

ABSTRACT
Candida albicans is a commensal yeast fungus of the human oral, gastrointestinal, and genital mucosal surfaces, and skin. Antibiotic-induced dysbiosis, iatrogenic immunosuppression, and/or medical interventions that impair the integrity of the mucocutaneous barrier and/or perturb protective host defense mechanisms enable C. albicans to become an opportunistic pathogen and cause debilitating mucocutaneous disease and/or life-threatening systemic infections. In this review, we synthesize our current knowledge of the tissue-specific determinants of C. albicans pathogenicity and host immune defense mechanisms.
KEYWORDS: Candida albicans, candidiasis, pathogenesis, virulence, immunity, host-pathogen interactions
Introduction
As one of the largest eukaryotic kingdoms, fungi have a variety of life cycle patterns with adaptations in metabolism and morphogenesis that enable them to adjust to the changing ecosystems [1]. Despite being estimated to have between 1.5 and 5 million fungal species, only a few hundred of those can cause clinical disease in humans [2]. The phylum Ascomycota contains some of the most successful human pathogens; these include virulent fungi that can infect individuals without immune compromise such as Histoplasma, Blastomyces, Coccidioides, and Paracoccidioides species as well as opportunistic fungi that cause disease primarily in immunosuppressed individuals such as Aspergillus, Fusarium, Scedosporium, and Candida species.
Candida species are responsible for the majority of human infections caused by fungal pathogens. Members of these species include the most frequent cause of opportunistic infections, Candida albicans, the drug-resistant Candida glabrata, the new global public health threat Candida auris, and other emerging species such as Candida tropicalis, Candida parapsilosis, and Candida krusei [3,4]. In this review, we focus on C. albicans and the reader is referred to excellent overviews of non-albicans Candida species elsewhere [4–8]. Existing as a commensal in a large proportion of the human population, C. albicans colonizes the oral, gastrointestinal, and genital tracts asymptomatically [9,10]. However, upon perturbation of barrier integrity and/or host immune responses, the fungus can migrate through the epithelium and access deep-seated anatomical niches to cause infection. Medically important infections caused by C. albicans can broadly be classified into two subtypes: mucosal and systemic (Figure 1). Mucocutaneous surfaces primarily affected by C. albicans are the vaginal (vulvovaginal candidiasis [VVC]), the oral (oropharyngeal candidiasis [OPC]), the esophageal (esophageal candidiasis [EPC]) and, less often, the nails (onychomycosis). Skin candidiasis is exceedingly uncommon and may rarely occur in a small proportion of patients with certain inborn errors of immunity (see below). Mucosal candidiasis, particularly in the form of VVC, can occur in people with intact immune functions, although immunocompromised individuals are at higher risk for increased frequency, severity and/or recurrence of mucosal infections. Systemic candidiasis affects sterile body sites such as the bloodstream and can involve the central nervous system (CNS), liver, spleen, heart, and/or kidneys. It can also involve the intra-abdominal compartment with or without bloodstream spread [4]. Systemic candidiasis is associated with high mortality despite the administration of antifungal therapy [4].
Figure 1.
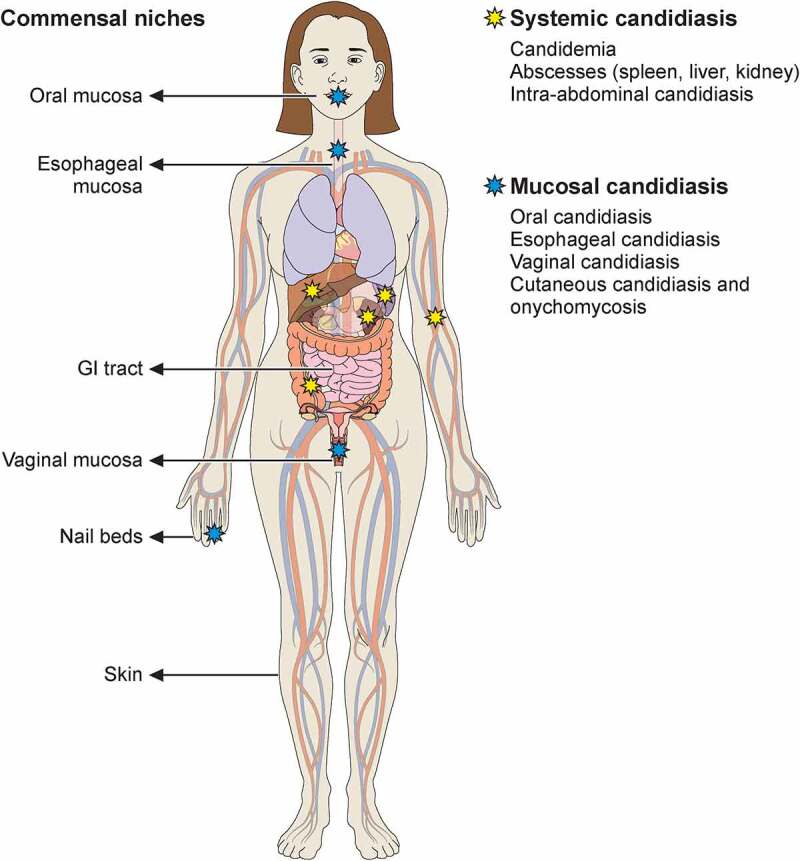
Commensal sites of C. albicans in the human body and clinical manifestations of C. albicans infection. Taking advantage of its commensal niches in the oral and genital mucosal surfaces and gastrointestinal tract, C. albicans can cause invasive disease (yellow star) and mucosal disease (blue star) in several tissues. Illustration created with BioRender.com.
To effectively cause mucosal and/or systemic disease and withstand the subsequent antifungal host response and antifungal drug treatment, C. albicans employs several virulence traits principal of which are: a) temperature adaptation; b) adhesion and invasion; c) nutrient acquisition; d) immune evasion; and e) drug tolerance. In this review, we will focus on C. albicans pathogenicity related to these five traits tracking the fungus route from being a mucosal commensal to becoming an opportunistic pathogen causing mucosal and/or systemic disease. We will also highlight the roles of the innate and adaptive immune systems and of the mucocutaneous barrier in preventing and curtailing disease and we will briefly discuss key antifungal drug targets and the countermeasures employed by C. albicans to achieve antifungal drug tolerance and resistance.
C. albicans: From commensal to opportunistic pathogen
C. albicans is a ubiquitous fungal organism residing in the mucosa of humans while also living in certain environmental reservoirs [11]. Human colonization of the mouth, vagina, and gut typically develops during infancy, primarily during vaginal delivery or breastfeeding [12,13]. In mice, which are not naturally colonized by C. albicans, colonization resistance relies on the presence of intact endogenous microbiota [14] and antimicrobial peptides (AMP) such as CRAMP, a peptide related to the human AMP cathelicidin LL-37 [15,16].
Recently, it was shown that immune selection by intestinal IgA against C. albicans filaments suppresses harmful fungal effectors while improving the competitive fitness of C. albicans yeast as a commensal [17]. Within the gut, C. albicans helps shape the composition of the healthy microbiota by inhibiting multiple dominant genera of gut bacteria [18] and local inflammation [19]. The presence of C. albicans in the gut is linked to an increase in splenic IgG-producing B cells and systemic antifungal IgG conferring protection against candidemia [20–22]. In mice, multiple passages of C. albicans resulted in fungal adaptation toward colonization and improved protection against subsequent nonspecific infections in a lymphocyte-independent manner [23].
C. albicans carriage is asymptomatic in most individuals and disease typically arises when perturbation of host homeostasis and/or endogenous microbiota occurs. It was recently shown that administration of β-lactam antibiotics causes the release of bacterial peptidoglycan subunits—including tracheal cytotoxin among others—that induces the invasive hyphal fungal program in the gut [24]. Such antibiotic-induced pertrubations, immune system abnormalities (see below), changes in the microbiome, and/or changes in mucocutaneous barrier integrity [25,26] enable C. albicans to become an opportunistic pathogen in the context of expression of an array of virulence determinants (Table 1).
Table 1.
Key C. albicans-associated virulence factors
| Immune resistance and adaptation | ||
|---|---|---|
| Adhesion/invasion | Associated genes | Function |
| ALS1 | Adhesin | |
| ALS3 | Adhesin | |
| SSA1 | AMP binding protein | |
| HWP | Hyphal-associated GPI-linked protein | |
| AlA1 | Fibronectin-binding adhesin | |
| MNT1 and MNT2 | Involved in O-glycosylation | |
| INT1 | Integrin-like protein involved in adhesion | |
| Phenotypic switch | Associated genes | Function |
| CPH1 | Transcription factor | |
| EFG1 | Transcription factor | |
| INT1 | Filamentation inducer | |
| TUP1 | Filamentation inducer | |
| CZF1 | Hyphal growth | |
| TEC1 | Filamentation inducer | |
| Proteases | Associated genes | Function |
| Secreted aspartyl proteinases (SAP1-10) | Secreted proteases | |
| Phospholipases (PLB1-4) | Phospholipases that cause disruption of host membranes | |
| Lipases (LIP1–10) | Lipases | |
| Nutrient acquisition | Associated genes | Function |
| RBT5 | Heme-binding protein | |
| CSA1-2 | Heme-binding proteins | |
| PGA7 | Heme-binding protein | |
| ZRT1 | Zinc transporter | |
| PRA1 | Zinc acquisition | |
| Environmental adaptation | ||
| Biofilm formation | Associated genes | Function |
| BCR1 | Transcription factor | |
| TEC1 | Transcription factor | |
| EFG1 | Transcription factor | |
| MKC1 | Maintains cellular integrity and cell wall biogenesis | |
| pH sensing | Associated genes | Function |
| PHR1-2 | pH regulated genes that contribute to cell wall assembly and morphogenesis | |
| RIM101 | pH response pathway | |
| DFG16 | Plasma membrane receptor | |
| RIM21 | Plasma membrane receptor | |
| Thigmotropism | Associated genes | Function |
| CCH1 | Calcium channel | |
| MID1 | Calcium channel | |
| RSR1 | Hyphal orientation | |
| Stress response | Associated genes | Function |
| GPP1-2 | Glycerol biosynthesis | |
| SOD | Superoxide dismutase | |
| CTA1 | Catalase | |
| YNB1 | Nitrosative stress response | |
| HOG1 | Osmotic, oxidative and thermal stress response | |
| HSP genes | Heat shock and oxidative stress | |
| Drug resistance | ||
| Associated genes | Function | |
| CDR1 and CDR2 | Transporter of the ATP binding cassette superfamily | |
| TAC1 | Involved in the regulation of CDR1 | |
| MRR1 | Multidrug resistance regulator involved in the control of MDR1 | |
| FKS | Encodes the β-(1,3)-d-glucan synthase involved in echinocandin resistance | |
| MSH2 | DNA mismatch repair ATPase | |
| HSP90 | Heat shock chaperone | |
| UPC2 | Transcription factor associated with azole resistance | |
| ERG3 | Sterol desaturase associated with azole and AMB resistance | |
| ERG11 | Cytochrome P450 protein involved in demethylation of lanosterol, which upon modification it confers resistance to azoles | |
| ERG6 | Methyltransferase, which converts zymosterol to fecosterol and is important for azole and AMB resistance | |
| FCY2 | Cytosine permease involved in 5-FC resistance | |
| FCY1 | Cytosine deaminase involved in 5-FC resistance | |
| Immune evasion | ||
| Associated genes | Function | |
| PRA1 | Complement binding protein | |
| CRZ1 | Transcription factor. Crz1-dependent pathway activation induces lactate-induced β-glucan masking | |
| ACE | Transcription factor. Ace2 activation is involved in the network of lactate-induced β-glucan masking | |
| XOG1 | Exoglucanase involved in immune evasion | |
| ECE1 | Expressed in association with hyphae. Protein precursor of candidalysin | |
Fungal virulence determinants in C. albicans
Plasticity in switching between different morphogenic states
C. albicans displays a range of cellular growth states that help it establish presence and persistence in different mammalian tissues [27]. These cell phenotypic variations are associated with different yeast-like or filament-like morphologies and colony features as well as distinct genetic profiles [27]. Especially important in the pathogenic life cycle of C. albicans is its ability to change morphology to and from the yeast and hyphal forms to breach mucosal barriers and establish invasive disease. Typically thought of as the commensal form, the yeast, allows colonization of superficial commensal niches [28,29]. By contrast, the hyphal form is typically thought of as the invasive form of the fungus allowing C. albicans to penetrate host barriers and to gain access into deep-seated tissues [30]. Several environmental stimuli affect the morphological state of C. albicans including host temperature, pH, nutrient availability, or quorum sensing mechanisms [31–34]. Of interest, both C. albicans yeast and hyphal morphotypes can be found in different anatomical areas across the mouse gut [35].
The importance of the morphogenic transition from yeast to hyphae is shown by the fact that nonfilamentous C. albicans strains are avirulent [36]. However, the evolutionary success and ability to cause life-threatening human disease of other non-albicans Candida species that are unable to filament such as C. glabrata and C. auris indicates that filamentation is not a prerequisite for pathogenesis in Candida species. In fact, hyphae-locked C. albicans strains are also hypovirulent in vivo, indicating that the transition between the yeast and filamentous forms is most critical for effective virulence, rather than each of the morphogenic states itself [37]. In addition, both C. tropicalis and C. parapsilosis strains engineered to exhibit hyper-filamentation phenotypes due to constitutively expression of the transcriptional regulator UME6 showed a dramatic reduction in organ fungal burden during in vivo infection [38]. Notably, a recent report showed that metabolic adaptability and improved fitness led to enhanced fungal proliferation, which increased the virulence of filament-deficient strains in a mouse model of systemic candidiasis, when low fungal inocula were used. Interestingly, filament-deficient strains remained attenuated during intraperitoneal mouse infection, highlighting the tissue-specific cues that may affect the role of C. albicans morphogenic state and its virulence [39]. The ability of C. albicans to produce filaments in vivo during systemic candidiasis in mice correlates with the ability of the tissue to control infection. Thus, C. albicans produces filaments in the mouse kidney during systemic candidiasis whereas filamentation is not observed in the liver or spleen; this tissue-specific propensity to filament correlates with the ability of spleen and liver to control the infection as opposed to the kidney that is unable to control fungal proliferation and inexorably loses function [40]. Taken together, these observations underscore the critical contribution of the yeast-to-hyphae switch in C. albicans virulence and reinforce the importance of cell-intrinsic and tissue-specific environmental factors in the determination of the C. albicans morphotype under various niches and conditions.
Adhesion, invasion, and host cell damage1
The importance of adhesion and invasion factors for the success of C. albicans as a pathogen is highlighted by the different types of surfaces, ranging from host mucosal tissues to medical devices and instruments, which C. albicans can colonize. In tissue, C. albicans yeast cells adhere to the epithelium and/or endothelium and trigger hyphal elongation with subsequent active penetration of host cells. The process is mediated by adhesin and invasin members of the Als and Hwp1 families (reviewed in detail elsewhere) [41–43].
During mucosal infection, the interaction between epithelial cells and fungal ligands leads to induced endocytosis and active penetration by C. albicans. During systemic infection, active penetration can give access to blood vessels via which fungal cells reach distant body sites. Thereafter, endothelial penetration initiates colonization and disseminated disease [44]. Induced endocytosis is mediated by the adhesins and invasins Als3 and Ssa1, which bind host cell N-cadherin on endothelial cells and E-cadherin on oral epithelial cells [45]. In oral epithelial cells, induced endocytosis activates platelet-derived growth factor BB (PDGF BB) and neural precursor cell expressed developmentally down-regulated protein 9 (NEDD9) signaling [46]. In addition, Als3 and Ssa1 interact with epidermal growth factor receptor (Egfr) and Her2 (also known as Erbb2) and both receptors function cooperatively to induce C. albicans hyphae endocytosis [47]. Adhesion of C. albicans to epithelial cells is followed by hyphal formation, which can then actively penetrate the plasma membrane of epithelial cells [48,49]. The formation of hyphae is accompanied by the expression of hypha-associated proteins with known damage and immune activation capabilities [50]. Moreover, secretion of hydrolases by C. albicans hyphae facilitates active penetration into epithelia contributing to extracellular nutrient acquisition [51]. C. albicans expresses three different classes of secreted hydrolases: proteinases, phospholipases, and lipases. Secreted aspartic proteinases (Saps) comprise ten members of which some are secreted (Sap1–8) and some remain bound to the cell surface (Sap9–10) [52]. Saps have been shown to play pleiotropic roles in vitro and in vivo including inducing damage to epithelial cells thus promoting fungal virulence, recruitment of neutrophils, and induction of pro-inflammatory responses such as IL-1β and TNF-α [53–56]. Phospholipases are extracellularly secreted and act via disruption of host cell membranes [57]. Lipases consist of 10 members (Lip1–10) and promote virulence in a mouse model of systemic candidiasis [58,59].
Candidalysin is a newly discovered hyphae-derived peptide toxin that has recently been shown to be a major virulence determinant of C. albicans. Hyphal filaments express ECE1, which encodes the Ece1p protein. Ece1p is processed by Kex2p and Kex1p to generate mature candidalysin that is then secreted. At high concentrations, candidalysin interacts with cell membranes to form pore-like structures resulting in membrane damage [60,61]. The resultant calcium influx and oxidative stress induced in host cells result in rapid necrotic—rather than apoptotic—cell death [62]. Additionally, Als3-mediated endocytosis of hyphal filaments leads to the formation of an endocytic vacuole with a high concentration of candidalysin potentiating the damage on oral epithelial cells [63,64]. A possible mechanism of immune protection against prolonged candidalysin damage involves neutralization of the toxin by albumin, which acts as an anti-toxin through hydrophobic interactions [65]. Notably, besides being essential for epithelial cell damage, candidalysin is also important for activation of mucosal and tissue-specific systemic immune responses (see below).
Metabolic adaptation and nutrient acquisition
In most settings, glucose is the preferred carbon source for C. albicans [66]. However, in most anatomical sites of colonization, fungal growth occurs under glucose-limiting conditions. Under those circumstances, C. albicans adapts by upregulating alternative carbon utilization pathways, using carboxylic acids such as lactate, amino acids, and N-acetylglucosamine (GlcNAc) [67]. C. albicans mutant strains in these pathways have attenuated virulence [68,69].
Tissues can be a limiting source of nutrients to the fungus. In addition, the host can withhold trace nutrients from microbes via nutritional immunity [70,71]. Thus, C. albicans has evolved strategies for acquisition of scarcely available micronutrients. Under zinc limitation, C. albicans produces a zinc-binding protein, encoded by PRA1, which scavenges zinc from host tissues [72]. Interestingly, competition for scarce micronutrients also influences the host immune response. Secretion of Pra1 disrupts host defense by blocking the complement component C3 and subsequent fungal clearance [73]. Zinc limitation in C. albicans induces a hyper-adherent phenotype termed Goliath cells [74,75]. In addition, dynamic changes in copper assimilation during systemic infection contribute to pathogenesis. Up-regulation of the copper efflux pump, Crp1, and the copper importer, Ctr1, is observed in a sequential, temporally regulated, manner during renal candidiasis and both factors are essential for fungal virulence [76,77]. During invasive infection, iron homeostasis is also perturbed triggering changes in the renal iron landscape. Thus, in the kidney medulla iron accumulates, whereas in the renal cortex leukocyte infiltrates form iron exclusion zones around fungal lesions [71]. Under low iron conditions, C. albicans FTR1 acts as a high-affinity iron permease to promote iron uptake from ferritin and transferrin and is essential for virulence during systemic candidiasis [78]. Moreover, the adhesin and invasin Als3 was shown to promote iron uptake from ferritin in the context of C. albicans interactions with oral epithelial cells [79].
Stress resistance
The multitude of stressors imposed on C. albicans by host immune cells and the various microenvironment cues require the induction of differential stress resistance responses by C. albicans. For example, to escape oxidative killing by immune cells, C. albicans possesses six superoxide dismutases (Sods), which are all involved in the detoxification of reactive oxygen species (ROS) by converting O2− into molecular oxygen and hydrogen peroxide [80]. Accordingly, Sod-deficient C. albicans strains have reduced pathogenicity [81]. Additional fungal antioxidant proteins and DNA damage repair genes help attenuate oxidative stress [82,83].
Other stressors include pH changes, thermal and osmolarity shifts, nitrosative stress, and antifungal drug treatment [84–86]. Many of the fungal stress responses are modulated by heat shock proteins (Hsps), particularly Hsp90. Hsp90 is a ubiquitous and conserved ATP-dependent molecular chaperone that acts to stabilize diverse signal transducers. C. albicans Hsp90 enables fungal virulence and drug resistance. This effect is mediated via modulation of the mitogen activated kinase Mck1, the stress activated protein kinase Hog1/Sty1, and/or the protein phosphatase calcineurin [87]. Additional effects of Hsp90 disruption include reduction of tolerance to stress responses and induction of morphological transition from yeast to hyphal growth [87].
Masking of cell wall components for immune evasion
The first step in the development of an immune response to C. albicans is the recognition of fungal pathogen-associated molecular patterns (PAMPs) by host epithelial and immune cell pattern recognition receptors (PRRs) (Figure 2). The C. albicans cell wall is composed of chitin, β-glucan, and mannoproteins [88], which are recognized by host cell PRRs and are important for induction of protective antifungal immune responses.
Figure 2.
