

Abstract
Correct protein folding is essential for the health and function of living organisms. Yet, it is not well understood how unfolded proteins reach their native state and avoid aggregation, especially within the cellular milieu. Some proteins, especially small, single-domain and apparent two-state folders, successfully attain their native state upon dilution from denaturant. Yet, many more proteins undergo misfolding and aggregation during this process, in a concentration-dependent fashion. Once formed, native and aggregated states are often kinetically trapped relative to each other. Hence, the early stages of protein life are absolutely critical for proper kinetic channeling to the folded state and for long-term solubility and function. This review summarizes current knowledge on protein folding/aggregation mechanisms in buffered solution and within the bacterial cell, highlighting early stages. Remarkably, teamwork between nascent chain, ribosome, trigger factor and Hsp70 molecular chaperones enables all proteins to overcome aggregation propensities and reach a long-lived bioactive state.
Keywords: Cotranslational folding, aggregation, ribosome, chaperones, kinetic trapping, free landscapes
Overview.
For most proteins, a well-folded native three-dimensional protein structure is a prerequisite for biological activity. While intrinsically disordered proteins (IDPs) are an exception, protein folding remains a fundamental process for life on earth [1]. Yet, it is not well understood how unfolded proteins achieve their functional native state, despite the enormous number of conformations that they can potentially populate. The cell machinery ensures that predominantly native states are generated and that thermodynamically stable – and often undesirable – aggregated states are not. After being generated, many native and aggregated states of bacterial proteins remain kinetically trapped from each other, under physiologically relevant conditions. Given that the early stages of protein life are absolutely critical for the success of this process, this review will summarize current knowledge on protein folding in buffered solution and in the cell, including the early stages of protein’s life. Successful folding in the complex cellular environment has clearly evolved as a team effort and is often achieved through the combined involvement of many molecular players, including the ribosome and a variety of chaperones. This review focuses on three of these major players in bacteria, namely the ribosome, and the molecular chaperones trigger factor and Hsp70.
Protein folding and misfolding are intimately connected processes.
The practical consequences of aberrant protein folding are often severe and undesirable. For instance, protein overexpression in bacteria frequently leads to the formation of insoluble aggregates known as inclusion bodies. The latter species are difficult and expensive to disaggregate and to convert to the native state. This challenge often renders protein production in the basic-science, biotechnology, pharmaceutical and biomaterials settings extremely costly and inefficient [2, 3]. In medicine, protein misfolding and aggregation in higher organisms is often associated with deadly maladies known as proteinopathies, including brain disorders like Parkinson’s, Huntington’s, and Alzheimer’s disease [4, 5]. In summary, understanding how proteins fold is necessary to advance basic science, biotechnology and human health.
Protein folding research encompasses two major topics: the prediction of native structure from amino acid sequence and the mechanism by which proteins attain their native state. Significant advances were recently made in protein structure prediction. For instance, in the 2020 protein structure prediction challenge known as Critical Assessment of Structure Prediction (CASP), the software AlphaFold 2 (from the DeepMind artificial-intelligence company) predicted structures that matched the experimental structure of nearly two-thirds of the target proteins. This result is comparable, yet even better, to the predictions achieved with other protein-structure prediction programs (e.g., RoseTTaFold) [6] and to an earlier AlphaFold version [7, 8]. The AlphaFold family is based on a large structural-database and on sophisticated deep learning tools [7, 9]. Conveniently, AlphaFold 2 predictions have recently been integrated into online resources devoted to protein sequence and biochemical/physical properties, e.g., UniProt [10, 11].
The second protein folding topic is the mechanism by which proteins attain their native state. In addition to enhancing basic knowledge, understanding protein folding mechanisms is a prerequisite for comprehending and controlling the relative flux through the parallel kinetic paths that lead to either folding or aggregation in Nature. Mechanistic insights into the overall folding/misfolding/aggregation process promise to yield invaluable insights to design and experimentally generate next-generation aggregation-free biomaterials, biosensors and drugs, as well as to devise better strategies to combat a variety of deadly proteinopathies [12–14].
Refolding of small purified proteins into buffer: experimental studies.
Soluble and correctly folded proteins typically bury a significant fraction (60%–80%) of their nonpolar residues inside the core to minimize exposure to the hydrophilic environment of typical intracellular media [15–17]. Thus, an essential function of protein folding is the intramolecular burial of most nonpolar amino-acid side chains, rendering them inaccessible to mostly nonpolar/nonpolar-type interactions with other proteins. The latter interactions would eventually lead to intermolecular aggregation via the hydrophobic effect [18]. Research over the past decades sought to explain how small proteins achieve their soluble functional native structure. More recently, investigations have also explored how native structure formation is coupled with the avoidance of the pervasive risk of protein aggregation.
In the 1960s, Christian Anfinsen showed that ribonuclease A and other proteins fold reversibly from a chemically denatured unfolded state [19]. These results led Anfinsen to propose the well-known “thermodynamic hypothesis,” which states that the native state has the lowest free energy, out of all possible conformations. Anfinsen showed that reversible protein folding is fully determined by amino-acid sequence and environmental conditions, and that the folding process is under thermodynamic control [20]. In 1969, Cyrus Levinthal argued that because there are so many possible protein conformations, thermodynamic control is not sufficient for proteins to attain a folded state on biologically relevant timescales via a random conformational search [21]. On the other hand, it soon became clear that Levinthal’s paradox [22, 23] could be resolved if proteins were to fold via specific single or multiple pathways, progressively narrowing the accessible conformational space.
More recent experimental work identified the folding pathways of a variety of single-domain globular monomeric proteins [24–28]. In vitro folding has typically been studied upon refolding proteins from chemically or thermally denatured states. Upon refolding from denaturant for instance, proteins typically begin as a fairly expanded unfolded state bearing little or no secondary structure, and finally attain a compact, folded state bearing native secondary and tertiary structure [29, 30]. This final structure buries the nonpolar residues within the protein hydrophobic core, enabling the protein to be soluble within the hydrophilic environment of the cell.
The simplest folding pathways follow two-state mechanisms and include only the unfolded and folded states and a single transition state. Small (50–60 residues) single-domain, α-helical proteins that experimentally show two-state folding can form secondary structure before chain collapse (framework mechanism) (Figure 1A). Alternatively, secondary structure formation and chain collapse may occur concurrently (nucleation-condensation mechanism) (Figure 1B) [27, 31]. For instance, engrailed homeodomain from Drosophila melanogaster (59 residues) folds via the framework mechanism, human TRF1 Myb domain (52 residues) folds via nucleation-condensation, and human c-Myb transforming protein (54 residues) folds with a mixed framework/nucleation-condensation mechanism [27, 32] In general, small proteins with higher local α-helical propensity are more likely to fold via the framework model (leading to considerable secondary structure formation preceding global chain collapse) [33, 34].
Figure 1. Overview of protein folding mechanisms upon dilution from denaturant or upon recovery from temperature jumps.
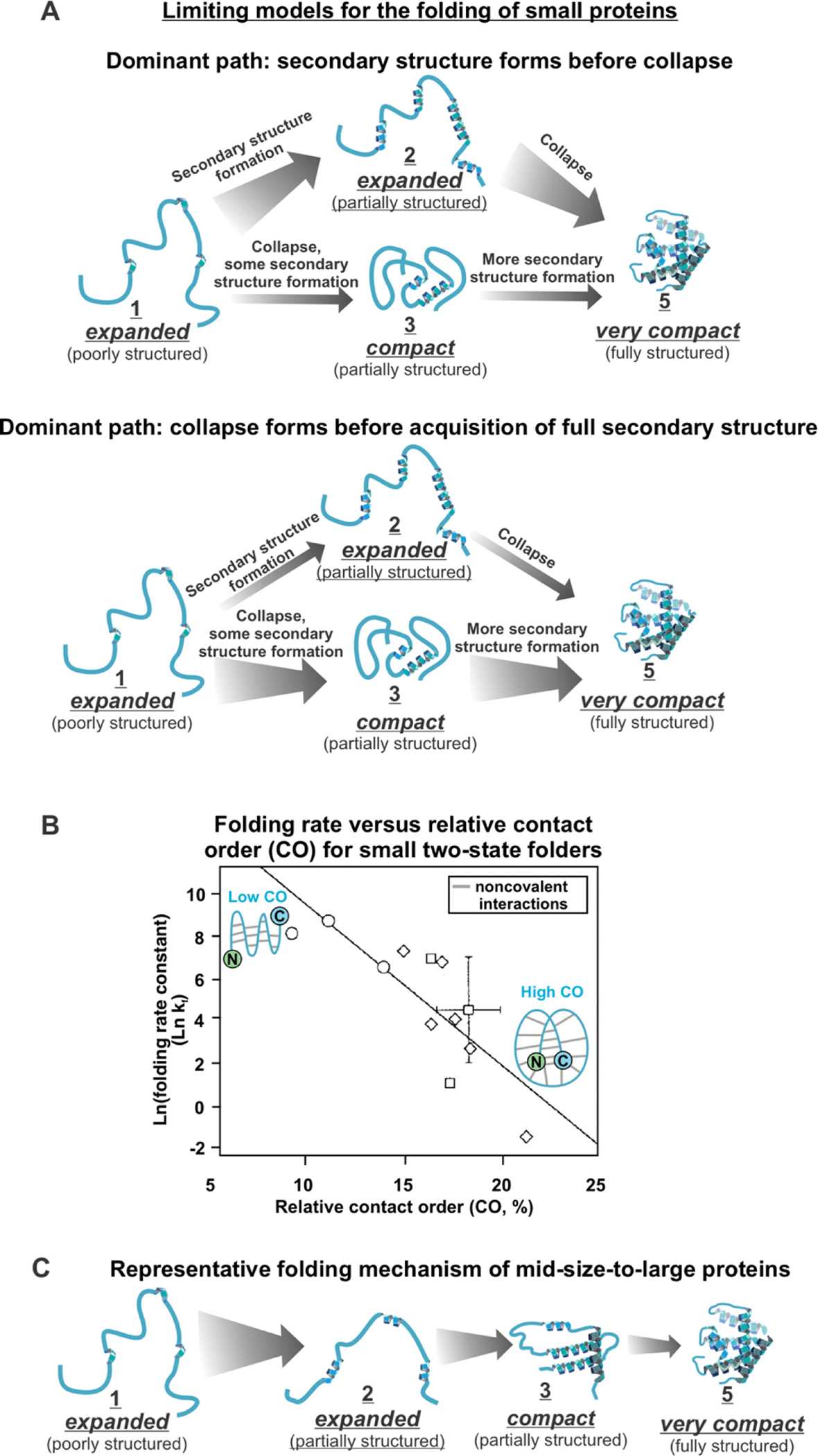
Some small (50–60 residues) proteins fold via (A) a mechanism dominated by secondary structure formation before chain collapse, or via (B) chain collapse preceding the formation of most secondary structure. (C) Plot of folding rate as a function of relative contact order (CO) for small (60–110 residues) two-state folding proteins. The graph is reprinted with permission from Figure 1A of J. Mol. Biol., 277, Plaxco, K. W.; Simons, K. T.; Baker, D., 985–994, Copyright (1998) [35]. (D) Larger proteins (> 60 residues) fold via more complex folding mechanisms, often including folding intermediates.
Remarkably, proteins were found to fold more slowly when their native state bears a greater number of long-range interactions. The latter are defined as noncovalent contacts between residues far away in sequence. This trend is described by the relative contact order (CO) parameter, which is defined as [35, 36]
| (1) |
where L is the total number of residues, N is the total number of noncovalent contacts between nonhydrogen atoms, and ΔSi,j is the sequence separation between interacting residues i and j [36]. As CO increases, the speed of protein folding decreases, for small two-state folding proteins (Figure 1C) [24, 35–37].
Refolding of mid-size to large purified proteins into buffer: experimental studies.
Most proteins in the cell are larger than 100 residues. For instance, the average protein size is 360 and 530 residues in prokaryotes and eukaryotes, respectively [38]. In addition, a significant fraction of proteins have multiple domains. For instance, 40–65% of proteins in prokaryotes and 65–80% of proteins in eukaryotes have multiple domains [39]. Larger single- and multi-domain proteins have, by definition, a large number of degrees of freedom and may experience more complex folding paths [40, 41].
Large proteins are more likely to have experimentally detectable folding intermediates [24, 40, 42]. While typical intermediates are on-path to the native state [43–55], a few off-path intermediates have also been identified [56]. In general, unequivocally identifying folding intermediates can be challenging. For instance, it was reported that transient aggregates can sometimes be mistaken for folding intermediates [57]. Two well-studied mid-size proteins are sperm whale and horse apomyoglobin, each of which has 153 residues and a multistate folding mechanism with experimentally-detectable compact intermediates (Figure 1D) [58–60]. Some of these intermediates are obligatory [61] and have a partially-folded structure bearing quasi-native features and lacking a few structural elements [58, 59].The major apomyoglobin folding intermediate later resolves to a fully native conformation via slight conformational rearrangements within the early-folding A, B, G and H helices, which then enable final native-structure formation during the later stages of folding [62].
One model for the folding of medium-to-large proteins (100–370 residues) is the foldon model, according to which proteins fold via progressively populating small independent cooperative units known as foldons (Figure 2) [28, 63–66]. Foldons are folding intermediates with some regions bearing native or quasi-native structure and other region unfolded or only partially folded. The presence of foldons naturally limits the dimensionality of the conformational search, providing a simple justification for how Nature avoids exhaustive sampling (Levinthal’s paradox) during protein folding [28, 67, 68]. Examples of proteins that fold via foldons include: cytochrome c (104 residues) [69], RnaseH (155 residues) [70], apoflavodoxin (179 residues) [71], apomyoglobin (153 residues) [72], staphylococcal nuclease (149 residues) [73] and the following two-domain proteins: maltose binding protein (370 residues) [74] and DapA (292 residues) [75].
Figure 2. Foldons and protein folding mechanisms.
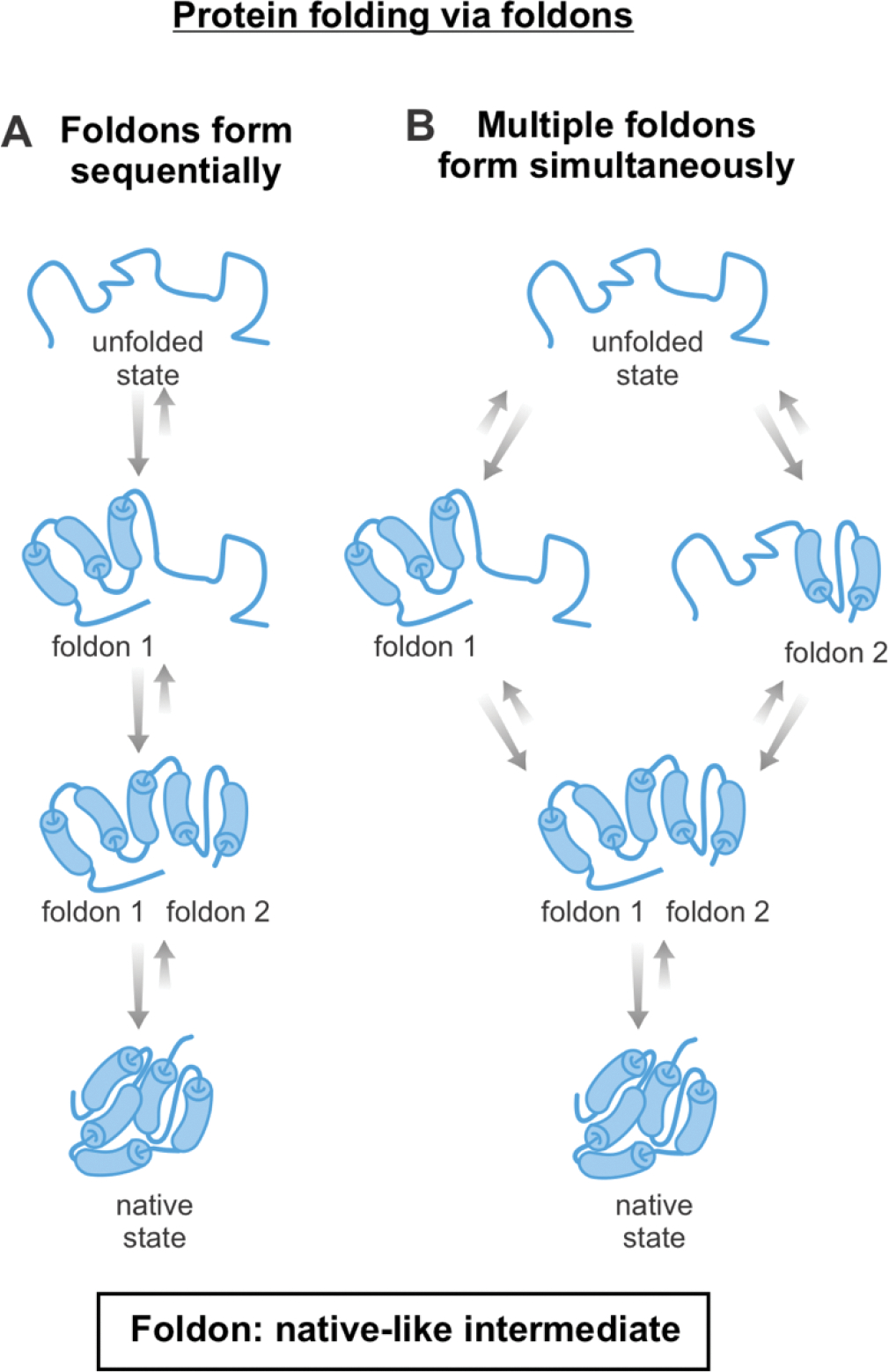
Scheme illustrating how proteins may fold via native-like intermediates denoted as foldons, which are generated either (A) sequentially or (B) in parallel.
Similar to foldons, some multi-domain proteins fold successfully when each domain folds independently [76]. Some multi-domain proteins show independent domain folding, including titin [77], fibronectin [78], and the double B domain of protein A (BBdpA) [79, 80]. Other proteins, including spectrin [81, 82], phosphoglycerate kinase [83], and the ankyrin repeat domains [80, 84] do not exhibit independently-folding domains, yet they are able to successfully refold from denaturant [39].
Folding of large purified proteins: experimental studies in vitro and in cell-like environments.
Many medium- to large-size proteins do not attain a 100% population of native state upon refolding from denaturant and give rise to some soluble or insoluble aggregates. Several examples of proteins that are known to form aggregates upon in vitro refolding from denaturant are shown in Table 1. This class of biomolecules includes proteins ranging from 153–550 residues, single and multi-domain proteins, monomeric proteins, and protein complexes. Without assistance from “folding helpers”, these and likely many other proteins are unable to fully populate their native state in solution, upon refolding into buffer at physiologically relevant temperature and pH.
Table 1.
List of proteins known to undergo aggregation upon in vitro refolding from denaturant.
| Protein name | Number of residues) | Size of monomer (kDa) | Number of domains | Monomer or complex | Reference |
|---|---|---|---|---|---|
| Luciferase | 550 | 60 | 2 | monomer | [259] |
| Rhodanese | 293 | 33 | 2 | monomer | [324] |
| Rubisco | 474 (large subunit), 122 (small subunit) | 520 total, 50 for large subunit, 15 for small subunit | 1 | hexadecamer (8 small subunits, 8 large subunits) | [325] |
| Apomyoglobin | 153 | 17 | 1 | monomer | [326] |
| Galactitol-1-phosphate 5-dehydrogenase | 346 | 37.4 | 2 | tetramer | [109] |
| Glutamate decarboxylase alpha | 466 | 52.7 | 3 | hexamer | [109] |
| Threonyl-tRNA synthetase (ThrRS) | 642 | 74 | 4 | dimer | [109] |
| 5,10-methylenetetrahydrofolate reductase | 296 | 33.1 | 1 | tetramer | [109] |
| S-adenosylmethionine synthetase | 384 | 41.8 | 3 | tetramer | [109] |
| Dihydrodipicolinate synthase (DHDPS) | 292 | 31.3 | 2 | tetramer | [109] |
| Tagatose-1,6-bisphosphate aldolase gatY (TBPA) | 286 | 30.8 | 1 | monomer | [109] |
| Tryptophanase | 471 | 52.8 | 2 | tetramer | [327] |
| α1-antitrypsin | 418 | 44.4 | 1 | monomer | [328] |
One key parameter that facilitates misfolding and aggregation over folding during refolding from denaturant and upon release from the ribosome in the cell is a slower folding rate than aggregation rate. Small (<50 residues) two-state folders tend to fold quickly, with folding rate constants (kf) greater than 12,000 s−1 (Figure 3A, Supplementary Table S1) [85]. Yet, many larger two-state proteins fold more slowly, and some two-state folders have observed kf values of less than 1 s−1. Large proteins (>200 residues) with multi-state folding mechanisms have the slowest folding rate constants, with kf values as low as 0.0004 s−1 (Figure 3B, Supplementary Table S1) [85]. Given that folding and aggregation pathways proceed in parallel, proteins that fold slowly are in general more likely to misfold and aggregate than proteins that fold rapidly [86–88]. While evolution has granted a few slow-folding proteins the ability to concurrently aggregate even more slowly (see well-behaved proteins in Figure 3) thereby defying aggregation, this is often not the case. For instance, many proteins undergo insoluble aggregate formation upon release from the ribosome in the absence of chaperones [89]. In addition, slow-folding globins undergo both folding and soluble aggregate formation upon release form the ribosome in the absence of the heme cofactor and chaperones [86].
Figure 3. Effect of size and folding mechanism on protein folding rates.
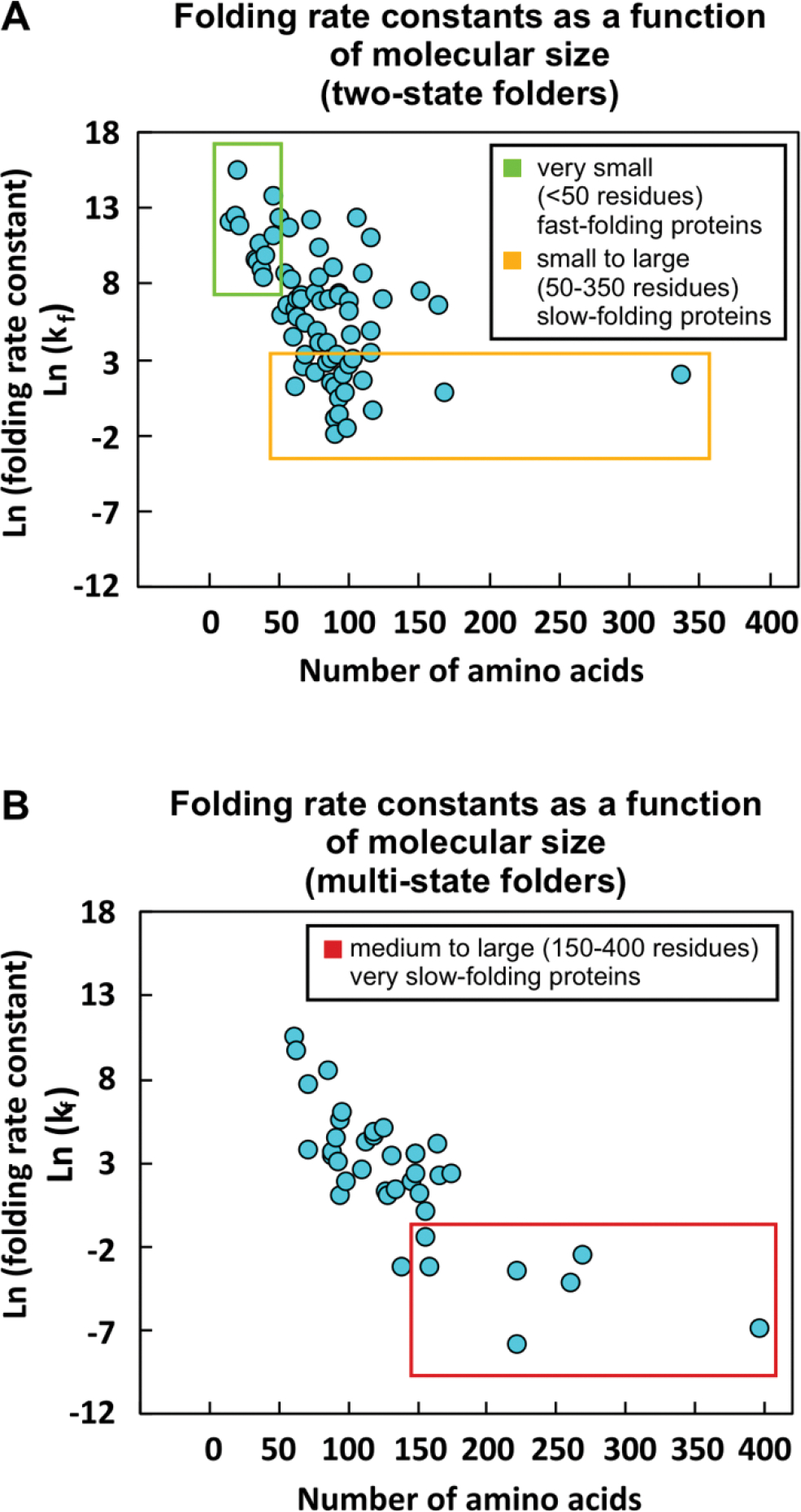
(A) Plot illustrating the dependence of protein folding rate constant (kf) on the number of residues for two-state folding proteins. Small (<50 residues) two-state proteins fold quickly with ln(kf) > 9.4 (green box). Many larger two-state folders fold more slowly (orange box). (B) Dependence of protein folding rate constant (kf) on the number of residues for multi-state folding proteins. Large (>200 residues) multi-state proteins have the slowest folding rates, with ln(kf) <−2.5 (red box). A list of the proteins and references for the data in this plot is available as Supplementary Information Table S1.
