

SUMMARY
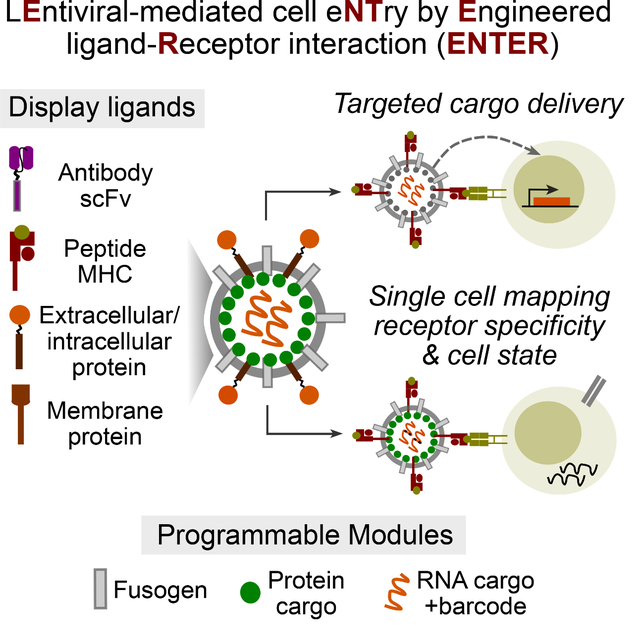
Cells communicate with each other via receptor-ligand interactions. Here we describe lentiviral-mediated cell entry by engineered receptor-ligand interaction (ENTER) to display ligand proteins, deliver payloads, and record receptor specificity. We optimize ENTER to decode interactions between T cell receptor (TCR)-MHC peptides, antibody-antigen, and other receptor-ligand pairs. A viral presentation strategy allows ENTER to capture interactions between B cell receptor and any antigen. We engineer ENTER to deliver genetic payloads to antigen-specific T or B cells to selectively modulate cellular behavior in mixed populations. Single-cell readout of ENTER by RNA-sequencing (ENTER-seq) enables multiplexed enumeration of antigen specificities, TCR clonality, cell-type and states of individual T cells. ENTER-seq of CMV-seropositive patient blood samples reveals the viral epitopes that drive effector memory T cell differentiation and inter- vs intra-clonal phenotypic diversity targeting the same epitope. ENTER technology enables systematic discovery of receptor specificity, linkage to cell fates, and antigen-specific cargo delivery.
Graphical Abstract
IN BRIEF
A modular, lentiviral-based display and delivery platform enables decoding of ligand-receptor interactions, receptor-specific genetic cargo delivery, and mapping of receptor specificity and cell states of single cells.
INTRODUCTION
Advances in single-cell genomics have provided unprecedented insights in deciphering molecular and cellular heterogeneity of biological systems. Despite substantial progress on deciphering the central dogma of gene expression, cell-extrinsic information such as intercellular ligand-receptor interaction is poorly explored at the single-cell level.
Cells communicate with each other through ligand-receptor interactions. For example, TCR on the surface of T cells can recognize and interact with the peptide-major histocompatibility complex (pMHC) on the surface of antigen-presenting cells (APCs) 1. TCR and B cell receptor (BCR) genes undergo somatic recombination to reach a diverse repertoire, allowing recognition of innumerable antigens 2. Many approaches have been developed to decipher the antigen specificity of TCRs including: (1) cell reporter assay using artificial APCs 3–6; (2) yeast display platform 7; (3) T cell based assay 8; and (4) DNA barcoded pMHC tetramer 9. Despite unique advantages for each technique, it is still challenging to rapidly screen pMHCs for primary T cells in clinical samples and simultaneously capture the antigen landscape, TCR repertoire, and transcriptome of individual T cells in a high-throughput manner. Similar challenges apply to study BCR-antigen interactions. Moreover, targeting T and B cells in an antigen-specific manner to selectively rewire their behavior is challenging. A recent method using pMHC presenting nanoparticles has enabled mRNA delivery in antigen-specific T cells for transient modulation 10. Another study using pMHC pseudotyped viruses allows genetic modification of antigen-specific T cells 11. However, there is still a lack of technology to selectively manipulate antigen-specific B cells beyond T cells.
Here we developed a modular viral display and delivery platform to decode ligand-receptor interactions, deliver cargos in target cells, and connect ligand-receptor interactions with cellular state. We termed this technology ENTER, which can systematically deorphanize pairs of interactions including TCR-pMHC, antibody-antigen, costimulatory ligand-receptors, and B cell antigen-BCR. ENTER permits gene delivery in a receptor-specific manner, allowing for the selective modulation in antigen-specific T and B cells. We further combined ENTER with droplet-based single-cell genomics profiling (ENTER-seq) to measure antigen specificity, TCR repertoire, gene expression and surface protein landscape in individual human primary T cells.
RESULTS
A viral display and delivery platform to capture ligand-receptor interactions
To achieve ligand-receptor interaction between lentiviruses and host cells, we reason that a user-defined ligand can complement a viral envelope protein with disrupted native receptor binding while maintaining fusion ability (Figure 1A). We focused on vesicular stomatitis virus G protein (VSV-G), a widely used viral envelope protein that interacts with low density lipoprotein receptor (LDLR) on host cells 12 13. In contrast to wild-type VSV-G pseudotyped virus, virus displaying mutant VSV-G (K47Q, R354A) that is defective on LDLR binding but retains fusion ability fails to infect Raji or Jurkat cells (Figure 1B) 14. When co-displaying a well-established anti-CD19 single-chain antibody variable fragment (sc-Fv) and the VSV-G mutant, viruses specifically infected Raji B cells with high levels of CD19 but not CD19-negative Jurkat T cells (Figure 1B). Thus, our platform reprograms the viral tropism to cell entry that is dependent on the interaction between user-defined ligand and paired host cell receptors.
Figure 1. A platform to display ligand proteins and fusogen on viral surface, deliver fluorescent proteins, and record ligand-receptor interaction by cell entry.

A. Schematic view of all-in-one viral platform. The lentiviruses are engineered in diverse modules including: (1) user-defined ligand proteins displayed on viral surface; (2) modified fusogen with intact fusion ability and defective binding to natural receptors; (3) cargo proteins fused with viral structure protein; and (4) barcoded viral RNA for tracing and gene delivery.
B. Schematic view of experimental set up and representative flow plot of GFP expression after 3 days of viral infection. Raji and Jurkat cells are infected by three groups of lentiviruses encoding GFP in the viral RNA: (1) viruses with wild-type VSV-G (left); (2) viruses with receptor-binding mutated VSV-G (middle); (3) viruses with VSV-G mutant and anti-CD19 scFv.
C. Schematic view (top) of experimental set up. GFP protein are fused with matrix protein (MA-GFP) or Nucleocapsid protein (NC-GFP), or viral protein R (VPR-GFP). scFv-CD19 displayed viruses carrying GFP protein fused with different viral proteins were incubated with Raji (CD19+) or Jurkat (CD19−) cells for 2 hours and then subjected to flow cytometry. Bar plot (bottom) showing the percentage of GFP+ cells upon incubation of viruses with different GFP fusion viral proteins.
D. Representative flow plots of GFP signal after transient viral incubation as in Figure 1C.
E. Schematic view of experimental set up and representative flow plot of GFP signal in primary human B cells with or without viral incubation. Naïve and activated human primary B cells were incubated with NC-GFP fused and scFv-CD19 displayed viruses for 2 hours followed by flow cytometry. B cells were gated on live CD20+ cells.
F. Histogram analysis of surface CD19 expression of groups from Figure 1E.
G. Bar plots showing scFv-CD19 virus binding and CD19 surface expression in naïve and activated human B cells
Data are represented as mean +/− SEM in C and G from triplicates.
P-values in Figure 1C and 1F are calculated by unpaired t-test. *** P<0.001, **** P<0.0001
Next, we fused the GFP protein with viral structural proteins to track transient virus binding and entry into host cells. After optimization (Method), nucleocapsid protein (NC) GFP fusion was found to perform the best using CD19-scFv displayed viruses and Raji cell model (Figure 3C–D). In addition, NC-GFP viruses displaying CD19-scFv can recognize and bind to primary CD19+ B cells from human blood. Similar to Raji B cells, 80% of activated human primary B cells were bound by NC-GFP labeled CD19-scFv displayed viruses whereas 60% of naïve B cells were GFP+ (Figure 1E). The discrepancy of binding of CD19-scFv viruses is likely due to the difference of CD19 expression between naïve and activated B cells (Figure 1F–G). Moreover, CD19 surface expression was dramatically decreased after incubation of CD19-scFv viruses (Figure 1G). Based on viral binding and fusion assay (Method, Figure S1A–B), the reduction of surface CD19 is mostly resulted from binding/masking of CD19-scFv displayed viruses rather than fusion/internalization. Consistently, specific binding and cell entry were observed when employing other ligand-receptor interaction such as CD40 ligands (Figure S1C–E). The incorporation of ligand protein and fusogen on virion surfaces were confirmed using an immunocapture assay (Figure S1F–G). These results suggest that ENTER can capture a highly specific ligand-receptor interaction in a transient viral binding assay and is applicable to multiple categories of receptor-ligand interactions.
Figure 3. Optimization of ENTER to present intracellular antigens on viral surface and decode interaction between BCR with antigens.

A. Schematic view of experimental design. During the assembly and budding of lentiviruses, certain host cell surface proteins can be incorporated into the surface of viruses. TM domains of host proteins selected from literature are fused with a B cell epitope derived from intracellular antigen HPV16 L2.
B. Representative flow plot from triplicates showing GFP signal in HPV-BCR+ B cells incubated with GFP viruses displaying HPV epitope fused with different TM domains. B cells without BCR expression serve as a negative control.
C. Bar plots showing the percentage of GFP+ B cells from Figure 3B.
D. Representative flow plot from triplicates showing GFP signal in RBD-BCR+ B cells upon incubation of ENTER viruses displaying SARS-CoV-2 Spike RBD antigen or HPV L2 antigen as a negative control (left). Bar plot showing the frequency of GFP+ cells upon incubation of on-target or off-target ENTER viruses (right).
E. Schematic view of experimental set up (top) and flow cytometry analysis (bottom) of B cell mixing experiment. Violet dye labeled RBD-BCR+ B cells were mixed with HPV-BCR+ B cells at different ratio, and then incubated with GFP viruses displaying RBD antigen fused with ICAM1 TM domain. Representative flow plot showing 1:1000 mixing of two B cell population and GFP signal of B cells gated on violet+ and violet− population.
F. Bar plot showing the signal-to-noise ratio of ENTER from Figure 3E and Figure S3C.
Data are represented as mean +/− SEM in C, D, and F from triplicates.
ENTER with pMHC displaying viruses maps TCR specificity
To investigate if ENTER can capture pMHC-TCR interactions, we engineered viruses to display a single chain of MHC fused with beta 2 microglobulin (B2M) and covalently linked peptide (Figure 2A). We observed a high surface expression of HLA-A*0201 (HLA-A2) when transfected into HEK293T HLA knockout cells, where all endogenous HLA class I alleles were knocked out by CRISPR-Cas9 (Method, Figure S2A).