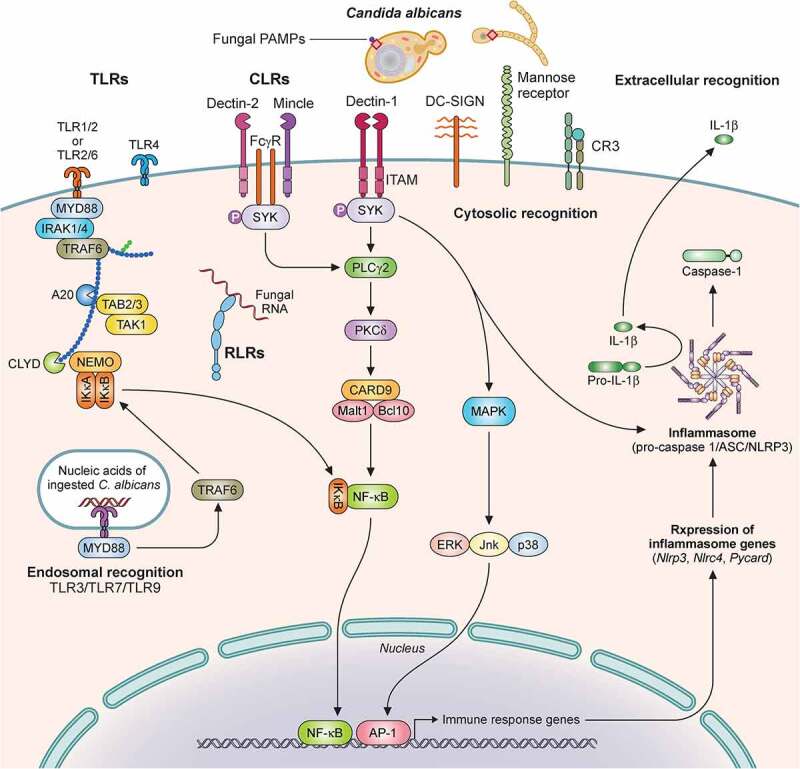
Recognition of C. albicans by immune cells is mediated by distinct pattern recognition receptor signaling pathways. Extracellular recognition of fungal ligands occurs via C-lectin receptors (CLRs) or by some of the Toll-like receptors (TLRs) such as TLR1, TLR2, TLR6, or TLR4. Recognition leads to the activation of intracellular signaling pathways dependent on several adaptor molecules inducing NF-κB activation and cytokine secretion. In addition, CLRs can activate AP-1 via MAPK also leading to cytokine secretion. Some TLRs (TLR3, TLR7, TLR9) recognize nucleic acids derived from C. albicans within the endosome. Within the cytosol, recognition is mediated by NOD-like Receptors (NLRs) with resultant inflammasome activation and IL‐1β processing. Recognition by TLRs and CLRs and secretion of pro-IL‐1β and pro-IL-18 triggers inflammasome assembly, activation of pro-caspase 1 to generate caspase-1, and cleavage of these two cytokines into their mature IL‐1β and IL-18 forms. Additional cytosolic recognition may occur via RIG-I-like receptors. Illustration created with BioRender.com. TLR, Toll like receptor; MyD88, Myeloid differentiation factor 88; IRAK1, Interleukin 1 receptor associated kinase 1; TAK1, transforming growth factor-β-activated kinase 1; TRAF, Tumor necrosis factor receptor-associated factor; TAB, TGF-beta-activated kinase; NEMO, nuclear factor-κB essential modulator; IκB kinase; CLYD, cylindromatosis tumor suppressor; RLR, RIG-I-like receptor; CLR, C-lectin receptor; Mincle, macrophage inducible Ca2+-dependent lectin receptor; FcγR, Fc receptors: SYK, Spleen tyrosine kinase; ITAM, immunoreceptor tyrosine based activation motif; PLCγ2 Phospholipase C gamma 2; PKCδ, Protein kinase C delta; CARD9, caspase recruitment domain-containing protein 9; Malt1, mucosa-associated lymphoid tissue lymphoma translocation 1; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; DC-SIGN, Dendritic cell-specific intercellular adhesion molecule-3-Grabbing non-integrin; CR3, Complement receptor 3; MAPK, mitogen-activated protein kinase; ERK, Extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; AP-1, activator protein-1; ASC, Apoptosis-associated speck-like protein containing a CARD; NLRP3, NLR family pyrin domain containing 3; NLRC4, NLR family CARD domain containing 4.
To avoid activating immune pathways that trigger these effector responses, C. albicans cells can minimize their PAMP exposure by masking cell wall components in response to metabolic cues. These protective responses circumvent the immune system by exploiting host signals to promote immune evasion [89]. Some of these signals include changes in oxygen availability, carbon source, or hormone levels [90–96]. The architecture of the fungal cell wall can also be altered by environmental stress during infection favoring immune recognition, as is the case for pH or iron changes [97,98]. For example, high iron decreases the levels of mannans and chitin and increases the levels of β-(1,3)-glucan by preventing activation of the fungal mitogen-activated protein kinase (MAPK) Choline/ethanolamine kinase 1 (Cek1), an effect mediated by lactate-induced Crz1 [98]. These changes reduce the susceptibility of C. albicans to cell wall-perturbing antifungal agents but also reduce survival upon phagocytosis by macrophages. Remarkably, treatment with sub-therapeutic doses of the antifungal drug caspofungin also causes exposure of C. albicans β-glucan both in vitro and in vivo and elicits pro-inflammatory cytokine secretion by primary macrophages [99]. This unmasking is modulated by changes in the regulatory gene network responsible for the cell wall architecture [100].
To avoid clearance within phagosomes, C. albicans activate programs to survive the nutrient-poor, acid environment. To adapt to this nutrient-poor niche, fungal cells induce metabolic starvation pathways, including gluconeogenesis, fatty acid degradation, and also downregulate translation [101]. To induce filamentation, yeast cells produce ammonia, which promotes neutralization of the acidic phagosomal pH and yeast-to-hyphae transition [102,103].
Initiation of host responses against C. albicans – the role of PRR systems
Central to the initiation of antifungal immune responses are the members of the C-type lectin (CLR) superfamily. Since the discovery of the role of DECTIN-1 in the recognition of fungal β-glucan [104,105], the characterization of other members of the CLR family, including—but not limited to—DECTIN-2 [106], DECTIN-3 [107], and MINCLE [108] has uncovered the importance of CLR-mediated signaling in innate immune sensing and control of pathogenic fungi, including C. albicans [109,110].
Fungal sensing via CLRs is the first step in the activation of the CLR–spleen tyrosine kinase (SYK)–caspase recruitment domain-containing protein 9 (CARD9) signaling pathway [109,110]. Following fungal ligand recognition by the corresponding CLR, its hemITAM or ITAM— depending on the CLR—is phosphorylated and SYK is activated, which leads to assembly of the CARD9-BCL10-MALT1 complex. This engagement activates the canonical nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) subunits c-Rel and p65 and induces innate and adaptive immune responses and cytokine secretion. Noncanonical NF-κB activation can also be induced in a SYK/NF-κB-inducing kinase (NIK)-dependent manner [110]. SYK-deficient mice are susceptible to systemic candidiasis (and other fungal infections) and SYK-deficient neutrophils are unable to control several Candida species associated with defects in ROS production, cytokine production, neutrophil extracellular trap (NET) formation, phagocytosis, and neutrophil swarming [111,112]. SYK integrates signals from multiple CLR-dependent and CLR-independent signaling pathways, thus, SYK activation requires a delicate balance whereby a suboptimal response can cause immunodeficiency, whereas an excessive response can lead to hyper-inflammatory disease and hematological malignancy [113,114].
The critical contribution of the CLR–SYK–CARD9 signaling pathway in human antifungal host defense is clearly portrayed by the observation that CARD9 deficiency is the only— among the >400 known to date—primary immunodeficiency disorder (PID) that promotes fungal-specific infection susceptibility without predisposition to bacterial, viral, or parasitic infections or noninfectious complications. In fact, CARD9 deficiency is the only known inherited condition that underlies susceptibility to both mucosal and systemic candidiasis, with the latter exhibiting a unique predilection for the CNS [115]. Aspergillosis—primarily extrapulmonary—, phaeohyphomycosis, and deep-seated dermatophytosis also occur in CARD9-deficient patients [116–119]. Critical CARD9-dependent fungal surveillance immune functions include Th17 cell differentiation, neutrophil recruitment to the CNS, pro-inflammatory cytokine and chemokine production, and phagocyte fungal killing [120–123]. The advent of the SYK inhibitor fostamatinib in clinical practice for the treatment of various inflammatory and neoplastic conditions may result in opportunistic mucosal and/or systemic fungal infections, as indicated by the early description of mucosal candidiasis and skin fungal disease in fostamatinib-treated individuals [114,124,125].
CARD9 relays signals from several upstream CLRs in a fungus- and tissue-specific manner, without a single CLR deficiency phenocopying CARD9 deficiency. For example, a few DECTIN-1–deficient patients who carry the p.Y238* CLEC7A mutation in homozygosity [126], which abolishes DECTIN-1–dependent signaling, were reported to develop recurrent VVC (RVVC) and onychomycosis. In addition, a single immunocompromised DECTIN-2–deficient patient carrying a homozygous deletion resulting in a frameshift and early stop codon in DECTIN-2 was recently reported to develop fatal invasive pulmonary aspergillosis [127]. As mentioned earlier, the C. albicans cell wall structure is dynamically altered during infection and therefore immune recognition often requires the concerted action of several PRRs to mount effective immune responses; among others, such interactions have been characterized between different CLRs, between CLRs and TLRs, and between TLRs and the complement C5a anaphylatoxin [4,128–130].
Toll-like receptors (TLRs) are expressed on both hematopoietic and non-hematopoietic cells and also participate in C. albicans sensing. Upon fungal PAMP recognition, MYD88 is recruited to the TLR and a signaling cascade is initiated that culminates in pro-inflammatory cytokine and chemokine production in a NF-κB dependent manner. MYD88 is required for activation of Langerhans cells and induction of the Th17 response during skin Candida infection (see below) [131]. Moreover, Myd88−/− mice are susceptible to systemic fungal infections—including systemic candidiasis—however MYD88-deficient patients do not develop candidiasis or other fungal disease, likely due to compensatory effects of other PRRs, primarily of CLRs [132,133].
TLR2 recognizes C. albicans phospholipomannans and TLR2 deficiency impairs neutrophil chemotaxis, phagocytic activity, and cytokine and chemokine production resulting in reduced survival during systemic candidiasis in mice [134,135]. However, other studies have shown a dispensable role for TLR2 during systemic candidiasis, which may be explained by the differential dependence of different C. albicans strains on TLR2 recognition [136]. Indeed, C. albicans strain-specific differential dependence on PRR recognition has also been documented for DECTIN-1 and TLR4, which recognizes C. albicans O-linked mannans [132,137–141]; in the setting of candidiasis in vivo, these differences underlie a wide variety of outcomes ranging from conferring survival benefit to promoting lethal immunopathology. In mice, TLR1 deficiency increased whereas TLR6 deficiency ameliorated intestinal inflammation and C. albicans burden in a colitis model [142]. In humans, polymorphisms in TLR1, TLR4, and TLR6 have been suggested to confer greater susceptibility to systemic candidiasis in acutely ill patients in the intensive care unit (ICU) in some studies [143–145]. C. albicans DNA sensing by TLR9 promotes IL-12p40 production [146], and TLR9—together with the mannose receptor and NOD2—has also been shown to recognize C. albicans chitin leading to the production of the anti-inflammatory cytokine IL-10 [147]. Moreover, the endosomal TLR7 and TLR3 have been implicated in C. albicans RNA sensing leading to the production of type I interferons and pro-inflammatory chemokines [148–150].
NOD-like receptors (NLRs) are cytoplasmic PRRs that enable the formation of inflammasomes, which are multiprotein complexes that process pro-IL-1β/pro-IL-18 into their mature forms. The NLRP3 inflammasome complex is formed by NLRP3, the adaptor protein ASC, and the effector caspase-1, although non-canonical caspase-8-dependent pro-IL-1β processing is also operational in C. albicans [151]. The morphological switch of C. albicans contributes to NLRP3 activation in a TLR2/DECTIN-1/SYK-dependent manner [152–154]. Mice deficient in NLRP3, ASC, or caspase-1 have increased fungal proliferation and decreased survival during systemic candidiasis [152,154]. Moreover, the NLR family member NLRP10 was critical for survival during systemic candidiasis in mice via promoting Th1 and Th17 responses, while being dispensable for pro-inflammatory cytokine production [155]. In addition, the NLRC4 inflammasome is important for the control of mucosal candidiasis in vivo. Specifically, NLRC4 is upregulated in oral mucosal tissues following C. albicans infection, particularly in the stromal compartment, it mediates—together with NLRP3—the induction of IL-1β, and NLRC4-deficient mice had reduced secretion of CRAMP, IL-17A, and IL-1β, and impaired ability to control mucosal fungal proliferation [156].
Other receptors that have been implicated in the initiation of immune responses against C. albicans include: a) the RIG-I-like receptor family receptor MDA5 (IFIHI), which plays a major role in viral RNA sensing [157], is induced in response to C. albicans hyphae, and may be dysfunctional during mucosal and systemic candidiasis in susceptible patients [158], and b) EphA2, a receptor tyrosine kinase present in epithelial cells, which mediates fungal β-glucan recognition and induces pro-inflammatory responses during OPC [159]. In addition, expression of EphA2 on neutrophils is important for immunity during OPC via MEK-ERK signaling and subsequent priming of nicotinamide adenine dinucleotide phosphate (NADPH)-subunit p47phox and ROS production, which results in fungal killing [160].
Following initial sensing by the innate immune system, host immune responses are deployed during mucocutaneous and systemic candidiasis; these responses are tissue-specific, compartmentalized, and distinct in the various forms of the infection. The cellular and molecular basis of these responses is briefly outlined in the next section.
Mucocutaneous candidiasis
OPC and EPC
Candidiasis of the mouth—primarily affecting the tongue, buccal mucosa, and gingivae—, throat, or esophagus is predominantly caused by C. albicans and is uncommon in healthy individuals. HIV/AIDS is a major risk factor for OPC and EPC, typically in patients with diminished CD4 T cell counts (<200 cells/mm3), whereas certain topical or systemic immunosuppressive agents also predispose to the infection such as corticosteroids [161], TNF-α inhibitors [162], and IL-17-targeted biologics (see below) [163]. Other local factors that contribute to OPC susceptibility include denture wearing and salivary hypofunction; salivary flow acts as a mechanical clearance mechanism by preventing adherence of C. albicans to oral epithelial cells and saliva contains potent AMPs with C. albicans-inhibitory properties (see below) [164].
The critical pathway mediating oral mucosal immune protection is IL-17 signaling [165]. Following the initial seminal reports of mice deficient in IL-17RA, IL-17RC, or the IL-17 receptor adaptor ACT1 being highly susceptible to OPC, subsequent studies in patients confirmed the critical contribution of this signaling axis in mucosal anti-Candida host defense [166–168]. Thus, patients with autosomal recessive complete deficiencies in IL-17RA, IL17RC, or ACT1/TRAF3IP2 develop fully penetrant, severe, treatment-refractory mucosal infections by Candida species, termed chronic mucocutaneous candidiasis (CMC) [169–172]. A single kindred carrying a heterozygous dominant-negative mutation in IL17F that impaired cellular responses to both IL17F and IL17AF was also reported to result in CMC, yet with incomplete penetrance [172]. Other inborn errors of immunity manifesting with CMC also map to defects in IL-17 signaling featuring varying degrees of decreased frequencies of circulating Th17 cells and/or impaired IL-17 cellular responses, further illustrating its importance for mucosal antifungal protection (Table 2) [173–178]. More recently, the use of IL-17 pathway-blocking monoclonal antibodies (mAbs) in patients with psoriasis and inflammatory bowel disease (IBD) has been associated with the development in some patients with mild, treatment-responsive OPC, but not CMC. Notably, the mean frequency of OPC in these patients is low (~1-10%), with a greater risk observed in patients receiving mAbs that target IL-17RA or combined IL-17A, IL-17F, and IL-17AF, followed by mAbs that target IL-17A, followed by mAbs that target IL-12p40 or IL-23p19 [163]. The resistance of these patients to CMC likely reflects the incomplete blockade of mucocutaneous IL-17 signaling by the administered mAbs [179,180]. Collectively, these data indicate that a complete absence of IL-17R responses promotes susceptibility to CMC in humans, whereas an mAb-induced blockade regimen that spares a fraction of mucosal IL-17R responses does not.2
Table 2.
Inborn errors of immunity underlying inherited susceptibility to C. albicans infections
| Primary immunodeficiency disorder | Associated gene | Mode of inheritance | Clinical presentation of infection |
|---|---|---|---|
| Mucosal candidiasis | |||
| APECED | AIRE | AR or AD | CMC |
| DOCK8 deficiency | DOCK8 | AR | CMC, viral infection (molluscum, HSV) |
| ZNF341 deficiency | ZNF341 | AR | CMC, bacterial infections (sinopulmonary and skin bacterial infections) |
| JNK1 haploinsufficiency | MAPK8 | AD | CMC, superficial skin bacterial infections |
| IRF8 deficiency | IRF8 | AR | CMC, disseminated NTM infection |
| RORγt deficiency | RORC | AR | CMC, disseminated NTM infection |
| IL-12p40 deficiency | IL12B | AR | CMC, intracellular bacterial and NTM infections |
| IL12Rβ1 deficiency | IL12RB1 | AR | CMC, intracellular bacterial and NTM infections |
| IL-17RA deficiency | IL17RA | AR | CMC, bacterial infections (superficial staphylococcal skin infections, bacterial pneumonias) |
| IL-17RC deficiency | IL17RC | AR | CMC |
| IL-17F deficiency | IL17F | AR | CMC |
| ACT1 deficiency | TRAF3IP2 | AR | CMC, bacterial infections (superficial staphylococcal skin infections, bacterial pneumonias) |
| Job’s syndrome | STAT3 | AD | CMC, onychomycosis, pulmonary mold infections, skin and pulmonary bacterial infections |
| STAT1 gain-of-function | STAT1 | AD | CMC, bacterial and NTM infections, viral infections, endemic fungal infections |
| SCID |
IL7RA IL2RG RAG1-2 JAK3 |
AR | CMC, bacterial infections, disseminated viral infections, PJP |
| EDA-ID |
KBKG IKBA |
AR | CMC, NTM infections |
| Systemic candidiasis | |||
| CGD |
CYBA CYBB NFC1 NFC2 |
AR or X-linked | invasive mold infections, systemic candidiasis (rare), invasive bacterial infections (Staphylococcus, Nocardia, Serratia) |
| LAD-1 | ITGB2 | AR | Systemic candidiasis, pyogenic bacterial infections (staphylococcal skin infections and gram negative bacteria, periodontitis) |
| Complete MPO deficiency | MPO | AR | Systemic candidiasis |
| Mucosal and systemic candidiasis | |||
| CARD9 deficiency | CARD9 | AR | CMC, Candida meningitis, colitis, endophthalmitis, and osteomyelitis, aspergillosis (including extrapulmonary), phaeohyphomycosis, protothecosis |
AD, autosomal dominant; AR, autosomal recessive; APECED, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; HSV, herpes simplex virus; NTM, nontuberculous mycobacteria; CMC, chronic mucocutaneous candidiasis; PJP, Pneumocystis jirovecii pneumonia; SCID, severe-combined immunodeficiency disorder; EDA-ID, anhidrotic ectodermal dysplasia with immunodeficiency: CGD chronic granulomatous disease; LAD-1, leukocyte adhesion deficiency type-1; MPO, myeloperoxidase; AIRE, autoimmune regulator; CARD9, caspase recruitment domain-containing protein 9; STAT, signal transducer and activator of transcription; DOCK8, dedicator of cytokinesis 8.
At the cellular level, CD4+ T cells, CD8+ T cells, γδ T cells, and type 3 innate lymphoid cells (ILC3) are the major sources of IL-17 during oral candidiasis [181,182]. Initial innate Th17 responses are regulated by Langerin-expressing dendritic cells (DCs) and are deployed by rapidly proliferating tissue-resident natural Th17 (nTh17) cells post-C. albicans challenge [182–184]. The development of C. albicans-specific Th17 cells following OPC involves antigen presentation and T cell priming by tissue-resident DCs in an Flt3L-dependent manner aided by monocyte-derived DCs in a CCR2-dependent manner [185]. C. albicans-specific IL-17-producing tissue-resident memory T cells (TRM) are efficient in maintaining prolonged colonization in a mouse model of C. albicans commensalism [186,187].
Of note, data from mice and humans suggest that the lack of IL-17 production by a certain lymphoid cell subset, may be compensated by other IL-17-producing lymphoid cells. For example, Tcrb−/− and Tcrgd−/− mice eventually control OPC whereas Rag1−/− mice that lack both αβ and γδ T cells are highly susceptible to the infection [188]. In agreement, patients with idiopathic CD4 lymphocytopenia who have diminished CD4+ T cells and patients with loss-of-expression mutations in the CD4 gene who also lack Th17 cell-derived IL-17 production do not manifest CMC [189–191]. Collectively, these data indicate that the susceptibility of HIV/AIDS patients to OPC may reflect defects in IL-17 production by both Th17 and non-Th17 cellular sources at the oral mucosa, as suggested by studies in SIV-infected non-human primates [192–194].