Kinetic trapping of the native state relative to aggregates: experimental studies in vitro and in cell-like environments.
After proteins attain their native state for the first time, they continue sampling thermally accessible conformational states. Structural dynamics is often important for protein function. For instance, as pictorially described in the plots of Figure 4A, some non-aggregation-prone proteins routinely fold and unfold in the cell, displaying Anfinsen-like behavior [19, 20]. On the other hand, other proteins have more complex energy landscapes, which include aggregates (Figure 4B–C). These proteins typically experience some degree of kinetic trapping to avoid aggregation under physiologically relevant conditions. Native states can be kinetically trapped relative to aggregates or aggregation-prone intermediates, as shown in Figure 4B [90–93]. Alternatively, native states can also be kinetically trapped relative to their unfolded states, hence rarely unfold during the lifetime of their host organism (Figure 4C) [94, 95].
Figure 4. Kinetic trapping of E. coli proteome relative to insoluble aggregates.
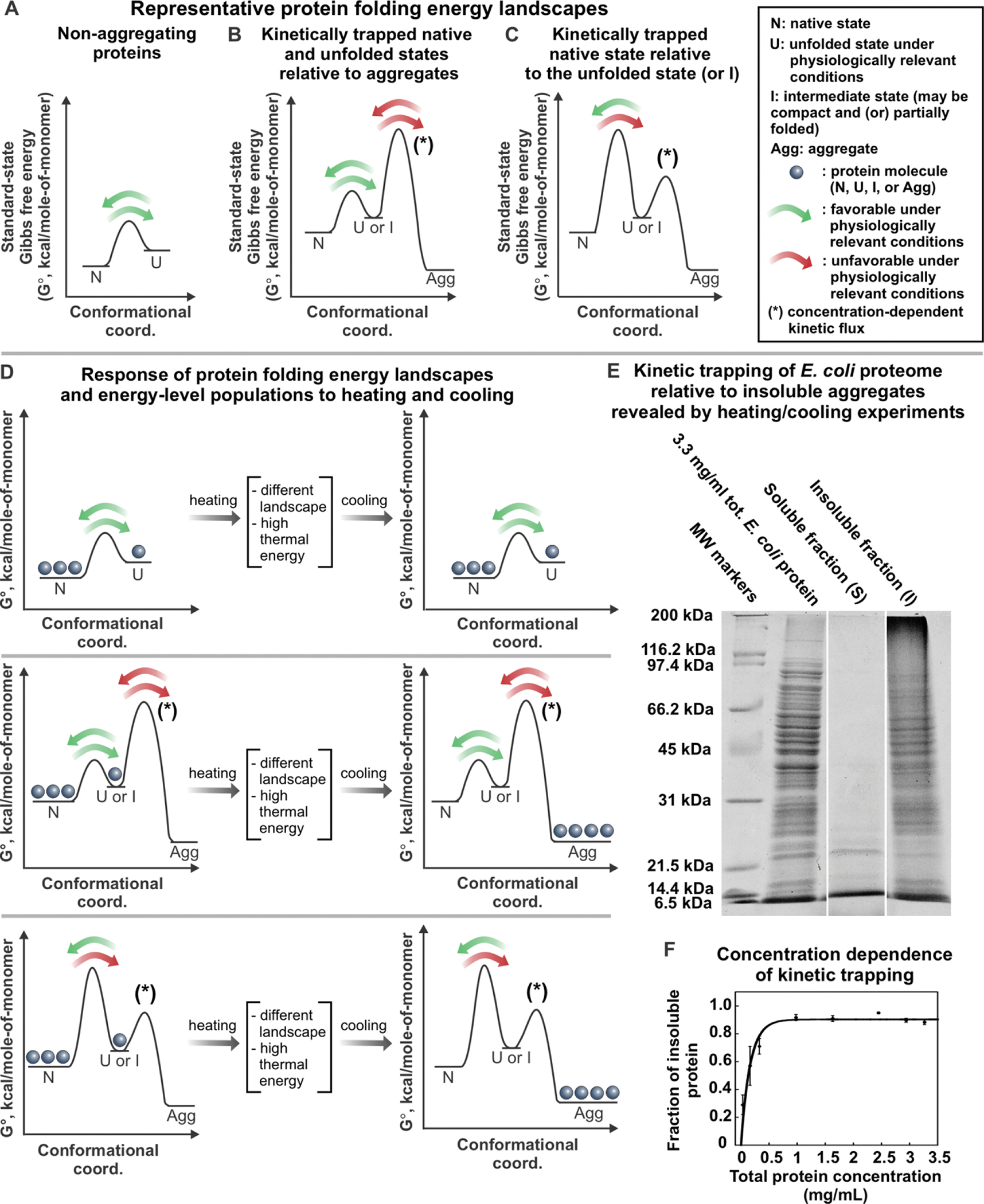
(A-C) Representative standard-state Gibbs free energy landscapes for (A) non-aggregating proteins, (B) proteins that have kinetically-trapped native and unfolded states relative to aggregated states, and (C) proteins that have kinetically-trapped native states relative to unfolded states or folding intermediates. (D) Variations in the population of proteins described in panels A, B and C, respectively, after heating and cooling. (E) SDS-Page analysis of soluble E. coli proteome upon heating for 20 hrs at 70 °C followed by slow cooling to room temperature. Sample centrifugation generated a supernatant (S) and an insoluble pellet (I), shown separately in the gel [92]. (F) Fraction of insoluble, aggregated E. coli proteome generated by procedure described above as a function of total protein concentration. The solid line is meant to guide the eye. Error bars denote the standard error for three independent experiments [92]. Panels E and F are adapted with permission from Varela, A. E.; Lang, J. F.; Wu, Y.; Dalphin, M. D.; Stangl, A. J.; Okuno, Y.; Cavagnero, S. J. Phys. Chem. B 2018, 122, 7682–7698. Copyright (2018) American Chemical Society.
Kinetic trapping of the native state relative to aggregates (Figure 4B) has been detected under physiologically relevant conditions for a number of proteins. A few eukaryotic proteins (including bovine insulin, and human β2-microgloblin, lysozyme and αβ-crystallin) are kinetically trapped and metastable relative to amyloid fibrils at pH 7 [90, 91, 96–98]. In addition, Varela et al. showed that sperm-whale apomyoglobin and most soluble proteins of the E. coli bacterium (ca. 2,246 – 2,545 proteins) are kinetically trapped relative to aggregates that are not necessarily amyloid in nature, under physiologically relevant conditions [92]. This result was found to hold at concentrations much lower than physiologically relevant concentrations [92]. In this study, kinetic trapping of bacterial proteins was demonstrated by heating the E. coli proteome, enabling it to transiently populate landscape regions inaccessible at physiological temperatures. The proteome was then slowly cooled, thus reverting back to physiologically relevant landscapes (Figure 4D). As shown in Figure 4D and 4E, after the heating-and-cooling process, most E. coli proteins form insoluble aggregates under both reducing and non-reducing conditions. Given the negligible extent of covalent protein modifications (assessed by mass spectrometry), this result demonstrates the presence of kinetic barriers that typically prevent conversion of several E. coli native proteins to the “aggregated region” of the landscape, under physiologically relevant conditions [92]. Kinetic trapping relative to insoluble aggregates occurs mostly for proteins larger than ca. 25 kDa at 0.5 – 3.5 mg/mL total protein concentration (Figure 4F) [92]. The above findings are significant because they show that many bacterial proteins have an energy landscape that includes aggregates. Native states are kinetically trapped relative to these aggregates under physiologically relevant conditions, hence they do not convert. It is common knowledge that thermally denatured proteins are particularly aggregation-prone. Indeed, some of this aggregation is known to be due to covalent protein modifications (e.g. new disulfide bridges upon boiling an egg). However, the study by Varela et al. (under reducing conditions) ruled out that the observed aggregation is a consequence of covalent modifications. The work was based on the analysis of an E. coli S100 protein mixture. Hence, its conclusions apply only within that mixture. While pure proteins like sperm whale apomyoglobin were also found to exhibit analogous behavior, future investigations on additional isolated proteins will contribute to establish the generality of the present findings.
Importantly, aggregation rates can be modulated by protein concentration and by environmental conditions that modify protein energy landscapes. Some changes in environmental conditions may even lead to protein covalent damage. For instance, an increase in protein concentration and kinetic-barrier curvature (for the aggregation rate-determining steps), as well as a decrease in barrier height, are sufficient to trigger pervasive aggregation. The latter phenomena are known to play a role in the case of deadly proteinopathies.
Major trends upon protein refolding from denaturants.
In summary, experimental studies to date show that there are some general trends in protein folding. These include: (a) a higher flux of folding via the framework model (leading to considerable secondary structure formation preceding global chain collapse) for small proteins with high local α-helical propensity, (b) highly populated folding intermediates, some of which are native-like (i.e., foldons), in proteins larger than ca. 100 residues, (c) an inverse correlation between relative contact order CO and folding rate constants for apparent 2-state folders. Finally, small (<50 residues) two-state folders fold quickly (kf > 12,000 s−1), many larger two-state folders fold more slowly (kf = 0.13 s−1 −12,000 s−1), and large (>200 residues) multi-state folders fold the slowest (kf = 0.0004–0.08 s−1) [85]. Yet, despite the above general trends, folding pathways upon refolding from denaturant and in the cell are overall quite diverse, for different protein folds [25, 27, 65].
Computational simulations of protein folding.
Computer simulations were extensively employed to characterize folding pathways and to define leading features of conformational energy landscapes. Computational approaches to simulate protein folding events, including molecular dynamics (MD) and Monte Carlo simulations and genetic algorithms, have been reviewed by Li et al [99]. The main challenge with simulating protein folding paths is that the typical 100 μs – ms timescale for this process is significantly longer than the capabilities of the traditional MD method. The first MD protein-folding simulation, carried out in 1998, required two months. This effort focused on the folding of villin, a 36-residue protein that folds on the microsecond timescale [100]. Advances in simulation algorithms and supercomputer technologies made it possible for MD methods based on unbiased empirical force fields to simulate the folding of ~100-residue proteins on the ms-timescale [99, 101]. Yet, most proteins from eukaryotes and prokaryotes are 300 amino acids or longer [38], so their folding cannot yet be simulated by unbiased MD techniques. One option is to simulate the folding of larger proteins with biased force fields that favor native contacts. This strategy is embraced by Gō models and structure-based models to simulate the folding of multi-domain proteins ranging from 150–400 residues [[76, 101, 102]. Gō models and other native-structure-based models greatly simplify folding landscapes by allowing only native interactions [101]. While the latter methods are successful at predicting the experimental behavior of small and mid-size well-behaved proteins [103, 104], they are likely unreliable in the case of less-well-behaved proteins. In addition, Gō models are typically inadequate to describe protein misfolding in the presence of concurrent aggregation leading to non-native-like self-associated states. Modified Gō models that incorporate misfolding have been developed [105]. These models require knowledge of high-resolution structures of both native and misfolded states. These structures are unfortunately not available in the case of most soluble misfolded states [105]. Another strategy to model the folding of large proteins is to use Markov-state models (MSM), which can model long timescale dynamics [106, 107]. MSMs partition the system into multiple states, and assume that transitions between states are memoryless. In other words, the probability of going from state x to state y only depends on states x and y, not any previously occupied states. Short MD simulations can be used to model small conformational changes within each state and can then be combined to predict long-timescale dynamics [106, 107]. Current challenges with using MSMs to model protein folding include correctly identifying the MSM states and interpreting folding mechanisms from MSMs. Recent machine learning advances can be employed to address the latter challenges [107]. In summary, while more work is necessary to develop simulations that can successfully model the experimentally observed folding and misfolding/aggregation of midsize to large proteins, the future of this area of research holds promise [89, 108, 109].
Energy landscapes.
Experimental data are often consistent with small single-domain proteins folding through a single pathway with either no or few intermediates [25, 110]. Yet, theoretical models suggest that the unfolded states reach the native state via multiple parallel pathways [42, 104, 111–114]. It is worth noting that experimentally observed 2- or 3-state refolding kinetics is not incompatible with multiple parallel folding paths [114]. Further, single-molecule experimental studies were able to unequivocally identify the existence of multiple parallel folding pathways for a single-domain protein that shows fast, two-state folding kinetics in bulk measurements [115]. Therefore, it is likely that many proteins fold via multiple parallel pathways, even when these paths cannot be explicitly resolved in bulk refolding experiments.
One theoretical model that promotes parallel folding pathways is based on the concept of folding funnel. The existence of folding funnels was initially suggested by Dill in 1987 [111], and then thoroughly detailed by Wolynes [103, 104, 112], Dill [41, 113] and coworkers. Folding funnels can be portrayed as two-dimensional diagrams, as shown in Figure 5A [103, 104, 112, 116, 117]. The y-axis describes the protein’s effective potential energy, which includes the potential energy of the protein chain and free-energy contributions arising from interactions with the solvent. The change in effective potential energy is proportional to the fraction of native contacts (Q). The horizontal axis represents the conformational entropy of the protein (Sprot, conf), so that the funnel width coincides with Sprot, conf. Note that the conformational-entropy term is distinct from the total entropy, and it does not include entropy contributions involving the solvent. The width of the funnel narrows as the effective potential energy decreases, showing that conformational entropy gets smaller (hence disfavoring folding), as the protein approaches the native state. In summary, the model postulates that, as each protein folds, both effective potential energy and conformational entropy decrease in concert, leading to an overall decrease in Helmholtz free energy. This process renders the overall landscape funnel-shaped [104, 116]. The diagram in Figure 5A qualitatively shows that there are typically very few conformations sharing the same effective potential energy and separated by significant local barriers. Hence the landscape is only very weakly “frustrated”. Multiple conformations that share the same energy contribute to increasing the density of states. Thus, entropy contributions due to both density of states and to solvent-related configurations need to be considered, in addition to the conformational entropy illustrated in the diagram of Figure 5A, to compute the total entropy.
Figure 5. Representative protein folding energy landscapes.
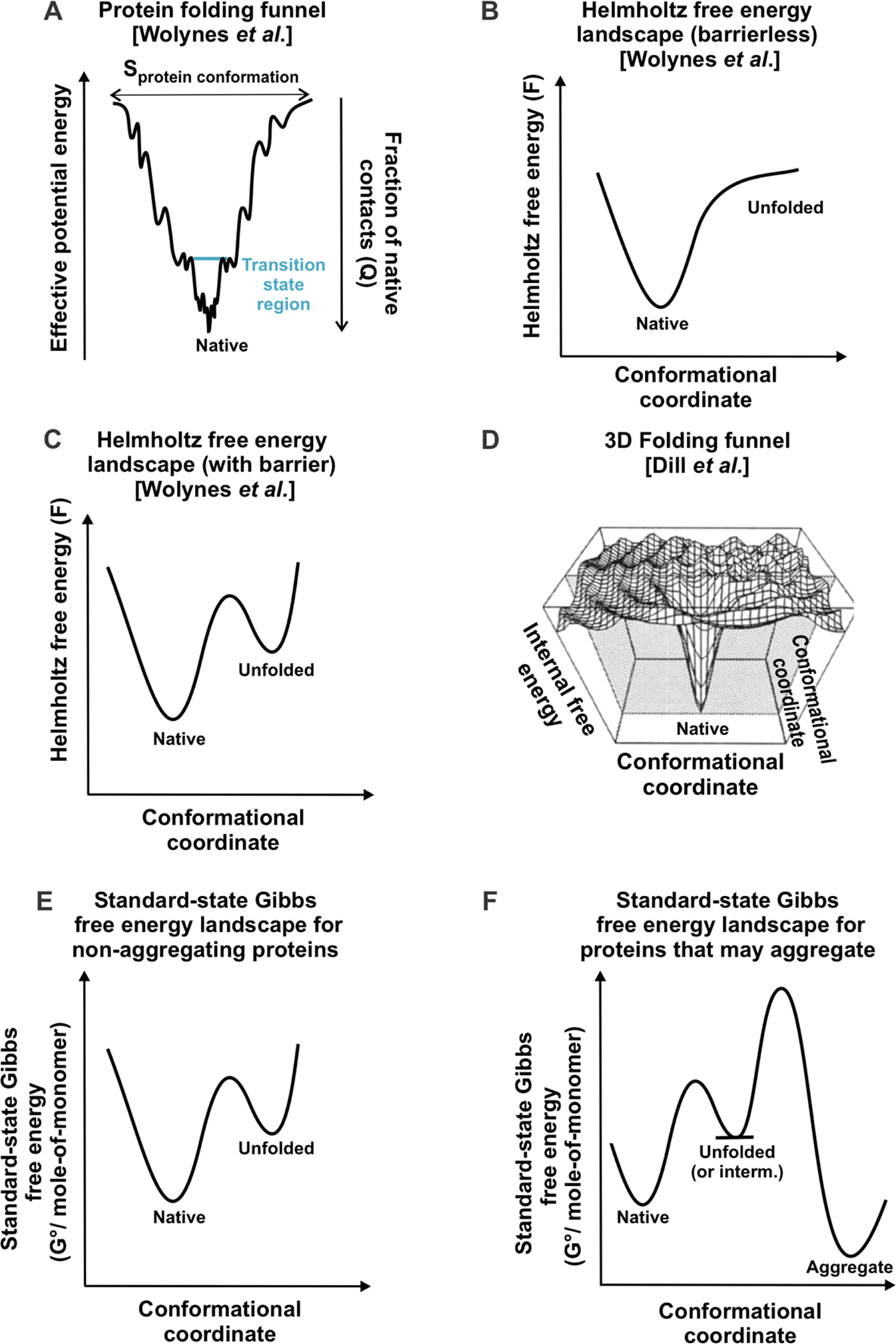
(A) Folding funnel proposed by Wolynes and coworkers, showing how protein conformational entropy decreases in concert with effective potential energy, as a protein folds to its native state [104, 116]. (B) Helmholtz free energy landscape for proteins that do not have a free-energy transition state for folding. (C) Helmholtz free energy landscape for proteins that have a free-energy transition state for folding. In panels A-C, the native state has 100% native contacts (Q = 1), and the unfolded state has Q = 0. (D) Multidimensional energy landscape. The vertical axis represents the potential energy of any given protein conformation plus the free energy of solvation [113]. Figure 1D is reprinted with permission from John Wiley and Sons [41] from figure 37C in Protein Science 4, Dill, K. A.; Bromberg, S.; Yue, K.; Chan, H. S.; Ftebig, K. M.; Yee, D. P.; Thomas, P. D. Principles of Protein Folding — a Perspective from Simple Exact Models. 4, 561–602. Copyright (1995). (E) Standard-state Gibbs free energy landscape of a protein that cannot form aggregates at a given temperature, pressure and solution conditions. (F) Gibbs free energy landscape for a protein that can form aggregates at a given temperature, pressure and solution conditions.
In the canonical ensemble, the change in total free energy of folding (ΔFtot) is defined as the difference between the changes in total internal energy (ΔUtot) and total entropy (ΔStot) terms according to
| (2) |
where T is temperature and ΔStot includes changes in both protein conformational entropy and entropy related to solvent molecules. The total internal energy is equal to the effective potential energy averaged over all microstates (i.e., protein conformations).
The negative change in conformational entropy as the protein folds is energetically unfavorable and must be compensated by favorable internal energy changes and/or favorable solvent entropy changes, so that the actual folding process ends up being thermodynamically favorable (ΔF < 0) [116]. If the energetically favorable internal-energy and solvent-entropy changes fully compensate or override the unfavorable conformational-entropy changes, the free energy landscape is barrierless, as shown in Figure 5B [112]. When the energetically unfavorable changes exceed the favorable contributions, the free energy landscape bears a barrier, as shown in Figure 5C. [117, 118]. Bryngelson et al. denote barrierless and barrier-containing free-energy folding scenarios as type 0 and type 1, respectively [112].
In the case of proteins whose folding free-energy landscapes bear a thermodynamic barrier (ΔG‡tot > 0), experimental studies showed that the thermodynamic activation parameters for folding, including activation enthalpy (ΔHf‡) and entropy changes (ΔSf‡), can be energetically favorable or unfavorable, as shown in Table 2. The Gibbs activation free energy for folding may be entropy or enthalpy driven, depending on whether TΔSf‡ or ΔHf‡ has a larger magnitude [119–121] (Table 2).
Table 2.
List of experimentally determined thermodynamic activation parameters for the folding of several proteins (unfolded, U → transition state, TS).
| Protein name | TΔSf‡ (U → TS) (kJ/mol) | ΔHf‡ (U → TS) (kJ/mol) | ΔGf‡ (U → TS) (kJ/mol) | Reaction is driven by: | Reference |
|---|---|---|---|---|---|
| Chymotrypsinogen | −155 | −66.7 | 88 | Entropy | [121] |
| Soybean trypsin inhibitor | −111 | −8.0 | 103 | Entropy | [121] |
| Chymotrypsin Inhibitor 2 (CI2) | 27.4 | 59.0 | 31.6 | Enthalpy | [119] |
| N-terminal domain of Ribosomal Protein L9 | 29.5 | 54.0 | 24.5 | Enthalpy | [119] |
| Ig binding domain of Y43W point mutation of protein L | 7.3 | 40.2 | 32.9 | Enthalpy | [119] |
| Immunophilin protein FKBP12 | 15.1 | 53.1 | 38.0 | Enthalpy | [119] |
| Transcriptional activator protein M2V GCN4-pl | 4.0 | 23.4 | 19.4 | Enthalpy | [119] |
| Cold shock protein B | 19.9 | 44.8 | 24.9 | Enthalpy | [119] |
| Cold shock protein B | −24 ± 2 | 31.6 ± 2.2 | 55.7 ± 1.0 | Enthalpy | [329] |
| Common-type acylphosphatase | −49.3 ± −4.9 | 23.6 ± 2.4 | 72.9 ± 0.4 | Entropy | [330] |
| Muscle acylphosphatase | −41.4 ± 4.1 | 40.7 ± 4.1 | 82.1 ± 0.4 | Neither | [330] |
| Apocytochrome b5 | −20.6 ± 5.5 | 42.4 ± 5.5 | 63 ± 8 | Enthalpy | [331] |
| Heart cytochrome c | 22 ± 9 | 59 ± 9 | 37 ± 13 | Enthalpy | [332] |
Protein-folding landscapes can also be visualized according to Dill et al. [41, 113] as three-dimensional curves, as shown in Figure 5D. In this case, the y axis is denoted as internal free energy, and is essentially equivalent to the effective potential energy of the landscapes by Wolynes et al. These landscapes are generated in the isothermal-isobaric ensemble, which is particularly relevant to biological systems, and assume constant temperature and pressure. It is worth noting that protein free energy landscapes are highly temperature [118], as well as pressure-dependent [122, 123].
