

ABSTRACT
A recent Perspective article described the “carbohydrate-insulin model (CIM)” of obesity, asserting that it “better reflects knowledge on the biology of weight control” as compared with what was described as the “dominant energy balance model (EBM),” which fails to consider “biological mechanisms that promote weight gain.” Unfortunately, the Perspective conflated and confused the principle of energy balance, a law of physics that is agnostic as to obesity mechanisms, with the EBM as a theoretical model of obesity that is firmly based on biology. In doing so, the authors presented a false choice between the CIM and a caricature of the EBM that does not reflect modern obesity science. Here, we present a more accurate description of the EBM where the brain is the primary organ responsible for body weight regulation operating mainly below our conscious awareness via complex endocrine, metabolic, and nervous system signals to control food intake in response to the body's dynamic energy needs as well as environmental influences. We also describe the recent history of the CIM and show how the latest “most comprehensive formulation” abandons a formerly central feature that required fat accumulation in adipose tissue to be the primary driver of positive energy balance. As such, the new CIM can be considered a special case of the more comprehensive EBM but with a narrower focus on diets high in glycemic load as the primary factor responsible for common obesity. We review data from a wide variety of studies that address the validity of each model and demonstrate that the EBM is a more robust theory of obesity than the CIM.
Keywords: obesity, food intake, energy balance, carbohydrates, insulin
Introduction
Theoretical models of the pathogenesis of obesity can help organize and synthesize observations to form hypotheses for experimental interrogation. The results of such experiments can be used to refine or refute models, thereby leading to a better understanding of the mechanistic drivers of common obesity. Therefore, an evidence-based model that increases our understanding of the factors responsible for obesity can be used to design more effective interventions for obesity prevention and therapy.
Two important questions regarding human obesity should be addressed by a successful model. First, what explains between-person variability in adiposity in a population? Second, what explains the global shifts in the population prevalence of obesity over the past several decades? As a partial answer to the first question, BMI is highly heritable and genetic differences explain ∼75% of BMI variability among individuals (1, 2). Regarding the second question, although changes in occupational physical activity and the built environment might have contributed to obesity by reducing overall physical activity (3), changes in the food environment are likely the primary driver of the increased obesity prevalence in recent decades (4). However, the specific aspects of the food environment that are most “obesogenic” and how they interact with the genetics of susceptible individuals to cause obesity are topics that are hotly debated, and competing theoretical models implicate different mechanisms.
A recent Perspective article described the theoretical “carbohydrate-insulin model (CIM)” of obesity, asserting that it “better reflects knowledge on the biology of weight control” as compared with what the authors described as the “dominant energy balance model (EBM),” which was claimed to conceptualize obesity “without considering the biological mechanisms promoting weight gain” (5). In doing so, the authors presented a false choice between the CIM and a caricature of the EBM as a theory of obesity that does not reflect modern obesity science.
Indeed, the recent Perspective conflated and confused the principle of energy balance, a law of physics that is agnostic to obesity mechanisms, with the EBM as a theoretical model of obesity. The Perspective described the EBM as an “inherent tautology” that “considers all calories to be metabolically alike for all practical purposes” (5). These statements more aptly refer to the law of physics and not the EBM as a theoretical model that is firmly based on biological mechanisms. To be clear, all theoretical models of obesity, including the CIM, must satisfy the principle of energy balance to avoid violating the laws of physics.
The Perspective also described a straw-man version of the theoretical EBM postulating that “energy-dense, tasty, modern processed foods drive a positive energy balance through increased intake” under “conscious control” (5). However, this is an inaccurate description of the EBM, which we clarify below. We also describe the recent history of the CIM and show how its latest “most comprehensive formulation” (5) constitutes abandonment of its formerly central feature (6–9) and can be considered as a special case of the EBM that focuses on diets with high glycemic load as the primary factor responsible for obesity. We review data from a wide variety of studies that address the validity of each model and demonstrate that the EBM is a more robust theory of obesity than the CIM.
The EBM of Obesity
The EBM proposes that the brain is the primary organ responsible for body weight regulation via integration of external signals from the food environment along with internal signals from peripheral organs to control food intake (Figure 1) (10). Specific brain regions, such as the hypothalamus, basal ganglia, and the brainstem modulate food intake below our conscious awareness via complex endocrine, metabolic, and nervous system signals (11–13) acting in response to the body's dynamic energy needs as well as environmental influences (14, 15). The orchestration of energy homeostasis occurs through short-term signals [e.g., ghrelin, peptide YY, glucagon-like peptide 1 (GLP-1), vagal afferents] controlling meal patterns (i.e., the initiation and cessation of feeding) and long-term signals (e.g., leptin) that modulate the activity of the short-term system thereby increasing or decreasing overall energy intake. Thus, whereas day-to-day energy intake and energy balance of an individual can be highly variable, neural regulation of energy balance is generally achieved over prolonged time scales (16–19).
FIGURE 1.
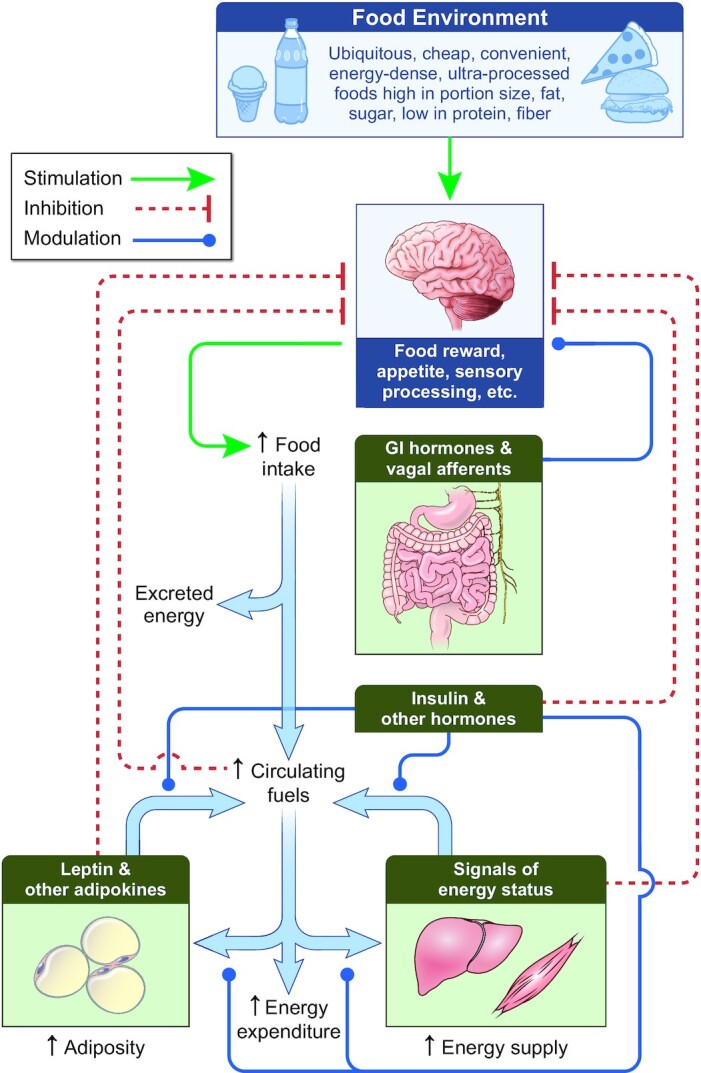
The energy balance model of obesity posits that body weight is regulated by the brain in response to external signals from the food environment that are integrated with internal signals to control food intake below our conscious awareness. Increased prevalence of obesity has resulted from changes in the food environment leading to increased food intake and circulating fuels. Hormones, including insulin, respond to nutrient intake and absorption to direct the flow of metabolic fluxes into and out of various organs and provide signals to the brain that control food intake. Energy supply to organs such as liver and muscle increases, which supports their increased growth during the development of obesity and can result in ectopic lipid accumulation. Signals indicating the energy status of various organs are sensed by the brain to control food intake by mechanisms that remain to be fully elucidated. Oxidation of carbohydrate, fat, and protein provides the body with its energy needs, which increase as obesity develops. Adaptations of metabolic fuel selection as well as changes in the endocrine milieu ensure that partitioning of overall energy imbalances are primarily reflected as changes in adipose tissue triglyceride storage regardless of diet composition. Inherited variation in the operation of these processes, particularly those in the brain, are responsible for a substantial proportion of the interindividual difference in susceptibility or resistance to developing obesity in a particular environment. Thick blue arrows indicate the flow of energy. GI, gastrointestinal.
The EBM proposes that the increasing population prevalence of obesity in recent decades is primarily due to changes in the food environment, including increased availability and marketing of a wide variety of inexpensive, convenient, energy-dense, ultraprocessed foods that are high in portion size, fat, and sugar, and low in protein and fiber. The CIM Perspective downplayed the potential role of conscious “liking” or palatability of food in driving obesity (5). However, palatability is only 1 dimension of the multifaceted concept of food reward that involves incentive salience, wanting, and motivation that primarily operate below our conscious awareness (11).