Mechanistically, IL-17 mediates a robust mucosal immune response to protect against C. albicans by acting on IL-17R-expressing epithelial cells to induce the production of potent AMPs such as β-defensins and S100A8/A9 [165,168,195]. Accordingly, Defb3−/− mice were susceptible to OPC [165]. Histatins are another important family of AMPs, which have been shown to prevent C. albicans colonization on epithelial cell surfaces, to protect the basal epithelial cell layer from apoptosis, and to alter C. albicans mitochondrial function resulting in fungal cell death [196–198]. IL-17 signaling also promotes the production of neutrophil-recruiting CXC chemokines (e.g. CXCL1, CXCL5) and has been shown to be indispensable for neutrophil recruitment in the C. albicans-infected oral mucosa in some—but not all—studies, potentially reflecting microbiome variations in the mouse colonies used in these different settings [195,199]. Mice lacking the CXCL1/CXCL5-targeted chemokine receptor CXCR2 were highly susceptible to OPC due to impaired neutrophil recruitment to the C. albicans-infected oral mucosa [200]. Besides IL-17R signaling, IL-1R signaling promotes mobilization of granulocytes from the bone marrow and neutrophil recruitment into the oral mucosa during OPC via endothelial cell production of granulocyte colony-stimulating factor (G-CSF) in response to keratinocyte-derived IL-1α [201].
IL-22 is another cytokine produced by type 17 innate and adaptive lymphoid cell subsets during OPC and plays an important role in antifungal resistance as shown by experiments in Il22r−/− mice, in wild-type mice administered IL-22-targeted mAbs, and in mice deficient in both IL-17R and IL-22R, which exhibit further increase in fungal proliferation compared to mice deficient in either IL-17R or IL-22R [199,202]. Mechanistically, IL-22 acts on its receptor on oral basal epithelial cells to provide survival and regeneration signals to the IL-17R-expressing oral suprabasal epithelial cell layer enabling its responsiveness to IL-17A [199]. In humans, no inborn errors of IL-22 immunity have thus far been reported to cause CMC. Patients with loss-of-function mutations in the IL-22 receptor subunit IL-10RB, who lack IL-22 (and IL-10, IL-26, IL-28, and IFNL1) responses, do not develop CMC but manifest with very early onset IBD [203,204]. These data collectively indicate that IL-22 deficiency appears to be tolerated in humans and that impaired IL-22 responses may act synergically with defective IL-17 responses to cooperatively impair mucosal anti-C. albicans host defense.
On the fungal front, as described above, C. albicans adherence to oral epithelial cells is achieved via the Als and Hwp families of adhesins/invasins [41–43,205]. Fungal recognition activates NF-κB and a biphasic MAPK innate response in oral epithelial cells. Triggered by C. albicans cell wall recognition and independent of fungal morphology, the first step in the signaling cascade involves NF-κB and MAPK/c-Jun activation. The second MAPK phase occurs in response to a greater C. albicans burden and filament formation, with c-Fos and MKP1 activation leading to induction of pro-inflammatory responses [206]. This complex response helps oral epithelial cells to discriminate between colonizing and invading C. albicans. During OPC, candidalysin acts as a driver of protective IL-17 responses as well as of IL-36 induction via synergistic interactions between IL-1α and EGFR signaling in oral epithelial cells [183,207]. In addition, in a PAMP-independent manner, candidalysin induces EGFR phosphorylation leading to secretion of neutrophil-targeted chemokines [208]. In candidalysin-exposed epithelial cells, blockade of IL-1α/IL-1R resulted in decreased IκBα phosphorylation, reduced induction of IκBζ, and impaired production of granulocyte-macrophage colony-stimulating factor (GM-CSF) and neutrophil-recruiting IL-8/CXCL8. Combined blockade of EGFR and IL-1R further suppressed pro-inflammatory cytokine production in candidalysin-exposed cells [209].
Although type 17 immunity is undoubtedly critical for protective mucosal anti-Candida host defense in mice and humans, we recently reported that, in certain settings, additional, IL-17R/IL-22-independent mechanisms can also promote mucosal fungal infection susceptibility. We studied mice and humans with Autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED), also known as Autoimmune polyglandular syndrome type 1 (APS-1), a monogenic autoimmune disorder characterized by loss-of-function mutations in the autoimmune regulator (AIRE) gene [210]. APECED patients feature selective infection susceptibility to CMC with a frequency of ~80-90%, associated with serum autoantibodies against IL-17F (frequency, ~20-85% depending on the cohort), IL-17A (frequency, ~35%), and IL-22 (frequency, ~70-90% depending on the cohort) [211–214]. However, the association between these autoantibodies and CMC in APECED is incompletely penetrant, and several patients who carry these autoantibodies do not develop CMC, while several other patients who lack these autoantibodies manifest CMC [211–214]. These data indicate that additional factors must contribute to susceptibility to CMC in APECED patients.
Indeed, we probed oral mucosal immune responses in Aire-deficient mice, which exhibited selective infection susceptibility to CMC despite the fact that they rarely develop type 17 cytokine-targeted autoantibodies (frequency, <10%) and they mount intact IL-17R/IL-22 mucosal immune responses during OPC [215]. These data indicate that impaired type 17 immunity is not the primary driver of OPC susceptibility in Aire-deficient mice. Instead, the OPC susceptibility in Aire-deficient mice was driven by the overproduction of interferon-γ (IFN-γ) by oral mucosal CD4+ and CD8+ T cells, which were both necessary and sufficient to promote infection in this setting via disrupting the oral epithelial barrier. Accordingly, genetic or pharmacological inhibition of IFN-γ or JAK-STAT signaling rescued the epithelial barrier defects and reversed OPC susceptibility in Aire-deficient mice [215]. Moreover, we found corroborative evidence of excessive type 1 and intact type 17 immune responses in the oral mucosa of APECED patients [215]. Taken together, these data indicate that, in certain settings, aberrant type 1 mucosal responses rather than impaired type 17 mucosal responses may promote mucosal fungal susceptibility and that T cell-driven immunopathology rather than impaired host resistance may underlie mucosal candidiasis. Together, these findings point to a novel conceptual framework for classifying CMC molecular subtypes across a spectrum of defective type 17 mucosal defense and/or immunopathology-promoting type 1 mucosal inflammation [188].
More studies are needed to further evaluate the relative contribution of these pathways in the initiation, persistence, and/or recurrence of CMC in additional APECED children and adults and to determine whether aberrant type 1 mucosal responses may contribute to CMC in other conditions with excessive type 1 inflammation such as a) trisomy 21 in which circulating Th17 cells are intact and b) STAT1 gain-of-function in which several patients develop CMC despite normal circulating Th17 cells and intact production of IL-17 by circulating T cells following fungal-specific stimulation and in which JAK-STAT inhibitors ameliorate CMC [216–218]. Of note, in a different setting, autoreactive T cells were shown to promote chronic mucosal fungal infection in mice leading to excessive inflammation, epithelial injury, and esophageal squamous cell carcinoma development, which is a feature of certain CMC-manifesting immune dysregulatory PIDs such as APECED and STAT1 gain-of-function [216,219–221].
VVC
VVC will occur at least once in ~75% of the women worldwide during their reproductive years with 6-10% of them developing >4 recurrent infections per year, a condition termed RVVC [222,223]. VVC, caused predominantly by C. albicans but also by C. glabrata, C. parapsilosis, C. krusei, and C. tropicalis, is a debilitating condition with substantial prevalence, economic burden, and morbidity [222]. VVC is associated with aberrant vaginal inflammation triggered by the presence of Candida in the setting of local immune dysregulation, hormonal changes, vaginal microbiome alterations, and/or damaged mucosa [223,224]. Accordingly, uncontrolled diabetes mellitus, sexual activity, increased estrogen states such as during pregnancy and oral contraceptive or hormone replacement therapy, and antibiotic use are among the most common risk factors for VVC [222,225]. Although beta-lactams are more frequently implicated in the development of VVC relative to other classes of antibiotics, the mechanisms by which specific antibiotics perturb the local microbiome to enable vaginal fungal colonization and infection remain elusive [225,226].
Notably, although HIV/AIDS patients are highly susceptible to OPC and EPC, they are not at a greater risk for developing VVC and, congruently, lymphocyte depletion does not impair fungal control during experimental VVC in mice [227–229]. In addition, although IL-17R responses are induced after infection, the control of fungal proliferation is not reliant on the IL-17 signaling axis during VVC, and patients receiving IL-17 pathway-targeted mAbs have not been reported to be at a significant risk for VVC [163,230]. Taken together, these observations highlight the differential oral and vaginal mucosa-specific host immune requirements for anti-Candida protection.
Vaginal epithelial cells avert C. albicans adhesion to and invasion of the mucosa via shedding of the superficial epithelial layer into the vaginal lumen and coating of epithelial cells with mucin [231]. Upon fungal sensing, vaginal epithelial cells activate early mitochondrial signaling characterized by a protective type I interferon response that is shared between C. albicans and non-albicans Candida species (i.e. C. glabrata, C. parapsilosis, and C. tropicalis). This is followed by a subsequent damage response that is specific to C. albicans and is directed by the secretion of candidalysin [232]. Moreover, similar to oral epithelial cells, vaginal epithelial cells employ NF-κB activation and a biphasic MAPK response to discriminate between C. albicans yeast and hyphal morphotypes, albeit with delayed c-Jun activation and differential pro-inflammatory responses characterized by reduced secretion of IL-6, CCL20, and G-CSF [206,233]. In addition, RNA-seq analysis of patient samples indicated that target genes of the PDGF BB and ERBB2 pathways were up-regulated during VVC whereas target genes of the NEDD9 pathway were not, in contrast to their induction in oral epithelial cells [46].
Investigations in mouse models and humans with VVC including studies of intravaginal challenge with live C. albicans in healthy adult women [234] have established that neutrophils drive immunopathology and underlie VVC symptoms while they are ineffective in mediating fungal clearance in the vaginal microenvironment. In mice with VVC, neutrophil depletion ameliorated inflammation without increasing vaginal fungal load [235]. C. albicans virulence factors that trigger neutrophil recruitment in the vagina include candidalysin and Sap1 through Sap6. [56,236–239,236,240]. The transepithelial migration of neutrophils into the vagina is promoted via the CXCL1-CXCR2 chemokine axis and via estradiol receptor alpha-dependent epithelial expression of CD44 and CD47, both of which are modulated differentially by estrogen and progesterone [241,242]. Notably, recent studies have shed light on the mechanisms of neutrophil dysfunction within the vaginal milieu, termed “neutrophil anergy”; specifically, vaginal heparan sulfate was shown to act as a competitive ligand for Mac-1 on neutrophils, which inhibits their Candida binding and killing properties [243,244].
Integral to the immunopathogenesis of VVC is also inflammasome-primarily NLRP3-activation, and IL-1β production, associated with C. albicans-derived candidalysin and Saps [238,245–247]. Nlrp3−/− mice have reduced neutrophil infiltration, alarmin production, and pro-inflammatory cytokine secretion in vaginal lavage fluid during VVC, and NLRP3 and caspase-1 are upregulated in women with VVC compared to asymptomatic women who were either C. albicans-colonized or non-colonized [238,248]. Correspondingly, the presence of the 12/9 genotype upon examination of a variable number tandem repeat polymorphism in the NLRP3 gene was associated with increased susceptibility to RVVC and a greater production of IL-1β in the vagina [249]. Moreover, a polymorphism in the SIGLEC15 gene, a lectin expressed by immune cells that binds sialic acid-containing structures, was associated with RVVC and correlated with increased IL1B and NLRP3 expression after Candida stimulation [250]. Additional polymorphisms in PRR (TLR2, CLEC7A) and cytokine (IL4) genes have also been associated with the development of RVVC [251–253]. Importantly, IL-22 curtails NLRP3 inflammasome activation and neutrophil recruitment during VVC by inducing the NLRC4 inflammasome, which promotes the production of the IL-1 receptor antagonist (IL-1Ra) [254]. In a mouse model of VVC, recombinant IL-1Ra reduced NLRP3-driven inflammation and protected against C. albicans [254], as did boosting of the protective effects of IL-22 via engaging the aryl hydrocarbon receptor with indole-3-aldehyde, thus providing potential translational avenues for therapeutic intervention [255,256]. In addition, an IL22 polymorphism that led to greater levels of IL-22 and decreased levels of pro-inflammatory cytokines in the vagina correlated with increased resistance to RVVC, as did an IDO1 polymorphism, which was associated with greater vaginal IDO1 expression, increased kynurenine levels, and higher IL-22 and decreased pro-inflammatory cytokine levels [253].
In the past decades, several groups have worked toward developing an anti-Candida vaccine, and VVC has been a major infection manifestation targeted for protection [257]. Promising preclinical data have been generated using vaccine candidates that target C. albicans β-glucan or Sap2 [258,259]. Yet, the most promising vaccine candidate to date, which has demonstrated efficacy in both preclinical models and in human clinical trials, is NDV-3A, which is based on the N-terminal portion of the C. albicans Als3 protein (rAls3p-N) with an alum adjuvant. In a mouse model of VVC, immunization with NDV-3A led to production of high-titer anti-rAls3p-N serum IgG and vaginal IgA antibodies, decreased neutrophil influx, and enhanced C. albicans killing by neutrophils, and protected against vaginal fungal proliferation in a manner dependent on both T and B lymphocytes [260]. In a Phase I clinical trial, administration of NDV-3A in healthy volunteers was safe and resulted in IgG and IgA antibody responses and in IFN-γ and IL-17A cellular responses [261]. In a Phase II, randomized, double-blinded, placebo-controlled clinical trial, administration of NDV-3A to women with RVVC was safe, highly immunogenic, and efficacious resulting in reduced frequency of symptomatic episodes of VVC, particularly in <40 year-old women [262]. Higher serum anti-rAls3p-N IgG titers—particularly of the IgG2 subclass—were observed in vaccinated women who did not experience VVC recurrence relative to those who recurred pointing to a potential surrogate immunological marker of vaccine efficacy [263].
Cutaneous C. albicans infections3
At the steady state, the human skin is colonized by diverse fungal species, predominantly Malassezia, whereas the abundance of Candida species increases dramatically in human skin with immune dysregulation and/or broad-spectrum antibiotic exposure [264–266]. The emerging multidrug-resistant C. auris is an efficient long-term colonizer of the mouse, porcine, and human skin—but not of the gastrointestinal tract in contrast to C. albicans [5,267,268]. C. albicans—but also C. tropicalis, C. parapsilosis, and other Candida species—can cause clinical mucocutaneous disease in the forms of onychomycosis, paronychia, diaper rash, balanitis—often in uncontrolled diabetes mellitus, or other cutaneous infections [269,270].
The outer layer of the epidermis—the stratum corneum—is a cornified envelope composed of dead keratinocytes, keratin, and lipids including ceramides with ultra-long-chain acyl moieties, which create a dense physical barrier against potential pathogens such as C. albicans. Mice deficient in ceramide synthase 3 have a defective cornified lipid envelope and disrupted cutaneous barrier function and are susceptible to C. albicans skin infection [271]. Underneath the stratum corneum, the granular, spinous, and basal layers of the skin epidermis contain live keratinocytes to which C. albicans adheres via interactions of fungal phosphoglycerate mutase (Gpm1) with epithelial cell vitronectin [272]. CLR- and TLR-expressing keratinocytes constitutively express IL-17R via which they respond to IL-17 to generate AMPs for achieving fungal clearance (see below). Moreover, melanocytes located in the basal layer of the epidermis synthesize melanin, which has antimicrobial properties, and recognize C. albicans via TLR4 to increase melanization and exert an inhibitory fungal effect [273,274].
Cutaneous nerve fibers in the epidermis and dermis, particularly those expressing the neuropeptide calcitonin gene-related peptide (CGRP) which is known to mediate pain signaling, have also been shown to participate in protective cutaneous responses against C. albicans through direct antifungal properties of CGRP, and by promoting keratinocyte proliferation and regulating IL-23 production by dermal DCs (see below) [275]. Sensory neurons are activated by C. albicans and their mechanical ablation or chemical denervation of TRPV1+ neurons impaired IL-23 and IL-17 responses and increased susceptibility to cutaneous C. albicans infection, which was rescued by the addition of CGRP [276]. In fact, activation of TRPV1+ neurons was shown to be sufficient to promote protective IL-17 responses during cutaneous C. albicans (and Staphylococcus aureus) infection, including eliciting anticipatory type 17 responses in adjacent uninfected skin [277]. The recent demonstration that MrgprD-expressing nonpeptidergic neurons promote cutaneous immune homeostasis and exert immunomodulatory functions during cutaneous S. aureus infection raises the possibility of their potential role during skin fungal challenge [278].
As with the oral mucosa, IL-23 produced by DCs and IL-17A produced by CD4+ T cells, CD8+ T cells, γδ T cells, and ILC3 are critical for protection against cutaneous C. albicans infection in vivo; instead, IL-22 is dispensable [279]. Three major DC subtypes exist in the skin: Langerhans cells are the only MHCII-expressing cell subset in the epidermis whereas CD11b+ DCs and CD103+ DCs constitute the dermal DC subsets [280]. Importantly, the morphology of C. albicans and the DC subset determine T-helper cell differentiation and fungal control in the skin [281,282]. Thus, yeast cells promote Th17 cell responses—which are critical for cutaneous fungal control— via DECTIN-1-and TLR/MYD88-mediated expression of IL-6 by Langerhans cells in the epidermis, whereas hyphae induce Th1 cell responses—which are dispensable for cutaneous fungal control—but not Th17 cell responses [131,281,283]. Thus, as opposed to Langerhans cells, CD11b+ DCs, which also express DECTIN-1, are not required for Th17 cell responses because DECTIN-1 ligation by hyphae does not occur in the dermis. Instead, CD103+ DCs, which lack DECTIN-1 expression, suppress Th17 cell development likely through the induction of the inhibitory cytokines IL-12 and IL-27 [282].
Although CD11b+ and CD103+ DCs are not required for Th17 cell differentiation, they are both important for the production of IL-17A by CD8+ T cells in the epidermis, which protects from C. albicans skin invasion [284]. Mice deficient in Langerhans cells (or in both Langerhans cells and CD103+ dermal DCs) do not exhibit defects in IL-23 production or fungal growth control during cutaneous candidiasis. Instead, CD11b+ dermal DCs are both necessary and sufficient for IL-23-mediated, IL-17-driven cutaneous protection against C. albicans. Specifically, IL-17-secreting dermal γδ T cells, particularly of the Vγ4 T cell receptor (TCR), constitutively express IL-23R and respond to IL-23 produced by CD301b+ dermal DCs to promote C. albicans clearance [276].
In a different mouse model of skin fungal abscess formation caused by injection of C. albicans hyphae into the deep dermis, a two-step process of initial fungal containment followed by fungal elimination ensues that depends on Nuclear factor of activated T cells (NFAT) signaling, which promotes IL-2 production by DCs and subsequent IFN-γ generation by NK cells. IFN-γ then acts to a) counteract the effects of TGF-β thus limiting myofibroblast differentiation and collagen deposition and to b) promote the generation of plasmin, which mediates collagen capsule digestion, skin ulceration, and elimination of C. albicans [285].
Candida skin colonization has also been associated with certain skin inflammatory diseases such as atopic dermatitis and psoriasis [286,287]. Recently, cutaneous recall responses to C. albicans were shown to promote psoriasiform skin inflammation in mice in a DECTIN-1-, Th17 cell-, neutrophil NET-, and Langerhans cell-dependent manner [288]. These findings support the notion that colonization and/or infection by C. albicans may predispose to or amplify psoriasis via the expansion of fungus-reactive Th17 cells.
Candidemia and systemic candidiasis
In addition to infections at barrier surfaces, C. albicans—together with emerging non-albicans Candida species—are a leading cause of life-threatening nosocomial bloodstream infections [289–291]. Certain underlying immunosuppressive conditions such as neutropenia and/or corticosteroid administration and medical interventions such as the use of central venous catheters or broad-spectrum antibiotics and chemotherapy- or abdominal surgery-induced gastrointestinal barrier disruption are major risk factors for candidemia and systemic candidiasis, especially in ICU patients [4]. Myeloid phagocytes including neutrophils, inflammatory monocytes, tissue-resident macrophages, and CD11b+ DCs are responsible for host defense against systemic candidiasis, whereas T and B lymphocytes and CD103+ DCs are dispensable; the only lymphoid cell subset that contributes to systemic anti-Candida immunity is innate NK cells [4,109,292–294].
Neutrophils represent the first line of innate defense against systemic candidiasis and neutropenic patients are at heightened risk for development of and suffering from poor outcomes after systemic candidiasis [4,295]. Early neutrophil recruitment and swarming at the site of infection is critical for effective fungal control though the precise host molecular signals that underlie early protective neutrophil responses remain poorly understood [296–299]. The organ-specific ability to rapidly recruit neutrophils correlates with fungal control in mice; thus, the spleen and liver rapidly recruit neutrophils and effectively control C. albicans, whereas the kidney exhibits sluggish neutrophil recruitment and is unable to curtail fungal proliferation [40]. During systemic candidiasis, candidalysin contributes to NLRP3 inflammasome activation with subsequent caspase-1-dependent IL-1β secretion and renal neutrophil recruitment [300,301]. In the C. albicans-infected CNS, neutrophil recruitment is also facilitated by candidalysin—while Saps are dispensable—which activates CARD9+ microglial cells to sequentially produce IL-1β and CXCL1 in a p38- and c-Fos-dependent manner for recruiting protective CXCR2+ neutrophils [121,302,303]. Recently, protection from C. albicans invasion of the CNS was surprisingly shown to also depend on meningeal IgA-secreting plasma cells that originate from the gut, are positioned adjacent to dural venous sinuses, and facilitate C. albicans entrapment in peri-sinus areas to restrict fungal spread in brain tissue [304]; whether meningeal IgA is impaired in CARD9 deficiency remains unknown. Immunization of mice with NDV-3A results in greater CXCL1 levels and improved neutrophil influx into infected tissues leading to decreased fungal burden after systemic C. albicans infection [305]. Future clinical studies will be needed to determine whether and how this vaccine may protect humans from systemic candidiasis.
