

Summary
Determining the effect of vaccine-induced immune response on disease risk is an important goal of vaccinology. Typically, immune correlates analyses are conducted prospectively with immune response measured shortly after vaccination and subsequent disease status regressed on immune response. In outbreaks and rare disease settings, collecting samples from all vaccinees is not feasible. The test negative design is a retrospective design used to measure vaccine efficacy where symptomatic individuals who present at a clinic are assessed for relevant disease (cases) or some other disease (controls) and vaccination status ascertained. This article proposes that test negative vaccinees have immune response to vaccine assessed both for relevant (e.g., Ebola) and irrelevant (e.g., vector) proteins. If the latter immune response is unaffected by active (Ebola) infection, and is correlated with the relevant immune response, it can serve as a proxy for the immune response of interest proximal to infection. We show that logistic regression using imputed immune response as the covariate and case disease as outcome can estimate the prospective immune response slope and detail the assumptions needed for unbiased inference. The method is evaluated by simulation under various scenarios including constant and decaying immune response. A simulated dataset motivated by ring vaccination for an ongoing Ebola outbreak is analyzed.
Keywords: Imputation; Likelihood, Logisitic regression; Regression calibration
1. Introduction
The test negative design is a kind of case–control study used to estimate vaccine efficacy (Fukushima and Hirota, 2017). Originally proposed for pneumococcal vaccination Broome and others (1980), it has been used extensively for influenza vaccine see Jackson and Nelson (2013) as well as rotavirus vaccine (Boom and others, 2010). The basic design uses symptomatic subjects who come to a clinic with symptoms that could be either the vaccine preventable case disease (say Ebola) or a different control disease (non-Ebola). Vaccination status is ascertained and vaccine efficacy (VE) calculated as one minus the odds of vaccine among cases divided by the odds of vaccine among controls. This equals the prospective vaccine efficacy due to the invariance of the odds ratio for prospective or case–control sampling. The test negative study requires that all symptomatic vaccinees have the same probability of a clinic visit, an analogous condition for the symptomatic unvaccinated subjects, and that exposure be the same for all vaccinated and unvaccinated subjects. Numerous authors have investigated the sensitivity of the test negative design to these assumptions Sullivan and others (2016), Westreich and Hudgens (2016), Fukushima and Hirota (2017), and Lewnard and others (2018).
An important goal in vaccine development is to evaluate how vaccine-induced immune response correlates with risk of disease, known as a “correlate of risk” (Qin and others, 2007). Identification of a specific immune response that is associated with low disease risk provides a target for vaccine development. A common approach to evaluate immune correlates is to measure immune response to the vaccine shortly after vaccination determine who gets infected during prospective follow-up and then use logistic regression to predict the probability of disease as a function of measured immune response. Ideally, such an approach would be used in outbreak settings, see Halloran and others (2020), but at times it may not be feasible to prospectively collect samples. In addition, for rare diseases in non-outbreak settings, an enormous number of samples may need to be collected for traditional correlates analysis. In other settings, the durability of a vaccine is of interest and thus the immune response at the time of exposure is of interest though this may be many years after vaccination.
This article details an approach to immune correlates analysis under a test negative design using samples collected from vaccinees proximal to exposure. To fix ideas, consider the rVSV vaccine for Ebola which induces an immune response to Ebola antigens say 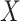 as well as to the Vesicular Stomatitis Vaccine (VSV) vector, say
as well as to the Vesicular Stomatitis Vaccine (VSV) vector, say 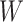 . Naively, one might measure
. Naively, one might measure 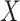 among the symptomatic vaccinees who visit a clinic and fit a logistic regression with outcome being Ebola and covariate
among the symptomatic vaccinees who visit a clinic and fit a logistic regression with outcome being Ebola and covariate 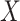 . The problem is that Ebola immune response is likely massive in those with Ebola infection. What we really want is the pure vaccine-induced
. The problem is that Ebola immune response is likely massive in those with Ebola infection. What we really want is the pure vaccine-induced 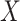 just prior to exposure in both Ebola cases and controls. As a proxy, we can use
just prior to exposure in both Ebola cases and controls. As a proxy, we can use 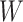 at the clinic visit which should be similar to
at the clinic visit which should be similar to 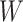 at exposure. Provided
at exposure. Provided 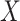 and
and 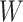 are correlated, we can use
are correlated, we can use 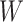 to impute the vaccine-induced
to impute the vaccine-induced 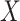 at exposure. We can then fit a logistic regression with Ebola as the outcome using the expectation of
at exposure. We can then fit a logistic regression with Ebola as the outcome using the expectation of 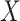 conditional on
conditional on 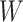 , say
, say 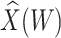 , as the covariate; also known as regression calibration. If the time interval from vaccination to exposure is relatively long, the substantial decay of the immune response over time may need to be addressed. This can be accomplished either by stratification, or flexible parametric modeling of risk over study time, provided the disease is rare. We evaluate the performance of the method via simulation and illustrate its use by a simulated dataset meant to reflect the recent Ebola outbreak in the Democratic Republic of the Congo where the rVSV Ebola vaccine was deployed using ring vaccination.
, as the covariate; also known as regression calibration. If the time interval from vaccination to exposure is relatively long, the substantial decay of the immune response over time may need to be addressed. This can be accomplished either by stratification, or flexible parametric modeling of risk over study time, provided the disease is rare. We evaluate the performance of the method via simulation and illustrate its use by a simulated dataset meant to reflect the recent Ebola outbreak in the Democratic Republic of the Congo where the rVSV Ebola vaccine was deployed using ring vaccination.
2. Formulation and models
2.1. Constant immune response
An important goal in vaccine development is to assess how immune response to the vaccine, 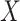 , is associated with the risk of infection/disease
, is associated with the risk of infection/disease 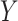 . In a randomized trial, vaccinees
. In a randomized trial, vaccinees 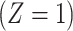 have an immune response
have an immune response 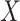 measured shortly after vaccination. For now, assume that
measured shortly after vaccination. For now, assume that 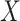 does not change. Volunteers are followed for a period of time
does not change. Volunteers are followed for a period of time 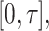 and disease status is recorded. With a test negative design, we need both the case disease and a different (control) disease for reference while the undiseased are not used. We thus define the disease of interest (case), a different disease (control), or undiseased which we denote by
and disease status is recorded. With a test negative design, we need both the case disease and a different (control) disease for reference while the undiseased are not used. We thus define the disease of interest (case), a different disease (control), or undiseased which we denote by 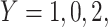 respectively. We need three possibilities for later use in a test negative design. If both diseases are acquired, we use the case disease. We assume that among vaccinees
respectively. We need three possibilities for later use in a test negative design. If both diseases are acquired, we use the case disease. We assume that among vaccinees
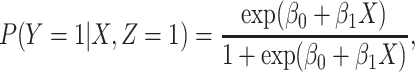 |
(2.1) |
and that
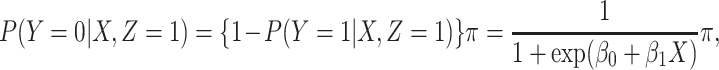 |
(2.2) |
where 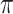 is the conditional probability that a vaccinee who avoids the case disease develops the control disease. Note that
is the conditional probability that a vaccinee who avoids the case disease develops the control disease. Note that 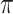 is assumed free of
is assumed free of 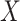 as it should not have an effect on acquiring the control disease. An immune correlates analysis relating risk of disease to immune response can be conducted by fitting (2.1) in vaccinees using as outcome the indicator that
as it should not have an effect on acquiring the control disease. An immune correlates analysis relating risk of disease to immune response can be conducted by fitting (2.1) in vaccinees using as outcome the indicator that 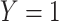 (versus
(versus 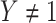 ). For developmental simplicity, we do not incorporate baseline covariates in (2.1) though one can.
). For developmental simplicity, we do not incorporate baseline covariates in (2.1) though one can.