Brain circuits controlling energy intake respond to a changing food environment in ways that are beginning to be elucidated, especially in mouse models (20, 21). For example, a bidirectional circuit was recently identified between hypothalamic neurons expressing agouti-related peptide (AgRP) that control homeostatic hunger and the midbrain dopamine system influencing food reward (22). Exposure to a high-fat diet alters the activity of this bidirectional circuit and results in excess energy intake, development of obesity, and devaluation of a low-fat diet that does not induce obesity (23). Distinct gut-brain pathways have been identified for sensing dietary fat and carbohydrate thereby modulating hypothalamic AgRP neuronal activity (24) and striatal dopamine release (11). Therefore, the EBM is consistent with the idea that diet composition, not simply its caloric content, could be an important factor in the central nervous system control of food intake.
The physical principle of energy balance does not specify the biological mechanisms determining how energy imbalances are partitioned within the body to result primarily in changes in adipose tissue fat stores compared with changes in energy stored or utilized in other body compartments—a fact that has been recently misinterpreted as being a deficiency in the EBM (25). However, based on extensive studies of human macronutrient balance in response to various dietary interventions (26–34), the EBM incorporates physiological mechanisms underlying energy partitioning whereby diet composition and amount affect whole-body net oxidation rates of carbohydrate, fat, and protein such that overall energy imbalances are primarily reflected as fat imbalances regardless of the composition of the diet (35–37). This regulation of macronutrient metabolism is the consequence of coordinated control of multiorgan metabolic fluxes by a variety of hormones, including but not limited to insulin, such that whole-body fat imbalances end up primarily reflected as changes in adipose tissue fat storage (38–40). The EBM therefore conceptualizes adipose tissue as an active endocrine organ evolved to dynamically coordinate the efficient storage and mobilization of energy (i.e., triglycerides) in response to energy surplus and deficit, respectively.
The EBM recognizes that individual differences in energy partitioning can result in different degrees of adiposity, even when energy intake is not different (41, 42). This can happen, in part, because accretion of lean body mass results in greater energy expenditure as compared with accumulation of body fat mass. Indeed, energy partitioning differences explain why females accumulate greater body fat than males during growth and development despite consuming fewer total calories (43). Also, body fat mass can be relatively constant or even decrease during periods of rapid growth when positive energy balance corresponds to increasing lean body mass (44). Furthermore, subtle differences in adipose tissue dynamics may affect energy partitioning and fuel utilization thereby influencing body composition over the long term (45–49), but the extent of such influences on common obesity are currently unclear.
The EBM allows for a role of decreased physical activity in the development of obesity, although not necessarily because of decreased energy expenditure per se but rather due to decreased precision of energy intake control (3, 50, 51). Furthermore, other factors can also play a role in the development of obesity within the EBM framework (52).
The above description of the EBM is inconsistent with the characterization by Ludwig et al. (5) in their Perspective and elsewhere (7) as a theoretical model that “essentially disregards knowledge about the biological influences on fat storage,” proposing that obesity results from “conscious control” of behaviors affecting energy intake or expenditure. Indeed, the EBM emphasizes that powerful internal and external signals influence the neural regulation of energy balance below our conscious awareness and explains why simple advice to “eat less and move more” is ineffective for sustained weight loss.
The CIM of Obesity
The CIM presented in the recent Perspective (5) is substantially different from its previous iterations (6–9). Box 1 provides a brief history of related theoretical concepts leading to Taubes’ 2007 proposal (9) of the adipocentric CIM whereby obesity results from increased dietary carbohydrates driving excess insulin secretion causing adipose tissue to accumulate and trap fat thereby starving nonadipose tissues of fuel. Thus, “by driving fat accumulation, carbohydrates also increase hunger and decrease the amount of energy we expend in metabolism and physical activity” (9) thereby resulting in positive energy balance with energy intake exceeding expenditure.
BOX 1.
Historical antecedents of the adipocentric CIM
In the early 20th century, the idea of “lipophilia” proposed adipose tissue was the primary site of dysregulation in obesity, although dietary carbohydrate-driven insulin secretion was not implicated in this pathophysiology (143). In the early 1950s, Pennington (144–148) proposed that people with obesity have a cellular defect in their ability to oxidize carbohydrate that resulted in increased de novo lipogenesis, suppression of adipose lipolysis, and thereby resulted in body fat accumulation along with reduced energy expenditure and increased appetite. Although such a cellular defect was never found, Pennington speculated that dietary carbohydrate-driven insulin secretion exacerbated the problem, which helped explain the apparent effectiveness of his low-carbohydrate diet regimen for treating obesity. In 1962, Astwood (149) expanded on the lipophilia concept by hypothesizing that increased appetite in some people with obesity could be due to aberrant action of insulin, cortisol, or other hormones to either trap fat in adipose tissue or prevent fat oxidation in peripheral tissues. Beginning in the mid-1970s and extending into the late 1990s, Mark Friedman (150–153) proposed that energy sensing in peripheral tissues, especially the liver, provides the primary signals to the brain controlling energy intake and noted that diets high in both fat and carbohydrate could be responsible for obesity due to aberrant fuel partitioning to adipose tissue leading to a decrement in liver energy status (150).
In the early 2000s, Ludwig (75, 154) hypothesized that overeating is the result of consuming high-glycemic-index foods that rapidly increase plasma glucose and insulin, resulting in uptake of nutrients in insulin-responsive tissues and subsequent decreases in circulating fuels in the late postprandial period that are sensed by the brain to promote hunger. In 2006, Lustig (155) linked increasing population obesity prevalence to “our current Western diet [that] is highly insulinogenic, as demonstrated by its increased energy density, high fat content, high glycemic index, increased fructose composition, decreased fiber, and decreased dairy content.” Lustig proposed that autonomic dysfunction potentiates diet-induced hyperinsulinemia, which was hypothesized to increase energy intake by antagonizing leptin signaling and increasing dopamine in the brain. Neither Ludwig nor Lustig emphasized a primary role of adipose tissue insulin signaling in their models of obesity. Rather, insulin was presumed to act on multiple organs in parallel, and the brain played a direct role in controlling energy intake either by sensing low concentrations of circulating fuels (75) or by altered central leptin and dopamine signaling as a result of hyperinsulinemia (155).
Taubes justified his adipocentric CIM by asserting that “by the mid-1960s four facts had been established beyond reasonable doubt: 1) carbohydrates are singularly responsible for prompting insulin secretion; 2) insulin is singularly responsible for inducing fat accumulation; 3) dietary carbohydrates are required for excess fat accumulation; and 4) both type 2 diabetics and the obese have abnormally elevated concentrations of circulating insulin” [emphasis added] (9). To explain the epidemiological observations of “the emergence of obesity in recently Westernized populations” Taubes stated that, “carbohydrates, and particularly refined carbohydrates – and perhaps the fructose content as well, and thus the amount of sugars consumed – are the prime suspects in chronic elevation of insulin; hence, they are the ultimate cause of common obesity” (9).
Unfortunately, the foundational “facts” of the adipocentric CIM are in error, particularly the claims of singularity and necessity about the roles of insulin and dietary carbohydrates on adipose tissue fat metabolism. Rather, adipose fat storage can occur in the absence of either dietary carbohydrate or an increase in insulin above basal concentrations (38, 53, 54). Thus, alternative physiological mechanisms allow body fat to be stored without the necessity of dietary carbohydrates—a feature of adipose tissue that likely had evolutionary advantages for omnivorous species. Furthermore, insulin secretion is determined by a variety of factors beyond dietary carbohydrate (55–57). Indeed, basal insulin concentrations are similarly affected by overall energy imbalance regardless of whether the imbalance is achieved by manipulating dietary carbohydrates or fat (58). Thus, there are potentially many paths to fat accumulation despite Taubes believing it to be an inescapable conclusion that “by stimulating insulin secretion, carbohydrates make us fat and cause obesity” (9).
Despite problems with the foundational logic of the adipocentric CIM, it was adopted more widely (6–8) with an emphasis that this “model considers fat cells as central to the etiology of obesity” such that “a high-carbohydrate diet…produces postprandial hyperinsulinemia, promotes deposition of calories in fat cells instead of oxidation in lean tissues, and thereby predisposes to weight gain through increased hunger, slowing metabolic rate, or both” (7). Accordingly, a popular diet book based on the adipocentric CIM claimed that insulin acts as “the ultimate fat cell fertilizer” that “ushers calories into fat cells, but restricts their passage back out”, and consequently “our fat cells make us overeat.” (6). Based on observations that insulin rapidly responds to changes in dietary carbohydrate and has expeditious effects on adipose tissue, readers were advised that “decreasing carbohydrate is the quickest and easiest way to lower insulin and jump-start weight loss” and low-carbohydrate diets result in “big declines in hunger, sometimes as early as day 1” (6).
Precisely how carbohydrate-driven insulin action on adipose tissue drives hunger or slows metabolic rate is not specified by the CIM, but low concentrations of circulating fuels in the late postprandial period after high- compared with low-glycemic-load meals have been proposed to be either sensed directly by the brain or via decreased energy status of peripheral organs like the liver. Of course, dips in blood glucose can increase hunger by mechanisms independent of those proposed by the CIM as described long ago in the glucostatic theories of Mayer (59, 60), LeMagnen (61), and Campfield (62, 63). Indeed, greater dips in blood glucose occurring 2–3 h after meals were recently associated with increased appetite in humans (64).