Figure 2. ENTER decodes interaction between pMHC with TCR.

A. Schematic view of pMHC displayed viruses and representative flow plot from two replicates showing GFP signal in specific TCR expressing Jurkat T cells upon incubation of pMHC displayed viruses. GFP fused viruses displaying either a 9-mer peptide (NY-ESO-1157–165) presented by HLA-A0201 allele or an 11-mer CMV peptide (pp65363–373) presented by HLA-A0101 allele were incubated with T cells expressing specific TCRs (e.g., NY-ESO-1157–165-TCR or CMV-pp65363–373-TCR) that recognize the cognate antigens. SP: signal peptide; Peptide: antigen peptide; B2M: Beta-2-Microglobulin.
B. Representative flow plot from two replicates showing GFP signal in Jurkat T cells expressing specific TCRs (e.g., NY-ESO-1157–165-TCR, CMV-pp65495–503-TCR, or Flu-m158–66-TCR) upon incubation of viruses displaying various HLA-A2 presented peptides. HPV16 E782–91 peptide displayed viruses serve as a negative control.
C. Representative flow plot from two replicates showing NY-ESO-1 TCR-T cells upon incubation of 2×108 ENTER viral particles displaying NY-ESO-1157–166 antigen (left) or with different amount of NY-ESO-1157–166 pMHC tetramers.
D. Comparison of binding efficiency (GFP+%) of viruses displaying antigen variants with different TCR affinity. 1G4wt-TCR T cells were incubated with ENTER displaying wild-type of mutant NY-ESO-1157–166 antigen variants for 2 hours. CMV-pp65495–503-TCR T cells were used as negative control. The viral titers were normalized using the p24 protein level.
E. Schematic view of experimental set up (top) and flow cytometry analysis (bottom) of TCR-T cell mixing experiment. Violet dye labeled Flu-m158–66-TCR T cells were mixed with CMV-pp65495–503-TCR T cells at different ratio, and then incubated with HLA-A2:m1 displayed GFP viruses for 2h followed by flow cytometry. Representative flow plot showing 1:1000 mixing of two T cell population and GFP signal of T cells gated on Violet+ and Violet− population.
F. Bar plot showing the signal-to-noise ratio of ENTER in Figure 2E and Figure S2D.
Data are represented as mean +/− SEM in D and F from triplicates.
Next, as a proof-of-concept, we generated GFP fused viruses displaying a well-known cancer-testis antigen NY-ESO-1 as a 9-mer peptide on HLA-A2, one of the most prevalent HLA alleles in humans 15 (Figure 2A). 88.2% of cognate NY-ESO-1 TCR-expressing T cells were labeled by NY-ESO-1 antigen-bearing GFP viruses, compared to 1.26% of T cells specific to a known human cytomegalovirus (CMV) epitope 5. Similarly, viruses displaying a CMV antigen as an 11-mer peptide on a different HLA allele HLA-A*01:01, specifically bound to CMV TCR-T cells rather than NY-ESO-1 TCR-T cells (Figure 2A). We further engineered viruses displaying diverse 9-mer antigen epitopes known presented on HLA-A2 allele 16,17. Over 87% of TCR-matching T cells are GFP+ after 2 hours of incubation with GFP fused viruses displaying cognate HLA-A2-peptide whereas only 1% of these T cells are labeled by negative control antigen displayed GFP viruses (Figure 2B). The highly specific binding of pMHC displaying viruses into TCR-matching T cells highlights the generality of the ENTER platform to present diverse pMHC antigens.
We further validated the specificity of our pMHC displayed viruses by co-staining with tetramers (Method, Figure S2B). Additionally, we observed that the pp65495–503 tetramer intensity and CD3 surface expression are significantly decreased after co-incubation with viruses displaying pp65495–503 (Figure S2C), similar to the observation of CD19 on Raji B cells (Figure 1G). This result indicated that pMHC displayed viruses specifically bind to and mask the TCR-CD3 complex, preventing later binding of pMHC tetramer and anti-CD3 antibody.
We found that pMHC displayed virus is more sensitive compared to pMHC tetramers on a molar basis per reagent (Method, Figure 2C). In addition, by varying a broad range of affinity between pMHC and TCR interaction, ENTER is able to detect TCR affinity as low as 10.8 uM when adding high titer of viruses (40ng p24) (Figure 2D). The TCR binding affinity is positively correlated with ENTER recognition efficiency, suggesting that ENTER may be applied to infer relative TCR affinity by measuring binding efficiency of pMHC displayed viruses (Figure 2D). Finally, we assessed the specificity and sensitivity of our pMHC viral display platform, by mixing on-target T cells and off-target T cells at different ratios (Method, Figure 2E, S2D). The signal-to-noise ratio was over 150-fold even when the frequency of on-target T cells is as low as 1 in 1000, further demonstrating a high specificity and sensitivity of ENTER platform (Figure 2F, S2D–E).
Decoding B cell specificity by ENTER viruses that display B cell antigens
B cells possess a high diversity of BCR that can specifically target foreign antigens from invading viruses and self-antigens from our own body 18. The main challenge of ENTER to decode B cell specificity is to display B cell antigens which do not contain their native transmembrane (TM) domains on the viral surface. To address it, we sought to engineer a TM domain for optimal surface display of B cell antigens (Figure 3A). We selected 10 candidate TM domains (Table S3, Method) to display a B cell antigen epitope derived from human papillomavirus (HPV) minor capsid antigen L2 (HPV16 L217–36) (Figure 3A–B). Next, we generated a BCR expressing B cell line that specifically targets HPV16 L217–36 19. The screen revealed ICAM1 TM domain as the top candidate since over 90% of HPV antigen-specific BCR+ B cells are GFP+ (Figure 3B–C). This is consistent with previous reports showing that ICAM1 is selectively acquired in budding viruses through interaction with viral matrix protein 20.
To test if the ICAM1 TM domain can be applied to present other B cell antigens, we engineered the viruses to display Receptor Binding Domain (RBD) from SARS-CoV-2 spike protein (Figure 3D). The result showed that 88% of spike-RBD BCR+ B cells were labeled by RBD displayed viruses, indicating that ENTER with optimized TM domain can be applied to decode B cell specificity for both linear epitopes (HPV16 L217–36 antigens) and full antigen domains (SARS-CoV-2 spike RBD). Additionally, we showed that ENTER can decode B cell specificity towards native cell surface B cell antigens such as HER2 21 (Figure S3A–B), highlighting the generalization of ENTER to display any B cell antigens from intracellular (HPV L2), extracellular (Spike RBD), and cell surface proteins (HER2). Finally, mixing experiments showed high specificity and sensitivity of ENTER to decipher interactions between BCR and B cell antigens (Figure 3E, S3C). The signal-to-noise ratio was around 100- to 200-fold (Figure 3F), indicating a profound specificity and sensitivity of our TM domain optimized viruses to display B cell antigen epitopes (Figure S3D–E).
ENTER cargo delivery to modulate the behavior of antigen-specific T and B cells
We investigated if ENTER is capable to deliver cargos into antigen-specific T or B cells. By utilizing GFP transgene as a cargo to measure the delivery efficiency and specificity, wild-type VSV-G pseudotyped virus transduced cells irrespective of TCR or BCR specificity (Figure S3F). When pp65495–503 pMHC viruses were used to transduce on-target CMV-pp65 TCR+ T cells and off-target NY-ESO-1 TCR+ T cells (Method), we observed 82 % of on-target TCR-T cells express GFP whereas only 0.22% of off-target TCR-T cells are GFP+ (Figure 4A–B). Similarly, we showed specific delivery of GFP transgene in on-target HER2 BCR+ B cells but not off-target RBD BCR+ B cells (Figure 4C–D).
Figure 4. ENTER permits cargo delivery in antigen-specific T or B cells.

A. Schematic view of cargo delivery in antigen-specific T cells (left). CMV-pp65 TCR+ T cells or NY-ESO-1 TCR+ T cells were individually infected with ENTER viruses that display pp65495–503 and carry GFP transgene as a cargo. Representative histogram plot showing GFP expression between CMV-pp65 TCR+ T cells and NY-ESO-1 TCR+ T cells after 2 days infection.
B. Bar plot showing the percentage of GFP+ cells as in Figure 4A.
C. Schematic view of cargo delivery in antigen-specific B cells (left). HER2 BCR+ B cells or RBD BCR+ B cells were individually infected with ENTER viruses that display HER2 and carry GFP transgene as a cargo. Representative histogram plot (right) showing GFP expression between HER2 BCR+ B cells and RBD BCR+ B cells after 2 days infection.
D. Bar plot showing the percentage of GFP+ cells as in Figure 4C.
E. Schematic view of suicide gene delivery in a pool of different antigen-specific T cells. CMV-pp65 TCR+ T cells and NY-ESO-1 TCR+ T cells expressing mScarlet were mixed together and then infected with ENTER viruses that display pp65495–503 and carry HSV-TK transgene. After 2 days of infection, GCV drug was added to kill HSV-TK expressing cells and cell survival was monitored for 4 days.
F. Bar plot showing the number of live T cells at day 4 post GCV treatment.
G. Kinetics analysis of normalized fold enrichment between the number of CMV-pp65 TCR+ T cells versus that of NY-ESO-1 TCR+ T cells that were either infected with ENTER carrying GFP gene or ENTER carrying HSV-TK gene upon GCV drug treatment.
H. Schematic view of suicide gene delivery in a pool of different antigen-specific B cells. HER2 BCR+ B cells and RBD BCR+ B cells expressing mScarlet were mixed together and then infected with ENTER viruses that display pp65495–503 and carry HSV-TK transgene. After 2 days of infection, GCV drug was added to kill HSV-TK expressing cells and cell survival was monitored for 4 days.
I. Bar plot showing the number of live B cells at day 4 post GCV treatment.
J. Kinetics analysis of normalized fold enrichment between the number of HER2 BCR+ B cells versus that of RBD BCR+ B cells that were either infected with ENTER carrying GFP gene or ENTER carrying HSV-TK gene upon GCV drug treatment.
K. Schematic view of shRNA delivery in a pool of different antigen-specific T cells. CMV-pp65 TCR+ T cells and NY-ESO-1 TCR+ T cells expressing mScarlet were mixed together and then infected with ENTER viruses that display pp65495–503 and carry FAS shRNA or control shRNA. Anti-FAS antibody was added to induce apoptosis.
L. Bar plot showing the surface expression of FAS in off-target NY-ESO-1 TCR+ T cells (uninfected group), and on-target CMV-pp65 TCR+ T cells transduced with control shRNA or FAS shRNA.
M. Bar plot showing the CMV-pp65 TCR+ T cells/NY-ESO-1 TCR+ T cells fold enrichment that is normalized by shCtrl group for the live cells (gated on Annexin V and 7-AAD double negative) as in K.
Data are represented as mean +/− SEM in bar plots from at least two biological replicates (4 replicates in M).