For certain settings, 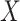 may not be measured shortly after vaccination. For rare diseases it may be prohibitively costly, or in outbreak settings it may not be logistically feasible. In other settings, the durability of a vaccine is unknown and thus
may not be measured shortly after vaccination. For rare diseases it may be prohibitively costly, or in outbreak settings it may not be logistically feasible. In other settings, the durability of a vaccine is unknown and thus 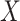 at exposure is of interest even though it may be many years since vaccination. Or in outbreak settings, it may not be logistically feasible to draw samples. In all these situations, a correlates analysis using the immune response proximal to exposure would be of great interest.
at exposure is of interest even though it may be many years since vaccination. Or in outbreak settings, it may not be logistically feasible to draw samples. In all these situations, a correlates analysis using the immune response proximal to exposure would be of great interest.
A design that collects information proximal to exposure is the test negative which takes symptomatic patients (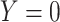 or
or 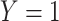 ) who present at a clinic and tests them for the disease of interest and records vaccination status. Those testing positive are classified as cases (
) who present at a clinic and tests them for the disease of interest and records vaccination status. Those testing positive are classified as cases (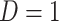 ) while those testing negative are classified as controls (
) while those testing negative are classified as controls (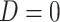 ). Formally,
). Formally, 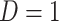 if
if 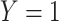 and
and 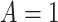 , while
, while 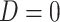 if
if 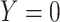 , and
, and 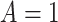 , where
, where 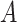 is the indicator a person with either disease (i.e.,
is the indicator a person with either disease (i.e., 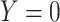 or 1) arrives at the clinic. Overall vaccine efficacy is estimated as
or 1) arrives at the clinic. Overall vaccine efficacy is estimated as
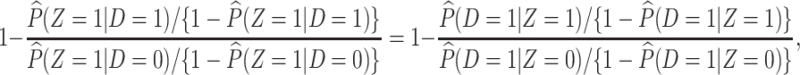 |
(2.3) |
where 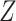 is the vaccine indicator see Guolo (2008). The second equality follows from the invariance of the odds to prospective and retrospective sampling. The test negative design can be much more efficient than a prospective study, but does require more assumptions.
is the vaccine indicator see Guolo (2008). The second equality follows from the invariance of the odds to prospective and retrospective sampling. The test negative design can be much more efficient than a prospective study, but does require more assumptions.
Suppose, we somehow knew 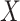 in all vaccinees who arrived at the clinic. Let
in all vaccinees who arrived at the clinic. Let 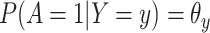 be the arrival probability for an symptomatic vaccinee with disease status
be the arrival probability for an symptomatic vaccinee with disease status 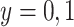 . Based on (2.1) and (2.2), we can derive the probability that a symptomatic vaccinee who arrives at the clinic has the case disease:
. Based on (2.1) and (2.2), we can derive the probability that a symptomatic vaccinee who arrives at the clinic has the case disease:
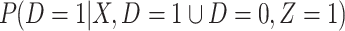 |
(2.4) |
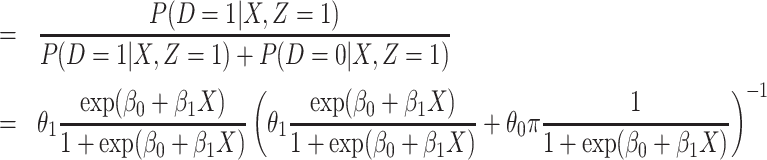 |
(2.5) |
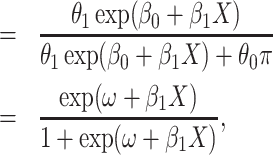 |
(2.6) |
where 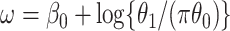 . Thus, if we fit a simple logistic regression model with
. Thus, if we fit a simple logistic regression model with 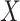 as covariate among vaccinees in the clinic, we can recover the slope
as covariate among vaccinees in the clinic, we can recover the slope 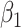 from (2.1). Note that this obtains even if we allow cases and controls to have different arrival probabilities. The usual test negative design requires
from (2.1). Note that this obtains even if we allow cases and controls to have different arrival probabilities. The usual test negative design requires 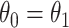 for all vaccinees. We next consider the actual setting where
for all vaccinees. We next consider the actual setting where 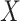 must be derived from measurements at arrival. Figuring out how to get
must be derived from measurements at arrival. Figuring out how to get 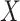 from test negative data is the major contribution of this article.
from test negative data is the major contribution of this article.
In practice, we can measure 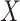 when subjects arrive at the clinic, say
when subjects arrive at the clinic, say 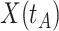 where
where 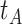 is the arrival time. Naively fitting (2.6) using the clinic
is the arrival time. Naively fitting (2.6) using the clinic 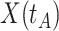 is problematic. For vaccinees who present with the control disease
is problematic. For vaccinees who present with the control disease 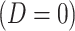 ,
, 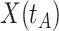 should be close to
should be close to 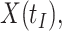 where
where 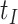 is just prior to infection as control disease should have little effect on the vaccine-induced immune response to the relevant vaccine antigen. But the vaccinated who present with the disease of interest
is just prior to infection as control disease should have little effect on the vaccine-induced immune response to the relevant vaccine antigen. But the vaccinated who present with the disease of interest 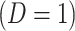 are likely to have quite high
are likely to have quite high 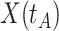 due to active infection. So
due to active infection. So 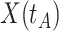 at arrival is different from
at arrival is different from 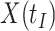 for vaccinees with
for vaccinees with 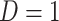 .
.
However, certain vaccines (e.g., vector based vaccines) also induce an immune response to irrelevant antigens (e.g., the vector), say 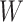 , which should be relatively unaffected by active infection. To fix ideas, suppose that
, which should be relatively unaffected by active infection. To fix ideas, suppose that 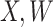 achieve stable values shortly after vaccination and remain constant in
achieve stable values shortly after vaccination and remain constant in 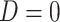 diseased controls. If
diseased controls. If 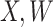 are correlated prior to exposure, then one could predict the unadulterated
are correlated prior to exposure, then one could predict the unadulterated 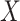 at exposure using the
at exposure using the 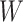 at presentation. One could then use
at presentation. One could then use 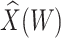 , the imputed immune response to relevant vaccine antigens at exposure, in lieu of
, the imputed immune response to relevant vaccine antigens at exposure, in lieu of 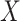 . This works if
. This works if 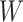 is unaffected by case or control disease and
is unaffected by case or control disease and 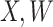 are unaffected by control disease. But these requirements can be weakened. Suppose that for both groups, we observe
are unaffected by control disease. But these requirements can be weakened. Suppose that for both groups, we observe 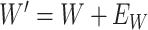 and for those with control disease we observe
and for those with control disease we observe 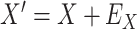 where
where 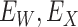 are errors (which can be correlated and have non-zero means). Thus
are errors (which can be correlated and have non-zero means). Thus 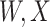 are the pre-infection values while
are the pre-infection values while 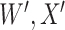 are the values observed at the clinic when subjects are diseased. If
are the values observed at the clinic when subjects are diseased. If 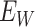 differs for case and control disease, however, the control model fitted on
differs for case and control disease, however, the control model fitted on 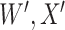 does not apply to cases and a more complex approach would be required.
does not apply to cases and a more complex approach would be required.
This strategy is displayed in Figure 1. The diamonds are actual 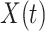 while the two solid bent lines are linear interpolations of the IgG antibody response to the outside of the Ebola virus i.e., Ebola glycoprotein (GP) following rVSV vaccination from two randomly selected subjects in Prevail 1, an immunogenicity trial of the rVSV vaccine Kennedy and others (2016). Immune response was measured at baseline, 1 week, 1 month, 6 months, and 1 year (diamonds). While IgG response to vector
while the two solid bent lines are linear interpolations of the IgG antibody response to the outside of the Ebola virus i.e., Ebola glycoprotein (GP) following rVSV vaccination from two randomly selected subjects in Prevail 1, an immunogenicity trial of the rVSV vaccine Kennedy and others (2016). Immune response was measured at baseline, 1 week, 1 month, 6 months, and 1 year (diamonds). While IgG response to vector 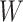 was not measured, we illustrate interpolated hypothetical values by the dashed bent lines. We pretend that these two subjects were infected, with, respectively, malaria and Ebola. They became symptomatic and arrived at a clinic a few days after productive exposure. The
was not measured, we illustrate interpolated hypothetical values by the dashed bent lines. We pretend that these two subjects were infected, with, respectively, malaria and Ebola. They became symptomatic and arrived at a clinic a few days after productive exposure. The 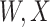 values for the malaria patient at arrival are similar to the values at exposure, but for the Ebola patient, only
values for the malaria patient at arrival are similar to the values at exposure, but for the Ebola patient, only 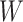 is similar at exposure and arrival while
is similar at exposure and arrival while 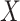 (dashed line) is massive from active Ebola infection., The bottom panel illustrates hypothetical data with a correlation of 0.70 between
(dashed line) is massive from active Ebola infection., The bottom panel illustrates hypothetical data with a correlation of 0.70 between 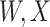 that might have been obtained from a sample of vaccinated controls who arrived at the clinic. From this relationship, we can impute
that might have been obtained from a sample of vaccinated controls who arrived at the clinic. From this relationship, we can impute 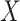 at infection for the Ebola patient.
at infection for the Ebola patient.