Depending on the size of C. albicans structures, recruited neutrophils employ different mechanisms to restrict the fungus [306]. These effector functions include phagocytosis and intracellular killing of C. albicans yeast cells via oxidative and non-oxidative cytotoxic mechanisms, degranulation of antimicrobial molecules and formation of NETs to counteract large extracellular fungal hyphae, generation of both pro- and anti-inflammatory cytokines and chemokines, and sequestration of trace elements [307–310]. Mechanisms of NET formation include β-glucan recognition by complement receptor 3 (CR3) in opsonized C. albicans whereas for unopsonized C. albicans, DECTIN-2 recognition and signaling via SYK and protein kinase delta (PKCδ) result in neutrophil elastase nuclear translocation, histone citrullination, and NETosis, while protein arginine deiminase 4 (PAD4) is dispensable [311–313].
One of the most important antifungal immune effector mechanisms in neutrophils is the generation of ROS via the sequential assembly of the NADPH oxidase complex at the phagosomal membrane and myeloperoxidase (MPO) activation [314]. NADPH oxidase-dependent potassium flux resulting in activation of neutrophil phagosomal proteases is thought to mediate oxidative burst-mediated fungal (including C. albicans) killing [315]. In mouse neutrophils, which differ from human neutrophils in their MPO and α-defensin content and activity [316], ROS generation was shown to be dependent on DECTIN-1 recognition leading to calcineurin and NFAT signaling and Mac-1/Vav/PKCδ activation [317]. The importance of ROS in antifungal defense is highlighted by human PIDs that impede ROS production and predispose to systemic fungal infections. For example, chronic granulomatous disease, caused by mutations in 4 out of 5 subunits of the NADPH oxidase complex—with the exception of p40phox—that abrogate oxidative burst, carries a ~40% lifetime risk of pulmonary aspergillosis [318,319], whereas systemic Candida infections occur less frequently (<5-10%) and often involve atypical anatomical niches such as the lymph nodes. Similarly, humans with complete MPO deficiency infrequently (~5%) suffer from systemic candidiasis, typically in the presence of additional predisposing factors such as diabetes mellitus [320]. Collectively, these observations highlight the critical contribution of compensatory non-oxidative mechanisms in Candida clearance.
Neutrophil non-oxidative fungal killing mechanisms include AMPs, hydrolases, and nutritional immunity. Two recently recognized molecular signals that mediate neutrophil granulogenesis, degranulation, and non-oxidative C. albicans killing include the endoplasmic reticulum transmembrane protein Jagunal homolog 1 (JAGN1) and the chemokine receptor CXCR1 [321,322]. In fact, the mutant CXCR1 allele CXCR1-T276 was shown to impair neutrophil degranulation and C. albicans killing and was associated with an increased risk of disseminated candidiasis in infected patients [321]. Moreover, using neutrophils from patients with various PIDs two independent signaling mechanisms were characterized that control phagolysosomal function and oxidative or non-oxidative burst-dependent killing in response to opsonized and unopsonized C. albicans yeast cells [323]. Specifically, killing of opsonized C. albicans occurs in a DECTIN-1-independent and SYK-dependent manner and relies on the NADPH oxidase system, Fcγ receptors, and protein kinase C (PKC). By contrast, killing of unopsonized C. albicans yeast cells by human neutrophils occurs independently of the NADPH oxidase system and relies on CR3, CARD9, and phosphoinositide-3-kinase (PI3K) [323].
Although crucial for fungal control, neutrophil-mediated immunity may also come at the cost of immunopathology and tissue injury, particularly in the renal tubules within which neutrophils and C. albicans invade [324]. The molecular mediators that underlie pathogenic neutrophil effects have been uncovered in the mouse model of systemic candidiasis. For example, excessive neutrophil recruitment during the late phase of infection is CCR1-dependent exerting detrimental effects on renal function and host survival [296,299,325]. Moreover, leukotriene B4-dependent neutrophil accumulation in the C. albicans-infected lung results in pulmonary capillaritis and hemorrhage and hypoxia [326]. Furthermore, the tyrosine kinase Tec, the suppressor of TCR signaling (Sts) phosphatases, the lectin galectin-3, the endoribonuclease MCPIP1, and IL-17C are also implicated in neutrophil-mediated immunopathology in C. albicans-infected tissues [327–330]. By contrast, DCs expressing dendritic cell natural killer lectin group receptor-1 (DNGR-1) inhibit renal CXCL2 expression and decrease neutrophil recruitment to ameliorate neutrophil-induced tissue damage during systemic candidiasis [331]. In addition, IL-17R signaling on renal tubular epithelial cells (RTECs) activates the Kallikrein-kinin system and protects RTEC from caspase-3-dependent apoptosis and ameliorates renal damage following systemic candidiasis [332]. Thus, although neutrophils play a critical role in defense against systemic candidiasis, their prolonged and/or excessive recruitment and activation may exert damaging effects. Additional studies are needed to delineate the complex tissue-specific regulatory networks that control the spatial and temporal regulation of neutrophil accumulation and function and to define the relevance of these pathways in humans with systemic candidiasis, in whom neutrophil-associated immunopathology has been observed in the settings of hepatosplenic candidiasis during neutrophil recovery and of renal candidiasis [333–335].
Besides neutrophils, mononuclear phagocytes also promote protective host defense during systemic candidiasis [293]. Specifically, inflammatory Ly6Chi monocytes, which migrate in infected tissues and differentiate into macrophages and monocyte-derived DCs, as well as tissue-resident macrophages, and DCs contribute to fungal clearance through both direct anti-Candida effector functions such as phagocytosis, fungal killing, cytokine production, antigen presentation, and inflammasome activation, and via boosting ROS generation and/or the candidacidal activity of neutrophils [112,336,337]. Inflammatory monocytes traffic into the C. albicans-infected kidney and CNS in a CCR2-dependent manner, can directly inhibit C. albicans growth, and are critical for fungal clearance in these tissues and host survival [338]. Inflammatory monocytes also promote the candidacidal activity of neutrophils. Specifically, splenic inflammatory monocytes produce IL-15 in a type I interferon-dependent manner and activate CCR5-recruited NK cells to produce GM-CSF, which in turn boosts the Candida killing capacity of renal neutrophils [337,339]; this NK function was shown to rely on IL-17R signaling [340]. Besides inflammatory monocytes, CD11b+ DCs depend on SYK signaling to generate IL-23, which represents another local renal molecular mechanism for augmenting GM-CSF production by NK cells and enhancing neutrophil candidacidal activity [112]. IL-23 also provides survival signals to neutrophils within the C. albicans-infected kidney acting in a partially autocrine, IL-17-independent manner to inhibit apoptosis and protect from infection [341]. Last, CD169+ renal macrophages represent another tissue-resident mononuclear phagocyte subset that contributes to priming neutrophil ROS production via IFN-γ and control of C. albicans renal proliferation [292].
In addition, renal tissue-resident macrophages form direct contacts with C. albicans yeast and hyphal forms within the first few hours following infection and exhibit candidacidal activity [324]. The chemokine receptor CX3CR1 is fundamental for control of C. albicans growth in the kidney and host survival by promoting renal macrophage accumulation, direct macrophage-C. albicans interactions, and macrophage killing. Mechanistically, CX3CR1 modulates macrophage survival by inhibiting caspase-3-dependent apoptosis associated with AKT activation [324]. In humans, the dysfunctional CX3CR1-M280 allele was associated with increased risk for developing candidemia and poor outcome after infection [324]. Mechanistically, individuals homozygous for the CX3CR1-M280 allele were shown to exhibit a defect in CX3CL1-mediated monocyte survival due to impaired AKT and ERK activation and had low blood monocyte counts at the steady state [342]. By contrast, CX3CR1-expressing macrophages are dispensable for OPC and VVC control in mice and humans [343]. However, in the gut, CX3CR1-expressing mononuclear phagocytes modulate the composition of and respond to gut fungal communities in a CLR/SYK-dependent manner and patients with IBD carrying the CX3CR1-M280 polymorphism have reduced antifungal antibody responses [344]. CX3CR1-expressing gut macrophages also respond to gut C. albicans to promote the expansion of germinal center-dependent B lymphocytes for the development of antifungal IgG responses that protect from systemic fungal challenge; this response is abrogated in the setting of CARD9 deficiency [20].
Upon encountering C. albicans, macrophages up-regulate signaling pathways involved in phagocytosis and inflammation [345]. The tetraspanin CD82 promotes clustering of DECTIN-1 in the phagocytic cup and DECTIN-1-dependent SYK signaling and mediates macrophage fungal killing and pro-inflammatory cytokine responses. Accordingly, Cd82−/− mice fail to control fungal growth and exhibit greater susceptibility in vivo and polymorphisms in the CD82 gene are associated with development of candidemia in patients [346]. To facilitate engulfment of long hyphal filaments, macrophages can fold fungal hyphae in a process that involves hyphal sensing by DECTIN-1 and β2-integrin and polymerization of the actin–myosin filaments of the phagosome [347]. To avoid rupture of the phagosome and maintain its integrity, macrophages increase the phagosome surface area by lysosome biosynthesis and fusion which is modulated by the transcriptional regulator TFEB [348]. C. albicans-mediated neutralization of the phagosome and yeast-to-hyphal transition trigger NLRP3-dependent lytic pyroptosis in macrophages [349–351]. CLR/SYK-mediated negative regulation of macrophage function during systemic candidiasis also occurs. Specifically, the E3-ubiquitin ligase CBLB targets DECTIN-1, DECTIN-2, and SYK for ubiquitination and degradation in macrophages (and DCs) and leads to impaired inflammasome activation, oxidative burst, and fungal killing and increased mortality during systemic candidiasis [352,353]. In addition, down-regulation of the CLR FcεRII (CD23) by engaging JNK1 signaling downstream of DECTIN-1 ligation in macrophages (and DCs) compromises FcεRII-mediated nitric oxide production and increases mortality during systemic candidiasis [354]. Thus, targeting CBLB and FcεRII may have therapeutic implications for systemic candidiasis.
Furthermore, recent studies have underscored the importance of modulating host metabolism in influencing phagocyte-mediated responses to systemic C. albicans infection. For example, CLR-mediated metabolic reprogramming of monocytes and macrophages mainly via induction of glucose metabolism and increased glycolysis is important for protection against systemic candidiasis [355]. To avoid clearance, C. albicans perturbs host glucose homeostasis by depleting glucose and triggering rapid macrophage death, which depend on glycolysis for energy [356]. Moreover, glutathione reductase (Gsr)-mediated redox regulation is necessary for C. albicans clearance by neutrophils and macrophages. Gsr−/− mice had increased kidney fungal burden, enhanced cytokine and chemokine responses, greater neutrophil infiltration in the infected kidney and heart, and increased mortality during systemic candidiasis. Mechanistically, Gsr deficiency led to defective phagocytosis, respiratory burst, and fungicidal activity in neutrophils and increased levels of pro-inflammatory cytokines and MAPK and SYK activities in macrophages [357]. In addition, restoration of glucose uptake in neutrophils by pharmacological inhibition of glycogen synthase kinase 3 beta (GSK3β) rescued ROS production and candidacidal function of neutrophils from uremic mice and patients with chronic kidney disease [358].
The ability of cells to exhibit immunological memory was thought of as an exclusive feature of the adaptive immune system, however activation of monocytes and macrophages can also result in enhanced responsiveness to subsequent triggers via a process termed trained immunity, which is mediated by epigenetic reprogramming [359]. First described in the setting of C. albicans infection in 2012, trained immunity promotes T and B lymphocyte-independent protection against systemic candidiasis following a first exposure to a non-lethal infectious dose. This protection is conferred by monocytes and macrophages via DECTIN-1/Raf-1/NF-κB activation after exposure to β-glucan [360,361]. Trained monocytes and macrophages exert enhanced pro-inflammatory responses and exhibit a metabolic shift toward aerobic glycolysis via AKT/mTOR/HIF-1α signaling [361].
In summary, the delineation of the molecular basis of antifungal host defense mechanisms against life-threatening systemic candidiasis holds promise for the identification of genetic variants in immune-related genes, which—either alone or in combination—may explain patient-specific susceptibility to infection. Besides the aforementioned genetic variation in the CXCR1, CX3CR1, CD82, and TLR gene loci [145,321,324,346], additional population studies have revealed increased susceptibility to systemic candidiasis in patients with certain genetic variants in the TNF, IL10, IL12B, CCL8, CD58, TAGAP, LCE4A-C1orf68, PSMB8, SP110, and STAT1 genes; strikingly, the combinatorial presence of certain genetic variants has been reported to lead to up to ~20-fold increases in host susceptibility to candidemia [144,362–365]. Collectively, these findings may eventually help devise personalized immunogenetics-based strategies that could allow for risk stratification, intensified diagnosis, targeted vaccination, antifungal prophylaxis, and/or prognostication of patients in the ICU, which may improve their outcomes.
Therapeutic interventions and drug resistance mechanisms
Factors contributing to the high morbidity and mortality of systemic candidiasis in patients include the poor performance of fungal diagnostics (reviewed elsewhere [366]) and the suboptimal efficacy of antifungal drugs in vivo. Currently available classes of antifungal drugs that are used to treat C. albicans infections include the polyenes, 5-flucytosine (5-FC), the azoles, and the echinocandins [367]. Terbinafine is an allylamine antifungal drug that inhibits ergosterol biosynthesis by targeting squalene epoxidase and blocking the conversion of squalene to squalene epoxide [368]. However, its use is restricted to the treatment of onychomycosis and cutaneous fungal infections due to its limited systemic bioavailability [369], and thus will not be further discussed here (Figure 3).
Figure 3.
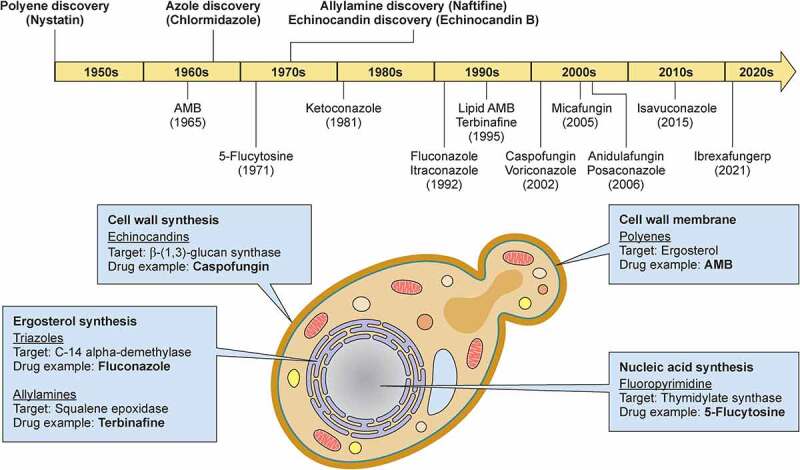
Milestones in antifungal drug development and fungal targets of the currently available antifungal agents. In the upper panel, the timeline depicts the date of discovery of the first indicated antifungal compound within each class of antifungal drugs and the date of FDA approval for the most common antifungal drugs with anti-Candida activity. In the lower panel, a C. albicans budding yeast is depicted and the targets of antifungal drugs are shown. Illustration created with BioRender.com. AMB, amphotericin B.
The oldest class of antifungal drugs available in the clinic are polyenes. Polyenes bind to ergosterol, a cell membrane sterol that is unique to fungi. Upon binding, polyenes form pores in the fungal cell membrane causing osmotic cell lysis. Besides pore formation, the mode of action of amphotericin B (AMB) also involves oxidative damage to fungal cells [370] and ergosterol sequestration, leading to extra-membranous aggregates [371]. The most recognizable member of the polyene family is AMB, which remains one of the most potent and broad-spectrum antifungal agents for the treatment of mucosal and systemic fungal infections, whereas nystatin, another polyene, is solely used topically for the treatment of OPC [372,373]. Nephrotoxicity is a common and limiting adverse effect to AMB, although the lipid drug formulations cause less renal damage [374]. The recent advent of the cochleated formulation of AMB shows promise for efficacious oral drug delivery without its nephrotoxic effects [375]. Despite the widespread use of AMB over decades, resistance of C. albicans to AMB is very uncommon and, when present, it is associated with mutations in the ERG3 or ERG6 genes of the ergosterol biosynthesis pathway, which result in decreased levels of ergosterol thus impeding the binding of AMB to the fungal cell membrane [376,377].
5-FC is a pyrimidine analog that is taken up by C. albicans and converted to 5-fluorouracil, which interferes with fungal DNA and RNA synthesis. The rapid development of C. albicans resistance to 5-FC when used as monotherapy, caused by mutations in cytosine permease and cytosine deaminase that decrease uptake or conversion of the drug to 5-fluorouracil, respectively, and the hematological, hepatic, and gastrointestinal adverse effects of the drug limit its use in the clinic [378]. Thus, 5-FC is typically administered in combination with AMB or triazoles in the setting of certain difficult-to-treat infections such as candidal endocarditis or meningitis [379].
The azoles target Erg11p/Cyp51 and inhibit the biosynthesis of ergosterol leading to the accumulation of toxic sterols on the fungal cell membranes such as 14α-methylergosta-8,24(28) dienol and increased levels of endogenous ROS [380,381], both of which contribute to fungal growth arrest. Azoles are divided into two subgroups based on their chemical structure: the imidazoles such as clotrimazole, ketoconazole, and miconazole, and the triazoles, which include fluconazole, itraconazole, voriconazole, posaconazole, and isavuconazole. The topical application of imidazoles is used for the treatment of mucosal candidiasis, whereas the triazoles are commonly used to treat mucosal and systemic infections by C. albicans, although triazole-specific toxicities and drug-drug interactions occasionally restrict their clinical use [382]. The emergence of azole resistance poses therapeutic challenges [383]. It is observed more often during treatment of patients with CMC compared to those with candidemia due to the recurrent nature of these infections and the repeated exposures to azoles [384–386]. The molecular mechanisms of azole resistance in C. albicans include: mutations in the ERG11 gene that result in ERG11 overexpression; gain-of-function mutations in the ergosterol biosynthesis pathway regulator UPC2 gene that also lead to increased ERG11 expression; mutations in the ERG3 or ERG6 genes that cause accumulation of toxic sterols; overexpression of drug efflux pumps including the ABC transporters CDR1 and CDR2 and the MFS transporter MDR1 caused by gain-of-function mutations in the transcription factors TAC1 and MRR1; genomic plasticity in the forms of aneuploidy, trisomy, loss of heterozygosity, and isochromosome formation; as well as enzymatic changes involved in the sphingolipid synthesis pathway [387,388]. Addition of a tetrazole group by substitution of the triazole metal-binding group has resulted in decreased drug-drug interactions and improved tolerability due to the greater specificity for the fungal ERG11 over the human CYP51 [389]. The tetrazole compounds VT-1161/oteseconazole and VT-1598 exhibit superior in vitro and in vivo activity against clinical C. albicans (including azole-resistant) strains from patients with CMC [390,391], and oteseconazole was safe and efficacious for the treatment of recurrent VVC in a Phase II randomized, double-blind, placebo-controlled clinical trial [392].
The most recently introduced class of antifungal drugs in the clinic, the parenterally used echinocandins, consist of the well-tolerated caspofungin, micafungin, and anidulafungin, which inhibit β-(1,3)-glucan synthase, an enzyme involved in the generation of β-(1,3)-glucan; in the absence of β-(1,3)-glucan, loss of fungal cell wall rigidity and cell lysis ensue [393]. The increasing incidence of echinocandin-resistant C. albicans clinical strains in recent years is concerning [394]. The most common mechanism of resistance relates to mutations in hotspot regions of the β-(1,3)-glucan synthase gene FKS [395], and are typically associated with prior echinocandin exposure [396,397]. In addition, although more prevalent in C. glabrata [398], mutations in the mismatch repair gene MSH2 can also result in echinocandin resistance in C. albicans; MSH2 mutations often confer cross-resistance to azoles and, alarmingly, may occur without prior exposure to echinocandins [399]. Recently, ibrexafungerp, an oral antifungal drug that belongs to a novel class of glucan synthase inhibitors termed triterpenoids, has shown significant activity against C. albicans (and multidrug-resistant C. glabrata and C. auris). Ibrexafungerp maintains its efficacy at low pH conditions which are often encountered in the vaginal mucosa and was recently FDA-approved for the treatment of VVC in women [400,401].
Another promising antifungal drug currently in Phase II clinical trials is the first-in-class fosmanogepix, an inhibitor of the fungal enzyme Gwt1, which is involved in glycosylphosphatidylinositol-anchored mannoprotein biosynthesis, trafficking, and anchoring to the cell membrane and outer cell wall [402]. Fosmanogepix exhibits a broad spectrum in vitro activity against C. albicans (including echinocandin-resistant) strains, other yeast, and mold fungi and has shown significant in vivo efficacy in mouse and rabbit models of OPC and disseminated C. albicans infections [403–405]. Although developing antifungal drugs is hindered by the evolutionary proximity between eukaryotic fungi and humans, additional novel antifungal agents are in different phases of development and promising candidates such as turbinmicin have recently emerged via metabolomic screens from the microbiome of marine animals [406–408]. For a more detailed review of the new antifungal agents in clinical development the reader is refered to [409].