The adipocentric CIM was expanded beyond dietary carbohydrates and a “comprehensive paradigm” was proposed whereby all obesogenic factors (e.g., amount of dietary protein, micronutrients, poor sleep, stress, physical inactivity, and environmental endocrine-disrupting chemicals) “affect insulin secretion or adipocyte biology directly” with increased energy intake and decreased energy expenditure as necessary downstream consequences (7). Hence, the adipocentric CIM achieves the much-touted reversal of the direction of causation whereby “positive energy balance does not cause increasing adiposity; rather, a shift in substrate partitioning favoring fat storage drives a positive energy balance” (5).
Abandonment of the Adipocentric CIM
Some of us (JRS and KDH) recently argued against the adipocentric CIM and suggested that insulin and other factors exert pleotropic actions on a variety of organs that influence energy balance (10). Interestingly, the latest formulation of the CIM (5) also de-emphasizes the formerly central role of adipose tissue and thereby abandons the “comprehensive paradigm” of the adipocentric CIM (7). The new CIM “considers that substrate partitioning and fat deposition are determined by the integrated actions of insulin, together with other hormones and autonomic inputs, in multiple organs, not just adipose tissue” (5). Unfortunately, the proposed mechanisms involved in this integrated, multiorgan, multihormone CIM remain unclear, including the mechanisms of “internal starvation” of nonadipose tissue and how this is sensed during the development of obesity.
Importantly, despite continued claims that the new “CIM proposes a reversal of causal direction” (5), this is no longer a necessary feature because all pathways to positive energy balance are not required to act downstream of adipose tissue fat accumulation. Indeed, the new CIM proposes the existence of parallel pathways influencing energy balance, but it is unclear exactly what these pathways are or how they might work. One such pathway involves a direct effect of dietary glycemic load on energy intake, presumably mediated by the brain (5). If this new direct pathway linking high-glycemic-load diets to increased energy intake dominates the proposed indirect effect downstream of adipose tissue, then the new CIM results in the usual causal direction of increased energy intake leading to adipose tissue fat accumulation. In that case, the new CIM can be considered an oversimplified version of the EBM with a focus on glycemic load as the main driver of excess energy intake.
Evaluation of the EBM and CIM
Although data might support various aspects of both the EBM and CIM, a valid model should withstand tests of its various predictions or be suitably modified or abandoned. Most importantly, theoretical models of obesity must explain between-person variability in adiposity as well as the recent global shift in its distribution. Below, we describe a wide body of evidence with implications for the validity of the CIM and EBM as plausible models that explain the heterogeneity of adiposity and the obesity pandemic.
Rodent studies
Rodent models are valuable for testing hypotheses of diet and body weight regulation because they are amenable to rigorous control of diet for extended periods and independent of potentially confounding factors such as the conscious desire to lose weight. Moreover, responses of different rodent strains to variable macronutrients in the diet can provide insights into the regulatory systems involved. However, the high level of control comes at the cost of questionable translational relevance to human obesity (10). For example, rodent studies have been critical in demonstrating the causal role of insulin affecting aspects of metabolism, food intake, and fat deposition (65–71); however, this evidence does not discriminate between EBM and CIM because both models recognize the importance of these processes (10).
Conversely, other rodent studies show that the role of dietary carbohydrates in determining body weight, as postulated by the CIM, is largely untenable. Most standard laboratory rodent diets are high in carbohydrates. Typical mouse unpurified diet (“chow”) consists of ∼70% carbohydrate, ∼10% fat, and ∼20% protein (by energy), which does not induce obesity. Shifts in the macronutrient distribution towards lower percentage carbohydrate and higher percentage fat, with protein constant, induces obesity in many strains, with a peak effect on body fatness observed at 20% carbohydrate, 60% fat, and 20% protein (72, 73). Although it has been argued that this is because the carbohydrates in such diets are not high-glycemic-index carbohydrates (74), this is incorrect. The main carbohydrate components of commercial mouse unpurified diet are corn starch, maltodextrin, and sucrose (75), which all have roughly equivalent glycemic indices in rodents (76). Even if one focuses on sucrose alone, feeding mice a diet with 73% calories as sucrose (82% of calories as carbohydrates, 8% as fat, and 10% as protein) was protective against obesity (77), consistent with the lower body weight gain of rats on a 51.3% glucose diet (by weight) (74). Alternative explanations for these findings include potential confounding by higher intakes of SFAs by rodents fed low-glycemic-index diets, such that saturated fats induce insulin resistance, hyperinsulinemia, and subsequent obesity (5, 74). As such, this argument underscores the importance of dietary factors other than carbohydrate per se in the pathogenesis of obesity, emphasizes the ability of some strains to resist diet-induced changes in fat storage, and directly refutes the notion that dietary carbohydrate is the main driver of body fat accumulation as proposed by the CIM.
Human genetics
The EBM implicates the brain as the primary organ responsible for obesity whereas the CIM implicates adipose tissue. Given the high heritability of obesity, the relative expression of common obesity genes in different organs provides evidence regarding the relative validity of these models. Other than rare mutations in the leptin gene that result in obesity due to the inability of the brain to sense adequate body fat stores, no genetic disorder primarily affecting the adipocyte or enhanced insulin action has been reproducibly reported to cause obesity. Humans who have homozygous mutations in genes encoding adipose triglyceride lipase (ATGL) or comparative gene identification-58 (CGI58) have a severe defect in adipocyte lipolysis yet do not develop obesity (78, 79). Nevertheless, adipose-enriched genes include variants influencing body fat distribution and features of the metabolic syndrome, such as insulin resistance (80).
Given that nervous systems have evolved to control energy intake (81), it is not surprising that every known monogenic disorder that causes human obesity involves a gene intimately involved in the control of energy intake by the hypothalamus (82). Furthermore, unbiased genome-wide association and gene expression studies have determined that variations in total adiposity between people are primarily due to differences in genes that are most highly expressed in the brain (82–84). For common variants, their effects on fat mass are so small that it is hard to definitively establish the impact of each individual variant on food intake, energy expenditure, or energy partitioning. An exception is the common variant with highest effect size, the fat mass and obesity-associated (FTO) gene, where carriers of the risk allele have consistently been reported to have increased appetite and/or objectively measured food intake (85, 86).
Epidemiological studies
There is heterogeneity both in the mean BMI among regions globally, by sex, and national income, and in BMI trends observed from 1980 to 2008 (87). The worldwide obesity epidemic has been attributed in large part to shifting toward industrialized Western diets (88, 89), characterized by refined grains, processed oils, sugar-sweetened beverages, animal products, and low intakes of nonstarchy vegetables and whole grains. Global trade and improved technologies have facilitated the rapid dissemination of food commodities both intentionally, to address crises of undernutrition, and via stealth expansion of the food industry (88–91). These have led to declines in whole grains, vegetables, and legumes, replaced by increases in caloric sweeteners and refined oils in the form of processed foods.
Despite such notable shifts in diet quality, US trends of average adult food intake indicate a remarkable stability in the macronutrient composition of calories from carbohydrate, fat, and protein (92). Further, whereas added sugar from foods has also been stable, its intake from beverages underwent a significant increase in the first half of the obesity epidemic (93). Since around 2000, modest improvements in diet quality have been observed, such as substituting from refined back to whole grains and a reduction in sugary beverages (94).
Global validated physical activity data are sparse relative to nutrition. There was an increase in the portion of the global population living in urban compared with rural areas, from 41% to 55% over 1985 to 2014 (95), suggesting a role for a sedentary lifestyle in obesity; however, an analysis from the NCD Risk Factor Collaboration indicated many countries experienced greater increases in BMI in rural rather than urban areas, suggesting trends in urbanization alone are insufficient to explain global obesity trends (96).
Overall, the accumulated trend data implicate a myriad of potential drivers of weight gain accompanying complex global nutrition, economic, and technological transitions (97). Their relative causal contributions, however, cannot be inferred on the population level. Nonetheless, evidence to suggest that carbohydrate intake explains between-country differences in body weight is nonexistent and recent trends do not support that dietary carbohydrate is the main driver of the US obesity epidemic.
Longitudinal cohort data have similarly identified several potential nutritional risk factors well beyond simple macronutrient composition for midlife weight gain. For example, an analysis of 121,335 healthy US adults related individual-level changes in dietary fats compared with carbohydrates with concomitant changes in body weight over 20 y of follow-up (98). Participants increasing calories from total fat at the expense of carbohydrate had modestly less weight gain over time, supporting a potential role for carbohydrate reduction in mitigating increases in weight gain in middle age. However, when the type of fat replacing the carbohydrate calories was considered, it was clear that changes in the quality of foods contributing dietary fat explained significant differences in weight gain above and beyond any contribution from a reduction in carbohydrate calories per se. A reduction in carbohydrates with increases in animal source fats was associated with significantly greater weight gain, whereas the same reduction in carbohydrates with increases in polyunsaturated fats was correlated with significantly less weight gain.
A large body of epidemiological evidence consistently finds meaningful differences in risk of overweight and obesity according to long-term adherence to a variety of healthful dietary patterns (99) and foods (100, 101), all with highly variable carbohydrate contents. For example, increasing intakes of potato chips, unprocessed red meat, and sugary beverages were significantly related to midlife weight gain, whereas increases in servings of whole grains, yogurt, and nuts related to weight loss (102). Thus, in addition to genetic factors, heterogeneity in weight gain is explained by variation in several nutritional factors and diet quality, independent of carbohydrate and glycemic index (103, 104). These data, consistent with the EBM, suggest a variety of potential dietary drivers of excess calorie intake but do not support the CIM proposition that dietary carbohydrates are the primary driver of obesity.