P-values are calculated by unpaired t-test. **** P<0.0001; *** P<0.001; **P<0.01; * P<0.05; n.s. P>=0.05
Next, we engineered pMHC displayed viruses to carry herpes simplex virus thymidine kinase (HSV-TK) gene, a well-established suicide gene in response to drug ganciclovir (GCV) 22, for targeted cell killing. After adding pp65495–503 displayed viruses carrying suicide gene to a mixture of CMV-pp65 TCR+ T cells and NY-ESO-1 TCR+ T cells, we observed a specific cargo delivery in on-target T cells (CMV-pp65 TCR+) (Figure S3G) and preferential depletion of on-target T cells compared to off-target T cells (Figure 4E–F). The killing was largely due to the specific delivery of suicide gene when compared to GFP control cargo (Figure 4G). Likewise, HER2 displayed viruses delivered HSV-TK to specific HER2 BCR+ B cells in a mixture with RBD BCR+ B cells (Figure 4H, Figure S3H), and resulted in efficient killing of targeted cells (Figure 4I–J). Our data suggest that ENTER enables selective depletion of one T or B cell clone among a pool of T or B cells by antigen-specific suicide gene delivery.
Conversely, ENTER enables selective survival of antigen-specific T cells. We delivered short hairpin RNA (shRNA) against the cell death receptor FAS (shFAS) in antigen-specific T cells to prevent the FAS-induced programmed cell death 23 (Method, Figure S3I). pp65495–503 displayed viruses carrying shFAS or control shRNA (shCtrl) were used to infect a pool of on-target (CMV-pp65 TCR+) and off-target (NY-ESO-1 TCR+) T cells (Figure 4K). A significant decrease of FAS protein surface expression was observed in on-target T cells infected with shFAS compared to shCtrl group or off-target uninfected T cells, indicating a targeted shRNA delivery (Figure 4L). When challenged with FAS activation to induce apoptosis (Figure 4K, Figure S3J, Method), we observed significant increase of on-target T cells among live cells after FAS knockdown compared to shCtrl group (Figure 4M). Together, our data showed that ENTER permits targeted cargo delivery to manipulate complex cellular populations with ligand-receptor specificity.
ENTER-seq captures pMHC specificity, TCR repertoire and gene expression profile with single-cell resolution
Given the unique feature of viral RNA that carries genetic information to encode surface-displayed ligand proteins, we combined ENTER platform with droplet-based single-cell RNA-seq to develop ENTER-seq. Specifically, we applied ENTER-seq to simultaneously map pMHC antigen specificity, TCR repertoire and gene expression of T cells at the single-cell level (Figure 5A). To efficiently capture the MHC-peptide information on viral RNA in each droplet, we inserted a capture tag in the linker region between B2M and MHC, and another PCR handle next to the CMV promoter (Figure 5B). This capture tag allows capture by commercially available 5’ GEM beads through hybridizing with the Template Switch Oligo (TSO) sequence conjugated on the beads. The insertion of the capture tag and PCR handle does not affect the display of pMHC on viruses and specific interaction with TCR-expressing T cells (Figure S4A–B).
Figure 5. ENTER-seq for massively parallel measurement of antigen peptide sequence, TCR sequence, and transcriptome.

A. Schematic view of ENTER-seq workflow. A library of pooled viruses displaying individual pMHC was incubated with T cells for 2 hours. GFP+ T cells were sorted for droplet-based single cell genomics profiling.
B. Viral RNA engineering strategy for droplet-based single cell capture. 10x genomics capture tag is inserted in the linker region between B2M and HLA-A2. 10x genomics PCR handle is inserted after CMV promoter. CMV: CMV promoter; SP: signal peptide sequence; Peptide: antigen peptide; B2M: Beta-2-Microglobulin; MHC Class I: HLA-A0201 allele; LTR: long terminal repeat; TSO: template switching oligo sequence.
C. Schematic view of T cell mixing experiment for ENTER-seq.
D. Number of Pp65495–503-TCR T cells UMI counts (x-axis) and NY-ESO-1157–165-TCR UMI counts (y-axis) associated with each cell barcode (dot). The colors are assigned as NY-ESO-1157–165-TCR+ T cells (light blue), Pp65495–503-TCR+ T cells (red), and doublets (green, with both NY-ESO-1157–165-TCR and Pp65495–503-TCR).
E. Scatter plots of TCR UMI counts after doublet removal, colored by enrichment ratio of pp65495–503-antigen UMI count among total UMI counts (left), and enrichment ratio of NY-ESO-1157–165-antigen UMI count among total UMI counts (right).
F. Number of pp65(495–503)-antigen UMI counts (x-axis) and NY-ESO-1157–165-antigen UMI counts (y-axis) associated with each cell barcode (dot) after doublet removal. The colors are assigned as NY-ESO-1157–165-TCR + T cells (light blue) and Pp65495–503-TCR + T cells (red).
To benchmark ENTER-seq for single-cell profiling of antigen specificity and TCR repertoire, we performed ENTER-seq on mixed TCR-expressing T cells with a pooled pMHC displaying viruses (Figure 5C). Analysis of the unique TCR sequence after filtering out doublets confirmed the mixing ratio of T cells at 9.4% (NY-ESO-1 TCR+) vs 90.6% (CMV-pp65 TCR+) (Figure 5D). We further recovered 4198 T cells with reliable antigen peptide information and TCR sequence after filtering the unique molecular identifier (UMI) count of TCRs and antigen peptides. We calculated the ratio of the UMI from the dominant antigen peptide among total peptides and observed a high concordance of antigen peptides to their paired TCR (Figure 5E). After matching TCR sequences to antigen peptides at the single-cell level, the result showed that 99.8% of pp65495–503+ cells and 97.4% of NY-ESO-1157–165 + cells are matched with their corresponding TCR sequences respectively (Figure 5F). Thus, ENTER-seq can sensitively and robustly capture the interaction of TCR repertoire and cognate HLA antigen peptide at the single-cell resolution.
ENTER-seq of peptide-enriched CMV-specific T cells uncover donor-specific immunogenic CMV epitopes and antigen-specific molecular phenotype
To apply ENTER-seq on human clinical samples, we chose ex vivo expanded anti-CMV T cells. Adoptive transfer of such T cells has shown great efficacy to control CMV infection in patients receiving transplantation 24–26. We first switched GFP to mNeon, a brighter reporter protein, and validated the ability of ENTER to detect antigen specific primary T cells (Method, Figure S4C–F). Next, to expand CMV-specific T cells, we cultured human peripheral mononuclear cells (PBMCs) from CMV seropositive donors with a pool of 12 CMV antigen peptides for 10 days 27–29 (Figure S4G). We confirmed the specificity of ENTER on peptide-stimulated T cells using tetramer co-staining (Figure S4H).
Using ENTER viruses displaying 12 CMV antigen epitopes, we observed dramatic expansion of CMV antigens-specific T cells in 2 out of 4 CMV seropositive HLA-A2+ donors (Figure S4G–I). Next, we performed ENTER-seq of expanded T cells from these two donors. Cells were also stained with hashtag antibodies that labelled each donor sample and CITE-seq antibodies that capture surface proteins (Figure 6A). The ENTER-seq result showed that expanded CMV-specific T cells (ENTER+) are phenotypically different from bystander T cells (ENTER−) (Figure 6B). To test if such phenotypic difference is induced by the binding of ENTER viruses, we compared RNA-seq data of CMV pp65 TCR-T cells with incubation of pp65495–503 displayed ENTER viruses or pp65495–503 tetramers, and identified only 28 differentially-expressed genes (Figure S4J). We further applied Leiden clustering on ENTER-seq data with or without ENTER-induced gene signature, and confirmed minimal impact of ENTER virus binding on cell clustering (Method, Figure S5A).
Figure 6. ENTER-seq of ex vivo expanded CMV-specific primary T cells.

A. Schematic view of CMV antigen peptide induced T cell expansion and ENTER-seq workflow.
B. UMAP plot showing cells with (ENTER+, colored in yellow) or without (ENTER−, colored in blue) CMV antigen displayed viruses binding.
C. UMAP plots showing CITE-seq of surface protein expression of CD45RA (naïve marker) and CD45RO (memory marker).
D. UMAP plot showing 10 clusters of human CD8+ T cell subsets labeled in different colors.
E. Bar plot showing the number of CMV antigen-specific T cells that recognize specific CMV antigen epitope in donor #1 (labeled in black) and donor #2 (labeled in gray).
F. UMAP plots showing the amount of CMV antigen epitopes per cell for the top 3 CMV antigen epitopes identified from E.
G. Heatmap showing column scaled expression of representative genes associated with effector function or Treg signature among different CMV antigen-specific T cells.
H. UMAP plot (left) showing the clonal expansion size of CMV antigen-specific T cells colored by the number of cells in each clonotype. Violin plot (right) showing the distribution of clone size in different CMV antigen-specific T cells colored by antigen epitopes.
I. Violin plots showing expression of cytokines and transcription factors in pp65495–503-specific TCR clones, separated and colored by CDR3 clones. A simplified model (right).
After integrating surface protein landscape and gene expression, we observed that peptide-enriched CMV-specific T cells are mainly effector memory T cells (TEM) (Figure 6C–D, Figure S5B–D). The single cell RNA-seq data clustered all T cells into 10 clusters (Figure 6D, Figure S5B–F). Our data showed that ENTER+ CMV-specific T cells are phenotypically different between two donors, indicating donor-specific immune response to CMV antigens (Figure S5G–H). Consistently, we observed a donor-specific viral epitope immunogenicity. The result showed a high frequency of US874–82- and UL100200–208- specific T cells specifically in donor #2 (Figure 6E). Interestingly, we observed 3 distinct cell clusters for the top 3 antigen epitopes, suggesting different epitopes may drive unique CD8+ T cell fates (Figure 6F). Indeed, we observed a high expression of regulatory T (Treg) gene signature enriched in UL100200–208- specific T cells 30–33, indicating that these cells resemble CD8+ Treg cells (Figure 6G) 34.
Inter-clonal phenotypic diversity underlying the same antigen specificity
An integrative analysis of TCR repertoire and antigen specificity showed higher clonal expansion of ENTER+ T cells than ENTER− T cells, and no overlap of expanded TCR clones between donors (Figure S6A–B). The clonal expansion size of pp65495–503 -specific T cells was substantially larger than that of UL100200–208-specific T cells (Figure 6H). Across all clonotypes, TCR clonal expansion was significantly correlated with the expression of genes associated with cytotoxicity but not exhaustion or activation (Figure S6C–D). We identified three dominant CDR3 clonotypes for pp65495–503 specific T cells, Two of them are identical with published pp65495–503-specific TCRs, validating the specificity of our platform (Figure S6E–F) 35. Combining CDR3 clonotypes with gene expression profiles in pp65495–503 specific T cells, we discovered that different clonotypes exhibit distinct gene expression program in cytokines and transcription factors, indicating an inter-clonal phenotypic diversity that targets the same antigen epitope (Figure 6I, S6G). Thus, ENTER-seq can functionally characterize both the TCR binding specificity and the TCR-associated cell states.