Outlook
The co-evolution of C. albicans with humans has established a complex balance between commensalism and pathogenicity for this yeast fungus. In recent decades, advances in modern medicine have enabled this pathobiont to become one of the most common human pathogens causing life-threatening healthcare-associated infections, the prevalence of which continues to be significant. Understanding the virulence traits of C. albicans, the tissue-specific mechanisms of anti-Candida host defense, and its mechanisms of resistance to the armamentarium of available antifungal drugs should enable the development of better strategies for the diagnosis and treatment of affected individuals, which may help improve patient outcomes.
Acknowledgments
We apologize to those colleagues whose papers could not be cited due to space limitations.
Funding Statement
This work was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, NIH; Division of Intramural Research, National Institute of Allergy and Infectious Diseases [ZIA AI001175].
Footnotes
In all subheadings, the PDF proof does not have a line to separate the previous from the next subsection. please add a line of separation to make it easy for the reader to discern when a segment changes-throughout the paper
In Table 2, in the PDF Proof (not in here), there are words that belong in the above sentence that are shown in the sentence below. Eg in IL-12p40 deficiency NTM and infections are separately by a line and they shouldn't. Same for LAD-1 where gram-negative and bacteria, periodontitis are separately by a line. Also there is an indent in the genes listed for SCID in the PDF and in the genes listed for CGD. they should all be listed without an indent.
as mentioned above, subsegments are separated well here but not on the PDF proof.
Abbreviations
Vulvovaginal candidiasis (VVC), Oropharyngeal candidiasis (OPC), Esophageal candidiasis (EPC), Central nervous system (CNS), Antimicrobial peptides (AMPs), Platelet-derived growth factor BB (PDGF BB), Neural precursor cell expressed developmentally down-regulated protein 9 (NEDD9), Epidermal growth factor receptor (EGFR), Secreted aspartic proteinases (Saps), N-acetylglucosamine (GlcNAc), Superoxide dismutases (Sods), Reactive oxygen species (ROS), Heat shock proteins (Hsps), Pathogen-associated molecular patterns (PAMPs), Pattern recognition receptors (PRR), MAPK (mitogen-activated protein kinase), CEK1 (Choline/ethanolamine kinase 1), C-type lectin receptors (CLRs), Spleen tyrosine kinase (SYK), Caspase recruitment domain-containing protein 9 (CARD9), Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), NF-κB-inducing kinase (NIK), Neutrophil extracellular traps (NETs), Primary immunodeficiency disorder (PID), Recurrent VVC (RVVC), Toll-like receptors (TLRs), Intensive care unit (ICU), NOD-like receptors (NLRs), Nicotinamide adenine dinucleotide phosphate (NADPH), Chronic mucocutaneous candidiasis (CMC), monoclonal antibodies (mAbs), Inflammatory bowel disease (IBD), type 3 innate lymphoid cells (ILC3), Dendritic cells (DCs), natural Th17 (nTh17), Tissue-resident memory T cells (TRM), Granulocyte colony-stimulating factor (G-CSF), Granulocyte-macrophage colony-stimulating factor (GM-CSF), Autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED), Autoimmune polyglandular syndrome type 1 (APS-1), Autoimmune regulator (AIRE), Interferon gamma (IFN-γ), IL-1 receptor antagonist (IL-1Ra), Phosphoglycerate mutase (Gpm1), Calcitonin gene-related peptide (CGRP), T cell receptor (TCR), Complement receptor 3 (CR3), PKCδ (protein kinase delta), NFAT (Nuclear factor of activated T cells), Protein arginine deiminase 4 (PAD4), Myeloperoxidase enzyme (MPO), Jagunal homolog 1 (JAGN1), Protein kinase C (PKC), Phosphoinositide-3-kinase (PI3K), Suppressor of TCR signaling (Sts), Dendritic cell natural killer lectin group receptor-1 (DNGR-1), Renal tubular epithelial cells (RTECs), Glutathione reductase (Gsr), Glycogen synthase kinase 3 beta (GSK3β), 5-flucytosine (5-FC), Amphotericin B (AMB).
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
No data sets were analyzed in this manuscript, and data availability is not applicable.
References
- [1].Choi J, Kim S-H.. A genome tree of life for the fungi kingdom. Proc Natl Acad Sci U S A. 2017. Aug 29; 114(35):9391–9396. 10.1073/pnas.1711939114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hawksworth DL. The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res. 2001. Dec;105:1422–1432. [Google Scholar]
- [3].Lamoth F, Lockhart SR, Berkow EL, et al. Changes in the epidemiological landscape of invasive candidiasis. J Antimicrob Chemother. [2018. Jan 1];73(suppl_1):i4–i13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pappas PG, Lionakis MS, Arendrup MC, et al. Invasive candidiasis. Nat Rev Dis Primers. 2018. May 11;4:18026. [DOI] [PubMed] [Google Scholar]
- [5].Huang X, Hurabielle C, Drummond RA, et al. Murine model of colonization with fungal pathogen Candida auris to explore skin tropism, host risk factors and therapeutic strategies. Cell Host Microbe. [2021. Feb 10];29(2):210–221 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Silva S, Negri M, Henriques M, et al. Candida glabrata, Candida parapsilosis and Candida tropicalis: biology, epidemiology, pathogenicity and antifungal resistance. FEMS Microbiol Rev. 2012. Mar;36(2):288–305. [DOI] [PubMed] [Google Scholar]
- [7].Silva S, Negri M, Henriques M, et al. Adherence and biofilm formation of non-Candida albicans Candida species. Trends Microbiol. 2011. May;19(5):241–247. [DOI] [PubMed] [Google Scholar]
- [8].Lionakis MS, Hohl TM. Call to action: how to tackle emerging nosocomial fungal infections comment. Cell Host Microbe. 2020. Jun 10;27(6):859–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bougnoux ME, Diogo D, Francois N, et al. Multilocus sequence typing reveals intrafamilial transmission and microevolutions of Candida albicans isolates from the human digestive tract. J Clin Microbiol. 2006. May;44(5):1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Beigi RH, Meyn LA, Moore DM, et al. Vaginal yeast colonization in nonpregnant women: a longitudinal study. Obstet Gynecol. 2004. Nov;104(5, Part 1):926–930. DOI: 10.1097/01.AOG.0000140687.51048.73. [DOI] [PubMed] [Google Scholar]
- [11].Opulente DA, Langdon QK, Buh KV, et al. Pathogenic budding yeasts isolated outside of clinical settings. FEMS Yeast Res. 2019. May 1;19(3). 10.1093/femsyr/foz032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chow BDW, Reardon JR, Perry EO, et al. Expressed breast milk as a predictor of neonatal yeast colonization in an intensive care setting. J Pediatr Infect Dis. 2014. Sep;3(3):213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ali GY, Algohary EHSS, Rashed KA, et al. Prevalence of Candida colonization in preterm newborns and VLBW in neonatal intensive care unit: role of maternal colonization as a risk factor in transmission of disease. J Matern-Fetal Neo M. 2012Jun; 251: 789–795 [DOI] [PubMed] [Google Scholar]
- [14].Mason KL, Downward JRE, Mason KD, et al. Candida albicans and bacterial microbiota interactions in the cecum during recolonization following broad-spectrum antibiotic therapy. Infect Immun. 2012. Oct;80(10):3371–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].McDonough L, Mishra AA, Tosini N, et al. Candida albicans isolates 529L and CHN1 exhibit stable colonization of the murine gastrointestinal tract. bioRxiv.2021. June, 27;450080. https://pubmed.ncbi.nlm.nih.gov/34724818/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fan D, Coughlin LA, Neubauer MM, et al. Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat Med. 2015. Jul;21(7):808–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ost KS, O’Meara TR, Stephens WZ, et al. Adaptive immunity induces mutualism between commensal eukaryotes. Nature. 2021 July 14; 596(7870):114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rao C, Coyte KZ, Bainter W, et al. Multi-kingdom ecological drivers of microbiota assembly in preterm infants. Nature. 2021 Feb 24;591(7851): 633–638 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kawakita M, Oyama T, Shirai I, et al. Cell wall N-glycan of Candida albicans ameliorates early hyper- and late hypo-immunoreactivity in sepsis. Commun Biol. [2021. March 16];4(1):342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Doron I, Leonardi I, Li XV, et al. Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell. [2021. Feb 18];184(4):1017–1031 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bacher P, Hohnstein T, Beerbaum E, et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against candida albicans. Cell. [2019. Mar 7];176(6):1340–1355 e15. [DOI] [PubMed] [Google Scholar]
- [22].Shao TY, Ang WXG, Jiang TT, et al. Commensal Candida albicans positively calibrates systemic Th17 immunological responses. Cell Host Microbe. [2019. Mar 13];25(3):404–417 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tso GHW, Reales-calderon JA, Tan ASM, et al. Experimental evolution of a fungal pathogen into a gut symbiont. Science. [2018. Nov 2];362(6414):589-+. [DOI] [PubMed] [Google Scholar]
- [24].Tan CT, Xu X, Qiao Y, et al. A peptidoglycan storm caused by beta-lactam antibiotic’s action on host microbiota drives Candida albicans infection. Nat Commun. [2021. May 7];12(1):2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhai B, Ola M, Rolling T, et al. High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat Med. 2020. Jan;26(1):59-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].McCarty TP, Pappas PG. Invasive Candidiasis. Infect Dis Clin North Am. 2016. Mar;30(1):103–124. [DOI] [PubMed] [Google Scholar]
- [27].Noble SM, Gianetti BA, Witchley JN. Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat Rev Microbiol. 2017. Feb;15(2):96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bohm L, Torsin S, Tint SH, et al. The yeast form of the fungus Candida albicans promotes persistence in the gut of gnotobiotic mice. PLoS Pathog. 2017. Oct;13(10):e1006699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pande K, Chen CB, Noble SM. Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat Genet. 2013. Sep;45(9):1088-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Desai JV. Candida albicans hyphae: from growth initiation to invasion. J Fungi (Basel). 2018. Jan 11;4(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lu Y, Su C, Liu H. Candida albicans hyphal initiation and elongation. Trends Microbiol. 2014. Dec;22(12):707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vylkova S, Carman AJ, Danhof HA, et al. The fungal pathogen Candida albicans autoinduces hyphal morphogenesis by raising extracellular pH. mBio. 2011;2(3):e00055–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Flanagan PR, Liu NN, Fitzpatrick DJ, et al. The Candida albicans TOR-activating GTPases Gtr1 and Rhb1 coregulate starvation responses and biofilm formation. mSphere. 2017. Nov-Dec;2(6):e00477–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lindsay AK, Deveau A, Piispanen AE, et al. Farnesol and cyclic AMP signaling effects on the hypha-to-yeast transition in Candida albicans. Eukaryot Cell. 2012. Oct;11(10):1219–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Witchley JN, Penumetcha P, Abon NV, et al. Candida albicans morphogenesis programs control the balance between gut commensalism and invasive infection. Cell Host Microbe. [2019. Mar 13];25(3):432–443 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lo HJ, Kohler JR, DiDomenico B, et al. Nonfilamentous C. albicans mutants are avirulent. Cell. [1997. Sep 5];90(5):939–949. [DOI] [PubMed] [Google Scholar]
- [37].Murad AM, Leng P, Straffon M, et al. NRG1 represses yeast-hypha morphogenesis and hypha-specific gene expression in Candida albicans. EMBO J. [2001. Sep 3];20(17):4742–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Banerjee M, Lazzell AL, Romo JA, et al. Filamentation is associated with reduced pathogenicity of multiple non-albicans Candida species. mSphere. 2019. Oct 16;4:(5):e00656–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dunker C, Polke M, Swidergall M, et al. Rapid proliferation due to better metabolic adaptation results in full virulence of a filament-deficient Candida albicans strain. Mycoses. 2020;63:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lionakis MS, Lim JK, Lee CC, et al. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J Innate Immun. 2011;3(2):180–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu Y, Filler SG. Candida albicans Als3, a multifunctional adhesin and invasin. Eukaryot Cell. 2011. Feb;10(2):168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nobile CJ, Nett JE, Andes DR, et al. Function of Candida albicans adhesin Hwp1 in biofilm formation. Eukaryot Cell. 2006. Oct;5(10):1604–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Swidergall M, Filler SG. Oropharyngeal Candidiasis: fungal invasion and epithelial cell responses. PLoS Pathog. 2017. Jan;13(1):e1006056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Filler SG, Swerdloff JN, Hobbs C, et al. Penetration and damage of endothelial-cells by Candida-albicans. Infect Immun. 1995. Mar;63(3):976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Phan QT, Myers CL, Fu Y, et al. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 2007. Mar;5(3):543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Liu YP, Shetty AC, Schwartz JA, et al. New signaling pathways govern the host response to C-albicans infection in various niches. Genome Res. 2015. May;25(5):679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhu WD, Phan QT, Boontheung P, et al. EGFR and HER2 receptor kinase signaling mediate epithelial cell invasion by Candida albicans during oropharyngeal infection. Proc Natl Acad Sci U S A. [2012. Aug 28];109(35):14194–14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].McCall AD, Pathirana RU, Prabhakar A, et al. Candida albicans biofilm development is governed by cooperative attachment and adhesion maintenance proteins. Npj Biofilms Microbi. 2019. Aug 23;5(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wachtler B, Wilson D, Haedicke K, et al. From attachment to damage: defined genes of Candida albicans mediate adhesion, invasion and damage during interaction with oral epithelial cells. PLoS One. [2011. Feb 23];6(2):e17046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Allert S, Forster TM, Svensson CM, et al. Candida albicans-induced epithelial damage mediates translocation through intestinal barriers. Mbio. 2018. May-Jun;9(3):e00915–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Naglik JR, Challacombe SJ, Hube B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev. 2003. Sep;67(3):400–428. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Albrecht A, Felk A, Pichova I, et al. Glycosylphosphatidylinositol- anchored proteases of Candida albicans target proteins necessary for both cellular processes and host-pathogen interactions. J Biol Chem. [2006. Jan 13];281(2):688–694. [DOI] [PubMed] [Google Scholar]
- [53].Schaller M, Korting HC, Schafer W, et al. Secreted aspartic proteinase (Sap) activity contributes to tissue damage in a model of human oral candidosis. Mol Microbiol. 1999. Oct;34(1):169–180. [DOI] [PubMed] [Google Scholar]
- [54].Hube B, Sanglard D, Odds FC, et al. Disruption of each of the secreted aspartyl proteinase genes SAP1, SAP2, and SAP3 of Candida albicans attenuates virulence. Infect Immun. 1997. Sep;65(9):3529–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pericolini E, Gabrielli E, Amacker M, et al. Secretory aspartyl proteinases cause vaginitis and can mediate vaginitis caused by candida albicans in mice. Mbio. 2015. May-Jun;6(3):e00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gabrielli E, Sabbatini S, Roselletti E, et al. In vivo induction of neutrophil chemotaxis by secretory aspartyl proteinases of Candida albicans. Virulence. [2016. Oct 2];7(7):819–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Leidich SD, Ibrahim AS, Fu Y, et al. Cloning and disruption of caPLB1, a phospholipase B gene involved in the pathogenicity of Candida albicans. J Biol Chem. [1998. Oct 2];273(40):26078–26086. [DOI] [PubMed] [Google Scholar]
- [58].Hube B, Stehr F, Bossenz M, et al. Secreted lipases of Candida albicans: cloning, characterisation and expression analysis of a new gene family with at least ten members. Arch Microbiol. 2000. Nov;174(5):362–374. [DOI] [PubMed] [Google Scholar]
- [59].Gacser A, Stehr F, Kroger C, et al. Lipase 8 affects the pathogenesis of Candida albicans. Infect Immun. 2007. Oct;75(10):4710–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Moyes DL, Wilson D, Richardson JP, et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. [2016. Apr 7];532(7597):64-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Naglik JR, Gaffen SL, Hube B. Candidalysin: discovery and function in Candida albicans infections. Curr Opin Microbiol. 2019. Dec;52:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Blagojevic M, Camilli G, Maxson M, et al. Candidalysin triggers epithelial cellular stresses that induce necrotic death. Cell Microbiol. 2021. Jun 16;23(10): e13371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Swidergall M, Solis NV, Millet N, et al. Activation of EphA2-EGFR signaling in oral epithelial cells by Candida albicans virulence factors. PLoS Pathog. 2021. Jan;17(1):e1009221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mogavero S, Sauer FM, Brunke S, et al. Candidalysin delivery to the invasion pocket is critical for host epithelial damage induced by Candida albicans. Cell Microbiol. 2021;23(10): e13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Austermeier S, Pekmezovic M, Porschitz P, et al. Albumin neutralizes hydrophobic toxins and modulates candida albicans pathogenicity. mBio. 2021. Jun 22;12(3): e0053121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Man A, Ciurea CN, Pasaroiu D, et al. New perspectives on the nutritional factors influencing growth rate of Candida albicans in diabetics. an in vitro study. Mem Inst Oswaldo Cruz. 2017. Sep;112(9):587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Williams RB, Lorenz MC. Multiple alternative carbon pathways combine to promote Candida albicans stress resistance, immune interactions, and virulence. mBio. 2020. Jan 14;11(1): e00357–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Vesely EM, Williams RB, Konopka JB, et al. N-acetylglucosamine metabolism promotes survival of Candida albicans in the phagosome. mSphere. 2017. Sep-Oct;2(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Alves R, Mota S, Silva S, et al. The carboxylic acid transporters Jen1 and Jen2 affect the architecture and fluconazole susceptibility of Candida albicans biofilm in the presence of lactate. Biofouling. 2017. Nov;33(10):943–954. [DOI] [PubMed] [Google Scholar]
- [70].Crawford AC, Lehtovirta-Morley LE, Alamir O, et al. Biphasic zinc compartmentalisation in a human fungal pathogen. PLoS Pathog. 2018. May;14(5):e1007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Potrykus J, Stead D, MacCallum DM, et al. Fungal iron availability during deep seated candidiasis is defined by a complex interplay involving systemic and local events. PLoS Pathog. 2013. Oct;9(10):e1003676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Citiulo F, Jacobsen ID, Miramon P, et al. Candida albicans scavenges host zinc via pra1 during endothelial invasion. PLoS Pathog. 2012;8(6):e1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Luo S, Dasari P, Reiher N, et al. The secreted Candida albicans protein pra1 disrupts host defense by broadly targeting and blocking complement C3 and C3 activation fragments. Mol Immunol. 2018;93:266–277. [DOI] [PubMed] [Google Scholar]
- [74].Malavia D, Lehtovirta-Morley LE, Alamir O, et al.Zinc limitation induces a hyper-adherent goliath phenotype in Candida albicans.Front Microbiol. 2017. Nov 14;8:2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Crawford A, Wilson D. Essential metals at the host-pathogen interface: nutritional immunity and micronutrient assimilation by human fungal pathogens. FEMS Yeast Res. 2015. Nov;15(7):fov071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mackie J, Szabo EK, Urgast DS, et al. Host-imposed copper poisoning impacts fungal micronutrient acquisition during systemic candida albicans infections. PLoS One. 2016;11(6):e0158683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Weissman Z, Berdicevsky I, Cavari BZ, et al. The high copper tolerance of Candida albicans is mediated by a P-type ATPase. Proc Natl Acad Sci U S A. [2000. Mar 28];97(7):3520–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ramanan N, Wang Y. A high-affinity iron permease essential for Candida albicans virulence. Science. 2000. May 12;288(5468):1062–1064. [DOI] [PubMed] [Google Scholar]
- [79].Almeida RS, Brunke S, Albrecht A, et al. the hyphal-associated adhesin and invasin Als3 of Candida albicans mediates iron acquisition from host ferritin. PLoS Pathog. 2008. Nov;4(11):e1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Frohner IE, Bourgeois C, Yatsyk K, et al. Candida albicans cell surface superoxide dismutases degrade host-derived reactive oxygen species to escape innate immune surveillance. Mol Microbiol. 2009. Jan;71(1):240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hwang CS, Rhie GE, Oh JH, et al. Copper- and zinc-containing superoxide dismutase (Cu/ZnSOD) is required for the protection of Candida albicans against oxidative stresses and the expression of its full virulence. Microbiol-Sgm. 2002;148:3705–3713. [DOI] [PubMed] [Google Scholar]
- [82].Loll-Krippleber R, d’Enfert C, Feri A, et al. A study of the DNA damage checkpoint in Candida albicans: uncoupling of the functions of Rad53 in DNA repair, cell cycle regulation and genotoxic stress-induced polarized growth. Mol Microbiol. 2014. Feb;91(3):452–471. [DOI] [PubMed] [Google Scholar]
- [83].Smith DA, Nicholls S, Morgan BA, et al. A conserved stress-activated protein kinase regulates a core stress response in the human pathogen Candida albicans. Mol Biol Cell. 2004. Sep;15(9):4179–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Danhof HA, Vylkova S, Vesely EM, et al. Robust extracellular pH modulation by candida albicans during growth in carboxylic acids. mBio. 2016. Nov 15;7(6):e01646–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Enjalbert B, Smith DA, Cornell MJ, et al. Role of the Hog1 stress-activated protein kinase in the global transcriptional response to stress in the fungal pathogen Candida albicans. Mol Biol Cell. 2006. Feb;17(2):1018–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Miramon P, Dunker C, Windecker H, et al. Cellular responses of Candida albicans to phagocytosis and the extracellular activities of neutrophils are critical to counteract carbohydrate starvation, oxidative and nitrosative stress. PLoS One. 2012;7(12):e52850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Leach MD, Klipp E, Cowen LE, et al. Fungal Hsp90: a biological transistor that tunes cellular outputs to thermal inputs. Nat Rev Microbiol. 2012. Oct;10(10):693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Gow NAR, Hube B. Importance of the Candida albicans cell wall during commensalism and infection. Curr Opin Microbiol. 2012. Aug;15(4):406–412. [DOI] [PubMed] [Google Scholar]