Human diet intervention studies
The CIM predicts that meaningful long-term weight loss is readily achieved through a reduction in dietary carbohydrate and glycemic load because such interventions directly address the fundamental cause of obesity and should thereby facilitate its reversal. Indeed, the CIM claims that “patients may experience less hunger and improved energy level, promoting spontaneous weight loss” and “weight reduction produced by carbohydrate restriction would…result in lower spontaneous food intake” (5). However, diet intervention trials have found that low-glycemic-load diets do not generally result in significantly greater long-term weight loss as compared with higher-glycemic-load diets (105–112).
Dietary protein can be a significant confounder in diet intervention studies and should be matched when evaluating the effects of glycemic load per se. For example, a study examining maintenance of lost weight over a 6 mo period found that only a high-protein, low-glycemic-index diet prevented weight regain whereas significant weight regain was observed for a lower-protein, low-glycemic-index diet and higher-glycemic-index diets with high or low protein (113). Interestingly, longer studies investigating maintenance of lost weight failed to show a benefit of low- compared with high-glycemic-index diets (114, 115). A meta-analysis of 53 randomized controlled trials of ≥1-y duration evaluated the effects of low-fat compared with higher-fat dietary interventions and found no significant difference in mean weight loss comparing the low-fat with higher-fat diets when dietary interventions were delivered with similar intensity (116). An updated network meta-analyses of 121 randomized controlled trials found no significant difference in mean weight loss at 6 mo comparing low-fat, low-carbohydrate, or moderate-carbohydrate with usual diet controls (117). For the individual diet types, Jenny Craig (55–60% carbohydrate) and Atkins (∼10% carbohydrate) were the most effective at 6 mo; however, at 12 mo, mean weight loss was significantly greater for Jenny Craig compared with Atkins.
The CIM Perspective admitted that “most participants in these studies have difficulty sustaining dietary change” (5), but this explanation is contrary to predictions of the CIM because low-glycemic-load diets should promote diet adherence by spontaneously decreasing hunger in comparison with higher-glycemic-load alternatives. In contrast, the EBM predicts that a variety of macronutrient compositions and eating patterns can result in long-term weight loss, so long as they ultimately confer a sustained reduction in energy intake. The EBM is not constrained by the degree of effort necessary to sustain adherence across diet types, and readily accommodates a direct influence of internal signals and the complex and dynamic food environment on the long-term control of energy intake that make it difficult to sustain dietary adherence (118–120).
To avoid the confounder of diet adherence, inpatient feeding studies prevent access to off-study food. Under such conditions, exposure to a high-glycemic-load diet is predicted by the CIM to lead to excess insulin secretion, accumulation of body fat, and downstream increases in appetite leading to greater ad libitum energy intake as compared with a lower-glycemic-load diet. However, a recent month-long inpatient study found that a 2 wk exposure to a high-glycemic-load diet resulted in ∼700 kcal/d lower ad libitum energy intake and body fat loss compared with the 2 wk spent by the same participants on a very-low-glycemic-load diet (121). Additionally, these results occurred despite the low-glycemic-load diet resulting in substantially lower insulin secretion. Although this study provides important evidence directly contradicting the CIM's predictions, it was wholly dismissed in the CIM Perspective as an example of “the pitfalls of extrapolating chronic macronutrient effects from studies of a few weeks’ duration” when the effects of the high-glycemic-load diet and excess postprandial insulin on appetite were apparently dominated by “factors of dubious relation to chronic energy balance—such as utensil size or plate color” (5). Interestingly, a recent outpatient controlled feeding study found that 10 to 15 wk of a high-carbohydrate diet significantly increased satiety compared with a low-carbohydrate diet despite resulting in significantly higher postprandial insulin and lower circulating fuels (122), which again refutes the CIM predictions and is consistent with the shorter inpatient study (121).
The CIM predicts that lower-carbohydrate diets decrease insulin and thereby mobilize fat from adipose tissue to result in greater fat loss compared with isocaloric higher-carbohydrate diets with matched protein. However, controlled feeding studies have produced results inconsistent with this prediction. For example, selective carbohydrate restriction led to substantial decreases in daily insulin secretion in patients with obesity but resulted in slightly less body fat loss compared with isocaloric selective fat restriction in the same patients (58). A meta-analysis of controlled feeding studies comparing isocaloric diets matched for protein found a small but significantly greater body fat loss with higher-carbohydrate diets (123). Furthermore, a recent 6-wk controlled feeding study found no significant differences in body fat loss between isocaloric very-low-carbohydrate compared with low-fat diets matched for protein (124). Finally, a 17-wk controlled feeding study employing isocaloric diets varying in glycemic load but matched for protein also failed to observe significant differences in body fat loss (109).
The EBM proposes that high dietary energy density is a potentially important driver of excess energy intake, and several trials have demonstrated significant long-term effects of diets differing in energy density (125–128). However, these data were ignored in the CIM Perspective, and the possibility of energy density being potentially important in long-term control of energy intake was rejected (5) on the basis of a single exploratory finding of no significant long-term weight loss in patients with breast cancer who were advised to eat more fruits and vegetables (129). Furthermore, the CIM Perspective conflated the concept of food palatability with ultraprocessed food, and it was claimed that there is a lack of relevant human intervention studies on the topic (5). But this ignored the results of a month-long inpatient human trial demonstrating excess ad libitum energy intake and weight gain when people were exposed to controlled food environments characterized by either ultraprocessed or unprocessed diets rated as similarly palatable and closely matched for available energy, macronutrients, sugar, sodium, fiber, and glycemic load (130). Such results demonstrate that factors other than dietary macronutrient composition and glycemic load can play important roles influencing human energy intake.
Human pharmacological intervention studies
Pharmacological manipulations can also be used to interrogate the validity of obesity models. An oft cited example in support of the CIM is that exogenous insulin treatment in people with diabetes often induces weight gain. But insulin therapy in treatment-naïve patients with type 1 diabetes normalizes their pathophysiological catabolic state and results in increased lean mass, reductions in ATP-requiring metabolic futile cycles, normalization of adipose tissue lipolysis, elimination of glycosuria, and, contrary to the CIM, resolution of polyphagia. Further, this all occurs despite insulin therapy decreasing circulating fuels. In type 2 diabetes, weight gain with insulin therapy can be due, in part, to regain of recently lost weight (131–133). Acute peripheral infusions of insulin have mixed effects on appetite and food intake in humans (134, 135), but intranasal insulin delivery to the brain inhibits food intake (136), consistent with rodent studies (137) and contrary to the CIM.
The CIM posits that a major reason why insulin induces weight gain is due to its effect on inhibiting adipose tissue lipolysis thereby trapping fat in fat cells (5). However, inhibiting adipose lipolysis with acipimox treatment for 6 mo had no significant effects on energy intake, resting energy expenditure, or body composition despite achieving a marked 38% reduction of plasma free fatty acid concentrations in adults with obesity (138). Finally, despite acutely increasing insulin secretion, GLP-1 receptor agonists are currently the most effective approved medications to treat obesity (139).
Conclusions
The principle of energy balance is a necessary constraint on all potentially viable models of obesity, including the CIM and EBM. Both models agree that diet quality and composition are important in prevention and treatment of obesity. Both models account for endocrine regulation, peripheral energy sensing, and energy partitioning. The latest iteration of the CIM retreats from its previous focus on postprandial insulin's direct effect on adipose tissue, but the new CIM differs from the EBM in that high-glycemic-load diets are identified as the main driver of increasing obesity prevalence. However, an overwhelming amount of evidence indicates that numerous variables in the food environment beyond high-glycemic foods can result in increased energy intake and the EBM posits that obesity can arise if any one or more of these factors are in play.
The EBM acknowledges the potential benefits of low-carbohydrate or low-glycemic-load diets in managing body weight or cardiometabolic outcomes in some individuals. Precision nutrition initiatives have stemmed from a hypothesis of inherent biological heterogeneity when an optimizing individuals’ diet (108, 140–142). Whereas such efforts are consistent with the multifactorial EBM, the CIM sets forth a single exposure as the primary determinant of common obesity and proposes a single “practical strategy” to treat obesity by prescribing low-glycemic-load diets (5) despite evidence that such interventions are no more effective than prescribing higher-glycemic-load alternatives (105–112, 114, 115).
Overall, we have documented a large body of evidence aligning with the EBM but inconsistent with the CIM. Further development of the EBM requires elucidation of the factors in the dynamic food environment that are most responsible for instigating obesity, the mechanisms by which these factors alter the brain circuits controlling food intake, and why some individuals are more susceptible to development of obesity than others. Answering these questions will result in improved public health and medical interventions for prevention and treatment of obesity.
Acknowledgments
We thank Stephan Guyenet for helpful comments on an earlier draft of the manuscript.