Fig. 1.

Top panel: IgG immune responses over time for two vaccinated volunteers from Prevail 1. The solid bent line is the linear interpolation of X = IgG to Ebola GP measured at weeks 0, 1, 4, 26, and 52 (red diamonds). The dotted red line is hypothetical X to reflect Ebola infection. The dashed bent line is hypothetical W=IgG to rVSV vector. The subject infected with malaria arrives with 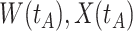 similar to those at infection. Only
similar to those at infection. Only 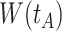 is similar to the value prior to infection for the Ebola infected volunteer. Bottom panel: a scatter plot of
is similar to the value prior to infection for the Ebola infected volunteer. Bottom panel: a scatter plot of 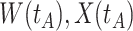 for the controls (circles) with imputation of
for the controls (circles) with imputation of 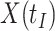 for the Ebola case (solid square).
for the Ebola case (solid square).
More formally, from the controls we can fit the model
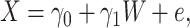 |
(2.7) |
where 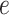 is an error term. Using the estimated parameters, we impute
is an error term. Using the estimated parameters, we impute 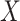 ,
,
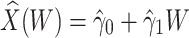 |
and fit the logistic regression model in the symptomatic vaccinees
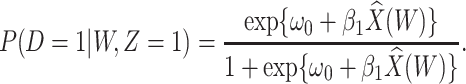 |
(2.8) |
This imputation is known as regression calibration and is a simple way to correct for measurement error in a covariate. Note that we impute 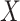 even in the vaccinated controls where
even in the vaccinated controls where 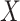 is known and thus treat both cases and controls in the same way. Using imputation in all those with
is known and thus treat both cases and controls in the same way. Using imputation in all those with 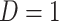 and
and 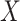 directly in all those with
directly in all those with 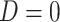 leads to substantial bias. Because we use
leads to substantial bias. Because we use 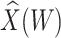 , which is estimated instead of
, which is estimated instead of 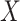 , which is fixed, standard errors from standard software fitting (2.8) are likely too small. A simple remedy is to use the bootstrap or use derived standard errors, see Rosner and others (1990).
, which is fixed, standard errors from standard software fitting (2.8) are likely too small. A simple remedy is to use the bootstrap or use derived standard errors, see Rosner and others (1990).
Instead of using an imputed 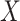 for regression calibration, one can perform a correlates analysis using
for regression calibration, one can perform a correlates analysis using 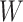 directly. While
directly. While 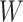 is not part of the mechanism of protection, “correlates” analyses are often done with immune responses that may not be causal but are presumably related to the causal mechanism. As an example, smallpox vaccination involves scraping the arm with antigen. A vaccine “take” is recorded if a pox forms. While the scar itself does not protect, it is a proxy for a robust relevant immune response to the vaccine and risk of disease by scar or “take” is of interest even though it is clearly a non-mechanistic correlate of risk using the nomenclature of Plotkin and Gilbert (2012). Use of
is not part of the mechanism of protection, “correlates” analyses are often done with immune responses that may not be causal but are presumably related to the causal mechanism. As an example, smallpox vaccination involves scraping the arm with antigen. A vaccine “take” is recorded if a pox forms. While the scar itself does not protect, it is a proxy for a robust relevant immune response to the vaccine and risk of disease by scar or “take” is of interest even though it is clearly a non-mechanistic correlate of risk using the nomenclature of Plotkin and Gilbert (2012). Use of 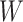 directly is simple, avoids the measurement error issue, requires no imputation, and has slightly better power than use of imputed
directly is simple, avoids the measurement error issue, requires no imputation, and has slightly better power than use of imputed 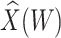 , as we will see. Use of
, as we will see. Use of 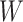 by itself, however, is not easily interpretable unless as a proxy for vaccine “take,” i.e.,
by itself, however, is not easily interpretable unless as a proxy for vaccine “take,” i.e., 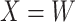 is binary and
is binary and 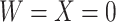 is like being unvaccinated.
is like being unvaccinated.
2.2. Waning immune response
The above development is appropriate if the immune responses 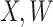 are stable during the followup period for the cohort study. In general, antibody decay over time and the above approach can lead to bias. To see this, suppose that nearly everyone was vaccinated prior to an outbreak which exploded and then waned so the risk of the case disease decreased over time, coinciding with the antibody decay. Further suppose that the control disease rate was constant over time. Then even if
are stable during the followup period for the cohort study. In general, antibody decay over time and the above approach can lead to bias. To see this, suppose that nearly everyone was vaccinated prior to an outbreak which exploded and then waned so the risk of the case disease decreased over time, coinciding with the antibody decay. Further suppose that the control disease rate was constant over time. Then even if 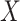 was unrelated to
was unrelated to 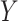 we would associate low
we would associate low 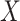 with low risk and we would tend to estimate a positive
with low risk and we would tend to estimate a positive 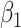 . And the problem is even more complicated as, in general, people will be vaccinated at various times and the case disease rate might vary with time.
. And the problem is even more complicated as, in general, people will be vaccinated at various times and the case disease rate might vary with time.
Let 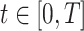 be the time since the start of the test negative study. To develop the time varying
be the time since the start of the test negative study. To develop the time varying 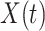 setting more formally, we will assume that the hazard for a case disease arriving at time
setting more formally, we will assume that the hazard for a case disease arriving at time 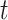 with covariate
with covariate 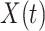 is given by
is given by
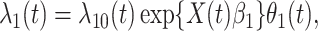 |
(2.9) |
where 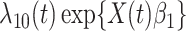 is the hazard for case disease a little before
is the hazard for case disease a little before 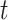 and
and 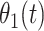 is the probability of arriving at the clinic at time
is the probability of arriving at the clinic at time 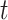 , given a case infection just prior to
, given a case infection just prior to 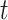 .
.
We analogously assume that the hazard for the control disease is independent of 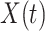 and arbitrary, as is the instantaneous probability of arriving. Thus, the hazard for a person with control disease arriving at the clinic is
and arbitrary, as is the instantaneous probability of arriving. Thus, the hazard for a person with control disease arriving at the clinic is
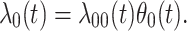 |
Recall that the probability of an event in a small interval [t,t+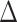 ), given no event prior to
), given no event prior to 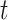 , is approximately
, is approximately 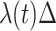 for an arbitrary hazard function
for an arbitrary hazard function 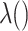 . Under a “rare” disease assumption, we can calculate the probability that a given vaccinee who showed up at time
. Under a “rare” disease assumption, we can calculate the probability that a given vaccinee who showed up at time 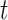 was a case as
was a case as
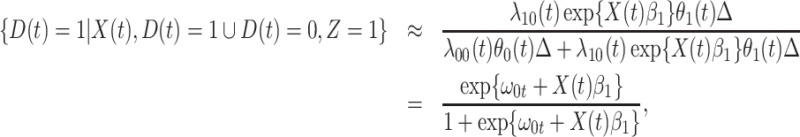 |
(2.10) |
where 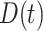 is the indicator of case disease for an arrival at time
is the indicator of case disease for an arrival at time 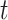 and
and
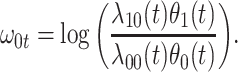 |
Now 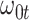 could be constant. One way this can happen is if
could be constant. One way this can happen is if  and
and 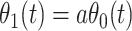 so
so 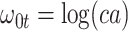 . If so, we can simply fit logistic regression to the
. If so, we can simply fit logistic regression to the 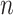 data points
data points 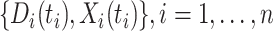 . In general,
. In general, 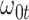 will not be constant so that
will not be constant so that
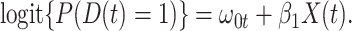 |
(2.11) |
We might specify 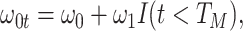 where
where 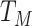 is say the median followup time. Or we could fit a quadratic function of log(t). In practice, different approaches to specify
is say the median followup time. Or we could fit a quadratic function of log(t). In practice, different approaches to specify 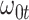 could be tried and the best one selected. Note that we only need know (or impute)
could be tried and the best one selected. Note that we only need know (or impute) 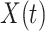 at the time of arrival to the clinic for each vaccinee.
at the time of arrival to the clinic for each vaccinee.