In the isothermal-isobaric ensemble, free-energy landscapes describe changes in the Gibbs free energy of the system (G) as a function of protein conformational coordinates. However, G varies before a chemical process reaches equilibrium (or before an irreversible reaction is complete), while standard-state free energy per mole (G°) does not [124]. Therefore, Cavagnero et al. proposed to plot protein folding free-energy landscapes as G° instead of G, as shown in Figure 5E [92, 93]. The standard-state free energy of the system G° is expressed on a per-mole-of-monomer basis, so that both monomeric and aggregated protein states can be reliably plotted within the same landscape [92, 93] (Figure 5, panels E and F). As discussed in a previous section, standard-state chemical-potential landscapes were recently employed to show that most bacterial proteins are kinetically trapped relative to a variety of aggregates [92, 93]. We have also adopted this type of representation in Figure 4 of the present review.
Getting back to effective-potential energy landscapes, the folding-funnel concept has also been employed to explain the folding of multi-domain proteins [76, 125]. Experimental studies show that some large multi-domain proteins are able to successfully and independently refold from denaturant. These proteins include titin [77], fibronectin [78], and the double B domain of protein A (BBdpA) [79, 80]. Computational studies proposed that these types of multi-domain proteins fold successfully via a “divide and conquer strategy,” according to which each domain folds independently [76, 125]. If each domain is able to fold independently, then several smaller folding funnels can be combined into a single large funnel. Therefore, the folding process is characterized by significantly fewer degrees of freedom than if interactions between domains were to play a role during the folding process (Figure 6) [76].
Figure 6. Funnel landscapes of multi-domain proteins.
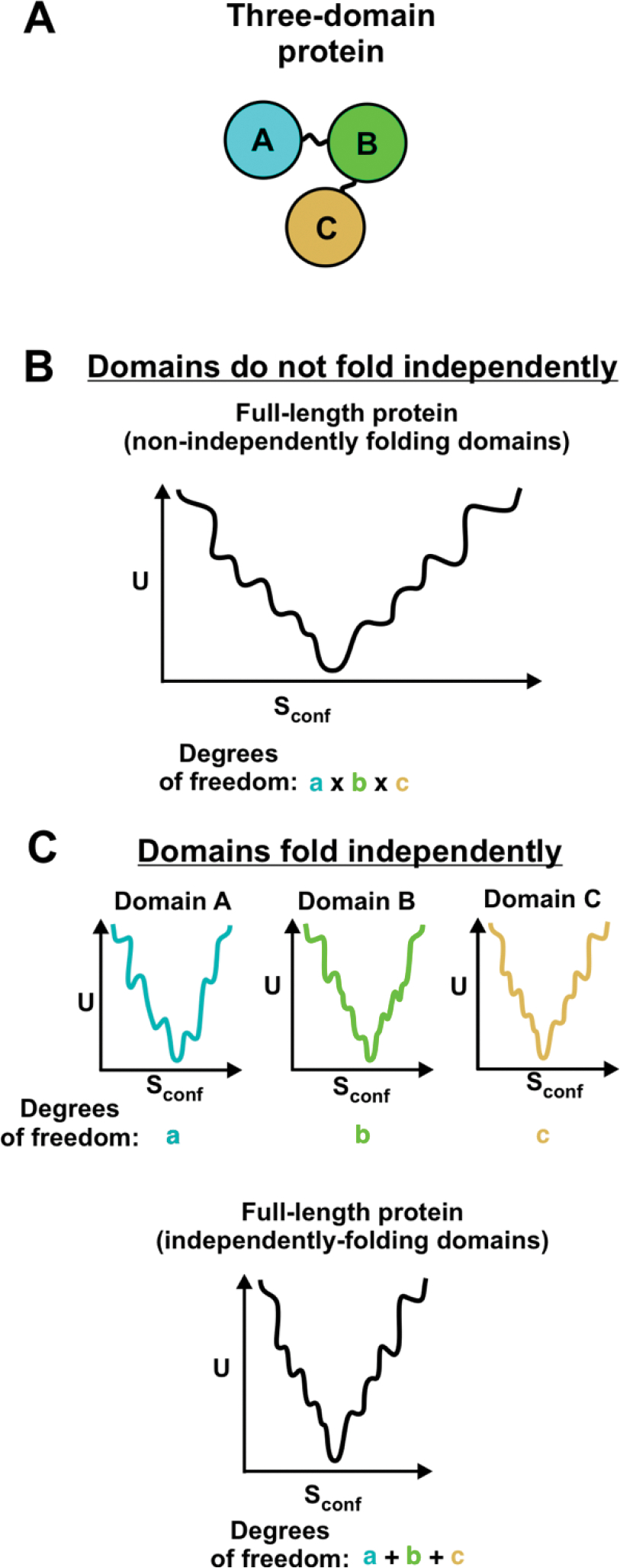
Some multi-domain proteins show independent folding of domains, and their folding can be described by combining the independent folding funnels for each domain [76]. (A) Cartoon of three-domain protein. (B) Folding funnel for multidomain protein with non-independently folding domains. The total number of degrees of freedom of the full-length protein is equal to the product of the degrees of freedom of each individual domain [76]. (C) Folding funnels for individual domains and combined folding funnel for multi-domain protein whose domains fold independently. In this case, the total number of degrees of freedom for the full-length protein is equal to the sum of the degrees of freedom for each individual domain [76].
Moving from simple model systems to more complex environments.
The studies described above focused on proteins refolding from denaturant. Physiologically relevant systems, however, not only include larger proteins but also involve more complex and crowded solution environments compared to buffered solutions. These environments can affect protein aggregation propensity.
In principle, crowded environments could decrease protein aggregation propensity. Molecular crowding from large inert cosolutes tends to stabilize the native state [117]. The crowding molecules decrease the volume available to the protein, pushing the protein toward a compact state, and thus reducing the total entropy of the system [1, 117] Yet, at high protein concentrations (> 100 g/L), interactions between the crowding molecules and proteins can decrease protein stability [1]. In some cases, crowding increases aggregation rate. For instance, the aggregation rate of α-synuclein is 10-fold greater in the presence of crowding agents than in plain buffer [126].
In general, crowding alone cannot fully explain the effect of many types of cosolutes on protein folding because it does not account for electrostatic and hydrophobic interactions. Some cosolutes, including osmolytes, stabilize the native state via repulsive interactions with the protein [117]. Site-specific ligand binding can also increase the stability of the native state. Other cosolutes, including denaturants, destabilize the native state via non-specific binding to the protein surface. Salt ions also affect protein stability, and their effect differs depending on the ion concentration as well as the location of charge in the folded and unfolded protein [117, 127–129]. Interestingly, the presence of other proteins with different sequences does not significantly change protein aggregation propensity, within complex mixtures. For instance, when bovine serum albumin and consensus tetratricopeptide repeat are mixed in solution, the solubility of each protein depends on its individual concentration and is minimally affected by the concentration of the other protein [130].
The refoldability of proteins after chemical denaturation within a complex protein collection from E. coli lysate was recently analyzed [108]. This study employed limited proteolysis via proteinase K to determine whether proteins refold to the native state. The results showed that 33% of E coli proteins do not refold to the native state after denaturation. Even more proteins may exhibit this characteristic behavior, upon taking soluble aggregates into account. Proteins with many domains are more likely to misfold than proteins with a single domain [108].
In a different study, Niwa et al. measured the solubility of all E. coli proteins within an E. coli cell-free system upon release from the ribosome in the absence of molecular chaperones [89]. Their results showed that only 28% of non-membrane proteins are ≥80% soluble (see eSol database: http://www.tanpaku.org/tp-esol/index.php?lang=en) [89, 131]. Larger proteins are more likely to form insoluble aggregates [89]. Namely, while 42% of small proteins (<30 kDa) are soluble, only 14% of large proteins are soluble [89]. Although the above experiments were carried out in the presence of the strong T7 promoter, the expressed-protein concentration range (2–100 μg/mL, average = 33 μg/mL) is comparable to the endogenous concentration range of most proteins in E. coli (4.7 – 153 μg/mL [0.11–4.30 μM], excluding outliers, median = 29.6 μg/mL [0.87 μM]) [132]. E. coli cellular concentrations were estimated from experimental copy numbers, assuming an E. coli cell volume of 10−12 mL. Proteins with concentrations larger than the third quartile value plus the quartile range (> 153 μg/mL, [>4.30 μM]) were considered outliers. This category included 17% of the 1,103 proteins quantified in this study [132]. In principle, higher concentration could decrease solubility relative to the results by Niwa et al. Apart from these selected high-abundance proteins, we expect the concentration of individual proteins generated via the above two methods to be similar, given the similar concentration ranges. Codon usage is not an issue, and any differences in translation rates in cell-free systems vs in vivo expression is would likely affect all proteins to a similar degree. The cell-free experiments performed by Niwa employ the strong T7 promoter, which leads to overexpression of bacterial proteins relative to conventional cellular production levels. However, cell-free systems have a lower percent of active ribosomes compared to live cells. Specifically, in the PURE system employed by Niwa et al., approximately 40% of ribosomes are active in protein synthesis at any given time, compared to 80% in E. coli cells [133, 134]. The lower ribosome activity of cell-free systems explains why the protein concentration in these experiments is comparable to cellular protein concentrations, despite the use of the T7 promoter. Note that the cell-free system employed in the study by Niwa et al. does not perfectly represent the cellular environment. This system lacks heme and other cofactors that may be required for correct folding of some proteins [135]. In addition, each protein was expressed individually. Therefore, proteins that give rise to hetero-complexes in live cells might show higher than regular aggregation levels in the work by Niwa et al. This outcome is likely in cases when the concentration of these proteins is higher than that of their complexation counterparts. Importantly, while the all aggregates detected by Niwa et al. were insoluble, it is known that some proteins can also form soluble aggregates upon release form the ribosome, if chaperones are not present [86]. Hence, the results by Niwa et al. may underestimate the actual extent of protein aggregation upon release from the ribosome, in the absence of molecular chaperones.
Once formed, soluble and insoluble aggregates are often kinetically trapped in E. coli, relative to the native state [92]. This phenomenon is responsible for the persistence of long-lived aggregation-free bioactive conformations. Further, amyloid aggregates are typically highly thermodynamically stable [42, 88]. While cellular quality-control systems can disaggregate and degrade misfolded proteins later in life, these processes are energetically costly [136–139]. Thus, correct folding in the early stages of protein life, including cotranslational and immediately post-translational folding, is critical for long-term protein solubility and function. The above experimental studies, performed in the absence of molecular chaperones, show significant levels of aggregation. Yet, in living cells molecular chaperones are present and enable the correct refolding of the numerous proteins that would otherwise aggregate, upon release from the ribosome.
Protein folding in the cell: The role of the ribosome.
The ribosome alters the folding energy landscape because many proteins begin folding cotranslationally, before they are fully synthesized and while they are still bound to the ribosome [24, 140–148]. Translation is vectorial and enables some proteins to fold sequentially, with N-terminal regions folding before C-terminal regions [149–152] [145], and sometimes enabling separate domains to fold independently. Independent folding of domains decreases the protein’s number of degrees of freedom (Figure 6) [76] and could prevent inter-domain misfolding interactions [153]. Rare codons clusters that slow down translation may provide more time for cotranslational folding [154, 155]. Rare codons often appear within protein domains and separate small structural units [154]. Synonymous codon substitution that alters translation rate can cause proteins to misfold, suggesting that Nature has optimized codon usage for correct folding [156].
The ribosome also reduces the number of accessible conformations by interacting with the nascent chain and spatially confining nascent chains motions. [141, 148, 157, 158] Nascent chains can interact with the ribosomal tunnel [159, 160] or surface [161–165]. The ribosomal tunnel is approximately 100 Å long and 10–20 Å wide [166, 167]. The tunnel can hold approximately 30–40 amino acids, depending on the nascent protein structure [158, 168–170], and it can fit more residues if the protein forms tertiary structure within the tunnel [171, 172]. Nascent proteins can form alpha-helical secondary structure [141, 158, 173, 174], tertiary interactions [142, 175], and even fully folded structures [171, 172, 176] within the tunnel. Nascent-chain compaction is a prerequisite for the folding of globular proteins and typically occurs after 54–59 nascent-chain residues have been synthesized [145, 171]. Larger tertiary structures can form within the vestibule [144, 177, 178] and outside the ribosomal tunnel [145, 179]. Ribosome-bound conformations may be dynamic and flexible [145, 175] The ribosome can destabilize full-length ribosome-bound protein structures outside the ribosome tunnel compared to released folded proteins [180].
Most single-domain proteins cannot fully fold into the native state until they are released from the ribosome and their C-terminal residues are available for folding. The C-terminal residues are usually important for folding because they bear key interaction counterparts, including sometimes residues expected to establish contacts with N-terminal regions of the protein chain [37, 181, 182]. Indeed, protein fragments lacking C-terminal residues are often insoluble [182]. Fortunately, the ribosome grants solubility to partially synthesized nascent chains [86]. Immediately post-translational folding sometimes involves structure formation by significant portions of the protein. For instance, apomyoglobin must incorporate at least 60 residues (40% of the total number of residues) into the native structure post-translationally [146]. Immediately after release from the ribosome, the nascent-protein region that becomes solvent-exposed may include a significant fraction of nonpolar residues. These nonpolar residues can either be buried intra- or inter-molecularly, giving rise to folding or aggregation, respectively. Therefore, the immediately post-translational steps are critical for the kinetic channeling of the nascent chain towards intramolecular folding, as opposed to intermolecular aggregation [86, 145, 146]. Once formed, most native and aggregated states in bacteria are kinetically trapped from each other, rendering later interconversion between these states highly unlikely [92].
Translation through the ribosome is sometimes sufficient to grant solubility to released proteins [86, 89]. Many proteins, however, require additional assistance from molecular chaperones to reach their soluble native structure [86, 89, 131].
Protein folding in the cell: The role of molecular chaperones.
Molecular chaperones act both co- and post-translationally and are able to prevent, and in some cases reverse, protein aggregation. Importantly, only 28% of the proteins synthesized with an E. coli cell-free system lacking molecular chaperones is soluble (excluding membrane proteins) [89]. Remarkably, molecular chaperones increase the solubility of 97% of these aggregation-prone proteins [131]. Correct folding and solubility are promoted by chaperones via a variety of mechanisms. Chaperones can catalyze conformational changes, including folding of unfolded states and unfolding of misfolded states, utilizing energy from ATP hydrolysis [183, 184]. Chaperones can also simply bind proteins and, in so doing, bury solvent exposed nonpolar regions and transiently decreases free-protein concentration. This chaperone action does not typically require ATP hydrolysis [183, 184]. Chaperones may be especially important to promote the folding of large and multi-domain proteins which tend to fold more slowly [36, 185–188], and are more likely to aggregate than two-state folding proteins [89, 108].
Bacterial cells contain a wide variety of chaperones that effectively mitigate the detrimental effect of misfolding and aggregation, including trigger factor (TF), the Hsp70 system, and GroEL/GroES [138, 189–191]. TF associates with nascent chains as they emerge from ribosomes, thus contributing to the prevention of aggregation and to the protein’s folding efficiency [192–196]. The affinity of TF for unfolded proteins is lower than the affinity of other chaperones. This thermodynamic property is accompanied by the rapid binding and release of client proteins [197], compatible with efficient translation [198]. DnaK, which is a prominent Hsp70 protein in prokaryotes, interacts with its substrates co- and post-translationally [199]. GroEL/GroES, a prokaryotic Hsp60 chaperone, acts downstream of DnaK upon de novo protein folding and facilitates correct folding through several functions including isolating proteins within its chamber (a.k.a. Anfinsen cage), catalyzing protein folding, and unfolding misfolded states [200–202]. ClpB, a prokaryotic heat-shock protein belonging to the Hsp100 class, solubilizes protein aggregates by threading protein chains through its central hexameric channel, thus facilitating disaggregation either alone [203, 204] or in combination with the Hsp70 chaperone network [205]. Figure 7 shows a graphical representation of the molecular chaperones that have currently been identified in E. coli. Given that the early stages of protein life are vital for long-term solubility and function, we focus on chaperones that act cotranslationally and immediately post-translationally. These chaperones include trigger factor and the Hsp70 system.
Figure 7. Pictorial representation of major pathways leading to protein native-structure formation in prokaryotes.
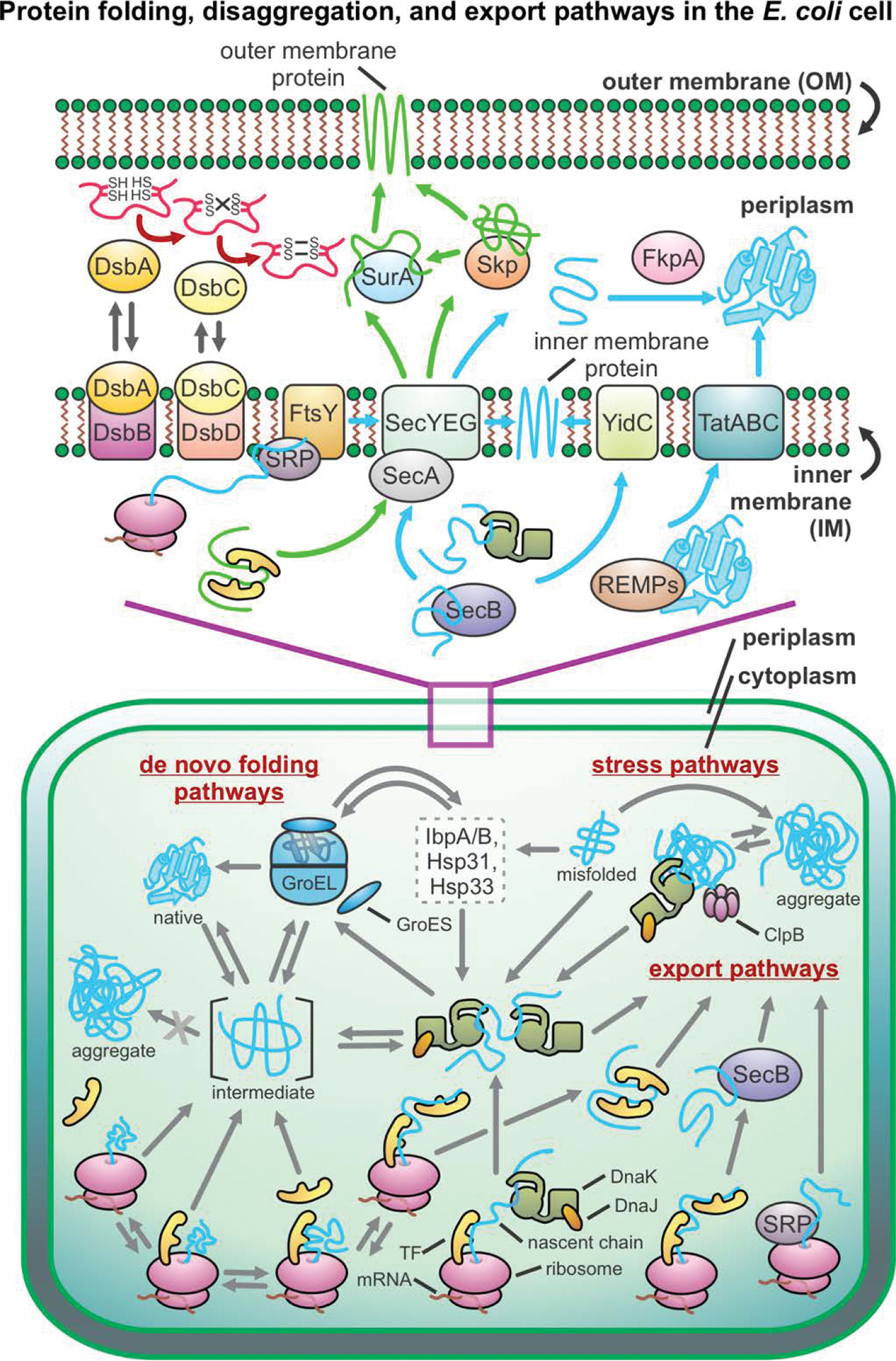
This scheme applies to gram-negative bacteria, e.g., E. coli. Abbreviations: SRP = signal recognition particle, REMPs = redox enzyme maturation protein, TatABC = twin-arginine translocation, TF = trigger factor.
Protein folding in the presence of the trigger factor chaperone.
Trigger factor (TF) is the only known ribosome-associated chaperone in bacteria (Figure 8). It was discovered by Crooke & Wickner who demonstrated that TF promotes the folding of the pro-OmpA protein to its membrane-assembly-competent form [206]. TF is both a chaperone and a cis/trans prolyl-isomerase [207], and it binds the ribosome with a 1:1 stoichiometry (Figure 8A). The cellular TF concentration is ~50 μM [208, 209]. This value is comparable to the ribosome concentration, though the latter varies as a function of cell growth rate. Ribosome-unbound TF undergoes a monomer-dimer equilibrium [209]. TF does not bind ATP and interacts with nascent chains cotranslationally (Figure 8B) [184, 192]. Deletion of TF in E. coli under regular growth conditions is not lethal, but the combined deletion of TF and DnaK causes protein aggregation and cell death [191, 199, 210]. TF binds to ribosome-bound nascent chains of most cytosolic proteins, outer membrane proteins, and periplasmic proteins [192, 211]. TF was also found to assist the refolding of some denatured proteins in vitro [197, 212–214]. Upon binding nascent proteins, TF delays acquisition of the fully native state and increases the ultimate yield of bioactive protein [137, 215]. Off the ribosome, TF binds client proteins in a predominantly unfolded conformation [216]. On the ribosome, TF reduces the force exerted by a cotranslationally folding chain, suggesting that it increases the population of unfolded nascent protein [217]. TF was also proposed to generate a “protected” space where nascent chains may be shielded from degradation and aggregation and may potentially fold cotranslationally (Figure 8B) [192, 218–220]. See additional comments in the section titled “Structure and dynamics of trigger factor client proteins”.
Figure 8. Protein folding in the presence of the trigger factor (TF) chaperone.