- [89].Pradhan A, Ma Q, de Assis LJ, et al. Anticipatory stress responses and immune evasion in fungal pathogens. Trends Microbiol. 2021. May;29(5):416–427. [DOI] [PubMed] [Google Scholar]
- [90].Ballou ER, Avelar GM, Childers DS, et al. Lactate signalling regulates fungal beta-glucan masking and immune evasion. Nat Microbiol. 2016. Dec 12;2:16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Pradhan A, Avelar GM, Bain JM, et al. Non-canonical signalling mediates changes in fungal cell wall PAMPs that drive immune evasion. Nat Commun. [2019. Nov 22];10(1):5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lopes JP, Stylianou M, Backman E, et al. Evasion of immune surveillance in low oxygen environments enhances Candida albicans virulence. mBio. 2018. Nov 6;9:(6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Pradhan A, Avelar GM, Bain JM, et al. Hypoxia promotes immune evasion by triggering beta-glucan masking on the Candida albicans cell surface via mitochondrial and cAMP-protein kinase A signaling. mBio. 2018. Nov 6;9(6):e02120–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kumwenda P, Cottier F, Keevan B, et al. Oestrogen promotes innate immune evasion of Candida albicans through inactivation of the alternative complement system. bioRxiv. 2020. July 22;207191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Childers DS, Avelar GM, Bain JM, et al. Epitope shaving promotes fungal immune evasion. mBio. 2020. Jul 7;11(4):e00984–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Cottier F, Sherrington S, Cockerill S, et al. Remasking of Candida albicans beta-glucan in response to environmental pH is regulated by quorum sensing. mBio. 2019. Oct 15;10(5):e02347–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Sherrington SL, Sorsby E, Mahtey N, et al. Adaptation of Candida albicans to environmental pH induces cell wall remodelling and enhances innate immune recognition. PLoS Pathog. 2017. May;13(5):e1006403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Tripathi A, Liverani E, Tsygankov AY, et al. Iron alters the cell wall composition and intracellular lactate to affect Candida albicans susceptibility to antifungals and host immune response. J Biol Chem. [2020. Jul 17];295(29):10032–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wheeler RT, Kombe D, Agarwala SD, et al. Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog. 2008. Dec;4(12):e1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Wheeler RT, Fink GR, Cormack B. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog. 2006. Apr;2(4):e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Lorenz MC, Bender JA, Fink GR. Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryot Cell. 2004. Oct;3(5):1076–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Westman J, Moran G, Mogavero S, et al. Candida albicans hyphal expansion causes phagosomal membrane damage and luminal alkalinization. mBio. 2018. Sep 11;9(5). 10.1128/mBio.01226-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Vylkova S, Lorenz MC, Krysan DJ. Modulation of phagosomal pH by Candida albicans promotes hyphal morphogenesis and requires Stp2p, a regulator of amino acid transport. PLoS Pathog. 2014. Mar;10(3):e1003995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Taylor PR, Tsoni SV, Willment JA, et al. Dectin-1 is required for β-glucan recognition and control of fungal infection. Nat Immunol. 2007. Jan;8(1):31–38. DOI: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Brown GD, Herre J, Williams DL, et al. Dectin-1 mediates the biological effects of β-Glucans. J Exp Med. 2003. May 5;197(9):1119–1124. 10.1084/jem.20021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Saijo S, Ikeda S, Yamabe K, et al. Dectin-2 recognition of α-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity. 2010. May 28;32(5):681–691. 10.1016/j.immuni.2010.05.001. [DOI] [PubMed] [Google Scholar]
- [107].Zhu -L-L, Zhao X-Q, Jiang C, et al. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity. 2013. Aug 22;39(2):324–334. 10.1016/j.immuni.2013.05.017. [DOI] [PubMed] [Google Scholar]
- [108].Wells CA, Salvage-Jones JA, Li X, et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J Immunol. 2008. Jun 1;180(11):7404–7413. 10.4049/jimmunol.180.11.7404. [DOI] [PubMed] [Google Scholar]
- [109].Lionakis MS, Levitz SM. Host control of fungal infections: lessons from basic studies and human cohorts. Annu Rev Immunol. 2018. Apr 26;36(1):157–191. 10.1146/annurev-immunol-042617-053318. [DOI] [PubMed] [Google Scholar]
- [110].Hardison SE, Brown GD. C-type lectin receptors orchestrate antifungal immunity. Nat Immunol. 2012. Sep;13(9):817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Negoro PE, Xu SY, Dagher Z, et al. Spleen tyrosine kinase is a critical regulator of neutrophil responses to Candida species. Mbio. 2020. May-Jun;11(3). doi: 10.1128/mBio.02043-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Whitney PG, Bar E, Osorio F, et al. Syk signaling in dendritic cells orchestrates innate resistance to systemic fungal infection. PLoS Pathog. 2014. Jul;10(7):e1004276. doi: 10.1371/journal.ppat.1004276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Wang L, Aschenbrenner D, Zeng Z, et al. Gain-of-function variants in SYK cause immune dysregulation and systemic inflammation in humans and mice. Nat Genet. 2021. Apr;53(4):500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Chamilos G, Lionakis MS, Kontoyiannis DP. Call for action: invasive fungal infections associated with ibrutinib and other small molecule kinase inhibitors targeting immune signaling pathways. Clin Infect Dis. 2018. Jan 6;66(1):140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Glocker EO, Hennigs A, Nabavi M, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. [2009. Oct 29];361(18):1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Rieber N, Gazendam RP, Freeman AF, et al. Extrapulmonary aspergillus infection in patients with CARD9 deficiency. JCI Insight. [2016. Oct 20];1(17):e89890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wang X, Zhang R, Wu W, et al. Impaired specific antifungal immunity in CARD9-deficient patients with phaeohyphomycosis. J Invest Dermatol. 2018. Mar;138(3):607–617. [DOI] [PubMed] [Google Scholar]
- [118].Lanternier F, Pathan S, Vincent QB, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med. [2013. Oct 31];369(18):1704–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Lanternier F, Mahdaviani SA, Barbati E, et al. Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species-induced meningoencephalitis, colitis, or both. J Allergy Clin Immunol. 2015. Jun;135(6):1558–68 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].LeibundGut-Landmann S, Gross O, Robinson MJ, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007. Jun;8(6):630–638. [DOI] [PubMed] [Google Scholar]
- [121].Drummond RA, Swamydas M, Oikonomou V, et al. CARD9(+) microglia promote antifungal immunity via IL-1beta- and CXCL1-mediated neutrophil recruitment. Nat Immunol. 2019. May;20(5):559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Drewniak A, Gazendam RP, Tool ATJ, et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood. [2013. Mar 28];121(13):2385–2392. [DOI] [PubMed] [Google Scholar]
- [123].Drummond RA, Franco LM, Lionakis MS. Human CARD9: a critical molecule of fungal immune surveillance. Front Immunol. 2018;9:1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. [2010. Apr 1];115(13):2578–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Zarakas MA, Desai JV, Chamilos G, et al. Fungal infections with ibrutinib and other small-molecule kinase inhibitors. Curr Fungal Infect Rep. 2019. Sep;13(3):86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Ferwerda B, Ferwerda G, Plantinga TS, et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med. [2009. Oct 29];361(18):1760–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Griffiths JS, White PL, Czubala MA, et al. A human Dectin-2 deficiency associated with invasive aspergillosis. J Infect Dis. 2021. Mar 18; 224(7):1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Vendele I, Willment JA, Silva LM, et al. Mannan detecting C-type lectin receptor probes recognise immune epitopes with diverse chemical, spatial and phylogenetic heterogeneity in fungal cell walls. PLoS Pathog. 2020. Jan;16(1):e1007927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Gantner BN, Simmons RM, Canavera SJ, et al. Collaborative induction of inflammatory responses by dectin-1 and toll-like receptor 2. J Exp Med. [2003. May 5];197(9):1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Netea MG, Gow NA, Munro CA, et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and toll-like receptors. J Clin Invest. 2006. Jun;116(6):1642–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Haley K, Igyarto BZ, Ortner D, et al. Langerhans cells require MyD88-dependent signals for candida albicans response but not for contact hypersensitivity or migration. J Immunol. [2012. May 1];188(9):4334–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Bellocchio S, Montagnoli C, Bozza S, et al. The contribution of the toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol. [2004. Mar 1];172(5):3059–3069. [DOI] [PubMed] [Google Scholar]
- [133].Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev. 2011. Jul;24(3):490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Tessarolli V, Gasparoto TH, Lima HR, et al. Absence of TLR2 influences survival of neutrophils after infection with Candida albicans. Med Mycol. 2010. Feb;48(1):129–140. [DOI] [PubMed] [Google Scholar]
- [135].Villamon E, Gozalbo D, Roig P, et al. Toll-like receptor-2 is essential in murine defenses against Candida albicans infections. Microbes Infect. 2004. Jan;6(1):1–7. [DOI] [PubMed] [Google Scholar]
- [136].Netea MG, Sutmuller R, Hermann C, et al. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol. [2004. Mar 15];172(6):3712–3718. [DOI] [PubMed] [Google Scholar]
- [137].Gasparoto TH, Tessarolli V, Garlet TP, et al. Absence of functional TLR4 impairs response of macrophages after Candida albicans infection. Med Mycol. 2010. Dec;48(8):1009–1017. [DOI] [PubMed] [Google Scholar]
- [138].Weindl G, Naglik JR, Kaesler S, et al. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J Clin Investig. 2007. Dec;117(12):3664–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Marakalala MJ, Vautier S, Potrykus J, et al. Differential adaptation of Candida albicans in vivo modulates immune recognition by dectin-1. PLoS Pathog. 2013;9(4):e1003315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Murciano C, Villamon E, Gozalbo D, et al. Toll-like receptor 4 defective mice carrying point or null mutations do not show increased susceptibility to Candida albicans in a model of hematogenously disseminated infection. Med Mycol. 2006. Mar;44(2):149–157. [DOI] [PubMed] [Google Scholar]
- [141].Netea MG, Gow NA, Joosten LA, et al. Variable recognition of Candida albicans strains by TLR4 and lectin recognition receptors. Med Mycol. 2010. Nov;48(7):897–903. [DOI] [PubMed] [Google Scholar]
- [142].Choteau L, Vancraeyneste H, Le Roy D, et al. Role of TLR1, TLR2 and TLR6 in the modulation of intestinal inflammation and Candida albicans elimination. Gut Pathog. 2017;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].der Graaf Ca V, Netea MG, Morre SA, et al. Toll-like receptor 4 Asp299Gly/Thr399Ile polymorphisms are a risk factor for Candida bloodstream infection. Eur Cytokine Netw. 2006. Mar;17(1):29–34. [PubMed] [Google Scholar]
- [144].Lionakis MS. Genetic susceptibility to fungal infections in humans. Curr Fungal Infect Rep. 2012. Mar 1;6(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Plantinga TS, Johnson MD, Scott WK, et al. Toll-like receptor 1 polymorphisms increase susceptibility to candidemia. J Infect Dis. [2012. Mar 15];205(6):934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Miyazato A, Nakamura K, Yamamoto N, et al. Toll-like receptor 9-dependent activation of myeloid dendritic cells by deoxynucleic acids from Candida albicans. Infect Immun. 2009. Jul;77(7):3056–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Wagener J, Malireddi RKS, Lenardon MD, et al. Fungal chitin dampens inflammation through IL-10 induction mediated by NOD2 and TLR9 activation. PLoS Pathog. 2014. Apr;10(4):e1004050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Biondo C, Signorino G, Costa A, et al. Recognition of yeast nucleic acids triggers a host-protective type I interferon response. Eur J Immunol. 2011. Jul;41(7):1969–1979. [DOI] [PubMed] [Google Scholar]
- [149].Bourgeois C, Majer O, Frohner IE, et al. Conventional dendritic cells mount a type I IFN response against Candida spp. requiring novel phagosomal TLR7-mediated IFN-beta signaling. J Immunol. [2011. Mar 1];186(5):3104–3112. [DOI] [PubMed] [Google Scholar]
- [150].Biondo C, Malara A, Costa A, et al. Recognition of fungal RNA by TLR7 has a nonredundant role in host defense against experimental candidiasis. Eur J Immunol. 2012. Oct;42(10):2632–2643. [DOI] [PubMed] [Google Scholar]
- [151].Gringhuis SI, Kaptein TM, Wevers BA, et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol. [2012. Jan 22];13(3):246–254. [DOI] [PubMed] [Google Scholar]
- [152].Joly S, Sutterwala FS. Cutting edge: candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. Virulence. 2010. Jul-Aug;1(4):276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Hise AG, Tomalka J, Ganesan S, et al. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe. [2009. May 8];5(5):487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Gross O, Poeck H, Bscheider M, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. [2009. May 21];459(7245):433–436. [DOI] [PubMed] [Google Scholar]
- [155].Joly S, Eisenbarth SC, Olivier AK, et al. Cutting edge: nlrp10 is essential for protective antifungal adaptive immunity against Candida albicans. J Immunol. [2012. Nov 15];189(10):4713–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Tomalka J, Ganesan S, Azodi E, et al. A novel role for the NLRC4 inflammasome in mucosal defenses against the fungal pathogen Candida albicans. PLoS Pathog. 2011. Dec;7(12):e1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Lamborn IT, Jing H, Zhang Y, et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med. [2017. Jul 3];214(7):1949–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Jaeger M, van der Lee R, Cheng SC, et al. The RIG-I-like helicase receptor MDA5 (IFIH1) is involved in the host defense against Candida infections. Eur J Clin Microbiol. 2015. May;34(5):963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Swidergall M, Solis NV, Lionakis MS, et al. EphA2 is an epithelial cell pattern recognition receptor for fungal beta-glucans. Nat Microbiol. 2018. Jan;3(1):53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Swidergall M, Solis NV, Wang Z, et al. EphA2 is a neutrophil receptor for Candida albicans that stimulates antifungal activity during oropharyngeal infection. Cell Rep. [2019. Jul 9];28(2):423–433 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Lionakis MS, Kontoyiannis DP. Glucocorticoids and invasive fungal infections. Lancet. 2003. Nov 29;362(9398):1828–1838. [DOI] [PubMed] [Google Scholar]
- [162].Smith JA, Kauffman CA. Endemic fungal infections in patients receiving tumour necrosis factor-alpha inhibitor therapy. Drugs. 2009. Jul 30;69(11):1403–1415. [DOI] [PubMed] [Google Scholar]
- [163].Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Brit J Dermatol. 2017. Jul;177(1):47–62. [DOI] [PubMed] [Google Scholar]
- [164].Ok SM, Ho D, Lynd T, et al. Candida infection associated with salivary gland-A narrative review. J Clin Med. 2020. Dec 30;10:(1): 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Conti HR, Bruno VM, Childs EE, et al. IL-17 receptor signaling in oral epithelial cells is critical for protection against oropharyngeal Candidiasis. Cell Host Microbe. [2016. Nov 09];20(5):606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Ho AW, Shen F, Conti HR, et al. IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J Immunol. [2010. Jul 15];185(2):1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Ferreira MC, Whibley N, Mamo AJ, et al. Interleukin-17-induced protein lipocalin 2 is dispensable for immunity to oral Candidiasis. Infect Immun. 2014. Mar;82(3):1030–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Conti HR, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. [2009. Feb 16];206(2):299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Ling Y, Cypowyj S, Aytekin C, et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med. [2015. May 04];212(5):619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Boisson B, Wang C, Pedergnana V, et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. [2013. Oct 17];39(4):676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Levy R, Okada S, Beziat V, et al. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci U S A. [2016. Dec 20];113(51):E8277–E8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. [2011. Apr 01];332(6025):65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Ochoa S, Constantine GM, Lionakis MS. Genetic susceptibility to fungal infection in children. Curr Opin Pediatr. 2020. Dec;32(6):780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Liu L, Okada S, Kong XF, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. [2011. Aug 1];208(8):1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Milner JD, Brenchley JM, Laurence A, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. [2008. Apr 10];452(7188):773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Okada S, Markle JG, Deenick EK, et al. IMMUNODEFICIENCIES. impairment of immunity to candida and mycobacterium in humans with bi-allelic RORC mutations. Science. [2015. Aug 7];349(6248):606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Puel A, Cypowyj S, Marodi L, et al. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol. 2012. Dec;12(6):616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Li J, Ritelli M, Ma CS, et al. Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-beta. Sci Immunol. 2019. Nov 29;4:(41): eaax7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Krueger JG, Fretzin S, Suarez-Farinas M, et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immun. 2012. Jul;130(1):145-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Krueger JG, Wharton KA Jr., Schlitt T, et al. IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J Allergy Clin Immunol. 2019. Sep;144(3):750–763. [DOI] [PubMed] [Google Scholar]
- [181].Gladiator A, Wangler N, Trautwein-Weidner K, et al. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol. [2013. Jan 15];190(2):521–525. [DOI] [PubMed] [Google Scholar]
- [182].Conti HR, Peterson AC, Brane L, et al. Oral-resident natural Th17 cells and gammadelta T cells control opportunistic Candida albicans infections. J Exp Med. [2014. Sep 22];211(10):2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Verma AH, Richardson JP, Zhou C, et al. Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci Immunol. 2017. Nov 3;2(17): eaam8834 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Sparber F, Dolowschiak T, Mertens S, et al. Langerin+ DCs regulate innate IL-17 production in the oral mucosa during Candida albicans-mediated infection. PLoS Pathog. 2018. May;14(5):e1007069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Trautwein-Weidner K, Gladiator A, Kirchner FR, et al. Antigen-specific Th17 cells are primed by distinct and complementary dendritic cell subsets in oropharyngeal Candidiasis. PLoS Pathog. 2015. Oct;11(10):e1005164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007. Jun;8(6):639–646. [DOI] [PubMed] [Google Scholar]
- [187].Kirchner FR, LeibundGut-Landmann S. Tissue-resident memory Th17 cells maintain stable fungal commensalism in the oral mucosa. Mucosal Immunol. 2021. Mar;14(2):455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Oikonomou V, Break TJ, Gaffen SL, et al. Infections in the monogenic autoimmune syndrome APECED. Curr Opin Immunol. 2021. Aug 18;72:286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Fernandes RA, Perez-Andres M, Blanco E, et al. Complete multilineage CD4 expression defect associated with warts due to an inherited homozygous CD4 gene mutation. Front Immunol. 2019;10:2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Zonios D, Sheikh V, Sereti I. Idiopathic CD4 lymphocytopenia: a case of missing, wandering or ineffective T cells. Arthritis Res Ther. 2012. Aug 31;14(4):222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Lisco A, Ye PY, Wong CS, et al. Lost in translation: lack of CD4 expression due to a novel genetic defect. J Infect Dis. [2021. Feb 15];223(4):645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Harris LD, Klatt NR, Vinton C, et al. Mechanisms underlying gammadelta T-cell subset perturbations in SIV-infected Asian rhesus macaques. Blood. [2010. Nov 18];116(20):4148–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Klatt NR, Estes JD, Sun X, et al. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol. 2012. Nov;5(6):646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Mudd JC, Busman-Sahay K, DiNapoli SR, et al. Hallmarks of primate lentiviral immunodeficiency infection recapitulate loss of innate lymphoid cells. Nat Commun. [2018. Sep 27];9(1):3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Trautwein-Weidner K, Gladiator A, Nur S, et al. IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol. 2015. Mar;8(2):221–231. [DOI] [PubMed] [Google Scholar]
- [196].Edgerton M, Koshlukova SE, Lo TE, et al. Candidacidal activity of salivary histatins. Identification of a histatin 5-binding protein on Candida albicans. J Biol Chem. [1998. Aug 7];273(32):20438–20447. [DOI] [PubMed] [Google Scholar]