3. Assumptions
The above approaches recover the prospective slope for 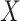 or
or 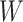 if the model assumptions are met. In this section, we delineate the assumptions when
if the model assumptions are met. In this section, we delineate the assumptions when 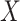 is known for all, constant after time
is known for all, constant after time 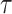 , the logistic-type model
, the logistic-type model 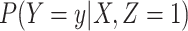 is correctly specified, and there is independence between exposure and
is correctly specified, and there is independence between exposure and 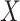 . The major issue is thus whether vaccinated cases (controls) arrive with fixed probability
. The major issue is thus whether vaccinated cases (controls) arrive with fixed probability 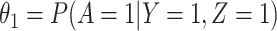 (
(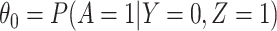 ). Or equivalently, whether those who arrive are a representative sample of cases and controls respectively. We allow
). Or equivalently, whether those who arrive are a representative sample of cases and controls respectively. We allow 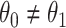 .
.
1. The vaccinees who arrive with the control disease provide a representative sample of the distribution of immune response among those vaccinated without the case disease.
This follows if 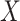 has no impact on the probability of acquiring the control disease and no impact on the probability of arriving at the clinic so
has no impact on the probability of acquiring the control disease and no impact on the probability of arriving at the clinic so 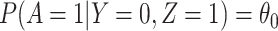 . Note we can allow
. Note we can allow 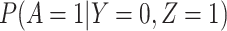 to depend on latent or measured disease stratus or baseline covariates as long as they independent of
to depend on latent or measured disease stratus or baseline covariates as long as they independent of 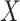 .
.
2. The vaccinees who arrive with the case disease provide a representative sample of the distribution of immune response among those vaccinated with the case disease.
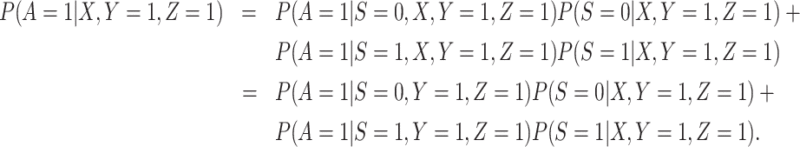 |
(3.12) |
The last equality follows if 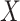 has no additional impact on
has no additional impact on 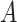 given severity which seems a reasonable assumption. Using the last equality, we can see that
given severity which seems a reasonable assumption. Using the last equality, we can see that 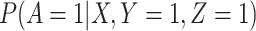 does not depend on
does not depend on 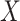 if either
if either 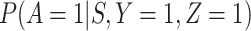 is free of
is free of 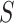 or
or 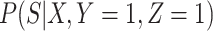 is free of
is free of 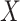 . We discuss each in turn.
. We discuss each in turn.
Now 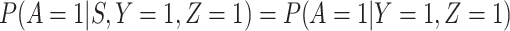 if severity does not impact health seeking behavior. This might occur if all cases are equally encouraged to arrive at a clinic and all cases are equally compliant, or if the severity gradient is modest enough that it does not change behavior.
if severity does not impact health seeking behavior. This might occur if all cases are equally encouraged to arrive at a clinic and all cases are equally compliant, or if the severity gradient is modest enough that it does not change behavior.
Next consider when 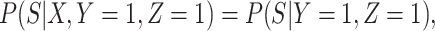 i.e.,
i.e., 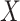 has no impact on severity given a vaccinee is infected. While this obtains if the vaccine’s mechanism of action is all-or-none, it is a weaker condition. An all-or-none vaccine implies that the distribution of severity is unchanged by vaccination so that
has no impact on severity given a vaccinee is infected. While this obtains if the vaccine’s mechanism of action is all-or-none, it is a weaker condition. An all-or-none vaccine implies that the distribution of severity is unchanged by vaccination so that  , where
, where 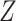 is the vaccine indicator. But we only require
is the vaccine indicator. But we only require 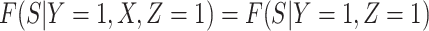 which allows
which allows 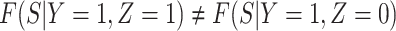 .
.
If neither assumption is plausible but case disease severity is observed, there is a remedy provided disease follows the prospective logistic regression model:
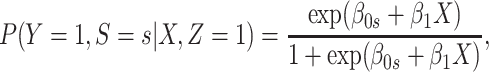 |
(3.13) |
for  Then even if
Then even if 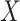 affects the arrival probability through the observable disease severity, recovery of
affects the arrival probability through the observable disease severity, recovery of 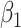 using data
using data 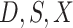 follows from arguments analogous to those used to derive (2.6). One can show that (3.13) implies the distribution of disease severity among the vaccinated infecteds,
follows from arguments analogous to those used to derive (2.6). One can show that (3.13) implies the distribution of disease severity among the vaccinated infecteds, 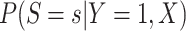 , varies with
, varies with 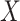 reflecting a kind of disease modifying vaccine.
reflecting a kind of disease modifying vaccine.
These conditions can be relaxed using baseline covariates 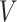 . If the prospective model (2.1) controls for confounding by specifying
. If the prospective model (2.1) controls for confounding by specifying 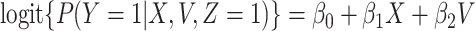 , then the above arguments go through, even if the arrival probabilities satisfy
, then the above arguments go through, even if the arrival probabilities satisfy 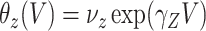 . One can show that
. One can show that 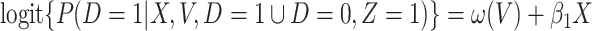 with
with 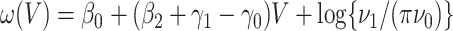 , and thus
, and thus 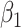 remains the coeffience for
remains the coeffience for 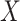 .
.
To summarize, assuming the verity of (3.12) and a correctly specified model for 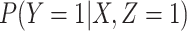 , we can recover
, we can recover 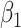 even if
even if 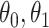 depend on
depend on 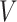 unless disease severity is unobservable,
unless disease severity is unobservable, 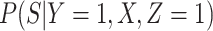 depends on
depends on 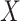 , and
, and 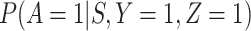 depends on
depends on 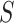 .
.
4. Simulations
4.1. Constant immune response
We illustrate performance for the simple case where antibody does not decay. We prospectively generate data for vaccinees which are then sampled retrospectively as in a test negative design. We assume a population of size 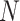 vaccinees and generate the case disease according to
vaccinees and generate the case disease according to
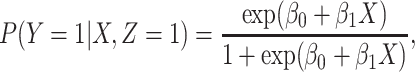 |
and sequentially generate control disease among the case uninfecteds as
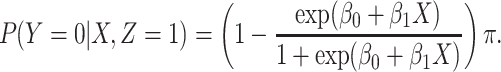 |
We specify 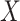 as Gaussian with mean= 3.00 and standard deviation =0.50 as in the Prevail 1 vaccine trial Kennedy and others (2016). We generate
as Gaussian with mean= 3.00 and standard deviation =0.50 as in the Prevail 1 vaccine trial Kennedy and others (2016). We generate
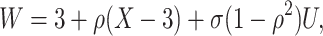 |
where 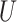 is standard normal. Thus,
is standard normal. Thus, 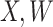 are bivariate normal with common mean 3.00, common standard deviation 0.50, and correlation
are bivariate normal with common mean 3.00, common standard deviation 0.50, and correlation 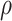 . We set
. We set 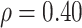 , 0.70, and 1.00. For reference, a study estimated a correlation of 0.70 between ELISA OD immune response readouts for VSV proteins (W) and Ebola proteins (X) at 56 days post vaccination Poetsch and others (2018).