ENTER-seq of primary CMV-specific T cells from patients reveals intra-clonal diversity in genes associated with cytotoxicity and type-I IFN response
To decode the anti-viral T cell memory in CMV seropositive patients, we generated ENTER viruses displaying top 3 CMV antigen epitopes identified above and performed ENTER-seq on primary T cells isolated directly from patient blood without ex vivo expansion (Figure 7A). Our data showed that CMV-specific T cells in patients are mainly terminally differentiated effector memory T cells (TEMRA) (Figure 7B–C). This is consistent with previous studies showing the accumulation of TEMRA CMV-specific T cells in CMV seropositive patients 36,37.
Figure 7. ENTER-seq of primary CMV-specific T cells isolated directly from CMV seropositive patient blood.

A. Schematic view of isolation of primary T cells from patient blood and ENTER-seq workflow.
B. UMAP plot (left) showing cells with (ENTER+, colored in yellow) or without (ENTER−, colored in blue) CMV antigen displayed viruses binding. UMAP plot (right) showing 13 clusters of human CD8+ T cell subsets labeled in different colors.
C. UMAP plots showing the surface protein expression of CD45RA and CD45RO from CITE-seq and expression of representative genes.
D. Heatmap showing the scaled z score of expression of genes associated diverse functions (e.g. type-I IFN, cytotoxicity etc) across different clusters.
E. UMAP plot showing the CMV antigen specificity colored by antigen epitope.
F. UMAP plot (left) showing the clonal expansion size of CMV antigen-specific T cells colored by the number of cells in each clonotype.
G. Violin plot showing the number of pMHC bound per cell in T cells with different clone size. P value was calculated by Mann-Whitney test. n.s. p>=0.05; * p<0.05; ** p<0.01, *** p<0.001, **** p<0.0001
H. CITE-seq density plot showing surface expression of CD45RA and CD45RO in pp65-specific T cells from donor #1 (colored by orange) and donor #2 (colored by blue) before and after peptide-induced expansion.
L. Flow cytometry plots showing CD45RA and CD45RO in pp65-specific T cells from donor #1 and donor #2 before and after peptide-induced expansion.
J. Density plot showing the distribution of type-I IFN ISG gene score and cytotoxicity gene score prior and post peptide-induced expansion in top 3 TCR clones of pp65-specific T cells (left). Density plot showing the expression of IL13 and EOMES prior and post peptide-induced expansion in top 3 TCR clones of pp65-specific T cells (right).
K. A proposed model of phenotypic transition of CMV-specific T cells upon ex vivo expansion.
After subset clustering, we observed heterogeneity of TEMRA populations in CMV-specific T cells with diverse patterns of gene expression associated with cytotoxic function, chemokines, costimulatory/coinhibitory molecules, and type-I IFN response (Figure 7D, Figure S7A). Such phenotypic heterogeneity was not induced by the binding of ENTER viruses (Figure S7B). Next, we observed that CMV-specific T cells are predominantly pp65495–503-specific T cells associated with a wide range of clonal expansion, confirming that pp65495–503 is a highly immunogenic CMV epitope (Figure 7E–F). We further observed a rare population of US874–82 specific T cells (43 cells) in patients sharing TCR with peptide expanded US874–82 specific T cells and there was a donor-specific expansion pattern (Figure S7C–F). Our data demonstrate the ability of ENTER-seq to detect multiple antigen specificities in the presence of highly dominant epitope from fresh PBMCs.
We quantified the number of pMHC bound per cell to measure the binding strength of pMHC displayed ENTER viruses. Our data showed significantly higher binding of pMHC in highly expanded T cell clones than lowly expanded T cells (Figure 7G), suggesting that high pMHC binding, a proxy of high TCR affinity is associated with and likely drives greater T cell clonal expansion. Finally, we found 3 dominant pp65-specific TCR clones in patients with same TCR sequence as pp65 peptide-expanded cells. Strikingly, we observed a phenotypic heterogeneity in the same TCR clones (Figure S7G–H). This data revealed an intra-clonal phenotypic diversity underlying the same TCR clones, suggesting that the T cell state may be impacted by local microenvironment beyond TCR specificity.
Phenotypic transition and clonal divergence of CMV-specific T cells upon ex vivo antigen peptide-induced expansion
To understand how antigen-specific expansion impacts the molecular phenotypes of anti-viral T cells, we compared the ENTER-seq of CMV-specific T cells prior and post peptide-induced expansion. We observed a phenotypic transition of pp65-specific T cells from TEMRA to TEM upon antigen-specific expansion, which was validated in both donors by flow cytometry (Figure 7H–I). Next, we utilized the TCR sequence as a “natural” barcode to track the cell state of each dominant T cell clone prior and post expansion. We found that the IFN-I ISG and cytotoxicity scores are highly heterogenous at rest in each T cell clone in patients, and there was a loss of type-I IFN ISG gene expression and upregulation of cytotoxic genes in all clones upon expansion (Figure 7J). Conversely, antigenic activation can also evoke inter-clonal phenotypic diversity in T helper 2 (Th2) cytokine gene (e.g. IL13 and IL4) expression (Figure 7J, Figure S7I). Thus, distinct TCR may recognize the same antigen differently to drive diverse transcriptional programs and cell states.
Together, our data revealed anti-CMV T cell state transition in cytotoxicity and type-I IFN response from TEMRA to TEM T cells, accompanied by upregulation of Th2 cytokine genes in specific T cell clones after peptide induced expansion (Figure 7K). Thus, ENTER-seq enabled a systematic dissection of T cell specificities, resting cell states, and antigen-evoked cell fate potentials.
DISCUSSION
Multifaceted viral platform for ligand display, cargo delivery, and interaction recording
Rather than collecting multiple single-purpose technologies, ENTER offers users one platform that solves many important problems. ENTER has advantages compared to platforms that decode TCR specificity (Table S1). The glycosylation pattern and protein folding in yeast/phage display platform is different from mammalian system, which may interfere with the correct MHC presentation and recognition of paired TCRs 38,39,40. ENTER is built in human cells and enables human glycosylation and protein folding patterns, as evidenced by our ability to present multiple HLA-peptide combinations. In addition, soluble recombinant TCR is required for screening in yeast/phage display platforms, thus making it challenging to test diverse TCRs in parallel 41. In contrast, ENTER allows users to screen clinical samples, opening the door to examine the vast diversity of human TCR repertoire. ENTER also has advantages over T cell reporter assay because the latter cannot record the pairing of pMHC-TCR at single cell level 4. ENTER can be engineered for targeted cargo delivery, and users may choose to transiently or stably deliver cargos with the flexibility of using integration-defective particles. ENTER may have applications in gene therapy or RNA medicine as ENTER can achieve exquisite cell type specificity compared to existing modalities like AAV 42,43.
Linking ligand-receptor interaction with molecular blueprints at single-cell resolution
ENTER-seq connects ligand-receptor interactions with cell states in single cells at a massive scale. ENTER-seq for pMHC is conceptually similar to DNA-barcoded pMHC tetramers but with several potential advantages 44. ENTER is more economical than commercial DNA barcoded pMHC tetramers, and can be easily implemented in any lab (Table S2). ENTER-seq library leverages lentiviral biology that ensures 2 copies of barcoded viral RNA for each viral particle 45, allowing quantification of pMHC binding strength which was not explored in studies using DNA barcoded pMHC tetramers. Our data revealed that highly expanded TCR clones are associated with higher pMHC binding, likely resulted from higher TCR affinity to the pMHC or higher TCR density in expanded clones. Finally, ENTER is more sensitive than pMHC tetramer on a molar basis per reagent. The superior sensitivity may arise from a high valency of displayed ligands. HIV-based lentiviral particle displays 14–100 molecules of envelope protein whereas pMHC tetramers are 4 linked molecules 46. ENTER-seq of T cells pre- and post-peptide stimulation unveiled cell state transitions upon expansion and inter-clonal phenotypic diversity in response to the same antigen. Such transitions and clonal divergence in Th2 cytokine expression might be impacted by the different TCR affinity/avidity/density to the same pMHC antigen, or different APC priming environment. Together, ENTER-seq allows investigators to record ligand-receptor specificity and read out the biological consequences of this interaction.
Translational application of ENTER in immunology and beyond
We envision several translational opportunities to build on the ENTER technology. ENTER may be used to screen immunogenic antigens or elite TCRs for the rational design of vaccine development or cancer immunotherapy 47–49. ENTER may also be applied to screen BCRs that target viral antigens, facilitating the development of therapeutic antibodies 50. Finally, ENTER enables targeted delivery of genetic payloads in antigen-specific T and B cells, which might be applied to reinvigorate exhausted anti-tumor T cells without triggering the immune-related adverse events, or deplete autoreactive T or B cells to treat autoimmunity 51–53. If ENTER may be extended to additional receptor-ligand pairs, such as G-protein coupled receptors, adhesion molecules, or protocadherins, ENTER may be used to address cell-cell connectivity beyond the immune system.
LIMITATIONS OF THE STUDY
In this work, ENTER presented pMHC as a single chain where any desired peptides are covalently linked with MHC. This presentation does not reflect endogenous antigen presentation pathway that can select and load optimal peptides on MHC. Computational prediction of peptide binding affinity to MHC and identification of MHC-bound peptides by mass spectrometry will facilitate the prioritization of optimal peptides to generate pMHC displayed ENTER viruses 54,55. In principle, ENTER may display any peptide antigen to decode a large pool of pMHC-TCR interactions from primary T cells. However, the current version is limited by its throughput in library generation. In the future, automation of ENTER cloning, transfection, and scalable lentiviral packaging strategy 56 are needed to increase the throughput. Future work on improving viral titer and pooled generation of ligand-displayed viruses will significantly expand the screening scale to uncover unknown ligand-receptor interactions. The largest protein ligand displayed on ENTER is ~120 KD (truncated HER2); larger ligands need to be tested. We engineered viruses as integration-defective and ligand-displayed viral like particles (VLP) for ENTER-seq thus we could not quantify them by conventional multiplicity of infection. Pooled VLP quantification could be achieved by viral RNA sequencing to quantify ligand containing reads. Lastly, antigen-specific gene delivery to edit immune receptor repertoire is not yet achieved in primary cells, which may be a fruitful application of this technology.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Howard Y. Chang (howchang@stanford.edu).
Materials Availability
The ENTER plasmids are available upon request to the lead contact with a completed Materials Transfer Agreement.
Experimental models and subject details
Cell lines
Raji, Ramos, and Jurkat related cell lines are cultured in RPMI supplemented with 10% FBS (Invitrogen) and 1X pen/strep. HEK 293T related cells are maintained in DMEM supplemented with 10% FBS and 1X pen/strep. HLA-KO HEK 293T cells were generated by electroporation of Cas9 RNP targeting HLA-A, HLA-B, and HLA-C allelles and further sorted HLA-KO cells based on surface expression of HLA-A/B/C. Ramos cells are obtained from Dr.Daniel Lingwood’s lab. Jurkat TCR negative −76 cells and Jurkat expressing CMV pp65 TCR, and flu-m1 TCR were obtained from Dr. Mark Davis’s lab. To generate stable cell lines including BCR and TCR expressing cells, Ramos or Jurkat cells were infected with viruses, and selected by sorting or using drug after 4–5 days. NY-ESO-1 TCR Jurkat and anti-RBD-BCR Ramos cells were infected with red mScarlet virus and selected with puromycin to generate mScarlet red fluorescent labeled cell lines
Human peripheral blood
Buffy coats from healthy donors were obtained from Stanford Blood Center with consent forms. LRS chambers from 4 HLA-A2+ CMV seropositive donors were obtained from Stanford Blood Center with consent forms.