- [197].Moffa EB, Mussi MC, Xiao Y, et al. Histatin 5 inhibits adhesion of C. albicans to reconstructed human oral epithelium. Front Microbiol. 2015;6:885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Kong EF, Tsui C, Boyce H, et al. Development and in vivo evaluation of a novel histatin-5 bioadhesive hydrogel formulation against oral Candidiasis. Antimicrob Agents Chemother. 2016. Feb;60(2):881–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Aggor FEY, Break TJ, Trevejo-Nunez G, et al. Oral epithelial IL-22/STAT3 signaling licenses IL-17-mediated immunity to oral mucosal candidiasis. Sci Immunol. 2020. Jun 5;5:(48):eaba0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Huppler AR, Conti HR, Hernandez-Santos N, et al. Role of neutrophils in IL-17-dependent immunity to mucosal candidiasis. J Immunol. [2014. Feb 15];192(4):1745–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Altmeier S, Toska A, Sparber F, et al. IL-1 coordinates the neutrophil response to C. albicans in the oral mucosa. PLoS Pathog. 2016. Sep;12(9):e1005882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Bichele R, Karner J, Truusalu K, et al. IL-22 neutralizing autoantibodies impair fungal clearance in murine oropharyngeal candidiasis model. Eur J Immunol. 2018. Mar;48(3):464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. [2009. Nov 19];361(21):2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Forbester JL, Lees EA, Goulding D, et al. Interleukin-22 promotes phagolysosomal fusion to induce protection against salmonella enterica typhimurium in human epithelial cells. Proc Natl Acad Sci U S A. [2018. Oct 2];115(40):10118–10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].de Groot Pw, Bader O, de Boer Ad, et al. Adhesins in human fungal pathogens: glue with plenty of stick. Eukaryot Cell. 2013. Apr;12(4):470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Moyes DL, Runglall M, Murciano C, et al. A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe. [2010. Sep 16];8(3):225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Verma AH, Zafar H, Ponde NO, et al. IL-36 and IL-1/IL-17 drive immunity to oral candidiasis via parallel mechanisms. J Immunol. [2018. Jul 15];201(2):627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Ho J, Yang X, Nikou SA, et al. Candidalysin activates innate epithelial immune responses via epidermal growth factor receptor. Nat Commun. [2019. May 24];10(1):2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Hanaoka M, Domae E. IL-1 alpha released from oral epithelial cells upon candidalysin exposure initiates an early innate epithelial response. Int Immunol. 2021. Mar;33(3):161–170. [DOI] [PubMed] [Google Scholar]
- [210].Constantine GM, Lionakis MS. Lessons from primary immunodeficiencies: autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol Rev. 2019. Jan;287(1):103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Ferre EM, Rose SR, Rosenzweig SD, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight. 2016. Aug 18;1(13): e88782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Puel A, Doffinger R, Natividad A, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. [2010. Feb 15];207(2):291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Kisand K, Boe Wolff AS, Podkrajsek KT, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med. [2010. Feb 15];207(2):299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Orlova EM, Sozaeva LS, Kareva MA, et al. Expanding the phenotypic and genotypic landscape of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. [2017. Sep 1];102(9):3546–3556. [DOI] [PubMed] [Google Scholar]
- [215].Break TJ, Oikonomou V, Dutzan N, et al. Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science. 2021. Jan 15;371(6526):eaay5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Toubiana J, Okada S, Hiller J, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. [2016. Jun 23];127(25):3154–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Higgins E, Al Shehri T, McAleer MA, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immun. 2015. Feb;135(2):551–U356. [DOI] [PubMed] [Google Scholar]
- [218].Kong XF, Worley L, Rinchai D, et al. Three copies of four interferon receptor genes underlie a mild type i interferonopathy in down syndrome. J Clin Immunol. 2020. Aug;40(6):807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Zhu F, Willette-Brown J, Song NY, et al. Autoreactive T cells and chronic fungal infection drive esophageal carcinogenesis. Cell Host Microbe. [2017. Apr 12];21(4):478–493 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Bruserud O, Costea DE, Laakso S, et al. Oral tongue malignancies in autoimmune polyendocrine syndrome type 1. Front Endocrinol (Lausanne). 2018;9:463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Sampaio EP, Ding L, Rose SR, et al. Novel signal transducer and activator of transcription 1 mutation disrupts small ubiquitin-related modifier conjugation causing gain of function. J Allergy Clin Immunol. 2018. May;141(5):1844–1853 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Denning DW, Kneale M, Sobel JD, et al. Global burden of recurrent vulvovaginal candidiasis: a systematic review. Lancet Infect Dis. 2018. Nov;18(11):e339–e347. [DOI] [PubMed] [Google Scholar]
- [223].Yano J, Sobel JD, Nyirjesy P, et al. Current patient perspectives of vulvovaginal candidiasis: incidence, symptoms, management and post-treatment outcomes. BMC Womens Health. [2019. Mar 29];19(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Bradford LL, Ravel J. The vaginal mycobiome: a contemporary perspective on fungi in women’s health and diseases. Virulence. 2017. Apr 3;8(3):342–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Xu J, Schwartz K, Bartoces M, et al. Effect of antibiotics on vulvovaginal candidiasis: a metroNet study. J Am Board Fam Med. 2008. Jul-Aug;21(4):261–268. [DOI] [PubMed] [Google Scholar]
- [226].Xu J, Sobel JD. Antibiotic-associated vulvovaginal Candidiasis. Curr Infect Dis Rep. 2003. Dec;5(6):481–487. [DOI] [PubMed] [Google Scholar]
- [227].White MH. Is vulvovaginal candidiasis an AIDS-related illness?. Clin Infect Dis. 1996. May;22 Suppl 2:S124–7. [DOI] [PubMed] [Google Scholar]
- [228].Fidel PL, Lynch ME, Sobel JD. Circulating Cd4 and Cd8 T-cells have little impact on host-defense against experimental vaginal Candidiasis. Infect Immun. 1995. Jul;63(7):2403–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Sobel JD. Vulvovaginal candidiasis: a comparison of HIV-positive and -negative women. Int J STD AIDS. 2002. Jun;13(6):358–362. [DOI] [PubMed] [Google Scholar]
- [230].Peters BM, Coleman BM, Willems HME, et al. The Interleukin (IL) 17R/IL-22R signaling axis is dispensable for vulvovaginal Candidiasis regardless of estrogen status. J Infect Dis. [2020. Apr 7];221(9):1554–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Ardizzoni A, Wheeler RT, Pericolini E. It takes two to tango: how a dysregulation of the innate immunity, coupled with candida virulence, triggers VVC onset. Front Microbiol. 2021. Jun 7;12: 692491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Pekmezovic M, Hovhannisyan H, Gresnigt MS, et al. Candida pathogens induce protective mitochondria-associated type I interferon signalling and a damage-driven response in vaginal epithelial cells. Nat Microbiol. 2021. May;6(5):643–657. [DOI] [PubMed] [Google Scholar]
- [233].Moyes DL, Murciano C, Runglall M, et al. Candida albicans yeast and hyphae are discriminated by MAPK signaling in vaginal epithelial cells. PLoS One. 2011;6(11):e26580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Fidel PL Jr., Barousse M, Espinosa T, et al. An intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect Immun. 2004. May;72(5):2939–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Black CA, Eyers FM, Russell A, et al. Acute neutropenia decreases inflammation associated with murine vaginal candidiasis but has no effect on the course of infection. Infect Immun. 1998. Mar;66(3):1273–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Richardson JP, Willems HME, Moyes DL, et al. Candidalysin drives epithelial signaling, neutrophil recruitment, and immunopathology at the vaginal mucosa. Infect Immun. 2018. Feb;86(2): e00645–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Pericolini E, Gabrielli E, Amacker M, et al. Secretory aspartyl proteinases cause vaginitis and can mediate vaginitis caused by Candida albicans in mice. mBio. [2015. Jun 2];6(3):e00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Bruno VM, Shetty AC, Yano J, et al. Transcriptomic analysis of vulvovaginal candidiasis identifies a role for the NLRP3 inflammasome. mBio. 2015. Apr 21;6(2):e00182–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Schaller M, Bein M, Korting HC, et al. The secreted aspartyl proteinases Sap1 and Sap2 cause tissue damage in an in vitro model of vaginal candidiasis based on reconstituted human vaginal epithelium. Infect Immun. 2003. Jun;71(6):3227–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Richardson JP, Willems HME, Moyes DL, et al. Candidalysin drives epithelial signaling, neutrophil recruitment, and immunopathology at the vaginal mucosa. Infect Immun. 2018. Feb;86(2):e00645–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Lasarte S, Samaniego R, Salinas-Munoz L, et al. Sex hormones coordinate neutrophil immunity in the vagina by controlling chemokine gradients. J Infect Dis. [2016. Feb 1];213(3):476–484. [DOI] [PubMed] [Google Scholar]
- [242].Salinas-Munoz L, Campos-Fernandez R, Mercader E, et al. Estrogen Receptor-Alpha (ESR1) governs the lower female reproductive tract vulnerability to Candida albicans. Front Immunol. 2018;9:1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Yano J, Noverr MC, Fidel PL Jr. Vaginal heparan sulfate linked to neutrophil dysfunction in the acute inflammatory response associated with experimental vulvovaginal Candidiasis. mBio. 2017. Mar 14;8(2): e00211–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Yano J, Peters BM, Noverr MC, et al. Novel mechanism behind the immunopathogenesis of vulvovaginal Candidiasis: “neutrophil anergy”. Infect Immun. 2018. Mar;86(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Lowes DJ, Hevener KE, Peters BM. Second-generation antidiabetic sulfonylureas inhibit Candida albicans and candidalysin-mediated activation of the NLRP3 inflammasome. Antimicrob Agents Chemother. 2020. ;64(2):e01777-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Roselletti E, Perito S, Gabrielli E, et al. NLRP3 inflammasome is a key player in human vulvovaginal disease caused by Candida albicans. Sci Rep. [2017. Dec 19];7(1):17877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Willems HME, Lowes DJ, Barker KS, et al. Comparative analysis of the capacity of the Candida species to elicit vaginal immunopathology. Infect Immun. 2018 Dec;86(12):e00527–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Roselletti E, Perito S, Gabrielli E, et al.NLRP3 inflammasome is a key player in human vulvovaginal disease caused by &ITCandida albicans&IT.Sci Rep-Uk. 2017. Dec 19;7(1): 17877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Jaeger M, Carvalho A, Cunha C, et al. Association of a variable number tandem repeat in the NLRP3 gene in women with susceptibility to RVVC. Eur J Clin Microbiol Infect Dis. 2016. May;35(5):797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Jaeger M, Pinelli M, Borghi M, et al. A systems genomics approach identifies SIGLEC15 as a susceptibility factor in recurrent vulvovaginal candidiasis. Sci Transl Med. 2019. Jun 12;11(496): eaar3558. [DOI] [PubMed] [Google Scholar]
- [251].Rosentul DC, Delsing CE, Jaeger M, et al. Gene polymorphisms in pattern recognition receptors and susceptibility to idiopathic recurrent vulvovaginal candidiasis. Front Microbiol. 2014;5:483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Babula O, Lazdane G, Kroica J, et al. Frequency of interleukin-4 (IL-4) −589 gene polymorphism and vaginal concentrations of IL-4, nitric oxide, and mannose-binding lectin in women with recurrent vulvovaginal candidiasis. Clin Infect Dis. [2005. May 1];40(9):1258–1262. [DOI] [PubMed] [Google Scholar]
- [253].De Luca A, Carvalho A, Cunha C, et al. IL-22 and IDO1 affect immunity and tolerance to murine and human vaginal candidiasis. PLoS Pathog. 2013;9(7):e1003486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Borghi M, De Luca A, Puccetti M, et al. Pathogenic NLRP3 inflammasome activity during Candida infection is negatively regulated by IL-22 via activation of NLRC4 and IL-1Ra. Cell Host Microbe. [2015. Aug 12];18(2):198–209. [DOI] [PubMed] [Google Scholar]
- [255].Zelante T, Iannitti RG, Cunha C, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. [2013. Aug 22];39(2):372–385. [DOI] [PubMed] [Google Scholar]
- [256].Borghi M, Pariano M, Solito V, et al. Targeting the aryl hydrocarbon receptor with Indole-3-aldehyde protects from vulvovaginal candidiasis via the IL-22-IL-18 cross-talk. Front Immunol. 2019;10:2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Tso GHW, Reales-Calderon JA, Pavelka N. The elusive anti-Candida vaccine: lessons from the past and opportunities for the future. Front Immunol. 2018;9:897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Sandini S, La Valle R, Deaglio S, et al. A highly immunogenic recombinant and truncated protein of the secreted aspartic proteases family (rSap2t) of Candida albicans as a mucosal anticandidal vaccine. FEMS Immunol Med Microbiol. 2011. Jul;62(2):215–224. [DOI] [PubMed] [Google Scholar]
- [259].Torosantucci A, Bromuro C, Chiani P, et al. A novel glyco-conjugate vaccine against fungal pathogens. J Exp Med. [2005. Sep 5];202(5):597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Ibrahim AS, Luo GPS, Gebremariam T, et al. NDV-3 protects mice from vulvovaginal candidiasis through T- and B-cell immune response. Vaccine. [2013. Nov 12];31(47):5549–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Schmidt CS, White CJ, Ibrahim AS, et al. NDV-3, a recombinant alum-adjuvanted vaccine for Candida and staphylococcus aureus, is safe and immunogenic in healthy adults. Vaccine. [2012. Dec 14];30(52):7594–7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Edwards JE Jr., Schwartz MM, Schmidt CS, et al. A fungal immunotherapeutic vaccine (NDV-3A) for treatment of recurrent vulvovaginal candidiasis-A phase 2 randomized, double-blind, placebo-controlled trial. Clin Infect Dis. [2018. Jun 1];66(12):1928–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Uppuluri P, Singh S, Alqarihi A, et al. Human anti-Als3p antibodies are surrogate markers of NDV-3A vaccine efficacy against recurrent vulvovaginal Candidiasis. Front Immunol. 2018;9:1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Findley K, Oh J, Yang J, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. [2013. Jun 20];498(7454):367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Oh J, Freeman AF, Program NCS, et al. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013. Dec;23(12):2103–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Timsit JF, Azoulay E, Schwebel C, et al. Empirical micafungin treatment and survival without invasive fungal infection in adults with ICU-acquired sepsis, Candida colonization, and multiple organ failure the empiricus randomized clinical trial. Jama-J Am Med Assoc. [2016. Oct 18];316(15):1555–1564. [DOI] [PubMed] [Google Scholar]
- [267].Proctor DM, Dangana T, Sexton DJ, et al. Integrated genomic, epidemiologic investigation of Candida auris skin colonization in a skilled nursing facility. Nat Med. 2021;27(8):1401-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Horton MV, Johnson CJ, Kernien JF, et al. Candida auris forms high-burden biofilms in skin niche conditions and on porcine skin. mSphere. 2020. Jan 22;5(1): e00910–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008. Sep;51(Suppl 4):2–15. [DOI] [PubMed] [Google Scholar]
- [270].Lisboa C, Santos A, Dias C, et al. Candida balanitis: risk factors. J Eur Acad Dermatol Venereol. 2010. Jul;24(7):820–826. [DOI] [PubMed] [Google Scholar]
- [271].Jennemann R, Rabionet M, Gorgas K, et al. Loss of ceramide synthase 3 causes lethal skin barrier disruption. Hum Mol Genet. [2012. Feb 1];21(3):586–608. [DOI] [PubMed] [Google Scholar]
- [272].Lopez CM, Wallich R, Riesbeck K, et al. Candida albicans uses the surface protein Gpm1 to attach to human endothelial cells and to keratinocytes via the adhesive protein vitronectin. PLoS One. 2014;9(3):e90796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Tapia CV, Falconer M, Tempio F, et al. Melanocytes and melanin represent a first line of innate immunity against Candida albicans. Med Mycol. 2014. Jul;52(5):445–454. [DOI] [PubMed] [Google Scholar]
- [274].Mackintosh JA. The antimicrobial properties of melanocytes, melanosomes and melanin and the evolution of black skin. J Theor Biol. 2001. Jul 21;211(2):101–113. [DOI] [PubMed] [Google Scholar]
- [275].Riol-Blanco L, Ordovas-Montanes J, Perro M, et al. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature. [2014. Jun 5];510(7503):157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Kashem SW, Riedl MS, Yao C, et al. Nociceptive sensory fibers drive interleukin-23 production from CD301b+ dermal dendritic cells and drive protective cutaneous immunity. Immunity. [2015. Sep 15];43(3):515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Cohen JA, Edwards TN, Liu AW, et al. Cutaneous TRPV1(+) neurons trigger protective innate type 17 anticipatory immunity. Cell. [2019. Aug 8];178(4):919–932 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Zhang S, Edwards TN, Chaudhri VK, et al. Nonpeptidergic neurons suppress mast cells via glutamate to maintain skin homeostasis. Cell. [2021. Apr 15];184(8):2151–2166 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Kagami S, Rizzo HL, Kurtz SE, et al. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J Immunol. [2010. Nov 1];185(9):5453–5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Kaplan DH. In vivo function of Langerhans cells and dermal dendritic cells. Trends Immunol. 2010. Dec;31(12):446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Kashem SW, Igyarto BZ, Gerami-Nejad M, et al. Candida albicans morphology and dendritic cell subsets determine T helper cell differentiation. Immunity. [2015. Feb 17];42(2):356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Igyarto BZ, Haley K, Ortner D, et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity. [2011. Aug 26];35(2):260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Park CO, Fu X, Jiang X, et al. Staged development of long-lived T-cell receptor alphabeta TH17 resident memory T-cell population to Candida albicans after skin infection. J Allergy Clin Immunol. 2018. Aug;142(2):647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Naik S, Bouladoux N, Linehan JL, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. [2015. Apr 2];520(7545):104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Santus W, Barresi S, Mingozzi F, et al. Skin infections are eliminated by cooperation of the fibrinolytic and innate immune systems. Sci Immunol. 2017. Sep 22;2(15): eaan2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Taheri Sarvtin M, Shokohi T, Hajheydari Z, et al. Evaluation of candidal colonization and specific humoral responses against Candida albicans in patients with psoriasis. Int J Dermatol. 2014. Dec;53(12):e555–60. [DOI] [PubMed] [Google Scholar]
- [287].Zhang E, Tanaka T, Tajima M, et al. Characterization of the skin fungal microbiota in patients with atopic dermatitis and in healthy subjects. Microbiol Immunol. 2011. Sep;55(9):625–632. [DOI] [PubMed] [Google Scholar]
- [288].Hurabielle C, Link VM, Bouladoux N, et al. Immunity to commensal skin fungi promotes psoriasiform skin inflammation. Proc Natl Acad Sci U S A. [2020. Jul 14];117(28):16465–16474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Strollo S, Lionakis MS, Adjemian J, et al. Epidemiology of hospitalizations associated with invasive Candidiasis, United States, 2002-2012(1). Emerg Infect Dis. 2016. Jan;23(1):7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Ricotta EE, Lai YL, Babiker A, et al. Invasive candidiasis species distribution and trends, United States, 2009-2017. J Infect Dis. [2021. Apr 8];223(7):1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Kullberg BJ, Arendrup MC. Invasive Candidiasis. N Engl J Med. 2015. Oct 8;373(15):1445–1456. [DOI] [PubMed] [Google Scholar]
- [292].Teo YJ, Ng SL, Mak KW, et al. Renal CD169(++) resident macrophages are crucial for protection against acute systemic candidiasis. Life Sci Alliance. 2021. May;4(5): e202000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Qian Q, Jutila MA, Van Rooijen N, et al. Elimination of mouse splenic macrophages correlates with increased susceptibility to experimental disseminated candidiasis. J Immunol. [1994. May 15];152(10):5000–5008. [PubMed] [Google Scholar]
- [294].Break TJ, Hoffman KW, Swamydas M, et al. Batf3-dependent CD103(+) dendritic cell accumulation is dispensable for mucosal and systemic antifungal host defense. Virulence. 2016;7(7):826–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Desai JV, Lionakis MS. The role of neutrophils in host defense against invasive fungal infections. Curr Clin Microbiol Rep. 2018. Sep;5(3):181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Lionakis MS, Fischer BG, Lim JK, et al. Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog. 2012;8(8):e1002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Hopke A, Scherer A, Kreuzburg S, et al. Neutrophil swarming delays the growth of clusters of pathogenic fungi. Nat Commun. [2020. Apr 27];11(1):2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Lammermann T, Afonso PV, Angermann BR, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. [2013. Jun 20];498(7454):371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Romani L, Mencacci A, Cenci E, et al. An immunoregulatory role for neutrophils in CD4+ T helper subset selection in mice with candidiasis. J Immunol. [1997. Mar 1];158(5):2356–2362. [PubMed] [Google Scholar]
- [300].Kasper L, Konig A, Koenig PA, et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat Commun. [2018. Oct 15];9(1):4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Swidergall M, Khalaji M, Solis NV, et al. Candidalysin is required for neutrophil recruitment and virulence during systemic Candida albicans infection. J Infect Dis. [2019. Sep 26];220(9):1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Drummond RA, Collar AL, Swamydas M, et al. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog. 2015. Dec;11(12):e1005293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Snarr BD, Drummond RA, Lionakis MS. It’s all in your head: antifungal immunity in the brain. Curr Opin Microbiol. 2020. Dec;58:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Fitzpatrick Z, Frazer G, Ferro A, et al. Gut-educated IgA plasma cells defend the meningeal venous sinuses. Nature. 2020. Nov;587(7834):472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Lin L, Ibrahim AS, Xu X, et al. Th1-Th17 cells mediate protective adaptive immunity against staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. 2009. Dec;5(12): e1000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Branzk N, Lubojemska A, Hardison SE, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014. Nov;15(11):1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Warnatsch A, Tsourouktsoglou TD, Branzk N, et al. Reactive oxygen species localization programs inflammation to clear microbes of different size. Immunity. [2017. Mar 21];46(3):421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Urban CF, Ermert D, Schmid M, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009. Oct;5(10):e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Urban CF, Backman E. Eradicating, retaining, balancing, swarming, shuttling and dumping: a myriad of tasks for neutrophils during fungal infection. Curr Opin Microbiol. 2020. Dec;58:106–115. [DOI] [PubMed] [Google Scholar]