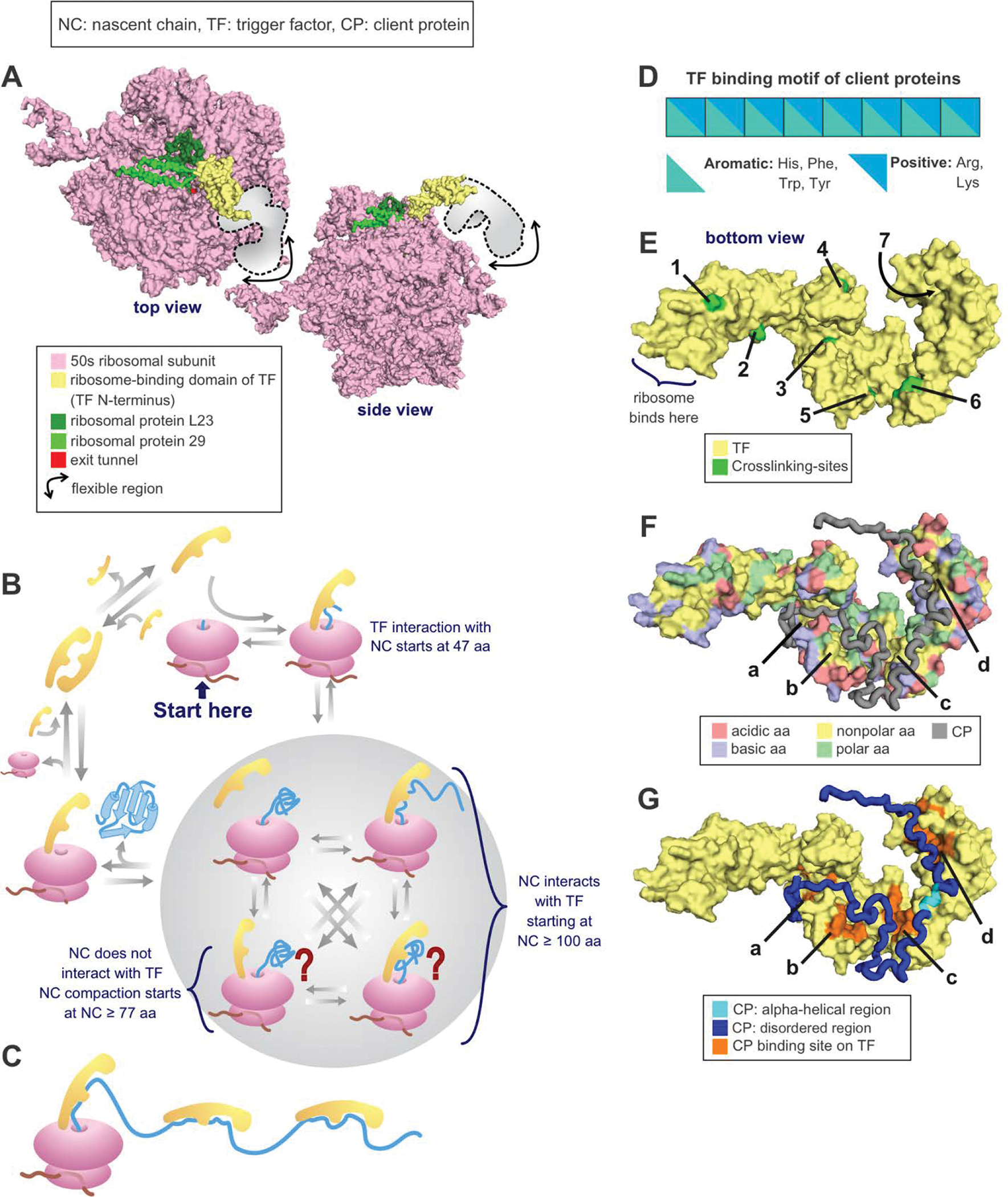
(A) Crystal structure of the RBD of TF bound to the 50s unit of the ribosome from eubacterium Deinococcus radiodurans. PDB: 2AAR [223]. (B) TF cycle. Note that TF is in rapid equilibrium with the ribosome. (C) Multiple TFs can be associated with the same nascent chain during translation or with the client protein in solution. (D) Nascent chain binding site for TF. (E) Crosslinking sites used to track the progression of the nascent chain as it travels throughout the TF [220]. PDB: 2MLX. (F) E. coli TF amino acids (aa) highlighted according to type. Note that the TF binding sites for PhoA, shown in the next panel, are all either nonpolar or polar. PDB: 2MLX. (G) E. coli TF associated with the 220–310 fragment of the PhoA client protein. PDB: 2MLX (Abbreviations: NC = nascent chain, TF = trigger factor, residues = amino acids).
Trigger factor structure and function.
TF is a 48 kDa (432 residues) protein comprising a ribosome-binding N-terminal domain, a peptidyl-prolyl isomerase (PPIase) domain, and a C-terminal domain [219, 221]. TF was described as having a dragon-shaped structure, with the N-terminal domain as the tail, the PPIase domain as the head, and the C-terminal domain, located in the central portion of the structure, forming the two arms [219]. The N-terminal domain binds ribosomal protein L23 and can also interact with L29 [219, 222–224]. TF’s PPIase activity has been demonstrated in vitro [207, 225]. Interestingly, this domain is not necessary for TF’s in vivo chaperone function [210, 213, 226]. The C-terminal domain performs the main chaperone function and TF fragments containing only the C-terminal domain prevent aggregation and promote folding in vitro [226] while fragments lacking the C-terminal domain show decreased chaperone activity [227].
Structure and dynamics of trigger factor client proteins.
Nascent proteins can interact with all three domains of TF [216]. TF typically binds nonpolar regions of nascent proteins [198, 228], though it can also interact with hydrophilic regions [192, 193]. The PPIase domain of TF binds eight-residue sequences enriched in aromatic and basic amino acids (Figure 8D) [229]. In vitro experiments featuring purified TF-client protein complexes revealed that this chaperone binds proteins with 40 or more residues [160, 220, 230]. On the other hand, in vivo investigations showed that TF binds ribosome-bound nascent proteins of 100 or more residues [211]. It has a higher affinity for ribosomes carrying nascent chains than for empty ribosomes, supporting its cotranslational role [198, 231]. TF’s affinity for the ribosome [145, 231] and nascent chains [232] increases with chain length, likely due to increased interactions between nascent chains and TF. Nascent proteins appear to move along the TF structure as they emerge from the ribosome. For instance, ribosome-bound isocitrate dehydrogenase interacts with the N-terminal domain first and, as the nascent chain elongates, it proceeds through the TF’s arms and then reaches the PPIase domain [220]. Some client proteins bind concurrently bind multiple TF proteins. For instance, PhoA can bind to up to three TF proteins (Figure 8C, E–G) [216].
Crystal [223, 224] and a cryoEM structures [232] show that, when TF binds the ribosome, its N-terminal ribosome binding domain undergoes a conformational change that exposes a nonpolar region to the ribosomal tunnel. This conformational transition enables TF to interact with nonpolar regions of unfolded nascent proteins [216, 217, 228, 233, 234]. TF was also proposed to create a shielded environment supporting aggregation-free cotranslational folding [218]. A crystal structure of an E. coli TF bound to Haloarcula marismortui ribosomes [219] show there is sufficient space between TF and the ribosome for a small to medium single-domain nascent protein to fold. This potential folding cavity was also shown in cryoEM structure of an E. coli TF and ribosome complex [220]. Yet, another crystal structure of the D. radiodurans TF and ribosome shows a much smaller space underneath TF that may not accommodate cotranslational folding [224].
Fluorescence anisotropy-decay showed that ribosome-bound nascent proteins form a compact structure both in the absence and presence of TF [145]. Hence TF is not necessary for nascent-chain compaction, though it could affect its population [145]. The average residence time for TF binding to ribosomes is at least 10 s [198, 209, 228, 231, 235]. This time is sufficient for the translation 100–200 amino acids in E. coli [192] and for the concurrent binding and unbinding of TF to nascent chains as they elongate, which occurs on the ms timescale [197, 236].
The two TF modes of action outline above, namely enhancing the population of unfolded clients and providing a protected environment supporting some nascent-chain compaction, are not mutually exclusive.
The known conformational flexibility of TF [219, 220, 223, 224, 232] is consistent with its ability to interact with a variety of nascent chains [237]. Finally, TF can also act post-translationally, by binding ribosome-released proteins [197] or by remaining bound to nascent chains after they are released from the ribosome [193, 228]. Post-translational interaction with TF may help stabilize protein monomers until they are assembled into complexes. This proposed role of TF in complex assembly is supported by the observation that cells lacking TF show a ribosome assembly defect under heat-stress conditions [193].
Protein folding in the presence of the Hsp70 chaperone.
In 1962, Ferruccio Ritossa observed that Drosophila larvae under heat stress show a “puffing pattern” around chromosomes that was later shown to result from an upregulation of the heat shock protein now known as Hsp70[238–245]. Later, the presence of Hsp70 chaperones was identified within wide a variety of organisms [244].
Hsp70 chaperones are ATP-dependent proteins that are routinely produced within the cell cytosol under non-stress conditions and that are also upregulated upon heat stress. Hsp70s are highly conserved and very important for maintaining cellular life [246, 247]. Hsp70s and Hsp70-like proteins are found across a wide variety of organisms, including prokaryotes, eukaryotes and even most archaea [149, 246], which are missing several other classes of chaperones (e.g., Hsp100 and Hsp90/83 [248]. The only chaperone more universally represented than Hsp70 is the Hsp60 chaperonin (known as GroEL in bacteria), which evolved first and is present in all living organisms [249]. While Hsp70 is widespread and generally ubiquitous, it is not universally represented and is missing from the genome of most hyperthermophiles [248, 250, 251] and two specific classes of bacteria [252].
The Hsp70 chaperone system includes Hsp70 and its cochaperones. In E. coli, this system includes DnaK (E. coli Hsp70) and cochaperones DnaJ (Hsp40) and GrpE. The latter is a nucleotide exchange factor (NEF). The Hsp70 chaperone system is considered a “central hub” in E. coli cells (Figure 7) due to its ability to interact with a wide variety of client proteins and due to its capability to influence a variety of cellular processes, spanning from de novo protein folding to protein transport and disaggregation [199, 253]. The concentration of Hsp70 within an E. coli cell is approximately 30–50 μM [254], and the total E. coli protein concentration is 5–8 mM [255]. Therefore, not all proteins in a cell can associate with the Hsp70 chaperone at once [256, 257]. DnaK displays a preference for 30–75 kDa client proteins and binds ~20% of newly synthesized proteins in the E. coli proteome (Figure 9A) [199, 258]. The Hsp70 chaperone system maintains cell homeostasis by holding unfolded proteins to prevent aggregation and by unfolding misfolded client proteins, so they can fold correctly [257, 259].
Figure 9. Key structural features required for the interactions of a client protein with the Hsp70 chaperone system.
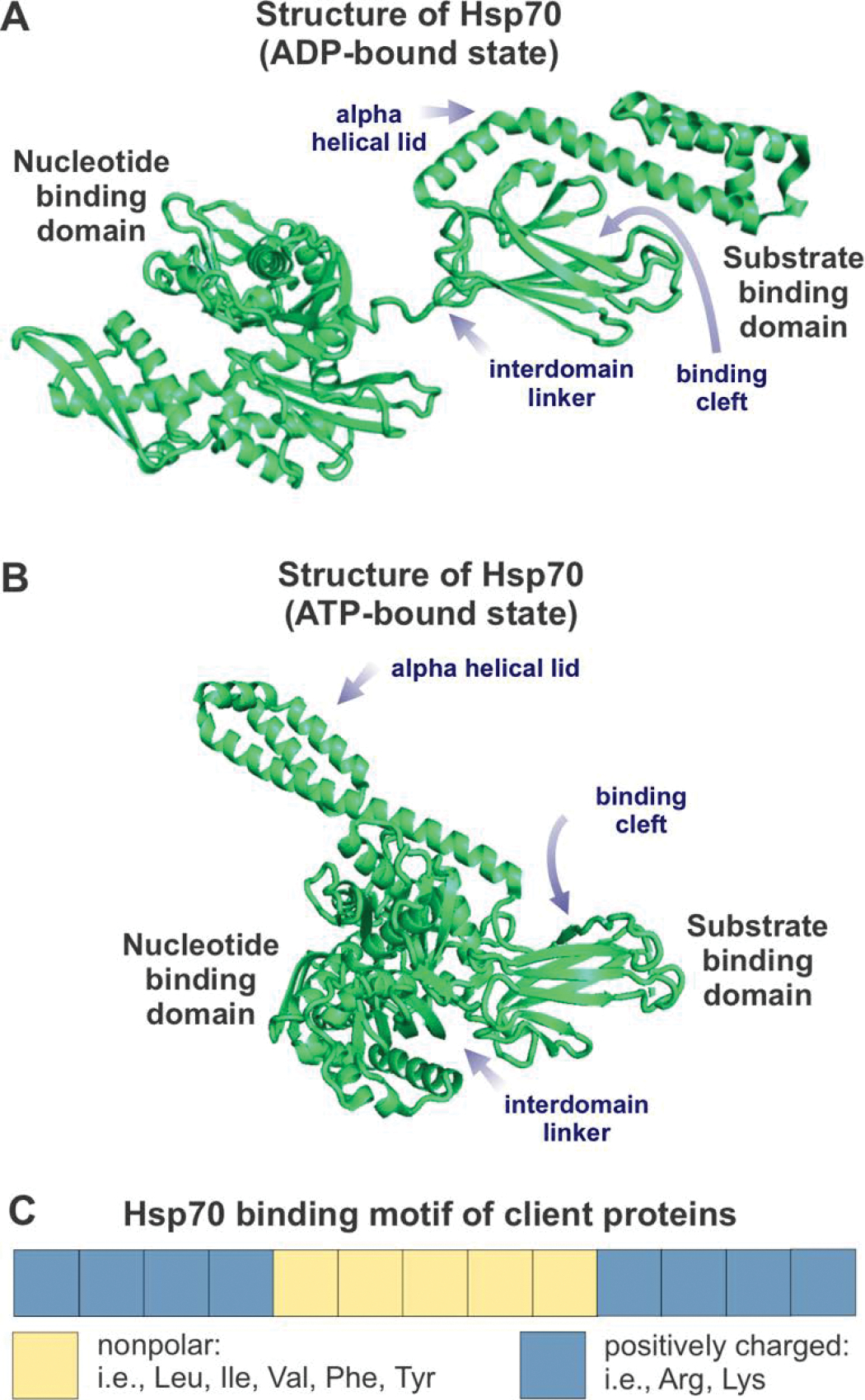
(A) Structure of ADP-bound (or nucleotide-free) Hsp70 chaperone (DnaK from E. coli). PDB ID: 2KHO. (B) Structure of ATP-bound DnaK chaperone. PDB ID: 4B9Q. (C) Client-protein binding motif for interaction with the E. coli Hsp70 chaperone DnaK, defined according to [284, 333]. Note that the positively charged residues flanking the central nonpolar core are progressively less important, as the sequence separation from the core increases.
The Hsp70 chaperone interacts with client proteins both co- and post-translationally to promote correct de novo folding of nascent chains [145, 191, 253]. Hsp70 assists the disassembly of protein complexes during bacteriophage replication [260], protein transport across membranes [261], and promotes the assembly of tail-anchored proteins within the cell membrane [262]. The Hsp70 chaperone system was reported to help disaggregating small aggregates and, in conjunction with other chaperones (e.g., ClpB), it was shown to assist the disaggregation of large aggregates [263–266]. The Hsp70 chaperone system can either take over client proteins from other chaperone systems and/or transfer them to other chaperone networks. Relevant chaperone networks include GroEL/ES [189], heat shock protein 90 (Hsp90) [267] and other small heat shock proteins like IbpA, IbpB, or inclusion-body binding proteins [268, 269].
Interestingly, Hsp70 is capable of preventing harm arising from deleterious mutations, thus granting key benefits to the parent organism in terms of both health and evolutionary rates [270, 271]. Therefore, Hsp70 and other chaperones allow organisms to experience greater genetic variation without harmful effects on fitness and could increase the species’ ability to evolve [270, 272]. In proteo-bacteria, client proteins with a high binding affinity for Hsp70 evolve faster than client proteins with low binding affinities for this chaperone [270, 271]. Given that Hsp70 increases protein evolution rate, overexpression of this chaperones may promote the efficiency of directed evolution [271]. Yet, chaperones do not always promote evolution, and other studies showed that Hsp70 and other chaperones sometimes decrease the client-protein evolution rate [273]. Additional future studies are necessary to fully understand the link between Hsp70 and evolutionary rates.
Not surprisingly, suboptimal Hsp70 function is linked to disease. If genes encoding the trigger factor (TF) and Hsp70 chaperones are concurrently knocked out, E. coli cells are no longer viable. This finding implies that the combination of TF and DnaK is essential for E. coli life. If only one of the two chaperone systems is knocked out, cells can survive but are more susceptible to stress [191, 210, 246]. Eukaryotic Hsp70 knockout or downregulation leads to increased levels of amyloid plaques in neurodegenerative diseases including Alzheimer’s and Huntington’s. Interestingly, while Hsp70 upregulation reduces the aggregation of plaque-forming proteins (a favorable effect), it also disfavors the apoptosis of cancerous cells (a deleterious effect) [274, 275]. Therefore, a carefully balanced chaperone concentration is required to support optimal health.
Hsp70 structure and function.
DnaK, the E. coli Hsp70 chaperone, consists of two domains comprising a 45 kDa nucleotide-binding domain (NBD) and a 25 kDa substrate-binding domain (SBD) [276]. The NBD contains two lobes that form a cleft that contains a binding site for a nucleotide (ATP or ADP) and specific cations (Mg2+ and 2 K+) [277–279]. The nucleotide state determines the conformation of Hsp70. If the chaperone is nucleotide-free or bound to ADP, then the two domains behave independently (Figure 9B). If Hsp70 is bound to ATP, then the chaperone lobes within the NBD rotate which subsequently cause the NBD and SBD domains dock to each other (Figure 9C) [279, 280].
The SBD contains two subdomains: the alpha-helical lid (SBDα), and the β-sheet pocket (SBDβ) [281]. SBDβ contains two beta sheets and two loops that form the pocket where the client protein binds [281, 282]. The conformation of the SBD varies between an open state when ATP is bound to the NBD and a closed state when the NBD is ADP-bound or nucleotide-free [278]. Crystal structures show that the binding pocket of ATP-bound DnaK exists in multiple open conformations and is likely dynamic and flexible [279, 282, 283]. The binding pocket preferentially interacts with a 4–5 residue long client-protein motif comprising aromatic (Phe, Tyr) or aliphatic (Val, Leu, Ile) nonpolar residues flanked by ca. four positively charged amino acids (Figure 9D). The characteristics of the amino acids towards the center of this motif are more important than the outer amino acids, for predicting binding to Hsp70 [284]. Interestingly, this binding motif occurs on average every 36 residues in most client proteins [284, 285].
The Hsp70 chaperone cycle.
Hsp70 chaperone activity proceeds via a functional cycle, which includes the Hsp40 (a J-domain protein) and nucleotide exchange factor (NEF) cochaperones. [286–288]. This cycle can be split into four main steps, as shown in Figure 10. The different stages of the Hsp70 chaperone cycle are briefly outlined below.
Figure 10. Scheme illustrating the major steps of the E. coli Hsp70 (a.k.a. DnaK) chaperone cycle.
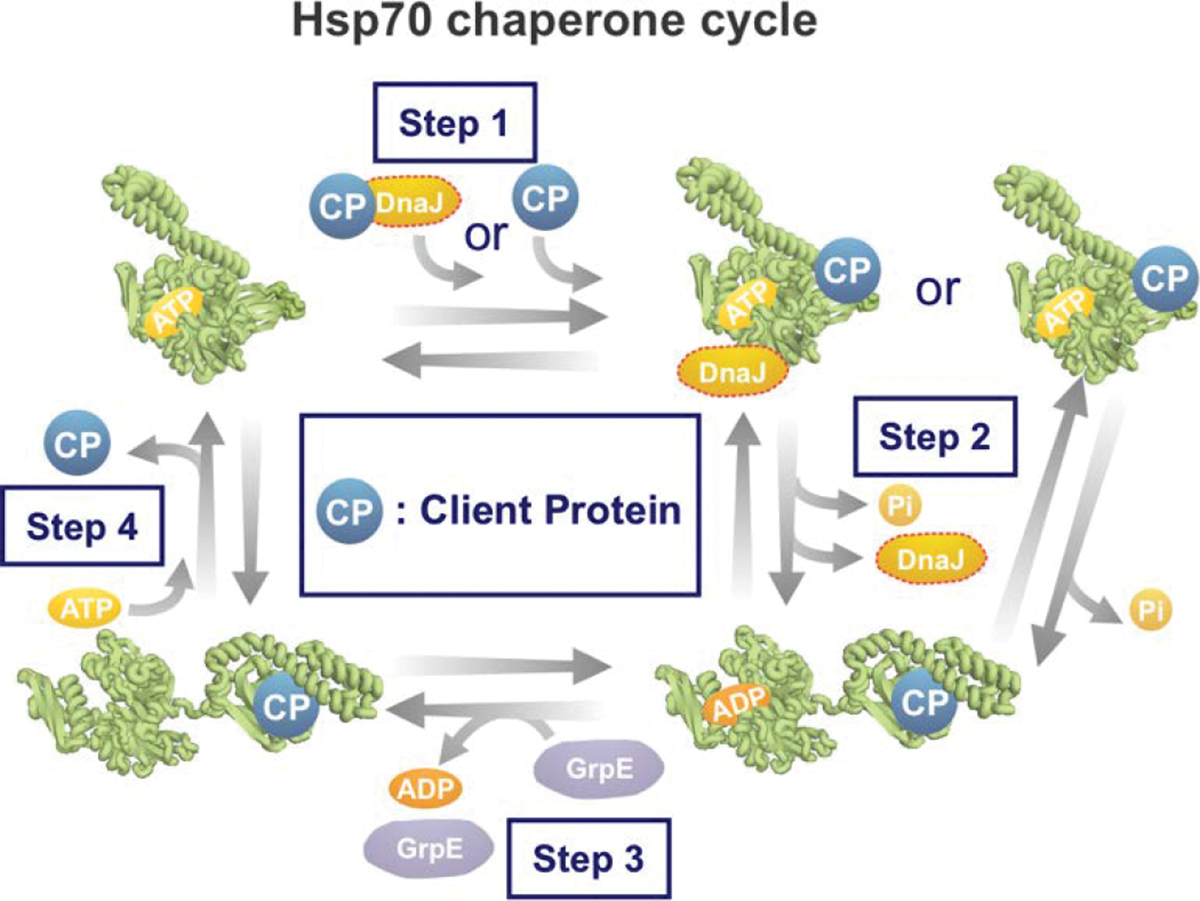
Hsp70 cooperates with co-chaperones DnaJ and GrpE through an ATP-dependent cycle to promote the folding of client proteins.
In the first step, the ATP-bound Hsp70 binds the client protein via hydrogen bonds and van der Waals interactions. However, that binding is very transient unless accompanied by ATP hydrolysis shown in step two [281]. ATP hydrolysis can occur in the absence and presence of client protein. However, the reaction is significantly slower in the absence of a bound client protein. To achieve maximal rate of ATP hydrolysis, a J protein (Hsp40s, DnaJ in E. coli) is needed. The J protein binds the client and transfers it to Hsp70 [247, 289–291]. J proteins have slightly different binding motifs to client proteins when compared to Hsp70. The J-protein binding motif enables the ultimate binding of Hsp70 to a wider range of misfolded or aggregated proteins, and likely targets this class of client proteins to Hsp70 because they may not be able to directly bind the Hsp70 binding motif described above (Figure 9D) [247, 290, 292, 293].
In the second step, ATP hydrolysis causes the SBD and NBD of ADP-Hsp70 to undock and behave independently, while staying covalently connected to the inter-domain linking region [294, 295]. The alpha-helical lid lowers towards the client protein, leading to increased client-protein affinity and slower client dissociation rate [247].
The third step involves departure of the ADP nucleotide from Hsp70. In bacteria, this step is rate-limiting and requires a nucleotide-exchange factor (NEF), e.g., GrpE (in E. coli) to promote the release of ADP, leaving Hsp70 in a nucleotide-free state [296, 297]. Upon nucleotide removal, NEF remains associated with Hsp70 and is thought to prevent ADP from rebinding [278, 298, 299]. While the α-helical lid is considered “closed” when DnaK is either ADP-bound or nucleotide-free, several studies showed that the lid is subject to slow dynamics and it occasionally reopens, thus allowing client proteins to bind/unbind the chaperone [279, 288, 300].
In the fourth step, ATP binds Hsp70 within the NBD cleft. The highly conserved nonpolar linker between the NBD and SBD upon ATP-binding, pulls the two domains together until they are firmly docked (Figure 9C) [280]. In addition, nucleotide-binding causes the α-helical lid to lift, enabling the client-protein to be released and the cycle to start anew [247, 278, 286, 295, 298, 299].