METHOD DETAILS
Plasmid cloning and construction
Primers were ordered from IDT DNA technologies, and gene fragments were synthesized by twist biosicence and IDT. All the constructs were made by gibson assembly (New England Biolabs) in general. Briefly pMD2.G (addgene#12259) was digested with EcoRI to remove wild-type VSV-g gene fragment. It assembled with mutated VSV-g (K37Q, R354Q) introduced by PCR primers to generate the VSV-g double mutant. psPax2 (addgene#12260) was digested with BsiWI and SphI to fuse eGFP after MA. To generate packaging vector with NC-eGFP/NC-mNeon fusion, psPAX2-D64V-NC-MS2 (addgene#122944) was digested with SphI and BspEI sequentially. Then part of gag and eGFP or mNeon were assembled together with backbone. GFP-VPR is obtained from Addgene (#83374)
To generate HPV16 L2 antigen specific BCR, light chain and heavy chain were amplified separately from vector JWW-1 (addgene#66748) and connected by a 2A peptide. Then it was inserted into a piggybac vector after CMV promoter (PB-CMV), after which, a PDGFR transmembrane (TM) domain and 2A-mCherry were added to express this antibody on cell surface. Anti-Her2 BCR was cloned in the same way from the source Trastuzumab vector (addgene#61883). To generate anti-SAR2-RBD BCR, DNA fragments encoding the light and heavy chain of a RBD antibody (Protein Data Bank under accession number 7BWJ, 50) were codon optimized and synthesized (Twist Bio). Afterward, a signal peptide was added to each chain and heavy chain was further extended to full length with a human IgG1 Fc and a PDGFR TM sequence. The BCR was then inserted into a lentiviral vector driven by a SFFV promoter with hygromycin resistance.
Single chain format of NY-ESO-1 TCR (Clone 1G4 wild-type (1G4wt) and its mutated version (a95:LY) with high affinity, alpha and beta chain in tandem linked by a 2A peptide, 65) was synthesized and inserted into a lentiviral vector with hygromycin resistance. TCR5, which binds to a p5 peptide from CMV virus was amplified from alpha (addgene#164999) and beta chain (addgene#165000) and made into single chain form as with NY-ESO-1 TCR above.
For displaying antigen and HLA peptide complex on viral surface, we first generated a cloning lentivirus vector with a strong CMV promoter, multiple cloning sites and the WPRE element to enhance the expression. CD19-CAR vector was generated by inserting a scFv CD19 (kindly provided by Mackall lab) with a CD8 stalking linker and TM into the lentiviral plasmid followed by 2A-puromycin and 2A-eGFP. We replaced scFv CD19 and TMs to generate other antigen candidates including HPV-L2 antigen, CD40L and CD40L mutant that contains two point-mutations (K142E, R202E), leading to decreased binding affinity towards CD4066. For TM domain screening, we swapped TM with 10 alternatives in the HPV-L2 antigen viral vector (Table S3). DNA fragment of SAR2 spike RBD domain was synthesized and inserted the lentiviral expression vector followed by CD8 stalking linker and TM domain similar to the above description. For Her2 display, truncated Her2 (a1-a700) fragment including its native TM and additional 55 aa cytoplasmic tail was amplified from WT HER2 (addgene#16257), and inserted into the above vector.
To display MHC-peptide complex, we built a single chain vector, which consists of a signal peptide, antigen peptide, a G4S linker, B2M, a second G4S linker and HLA allele in tandem. DNA that encodes human growth hormone signal peptide to beta2 microglobulin was synthesized and inserted into lentiviral vector together with HLA allele. Here HLA allele A0201 was amplified from addgene vector #119052, and allele A0101 was from addgene #165009. Two cysteine mutations were introduced to stabilize the peptide binding by a bisulfide bond between Y84C of HLA allele and G2C that lies in the G4S linker after peptide. To adapt it to 10xgenomics sequencing planform, we further inserted a 10xTSO sequence (Table S5) in the linker between B2M and HLA encoding amino acids SHIRN and a 10xPCR handle in 5’UTR after CMV promoter (Table S5). A cloning vector was built by replace antigen peptide with 2 esp3I sites, where various HLA peptides (Table S4) can be inserted conveniently.
Various vectors were generated for delivery purpose. We first replaced the VSV-G in pMD vector with different envelope proteins such as RBD, HER2, pp65-HLA-A2 in the same approach as VSV-g mutant. Next, we made cargo delivery vector, where cargos such as HSV-TK-2A-egfp, and eGFP only were driven by Ef1a promoter in a lentiviral vector. For shRNA delivery, we put different shRNA under human U6 promoter in a lentiviral vector containing eGFP and puromycin as fluorescent and selection markers. mScarlet transgene was inserted after EF1 short promoter in a lentiviral vector with puromycin resistance for labeling cells with a red fluorescent protein.
Transfection and lentiviral production
To generate regular lentivirus for cell line infection and production, per 6-well, HEK293T were transfected with a viral expression vector (2ug), pMD2.G (VSV-G wild type) (1ug), and psPax2 (2ug) with lipofectamine 3000. The media was changed one next day, and viral supernatant were collected twice at 48 hr and 72hr respectively. The virus was concentrated with 4x Lenti-X according to manufacturer’s protocol, and stored at 20x concentrated in −80°C. For making specific receptor targeting and integrating virus, VSV-G mutant was used instead. To produce antigen displaying virus that can be detected with fluorescence without integration, VSV-G mutant and fluorescent protein fused version (NC-eGFP or NC-mNeon) of psPAX2-D64V (D64V mutation on integrase) vectors were mixed with antigen expressing vector according to above ratio to transfect the HEK293T cells. For pMHC displayed viruses, HLA-KO HEK293T cells were used for transfection. Viruses were collected, concentrated to 40x, and stored in −80°C. To generate lentivirus for cargo delivery, per 6-well, HEK293T were transfected with a cargo expression vector (1.6ug), pMD2.G VSV-g mut (0.8ug), psPax2 (1.6ug), and envelope plasmid (1ug) with lipofectamine 3000. virus was collected as described above, concentrated to 40x, and stored in −80°C before use. Lentiviral titer was determined by Lenti-X GoStix Plus kit (Takarabio) according to the manufacture’s protocol.
Lentiviral infection and viral incubation assay
30uL concentrated lentiviruses are added into 250K target cells in 12 well plate. 3 days later, GFP signal is measured by flow cytometry. For viral incubation assay, 200K target cells were collected in tubes and the supernatant is removed after centrifugation. The cell pellet was resuspended in 30uL concentrated GFP fused lentiviruses and incubated at 37°C. After 2hr incubation, cells are stained with flow cytometry antibodies for 10 min at 4°C (if needed), washed by RPMI medium for twice, and finally subjected to flow cytometry.
To determine if the TCR affinity to pMHC impact the binding of pMHC display viruses to TCR expressing T cells, a TCR-T cells line (1G4 wild-type) that recognize NY-ESO-1 antigen variants with different known TCR affinities (Kd range from 7–85 uM) 67 was generated. The NY-ESO-1 TCR T cell line utilized in previous experiments (Figure 2A–C) is very similar to 1G4wt TCR T cell line except mutations on its TCR (a95:LY mutation), resulting in very high binding affinity (Kd=0.7uM) to NY-ESO-1157–165 antigen 65. ENTER virus was generated to display different antigen peptide variants such as wild-type peptide (SLLMWITQC), L3A mutant (SLAMWITQC), and T7A mutant (SLLMWIAQC). To quantify binding of ENTER viruses displaying pMHC antigen variants with different TCR affinity in Figure 2D, we normalized the viral titer and incubated 100K TCR T cells (1G4 wild-type or a95:LY mutant) or off-target CMV-pp65 TCR+T cells with a titration (4ng, 20ng, 40ng p24 level) of ENTER viruses displaying antigen variants. Upon 2-hour incubation, cells were washed and subjected to flow cytometry to quantify GFP+ cells.
To compare the sensitivity of pMHC displayed ENTER viruses and pMHC tetramers per molar basis of each reagent in Figure 2C, we incubated 100K NY-ESO-1 TCR+ T cells with 2×108 ENTER viruses (20ng p24) displaying NY-ESO-1157–165 antigen or a range of NY-ESO-1157–165 pMHC tetramers (2×108 to 8×109) for 2 hours. The calculation of molar number for each reagent is shown below. Since 104 viral particles contain 1 pg p24 protein, viruses with 20ng p24 = 20*1000*104= 2×108 viral particles. For pMHC tetramer, the molecular weight is around 500 KD (PE-streptavidin:~300KD, pMHC tetramer:~200 KD (50KD monomer*4)). Thus, 1 ug pMHC tetramer = 1ug*106*(6.02×1023)/500/1000 = 1.2×1012 tetramers.
Viral structural protein-GFP fusion optimization
To fuse GFP protein to the viral structural proteins, we selected viral proteins including matrix protein (MA), nucleocapsid protein (NC), and HIV accessory protein named viral protein R (VPR) given following reasons. During viral assembly and synthesis, MA and NC are processed from Gag precursor protein which can be assembled in cis as 3000 copies of MA or NC per viral particle 68,69. VPR can be incorporated in trans into the viral particle via interaction with Gag protein as 500 copies of VPR per viral particle 70. We fused the GFP protein with viral structural proteins to track ligand displayed viruses. We measured the transient viral binding/entry into CD19+ Raji cells using CD19-scFv displayed viruses carrying GFP protein instead of viral integration to express GFP by flow cytometry.
Viral binding and fusion assay
20uL CD19-scFv displayed GFP viruses were incubated with 200K Raji B cells at 4°C or 37°C for 2 hours. Cells were washed twice and subjected to 0.5 mg/mL proteinase K treatment for 15 min at 37°C which will digest cell surface binding viruses. Cells prior and post proteinase K treatment were subjected to flow cytometry to quantify the percentage of GFP positive cells.
Immunocapture assay
10uL Protein G Dynabeads were incubated in 1mL blocking buffer (PBS with 0.1%BSA) for 20 min at room temperature. 2ug anti-CD40L antibody (Cat#157009, Biolegend) or anti-VSV-G antibody (clone 8G5F11, Millipore sigma), or IgG antibody were added into beads with 100uL blocking buffer and rotated for 30 min at 4°C. The antibody conjugated beads were washed three times and the supernatant was removed. 30uL CD40L displayed viruses were added into beads with 30uL blocking buffer and rotated for 1 hour at room temperature. 5uL CD40L displayed virus from the same batch was prepared as input samples. The beads were washed three times and the supernatant was removed. 100uL Trizol was added into beads or input sample and subjected to RNA extraction by Zymo Quick-RNA Miniprep Kit. RT-qPCR was performed using Stratagene Brilliant II SYBR Green QRT-PCR Master Mix (Agilent).