- [310].Lehman HK, Segal BH. The role of neutrophils in host defense and disease. J Allergy Clin Immunol. 2020. Jun;145(6):1535–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Byrd AS, O’Brien XM, Johnson CM, et al. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol. [2013. Apr 15];190(8):4136–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Wu SY, Weng CL, Jheng MJ, et al. Candida albicans triggers NADPH oxidase-independent neutrophil extracellular traps through dectin-2. PLoS Pathog. 2019. Nov;15(11): e1008096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Guiducci E, Lemberg C, Kung N, et al. Candida albicans-induced netosis is independent of peptidylarginine deiminase 4. Front Immunol. 2018. Jul 9;9:1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Warris A, Ballou ER. Oxidative responses and fungal infection biology. Semin Cell Dev Biol. 2019. May;89:34–46. [DOI] [PubMed] [Google Scholar]
- [315].Reeves EP, Lu H, Lortat-Jacob H, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. [2002. Mar 21];416(6878):291–297. [DOI] [PubMed] [Google Scholar]
- [316].Ermert D, Niemiec MJ, Rohm M, et al. Candida albicans escapes from mouse neutrophils. J Leukoc Biol. 2013. Aug;94(2):223–236. [DOI] [PubMed] [Google Scholar]
- [317].Li X, Utomo A, Cullere X, et al. The beta-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe. [2011. Dec 15];10(6):603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Henriet S, Verweij PE, Holland SM, et al. Invasive fungal infections in patients with chronic granulomatous disease. Adv Exp Med Biol. 2013;764:27–55. [DOI] [PubMed] [Google Scholar]
- [319].van de Geer A, Nieto-Patlan A, Kuhns DB, et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest. [2018. Aug 31];128(9):3957–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Lehrer RI, Cline MJ. Leukocyte myeloperoxidase deficiency and disseminated candidiasis: the role of myeloperoxidase in resistance to Candida infection. J Clin Invest. 1969. Aug;48(8):1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Swamydas M, Gao JL, Break TJ, et al. CXCR1-mediated neutrophil degranulation and fungal killing promote Candida clearance and host survival. Sci Transl Med. [2016. Jan 20];8(322):322ra10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Wirnsberger G, Zwolanek F, Stadlmann J, et al. Jagunal homolog 1 is a critical regulator of neutrophil function in fungal host defense. Nat Genet. 2014. Sep;46(9):1028-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Gazendam R, Hamme JL, Tool A, et al. Two independent killing mechanisms of Candida albicans by human neutrophils: evidence from innate immunity defects. J Clin Immunol. 2014;34:S142–S142. [DOI] [PubMed] [Google Scholar]
- [324].Lionakis MS, Swamydas M, Fischer BG, et al. CX3CR1-dependent renal macrophage survival promotes Candida control and host survival. J Clin Invest. 2013. Dec;123(12):5035–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Lionakis MS, Albert ND, Swamydas M, et al. Pharmacological blockade of the chemokine receptor CCR1 protects mice from systemic candidiasis of hematogenous origin. Antimicrob Agents Chemother. 2017. Mar;61(3): e02365–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Lee EKS, Gillrie MR, Li L, et al. Leukotriene B4-mediated neutrophil recruitment causes pulmonary capillaritis during lethal fungal sepsis. Cell Host Microbe. [2018. Jan 10];23(1):121–133 e4. [DOI] [PubMed] [Google Scholar]
- [327].Zwolanek F, Riedelberger M, Stolz V, et al. The non-receptor tyrosine kinase tec controls assembly and activity of the noncanonical caspase-8 inflammasome. PLoS Pathog. 2014. Dec;10(12):e1004525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].Garg AV, Amatya N, Chen K, et al. MCPIP1 endoribonuclease activity negatively regulates interleukin-17-mediated signaling and inflammation. Immunity. [2015. Sep 15];43(3):475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Naseem S, Frank D, Konopka JB, et al. Protection from systemic Candida albicans infection by inactivation of the Sts phosphatases. Infect Immun. 2015. Feb;83(2):637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Huang JL, Meng SS, Hong SJ, et al. IL-17C is required for lethal inflammation during systemic fungal infection. Cell Mol Immunol. 2016. Jul;13(4):474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Del Fresno C, Saz-Leal P, Enamorado M, et al. DNGR-1 in dendritic cells limits tissue damage by dampening neutrophil recruitment. Science. [2018. Oct 19];362(6412):351–356. [DOI] [PubMed] [Google Scholar]
- [332].Ramani K, Jawale CV, Verma AH, et al. Unexpected kidney-restricted role for IL-17 receptor signaling in defense against systemic Candida albicans infection. JCI Insight. 2018. May 3;3(9): e98241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [333].Candon S, Rammaert B, Foray AP, et al. Chronic disseminated candidiasis during hematological malignancies: an immune reconstitution inflammatory syndrome with expansion of pathogen-specific t helper type 1 cells. J Infect Dis. [2020. May 11];221(11):1907–1916. [DOI] [PubMed] [Google Scholar]
- [334].Bryant K, Maxfield C, Rabalais G. Renal candidiasis in neonates with candiduria. Pediatr Infect Dis J. 1999. Nov;18(11):959–963. [DOI] [PubMed] [Google Scholar]
- [335].Tomashefski JF Jr., Abramowsky CR. Candida-associated renal papillary necrosis. Am J Clin Pathol. 1981. Feb;75(2):190–194. [DOI] [PubMed] [Google Scholar]
- [336].Heung LJ. Monocytes and the host response to fungal pathogens. Front Cell Infect Microbiol. 2020;10:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Dominguez-Andres J, Feo-Lucas L, Minguito de La Escalera M, et al. Inflammatory Ly6C(high) monocytes protect against candidiasis through IL-15-driven nk cell/neutrophil activation. Immunity. [2017. Jun 20];46(6):1059–1072 e4. [DOI] [PubMed] [Google Scholar]
- [338].Ngo LY, Kasahara S, Kumasaka DK, et al. Inflammatory monocytes mediate early and organ-specific innate defense during systemic candidiasis. J Infect Dis. [2014. Jan 1];209(1):109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [339].Nguyen NZN, Tran VG, Lee S, et al. CCR5-mediated recruitment of nk cells to the kidney is a critical step for host defense to systemic Candida albicans infection. Immune Netw. 2020. Dec;20(6): e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [340].Bar E, Whitney PG, Moor K, et al. IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity. [2014. Jan 16];40(1):117–127. [DOI] [PubMed] [Google Scholar]
- [341].Nur S, Sparber F, Lemberg C, et al. IL-23 supports host defense against systemic Candida albicans infection by ensuring myeloid cell survival. PLoS Pathog. 2019. Dec;15(12):e1008115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [342].Collar AL, Swamydas M, O’Hayre M, et al. The homozygous CX3CR1-M280 mutation impairs human monocyte survival. JCI Insight. 2018. Feb 8;3(3):e95417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Break TJ, Jaeger M, Solis NV, et al. CX3CR1 is dispensable for control of mucosal Candida albicans infections in mice and humans. Infect Immun. 2015. Mar;83(3):958–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].Leonardi I, Li X, Semon A, et al. CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science. [2018. Jan 12];359(6372):232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [345].Munoz JF, Delorey T, Ford CB, et al.Coordinated host-pathogen transcriptional dynamics revealed using sorted subpopulations and single macrophages infected with Candida albicans. Nat Commun. 2019. Apr 8;10(01): 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [346].Tam JM, Reedy JL, Lukason DP, et al. Tetraspanin CD82 organizes Dectin-1 into signaling domains to mediate cellular responses to Candida albicans. J Immunol. [2019. Jun 1];202(11):3256–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [347].Bain JM, Alonso MF, Childers DS, et al. Immune cells fold and damage fungal hyphae. Proc Natl Acad Sci U S A. 2021. Apr 13;118(15): e2020484118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [348].Westman J, Walpole GFW, Kasper L, et al. Lysosome fusion maintains phagosome integrity during fungal infection. Cell Host Microbe. [2020. Dec 9];28(6):798–812 e6. [DOI] [PubMed] [Google Scholar]
- [349].Wellington M, Koselny K, Sutterwala FS, et al. Candida albicans triggers NLRP3-mediated pyroptosis in macrophages. Eukaryot Cell. 2014. Feb;13(2):329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [350].Uwamahoro N, Verma-Gaur J, Shen HH, et al. The pathogen Candida albicans hijacks pyroptosis for escape from macrophages. mBio. [2014. Mar 25];5(2):e00003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [351].Vylkova S, Lorenz MC. Phagosomal neutralization by the fungal pathogen Candida albicans induces macrophage pyroptosis. Infect Immun. 2017. Feb;85(2): e00832–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [352].Xiao Y, Tang J, Guo H, et al. Targeting CBLB as a potential therapeutic approach for disseminated candidiasis. Nat Med. 2016. Aug;22(8):906–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [353].Wirnsberger G, Zwolanek F, Asaoka T, et al. Inhibition of CBLB protects from lethal Candida albicans sepsis. Nat Med. 2016. Aug;22(8):915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [354].Zhao X, Guo Y, Jiang C, et al. JNK1 negatively controls antifungal innate immunity by suppressing CD23 expression. Nat Med. 2017. Mar;23(3):337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [355].Dominguez-Andres J, Arts RJW, Ter Horst R, et al. Rewiring monocyte glucose metabolism via C-type lectin signaling protects against disseminated candidiasis. PLoS Pathog. 2017. Sep;13(9): e1006632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [356].Tucey TM, Verma J, Harrison PF, et al. Glucose homeostasis is important for immune cell viability during candida challenge and host survival of systemic fungal infection. Cell Metab. [2018. May 1];27(5):988-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [357].Kim VY, Batty A, Li J, et al. Glutathione reductase promotes fungal clearance and suppresses inflammation during systemic Candida albicans infection in mice. J Immunol. [2019. Oct 15];203(8):2239–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [358].Jawale CV, Ramani K, Li DD, et al. Restoring glucose uptake rescues neutrophil dysfunction and protects against systemic fungal infection in mouse models of kidney disease. Sci Transl Med. 2020. Jun 17;12(548: eaay5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [359].Netea MG, Dominguez-Andres J, Barreiro LB, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. 2020. Jun;20(6):375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [360].Quintin J, Saeed S, Martens JHA, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. [2012. Aug 16];12(2):223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [361].Saeed S, Quintin J, Kerstens HH, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. [2014. Sep 26];345(6204):1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [362].Johnson MD, Plantinga TS, van de Vosse E, et al. Cytokine gene polymorphisms and the outcome of invasive candidiasis: a prospective cohort study. Clin Infect Dis. [2012. Feb 15];54(4):502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [363].Kumar V, Cheng SC, Johnson MD, et al. Immunochip SNP array identifies novel genetic variants conferring susceptibility to candidaemia. Nat Commun. 2014. Sep 8;5:4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [364].Smeekens SP, Ng A, Kumar V, et al. Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. Nat Commun. 2013;4:1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [365].Wojtowicz A, Tissot F, Lamoth F, et al. Polymorphisms in tumor necrosis factor-alpha increase susceptibility to intra-abdominal Candida infection in high-risk surgical ICU patients*. Crit Care Med. 2014. Apr;42(4):e304–8. [DOI] [PubMed] [Google Scholar]
- [366].Arvanitis M, Anagnostou T, Fuchs BB, et al. Molecular and nonmolecular diagnostic methods for invasive fungal infections. Clin Microbiol Rev. 2014. Jul;27(3):490–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [367].Roemer T, Krysan DJ. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med. 2014. May 1; 4(5): a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [368].Odds FC, Brown AJ, Gow NA. Antifungal agents: mechanisms of action. Trends Microbiol. 2003. Jun;11(6):272–279. [DOI] [PubMed] [Google Scholar]
- [369].Balfour JA, Faulds DT. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in superficial mycoses. Drugs. 1992. Feb;43(2):259–284. [DOI] [PubMed] [Google Scholar]
- [370].Kobayashi D, Kondo K, Uehara N, et al. Endogenous reactive oxygen species is an important mediator of miconazole antifungal effect. Antimicrob Agents Chemother. 2002. Oct;46(10):3113–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [371].Anderson TM, Clay MC, Cioffi AG, et al. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat Chem Biol. 2014. May;10(5):400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [372].Felton T, Troke PF, Hope WW. Tissue penetration of antifungal agents. Clin Microbiol Rev. 2014. Jan;27(1):68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [373].Donovick R, Gold W, Pagano JF, et al. Amphotericins A and B, antifungal antibiotics produced by a streptomycete. I. In vitro studies. Antibiot Annu. 1955;3:579–586. [PubMed] [Google Scholar]
- [374].Cavassin FB, Bau-Carneiro JL, Vilas-Boas RR, et al. Sixty years of amphotericin b: an overview of the main antifungal agent used to treat invasive fungal infections. Infect Dis Ther. 2021. Mar;10(1):115–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [375].Lu R, Hollingsworth C, Qiu J, et al. Efficacy of oral encochleated amphotericin b in a mouse model of cryptococcal meningoencephalitis. mBio. 2019. May 28;10(3): e00724–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [376].Sanglard D, Ischer F, Parkinson T, et al. Candida albicans mutations in the ergosterol biosynthetic pathway and resistance to several antifungal agents. Antimicrob Agents Chemother. 2003. Aug;47(8):2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [377].Miyazaki Y, Geber A, Miyazaki H, et al. Cloning, sequencing, expression and allelic sequence diversity of ERG3 (C-5 sterol desaturase gene) in Candida albicans. Gene. [1999. Aug 5];236(1):43–51. [DOI] [PubMed] [Google Scholar]
- [378].Vermes A, Guchelaar HJ, Dankert J. Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J Antimicrob Chemother. 2000. Aug;46(2):171–179. [DOI] [PubMed] [Google Scholar]
- [379].Vermes A, Guchelaar HJ, Dankert J. Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J Antimicrob Chemoth. 2000. Aug;46(2):171–179. [DOI] [PubMed] [Google Scholar]
- [380].Bhattacharya S, Esquivel BD, White TC. Overexpression or deletion of ergosterol biosynthesis genes alters doubling time, response to stress agents, and drug susceptibility in saccharomyces cerevisiae. mBio. 2018. Jul 24;9(4): e01291–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [381].Kobayashi D, Kondo K, Uehara N, et al. Endogenous reactive oxygen species is an important mediator of miconazole antifungal effect. Antimicrob Agents Ch. 2002. Oct;46(10):3113–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [382].Nett JE, Andes DR. Antifungal agents: spectrum of activity, pharmacology, and clinical indications. Infect Dis Clin North Am. 2016. Mar;30(1):51–83. [DOI] [PubMed] [Google Scholar]
- [383].Lortholary O, Desnos-Ollivier M, Sitbon K, et al. Recent exposure to caspofungin or fluconazole influences the epidemiology of candidemia: a prospective multicenter study involving 2,441 patients. Antimicrob Agents Chemother. 2011. Feb;55(2):532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [384].Castanheira M, Messer SA, Rhomberg PR, et al. Isavuconazole and nine comparator antifungal susceptibility profiles for common and uncommon Candida species collected in 2012: application of new CLSI clinical breakpoints and epidemiological cutoff values. Mycopathologia. 2014. Aug;178(1–2):1–9. [DOI] [PubMed] [Google Scholar]
- [385].Enwuru CA, Ogunledun A, Idika N, et al. Fluconazole resistant opportunistic oro-pharyngeal Candida and non-Candida yeast-like isolates from HIV infected patients attending ARV clinics in Lagos, Nigeria. Afr Health Sci. 2008. Sep;8(3):142–148. [PMC free article] [PubMed] [Google Scholar]
- [386].Martinez M, Lopez-Ribot JL, Kirkpatrick WR, et al. Heterogeneous mechanisms of azole resistance in Candida albicans clinical isolates from an HIV-infected patient on continuous fluconazole therapy for oropharyngeal candidosis. J Antimicrob Chemother. 2002. Mar;49(3):515–524. [DOI] [PubMed] [Google Scholar]
- [387].Gao J, Wang H, Li Z, et al. Candida albicans gains azole resistance by altering sphingolipid composition. Nat Commun. [2018. Oct 29];9(1):4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [388].Bhattacharya S, Sae-Tia S, Fries BC. Candidiasis and mechanisms of antifungal resistance. Antibiotics (Basel). 2020. Jun 9;9(6): 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [389].Wiederhold NP. The antifungal arsenal: alternative drugs and future targets. Int J Antimicrob Agents. 2018. Mar;51(3):333–339. [DOI] [PubMed] [Google Scholar]
- [390].Break TJ, Desai JV, Healey KR, et al. VT-1598 inhibits the in vitro growth of mucosal Candida strains and protects against fluconazole-susceptible and -resistant oral candidiasis in IL-17 signalling-deficient mice. J Antimicrob Chemother. [2018. Aug 1];73(8):2089–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [391].Break TJ, Desai JV, Natarajan M, et al. VT-1161 protects mice against oropharyngeal candidiasis caused by fluconazole-susceptible and -resistant Candida albicans. J Antimicrob Chemother. [2018. Jan 1];73(1):151–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [392].Brand SR, Degenhardt TP, Person K, et al. A phase 2, randomized, double-blind, placebo-controlled, dose-ranging study to evaluate the efficacy and safety of orally administered VT-1161 in the treatment of recurrent vulvovaginal candidiasis. Am J Obstet Gynecol. 2018. Jun;218(6):624 e1–624 e9. [DOI] [PubMed] [Google Scholar]
- [393].Onishi J, Meinz M, Thompson J, et al. Discovery of novel antifungal (1,3)-beta-D-glucan synthase inhibitors. Antimicrob Agents Chemother. 2000. Feb;44(2):368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [394].Beyda ND, Lewis RE, Garey KW. Echinocandin resistance in Candida species: mechanisms of reduced susceptibility and therapeutic approaches. Ann Pharmacother. 2012. Jul-Aug;46(7–8):1086–1096. [DOI] [PubMed] [Google Scholar]
- [395].Douglas CM, D’Ippolito JA, Shei GJ, et al. Identification of the FKS1 gene of Candida albicans as the essential target of 1,3-beta-D-glucan synthase inhibitors. Antimicrob Agents Chemother. 1997. Nov;41(11):2471–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [396].Shields RK, Nguyen MH, Press EG, et al. Rate of FKS mutations among consecutive Candida isolates causing bloodstream infection (vol 59, pg 7465, 2015). Antimicrob Agents Ch. 2016. Mar;60(3):1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [397].Arendrup MC, Cuenca-Estrella M, Lass-Florl C, et al. Breakpoints for antifungal agents: an update from EUCAST focussing on echinocandins against Candida spp. and triazoles against aspergillus spp. Drug Resist Updat. 2013. Dec;16(6):81–95. [DOI] [PubMed] [Google Scholar]
- [398].Healey KR, Zhao Y, Perez WB, et al. Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat Commun. 2016. Mar 29;7:11128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [399].Legrand M, Chan CL, Jauert PA, et al. Role of DNA mismatch repair and double-strand break repair in genome stability and antifungal drug resistance in Candida albicans. Eukaryot Cell. 2007. Dec;6(12):2194–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [400].Arendrup MC, Jorgensen KM, Hare RK, et al. In vitro activity of ibrexafungerp (SCY-078) against candida auris isolates as determined by EUCAST methodology and comparison with activity against C. albicans and C. glabrata and with the activities of six comparator agents. Antimicrob Agents Chemother. 2020. Feb 21;64:(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [401].Sobel JD, Borroto-Esoda K, Azie N, et al. In vitro pH activity of ibrexafungerp against fluconazole-susceptible and -resistant candida isolates from women with vulvovaginal Candidiasis. Antimicrob Agents Chemother. [2021. Jul 16];65(8):e0056221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [402].Umemura M, Okamoto M, Nakayama K, et al. GWT1 gene is required for inositol acylation of glycosylphosphatidylinositol anchors in yeast. J Biol Chem. [2003. Jun 27];278(26):23639–23647. [DOI] [PubMed] [Google Scholar]
- [403].Hata K, Horii T, Miyazaki M, et al. Efficacy of oral E1210, a new broad-spectrum antifungal with a novel mechanism of action, in murine models of candidiasis, aspergillosis, and fusariosis. Antimicrob Agents Chemother. 2011. Oct;55(10):4543–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [404].Petraitiene R, Petraitis V, Maung BBW, et al. Efficacy and pharmacokinetics of fosmanogepix (APX001) in the treatment of candida endophthalmitis and hematogenous meningoencephalitis in nonneutropenic rabbits. Antimicrob Agents Chemother. 2021. Feb 17;65(3):e01795–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [405].Shaw KJ, Ibrahim AS. Fosmanogepix: a review of the first-in-class broad spectrum agent for the treatment of invasive fungal infections. J Fungi (Basel). 2020. Oct 22;6(4):239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [406].Zhang F, Zhao M, Braun DR, et al. A marine microbiome antifungal targets urgent-threat drug-resistant fungi. Science. [2020. Nov 20];370(6519):974–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [407].Zhao M, Zhang F, Zarnowski R, et al. Turbinmicin inhibits Candida biofilm growth by disrupting fungal vesicle-mediated trafficking. J Clin Investig. 2021. Mar 1;131(5):e145123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [408].Perfect JR. The antifungal pipeline: a reality check. Nat Rev Drug Discov. 2017. Sep;16(9):603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [409].Hoenigl M, Sprute R, Egger M, et al. The antifungal pipeline: fosmanogepix, ibrexafungerp, olorofim, opelconazole, and rezafungin. Drugs. 2021. Oct;81(15):1703–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data sets were analyzed in this manuscript, and data availability is not applicable.