Structure and dynamics of Hsp70-bound client proteins.
The client protein can also change conformation during the Hsp70 cycle. The first NMR study of Hsp70-bound peptides showed that client proteins bound to nucleotide-free bacterial Hsp70 have a more extended conformation than in chaperone-free solution [301]. Later studies using electron paramagnetic resonance spectroscopy (EPR) confirmed these results for peptides bound to nucleotide-free, ADP-bound, or ATP-bound Hsp70 [302]. NMR studies on N-terminal fragments of apoMb alone showed that this protein has some helical structure [303]. Binding to Hsp70 unwinds the local helix structure of residues in the Hsp70 binding site [303]. Yet, regions distant from the binding site form non-native α-helical structure [304]. Single-molecule Főrster resonance energy transfer (FRET) experiments showed that the protein rhodanese (296 residues) lacks stable tertiary structure when bound to bacterial ADP-Hsp70 [305].
NMR studies by Lee and coworkers [306] showed that the drkN SH3 client protein, an N-terminal SH3 domain from Drosophila, populates multiple globally unfolded interconverting states while bound to ADP-Hsp70. The bound protein also populates additional spectroscopically undetectable states that account for 43% of the entire chaperone-bound population. This result is important because it shows that conformational sampling takes place while the client protein is bound to the ADP-Hsp70 chaperone [306]. Therefore, Hsp70-bound client proteins are dynamic chains that are able to sample distinct conformational states (and potentially fold, partially fold or unfold) while chaperone bound. Clearly, additional research needs to be performed to define the nature of the Hsp70-bound client more accurately-protein states and how they depend on client-protein amino-acid sequence.
Hsp70 promotes folding and prevents aggregation via two mechanisms. First, this chaperone “holds” (i.e., binds) client proteins in a predominantly unfolded or partially folded state, thus preventing aggregation by effectively lowering the concentration of client-protein conformations in solution. Second, Hsp70 promotes conformational changes within the bound client proteins that enable the conversion of misfolded client proteins to the folded state. In this review, we denote the first mechanism as “hold-only” and the second as “fold-promoting” behavior (Figure 11). Note that we use these terms instead of the more common “holdase” and “foldase” descriptors. A brief justification follows. In the biochemical literature, the “ase” suffix is typically employed to denote enzymes that catalyze reactions that lead to bond breaking or covalent scission of substrates into smaller components. While the Hsp70 chaperone system can lead to faster generation of the client-protein native state [259], in some cases, Hsp70 slows down native-state formation [307]. In both cases no covalent cuts are introduced. Therefore, we opted not to use the “ase” terminology.
Figure 11. Simplified schemes illustrating chaperone-assisted protein folding.
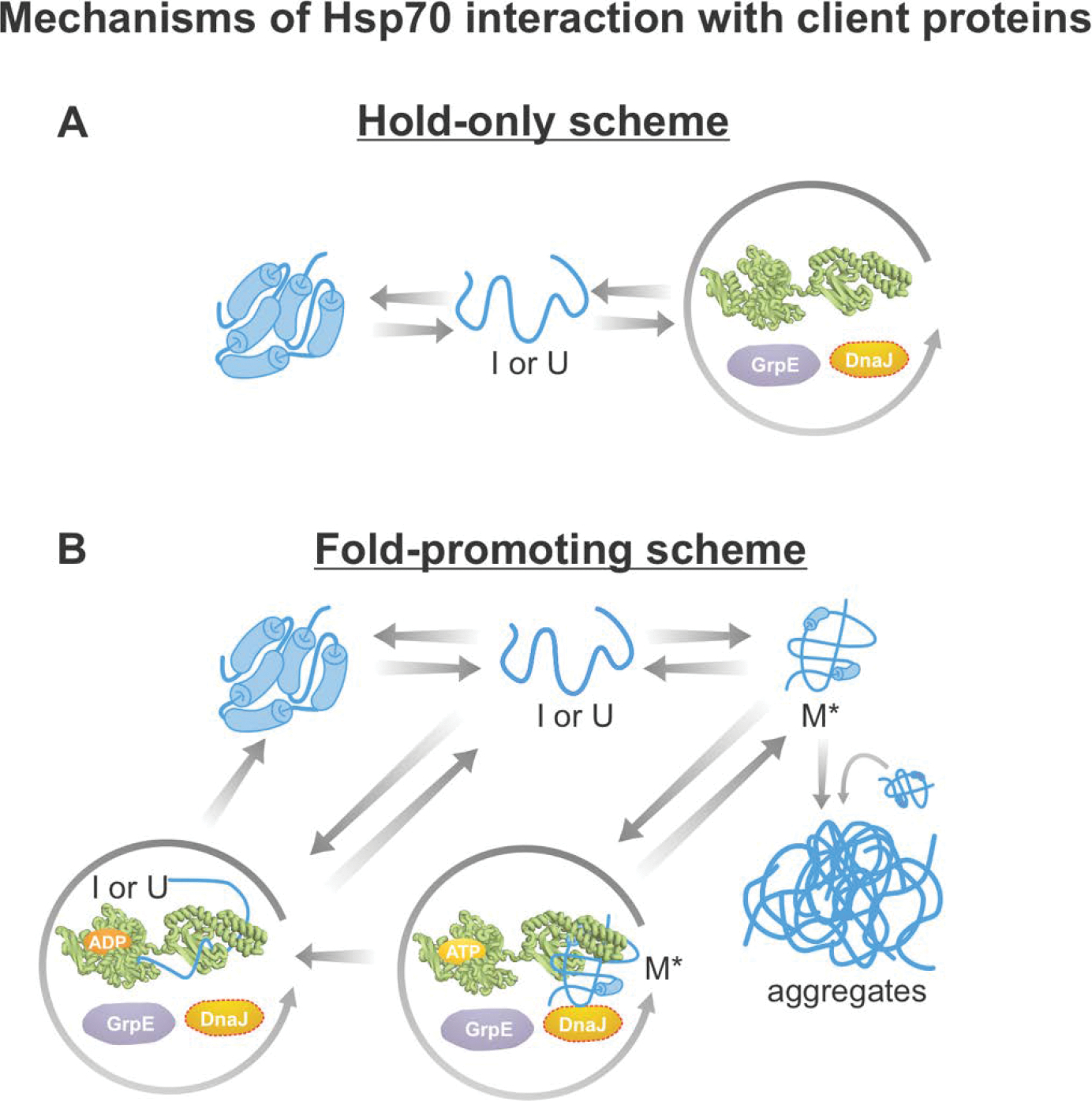
The diagrams in this figure are consistent with experimental results achieved with distinct classes of client proteins. (A) Hold-only model consistent with both computational and experimental results on non-aggregation-prone proteins bearing one Hsp70 binding site [307, 315]. (B) Fold-promoting models consistent with experimental results obtained with aggregation-prone client proteins bearing multiple chaperone binding sites per molecule. For instance, firefly luciferase (fluc) populate their native states more quickly and avoid generating aggregates in the presence of the Hsp70 chaperone system [259]. According to this fold-promoting model, the Hsp70 chaperone system catalyzes the conversion of misfolded monomers (M*) to the native state and, in so doing, increases the yields and observed rates of native-structure formation.
According to the “hold-only” mechanism, Hsp70 transiently binds client proteins whether or not they are aggregation-prone, thus decreasing the concentration of free proteins in solution (Figure 11A) [88, 246, 275]. This mode of action prevents aggregation because the nucleation and elongation rates of individual molecules undergoing nucleated-polymerization-like aggregation are concentration dependent. On the other hand, the rate of folding of monomeric proteins is not concentration dependent [308–310].
Several chaperone systems can adopt a hold-only-type mechanism, including the Hsp40, Hsp70 and GroEL/ES machineries [311–314]. Note that the Hsp70 and GroEL/ES chaperones may also facilitate folding and prevent aggregation via a fold-promoting-type mechanism [200]. The hold-only mode of Hsp70 action is supported by size-exclusion chromatography experiments that determined the degree of chaperone association of three distinct non-aggregation-prone model client proteins [315]. This study showed that, during folding away from equilibrium, Hsp70 interacts mostly with slow-folding proteins. In contrast, at equilibrium Hsp70 interacts preferentially with thermodynamically unstable proteins [315]. Similar conclusions were reached in separate experimental studies focusing on the apparent folding rate of RNAse H in the absence and presence of the Hsp70 chaperone system [307].
Optical tweezer experiments showed that Hsp70 binds and stabilizes unfolded maltose binding protein [316]. Similar results were obtained for Hsp70-bound drkN SH3 by NMR, except that additional conformations were found [306]. Earlier NMR studies with client peptides showed an effectively unfolded (conformationally expanded, β-sheet-like) population of Hsp70-bound peptides [301]. Similar results were later obtained by electron paramagnetic resonance (EPR) [302]. Single molecule FRET studies showed that the large client protein rhodanese concurrently binds several Hsp70 molecules [305]. Hsp70 binding to unfolded client proteins (e.g., RNAse H) slows down the observed rate of native-state acquisition [307]. Chaperone binding can occur directly to the ADP-bound state of Hsp70, suggesting that the hold-only mechanism does not require ATP hydrolysis for client binding [302, 306]. Therefore, interestingly, Hsp70 can both accelerate or slow-down acquisition of native structure via the fold-promoting and hold-only mechanisms, respectively. The experimental evidence available so far is consistent with the fact that Hsp70 uses the fold-promoting mechanism when interacting with aggregation-prone proteins that proceed via one or more misfolded intermediates [259]. Conversely, Hsp70 employs the hold-only mechanism upon interacting with non-aggregation-prone proteins [315, 317], which are characterized by free-energy landscapes similar to those of Figure 4A. The latter scenario is facilitated in the case of thermodynamic unstable and(or) slow-folding client proteins [315]. Additional work in this area is necessary, to more comprehensively characterize all viable scenarios. For instance, aggregation-prone client proteins that do not significantly populate misfolded intermediates may interact with Hsp70 via the hold-only mode.
The second mechanism adopted by Hsp70 to prevent client protein aggregation is denoted here as “fold-promoting” behavior. According to this mechanism, Hsp70 binds and unfolds misfolded proteins. In this way, Hsp70 enables misfolded states to bypass kinetic trapping relative to the native state give rise to the native conformation [318]. Fluorescence studies in the bulk and at the single-molecule level showed that ATP- and client-protein-bound Hsp70 undergoes ATP hydrolysis concurrently with unfolding of misfolded client proteins. Further, the unfolding of the misfolded state does not take place in the absence of ATP [259, 318]. This combined evidence strongly suggests that Hsp70 uses energy from ATP hydrolysis to unfold misfolded luciferase [259, 318]. Upon release from the chaperone, luciferase can then fold to the native state. Hydrolysis of five ATPs is required to enable the correct folding of one single luciferase protein, suggesting that one out of five unfolded proteins folds correctly to the native state, while the others misfold. Therefore, multiple cycles of chaperone binding and release are required for the client protein to fold correctly [259, 318].
Client proteins bearing more than one Hsp70 binding site may interact with multiple Hsp70 chaperones at once [305, 319]. Binding multiple chaperones causes steric repulsion that causes client proteins to adopt an expanded state [259, 305]. When multiple chaperones are bound to a single client protein, release from the chaperone is likely asynchronous. This causes the client protein to spend more time in a chaperone-associated state (bearing at least one bound chaperone) than if it were bound to one single chaperone [305, 320, 321]. Asynchronous release may allow different regions of the client protein to fold independently, similar to the foldon mechanism, and prevent misfolding interactions.
It was reported that Hsp70 can also assist protein disaggregation. This process has low efficiency in the presence of Hsp70 alone [265] and is much more effective when Hsp70 cooperates with other chaperones including Hsp100-type disaggregases including bacterial ClpB [138, 322, 323].
Conclusions.
A critical event in protein folding is the burial of most nonpolar residues away from the hydrophilic solvent. This process is accompanied by the formation of a spatially organized 3D structure, which is in most cases kinetically trapped from a variety of aggregated states, under physiologically relevant conditions. Some proteins successfully fold fast and independently. This category includes small, mid-size and large proteins. On the other hand, many proteins, especially some mid-size and most large multi-state-folding proteins, fold independently but slowly. Many mid-size and large proteins, bearing molecular-weight ranges highly represented in bacteria, are very aggregation-prone upon refolding from denaturant or as they emerge from the ribosome. These proteins require the cellular machinery in the early stages of their life, so that they populate bioactive states that remain kinetically trapped from misfolded aggregates under physiologically relevant conditions. In bacteria, the relevant machinery responsible for the formation of the native state at birth includes the ribosome and the molecular chaperones trigger factor and Hsp70. It is therefore clear that the early steps in protein folding in the cell, including co- and immediately post-translational folding, are essential for long-term protein solubility and function. They key aspects of this process are schematically illustrated in Figure 12.
Figure 12. Summary of protein folding standard-state Gibbs free energy landscapes.
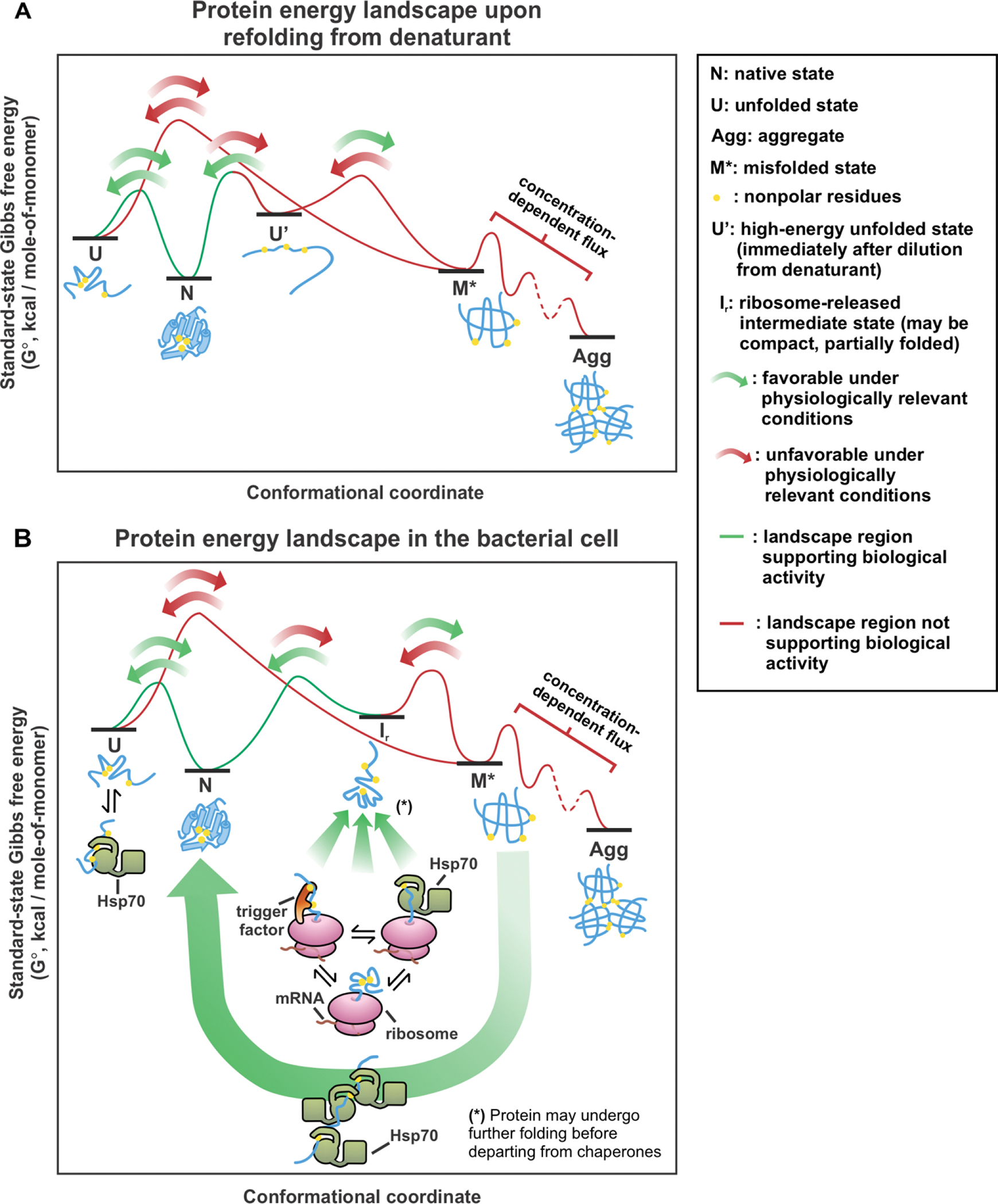
(A) Protein folding in vitro upon dilution from denaturant, and (B) ribosome and chaperone-assisted protein folding within the bacterial cell.
In conclusion, while some proteins are capable of folding independently when diluted from denaturant or upon release from the ribosome, many proteins need assistance from the cellular machinery in the early stages of their life. In this way, they remain bioactive and kinetically trapped from harmful aggregates over extremely long time spans.
Supplementary Material
HIGHLIGHTS.
Many proteins misfold and aggregate upon refolding from denaturant
Ribosome and chaperones kinetically channel aggregation-prone clients to native state
This review highlights the roles of ribosome, trigger factor and Hsp70 chaperones
The native state of bacterial proteins is often kinetically trapped relative to aggregates
ACKNOWLEDGMENTS
We are thankful to Xuhui Huang for helpful discussions, and we are grateful to Dmitry Ivankov for sharing the compiled data for protein folding rates that we used to generate Figure 3, and Supplementary Tables S1 and S2. We thank the National Science Foundation (NSF) for funding (grants MCB-2124672 and CBET 1912259 to S.C.). In addition, R.H. thanks the National Institute of General Medical Sciences of the National Institutes of Health for a TEAM-Science Fellowship (award number R25GM083252).