Transmembrane domain optimization for viral display of B cell antigens
To display B cell antigens which do not contain their native TM domains on the viral surface, we sought to engineer a heterologous TM domain for optimal surface display of B cell antigens. To select candidate TM domains for viral surface display, we took advantage of the unique ability of HIV-1 viruses to incorporate host proteins on the viral surface during virus budding. Nascent HIV-1 viruses can selectively incorporate certain host TM proteins while excluding other abundant host surface proteins during viral assembly and budding 71. A list of highly abundant host TM proteins that are incorporated into viral surface from previous literature using mass spectrometry of viruses, immunocapture assay, and flow virometry method 20,72–75 was selected. This list (Table S3) of host TM proteins includes MHC class I and II molecules (HLA-DRA, HLA-DRB, HLA-A2), adhesion molecules (ICAM1, CD43, CD162, CD62L), and integrin family members (CD49d, LFA-1) along with TM from VSV-G. To determine the specificity and efficiency of viral display of B cell antigens with these diverse TM domains, ENTER viruses was generated expressing a B cell antigen epitope derived from human papillomavirus (HPV) minor capsid antigen L2 (HPV16 L217–36) and fused with TM domains from the prioritized list. Next, a BCR expressing B cell line that specifically targets this HPV16 L2 B cell epitope19 was made. After incubating the TM domain fused and B cell epitope displayed viruses with B cells either expressing HPV-BCR (on-target) or without any BCR (off-target), the percentage of GFP+ B cells was quantified with flow cytometry to measure the efficiency and specificity.
Targeted cargo delivery by ENTER virus
Cells were incubated with virus in media with 6ug/ml of polybrene as described above. Delivery efficiency and specificity were assessed after 3 days with flow-cytometry (Attune NxT). When needed, the cells were first stained with PeCy7 anti-human IgG (for B-cells, clone G18–145, BD bioscience), or APC anti-human CD3 (for T-cells, clone HIT3a, Biolegend) before flow cytometry analysis.
For HSV-TK cell killing assay, two population of cells with one labeled by mScarlet (off-target) and one non-fluorescent (on-target) was mixed at 1:1 ratio and incubated with virus. Here CMV-pp65 TCR+ T cells (on-target) and NY-ESO-1 TCR+ T cells (off-target) that express mScarlet were mixed, while for B-cell delivery, HER2 BCR+ B cells as on-target cells and RBD BCR+ B (mScarlet+) as off-target cells were mixed. After 3 days, ganciclovir (GCV, Invivogen) was added to a final concentration of 0.1ug/ml, which was counted as day 0. Cell media and drug were refreshed every 3 days. After 2 days, 300ul of cell culture were taken every day, stained with IgG or CD3 before analyzed by flow cytometry. The ratio of live on-target cells over off-target cells were calculated and plotted over the days (normalized to Day 0). Alternatively, raw count of live cells for targeted or NT population at day 4 of treatment were also compared between TK and eGFP only delivery.
For apoptosis assay with FAS shRNA delivery, Jurkat T cells were infected different shRNAs, stained with PE-FAS/CD95 (Biolegend) to compare the effect of shRNA knockdown. Mixture of CMV-Jurkat (on-target) and mScarlet+ NY-ESO-Jurkat (off-target) were incubated with shRNA virus. After 5 days, anti-FAS antibody (Clone CH11, Millipore Sigma) was added at 0.25ug/ml to induce apoptosis. The cells were collected after 14 hr, and stained with APC anti-Annexin V (Biolegend) and 7-AAD according to manufacturer’s protocol. Then the samples were analyzed with a flow cytometry (BD LSR II). We first gated on 7-AAD-low and Annexin V-low population. Then the ratio of transduced on-target cells over off-targeted cells were compared between FAS shRNA and control shRNA to generate a bar graph with normalization.
ENTER virus incubation with mixed cell population
For T cell mixing experiment, Flu-m1 TCR expressing Jurkat T cells were labeled by CellTrace Violet dye (#C34571, Thermofisher) according to manufacturer’s protocol. Violet labeled Flu-m1 TCR+ T cells were mixed with NY-ESO-1 TCR+ T cells at diverse ratios including 1:1, 1:10, 1:100, 1:1000. The mixed T cells were incubated with 40uL concentrated HLA-A2-Flu antigen displayed GFP viruses for 2 hr at 37°C. T cells were stained with CD3-APC (clone HIT3a, BioLegend) antibody, washed twice, and subjected to flow cytometry. For B cell mixing experiment, Spike-RBD BCR expressing Ramos B cells were labeled by CellTrace Violet dye and mixed with HPV-L2 BCR expressing Ramos B cells at diverse ratios including 1:1, 1:10, 1:100, 1:1000. The mixed cells were incubated with 40uL concentrated RBD-antigen displayed GFP viruses for 2hr at 37°C. B cells were stained with IgG-PE-Cy7 antibody (clone G18–145, BD Biosciences), washed twice, and subjected to flow cytometry. The metrics are calculated below:
Human primary immune cell isolation and activation
Peripheral blood mononuclear cells (PBMC) from healthy donors were isolated using Lymphoprep (Cat# 07811, STEMCELL Technologies) density-gradient centrifugation and cryopreserved and stored in −80°C. B cells were purified from thawed PBMCs by negative selection using EasySep Human B Cell Enrichment Kit (Cat#19844, STEMCELL Technologies) according to the manufacturer’s protocol. Isolated B cells were cultured in IMDM medium supplemented with 10% FBS and 55 mM beta-mercaptoethanol at 1×106 cell/mL and activated by CellXVivo Human B cell expander (1:250 dilution, R&D system) and 50 ng/mL IL2 (Cat#200–02-10ug, PeproTech) for two days. PBMCs from CMV seropositive patients were isolated and stored as above. CD8+ T cells were purified from thawed PBMCs by negative selection using EasySep Human CD8+ T Cell Enrichment Kit (Cat#19053, STEMCELL Technologies) according to the manufacturer’s protocol.
Peptide enrichment of antigen-specific T cells
Short 9-mer peptides encoding CMV epitopes (compatible to HLA-A2 allele, Table S4) were synthesized by Elimbio in lyophilized powders. The peptides were dissolved in DMSO in 10mg/mL. PBMC were isolated from donor blood described as above. PBMC were cultured in T cell medium (RPMI medium supplemented with 10% FBS, 1X penstrep, 100mM HEPES, 55 mM beta-mercaptoethanol). Individual peptide (10ug/mL) or pooled peptides (1ug/mL for each peptide) were added into PBMC for culturing 10 days in T cell medium. 50ng/mL IL-2 were added every two days. The peptides are processed and presented by autologous antigen-presenting cells which will stimulate CMV antigen-specific T cells for later expansion. To test the specificity of our ENTER viruses on peptide-enriched T cells, we expanded T cells using peptide pp65495–503 and then stained these T cells with pp65495–503 displayed mNeon viruses followed by pp65495–503 PE-tetramer. After peptide enrichment, PBMCs were incubated with viruses and/or PE-tetramer and then analyzed by flow cytometry.
Flow cytometry
B cells were incubated with viruses for 2 hr and then stained with Human TruStain FcXTM (Fc Block, BioLegend), CD19-APC (clone HIB19), IgG (clone G18–145, BD Biosciences) and CD20-V450 (clone L27) antibody in cell staining buffer (BioLegend) for 10 min at 4°C. Jurkat TCR-T cells were incubated with viruses for 2 hr and then stained with CD3-APC (clone HIT3a) and PE labeled pMHC tetramers (NIH tetramer core) for 30 min at 4°C. For ENTER-seq of primary T cells, cells were incubated with viruses for 2 hr and then stained with human Fc Block, CD3-APC, CD8-BV711 (clone SK1), tetramer-PE if needed, and viability dye for 30 min at 4°C. After staining, cells were washed twice by cell straining buffer and analyzed by flow cytometry (Attune, Thermofisher) or sorted by Aria FACS machine. All antibodies are from BioLegend if not specified. Tetramers are from NIH tetramer core.
RNA-seq experiment and analysis
CMV pp65-TCR+ T cells were incubated with 30uL pp65495–503 displayed ENTER viruses or 1ug pp65495–503 tetramer for 2 hours. Cells were washed twice and subject to RNA extraction. RNA was extracted using Quick-RNA Miniprep Kit with on-column Dnase digestion (Zymo Research). At least 100ng RNA was used to prepare the RNA-seq library using TruSeq® Stranded mRNA Library Prep Kit (Cat# 20020594, Illumina) for each sample following the manufacturer’s instruction. The library was sequenced on an Illumina Nextseq to generate 2X150 paired-end reads. RNA-seq reads were mapped to the human genome (hg19) using STAR with default parameters (--outFilterMultimapNmax 1 --alignEndsType EndToEnd --outSAMattributes NH HI NM MD). Quantification of aligned reads at the gene level was performed by HTseq count with default parameters (--stranded=reverse –additional-attr=gene_name). Raw counts were used to identify differentially expressed genes (DEG) using DESeq2 with size factor normalization and DEGs were identified if Benjamini & Hochberg adjusted p-value<0.01 and over 2-fold change difference of gene expression. We identified 28 genes that are differentially expressed between ENTER group and tetramer group (2-fold change and adjusted P value<0.01). Notably, all 28 genes are significantly upregulated in ENTER virus-treated cells and are mainly associated with TCR activation (CD69) and transcription factors induced by TCR signaling (FOS, NR4A1, NR4A3, EGR1 etc).
ENTER-seq workflow of mixed TCR-expressed Jurkat T cells
To mimic a real-life T cell population, 10% of T cells with TCR recognizing NY-ESO-1157-165 antigen were mixed with 90% of T cells with TCR recognizing CMV pp65495–503 antigen, and then incubated with pooled viruses displaying NY-ESO-1157–165 antigen or pp65495–503 antigen. After 2 hours incubation, the GFP+ cells were sorted on BD Aria II afterward. Commercial 10xgenomics 5’ RNA kit was customized to read out HLA peptide, TCR, and transcriptome simultaneously per single cell. Immediately after sorting, the cells were washed once at 4°C with PBS+0.4% BSA, mixed with RT (reverse transcription) mixture spiked in with customized TCRalpha RT primers at 0.1uM, and loaded to 10x chromium. The cDNA was amplified and cleaned up to generate the transcriptome according to manufacturer’s protocol.
During cDNA cleanup, the supernatant that contains shorter fragment of HLA peptide and TCR information was further mixed with SPRISelect beads to 0.9x, and cleaned up. The library that encodes HLA peptide were generated through 2 round of nested PCRs and a final round of indexing PCR. First, we enriched the HLA peptide cDNA by 8 cycles of PCR (98°C for 45 min, then 8x of 20sec at 98°C 20 sec, 20 sec at 59°C and 30sec at 72°C) with 0.5uM 10x_5pRNA_Fw, and HLA_nested_Fw. After cleanup, 5ul of elution was used the second round of PCR with 0.5um nested primer and Illumina adapters P7_Tru_HLA_fw and P5_adapter primer as above. Last, take 5ul of elution to generate final library with Illumina Truseq based index primers. The above primers were designed in a way compatible with dual index, thus either customized index primer or 10x dual index primers can be used here.