, 0.70, and 1.00. For reference, a study estimated a correlation of 0.70 between ELISA OD immune response readouts for VSV proteins (W) and Ebola proteins (X) at 56 days post vaccination Poetsch and others (2018).
We specify 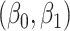 as (
as ( 5.51, 0.00), (
5.51, 0.00), ( 4.05,
4.05,  0.60), and (
0.60), and ( 1.12,
1.12,  1.81). These correspond to no, modest, and strong effects of
1.81). These correspond to no, modest, and strong effects of 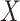 . To help interpret
. To help interpret 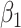 , the ratios of risk of case disease for the 1st versus 8th octile of
, the ratios of risk of case disease for the 1st versus 8th octile of 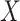 are 1, 2, and 10, respectively, for
are 1, 2, and 10, respectively, for 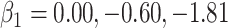 . We set the conditional risk of control disease as
. We set the conditional risk of control disease as 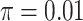 ,
, 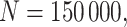 and
and 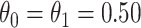 so that vaccinated cases and controls arrive with equal probability.
so that vaccinated cases and controls arrive with equal probability.
We evaluate estimation of the model
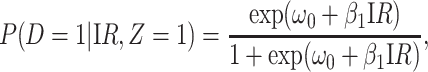 |
(4.14) |
where IR, the immune response, is 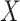 ,
, 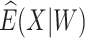 , or
, or 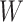
Table 1 presents the results using 10 000 simulated studies per scenario. Columns 4 and 5 provide the sample mean and variance of 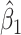 . Column 6 reports the rejection rate for the two-sided
. Column 6 reports the rejection rate for the two-sided 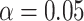 Wald test using the Monte Carlo standard error over the 10 000 simulated studies. We see that we have power of around 100% to test the effect of IR when
Wald test using the Monte Carlo standard error over the 10 000 simulated studies. We see that we have power of around 100% to test the effect of IR when 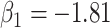 for all scenarios, while for
for all scenarios, while for 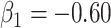 we need a correlation of 0.70 to approach 80% power. The type I error rate seems consistent with the
we need a correlation of 0.70 to approach 80% power. The type I error rate seems consistent with the 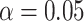 used for testing.
used for testing.
Table 1.
Simulated performance of logistic regression using different covariates for IR immune response; 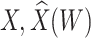 , or
, or 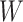 . Sample statistics for different parameters estimates are presented.
. Sample statistics for different parameters estimates are presented. 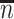 is the average number of symptomatic vaccinees who arrive and
is the average number of symptomatic vaccinees who arrive and 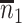 the average number of vaccinees who arrive with the case disease. 1000 test negative designs are simulated for each row. The last row has
the average number of vaccinees who arrive with the case disease. 1000 test negative designs are simulated for each row. The last row has 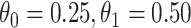 all other rows have
all other rows have 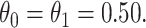 Each row is based on 10 000 simulated studies.
Each row is based on 10 000 simulated studies.
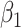
|
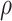
|
IR |
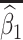
|
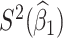
|
% Reject |
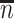
|
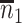
|
|---|---|---|---|---|---|---|---|
| 0.000 | 1.000 |
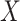
|
 0.002
0.002 |
0.019 | 0.053 | 1049 | 302 |
 0.600
0.600 |
1.000 |
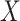
|
 0.602
0.602 |
0.024 | 0.973 | 972 | 225 |
 1.810
1.810 |
1.000 |
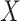
|
 1.821
1.821 |
0.040 | 1.000 | 908 | 160 |
| 0.000 | 0.700 |
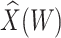
|
 0.001
0.001 |
0.040 | 0.048 | 1048 | 302 |
 0.600
0.600 |
0.700 |
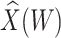
|
 0.605
0.605 |
0.049 | 0.786 | 973 | 225 |
 1.810
1.810 |
0.700 |
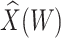
|
 1.826
1.826 |
0.076 | 1.000 | 909 | 160 |
| 0.000 | 0.400 |
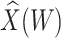
|
0.003 | 0.122 | 0.049 | 1049 | 302 |
 0.600
0.600 |
0.400 |
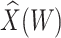
|
 0.603
0.603 |
0.152 | 0.332 | 973 | 225 |
 1.810
1.810 |
0.400 |
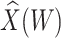
|
 1.827
1.827 |
0.232 | 0.974 | 909 | 160 |
| 0.000 | 0.700 |
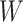
|
 0.001
0.001 |
0.020 | 0.048 | 1048 | 302 |
 0.600
0.600 |
0.700 |
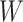
|
 0.423
0.423 |
0.024 | 0.790 | 973 | 225 |
 1.810
1.810 |
0.700 |
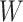
|
 1.274
1.274 |
0.035 | 1.000 | 909 | 160 |
| 0.000 | 0.400 |
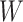
|
0.001 | 0.019 | 0.048 | 1049 | 302 |
 0.600
0.600 |
0.400 |
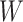
|
 0.239
0.239 |
0.023 | 0.344 | 973 | 225 |
 1.810
1.810 |
0.400 |
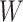
|
 0.725
0.725 |
0.032 | 0.984 | 909 | 160 |
 0.600
0.600 |
1.000 |
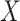
|
 0.604
0.604 |
0.031 | 0.938 | 599 | 225 |
Regression calibration recovers the true 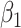 even with
even with 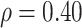 . In the Appendix, we derive the correct (marginalized) probability of
. In the Appendix, we derive the correct (marginalized) probability of 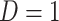 given
given 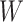 under our assumptions. We show that this curve as a function of
under our assumptions. We show that this curve as a function of 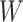 (with slope
(with slope 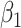 ) is virtually identical to the regression calibration curve (with the same slope
) is virtually identical to the regression calibration curve (with the same slope 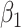 ), irrespective of
), irrespective of 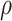 . This fidelity explains the excellent recovery of
. This fidelity explains the excellent recovery of 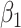 with regression calibration. Regression calibration does become more biased as
with regression calibration. Regression calibration does become more biased as 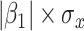 increases, but for these simulations,
increases, but for these simulations, 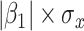 is relatively small, Follmann and others (1999).
is relatively small, Follmann and others (1999).
Intuitively, as 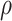 decreases estimation becomes more difficult. For
decreases estimation becomes more difficult. For 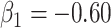 , the variance for
, the variance for 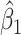 under regression calibration is 0.024, 0.049, and 0.152 for
under regression calibration is 0.024, 0.049, and 0.152 for 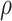 = 1.00, 0.70, and 0.40, respectively. So the variance roughly doubles as
= 1.00, 0.70, and 0.40, respectively. So the variance roughly doubles as 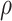 goes from 1.00 to 0.70 and triples as
goes from 1.00 to 0.70 and triples as 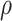 goes from 0.70 to 0.40. In contrast, with direct use of
goes from 0.70 to 0.40. In contrast, with direct use of 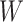 the variance of
the variance of 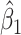 stays constant at about 0.023 or 0.024 for all
stays constant at about 0.023 or 0.024 for all 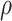 with
with 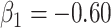 . The use of
. The use of 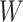 in lieu of
in lieu of 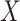 , of course, leads to a smaller estimate of
, of course, leads to a smaller estimate of 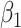 but this is counterbalanced by the smaller variance. The upshot is that the power for testing
but this is counterbalanced by the smaller variance. The upshot is that the power for testing 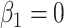 is nearly identical for regression calibration and direct use of
is nearly identical for regression calibration and direct use of 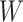 for all scenarios.
for all scenarios.
In the Appendix, we provide additional simulations with sample sizes 1/10th and 10 times that of Table 1. The three approaches have less power and more variability with the smaller sample size and the opposite with larger sample size. Regression calibration continues to be nearly unbiased for the larger sample size, though it has some bias for the smaller sample size.