The authors’ responsibilities were as follows—KDH, JMF, SK, SO'R, JRS, ER, DKT: wrote the first draft of the manuscript; KDH: takes responsibility for design, writing, and the final content; and all authors: participated in manuscript revision, and read and approved the final manuscript. KDH has participated in several debates with David S. Ludwig on the merits and demerits of the CIM and has received funding from the Nutrition Science Initiative to test predictions of the CIM. JF reports royalty payments for leptin for the treatment of lipodystrophy. SK serves as a scientific consultant for Altimmune, Janssen, and B2M and has a sponsored research agreement with Janssen. SO'R receives remuneration for scientific advice from Pfizer, AstraZeneca and ThirdRock Ventures. DHR reports personal fees from Altimmune, personal fees from Amgen, personal fees and non-financial support from Boeringer Ingelheim, personal fees and non-financial support from Calibrate, personal fees and non-financial support from Epitomee, personal fees from Gila Therapeutics, personal fees and non-financial support from IFA Celtic, personal fees and non-financial support from Janssen, personal fees and non-financial support from Novo Nordisk, personal fees from Phenomix, personal fees from Quintiles, personal fees and non-financial support from real appeal (United Health Care), personal fees from Rhythm, personal fees and non-financial support from Sanofi, personal fees and non-financial support from Scientific Intake, personal fees and non-financial support from Xeno Bioscience, personal fees from YSOPIA, personal fees from Zealand, personal fees from Lilly, personal fees from Wondr Health, personal fees from Roman Health, outside the submitted work. All other authors report no conflicts of interest.
Notes
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases.
DKT is the Academic Editor for the American Journal of Clinical Nutrition but played no role in the evaluation of this manuscript.
Abbreviations used: AgRP, agouti-related peptide; CIM, carbohydrate-insulin model; EBM, energy balance model; FTO, fat mass and obesity-associated gene; GLP-1, glucagon-like peptide 1.
Contributor Information
Kevin D Hall, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
I Sadaf Farooqi, Wellcome-MRC Institute of Metabolic Science, University of Cambridge.
Jeffery M Friedman, The Rockefeller University.
Samuel Klein, Washington University School of Medicine in St Louis.
Ruth J F Loos, Washington University School of Medicine in St Louis; Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen.
David J Mangelsdorf, University of Texas Southwestern Medical Center.
Stephen O'Rahilly, Wellcome-MRC Institute of Metabolic Science, University of Cambridge.
Eric Ravussin, Pennington Biomedical Research Center.
Leanne M Redman, Pennington Biomedical Research Center.
Donna H Ryan, Pennington Biomedical Research Center.
John R Speakman, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzen, China, and the University of Aberdeen, Aberdeen, United Kingdom.
Deirdre K Tobias, Brigham and Women's Hospital and Harvard Medical School.
References
- 1. Elks CE, den Hoed M, Zhao JH, Sharp SJ, Wareham NJ, Loos RJ, Ong KK. Variability in the heritability of body mass index: a systematic review and meta-regression. Front Endocrinol. 2012;3:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA. 1986;256(1):51–4. [PubMed] [Google Scholar]
- 3. Church T, Martin CK. The obesity epidemic: a consequence of reduced energy expenditure and the uncoupling of energy intake?. Obesity. 2018;26(1):14–16. [DOI] [PubMed] [Google Scholar]
- 4. Hall KD. Did the food environment cause the obesity epidemic?. Obesity. 2018;26(1):11–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ludwig DS, Aronne LJ, Astrup A, de Cabo R, Cantley LC, Friedman MI, Heymsfield SB, Johnson JD, King JC, Krauss RMet al. The carbohydrate-insulin model: a physiological perspective on the obesity pandemic. Am J Clin Nutr. 2021;114(6):1873–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ludwig DS. Always hungry? Conquer cravings, retrain your fat cells and lose weight permanently. New York: Grand Central Life & Style; 2016. [Google Scholar]
- 7. Ludwig DS, Ebbeling CB. The carbohydrate-insulin model of obesity: beyond “calories in, calories out”. JAMA Intern Med. 2018;178(8):1098–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ludwig DS, Friedman MI. Increasing adiposity: consequence or cause of overeating?. JAMA. 2014;311(21):2167–8. [DOI] [PubMed] [Google Scholar]
- 9. Taubes G. Good calories, bad calories: challenging the conventional wisdom on diet, weight control, and disease. New York: Alfred A. Knopf; 2007. [Google Scholar]
- 10. Speakman JR, Hall KD. Carbohydrates, insulin, and obesity. Science. 2021;372(6542):577–8. [DOI] [PubMed] [Google Scholar]
- 11. de Araujo IE, Schatzker M, Small DM. Rethinking food reward. Annu Rev Psychol. 2020;71(1):139–64. [DOI] [PubMed] [Google Scholar]
- 12. Schwartz MW, Seeley RJ, Zeltser LM, Drewnowski A, Ravussin E, Redman LM, Leibel RL. Obesity pathogenesis: an Endocrine Society scientific statement. Endocr Rev. 2017;38(4):267–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Watts AG, Kanoski SE, Sanchez-Watts G, Langhans W. The physiological control of eating: signals, neurons, and networks. Physiol Rev. 2022;102:689–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blundell JE, Gibbons C, Beaulieu K, Casanova N, Duarte C, Finlayson G, Stubbs RJ, Hopkins M. The drive to eat in homo sapiens: energy expenditure drives energy intake. Physiol Behav. 2020;219:112846. [DOI] [PubMed] [Google Scholar]
- 15. Dulloo AG, Jacquet J, Miles-Chan JL, Schutz Y. Passive and active roles of fat-free mass in the control of energy intake and body composition regulation. Eur J Clin Nutr. 2017;71(3):353–7. [DOI] [PubMed] [Google Scholar]
- 16. Bray GA, Flatt JP, Volaufova J, Delany JP, Champagne CM. Corrective responses in human food intake identified from an analysis of 7-d food-intake records. Am J Clin Nutr. 2008;88(6):1504–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chow CC, Hall KD. Short and long-term energy intake patterns and their implications for human body weight regulation. Physiol Behav. 2014;134:60–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Edholm OG, Adam JM, Healy MJ, Wolff HS, Goldsmith R, Best TW. Food intake and energy expenditure of army recruits. Br J Nutr. 1970;24(4):1091–107. [DOI] [PubMed] [Google Scholar]
- 19. Flatt JP. How NOT to approach the obesity problem. Obes Res. 1997;5(6):632–3. [DOI] [PubMed] [Google Scholar]
- 20. Berthoud HR, Morrison CD, Ackroff K, Sclafani A. Learning of food preferences: mechanisms and implications for obesity and metabolic diseases. Int J Obes (Lond). 2021;45(10):2156–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sternson SM, Eiselt AK. Three pillars for the neural control of appetite. Annu Rev Physiol. 2017;79(1):401–23. [DOI] [PubMed] [Google Scholar]
- 22. Alhadeff AL, Goldstein N, Park O, Klima ML, Vargas A, Betley JN. Natural and drug rewards engage distinct pathways that converge on coordinated hypothalamic and reward circuits. Neuron. 2019;103(5):891–908..e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mazzone CM, Liang-Guallpa J, Li C, Wolcott NS, Boone MH, Southern M, Kobzar NP, Salgado IA, Reddy DM, Sun Fet al. High-fat food biases hypothalamic and mesolimbic expression of consummatory drives. Nat Neurosci. 2020;23(10):1253–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goldstein N, McKnight AD, Carty JRE, Arnold M, Betley JN, Alhadeff AL. Hypothalamic detection of macronutrients via multiple gut-brain pathways. Cell Metab. 2021;33(3):676–87.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Torres-Carot V, Suárez-González A, Lobato-Foulques C. The energy balance hypothesis of obesity: do the laws of thermodynamics explain excessive adiposity?. Eur J Clin Nutr. 2022. doi: 10.1038/s41430-021-01064-4. [DOI] [PubMed] [Google Scholar]
- 26. Abbott WG, Howard BV, Christin L, Freymond D, Lillioja S, Boyce VL, Anderson TE, Bogardus C, Ravussin E. Short-term energy balance: relationship with protein, carbohydrate, and fat balances. Am J Physiol. 1988;255(3 Pt 1):E332–7. [DOI] [PubMed] [Google Scholar]
- 27. Flatt JP. Importance of nutrient balance in body weight regulation. Diabetes Metab Rev. 1988;4(6):571–81. [DOI] [PubMed] [Google Scholar]
- 28. Flatt JP. Body composition, respiratory quotient, and weight maintenance. Am J Clin Nutr. 1995;62(5):1107S–17S. [DOI] [PubMed] [Google Scholar]
- 29. Flatt JP. McCollum Award Lecture, 1995: diet, lifestyle, and weight maintenance. Am J Clin Nutr. 1995;62(4):820–36. [DOI] [PubMed] [Google Scholar]
- 30. Flatt JP. Use and storage of carbohydrate and fat. Am J Clin Nutr. 1995;61(4):952S–9S. [DOI] [PubMed] [Google Scholar]