To read out TCR information in Jurkat cell lines, we generated the library that covers VDJ part of TCR alpha to infer cell’s TCR identity. First, the TCR DNA were enriched by nested PCR (specifically, 98°C for 45 min, then 8x of 20sec at 98°C 20 sec, 20 sec at 59°C and 30sec at 72°C) with 0.5uM 10x_5pRNA_Fw and 0.5uM mix of nyeso_TRAc_rev and hs_TRAc_rev targeting two different TCR. Next, take 5ul of elution to run the second round of nested PCR with Illumina adapter (a mixture of P7_TRAc_nyeso_Rev and P7_TRAc_hs_Rev) and P5_adapter primer for 8 cycles, followed by a final index PCR similar to HLA libraries.
ENTER-seq analysis of mixed TCR-expressed Jurkat T cells
The libraries are sequenced using Illumina’s Novaseq and Miseq platforms. Transcriptiome fastq files were analyzed using 10X’s cellranger to provide single cell barcodes. The fastq files of TCR libraries were mapped to TCR alpha chain with custom python script. The UMI count of each type TCR per cells’ barcodes was calculated. To exclude doublet, we require, per gem barcode, the UMI count for the dominant TCR is at least 10 times more than those non-dominant TCR species. Next, HLA peptide reads were processed using cellranger count with peptide sequence as feature reference. Downstream analysis and plots were generated with matplotlib package in python.
ENTER-seq workflow of primary T cells prior or post CMV antigen peptide-induced expansion ex vivo
Peptide stimulated donor PBMC were collected and stain with a mixture of 12 viruses displaying CMV antigen epitopes including IE181–89, IE1316–324, US150A152–161, US874–82, UL100200–208, UL46100–108, pp65417–425, pp65325–333, pp65188–196, pp65120–128, pp65495–503, and pp6514–22. The peptide sequences for CMV antigens were listed in Table S4. After 2 hr, cells were also stained with barcoded antibodies CD45RA, CD45RO, and IL7R (Biolegend totalseq-C, cat# 304163, cat# 304259, cat# 351356), live dead dye, CD3-APC and CD8-BV711 for 20 min on ice. Sample from each donor is also stained with unique hash-tag barcoded antibody (Biolegend totalseq-C, cat# 394661, cat# 394663). After two washes, CD8+ CD3+ GFP+ cells were sorted to run on 10x genomics platform using 5’ RNA and VDJ kits according to manufacturer’s protocol. Here per sample, we obtained 10x gene expression library, VDJ library, and feature barcoded CITE-seq library according to manufacturer’s protocol. In addition, HLA peptide library was generated in the same way as described above. Final libraries are sequenced on either Illumina Miseq, Nextseq 550 or Novaseq 6000. For ENTER-seq of primary T cells isolated directly from patient blood samples without peptide stimulation/expansion, we first purified total CD8+ T cells from cryopreserved patient PBMC samples using EasySep human CD8+ T cell isolation kit (Cat#17953, STEMCELL Technologies) following the user’s protocol. Next, we prepared a mixture of top 3 ENTER pMHC viruses (pp65495–503, US874–82 and UL100200–208) for 2 hours at 37°C. Later steps of antibody staining, flow cytometry sorting, and10x genomics library generation are the same as ex vivo expanded T cells described above.
ENTER-seq analysis of primary T cells from CMV seropositive donors
The scRNA-seq reads were aligned to GRCh38 genome and quantified using cellranger count (10x genomics). The CITE-seq reads were processed using cellranger count with antibody oligo barcode as feature reference. The TCR-seq reads were mapped to VDJ compatible reference (refdata-cellranger-vdj-GRCh38-alts-ensembl-5.0.0) using cellranger vdj (10x genomics). HLA peptide reads were processed using cellranger count with peptide sequence as feature reference.
The later analyses for single cell RNA-seq and CITE-seq were performed using SCANPY63. Cells with less than 200 genes detected or greater than 10% mitochondrial RNA reads were excluded from analysis. Doublet cells were removed using CITE-seq analysis of barcoded hashtag antibody labeling donor origins. For cell clustering, raw UMI counts were first normalized by total counts to correct library size and then log-normalized. Variable genes were called using scanpy.pp.highly_variable_genes() with default parameters. Variable TCR genes were removed before principal component analysis (PCA) to prevent clustering bias from variable TCR transcripts. Next, we regress out effects of total counts per cell and the percentage of mitochondrial genes, and then scale the data to unit variance. Scaled data were used as input into PCA analysis on the basis of variable genes (without TCR genes). Clusters were identified using Leiden graph-clustering method with the first 40 principal components. To determine if ENTER virus-induced gene expression can impact T cell state and clustering, we performed Leiden graph-clustering before and after removal of 28 ENTER virus induced genes that are identified from bulk RNA-seq data. UMAP plots were generated using scanpy.tl.umap() and scanpy.pl.umap() with default parameters. Heatmap plots were generated using scanpy.tl.heatmap() with raw value or z score scaled gene expression.
Initial clusters were annotated using expression of known markers including CD3E, CD4, CD8A, CD45RA, CD45RO, CCR7, GZMB, and KLRB1. All CD8+T cells are CD3E+CD8A+CD4−. Naïve T cells are CD45RA+CCR7+. Central memory T cells (TCM) are CD45RA−CCR7+. Effector memory T (TEM) cells are CD45RO+CCR7−. Terminal effector cells re-expressing CD45RA (TEMRA) are CD45RA+CCR7−CD45RO−, MAIT cells are KLRB1+CXCR6+TRAV1–2+. The gene score was calculated using scanpy.tl.score_genes() with ctrl_size=500 and use_raw=True. The gene set of cytotoxic genes were curated from well-established cytotoxic molecules. The T cell exhaustion genes, T cell activation genes, and type-I IFN response genes were selected from previous literature 76. For primary CMV-specific T cells in patients without peptide stimulation, further subset clustering showed heterogeneity of TEMRA populations in CMV-specific T cells. For example, in CMV-specific T cells, TEMRA #1 cluster contains high expression of cytotoxic genes like IFNG, TNF, and PRF1 but not GZMK whereas TEMRA #4 cluster is IFNG−TNF−PRF1+GZMK+ (Figure 7C, S7A). Notably, TEMRA #2 cluster in CMV-specific T cells contains low expression of all cytotoxic genes but high expression of type-I IFN stimulated genes (ISG) such as ISG15, ISG20, IFIT1, and OASL etc (Figure 7C, S7A). Such upregulation of ISG genes reflected a specific induction of type-I IFN response in a small subset of CMV-specific T cells, which might be stimulated by local production of type-I IFN responding to CMV viruses or bystander production of type-I IFN from other pathogens in patients.
TCR relevant analyses were performed using Scirpy 64. The contig annotation files generated by cellranger vdj were used as input for TCR analysis. TCR qualities were analyzed using scirpy.tl.chain_qc(). The TCR clonotypes were defined using scirpy.pp.ir_dist() and scirpy.tl.define_clonotypes() with default parameters based on CDR3 nucleotide sequence similarity. The TCR clonotypes were visualized on a network using scirpy. tl.clonotype_network() with min_cells=3. The CDR3 amino acid compositions were generated using weblogo 77. Cells with more than 5 raw count of HLA peptides for any individual antigens were labeled as antigen-specific T cells. We quantified the antigen peptide count per cell using log(count+1) transformation. We divided the pp65-specific T cells into diverse clonally expanded cells with clone size >50, or > 10, or >=1. The distribution of antigen peptide count bound per cell in different clonally expanded T cells were showed in a violin plot. Using dominant TCR clonotypes as a barcode, we generated 2D density plots using kdeplot() function to show cytotoxic gene score and type-I IFN gene score of T cells with same TCR sequence before and after antigen-induced expansion. All plots (e.g. violin plots, scatter plots, density plots and bar plots) were generated by Python matplotlib and seaborn.
Using TCR as a barcode, we identify false negative T cells in ENTER negative population who share the same TCR sequences with the dominant antigen-specific T cells. Similarly, we can identify false positive antigen-specific T cells such as pp65-specific T cells by tracking TCR sequences between pp65-specific T cells with dominant clones and other CMV-antigen specific T cells or ENTER-negative T cells. We further calculated the false negative rate (FNR) and false positive rate (FPR) for top 3 antigen epitopes. For pp65495–503-specific T cells, FNR=0.19%; FDR= 0.36%. For US874–82 -specific T cells, FNR=2.73%; FDR=0.18%. For UL100200–208 -specific T cells, FNR=0%; FDR=0.07%.
QUANTIFICATION AND STATISTICAL ANALYSES
Statistical analyses are performed by Python or Graphpad Prism. Information on specific statistical tests is listed in the figure legends and/or method details. Data are represented by mean+/−SEM in bar plots as indicated in the figure legends. P values are listed on figures or figure legends.
DATA AND CODE AVAILABILITY
All sequencing data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of publication. Accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Supplementary Material
A. Flow cytometry analysis of the viral binding and fusion of scFv-CD19 displayed viruses on Raji B cells at different temperature before and after proteinase K treatment.
B. Bar plot showing the percentage of GFP+ cells from Figure S1A.
C. Schematic view of experimental set up. CD40 expressing Raji B cells were incubated with GFP viruses displaying either wild-type CD40 ligand (CD40L) or CD40L mutant (K142E, R202E) with decreased binding to its cognate receptor CD40.
D. Flow cytometry analysis of GFP signal in Raji B cells upon incubation with either wild-type CD40L or mutant CD40L displayed GFP viruses.
E. Bar plot showing the percentage of GFP+ cells from Figure S1C–D.
F. Schematic view of immunocapture assay. Magnetic beads were conjugated with anti-CD40L, anti-VSVG, and IgG antibodies and then incubated with CD40L displayed viruses. Viruses that were immunocaptured were subjected to viral RNA isolation and qRT-PCR of CD40L.
G. Bar plot showing the qRT-PCR result of enrichment of ENTER viruses by different antibody conjugated beads as in Figure S1F.
Data are represented as mean +/− SEM in B and G from two biological replicates, and in E from triplicates.
P-values were calculated by unpaired t-test. n.s. not significant p>0.05; * p<0.05; *** p<0.001
A. Flow cytometry analysis of HLA-A2 and B2M surface expression in wild-type HEK293T, HLA-KO HEK293T, and HLA-A2 reconstituted HLA-KO HEK293T cells.
B. Flow cytometry plots of CMV-pp65495–503-TCR T cells upon 2-hour incubation of HLA-A2-peptide displayed GFP viruses followed by staining of PE-tetramers. M1(58–66) (flu antigen) displayed viruses and M1(58–66) tetramers are negative controls.
C. Histogram plots (left) and Bar plots (right) showing tetramer intensity and CD3 surface expression of CMV-pp65495–503-TCR T cells.