The above simulations were conducted under the standard assumptions of the test negative design. In the final row, we set 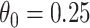 while
while 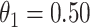 so that vaccinees with the control disease have half the arrival probability of the vaccinated cases, which violates the standard test negative assumption. We set
so that vaccinees with the control disease have half the arrival probability of the vaccinated cases, which violates the standard test negative assumption. We set 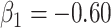 and evaluate
and evaluate 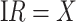 so that the second line of Table 1, which has
so that the second line of Table 1, which has 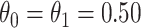 provides a reference. As expected, we see the estimate of
provides a reference. As expected, we see the estimate of 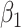 is still unbiased, though the variance for
is still unbiased, though the variance for 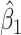 is slightly increased due to fewer control vaccinees.
is slightly increased due to fewer control vaccinees.
4.2. Waning immune response
In this subsection, we provide a brief evaluation of the model (2.11). To focus on the issue of antibody decay, we assume that  is measured without error, and that everyone with either disease arrives at a clinic. We specify the instantaneous conditional risk for control disease using a Weibull hazard,
is measured without error, and that everyone with either disease arrives at a clinic. We specify the instantaneous conditional risk for control disease using a Weibull hazard,
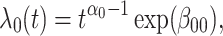 |
where 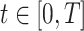 is the time since start of the test negative study. When
is the time since start of the test negative study. When 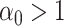 (
(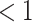 ) the hazard is increasing (decreasing). The instantaneous conditional risk of case disease is specified as
) the hazard is increasing (decreasing). The instantaneous conditional risk of case disease is specified as
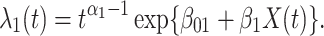 |
For 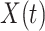 , we specify linear antibody decay according to a random effects model. For person
, we specify linear antibody decay according to a random effects model. For person 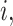 the decay is
the decay is
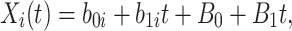 |
where 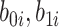 are independent normals with mean 0 and standard deviations
are independent normals with mean 0 and standard deviations 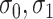 , respectively. The parameter
, respectively. The parameter 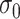 reflects both natural variation in immune response to vaccination as well as variation in vaccination times as those vaccinated long ago would tend to have smaller
reflects both natural variation in immune response to vaccination as well as variation in vaccination times as those vaccinated long ago would tend to have smaller 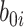 .
.
We generate times to case and control disease by the inverse cumulative distribution function method where we generate 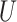 , a uniform [0,1] random variable and then determine the disease arrival time
, a uniform [0,1] random variable and then determine the disease arrival time 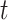 that solves
that solves 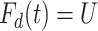 for
for 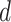 = 0,1. For the case disease, this is solved by numerically minimizing
= 0,1. For the case disease, this is solved by numerically minimizing 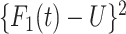 . For subjects who experience both case and control events, we use the first and discard the second. We set
. For subjects who experience both case and control events, we use the first and discard the second. We set 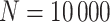 vaccinees and follow subjects until we observe 1000 first events.
vaccinees and follow subjects until we observe 1000 first events.
For all scenarios, we set 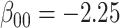 , (
, (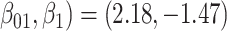 , and
, and 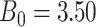 . Our reference case is
. Our reference case is 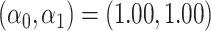 , or constant risk of each disease and
, or constant risk of each disease and 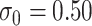 ,
,  . We then vary these parameters to see their impact on performance. The hazards and distribution of arrival times are graphed in the Supplementary material available at Biostatistics online.
. We then vary these parameters to see their impact on performance. The hazards and distribution of arrival times are graphed in the Supplementary material available at Biostatistics online.
We evaluate two different modeling strategies for (2.11). The first is to treat 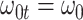 as fixed with no dependence on time. The second is to evaluate five models and pick the one with the best Akaike Information Criterion (AIC). The five models are to treat
as fixed with no dependence on time. The second is to evaluate five models and pick the one with the best Akaike Information Criterion (AIC). The five models are to treat 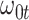 as constant (1), linear in
as constant (1), linear in 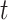 (2), quadratic in
(2), quadratic in 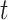 (3), linear in
(3), linear in 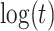 (4), and quadratic in
(4), and quadratic in 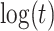 . This second strategy is meant to mimic how a data analyst might address a non-constant
. This second strategy is meant to mimic how a data analyst might address a non-constant 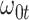 . To summarize performance, we calculate the sample mean and variance for the estimate of
. To summarize performance, we calculate the sample mean and variance for the estimate of 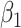 under modeling strategies 1 and 2. We also report the modal model choice of strategy 2 and the number of times that modal choice was chosen. Additionally, we report the number of cases and controls for the last data set.
under modeling strategies 1 and 2. We also report the modal model choice of strategy 2 and the number of times that modal choice was chosen. Additionally, we report the number of cases and controls for the last data set.
Table 2 presents the summary results. We see that both modeling strategies 1 and 2 recover 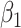 very well for scenario 1, our reference, and scenario 2, where
very well for scenario 1, our reference, and scenario 2, where 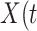 ) has more variability and a steeper mean decline. Both scenarios 1 and 2 have constant hazards for both case and control disease. For non-constant hazards, modeling strategy 1 is biased with bias of around
) has more variability and a steeper mean decline. Both scenarios 1 and 2 have constant hazards for both case and control disease. For non-constant hazards, modeling strategy 1 is biased with bias of around 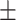 10%. Modeling strategy 2 has minimal bias for non-constant hazards and picks
10%. Modeling strategy 2 has minimal bias for non-constant hazards and picks 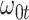 as a linear function of
as a linear function of 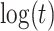 in about 70–80% of the simulations using the AIC criterion.
in about 70–80% of the simulations using the AIC criterion.
Table 2.
Performance of logistic regression based strategies for dealing with the nuisance function 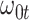 . Strategy 1: Fix
. Strategy 1: Fix 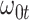 at
at 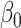 . Strategy 2: Choose
. Strategy 2: Choose 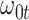 based on AIC. The sample average and variance over the simulations are given for
based on AIC. The sample average and variance over the simulations are given for 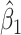 under the two strategies. AIC choice reports the modal choice for strategy 2 while
under the two strategies. AIC choice reports the modal choice for strategy 2 while 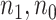 are the number of cases and controls for the last simulated dataset. The true value of
are the number of cases and controls for the last simulated dataset. The true value of 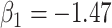 . Each row is based on 1000 simulated studies.
. Each row is based on 1000 simulated studies.
| Strategy 1 | Strategy 2 | AIC | # times | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Scenario |
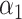
|
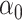
|
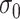
|
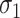
|
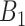
|
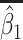
|
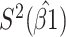
|
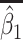
|
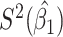
|
Choice | Chosen |
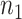
|
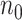
|
| 1 | 1.00 | 1.00 | 0.5 | 0.1 |
 0.5
0.5 |
 1.47
1.47 |
0.0204 |
 1.47
1.47 |
0.0209 | 1 | 735 | 426 | 574 |
| 2 | 1.00 | 1.00 | 1.0 | 0.5 |
 1.0
1.0 |
 1.48
1.48 |
0.0100 |
 1.48
1.48 |
0.0102 | 1 | 693 | 629 | 371 |
| 3 | 1.25 | 0.75 | 0.5 | 0.1 |
 0.5
0.5 |
 1.59
1.59 |
0.0268 |
 1.48
1.48 |
0.0271 | 4 | 738 | 235 | 765 |
| 4 | 0.75 | 1.25 | 0.5 | 0.1 |
 0.5
0.5 |
 1.35
1.35 |
0.0213 |
 1.48
1.48 |
0.0239 | 4 | 780 | 618 | 382 |
| 5 | 1.50 | 0.50 | 0.5 | 0.1 |
 0.5
0.5 |
 1.61
1.61 |
0.0858 |
 1.49
1.49 |
0.0927 | 4 | 770 | 66 | 934 |
| 6 | 0.50 | 1.50 | 0.5 | 0.1 |
 0.5
0.5 |
 1.29
1.29 |
0.0324 |
 1.48
1.48 |
0.0392 | 4 | 828 | 829 | 171 |
Strictly speaking, (2.10) obtains under a “rare” disease assumption. In the above simulations, 10% of the vaccinees acquired disease with no noticeable bias. We did further simulations and did observe about 7% bias for scenario 2 with 30% disease acquisition.