REFERENCES
- [1].Theillet F-X, Binolfi A, Frembgen-Kesner T, Hingorani K, Sarkar M, Kyne C, Li C, Crowley PB, Gierasch L, Pielak GJ, Elcock AH, Gershenson A, Selenko P, Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs), Chem. Rev. 114 (2014) 6661–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yamaguchi H, Miyazaki M, Refolding techniques for recovering biologically active recombinant proteins from inclusion bodies, Biomolecules 4 (2014) 235–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Betts SD, King J, Cold rescue of the thermolabile tailspike intermediate at the junction between productive folding and off-pathway aggregation, Protein Sci. 7 (1998) 1516–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Speed MA, Wang DI, King J, Specific aggregation of partially folded polypeptide chains: The molecular basis of inclusion body composition, Nat. Biotechnol. 14 (1996) 1283–1287. [DOI] [PubMed] [Google Scholar]
- [5].Tycko R, Amyloid polymorphism: structural basis and neurobiological relevance, Neuron 86 (2015) 632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, Millán C, Park H, Adams C, Glassman CR, DeGiovanni A, Pereira JH, Rodrigues AV, Dijk A.A.v., Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Garcia KC, Grishin NV, Adams PD, Read RJ, Baker D, Accurate prediction of protein structures and interactions using a three-track neural network, Science 373 (2021) 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold, Nature 596 (2021) 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin CL, Zidek A, Nelson AWR, Bridgland A, Penedones H, Petersen S, Simonyan K, Crossan S, Kohli P, Jones DT, Silver D, Kavukcuoglu K, Hassabis D, Improved protein structure prediction using potentials from deep learning, Nature 577 (2020) 706–710. [DOI] [PubMed] [Google Scholar]
- [9].Miller JL, Deep learning opens up protein science’s next frontiers, Phys. Today 74 (2021). [Google Scholar]
- [10].Bateman A, Martin MJ, O’Donovan C, Magrane M, Apweiler R, Alpi E, Antunes R, Ar-Ganiska J, Bely B, Bingley M, Bonilla C, Britto R, Bursteinas B, Chavali G, Cibrian-Uhalte E, Da Silva A, De Giorgi M, Dogan T, Fazzini F, Gane P, Cas-Tro LG, Garmiri P, Hatton-Ellis E, Hieta R, Huntley R, Legge D, Liu WD, Luo J, MacDougall A, Mutowo P, Nightin-Gale A, Orchard S, Pichler K, Poggioli D, Pundir S, Pureza L, Qi GY, Rosanoff S, Saidi R, Sawford T, Shypitsyna A, Turner E, Volynkin V, Wardell T, Watkins X, Watkins, Cowley A, Figueira L, Li WZ, McWilliam H, Lopez R, Xenarios I, Bougueleret L, Bridge A, Poux S, Redaschi N, Aimo L, Argoud-Puy G, Auchincloss A, Axelsen K, Bansal P, Baratin D, Blatter MC, Boeckmann B, Bolleman J, Boutet E, Breuza L, Casal-Casas C, De Castro E, Coudert E, Cuche B, Doche M, Dornevil D, Duvaud S, Estreicher A, Famiglietti L, Feuermann M, Gasteiger E, Gehant S, Gerritsen V, Gos A, Gruaz-Gumowski N, Hinz U, Hulo C, Jungo F, Keller G, Lara V, Lemercier P, Lieberherr D, Lombardot T, Martin X, Masson P, Morgat A, Neto T, Nouspikel N, Paesano S, Pedruzzi I, Pilbout S, Pozzato M, Pruess M, Rivoire C, Roechert B, Schneider M, Sigrist C, Sonesson K, Staehli S, Stutz A, Sundaram S, Tognolli M, Verbregue L, Veuthey AL, Wu CH, Arighi CN, Arminski L, Chen CM, Chen YX, Garavelli JS, Huang HZ, Laiho KT, McGarvey P, Natale DA, Suzek BE, Vinayaka CR, Wang QH, Wang YQ, Yeh LS, Yerramalla MS, Zhang J, UniProt C, UniProt: a hub for protein information, Nucleic Acids Res. 43 (2015) D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bateman A, Martin MJ, Orchard S, Magrane M, Agivetova R, Ahmad S, Alpi E, Bowler-Barnett EH, Britto R, Bursteinas B, Bye-A-Jee H, Coetzee R, Cukura A, Da Silva A, Denny P, Dogan T, Ebenezer T, Fan J, Castro LG, Garmiri P, Georghiou G, Gonzales L, Hatton-Ellis E, Hussein A, Ignatchenko A, Insana G, Ishtiaq R, Jokinen P, Joshi V, Jyothi D, Lock A, Lopez R, Luciani A, Luo J, Lussi Y, Mac-Dougall A, Madeira F, Mahmoudy M, Menchi M, Mishra A, Moulang K, Nightingale A, Oliveira CS, Pundir S, Qi GY, Raj S, Rice D, Lopez MR, Saidi R, Sampson J, Sawford T, Speretta E, Turner E, Tyagi N, Vasudev P, Volynkin V, Warner K, Watkins X, Zaru R, Zellner H, Bridge A, Poux S, Redaschi N, Aimo L, Argoud-Puy G, Auchincloss A, Axelsen K, Bansal P, Baratin D, Blatter MC, Bolleman J, Boutet E, Breuza L, Casals-Casas C, de Castro E, Echioukh KC, Coudert E, Cuche B, Doche M, Dornevil D, Estreicher A, Famiglietti ML, Feuermann M, Gasteiger E, Gehant S, Gerritsen V, Gos A, Gruaz-Gumowski N, Hinz U, Hulo C, Hyka-Nouspikel N, Jungo F, Keller G, Kerhornou A, Lara V, Le Mercier P, Lieberherr D, Lombardot T, Martin X, Masson P, Morgat A, Neto TB, Paesano S, Pedruzzi I, Pilbout S, Pourcel L, Pozzato M, Pruess M, Rivoire C, Sigrist C, Sonesson K, Stutz A, Sundaram S, Tognolli M, Verbregue L, Wu CH, Arighi CN, Arminski L, Chen CM, Chen YX, Garavelli JS, Huang HZ, Laiho K, McGarvey P, Natale DA, Ross K, Vinayaka CR, Wang QH, Wang YQ, Yeh LS, Zhang J, UniProt C, UniProt: the universal protein knowledgebase in 2021, Nucleic Acids Res. 49 (2021) D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Leader B, Baca QJ, Golan DE, Protein therapeutics: A summary and pharmacological classification, Nat. Rev. Drug Discov. 7 (2008) 21–39. [DOI] [PubMed] [Google Scholar]
- [13].Greenwald EC, Mehta S, Zhang J, Genetically encoded fluorescent biosensors illuminate the spatiotemporal regulation of signaling networks, Chem. Rev. 118 (2018) 11707–11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gebauer M, Skerra A, Engineered protein scaffolds as next-generation therapeutics, Annual Review of Pharmacology and Toxicology 60 (2020) 391–415. [DOI] [PubMed] [Google Scholar]
- [15].Richards FM, Structure of proteins, Annu. Rev. Biochem. 32 (1963) 269–300. [DOI] [PubMed] [Google Scholar]
- [16].Chothia C, Hydrophobic bonding and accessible surface area in proteins, Nature 248 (1974) 338–339. [DOI] [PubMed] [Google Scholar]
- [17].Chothia C, The nature of the accessible and buried surfaces in proteins, J. Mol. Biol. 105 (1976) 1–12. [DOI] [PubMed] [Google Scholar]
- [18].Southall NT, Dill KA, Haymet ADJ, A view of the hydrophobic effect, J. Phys. Chem. B 106 (2002) 521–533. [Google Scholar]
- [19].Anfinsen CB, Haber E, Sela M, White FH, The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain, Proc. Natl. Acad. Sci. U.S.A. 47 (1961) 1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Anfinsen CB, Principles that govern the folding of protein chains, Science 181 (1973) 223–230. [DOI] [PubMed] [Google Scholar]
- [21].Levinthal C, How to fold graciously, Mossbauer spectroscopy in biological systems 67 (1969) 22–24. [Google Scholar]
- [22].Dill KA, Theory for the folding and stability of globular proteins, Biochemistry 24 (1985) 1501–1509. [DOI] [PubMed] [Google Scholar]
- [23].Zwanzig R, Szabo A, Bagchi B, Levinthal’s paradox, Proc. Natl. Acad. Sci. U.S.A. 89 (1992) 20–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fedyukina DV, Cavagnero S, Protein folding at the exit tunnel, Annu. Rev. Biophys. 40 (2011) 337–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sosnick TR, Barrick D, The folding of single domain proteins—have we reached a consensus?, Curr. Opin. Struct. Biol. 21 (2011) 12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Englander SW, Mayne L, The nature of protein folding pathways, Proc. Natl. Acad. Sci. U.S.A. 111 (2014) 15873–15880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gianni S, Guydosh NR, Khan F, Caldas TD, Mayor U, White GWN, DeMarco ML, Daggett V, Fersht AR, Unifying features in protein-folding mechanisms, Proc. Natl. Acad. Sci. U.S.A. 100 (2003) 13286–13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Englander SW, Protein folding intermediates and pathways studied by hydrogen exchange, Annual Review of Biophysics and Biomolecular Structure 29 (2000) 213–238. [DOI] [PubMed] [Google Scholar]
- [29].Radford SE, Protein folding: progress made and promises ahead, Trends Biochem. Sci. 25 (2000) 611–618. [DOI] [PubMed] [Google Scholar]
- [30].Nelson DL, Lehninger AL, Cox MM, Lehninger principles of biochemistry, W. H. Freeman, New York, 2008. [Google Scholar]
- [31].Daggett V, Fersht AR, Is there a unifying mechanism for protein folding?, Trends Biochem. Sci. 28 (2003) 18–25. [DOI] [PubMed] [Google Scholar]
- [32].Mayor U, Guydosh NR, Johnson CM, Grossmann JG, Sato S, Jas GS, Freund SMV, Alonso DOV, Daggett V, Fersht AR, The complete folding pathway of a protein from nanoseconds to microseconds, Nature 421 (2003) 863–867. [DOI] [PubMed] [Google Scholar]
- [33].White GWN, Gianni S, Grossmann JG, Jemth P, Fersht AR, Daggett V, Simulation and experiment conspire to reveal cryptic intermediates and a slide from the nucleation-condensation to framework mechanism of folding, J. Mol. Biol 350 (2005) 757–775. [DOI] [PubMed] [Google Scholar]
- [34].Arai M, Unified understanding of folding and binding mechanisms of globular and intrinsically disordered proteins, Biophys. Rev 10 (2018) 163–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Plaxco KW, Simons KT, Baker D, Contact order, transition state placement and the refolding rates of single domain proteins, J. Mol. Biol. 277 (1998) 985–994. [DOI] [PubMed] [Google Scholar]
- [36].Ivankov DN, Garbuzynskiy SO, Alm E, Plaxco KW, Baker D, Finkelstein AV, Contact order revisited: Influence of protein size on the folding rate, Protein Sci. 12 (2003) 2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kurt N, Mounce BC, Ellison PA, Cavagnero S, Residue-specific contact order and contact breadth in single-domain proteins: implications for folding as a function of chain elongation, Biotechnol. Prog. 24 (2008) 570–575. [DOI] [PubMed] [Google Scholar]
- [38].Wang M, Kurland CG, Caetano-Anollés G, Reductive evolution of proteomes and protein structures, Proc. Natl. Acad. Sci. U.S.A. 108 (2011) 11954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Han J-H, Batey S, Nickson AA, Teichmann SA, Clarke J, The folding and evolution of multidomain proteins, Nat. Rev. Mol. Cell Biol. 8 (2007) 319–330. [DOI] [PubMed] [Google Scholar]
- [40].Gianni S, Ivarsson Y, Jemth P, Brunori M, Travaglini-Allocatelli C, Identification and characterization of protein folding intermediates, Biophys. Chem. 128 (2007) 105–113. [DOI] [PubMed] [Google Scholar]
- [41].Dill KA, Bromberg S, Yue K, Chan HS, Ftebig KM, Yee DP, Thomas PD, Principles of protein folding — A perspective from simple exact models, Protein Sci. 4 (1995) 561–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Baldwin RL, The nature of protein folding pathways: The classical versus the new view, J. Biomol. NMR 5 (1995) 103–109. [DOI] [PubMed] [Google Scholar]
- [43].Cecconi C, Shank EA, Bustamante C, Marqusee S, Direct observation of the three-state folding of a single protein molecule, Science 309 (2005) 2057–2060. [DOI] [PubMed] [Google Scholar]
- [44].Chamberlain AK, Marqusee S, Molten globule unfolding monitored by hydrogen exchange in urea, Biochemistry 37 (1998) 1736–1742. [DOI] [PubMed] [Google Scholar]
- [45].Griko YV, Freire E, Privalov PL, Energetics of the alpha-lactalbumin states - a calorimetric and statistical thermodynamic study, Biochemistry 33 (1994) 1889–1899. [DOI] [PubMed] [Google Scholar]
- [46].Jha SK, Udgaonkar JB, Direct evidence for a dry molten globule intermediate during the unfolding of a small protein, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 12289–12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rami BR, Udgaonkar JB, Mechanism of formation of a productive molten globule form of barstar, Biochemistry 41 (2002) 1710–1716. [DOI] [PubMed] [Google Scholar]
- [48].Schulman BA, Kim PS, Dobson CM, Redfield C, A residue-specific NMR view of the non-cooperative unfolding of a molten globule, Nat. Struct. Biol. 4 (1997) 630–634. [DOI] [PubMed] [Google Scholar]
- [49].Shimizu A, Ikeguchi M, Sugai S, Unfolding of the molten globule state of alpha-lactalbumin studied by H-1-NMR, Biochemistry 32 (1993) 13198–13203. [DOI] [PubMed] [Google Scholar]
- [50].Feng H, Zhou Z, Bai Y, A protein folding pathway with multiple folding intermediates at atomic resolution, Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Laurents DV, Bruix M, Jamin M, Baldwin RL, A pulse-chase-competition experiment to determine if a folding intermediate is on or off-pathway: Application to ribonuclease A, J. Mol. Biol. 283 (1998) 669–678. [DOI] [PubMed] [Google Scholar]
- [52].Bai YW, Kinetic evidence of an on-pathway intermediate in the folding of lysozyme, Protein Sci. 9 (2000) 194–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bai YW, Kinetic evidence for an on-pathway intermediate in the folding of cytochrome c, Proc. Natl. Acad. Sci. U.S.A. 96 (1999) 477–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Heidary DK, O’Neill JC, Roy M, Jennings PA, An essential intermediate in the folding of dihydrofolate reductase, Proc. Natl. Acad. Sci. U. S. A. 97 (2000) 5866–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Spudich GM, Miller EJ, Marqusee S, Destabilization of the Escherichia coli RNase H kinetic intermediate: Switching between a two-state and three-state folding mechanism, J. Mol. Biol. 335 (2004) 609–618. [DOI] [PubMed] [Google Scholar]
- [56].Lindhoud S, Pirchi M, Westphal AH, Haran G, van Mierlo CPM, Gradual folding of an off-pathway molten globule detected at the single-molecule level, J. Mol. Biol. 427 (2015) 3148–3157. [DOI] [PubMed] [Google Scholar]
- [57].Silow M, Oliveberg M, Transient aggregates in protein folding are easily mistaken for folding intermediates, Proc. Natl. Acad. Sci. U.S.A. 94 (1997) 6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dyson HJ, Wright PE, How does your protein fold? Elucidating the apomyoglobin folding pathway, Acc. Chem. Res. 50 (2017) 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Uzawa T, Akiyama S, Kimura T, Takahashi S, Ishimori K, Morishima I, Fujisawa T, Collapse and search dynamics of apomyoglobin folding revealed by submillisecond observations of α-helical content and compactness, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Eliezer D, Jennings PA, Wright PE, Doniach S, Hodgson KO, Tsuruta H, The radius of gyration of an apomyoglobin folding intermediate, Science 270 (1995) 487–487. [DOI] [PubMed] [Google Scholar]
- [61].Tsui V, Garcia C, Cavagnero S, Siuzdak G, Dyson HJ, Wright PE, Quench-flow experiments combined with mass spectrometry show apomyoglobin folds through an obligatory intermediate, Protein Sci. 8 (1999) 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Aoto PC, Nishimura C, Dyson HJ, Wright PE, Probing the non-native H helix translocation in apomyoglobin folding intermediates, Biochemistry 53 (2014) 3767–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bédard S, Krishna MM, Mayne L, Englander SW, Protein folding: Independent unrelated pathways or predetermined pathway with optional errors, Proc. Natl. Acad. Sci. U.S.A. 105 (2008) 7182–7187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Englander SW, Mayne L, The case for defined protein folding pathways, Proc. Natl. Acad. Sci. U.S.A. 114 (2017) 8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Englander SW, Mayne L, Krishna MM, Protein folding and misfolding: mechanism and principles, Q. Rev. Biophys. 40 (2007) 1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Englander SW, Mayne L, Kan Z-Y, Hu W, Protein folding—how and why: By hydrogen exchange, fragment separation, and mass spectrometry, Annu. Rev. Biophys. 45 (2016) 135–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chamberlain AK, Marqusee S, Comparison of equilibrium and kinetic approaches for determining protein folding mechanisms, Adv. Protein Chem. 53 (2000) 283–328. [DOI] [PubMed] [Google Scholar]
- [68].Krishna MM, Englander SW, A unified mechanism for protein folding: Predetermined pathways with optional errors, Protein Sci. 16 (2007) 449–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bai YW, Sosnick TR, Mayne L, Englander SW, Protein-folding intermediates - native-state hydrogen-exchange, Science 269 (1995) 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hu WB, Walters BT, Kan ZY, Mayne L, Rosen LE, Marqusee S, Englander SW, Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry, Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 7684–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bollen YJM, Kamphuis MB, van Mierlo CPM, The folding energy landscape of apoflavodoxin is rugged: Hydrogen exchange reveals nonproductive misfolded intermediates, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 4095–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Uzawa T, Nishimura C, Akiyama S, Ishimori K, Takahashi S, Dyson HJ, Wright PE, Hierarchical folding mechanism of apomyoglobin revealed by ultra-fast H/D exchange coupled with 2D NMR, Proc. Natl. Acad. Sci. U.S.A. 105 (2008) 13859–13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bedard S, Mayne LC, Peterson RW, Wand AJ, Englander SW, The foldon substructure of staphylococcal nuclease, J. Mol. Biol. 376 (2008) 1142–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Walters BT, Mayne L, Hinshaw JR, Sosnick TR, Englander SW, Folding of a large protein at high structural resolution, Proc. Natl. Acad. Sci. U.S.A. 110 (2013) 18898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Georgescauld F, Popova K, Gupta AJ, Bracher A, Engen JR, Hayer-Hartl M, Hartl FU, GroEL/ES chaperonin modulates the mechanism and accelerates the rate of TIM-barrel domain folding, Cell 157 (2014) 922–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang Y, Chu X, Suo Z, Wang E, Wang J, Multidomain protein solves the folding problem by multifunnel combined landscape: Theoretical investigation of a y-family DNA polymerase, J. Am. Chem. Soc. 134 (2012) 13755–13764. [DOI] [PubMed] [Google Scholar]
- [77].Scott KA, Steward A, Fowler SB, Clarke J, Titin; a multidomain protein that behaves as the sum of its parts, J. Mol. Biol. 315 (2002) 819–829. [DOI] [PubMed] [Google Scholar]
- [78].Steward A, Adhya S, Clarke J, Sequence conservation in lg-like domains: The role of highly conserved proline residues in the fibronectin type III superfamily, J. Mol. Biol. 318 (2002) 935–940. [DOI] [PubMed] [Google Scholar]
- [79].Arora P, Hammes GG, Oas TG, Folding Mechanism of a Multiple Independently-Folding Domain Protein: Double B Domain of Protein A, Biochemistry 45 (2006) 12312–12324. [DOI] [PubMed] [Google Scholar]
- [80].Petersen M, Barrick D, Analysis of Tandem Repeat Protein Folding Using Nearest-Neighbor Models, Annu. Rev. Biophys 50 (2021) 245–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Batey S, Clarke J, Apparent cooperativity in the folding of multidomain proteins depends on the relative rates of folding of the constituent domains, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 18113–18118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Batey S, Randles LG, Steward A, Clarke J, Cooperative Folding in a Multi-domain Protein, J. Mol. Biol. 349 (2005) 1045–1059. [DOI] [PubMed] [Google Scholar]
- [83].Osvath S, Kohler G, Zavodszky P, Fidy J, Asymmetric effect of domain interactions on the kinetics of folding in yeast phosphoglycerate kinase, Protein Sci. 14 (2005) 1609–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mello Cecilia C, Barrick D, An experimentally determined protein folding energy landscape, Proc. Natl. Acad. Sci. U.S.A. 101 (2004) 14102–14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Corrales M, Cuscó P, Usmanova DR, Chen H-C, Bogatyreva NS, Filion GJ, Ivankov DN, Machine learning: How much does it tell about protein folding rates?, PLoS One 10 (2015) e0143166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Addabbo RM, Dalphin MD, Mecha MF, Liu Y, Staikos A, Guzman-Luna V, Cavagnero S, Complementary role of co- and post-translational events in de novo protein biogenesis, J. Phys. Chem. B 124 (2020) 6488–6507. [DOI] [PubMed] [Google Scholar]
- [87].Cox BG, Modern liquid phase kinetics, University Press, Oxford; New York, 1994. [Google Scholar]
- [88].Chiti F, Taddei N, Baroni F, Capanni C, Stefani M, Ramponi G, Dobson CM, Kinetic partitioning of protein folding and aggregation, Nat. Struct. Biol. 9 (2002) 137–143. [DOI] [PubMed] [Google Scholar]
- [89].Niwa T, Ying B-W, Saito K, Jin W, Takada S, Ueda T, Taguchi H, Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins, Proc. Natl. Acad. Sci. U.S.A. 106 (2009) 4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Baldwin AJ, Knowles TPJ, Tartaglia GG, Fitzpatrick AW, Devlin GL, Shammas SL, Waudby CA, Mossuto MF, Meehan S, Gras SL, Christodoulou J, Anthony-Cahill SJ, Barker PD, Vendruscolo M, Dobson CM, Metastability of native proteins and the phenomenon of amyloid formation, J. Am. Chem. Soc. 133 (2011) 14160–14163. [DOI] [PubMed] [Google Scholar]
- [91].Yoshimura Y, Lin YX, Yagi H, Lee YH, Kitayama H, Sakurai K, So M, Ogi H, Naiki H, Goto Y, Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 14446–14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Varela AE, Lang JF, Wu Y, Dalphin MD, Stangl AJ, Okuno Y, Cavagnero S, Kinetic trapping of folded proteins relative to aggregates under physiologically relevant conditions, J. Phys. Chem. B 122 (2018) 7682–7698. [DOI] [PubMed] [Google Scholar]
- [93].Varela AE, England KA, Cavagnero S, Kinetic trapping in protein folding, Protein Eng. Des. Sel. 32 (2019) 103–108. [DOI] [PubMed] [Google Scholar]
- [94].Manning M, Colón W, Structural Basis of Protein Kinetic Stability: Resistance to Sodium Dodecyl Sulfate Suggests a Central Role for Rigidity and a Bias Toward β-Sheet Structure, Biochemistry 43 (2004) 11248–11254. [DOI] [PubMed] [Google Scholar]
- [95].Xia K, Zhang S, Solina BA, Barquera B, Colón W, Do Prokaryotes Have More Kinetically Stable Proteins Than Eukaryotic Organisms?, Biochemistry 49 (2010) 7239–7241. [DOI] [PubMed] [Google Scholar]
- [96].Ciryam P, Antalek M, Cid F, Tartaglia GG, Dobson CM, Guettsches AK, Eggers B, Vorgerd M, Marcus K, Kley RA, Morimoto RI, Vendruscolo M, Weihl CC, A metastable subproteome underlies inclusion formation in muscle proteinopathies, Acta Neuropathol. Commun. 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kundra R, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M, Protein homeostasis of a metastable subproteome associated with Alzheimer’s disease, Proc. Natl. Acad. Sci. U. S. A. 114 (2017) E5703–E5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Noji M, Samejima T, Yamaguchi K, So M, Yuzu K, Chatani E, Akazawa-Ogawa Y, Hagihara Y, Kawata Y, Ikenaka K, Mochizuki H, Kardos J, Otzen DE, Bellotti V, Buchner J, Goto Y, Breakdown of supersaturation barrier links protein folding to amyloid formation, Commun. Biol 4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Li B, Fooksa M, Heinze S, Meiler J, Finding the needle in the haystack: towards solving the protein-folding problem computationally, Critical Reviews in Biochemistry and Molecular Biology 53 (2018) 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Duan Y, Kollman PA, Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution, Science 282 (1998) 740–744. [DOI] [PubMed] [Google Scholar]
- [101].Gershenson A, Gosavi S, Faccioli P, Wintrode PL, Successes and challenges in simulating the folding of large proteins, J. Biol. Chem 295 (2020) 15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Li W, Terakawa T, Wang W, Takada S, Energy landscape and multiroute folding of topologically complex proteins adenylate kinase and 2ouf-knot, Proc. Natl. Acad. Sci. U.S.A. 109 (2012) 17789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Onuchic JN, Wolynes PG, Luthey-Schulten Z, Socci ND, Toward an outline of the topography of a realistic protein-folding funnel, Proc. Natl. Acad. Sci. U.S.A. 92 (1995) 3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Wolynes PG, Onuchic JN, Thirumalai D, Navigating the folding routes, Science 267 (1995) 1619–1620. [DOI] [PubMed] [Google Scholar]
