐39
Supplementary table S156: Pathways enriched in Module‐39
Supplementary table S157: BP terms enriched in Module‐40
Supplementary table S158: CC terms enriched in Module‐40
Supplementary table S159: MF terms enriched in Module‐40
Supplementary table S160: Pathways enriched in Module‐40
Supplementary table S161: BP terms enriched in Module‐41
Supplementary table S162: CC terms enriched in Module‐41
Supplementary table S163: MF terms enriched in Module‐41
Supplementary table S164: Pathways enriched in Module‐41
Supplementary table S165: BP terms enriched in Module‐42
Supplementary table S166: CC terms enriched in Module‐42
Supplementary table S167: MF terms enriched in Module‐42
Supplementary table S168: Pathways enriched in Module‐42
Supplementary table S169: BP terms enriched in Module‐43
Supplementary table S170: CC terms enriched in Module‐43
Supplementary table S171: MF terms enriched in Module‐43
Supplementary table S172: Pathways enriched in Module‐43
Supplementary table S173: BP terms enriched in Module‐44
Supplementary table S174: CC terms enriched in Module‐44
Supplementary table S175: MF terms enriched in Module‐44
Supplementary table S176: Pathways enriched in Module‐44
Supplementary table S177: BP terms enriched in Module‐45
Supplementary table S178: CC terms enriched in Module‐45
Supplementary table S179: MF terms enriched in Module‐45
Supplementary table S180: Pathways enriched in Module‐45
Supplementary table S181: BP terms enriched in Module‐46
Supplementary table S182: CC terms enriched in Module‐46
Supplementary table S183: MF terms enriched in Module‐46
Supplementary table S184: Pathways enriched in Module‐46
Supplementary table S185: BP terms enriched in Module‐47
Supplementary table S186: CC terms enriched in Module‐47
Supplementary table S187: MF terms enriched in Module‐47
Supplementary table S188: Pathways enriched in Module‐47
Supplementary table S189: BP terms enriched in Module‐48
Supplementary table S190: CC terms enriched in Module‐48
Supplementary table S191: MF terms enriched in Module‐48
Supplementary table S192: Pathways enriched in Module‐48
Supplementary table S193: BP terms enriched in Module‐49
Supplementary table S194: CC terms enriched in Module‐49
Supplementary table S195: MF terms enriched in Module‐49
Supplementary table S196: Pathways enriched in Module‐49
Supplementary table S197: BP terms enriched in Module‐50
Supplementary table S198: CC terms enriched in Module‐50
Supplementary table S199: MF terms enriched in Module‐50
Supplementary table S200: Pathways enriched in Module‐50
Supplementary table S201: BP terms enriched in Module‐51
Supplementary table S202: CC terms enriched in Module‐51
Supplementary table S203: MF terms enriched in Module‐51
Supplementary table S204: Pathways enriched in Module‐51
Supplementary table S205: BP terms enriched in Module‐52
Supplementary table S206: CC terms enriched in Module‐52
Supplementary table S207: MF terms enriched in Module‐52
Supplementary table S208: Pathways enriched in Module‐52
Supplementary table S209: BP terms enriched in Module‐53
Supplementary table S210: CC terms enriched in Module‐53
Supplementary table S211: MF terms enriched in Module‐53
Supplementary table S212: Pathways enriched in Module‐53
Supplementary table S213: BP terms enriched in Module‐54
Supplementary table S214: CC terms enriched in Module‐54
Supplementary table S215: MF terms enriched in Module‐54
Supplementary table S216: Pathways enriched in Module‐54
Supplementary table S217: List of novel genes identified as the first neighbors of RBOHD.
Supplementary table S218: List of novel genes identified as the first neighbors of AtNEET.
Supplementary table S219: List of novel genes identified as the first neighbors of HSFA1D.
Data Availability Statement
All data have been made publicly available at https://doi.org/10.6084/m9.figshare.16752733.v4, and the associated source codes have been provided at the project's GitHub site: https://github.com/sohamsg90/WGCNA_Arabidopsis-main.