5. Example
In the 2018–2020, outbreak of Ebola in the Democratic Republic of the Congo a ring vaccination campaign was conducted. Local surveillance teams kept track of new Ebola cases which were then relayed to a WHO vaccination team that vaccinated the contacts and contacts of contacts of the index case. Several hundred thousand at risk people were thus vaccinated. Following vaccination, the vaccinees were monitored and encouraged to visit an Ebola transit/treatment center once any symptoms developed. The ring campaign used the rVSV vaccine which is estimated to have quite high efficacy though breakthrough infections occur, WHO (2019) and Henao-Restrepo and others (2015). The durability of the vaccine and the impact of immune response on risk of disease are unknown. Prospectively collecting samples in all vaccinees for correlates analysis has not been done.
To illustrate our approach, we simulate a dataset meant to loosely approximate a large Ebola ring vaccination campaign with a vaccine whose substantial efficacy is modulated by immune response. We assume that each person’s immune response is relatively stable over time. We assume 150 000 at risk subjects are vaccinated and generate case and control infections as in the previous section with 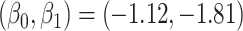 . We set
. We set 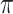 , the conditional risk of control disease (e.g., malaria) equal to 0.005 and assume that vaccinated subjects arrive at the transit center with fixed probability
, the conditional risk of control disease (e.g., malaria) equal to 0.005 and assume that vaccinated subjects arrive at the transit center with fixed probability 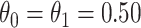 , as one might expect from regular monitoring and encouragement. Note that with regular monitoring of all subjects in a ring, the assumption that
, as one might expect from regular monitoring and encouragement. Note that with regular monitoring of all subjects in a ring, the assumption that 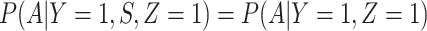 for vaccinees may hold, thus even if rVSV does modify disease, the estimated risk of disease as a function of immune response should be unbiased.
for vaccinees may hold, thus even if rVSV does modify disease, the estimated risk of disease as a function of immune response should be unbiased.
To provide additional context, we assume 300 000 unvaccinated subjects are at risk (e.g., have first or second degree contact with an Ebola case) and generate case infections with probability 0.01. We generate control infections from this set with conditional probability 0.005 just as for the vaccinated. We assume that the unvaccinated contacts arrive with probability 0.25 for both case and control disease, irrespective of severity. Finally, we assume that 20% of the arrivals occur within 35 days of contact with an Ebola case. For vaccinees, this should ensure a relatively stable vaccine-induced immune response at arrival.
For this single simulated dataset, a total of 114 unvaccinated contacts of Ebola cases arrived at least 35 days post contact. The proportion with Ebola Virus Disease (EVD) was 0.69. In contrast, 127 vaccinated contacts of an Ebola case arrived at least 35 days post contact with an EVD proportion of 0.28. Thus the overall test negative VE for these late arrivals is estimated as
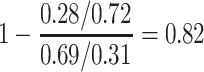 |
The above VE estimate is unbiased under the standard test negative assumptions that vaccinees (unvaccinated) have fixed probability 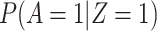 (
(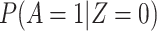 ) of arriving at a transit center, irrespective of true disease status, disease severity, and equal exposure of vaccinees and unvaccinated. This latter assumption may be questioned in a ring study where the vaccinees are all known contacts of a case.
) of arriving at a transit center, irrespective of true disease status, disease severity, and equal exposure of vaccinees and unvaccinated. This latter assumption may be questioned in a ring study where the vaccinees are all known contacts of a case.
Figure 2 displays the data, jittered for the unvaccinated for whom we set 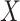 to 0, along with the fitted logistic regression model
to 0, along with the fitted logistic regression model 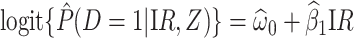 where
where 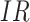 is the immune response covariate,
is the immune response covariate, 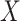 ,
, 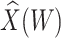 , or
, or 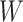 . For the unvaccinated, we draw a dashed line at the observed Ebola disease rate of 0.69 for reference. Table 3 provides the estimated parameters. We see that, as expected, using
. For the unvaccinated, we draw a dashed line at the observed Ebola disease rate of 0.69 for reference. Table 3 provides the estimated parameters. We see that, as expected, using 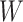 results in a smaller
results in a smaller 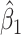 than use of
than use of 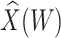 and that the Wald statistic for testing
and that the Wald statistic for testing 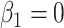 for
for 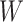 is identical to the Wald statistic based on
is identical to the Wald statistic based on 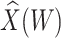 , when we use the naive standard error. We also provide the bootstrap standard error and Wald statistic for regression calibration based on 1000 resamples. The resultant standard errors are slightly larger than the naive standard errors. The two-sided p-values for testing an effect of
, when we use the naive standard error. We also provide the bootstrap standard error and Wald statistic for regression calibration based on 1000 resamples. The resultant standard errors are slightly larger than the naive standard errors. The two-sided p-values for testing an effect of 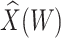 and
and 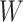 on disease are, respectively, 0.02 and 0.01. Recall that the estimates of
on disease are, respectively, 0.02 and 0.01. Recall that the estimates of 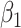 are unbiased even if the arrival probability for vaccinees depends on latent severity, provided the vaccine has an all-or-none mechanism. Estimates of
are unbiased even if the arrival probability for vaccinees depends on latent severity, provided the vaccine has an all-or-none mechanism. Estimates of 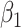 are also unbiased even if the vaccine modifies latent disease severity, provided the arrival probability is the same for all vaccinees with the case disease.
are also unbiased even if the vaccine modifies latent disease severity, provided the arrival probability is the same for all vaccinees with the case disease.
Fig. 2.

Simulated data to illustrate an immune correlates analysis based on a test negative design for the 2018–2020 Ebola outbreak in the DRC. Unvaccinated subjects’ data are given by jittered open circles near x-axis near 0 and whose Ebola disease rate of 0.69 is given by the dashed line. Vaccinated subjects’ data are given by diamonds. The estimated probability of disease for vaccinees is given by the solid curve. The three panels correspond to using 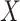 ,
, 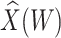 , and
, and 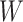 as the immune response, respectively.
as the immune response, respectively.
Table 3.
Parameter estimates for a simulated data set with 114 unvaccinated and 127 vaccinated subjects. We evaluate 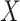 ,
, 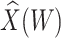 , and
, and 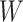 as the immune response covariate.
as the immune response covariate.
IR = 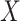
|
IR=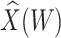
|
IR= 
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| parm | est | se | Wald | est | se
|
Wald
|
se
|
Wald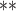
|
est | se | wald |
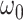
|
3.68 | 1.28 | 2.86 | 3.28 | 1.68 | 1.95 | 1.70 | 1.93 | 2.07 | 1.20 | 1.72 |
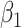
|
 1.64
1.64 |
0.46 |
 3.54
3.54 |
 1.45
1.45 |
0.58 |
 2.49
2.49 |
0.60 |
 2.40
2.40 |
 1.04
1.04 |
0.42 |
 2.49
2.49 |
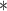 Naive standard errors,
Naive standard errors,  Bootstrap standard errors
Bootstrap standard errors
To help interpret the magnitude of the effect of the imputed immune response on risk of disease, we can compare the ratio of the risk of disease at the 10th versus 90th percentile of the observed distribution of 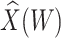 . These percentiles are 2.51 and 3.39, respectively, with associated probabilities of disease of 0.42 and 0.16 for these late vaccinated arrivals. Thus the risk is roughly 2.5 times greater at the 10th versus 90th percentile of
. These percentiles are 2.51 and 3.39, respectively, with associated probabilities of disease of 0.42 and 0.16 for these late vaccinated arrivals. Thus the risk is roughly 2.5 times greater at the 10th versus 90th percentile of 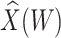 indicating a substantial effect of antibody on EVD risk.
indicating a substantial effect of antibody on EVD risk.