- 31. Schutz Y. The adjustment of energy expenditure and oxidation to energy intake: the role of carbohydrate and fat balance. Int J Obes Relat Metab Disord. 1993;17(Suppl 3):S23–7.; discussion S41 –2. [PubMed] [Google Scholar]
- 32. Schutz Y, Flatt JP, Jequier E. Failure of dietary fat intake to promote fat oxidation: a factor favoring the development of obesity. Am J Clin Nutr. 1989;50(2):307–14. [DOI] [PubMed] [Google Scholar]
- 33. Schutz Y, Tremblay A, Weinsier RL, Nelson KM. Role of fat oxidation in the long-term stabilization of body weight in obese women. Am J Clin Nutr. 1992;55(3):670–4. [DOI] [PubMed] [Google Scholar]
- 34. Stubbs RJ. Nutrition Society Medal Lecture. Appetite, feeding behaviour and energy balance in human subjects. Proc Nutr Soc. 1998;57(3):341–56. [DOI] [PubMed] [Google Scholar]
- 35. Frayn KN. Physiological regulation of macronutrient balance. Int J Obes Relat Metab Disord. 1995;19(Suppl 5):S4–10. [PubMed] [Google Scholar]
- 36. Hall KD. Modeling metabolic adaptations and energy regulation in humans. Annu Rev Nutr. 2012;32(1):35–54. [DOI] [PubMed] [Google Scholar]
- 37. Hall KD, Bain HL, Chow CC. How adaptations of substrate utilization regulate body composition. Int J Obes. 2007;31(9):1378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carpentier AC. 100(th) anniversary of the discovery of insulin perspective: insulin and adipose tissue fatty acid metabolism. Am J Physiol Endocrinol Metab. 2021;320(4):E653–E670. [DOI] [PubMed] [Google Scholar]
- 39. Frayn KN. Turning over our fat stores: the key to metabolic health. Blaxter Award Lecture 2018. Proc Nutr Soc. 2019;78(3):398–406. [DOI] [PubMed] [Google Scholar]
- 40. Frayn KN, Karpe F, Fielding BA, Macdonald IA, Coppack SW. Integrative physiology of human adipose tissue. Int J Obes. 2003;27(8):875–88. [DOI] [PubMed] [Google Scholar]
- 41. Payne PR, Dugdale AE. Mechanisms for the control of body-weight. Lancet. 1977;309(8011):583–6. [DOI] [PubMed] [Google Scholar]
- 42. Payne PR, Dugdale AE. A model for the prediction of energy balance and body weight. Ann Hum Biol. 1977;4(6):525–35. [DOI] [PubMed] [Google Scholar]
- 43. Hall KD, Butte NF, Swinburn BA, Chow CC. Dynamics of childhood growth and obesity: development and validation of a quantitative mathematical model. Lancet Diabetes Endocrinol. 2013;1(2):97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jordan PN, Hall KD. Dynamic coordination of macronutrient balance during infant growth: insights from a mathematical model. Am J Clin Nutr. 2008;87(3):692–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arner P, Bernard S, Salehpour M, Possnert G, Liebl J, Steier P, Buchholz BA, Eriksson M, Arner E, Hauner Het al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature. 2011;478(7367):110–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hall KD. Predicted impact of adipose insulin resistance on long-term human body composition. Obesity. 2006;14(S9):A212. [Google Scholar]
- 47. Ryden M, Andersson DP, Bernard S, Spalding K, Arner P. Adipocyte triglyceride turnover and lipolysis in lean and overweight subjects. J Lipid Res. 2013;54(10):2909–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, Naslund E, Britton Tet al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–7. [DOI] [PubMed] [Google Scholar]
- 49. Spalding KL, Bernard S, Naslund E, Salehpour M, Possnert G, Appelsved L, Fu KY, Alkass K, Druid H, Thorell Aet al. Impact of fat mass and distribution on lipid turnover in human adipose tissue. Nat Commun. 2017;8(1):15253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mayer J, Roy P, Mitra KP. Relation between caloric intake, body weight, and physical work: studies in an industrial male population in West Bengal. Am J Clin Nutr. 1956;4(2):169–75. [DOI] [PubMed] [Google Scholar]
- 51. Melby CL, Paris HL, Foright RM, Peth J. Attenuating the biologic drive for weight regain following weight loss: must what goes down always go back up?. Nutrients. 2017;9(5):468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McAllister EJ, Dhurandhar NV, Keith SW, Aronne LJ, Barger J, Baskin M, Benca RM, Biggio J, Boggiano MM, Eisenmann JCet al. Ten putative contributors to the obesity epidemic. Crit Rev Food Sci Nutr. 2009;49(10):868–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Evans K, Clark ML, Frayn KN. Effects of an oral and intravenous fat load on adipose tissue and forearm lipid metabolism. Am J Physiol. 1999;276(2 Pt 1):E241–8. [DOI] [PubMed] [Google Scholar]
- 54. Samra JS, Giles SL, Summers LK, Evans RD, Arner P, Humphreys SM, Clark ML, Frayn KN. Peripheral fat metabolism during infusion of an exogenous triacylglycerol emulsion. Int J Obes. 1998;22(8):806–12. [DOI] [PubMed] [Google Scholar]
- 55. Corkey BE, Deeney JT, Merrins MJ. What regulates basal insulin secretion and causes hyperinsulinemia?. Diabetes. 2021;70(10):2174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Henquin JC. Non-glucose modulators of insulin secretion in healthy humans: (dis)similarities between islet and in vivo studies. Metabolism. 2021;122:154821. [DOI] [PubMed] [Google Scholar]
- 57. Newsholme P, Krause M. Nutritional regulation of insulin secretion: implications for diabetes. Clin Biochem Rev. 2012;33(2):35–47. [PMC free article] [PubMed] [Google Scholar]
- 58. Hall KD, Bemis T, Brychta R, Chen KY, Courville A, Crayner EJ, Goodwin S, Guo J, Howard L, Knuth NDet al. Calorie for calorie, dietary fat restriction results in more body fat loss than carbohydrate restriction in people with obesity. Cell Metab. 2015;22(3):427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mayer J. Glucostatic mechanism of regulation of food intake. N Engl J Med. 1953;249(1):13–16. [DOI] [PubMed] [Google Scholar]
- 60. Mayer J. Regulation of energy intake and the body weight: the glucostatic theory and the lipostatic hypothesis. Ann NY Acad Sci. 1955;63(1):15–43. [DOI] [PubMed] [Google Scholar]
- 61. Louis-Sylvestre J, Le Magnen J. Fall in blood glucose level precedes meal onset in free-feeding rats. Neurosci Biobehav Rev. 1980;4:13–15. [DOI] [PubMed] [Google Scholar]
- 62. Campfield LA, Smith FJ. Blood glucose dynamics and control of meal initiation: a pattern detection and recognition theory. Physiol Rev. 2003;83(1):25–58. [DOI] [PubMed] [Google Scholar]
- 63. Campfield LA, Smith FJ, Rosenbaum M, Hirsch J. Human eating: evidence for a physiological basis using a modified paradigm. Neurosci Biobehav Rev. 1996;20(1):133–7. [DOI] [PubMed] [Google Scholar]
- 64. Wyatt P, Berry SE, Finlayson G, O'Driscoll R, Hadjigeorgiou G, Drew DA, Khatib HA, Nguyen LH, Linenberg I, Chan ATet al. Postprandial glycaemic dips predict appetite and energy intake in healthy individuals. Nature Metab. 2021;3(4):523–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Müller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289(5487):2122–5. [DOI] [PubMed] [Google Scholar]
- 66. Cusin I, Rohner-Jeanrenaud F, Terrettaz J, Jeanrenaud B. Hyperinsulinemia and its impact on obesity and insulin resistance. Int J Obes Relat Metab Disord. 1992;16(Suppl 4):S1–11. [PubMed] [Google Scholar]
- 67. Dallon BW, Parker BA, Hodson AE, Tippetts TS, Harrison ME, Appiah MMA, Witt JE, Gibbs JL, Gray HM, Sant TMet al. Insulin selectively reduces mitochondrial uncoupling in brown adipose tissue in mice. Biochem J. 2018;475(3):561–9. [DOI] [PubMed] [Google Scholar]
- 68. Mehran AE, Templeman NM, Brigidi GS, Lim GE, Chu KY, Hu X, Botezelli JD, Asadi A, Hoffman BG, Kieffer TJet al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012;16(6):723–37. [DOI] [PubMed] [Google Scholar]
- 69. Page MM, Skovsø S, Cen H, Chiu AP, Dionne DA, Hutchinson DF, Lim GE, Szabat M, Flibotte S, Sinha Set al. Reducing insulin via conditional partial gene ablation in adults reverses diet-induced weight gain. FASEB J. 2018;32(3):1196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Templeman NM, Skovsø S, Page MM, Lim GE, Johnson JD. A causal role for hyperinsulinemia in obesity. J Endocrinol. 2017;232(3):R173–R183. [DOI] [PubMed] [Google Scholar]
- 71. Torbay N, Bracco EF, Geliebter A, Stewart IM, Hashim SA. Insulin increases body fat despite control of food intake and physical activity. Am J Physiol. 1985;248(1 Pt 2):R120–4. [DOI] [PubMed] [Google Scholar]
- 72. Hu S, Wang L, Togo J, Yang D, Xu Y, Wu Y, Douglas A, Speakman JR. The carbohydrate-insulin model does not explain the impact of varying dietary macronutrients on the body weight and adiposity of mice. Mol Metab. 2020;32:27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hu S, Wang L, Yang D, Li L, Togo J, Wu Y, Liu Q, Li B, Li M, Wang Get al. Dietary fat, but not protein or carbohydrate, regulates energy intake and causes adiposity in mice. Cell Metab. 2018;28(3):415–31.e4..] [DOI] [PubMed] [Google Scholar]
- 74. Ludwig DS, Ebbeling CB, Bikman BT, Johnson JD. Testing the carbohydrate-insulin model in mice: the importance of distinguishing primary hyperinsulinemia from insulin resistance and metabolic dysfunction. Mol Metab. 2020;35:100960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ludwig DS. The glycemic index: physiological mechanisms relating to obesity, diabetes, and cardiovascular disease. JAMA. 2002;287(18):2414–23. [DOI] [PubMed] [Google Scholar]