- [105].Trovato F, O’Brien EP, Fast Protein Translation Can Promote Co- and Posttranslational Folding of Misfolding-Prone Proteins, Biophys. J. 112 (2017) 1807–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Husic BE, Pande VS, Markov state models: From an art to a science, J. Am. Chem. Soc. 140 (2018) 2386–2396. [DOI] [PubMed] [Google Scholar]
- [107].Konovalov KA, Unarta IC, Cao S, Goonetilleke EC, Huang X, Markov state models to study the functional dynamics of proteins in the wake of machine learning, JACS Au 1 (2021) 1330–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].To P, Whitehead B, Tarbox HE, Fried SD, Nonrefoldability is pervasive across the E. coli proteome, J. Am. Chem. Soc. 143 (2021) 11435–11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kerner MJ, Naylor DJ, Ishihama Y, Maier T, Chang H-C, Stines AP, Georgopoulos C, Frishman D, Hayer-Hartl M, Mann M, Hartl FU, Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli, Cell 122 (2005) 209–220. [DOI] [PubMed] [Google Scholar]
- [110].Jackson SE, How do small single-domain proteins fold?, Folding and Design 3 (1998) R81–R91. [DOI] [PubMed] [Google Scholar]
- [111].Dill KA, The stabilities of globular proteins, in: Oxender DL, Fox CF (Eds.) Protein Eng, Alan R. Liss, Inc, New York, 1987, pp. 187–192. [Google Scholar]
- [112].Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG, Funnels, pathways, and the energy landscape of protein folding: a synthesis, Proteins 21 (1995) 167–195. [DOI] [PubMed] [Google Scholar]
- [113].Dill KA, Chan HS, From Levinthal to pathways to funnels, Nat. Struct. Biol. 4 (1997) 10–19. [DOI] [PubMed] [Google Scholar]
- [114].Ellison PA, Cavagnero S, Role of unfolded state heterogeneity and en-route ruggedness in protein folding kinetics, Protein Sci. 15 (2006) 564–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Schönfelder J, Perez-Jimenez R, Muñoz V, A simple two-state protein unfolds mechanically via multiple heterogeneous pathways at single-molecule resolution, Nat. Commun. 7 (2016) 11777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Karplus M, Behind the folding funnel diagram, Nat. Chem. Biol. 7 (2011) 401–404. [DOI] [PubMed] [Google Scholar]
- [117].Gruebele M, Dave K, Sukenik S, Globular protein folding in vitro and in vivo, Annu. Rev. Biophys 45 (2016) 233–251. [DOI] [PubMed] [Google Scholar]
- [118].Becktel WJ, Schellman JA, Protein stability curves, Biopolymers 26 (1987) 1859–1877. [DOI] [PubMed] [Google Scholar]
- [119].Akmal A, Muñoz V, The nature of the free energy barriers to two-state folding, Proteins 57 (2004) 142–152. [DOI] [PubMed] [Google Scholar]
- [120].Liu Z, Chan HS, Desolvation is a Likely Origin of Robust Enthalpic Barriers to Protein Folding, J. Mol. Biol. 349 (2005) 872–889. [DOI] [PubMed] [Google Scholar]
- [121].Lumry R, Eyring H, CONFORMATION CHANGES OF PROTEINS, J. Phys. Chem. 58 (1954) 110–120. [Google Scholar]
- [122].Zipp A, Kauzmann W, Pressure denaturation of metmyoglobin, Biochemistry 12 (1973) 4217–4228. [DOI] [PubMed] [Google Scholar]
- [123].Roche J, Caro JA, Norberto DR, Barthe P, Roumestand C, Schlessman JL, Garcia AE, García-Moreno E. B, Royer CA, Cavities determine the pressure unfolding of proteins, Proc. Natl. Acad. Sci. U.S.A. 109 (2012) 6945–6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Levine IN, Physical Chemistry, 6th edition ed., McGraw-Hill, Boston, 2009. [Google Scholar]
- [125].Wolynes PG, Folding funnels and energy landscapes of larger proteins within the capillarity approximation, Proc. Natl. Acad. Sci. U.S.A. 94 (1997) 6170–6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Horvath I, Kumar R, Wittung-Stafshede P, Macromolecular crowding modulates α-synuclein amyloid fiber growth, Biophys. J. 120 (2021) 3374–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Record MT, Anderson CF, Lohman TM, Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: the roles of ion association or release, screening, and ion effects on water activity, Q. Rev. Biophys. 11 (1978) 103–178. [DOI] [PubMed] [Google Scholar]
- [128].Record MT Jr., Zhang W, Anderson CF, Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: a practical guide to recognizing and interpreting polyelectrolyte effects, Hofmeister effects, and osmotic effects of salts, Adv. Protein Chem. 51 (1998) 281–353. [DOI] [PubMed] [Google Scholar]
- [129].Sukenik S, Sapir L, Gilman-Politi R, Harries D, Diversity in the mechanisms of cosolute action on biomolecular processes, Faraday Discuss. 160 (2013) 225–237. [DOI] [PubMed] [Google Scholar]
- [130].Bianco V, Alonso-Navarro M, Di Silvio D, Moya S, Cortajarena AL, Coluzza I, Proteins are Solitary! Pathways of Protein Folding and Aggregation in Protein Mixtures, J. Phys. Chem. Lett 10 (2019) 4800–4804. [DOI] [PubMed] [Google Scholar]
- [131].Niwa T, Kanamori T, Ueda T, Taguchi H, Global analysis of chaperone effects using a reconstituted cell-free translation system, Proc. Natl. Acad. Sci. U.S.A. 109 (2012) 8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ishihama Y, Schmidt T, Rappsilber J, Mann M, Hartl FU, Kerner MJ, Frishman D, Protein abundance profiling of the Escherichia coli cytosol, BMC Genomics 9 (2008) 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Forchhammer J, Lindahl L, Growth rate of polypeptide chains as a function of the cell growth rate in a mutant of Escherichia coli 15, J. Mol. Biol. 55 (1971) 563–568. [DOI] [PubMed] [Google Scholar]
- [134].Kempf N, Remes C, Ledesch R, Züchner T, Höfig H, Ritter I, Katranidis A, Fitter J, A Novel Method to Evaluate Ribosomal Performance in Cell-Free Protein Synthesis Systems, Sci. Rep. 7 (2017) 46753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Kuruma Y, Ueda T, The PURE system for the cell-free synthesis of membrane proteins, Nat. Protoc. 10 (2015) 1328–1344. [DOI] [PubMed] [Google Scholar]
- [136].Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Ulrich Hartl F, Molecular chaperone functions in protein folding and proteostasis, Annu. Rev. Biochem. 82 (2013) 323–355. [DOI] [PubMed] [Google Scholar]
- [137].Balchin D, Hayer-Hartl M, Hartl FU, In vivo aspects of protein folding and quality control, Science 353 (2016) aac4354. [DOI] [PubMed] [Google Scholar]
- [138].Mogk A, Bukau B, Kampinga HH, Cellular handling of protein aggregates by disaggregation machines, Mol. Cell 69 (2018) 214–226. [DOI] [PubMed] [Google Scholar]
- [139].Doyle SM, Genest O, Wickner S, Protein rescue from aggregates by powerful molecular chaperone machines, Nat. Rev. Mol. Cell Biol. 14 (2013) 617. [DOI] [PubMed] [Google Scholar]
- [140].Waudby CA, Dobson CM, Christodoulou J, Nature and regulation of protein folding on the ribosome, Trends Biochem. Sci. 44 (2019) 914–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Lu J, Deutsch C, Folding zones inside the ribosomal exit tunnel, Nat. Struct. Mol. Biol. 12 (2005) 1123–1129. [DOI] [PubMed] [Google Scholar]
- [142].Kosolapov A, Deutsch C, Tertiary interactions within the ribosomal exit tunnel, Nat. Struct. Mol. Biol. 16 (2009) 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Tu LW, Deutsch C, A folding zone in the ribosomal exit tunnel for Kv1.3 helix formation, J. Mol. Biol. 396 (2010) 1346–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Tu L, Khanna P, Deutsch C, Transmembrane segments form tertiary hairpins in the folding vestibule of the ribosome, J. Mol. Biol. 426 (2014) 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Ellis JP, Bakke CK, Kirchdoerfer RN, Jungbauer LM, Cavagnero S, Chain dynamics of nascent polypeptides emerging from the ribosome, ACS Chem. Biol. 3 (2008) 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Hutchinson RB, Chen X, Zhou N, Cavagnero S, Fluorescence anisotropy decays and microscale-volume viscometry reveal the compaction of ribosome-bound nascent proteins, J. Phys. Chem. B 125 (2021) 6543–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Hardesty B, Kramer G, Folding of a nascent peptide on the ribosome, Prog. Nucleic Acid Res. Mol. Biol. 66 (2001) 41–66. [DOI] [PubMed] [Google Scholar]
- [148].Ellis JP, Culviner PH, Cavagnero S, Confined dynamics of a ribosome-bound nascent globin: Cone angle analysis of fluorescence depolarization decays in the presence of two local motions, Protein Sci. 18 (2009) 2003–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Frydman J, Erdjument-Bromage H, Tempst P, Hartl FU, Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase, Nat. Struct. Biol. 6 (1999) 697–705. [DOI] [PubMed] [Google Scholar]
- [150].Evans MS, Sander IM, Clark PL, Cotranslational Folding Promotes β-Helix Formation and Avoids Aggregation In Vivo, J. Mol. Biol. 383 (2008) 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Kim SJ, Yoon JS, Shishido H, Yang Z, Rooney LA, Barral JM, Skach WR, Translational tuning optimizes nascent protein folding in cells, Science 348 (2015) 444. [DOI] [PubMed] [Google Scholar]
- [152].Liu K, Maciuba K, Kaiser CM, The ribosome cooperates with a chaperone to guide multi-domain protein folding, Mol. Cell 74 (2019) 310–319.e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Bitran A, Jacobs WM, Zhai X, Shakhnovich E, Cotranslational folding allows misfolding-prone proteins to circumvent deep kinetic traps, Proc. Natl. Acad. Sci. U.S.A. 117 (2020) 1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Chaney JL, Steele A, Carmichael R, Rodriguez A, Specht AT, Ngo K, Li J, Emrich S, Clark PL, Widespread position-specific conservation of synonymous rare codons within coding sequences, PLoS Comput. Biol. 13 (2017) e1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Zhao V, Jacobs WM, Shakhnovich E, Effect of protein structure on evolution of cotranslational folding, Biophys. J. 119 (2020) 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Buhr F, Jha S, Thommen M, Mittelstaet J, Kutz F, Schwalbe H, Rodnina MV, Komar AA, Synonymous codons direct cotranslational folding toward different protein conformations, Mol. Cell 61 (2016) 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Ziv G, Haran G, Thirumalai D, Ribosome exit tunnel can entropically stabilize α-helices, Proc. Natl. Acad. Sci. U.S.A. 102 (2005) 18956–18961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Woolhead CA, McCormick PJ, Johnson AE, Nascent membrane and secretory proteins differ in FRET-detected folding far inside the ribosome and in their exposure to ribosomal proteins, Cell 116 (2004) 725–736. [DOI] [PubMed] [Google Scholar]
- [159].Cruz-Vera LR, Rajagopal S, Squires C, Yanofsky C, Features of ribosome-peptidyl-tRNA interactions essential for tryptophan induction of tna operon expression, Mol. Cell 19 (2005) 333–343. [DOI] [PubMed] [Google Scholar]
- [160].Houben ENG, Zarivach R, Oudega B, Luirink J Early encounters of a nascent membrane protein : specificity and timing of contacts inside and outside the ribosome, J. Cell Biol. 170 (2005) 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Ullers RS, Houben EN, Raine A, ten Hagen-Jongman CM, Ehrenberg M, Brunner J, Oudega B, Harms N, Luirink J, Interplay of signal recognition particle and trigger factor at L23 near the nascent chain exit site on the Escherichia coli ribosome, J. Cell Biol. 161 (2003) 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Peterson JH, Woolhead CA, Bernstein HD, The conformation of a nascent polypeptide inside the ribosome tunnel affects protein targeting and protein folding, Mol. Microbiol. 78 (2010) 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Wang S, Jomaa A, Jaskolowski M, Yang C, Ban N, Shan S, The molecular mechanism of cotranslational membrane protein recognition and targeting by SecA, Nat. Struct. Mol. Biol. 26 (2019) 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Guzman-Luna V, Fuchs AM, Allen AJ, Staikos A, Cavagnero S, An intrinsically disordered nascent protein interacts with specific regions of the ribosomal surface near the exit tunnel, Commun. Biol. 4 (2021) 1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Cassaignau AME, Włodarski T, Chan SHS, Woodburn LF, Bukvin IV, Streit JO, Cabrita LD, Waudby CA, Christodoulou J, Interactions between nascent proteins and the ribosome surface inhibit co-translational folding, Nat. Chem (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Dao Duc K, Batra SS, Bhattacharya N, Jamie HD Cate, Yun S. Song, Differences in the path to exit the ribosome across the three domains of life, Nucleic Acids Res. 47 (2019) 4198–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Noeske J, Wasserman MR, Terry DS, Altman RB, Blanchard SC, Cate JHD, High-resolution structure of the Escherichia coli ribosome, Nat. Struct. Mol. Biol. 22 (2015) 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Tsalkova T, Odom OW, Kramer G, Hardesty B, Different conformations of nascent peptides on ribosomes, J. Mol. Biol. 278 (1998) 713–723. [DOI] [PubMed] [Google Scholar]
- [169].Kramer G, Ramachandiran V, Hardesty B, Cotranslational folding — omnia mea mecum porto?, Int. J. Biochem. Cell B 33 (2001) 541–553. [DOI] [PubMed] [Google Scholar]
- [170].Malkin LI, Rich A, Partial resistance of nascent polypeptide chains to proteolytic digestion due to ribosomal shielding, J. Mol. Biol. 26 (1967) 329–346. [DOI] [PubMed] [Google Scholar]
- [171].Nilsson Ola B., Hedman R, Marino J, Wickles S, Bischoff L, Johansson M, Müller-Lucks A, Trovato F, Puglisi Joseph D., O’Brien Edward P., Beckmann R, von Heijne G, Cotranslational protein folding inside the ribosome exit tunnel, Cell Rep. 12 (2015) 1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Marino J, von Heijne G, Beckmann R, Small protein domains fold inside the ribosome exit tunnel, FEBS Lett. 590 (2016) 655–660. [DOI] [PubMed] [Google Scholar]
- [173].Bhushan S, Gartmann M, Halic M, Armache J-P, Jarasch A, Mielke T, Berninghausen O, Wilson DN, Beckmann R, α-Helical nascent polypeptide chains visualized within distinct regions of the ribosomal exit tunnel, Nat. Struct. Mol. Biol. 17 (2010) 313–317. [DOI] [PubMed] [Google Scholar]
- [174].Lu J, Deutsch C, Secondary structure formation of a transmembrane segment in Kv channels, Biochemistry 44 (2005) 8230–8243. [DOI] [PubMed] [Google Scholar]
- [175].Liutkute M, Maiti M, Samatova E, Enderlein J, Rodnina MV, Gradual compaction of the nascent peptide during cotranslational folding on the ribosome, eLife 9 (2020) e60895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Farías-Rico JA, Ruud Selin F, Myronidi I, Frühauf M, von Heijne G, Effects of protein size, thermodynamic stability, and net charge on cotranslational folding on the ribosome, Proc. Natl. Acad. Sci. U.S.A. 115 (2018) E9280–E9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Tian P, Steward A, Kudva R, Su T, Shilling PJ, Nickson AA, Hollins JJ, Beckmann R, von Heijne G, Clarke J, Best RB, Folding pathway of an Ig domain is conserved on and off the ribosome, Proc. Natl. Acad. Sci. U.S.A. 115 (2018) E11284–E11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Nilsson OB, Nickson AA, Hollins JJ, Wickles S, Steward A, Beckmann R, von Heijne G, Clarke J, Cotranslational folding of spectrin domains via partially structured states, Nat. Struct. Mol. Biol. 24 (2017) 221–225. [DOI] [PubMed] [Google Scholar]
- [179].Mercier E, Rodnina MV, Co-translational folding trajectory of the HemK helical domain, Biochemistry (2018). [DOI] [PubMed] [Google Scholar]
- [180].Samelson AJ, Jensen MK, Soto RA, Cate JHD, Marqusee S, Quantitative determination of ribosome nascent chain stability, Proc. Natl. Acad. Sci. U.S.A. 113 (2016) 13402–13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Mounce BC, Kurt N, Ellison PA, Cavagnero S, Nonrandom distribution of intramolecular contacts in native single-domain proteins, Proteins 75 (2009) 404–412. [DOI] [PubMed] [Google Scholar]
- [182].Kurt N, Cavagnero S, The burial of solvent-accessible surface area is a predictor of polypeptide folding and misfolding as a function of chain elongation, J. Am. Chem. Soc. 127 (2005) 15690–15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Frydman J, Folding of newly translated proteins in vivo: The role of molecular chaperones, Annu. Rev. Biochem. 70 (2001) 603–647. [DOI] [PubMed] [Google Scholar]
- [184].Koubek J, Schmitt J, Galmozzi CV, Kramer G, Mechanisms of cotranslational protein maturation in bacteria, Front. Mol. Biosci 8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Finkelstein AV, Badretdinov AY, Physical reason for fast folding of the stable spatial structure of proteins: A solution of the Levinthal paradox, Mol. Biol. 31 (1997) 391–398. [Google Scholar]
- [186].Finkelstein AV, Badretdinov AY, Rate of protein folding near the point of thermodynamic equilibrium between the coil and the most stable chain fold, Fold Des. 2 (1997) 115–121. [DOI] [PubMed] [Google Scholar]
- [187].Gutin AM, Abkevich VI, Shakhnovich EI, Chain length scaling of protein folding time, Phys. Rev. Lett. 77 (1996) 5433–5436. [DOI] [PubMed] [Google Scholar]
- [188].Thirumalai D, From minimal models to real proteins - time scales for protein-folding kinetics, J. Phys. I 5 (1995) 1457–1467. [Google Scholar]
- [189].Langer T, Lu C, Echols H, Flanagan J, Hayer MK, Hartl FU, Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding, Nature 356 (1992) 683–689. [DOI] [PubMed] [Google Scholar]
- [190].Martin J, Langer T, Boteva R, Schramel A, Horwich AL, Hartl FU, Chaperonin-mediated protein folding at the surface of groEL through a 'molten globule'-like intermediate, Nature 352 (1991) 36. [DOI] [PubMed] [Google Scholar]
- [191].Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B, Trigger Factor and DnaK Cooperate in Folding of Newly Synthesized Proteins, Nature 400 (1999) 693–696. [DOI] [PubMed] [Google Scholar]
- [192].Hoffmann A, Bukau B, Kramer G, Structure and function of the molecular chaperone Trigger Factor, Biochim. Biophys. Acta Mol. Cell Res 1803 (2010) 650–661. [DOI] [PubMed] [Google Scholar]
- [193].Martinez-Hackert E, Hendrickson WA, Promiscuous substrate recognition in folding and assembly activities of the trigger factor chaperone, Cell 138 (2009) 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Valent QA, Kendall DA, High S, Kusters R, Oudega B, Lurink J, Early events in preprotein recognition in E.coli: interaction of SRP and trigger factor with nascent polypeptides, EMBO J. 14 (1995) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Schaffitzel E, Rudiger S, Bukau B, Deuerling E, Functional dissection of trigger factor and DnaK: Interactions with nascent polypeptides and thermally denatured proteins, Biol. Chem. 382 (2001) 9. [DOI] [PubMed] [Google Scholar]
- [196].Bukau B, Deuerling E, Pfund C, Craig EA, Getting newly synthesized proteins into shape, Cell 101 (2000) 119–122. [DOI] [PubMed] [Google Scholar]
- [197].Maier R, Scholz C, Schmid FX, Dynamic association of trigger factor with protein substrates1, J. Mol. Biol. 314 (2001) 1181–1190. [DOI] [PubMed] [Google Scholar]
- [198].Rutkowska A, Mayer MP, Hoffmann A, Merz F, Zachmann-Brand B, Schaffitzel C, Ban N, Deuerling E, Bukau B, Dynamics of trigger factor interaction with translating ribosomes, J. Biol. Chem. 283 (2008) 4124–4132. [DOI] [PubMed] [Google Scholar]
- [199].Teter SA, Houry WA, Ang D, Tradler T, Rockabrand D, Fischer G, Blum P, Georgopoulos C, Hartl FU, Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains, Cell 97 (1999) 755–765. [DOI] [PubMed] [Google Scholar]
- [200].Balchin D, Hayer-Hartl M, Hartl FU, Recent advances in understanding catalysis of protein folding by molecular chaperones, FEBS Lett. 594 (2020) 2770–2781. [DOI] [PubMed] [Google Scholar]
- [201].Ewalt KL, Hendrick JP, Houry WA, Hartl FU, In vivo observation of polypeptide flux through the bacterial chaperonin system, Cell 90 (1997) 491–500. [DOI] [PubMed] [Google Scholar]
- [202].Houry WA, Frishman D, Eckerskorn C, Lottspeich F, Hartl FU, Identification of in vivo substrates of the chaperonin GroEL, Nature 402 (1999) 147–154. [DOI] [PubMed] [Google Scholar]
- [203].Haslberger T, Zdanowicz A, Brand I, Kirstein J, Turgay K, Mogk A, Bukau B, Protein disaggregation by the AAA+ chaperone ClpB involves partial threading of looped polypeptide segments, Nat. Struct. Mol. Biol. 15 (2008) 641–650. [DOI] [PubMed] [Google Scholar]
- [204].Baneyx F, Mujacic M, Recombinant protein folding and misfolding in Escherichia coli, Nat. Biotechnol. 22 (2004) 1399–1408. [DOI] [PubMed] [Google Scholar]
- [205].Rosenzweig R, Moradi S, Zarrine-Afsar A, Glover JR, Kay LE, Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction, Science 339 (2013) 1080–1083. [DOI] [PubMed] [Google Scholar]
- [206].Crooke E, Wickner W, Trigger Factor: A soluble protein that folds pro-OmpA into a membrane-assembly-competent form, Proc. Natl. Acad. Sci. U.S.A. 84 (1987) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Stoller G, Rucknagel KP, Nierhaus KH, Schmid FX, Fischer G, Rahfeld J-U, A ribosome-associated peptidyl-prolyl cis/trans isomerse identified as the trigger factor, EMBO J. 14 (1995) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Lill R, Crooke E, Guthrie B, Wickner W, The “trigger factor cycle” includes ribosomes, presecretory proteins, and the plasma membrane, Cell 54 (1988) 1013–1018. [DOI] [PubMed] [Google Scholar]
- [209].Patzelt H, Kramer G, Rauch T, Schönfeld H-J, Bukau B, Deuerling E, Three-state equilibrium of Escherichia coli trigger factor, 383 (2002) 1611–1619. [DOI] [PubMed] [Google Scholar]
- [210].Genevaux P, Keppel F, Schwager F, Langendijk-Genevaux PS, Hartl FU, Georgopoulos C, In vivo analysis of the overlapping functions of DnaK and trigger factor, EMBO Rep. 5 (2004) 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Oh E, Becker Annemarie H., Sandikci A, Huber D, Chaba R, Gloge F, Nichols Robert J., Typas A, Gross Carol A., Kramer G, Weissman Jonathan S., Bukau B, Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo, Cell 147 (2011) 1295–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Kramer G, Rutkowska A, Wegrzyn RD, Patzelt H, Kurz TA, Merz F, Rauch T, Vorderwulbecke S, Deuerling E, Bukau B, Functional dissection of Escherichia coli trigger factor: Unraveling the function of individual domains, J. Bacteriol. 186 (2004) 3777–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Kramer G, Patzelt H, Rauch T, Kurz TA, Vorderwulbecke S, Bukau B, Deuerling E, Trigger factor peptidyl-prolyl cis/trans isomerase activity is not essential for the folding of cytosolic proteins in Escherichia coli, J. Biol. Chem. 279 (2004) 14165–14170. [DOI] [PubMed] [Google Scholar]
- [214].Huang G-C, Li Z-Y, Zhou J-M, Fischer G, Assisted folding of D-glyceraldehyde-3-phosphate dehydrogenase by trigger factor, Protein Sci. 9 (2000) 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Agashe VR, Guha S, Chang HC, Genevaux P, Hayer-Hartl M, Stemp M, Georgopoulos C, Hartl FU, Barral JM, Function of trigger factor and DnaK in multidomain protein folding: Increase in yield at the expense of folding speed, Cell 117 (2004) 199–209. [DOI] [PubMed] [Google Scholar]
- [216].Saio T, Guan X, Rossi P, Economou A, Kalodimos CG, Structural basis for protein antiaggregation activity of the trigger factor chaperone, Science 344 (2014) 1250494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Nilsson OB, Muller-Lucks A, Kramer G, Bukau B, von Heijne G, Trigger factor reduces the force exerted on the nascent chain by a cotranslationally folding protein, J. Mol. Biol. 428 (2016) 1356–1364. [DOI] [PubMed] [Google Scholar]
- [218].Hoffmann A, Merz F, Rutkowska A, Zachmann-Brand B, Deuerling E, Bukau B, Trigger factor forms a protective shield for nascent polypeptides at the ribosome, J. Biol. Chem. 281 (2006) 6539–6545. [DOI] [PubMed] [Google Scholar]
- [219].Ferbitz L, Maier T, Patzelt H, Bukau B, Deuerling E, Ban N, Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins, Nature 431 (2004) 590–596. [DOI] [PubMed] [Google Scholar]
- [220].Merz F, Boehringer D, Schaffitzel C, Preissler S, Hoffmann A, Maier T, Rutkowska A, Lozza J, Ban N, Bukau B, Deuerling E, Molecular mechanism and structure of Trigger Factor bound to the translating ribosome, EMBO J. 27 (2008) 1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Zarnt T, Tradler T, Stoller G, Scholz C, Schmid FX, Fischer G, Modular structure of the trigger factor required for high activity in protein folding11Edited by A. R. Fersht, J. Mol. Biol. 271 (1997) 827–837. [DOI] [PubMed] [Google Scholar]