6. Discussion
This research arose from the design of an immune correlates study using test negative data for the 2018–2020 Democratic Republic of the Congo (DRC) Ebola outbreak. The main contribution of this article is the idea that an estimate of the effect of the relevant immune response 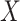 on the risk of disease can be estimated by using an irrelevant immune response
on the risk of disease can be estimated by using an irrelevant immune response 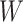 . We use the proxy
. We use the proxy 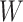 to impute
to impute 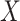 based on a prediction model that is estimated in vaccinated controls using samples collected upon arrival at a clinic. The method extends to settings where antibody substantially decays over time. Simulations demonstrated the feasibility of the proposed methods.
based on a prediction model that is estimated in vaccinated controls using samples collected upon arrival at a clinic. The method extends to settings where antibody substantially decays over time. Simulations demonstrated the feasibility of the proposed methods.
This approach applies beyond Ebola. The key requirement is that the vaccine of interest produce irrelevant immune responses that will be unaffected by the disease of interest, and that these immune responses are correlated with the immune response of interest. One such application is for extremely rare diseases such as the prevention of Zika-induced congenital birth defects. Suppose Puerto Rico begins Zika vaccination in 2025 and an outbreak sweeps the island in 2040. Since Zika-induced birth defects are rare, a prospective immune correlates design would require long-term storage of samples from hundreds of thousands of women just after vaccination. Furthermore, the values collected in 2025 onward would not address the question of the immune response at Zika exposure in 2040. But using the methods of this article we could sample cases (mothers of children with birth defects who are antibody positive for non-vaccine Zika antigens) and controls (mothers of children with birth defects who are antibody negative for non-vaccine Zika antigens). We could then apply a test negative design to assess overall VE and also perform an immune correlates analysis with 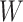 and
and 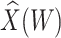 , providing that a
, providing that a 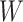 that predicts
that predicts 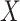 were available.
were available.
We focused on 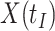 or the immune response proximal to infection. However, traditional immune correlates analyses focus on
or the immune response proximal to infection. However, traditional immune correlates analyses focus on 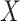 shortly after vaccination. Our methods for time constant immune response directly apply as here
shortly after vaccination. Our methods for time constant immune response directly apply as here 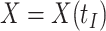 . If antibody decay occurs, one could in principle apply (2.10) with
. If antibody decay occurs, one could in principle apply (2.10) with 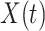 replaced by
replaced by 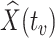 , the predicted value of
, the predicted value of 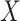 shortly after vaccination. This would require the development of models to predict
shortly after vaccination. This would require the development of models to predict 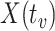 .
.
The biggest challenge in this approach is whether the biology is amenable. Some vaccines won’t induce a 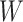 at all. Other times, even if
at all. Other times, even if 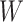 is induced the correlation may be too weak for this method to work. This may be more of a problem with pre-existing immunity to either the antigens of the vaccine or the vector. On the other hand, it is not really required that
is induced the correlation may be too weak for this method to work. This may be more of a problem with pre-existing immunity to either the antigens of the vaccine or the vector. On the other hand, it is not really required that 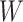 be an immune response to the vector. What is required is the existence of covariates
be an immune response to the vector. What is required is the existence of covariates 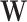 that predict
that predict 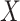 among vaccinees and that
among vaccinees and that 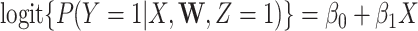 . Development of such methods is left to future work.
. Development of such methods is left to future work.
Supplementary Material
Acknowledgments
This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). We thank Jing Qin and Michael Fay for helpful comments.
Conflict of Interest: None declared.
7. Software
The code to reproduce the example is given at https://github.com/follmand/Test-Negative-Immune-Correlates.
Supplementary materials
Supplementary material is available at http://biostatistics.oxfordjournals.org.
References
- Boom, J. A., Tate, J. E., Sahni, L. C., Rench, M. A., Hull, J. J., Gentsch, J. R., Patel, M. M., Baker, C. J. and Parashar, U. D. (2010). Effectiveness of pentavalent rotavirus vaccine in a large urban population in the United States. Pediatrics 125, e199–e207. [DOI] [PubMed] [Google Scholar]
- Broome, C. V., Facklam, R. R. and Fraser, D. W. (1980). Pneumococcal disease after pneumococcal vaccination: an alternative method to estimate the efficacy of pneumococcal vaccine. New England Journal of Medicine 303, 549–552. [DOI] [PubMed] [Google Scholar]
- Follmann, D. A., Hunsberger, S. A. and Albert, P. S. (1999). Repeated probit regression when covariates are measured with error. Biometrics, 55, 403–409. [DOI] [PubMed] [Google Scholar]
- Fukushima, W. and Hirota, Y. (2017). Basic principles of test-negative design in evaluating influenza vaccine effectiveness. Vaccine 35, 4796–4800. [DOI] [PubMed] [Google Scholar]
- Guolo, A. (2008). A flexible approach to measurement error correction in case–control studies. Biometrics 64, 1207–1214. [DOI] [PubMed] [Google Scholar]
- Halloran, M. E., Longini, I. M. and Gilbert, P. B. (2020). Designing a study of correlates of risk for Ebola vaccination. American Journal of Epidemiology 189, 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Restrepo, A. M., Longini, I. M., Egger, M., Dean, N. E., Edmunds, W. J., Camacho, A., Carroll, M. W., Doumbia, M, Draguez, B., Duraffour, S.et al. (2015). Efficacy and effectiveness of an RVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the guinea ring vaccination cluster-randomised trial. The Lancet 386, 857–866. [DOI] [PubMed] [Google Scholar]
- Jackson, M. L. and Nelson, J. C. (2013). The test-negative design for estimating influenza vaccine effectiveness. Vaccine 31, 2165–2168. [DOI] [PubMed] [Google Scholar]
- Kennedy, S. B., Neaton, J. D., Lane, H. C., Kieh, M. W. S., Massaquoi, M. B. F., Touchette, N. A., Nason, M. C., Follmann, D. A., Boley, F. K., Johnson, M. P.et al. (2016). Implementation of an Ebola virus disease vaccine clinical trial during the Ebola epidemic in Liberia: design, procedures, and challenges. Clinical Trials, 13, 49–56. [DOI] [PubMed] [Google Scholar]
- Lewnard, J. A., Tedijanto, C., Cowling, B. J. and Lipsitch, M. (2018). Measurement of vaccine direct effects under the test-negative design. American Journal of Epidemiology 187, 2686–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin, S. A. and Gilbert, P. B. (2012). Nomenclature for immune correlates of protection after vaccination. Clinical Infectious Diseases 54, 1615–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poetsch, J. H., Dahlke, C., Zinser, M. E., Kasonta, R., Lunemann, S., Rechtien, A., Ly, M. L., Stubbe, H. C., Krähling, V., Biedenkopf, N.et al. (2018). Detectable vesicular stomatitis virus (VSV)-specific humoral and cellular immune responses following VSV-Ebola virus vaccination in humans. The Journal of Infectious Diseases 219, 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, L., Gilbert, P. B., Corey, L., McElrath, M. J. and Self, S. G. (2007). A framework for assessing immunological correlates of protection in vaccine trials. The Journal of Infectious Diseases 196, 1304–1312. [DOI] [PubMed] [Google Scholar]
- Rosner, B., Spiegelman, D. and Willett, W. C. (1990). Correction of logistic regression relative risk estimates and confidence intervals for measurement error: the case of multiple covariates measured with error. American Journal of Epidemiology 132, 734–745. [DOI] [PubMed] [Google Scholar]
- Sullivan, S. G., Tchetgen Tchetgen, E. J. and Cowling, B. J. (2016). Theoretical basis of the test-negative study design for assessment of influenza vaccine effectiveness. American Journal of Epidemiology 184, 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westreich, D. and Hudgens, M. G. (2016). Invited commentary: beware the test-negative design. American Journal of Epidemiology 184, 354–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. (2019). Preliminary Results on the Efficacy of rVSV-ZEBOV-GP Ebola Vaccine using the Ring Vaccination Strategy in the Control of an Ebola Outbreak in the Democratic Republic of the Congo: An Example of Integration of Research into Epidemic Response. https://reliefweb.int/report/democratic-republic-congo/preliminary-results-efficacy-rvsv-zebov-gp-ebola-vaccine-using-ring.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.