- 76. Campbell GJ, Belobrajdic DP, Bell-Anderson KS. Determining the glycaemic index of standard and high-sugar rodent diets in C57BL/6 mice. Nutrients. 2018;10(7):856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Togo J, Hu S, Li M, Niu C, Speakman JR. Impact of dietary sucrose on adiposity and glucose homeostasis in C57BL/6J mice depends on mode of ingestion: liquid or solid. Mol Metab. 2019;27:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Reilich P, Horvath R, Krause S, Schramm N, Turnbull DM, Trenell M, Hollingsworth KG, Gorman GS, Hans VH, Reimann Jet al. The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene. J Neurol. 2011;258(11):1987–97. [DOI] [PubMed] [Google Scholar]
- 79. Lefèvre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach Jet al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet. 2001;69(5):1002–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, Gaulton KJ, Eicher JD, Sharp SJ, Luan Jet al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Musser JM, Schippers KJ, Nickel M, Mizzon G, Kohn AB, Pape C, Ronchi P, Papadopoulos N, Tarashansky AJ, Hammel JUet al. Profiling cellular diversity in sponges informs animal cell type and nervous system evolution. Science. 2021;374(6568):717–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Loos RJF, Yeo GSH. The genetics of obesity: from discovery to biology. Nat Rev Genet. 2022;23:120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, Gazal S, Loh PR, Lareau C, Shoresh Net al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet. 2018;50(4):621–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang Jet al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Speakman JR. The ‘fat mass and obesity related’ (FTO) gene: mechanisms of impact on obesity and energy balance. Curr Obes Rep. 2015;4(1):73–91. [DOI] [PubMed] [Google Scholar]
- 86. Melhorn SJ, Askren MK, Chung WK, Kratz M, Bosch TA, Tyagi V, Webb MF, De Leon MRB, Grabowski TJ, Leibel RLet al. FTO genotype impacts food intake and corticolimbic activation. Am J Clin Nutr. 2018;107(2):145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, Singh GM, Gutierrez HR, Lu Y, Bahalim ANet al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9·1 million participants. Lancet. 2011;377(9765):557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Popkin BM, Adair LS, Ng SW. Global nutrition transition and the pandemic of obesity in developing countries. Nutr Rev. 2012;70(1):3–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Popkin BM, Ng SW. The nutrition transition to a stage of high obesity and noncommunicable disease prevalence dominated by ultra-processed foods is not inevitable. Obes Rev. 2022;23(1):e13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Monteiro CA, Moubarac JC, Cannon G, Ng SW, Popkin B. Ultra-processed products are becoming dominant in the global food system. Obes Rev. 2013;14:21–8. [DOI] [PubMed] [Google Scholar]
- 91. Roberts P. The end of food. New York: Houghton Mifflin Harcourt Publishing Company; 2008. [Google Scholar]
- 92. Shan Z, Rehm CD, Rogers G, Ruan M, Wang DD, Hu FB, Mozaffarian D, Zhang FF, Bhupathiraju SN. Trends in dietary carbohydrate, protein, and fat intake and diet quality among US adults, 1999-2016. JAMA. 2019;322(12):1178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Popkin BM, Hawkes C. Sweetening of the global diet, particularly beverages: patterns, trends, and policy responses. Lancet Diabetes Endocrinol. 2016;4(2):174–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Rehm CD, Peñalvo JL, Afshin A, Mozaffarian D. Dietary intake among US adults, 1999-2012. JAMA. 2016;315(23):2542–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. United Nations Department of Economic and Social Affairs Population Division . World urbanization prospects: the 2014 revision. New York: United Nations;2015. [Google Scholar]
- 96.NCD Risk Factor Collaboration (NCD-RisC). Rising rural body-mass index is the main driver of the global obesity epidemic in adults. Nature. 2019;569(7755):260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bleich S, Cutler D, Murray C, Adams A. Why is the developed world obese?. Annu Rev Public Health. 2008;29(1):273–95. [DOI] [PubMed] [Google Scholar]
- 98. Liu X, Li Y, Tobias DK, Wang DD, Manson JE, Willett WC, Hu FB. Changes in types of dietary fats influence long-term weight change in US women and men. J Nutr. 2018;148(11):1821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lotfi K, Saneei P, Hajhashemy Z, Esmaillzadeh A. Adherence to the Mediterranean diet, five-year weight change, and risk of overweight and obesity: a systematic review and dose-response meta-analysis of prospective cohort studies. Adv Nutr. 2022;13:152–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Maki KC, Palacios OM, Koecher K, Sawicki CM, Livingston KA, Bell M, Nelson Cortes H, McKeown NM. The relationship between whole grain intake and body weight: results of meta-analyses of observational studies and randomized controlled trials. Nutrients. 2019;11(6):1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Schlesinger S, Neuenschwander M, Schwedhelm C, Hoffmann G, Bechthold A, Boeing H, Schwingshackl L. Food groups and risk of overweight, obesity, and weight gain: a systematic review and dose-response meta-analysis of prospective studies. Adv Nutr. 2019;10(2):205–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. He K, Hu FB, Colditz GA, Manson JE, Willett WC, Liu S. Changes in intake of fruits and vegetables in relation to risk of obesity and weight gain among middle-aged women. Int J Obes. 2004;28(12):1569–74. [DOI] [PubMed] [Google Scholar]
- 103. Mozaffarian D, Hao T, Rimm EB, Willett WC, Hu FB. Changes in diet and lifestyle and long-term weight gain in women and men. N Engl J Med. 2011;364(25):2392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wang T, Heianza Y, Sun D, Zheng Y, Huang T, Ma W, Rimm EB, Manson JE, Hu FB, Willett WCet al. Improving fruit and vegetable intake attenuates the genetic association with long-term weight gain. Am J Clin Nutr. 2019;110(3):759–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Das SK, Gilhooly CH, Golden JK, Pittas AG, Fuss PJ, Cheatham RA, Tyler S, Tsay M, McCrory MA, Lichtenstein AHet al. Long-term effects of 2 energy-restricted diets differing in glycemic load on dietary adherence, body composition, and metabolism in CALERIE: a 1-y randomized controlled trial. Am J Clin Nutr. 2007;85(4):1023–30. [DOI] [PubMed] [Google Scholar]
- 106. Ebbeling CB, Leidig MM, Feldman HA, Lovesky MM, Ludwig DS. Effects of a low-glycemic load vs low-fat diet in obese young adults: a randomized trial. JAMA. 2007;297(19):2092–102. [DOI] [PubMed] [Google Scholar]
- 107. Gaesser GA, Miller Jones J, Angadi SS. Perspective: does glycemic index matter for weight loss and obesity prevention? Examination of the evidence on “fast” compared with “slow” carbs. Adv Nutr. 2021;12(6):2076–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gardner CD, Trepanowski JF, Del Gobbo LCet al. Effect of low-fat vs low-carbohydrate diet on 12-month weight loss in overweight adults and the association with genotype pattern or insulin secretion: the dietfits randomized clinical trial. JAMA. 2018;319(7):667–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Karl JP, Roberts SB, Schaefer EJ, Gleason JA, Fuss P, Rasmussen H, Saltzman E, Das SK. Effects of carbohydrate quantity and glycemic index on resting metabolic rate and body composition during weight loss. Obesity. 2015;23(11):2190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mirza NM, Palmer MG, Sinclair KB, McCarter R, He J, Ebbeling CB, Ludwig DS, Yanovski JA. Effects of a low glycemic load or a low-fat dietary intervention on body weight in obese Hispanic American children and adolescents: a randomized controlled trial. Am J Clin Nutr. 2013;97(2):276–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Schwingshackl L, Hoffmann G. Long-term effects of low glycemic index/load vs. high glycemic index/load diets on parameters of obesity and obesity-associated risks: a systematic review and meta-analysis. Nutr Metab Cardiovasc Dis. 2013;23(8):699–706. [DOI] [PubMed] [Google Scholar]
- 112. Zafar MI, Mills KE, Zheng J, Peng MM, Ye X, Chen LL. Low glycaemic index diets as an intervention for obesity: a systematic review and meta-analysis. Obes Rev. 2019;20(2):290–315. [DOI] [PubMed] [Google Scholar]