- [222].Kramer G, Rauch T, Rist W, Vorderwülbecke S, Patzelt H, Schulze-Specking A, Ban N, Deuerling E, Bukau B, L23 protein functions as a chaperone docking site on the ribosome, Nature 419 (2002) 171–174. [DOI] [PubMed] [Google Scholar]
- [223].Baram D, Pyetan E, Sittner A, Auerbach-Nevo T, Bashan A, Yonath A, Structure of trigger factor binding domain in biologically homologous complex with eubacterial ribosome reveals its chaperone action, Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Schlünzen F, Wilson DN, Tian P, Harms JM, McInnes SJ, Hansen HAS, Albrecht R, Buerger J, Wilbanks SM, Fucini P, The Binding Mode of the Trigger Factor on the Ribosome: Implications for Protein Folding and SRP Interaction, Structure 13 (2005) 1685–1694. [DOI] [PubMed] [Google Scholar]
- [225].Hesterkamp T, Hauser S, Lutcke H, Bukau B, Escherichia coli trigger factoris a prolyl isomerase that associates with nascent polypeptide chains, Proc. Natl. Acad. Sci. U.S.A. 93 (1996) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Merz F, Hoffmann A, Rutkowska A, Zachmann-Brand B, Bukau B, Deuerling E, The C-terminal domain of Escherichia coli trigger factor represents the central module of its chaperone activity, J. Biol. Chem. 281 (2006) 31963–31971. [DOI] [PubMed] [Google Scholar]
- [227].Zeng L-L, Yu L, Li Z-Y, Perrett S, Zhou J-M, Effect of C-terminal truncation on the molecular chaperone function and dimerization of Escherichia coli trigger factor, Biochimie 88 (2006) 613–619. [DOI] [PubMed] [Google Scholar]
- [228].Kaiser CM, Chang HC, Agashe VR, Lakshmipathy SK, Etchells SA, Hayer-Hartl M, Hartl FU, Barral JM, Real-time observation of trigger factor function on translating ribosomes, Nature 444 (2006) 455–460. [DOI] [PubMed] [Google Scholar]
- [229].Patzelt H, Rudiger S, Brehmer D, Kramer G, Vorderwulbecke S, Schaffitzel E, Waitz A, Hesterkamp T, Dong L, Schneider-Mergener J, Bukau B, Deuerling E, Binding specificity of Escherichia coli trigger factor, Proc. Natl. Acad. Sci. U.S.A. 98 (2001) 14244–14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Lakshmipathy SK, Tomic S, Kaiser CM, Chang HC, Genevaux P, Georgopoulos C, Barral JM, Johnson AE, Hartl FU, Etchells SA, Identification of nascent chain interaction sites on trigger factor, J. Biol. Chem. 282 (2007) 12186–12193. [DOI] [PubMed] [Google Scholar]
- [231].Raine A, Lovmar M, Wikberg J, Ehrenberg M, Trigger factor binding to ribosomes with nascent peptide chains of varying lengths and sequences, J. Biol. Chem. 281 (2006) 28033–28038. [DOI] [PubMed] [Google Scholar]
- [232].Deeng J, Chan KY, van der Sluis EO, Berninghausen O, Han W, Gumbart J, Schulten K, Beatrix B, Beckmann R, Dynamic behavior of trigger factor on the ribosome, J. Mol. Biol. 428 (2016) 3588–3602. [DOI] [PubMed] [Google Scholar]
- [233].Hoffmann A, Becker AH, Zachmann-Brand B, Deuerling E, Bukau B, Kramer G, Concerted Action of the Ribosome and the Associated Chaperone Trigger Factor Confines Nascent Polypeptide Folding, Mol. Cell 48 (2012) 63–74. [DOI] [PubMed] [Google Scholar]
- [234].Mashaghi A, Kramer G, Bechtluft P, Zachmann-Brand B, Driessen AJM, Bukau B, Tans SJ, Reshaping of the conformational search of a protein by the chaperone trigger factor, Nature 500 (2013) 98. [DOI] [PubMed] [Google Scholar]
- [235].Maier R, Eckert B, Scholz C, Lilie H, Schmid F-X, Interaction of trigger factor with the ribosome, J. Mol. Biol. 326 (2003) 585–592. [DOI] [PubMed] [Google Scholar]
- [236].Scholz C, Stoller G, Zarnt T, Fischer G, Schmid FX, Cooperation of enzymatic and chaperone functions of trigger factor in the catalysis of protein folding, EMBO J. 16 (1997) 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Lakshmipathy SK, Gupta R, Pinkert S, Etchells SA, Hartl FU, Versatility of trigger factor interactions with ribosome-nascent chain complexes, J. Biol. Chem. 285 (2010) 27911–27923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Ritossa F, A new puffing pattern induced by temperature shock and DNP in drosophila, Experientia 18 (1962) 571–573. [Google Scholar]
- [239].Ritossa F, New puffs induced by temperature shock, DNP and salicylate in salivary chromosomes of Drosophila melanogaster, Drosoph. Inf. Serv 37 (1963) 122–123. [Google Scholar]
- [240].Ritossa F, Experimental activation of specific loci in polytene chromosomes of Drosophila, Exp. Cell Res. 35 (1964) 601–607. [DOI] [PubMed] [Google Scholar]
- [241].Ritossa F, Discovery of the heat shock response, Cell Stress Chaperones 1 (1996) 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Craig EA, Gross CA, Is Hsp70 the cellular thermometer?, Trends Biochem. Sci. 16 (1991) 135–140. [DOI] [PubMed] [Google Scholar]
- [243].Tissiéres A, Mitchell HK, Tracy UM, Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs, J. Mol. Biol. 84 (1974) 389–398. [DOI] [PubMed] [Google Scholar]
- [244].Lindquist S, Craig EA, The heat-shock proteins, Annu. Rev. Genet. 22 (1988) 631–677. [DOI] [PubMed] [Google Scholar]
- [245].Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B, Hightower LE, Guidelines for the nomenclature of the human heat shock proteins, Cell Stress and Chaperones 14 (2009) 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Goloubinoff P, De Los Rios P, The mechanism of Hsp70 chaperones:(entropic) pulling the models together, Trends Biochem. Sci. 32 (2007) 372–380. [DOI] [PubMed] [Google Scholar]
- [247].Mayer MP, Bukau B, Hsp70 chaperones: Cellular functions and molecular mechanism, Cellular and Molecular Life Sciences 62 (2005) 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Laksanalamai P, Whitehead TA, Robb FT, Minimal protein-folding systems in hyperthermophilic archaea, Nat. Rev. Microbiol. 2 (2004) 315–324. [DOI] [PubMed] [Google Scholar]
- [249].Rebeaud ME, Mallik S, Goloubinoff P, Tawfik DS, On the evolution of chaperones and cochaperones and the expansion of proteomes across the Tree of Life, Proc. Natl. Acad. Sci. U.S.A. 118 (2021) e2020885118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Macario AJ, Lange M, Ahring BK, De Macario EC, Stress genes and proteins in the archaea, Microbiology and Molecular Biology Reviews 63 (1999) 923–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Gribaldo S, Lumia V, Creti R, Conway de Macario E, Sanangelantoni A, Cammarano P, Discontinuous occurrence of the hsp70 (dnaK) gene among Archaea and sequence features of HSP70 suggest a novel outlook on phylogenies inferred from this protein, J. Bacteriol. 181 (1999) 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Warnecke T, Loss of the DnaK-DnaJ-GrpE chaperone system among the Aquificales, Molecular biology and evolution 29 (2012) 3485–3495. [DOI] [PubMed] [Google Scholar]
- [253].Calloni G, Chen T, Schermann SM, Chang HC, Genevaux P, Agostini F, Tartaglia GG, Hayer-Hartl M, Hartl FU, DnaK functions as a central hub in the E. coli chaperone network, Cell Reports 1 (2012) 251–264. [DOI] [PubMed] [Google Scholar]
- [254].Hesterkamp T, Bukau B, Role of the DnaK and HscA homologs of Hsp70 chaperones in protein folding in E.coli, EMBO J. 17 (1998) 4818–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Zimmerman SB, Trach SO, Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli, J. Mol. Biol. 222 (1991) 599–620. [DOI] [PubMed] [Google Scholar]
- [256].Hartl FU, Hayer-Hartl M, Molecular Chaperones in the Cytosol: from Nascent Chain to Folded Protein, Science 295 (2002) 8. [DOI] [PubMed] [Google Scholar]
- [257].Mogk A, Tomoyasu T, Goloubinoff P, Rüdiger S, Röder D, Langen H, Bukau B, Identification of thermolabile Escherichia coli proteins: Prevention and reversion of aggregation by DnaK and ClpB, EMBO J. 18 (1999) 6934–6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Hartl FU, Hayer-Hartl M, Converging concepts of protein folding in vitro and in vivo, Nat. Struct. Mol. Biol. 16 (2009) 574–581. [DOI] [PubMed] [Google Scholar]
- [259].Imamoglu R, Balchin D, Hayer-Hartl M, Hartl FU, Bacterial Hsp70 resolves misfolded states and accelerates productive folding of a multi-domain protein, Nat. Commun. 11 (2020) 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Zylicz M, Ang D, Liberek K, Georgopoulos C, Initiation of lambda DNA replication with purified host-and bacteriophage-encoded proteins: the role of the DnaK, DnaJ and GrpE heat shock proteins, EMBO J. 8 (1989) 1601–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Meimaridou E, Gooljar SB, Chapple JP, From hatching to dispatching: the multiple cellular roles of the Hsp70 molecular chaperone machinery, Journal of molecular endocrinology 42 (2009) 1–9. [DOI] [PubMed] [Google Scholar]
- [262].Peschke M, Le Goff M, Koningstein GM, Karyolaimos A, de Gier J-W, van Ulsen P, Luirink J, SRP, FtsY, DnaK and YidC are required for the biogenesis of the E. coli tail-anchored membrane proteins DjlC and Flk, J. Mol. Biol. 430 (2018) 389–403. [DOI] [PubMed] [Google Scholar]
- [263].Mogk A, Bukau B, Molecular chaperones: Structure of a protein disaggregase, Curr. Biol. 14 (2004) R78–80. [DOI] [PubMed] [Google Scholar]
- [264].Aguado A, Fernandez-Higuero JA, Moro F, Muga A, Chaperone-assisted protein aggregate reactivation: Different solutions for the same problem, Arch Biochem Biophys 580 (2015) 121–134. [DOI] [PubMed] [Google Scholar]
- [265].Diamant S, Ben-Zvi AP, Bukau B, Goloubinoff P, Size-dependent disaggregation of stable protein aggregates by the DnaK chaperone machinery, J. Biol. Chem. 275 (2000) 21107–21113. [DOI] [PubMed] [Google Scholar]
- [266].Zolkiewski M, ClpB cooperates with DnaK, DnaJ, and GrpE in suppressing protein aggregation: a novel multi-chaperone system from Escherichia coli, J. Biol. Chem. 274 (1999) 28083–28086. [DOI] [PubMed] [Google Scholar]
- [267].Goloubinoff P, Mogk A, Zvi APB, Tomoyasu T, Bukau B, Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network, Proc. Natl. Acad. Sci. U.S.A. 96 (1999) 13732–13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Veinger L, Diamant S, Buchner J, Goloubinoff P, The small heat-shock protein IbpB from Escherichia coli stabilizes stress-denatured proteins for subsequent refolding by a multichaperone network, J. Biol. Chem. 273 (1998) 11032–11037. [DOI] [PubMed] [Google Scholar]
- [269].Żwirowski S, Kłosowska A, Obuchowski I, Nillegoda NB, Piróg A, Ziętkiewicz S, Bukau B, Mogk A, Liberek K, Hsp70 displaces small heat shock proteins from aggregates to initiate protein refolding, EMBO J. 36 (2017) 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Aguilar-Rodríguez J, Sabater-Muñoz B, Montagud-Martínez R, Berlanga V, Alvarez-Ponce D, Wagner A, Fares MA, The molecular chaperone DnaK is a source of mutational robustness, Genome Biol. Evol. 8 (2016) 2979–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Kadibalban AS, Bogumil D, Landan G, Dagan T, DnaK-dependent accelerated evolutionary rate in prokaryotes, Genome Biol. Evol. 8 (2016) 1590–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Rutherford SL, Between genotype and phenotype: protein chaperones and evolvability, Nat. Rev. Genet. 4 (2003) 263–274. [DOI] [PubMed] [Google Scholar]
- [273].Victor MP, Acharya D, Chakraborty S, Ghosh TC, Chaperone client proteins evolve slower than non-client proteins, Funct. Integr. Genomics 20 (2020) 621–631. [DOI] [PubMed] [Google Scholar]
- [274].Brodsky JL, Chiosis G, Hsp70 molecular chaperones: emerging roles in human disease and identification of small molecule modulators, Curr. Top. Med. Chem. 6 (2006) 1215–1225. [DOI] [PubMed] [Google Scholar]
- [275].Monsellier E, Chiti F, Prevention of amyloid-like aggregation as a driving force of protein evolution, EMBO Rep. 8 (2007) 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER, Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate, Proc. Natl. Acad. Sci. U.S.A. 106 (2009) 8471–8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Sondermann H, Scheufler C, Schneider C, Höhfeld J, Hartl F-U, Moarefi I, Structure of a Bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors, Science 291 (2001) 1553–1557. [DOI] [PubMed] [Google Scholar]
- [278].Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F-U, Kuriyan J, Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK, Science 276 (1997) 431–435. [DOI] [PubMed] [Google Scholar]
- [279].Kityk R, Kopp J, Sinning I, Mayer MP, Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones, Mol. Cell 48 (2012) 863–874. [DOI] [PubMed] [Google Scholar]
- [280].Buchberger A, Theyssen H, Schröder H, McCarty JS, Virgallita G, Milkereit P, Reinstein J, Bukau B, Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication, J. Biol. Chem. 270 (1995) 16903–16910. [DOI] [PubMed] [Google Scholar]
- [281].Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA, Structural analysis of substrate binding by the molecular chaperone DnaK, Science 272 (1996) 1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Liu QL, Liang C, Zhou L, Structural and functional analysis of the Hsp70/Hsp40 chaperone system, Protein Sci. 29 (2020) 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Qi R, Sarbeng EB, Liu Q, Le KQ, Xu X, Xu H, Yang J, Wong JL, Vorvis C, Hendrickson WA, Zhou L, Liu Q, Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP, Nat. Struct. Mol. Biol. 20 (2013) 900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Rüdiger S, Germeroth L, Schneider-Mergener J, Bukau B, Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries, EMBO J. 16 (1997) 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Vega CA, Kurt N, Chen Z, Rüdiger S, Cavagnero S, Binding specificity of an α-helical protein sequence to a full-length Hsp70 chaperone and its minimal substrate-binding domain, Biochemistry 45 (2006) 13835–13846. [DOI] [PubMed] [Google Scholar]
- [286].Mayer MP, Rudiger S, Bukau B, Molecular basis for interactions of the DnaK chaperone with substrates, Biol. Chem. 381 (2000) 9. [DOI] [PubMed] [Google Scholar]
- [287].Schmid D, Baici A, Gehring H, Christen P, Kinetics of molecular chaperone action, Science 263 (1994) 971–973. [DOI] [PubMed] [Google Scholar]
- [288].Mayer MP, Schröder H, Rüdiger S, Paal K, Laufen T, Bukau B, Multistep mechanism of substrate binding determines chaperone activity of Hsp70, Nat. Struct. Biol. 7 (2000) 586–593. [DOI] [PubMed] [Google Scholar]
- [289].McCarty JS, Buchberger A, Reinstein J, Bukau B, The role of ATP in the functional cycle of the DnaK chaperone system, J. Mol. Biol. 249 (1995) 126–137. [DOI] [PubMed] [Google Scholar]
- [290].Clerico EM, Tilitsky JM, Meng W, Gierasch LM, How Hsp70 molecular machines interact with their substrates to mediate diverse physiological functions, J. Mol. Biol. 427 (2015) 1575–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B, Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones, Proc. Natl. Acad. Sci. U.S.A. 96 (1999) 5452–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Mayer MP, The Hsp70-chaperone machines in bacteria, Front. Mol. Biosci 8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Barends TR, Brosi RW, Steinmetz A, Scherer A, Hartmann E, Eschenbach J, Lorenz T, Seidel R, Shoeman RL, Zimmermann S, Bittl R, Schlichting I, Reinstein J, Combining crystallography and EPR: Crystal and solution structures of the multidomain cochaperone DnaJ, Acta Crystallogr. D Biol. Crystallogr 69 (2013) 1540–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Vogel M, Mayer MP, Bukau B, Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker, J. Biol. Chem. 281 (2006) 38705–38711. [DOI] [PubMed] [Google Scholar]
- [295].Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM, Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker, Mol. Cell 26 (2007) 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Gething M-J, Molecular chaperones: Clasping the prize, Curr. Biol. 6 (1996) 1573–1576. [DOI] [PubMed] [Google Scholar]
- [297].Kuriyan J, Wilz S, Karplus M, Petsko GA, X-ray structure and refinement of carbon-monoxy (Fe II)-myoglobin at 1.5 Å resolution, J. Mol. Biol. 192 (1986) 133–154. [DOI] [PubMed] [Google Scholar]
- [298].Buchberger A, Schröder H, Büttner M, Valencia A, Bukau B, A conserved loop in the ATPase domain of the DnaK chaperone is essential for stable binding of GrpE, Nat. Struct. Biol. 1 (1994) 95–101. [DOI] [PubMed] [Google Scholar]
- [299].Schönfeld H-J, Schmidt D, Schröder H, Bukau B, The DnaK chaperone system of Escherichia coli: quaternary structures and interactions of the DnaK and GrpE components, J. Biol. Chem. 270 (1995) 2183–2189. [DOI] [PubMed] [Google Scholar]
- [300].Lai AL, Clerico EM, Blackburn ME, Patel NA, Robinson CV, Borbat PP, Freed JH, Gierasch LM, Key features of an Hsp70 chaperone allosteric landscape revealed by ion-mobility native mass spectrometry and double electron-electron resonance, J. Biol. Chem. 292 (2017) 8773–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Landry SJ, Jordan R, McMacken R, Gierasch LM, Different conformations for the same polypeptide bound to chaperones DnaK and GroEL, Nature 355 (1992) 455. [DOI] [PubMed] [Google Scholar]
- [302].Popp S, Packschies L, Radzwill N, Vogel KP, Steinhoff H-J, Reinstein J, Structural dynamics of the DnaK–peptide complex, J. Mol. Biol. 347 (2005) 1039–1052. [DOI] [PubMed] [Google Scholar]
- [303].Chen Z, Kurt N, Rajagopalan S, Cavagnero S, Secondary structure mapping of DnaK-bound protein fragments: chain helicity and local helix unwinding at the binding site, Biochemistry 45 (2006) 12325–12333. [DOI] [PubMed] [Google Scholar]
- [304].Kurt N, Cavagnero S, Nonnative helical motif in a chaperone-bound protein fragment, Biophys. J. 94 (2008) L48–L50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Kellner R, Hofmann H, Barducci A, Wunderlich B, Nettels D, Schuler B, Single-molecule spectroscopy reveals chaperone-mediated expansion of substrate protein, Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 13355–13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Lee JH, Zhang D, Hughes C, Okuno Y, Sekhar A, Cavagnero S, Heterogeneous binding of the SH3 client protein to the DnaK molecular chaperone, Proc. Natl. Acad. Sci. U.S.A. 112 (2015) E4206–E4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Sekhar A, Santiago M, Lam HN, Lee JH, Cavagnero S, Transient interactions of a slow-folding protein with the Hsp70 chaperone machinery, Protein Sci. 21 (2012) 1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Cohen SI, Vendruscolo M, Dobson CM, Knowles TP, From macroscopic measurements to microscopic mechanisms of protein aggregation, J. Mol. Biol. 421 (2012) 160–171. [DOI] [PubMed] [Google Scholar]
- [309].Ciryam P, Kundra R, Morimoto RI, Dobson CM, Vendruscolo M, Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases, Trends Pharmacol. Sci. 36 (2015) 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Hayashi H, Kimura N, Yamaguchi H, Hasegawa K, Yokoseki T, Shibata M, Yamamoto N, Michikawa M, Yoshikawa Y, Terao K, A seed for Alzheimer amyloid in the brain, J. Neurosci. 24 (2004) 4894–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Jewett A, Shea J-E, Folding on the chaperone: yield enhancement through loose binding, J. Mol. Biol. 363 (2006) 945–957. [DOI] [PubMed] [Google Scholar]
- [312].Jakob U, Gaestel M, Engel K, Buchner J, Small heat shock proteins are molecular chaperones, J. Biol. Chem. 268 (1993) 1517–1520. [PubMed] [Google Scholar]
- [313].Buchner J, Schmidt M, Fuchs M, Jaenicke R, Rudolph R, Schmid FX, Kiefhaber T, GroE facilitates refolding of citrate synthase by suppressing aggregation, Biochemistry 30 (1991) 6. [DOI] [PubMed] [Google Scholar]
- [314].Cry DM, Cooperation of the molecular chaperone Ydj1 with specific Hsp70 homologs to suppress protein aggregation, FEBS Lett. 359 (1995) 129–132. [DOI] [PubMed] [Google Scholar]
- [315].Sekhar A, Lam HN, Cavagnero S, Protein folding rates and thermodynamic stability are key determinants for interaction with the Hsp70 chaperone system, Protein Sci. 21 (2012) 1489–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Mashaghi A, Bezrukavnikov S, Minde DP, Wentink AS, Kityk R, Zachmann-Brand B, Mayer MP, Kramer G, Bukau B, Tans SJ, Alternative modes of client binding enable functional plasticity of Hsp70, Nature 539 (2016) 448–451. [DOI] [PubMed] [Google Scholar]
- [317].Mecha MF, Macchi JK, Winkler GLW, Yang H, Chen X, Hutchinson RB, Cavagnero S, In praise of imperfection: The Hsp70 chaperone prevents aggregation of a predictable class of proteins over a limited time span, In preparation. [Google Scholar]
- [318].Sharma SK, De Los Rios P, Christen P, Lustig A, Goloubinoff P, The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase, Nat. Chem. Biol. 6 (2010) 914–920. [DOI] [PubMed] [Google Scholar]
- [319].Noguchi A, Ikeda A, Mezaki M, Fukumori Y, Kanemori M, DnaJ-promoted binding of DnaK to multiple sites on σ32 in the presence of ATP, J. Bacteriol. 196 (2014) 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Sekhar A, Nagesh J, Rosenzweig R, Kay LE, Conformational heterogeneity in the Hsp70 chaperone-substrate ensemble identified from analysis of NMR-detected titration data, Protein Sci. 26 (2017) 2207–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Rosenzweig R, Sekhar A, Nagesh J, Kay LE, Promiscuous binding by Hsp70 results in conformational heterogeneity and fuzzy chaperone-substrate ensembles, eLife 6 (2017) e28030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Clerico EM, Meng WL, Pozhidaeva A, Bhasne K, Petridis C, Gierasch LM, Hsp70 molecular chaperones: Multifunctional allosteric holding and unfolding machines, Biochem. J. 476 (2019) 1653–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B, The Hsp70 chaperone network, Nat. Rev. Mol. Cell Biol. 20 (2019) 665–680. [DOI] [PubMed] [Google Scholar]
- [324].Mendoza JA, Rogers E, Lorimer GH, Horowitz PM, Chaperonins facilitate the in vitro folding of monomeric mitochondrial rhodanese, J. Biol. Chem. 266 (1991) 13044–13049. [PubMed] [Google Scholar]
- [325].Liu C, Young AL, Starling-Windhof A, Bracher A, Saschenbrecker S, Rao BV, Rao KV, Berninghausen O, Mielke T, Hartl FU, Beckmann R, Hayer-Hartl M, Coupled chaperone action in folding and assembly of hexadecameric Rubisco, Nature 463 (2010) 197–202. [DOI] [PubMed] [Google Scholar]
- [326].Chow C, Kurt N, Murphy RM, Cavagnero S, Structural characterization of apomyoglobin self-associated species in aqueous buffer and urea solution, Biophys. J. 90 (2006) 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].London J, Skrzynia C, Goldberg ME, Renaturation of Escherichia coli tryptophanase after exposure to 8 m urea, Eur. J. Biochem. 47 (1974) 409–415. [DOI] [PubMed] [Google Scholar]
- [328].Kim D, Yu M-H, Folding pathway of human α1-antitrypsin: characterization of an intermediate that is active but prone to aggregation, Biochem. Biophys. Res. Commun. 226 (1996) 378–384. [DOI] [PubMed] [Google Scholar]
- [329].Schindler T, Schmid FX, Thermodynamic properties of an extremely rapid protein folding reaction, Biochemistry 35 (1996) 16833–16842. [DOI] [PubMed] [Google Scholar]
- [330].Taddei N, Chiti F, Paoli P, Fiaschi T, Bucciantini M, Stefani M, Dobson CM, Ramponi G, Thermodynamics and kinetics of folding of common-type acylphosphatase: Comparison to the highly homologous muscle isoenzyme, Biochemistry 38 (1999) 2135–2142. [DOI] [PubMed] [Google Scholar]
- [331].Manyusa S, Whitford D, Defining Folding and Unfolding Reactions of Apocytochrome b5 Using Equilibrium and Kinetic Fluorescence Measurements, Biochemistry 38 (1999) 9533–9540. [DOI] [PubMed] [Google Scholar]
- [332].Pascher T, Temperature and Driving Force Dependence of the Folding Rate of Reduced Horse Heart Cytochrome c, Biochemistry 40 (2001) 5812–5820. [DOI] [PubMed] [Google Scholar]
- [333].Rudiger S, Schneider-Mergener J, Bukau B, Its substrate specificity characterizes the DnaJ co-chaperone as a scanning factor for the DnaK chaperone, EMBO J. 20 (2001) 1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.