- 113. Larsen TM, Dalskov SM, van Baak M, Jebb SA, Papadaki A, Pfeiffer AF, Martinez JA, Handjieva-Darlenska T, Kunesova M, Pihlsgard Met al. Diets with high or low protein content and glycemic index for weight-loss maintenance. N Engl J Med. 2010;363(22):2102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Due A, Larsen TM, Mu H, Hermansen K, Stender S, Toubro S, Allison DB, Astrup A. The effect of three different ad libitum diets for weight loss maintenance: a randomized 18-month trial. Eur J Nutr. 2017;56(2):727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Aller EE, Larsen TM, Claus H, Lindroos AK, Kafatos A, Pfeiffer A, Martinez JA, Handjieva-Darlenska T, Kunesova M, Stender Set al. Weight loss maintenance in overweight subjects on ad libitum diets with high or low protein content and glycemic index: the DIOGENES trial 12-month results. Int J Obes. 2014;38(12):1511–17. [DOI] [PubMed] [Google Scholar]
- 116. Tobias DK, Chen M, Manson JE, Ludwig DS, Willett W, Hu FB. Effect of low-fat vs. other diet interventions on long-term weight change in adults: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2015;3(12):968–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ge L, Sadeghirad B, Ball GDC, da Costa BR, Hitchcock CL, Svendrovski A, Kiflen R, Quadri K, Kwon HY, Karamouzian Met al. Comparison of dietary macronutrient patterns of 14 popular named dietary programmes for weight and cardiovascular risk factor reduction in adults: systematic review and network meta-analysis of randomised trials. BMJ. 2020;369:m696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Freedhoff Y, Hall KD. Weight loss diet studies: we need help not hype. Lancet. 2016;388(10047):849–51. [DOI] [PubMed] [Google Scholar]
- 119. Guo J, Robinson JL, Gardner CD, Hall KD. Objective versus self-reported energy intake changes during low-carbohydrate and low-fat diets. Obesity. 2019;27(3):420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Polidori D, Sanghvi A, Seeley RJ, Hall KD. How strongly does appetite counter weight loss? Quantification of the feedback control of human energy intake. Obesity. 2016;24(11):2289–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hall KD, Guo J, Courville AB, Boring J, Brychta R, Chen KY, Darcey V, Forde CG, Gharib AM, Gallagher Iet al. Effect of a plant-based, low-fat diet versus an animal-based, ketogenic diet on ad libitum energy intake. Nat Med. 2021;27(2):344–53. [DOI] [PubMed] [Google Scholar]
- 122. Shimy KJ, Feldman HA, Klein GL, Bielak L, Ebbeling CB, Ludwig DS. Effects of dietary carbohydrate content on circulating metabolic fuel availability in the postprandial state. J Endocr Soc. 2020;4(7):bvaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hall KD, Guo J. Obesity energetics: body weight regulation and the effects of diet composition. Gastroenterology. 2017;152(7):1718–27.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Buga A, Kackley ML, Crabtree CD, Sapper TN, McCabe L, Fell B, LaFountain RA, Hyde PN, Martini ER, Bowman Jet al. The effects of a 6-week controlled, hypocaloric ketogenic diet, with and without exogenous ketone salts, on body composition responses. Front Nutr. 2021;8:618520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ello-Martin JA, Roe LS, Ledikwe JH, Beach AM, Rolls BJ. Dietary energy density in the treatment of obesity: a year-long trial comparing 2 weight-loss diets. Am J Clin Nutr. 2007;85(6):1465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ledikwe JH, Rolls BJ, Smiciklas-Wright H, Mitchell DC, Ard JD, Champagne C, Karanja N, Lin PH, Stevens VJ, Appel LJ. Reductions in dietary energy density are associated with weight loss in overweight and obese participants in the PREMIER trial. Am J Clin Nutr. 2007;85(5):1212–21. [DOI] [PubMed] [Google Scholar]
- 127. Lowe MR, Butryn ML, Thomas JG, Coletta M. Meal replacements, reduced energy density eating, and weight loss maintenance in primary care patients: a randomized controlled trial. Obesity. 2014;22(1):94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rolls BJ, Roe LS, Beach AM, Kris-Etherton PM. Provision of foods differing in energy density affects long-term weight loss. Obes Res. 2005;13(6):1052–60. [DOI] [PubMed] [Google Scholar]
- 129. Saquib N, Natarajan L, Rock CL, Flatt SW, Madlensky L, Kealey S, Pierce JP. The impact of a long-term reduction in dietary energy density on body weight within a randomized diet trial. Nutr Cancer. 2007;60(1):31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Hall KD, Ayuketah A, Brychta R, Cai H, Cassimatis T, Chen KY, Chung ST, Costa E, Courville A, Darcey Vet al. Ultra-processed diets cause excess calorie intake and weight gain: an inpatient randomized controlled trial of ad libitum food intake. Cell Metab. 2019;30(1):67–77.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Edens MA, van Dijk PR, Hak E, Bilo HJG. Course of body weight before and after the initiation of insulin therapy in type 2 diabetes mellitus: retrospective inception cohort study (ZODIAC 58). Endocrinol Diabetes Metab. 2021;4(2):e00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Larger E, Rufat P, Dubois-Laforgue D, Ledoux S. Insulin therapy does not itself induce weight gain in patients with type 2 diabetes. Diabetes Care. 2001;24(10):1849–50. [DOI] [PubMed] [Google Scholar]
- 133. Larger E. Weight gain and insulin treatment. Diabetes Metab. 2005;31(4 Pt 2):4s51–4s6. [DOI] [PubMed] [Google Scholar]
- 134. Rodin J, Wack J, Ferrannini E, DeFronzo RA. Effect of insulin and glucose on feeding behavior. Metabolism. 1985;34(9):826–31. [DOI] [PubMed] [Google Scholar]
- 135. Chapman IM, Goble EA, Wittert GA, Morley JE, Horowitz M. Effect of intravenous glucose and euglycemic insulin infusions on short-term appetite and food intake. Am J Physiol. 1998;274(3):R596–603. [DOI] [PubMed] [Google Scholar]
- 136. Spetter MS. Current state of the use of neuroimaging techniques to understand and alter appetite control in humans. Curr Opin Clin Nutr Metab Care. 2018;21(5):329–35. [DOI] [PubMed] [Google Scholar]
- 137. Porte D Jr, Baskin DG, Schwartz MW. Leptin and insulin action in the central nervous system. Nutr Rev. 2002;60(Suppl 10):S20–9.; discussion S68–84, 85–7. [DOI] [PubMed] [Google Scholar]
- 138. Makimura H, Stanley TL, Suresh C, De Sousa-Coelho AL, Frontera WR, Syu S, Braun LR, Looby SE, Feldpausch MN, Torriani Met al. Metabolic effects of long-term reduction in free fatty acids with acipimox in obesity: a randomized trial. J Clin Endocrinol Metab. 2016;101(3):1123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Drucker DJ. GLP-1 physiology informs the pharmacotherapy of obesity. Mol Metab. 2021;57:101351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Astrup A, Hjorth MF. Classification of obesity targeted personalized dietary weight loss management based on carbohydrate tolerance. Eur J Clin Nutr. 2018;72(9):1300–4. [DOI] [PubMed] [Google Scholar]
- 141. Hjorth MF, Astrup A, Zohar Y, Urban LE, Sayer RD, Patterson BW, Herring SJ, Klein S, Zemel BS, Foster GDet al. Personalized nutrition: pretreatment glucose metabolism determines individual long-term weight loss responsiveness in individuals with obesity on low-carbohydrate versus low-fat diet. Int J Obes (Lond). 2019;43:2037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hjorth MF, Zohar Y, Hill JO, Astrup A. Personalized dietary management of overweight and obesity based on measures of insulin and glucose. Annu Rev Nutr. 2018;38(1):245–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Bauer J. Obesity: its pathogenesis, etiology and treatment. Arch Intern Med. 1941;67(5):968–94. [Google Scholar]
- 144. Pennington AW. Obesity. Med Times. 1952;80(7):389–98. [PubMed] [Google Scholar]
- 145. Pennington AW. An alternate approach to the problem of obesity. Am J Clin Nutr. 1953;1(2):100–6. [DOI] [PubMed] [Google Scholar]
- 146. Pennington AW. A reorientation on obesity. N Engl J Med. 1953;248(23):959–64. [DOI] [PubMed] [Google Scholar]
- 147. Pennington AW. Obesity: overnutrition or disease of metabolism?. Am J Dig Dis. 1953;20(9):268–74. [DOI] [PubMed] [Google Scholar]
- 148. Pennington AW. Pathophysiology of obesity. Am J Dig Dis. 1954;21(3):69–73. [DOI] [PubMed] [Google Scholar]
- 149. Astwood EB. The heritage of corpulence. Endocrinology. 1962;71(2):337–41. [DOI] [PubMed] [Google Scholar]
- 150. Friedman MI. Body fat and the metabolic control of food intake. Int J Obes. 1990;14(Suppl 3):53–66.; discussion 66–7. [PubMed] [Google Scholar]
- 151. Friedman MI. An energy sensor for control of energy intake. Proc Nutr Soc. 1997;56(1A):41–50. [DOI] [PubMed] [Google Scholar]
- 152. Friedman MI. Fuel partitioning and food intake. Am J Clin Nutr. 1998;67(3):513S–18S. [DOI] [PubMed] [Google Scholar]
- 153. Friedman MI, Stricker EM. The physiological psychology of hunger: a physiological perspective. Psychol Rev. 1976;83(6):409–31. [PubMed] [Google Scholar]
- 154. Ludwig DS. Dietary glycemic index and obesity. J Nutr. 2000;130(2):280S–3S. [DOI] [PubMed] [Google Scholar]
- 155. Lustig RH. Childhood obesity: behavioral aberration or biochemical drive? Reinterpreting the first law of thermodynamics. Nat Clin Pract Endocrinol Metab. 2006;2(8):447–58. [DOI] [PubMed] [Google Scholar]