D. Schematic view of experimental design (top) and flow cytometry analysis (bottom) of T cell mixing experiments. Flu-m158–66-TCR T cells were labeled by Cell Trace Violet dye and then mixed with NY-ESO-1157–165-TCR T cells at different ratio. Mixed T cell population was incubated with HLA-A2:m1 displayed GFP viruses for 2h and then subjected to flow cytometry. Representative flow cytometry plot showing mixing of two T cell population and GFP signal of T cells gated on Violet+ and Violet− population.
E. Bar plots showing sensitivity (left) and specificity (right) of ENTER from Figure S2D.
Data are represented as mean +/− SEM in C from two biological replicates and in E from triplicates.
A. Flow cytometry analysis of GFP+ cells among HER2-BCR+ B cells that were incubated with HER2 displayed viruses or RBD displayed viruses.
B. Bar plot showing the percentage of GFP+ cells as in Figure S3A.
C. Schematic view of experimental design (top) and flow cytometry analysis (bottom) of B cell mixing experiments. RBD-BCR+ B cells were labeled by Cell Trace violet and mixed with HPV-BCR+ B cells at different ratios. Mixed B cell population was incubated with GFP viruses displaying RBD antigen fused with ICAM1 TM domain and then subjected to flow cytometry. Representative flow cytometry plot showing mixing of two B cell population and GFP signal of B cells gated on Violet+ and Violet− population.
D. Bar plot showing the sensitivity of ENTER from Figure S3C.
E. Bar plot showing the specificity of ENTER from Figure S3C.
F. Bar plot showing the delivery efficiency of viruses with wild-type VSV-G in cell types with different antigen specificity. P-values were calculated by unpaired t-test. n.s. not significant P>=0.05.
G. Flow cytometry analysis of cargo delivery in T cell mixing experiment. mScarlet expressed NY-ESO-1 TCR+ T cells were mixed with CMV-pp65 TCR T cells and then infected with ENTER viruses displaying pp65495–503 antigen and carrying transgene containing HSV-TK and GFP.
H. Flow cytometry analysis of cargo delivery in T cell mixing experiment. mCherry expressed RBD BCR+ B cells were mixed with HER2 BCR+ B cells and then infected with ENTER viruses displaying HER2 antigen and carrying transgene containing HSV-TK and GFP.
I. Histogram plot showing the surface expression of FAS in T cells transduced with diverse FAS shRNAs or control shRNA.
J. Representative flow plot showing the Annexin V and 7-AAD gating of T cells upon anti-FAS induced apoptosis and cell death.
Data are represented as mean +/− SEM in B, D and E from triplicates, and in F from two replicates.
A. Flow cytometry analysis of GFP signal in T cells expressing TCRs (NY-ESO-1 TCR or CMV pp65-TCR) upon incubation with NY-ESO-1157–165 antigen peptide displayed GFP viruses whose viral RNA are either intact or inserted with sequencing capture tag.
B. Bar plot showing the percentage of GFP+ cells from Figure S4A.
C. Schematic view of experimental design. Donor isolated T cells were incubated with pp65495–503 displayed viruses carrying either GFP or mNeon fluorescence proteins and then stained with pp65495–503 tetramer and other antibodies followed by flow cytometry. We first gated on CD3+ CD8+ T cells to measure pp65-specific T cells using tetramer as a positive control, and then monitored GFP signals in pp65 tetramer+ T cells.
D. Flow cytometry analysis of tetramer and GFP signal in primary human T cells from donors with CMV infection as in Figure S4C.
E. Bar plot showing the percentage of GFP+ and GFP− cells among pp65 tetramer+ T cells from Figure S4D.
F. Representative flow plot showing the GFP+ cells among pp65 tetramer+ T cells after incubation with pp65495–503 viruses or negative control viruses (NY-ESO-1157–165)
G. Schematic view of experimental design. PBMCs were isolated from CMV seropositive HLA-A2+ donors and cultured with a pool of twelve different CMV antigen peptides (10 ug/mL) for 10 days. Primary T cells prior or post peptide-induced expansion were incubated with pp65 antigen displayed mNeon viruses for 2 hours and then stained with antibodies followed by flow cytometry.
H. Representative flow plot showing co-staining of pp65495–503 tetramer and pp65495–503 displayed mNeon viruses in pp65495–503 peptide enriched T cells post 15 days expansion. These T cells are gated on live CD8+ CD3+ T cells.
I. Representative flow cytometry plot showing the percentage of GFP+ T cells (stained by 12 pooled HLA-A2:CMV-antigen mNeon viruses) from 4 different CMV seropositive donors prior or post pooled CMV peptide-induced expansion as in Figure S4G.
J. MA plot showing the bulk RNA-seq analysis of CMV-pp65 TCR T cells incubated with pp65495–503 tetramer or pp65495–503 displayed ENTER viruses. Genes with log2 fold change and adjusted p value <0.01 are highlighted in red.
Data are represented as mean +/− SEM in B from two replicates and E from triplicates.
P-values were calculated by unpaired t-test. n.s. P>=0.05 * p<0.05
A. UMAP plots showing CMV-specific T cells (yellow) and bystander T cells (blue) via clustering before or after removal of ENTER-induced gene signature.
B. UMAP plot showing 10 clusters of human CD8 T cell subsets
C. UMAP plots showing amount of surface protein CD127 from CITE-seq, and expression of genes for naïve T cells (CCR7, SELL, and LEF1).
D. UMAP plots showing gene expression of cytolytic molecules.
E. UMAP plots showing gene expression of markers for MAIT cells
F. UMAP plots showing expression of marker genes for each subset/cluster.
G. UMAP plot showing donor origin.
H. Fraction of T cell subsets from 10 clusters in CMV antigen-specific T cells (ENTER+) and bystander T cells (ENTER−), separated by donor origin.
A. UMAP plots showing the clonal expansion size of CMV antigen-specific T cells (ENTER+) and bystander T cells (ENTER−).
B. TCR clonotypes of CMV antigen-specific T cells colored by donor. Each circle represents a clonotype with identical CDR3 nucleotide sequences. The size of circle represents the number of cells in each clonotype.
C. Scatter plots showing the correlation of clonal expansion size with cytotoxic gene score.
D. Scatter plots showing the correlation of clonal expansion size with exhaustion gene score or activation gene score.
E. A summary table of pp65495–503-specific TCR clones showing convergent TCR clonotypes with identical CDR3 amino acid sequences
F. Summary tables of US874–82 - and UL100200–208 -specific TCR clones.
G. Fraction of T cell subsets in pp65495–503-specific TCR clones, separated by CDR3 clones and colored by 10 clusters.
Coefficient r and p values in Figure S6C–D are calculated by Pearson correlation.
A. UMAP plots showing the expression of key genes associated with diverse functions (type-I IFN ISG, cytotoxicity, chemokine, and transcription factors)
B. UMAP plots showing the subset clustering of primary T cells directly isolated from patient blood sample before and after removal of ENTER-induced gene signature
C. UMAP embedding density plots showing the density of CMV antigen-specific T cells for different CMV antigen epitopes.
D. Bar plot showing the percentage of US874–82-specific T cells with shared TCR before and after peptide induced expansion.
E. The number of US874–82-specific T cells isolated from fresh PBMC before expansion, separated by donor and colored by TCR CDR3 sequence. TCR sequence shared before and after expansion are circled in black.
F. A summary table of top TCR clones of US874–82-specific T cells (identified from E) with cell number and frequency metrics before and after expansion.
G. The number of T cell subsets in pp65495–503-specific TCR clones, separated by CDR3 clones and colored by 13 clusters.
H. Heatmap showing the expression of genes associated with diverse functions among different TCR clone-specific T cells.
I. Density plots showing the expression of IL13 and IL4 in different TCR clone-specific T cells before and after peptide-induced expansion.
KEY RESOURCES TABLE.
| REAGENT or | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC-anti-mouse CD40L (clone SA047C3) | Biolegend | Cat#157009; RRID: AB_2890718 |
| Anti-VSV-G (clone 8G5F11) | Millipore-Sigma | Cat# MABF2337-100UG |
| PE-Cy™7 Mouse Anti-Human IgG, Clone G18-145 (RUO) | BD Biosciences | Cat# 561298;RRID: AB_10611712 |
| APC anti-human CD3 (clone HIT3a) | Biolegend | Cat#300312; RRID: AB_314048 |
| PE anti-human CD95 (Fas) | Biolegend | Cat#305607; RRID: AB_314545 |
| Anti-Fas Antibody (human, activating), clone CH11 | Millipore-Sigma | Cat#05-201; RRID:AB_309653 |
| APC Annexin V | Biolegend | Cat#640920; RRID: AB_2561515 |
| Human TruStain FcX | Biolegend | Cat#422302; RRID: AB_2818986 |
| APC anti-human CD19 (clone HIB19) | Biolegend | Cat#302212; RRID: AB_314242 |
| V450-CD20 (clone L27) | BD Biosciences | Cat#561163; RRID: AB_10563614 |
| BV711 anti-human CD8 (clone Sk1) | Biolegend | Cat#344734; RRID: AB_2565243 |
| TotalSeq™-C0063 anti-human CD45RA (clone HI100) | Biolegend | Cat# 304163; RRID: AB_2800764 |
| TotalSeq™-C0087 anti-human CD45RO (clone UCHL1) | Biolegend | Cat# 304259; RRID: AB_2800766 |
| TotalSeq™-C0390 anti-human CD127 (IL-7Rα) (clone A019D5) | Biolegend | Cat# 351356; RRID: AB_2800937 |
| TotalSeq™-C0251 anti-human Hashtag 1 | Biolegend | Cat# 394661; RRID: AB_2801031 |
| TotalSeq™-C0252 anti-human Hashtag 2 | Biolegend | Cat# 394663; RRID: AB_2801032 |
| Bacterial and virus strains | ||
| Stbl3 chemical competent cells | Thermofisher | Cat# C737303 |
| Biological samples | ||
| Human Blood buffy coat from LRS chambers | Stanford Blood Center | https://stanfordbloodcenter.org/research/buffy-coat-from-the-lrs-chamber/ |
| Chemicals, peptides, and recombinant proteins | ||
| Ganciclovir (GCV) | InvivoGen | Cat# sud-gcv |
| CellTrace Violet dye | Thermofisher | Cat#C34571 |
| IL2 | PeproTech | Cat#200-02-10ug |
| Synthesized peptides | Elimbio | Table S4 |
| Critical commercial assays | ||
| NEBuilder HiFi DNA Assembly Master Mix | NEB | Cat# E2621L |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | Cat# M0541L |
| Lenti-X GoStix Plus kit | Takara | Cat#631281 |
| Lenti-X Concentrator | Takara | Cat#631232 |
| Brilliant II SYBR Green qRT-PCR 1-Step Master Mix | Agilent | Cat#600825 |
| Quick-RNA miniprep kit | Zymo | Cat#R1055 |
| Annexin V Apoptosis Detection Kit | Invitrogen | Cat# 88-8006-72 |
| Lymphoprep | STEMCELL Technologies | Cat# 07811 |
| EasySep Human B Cell Enrichment Kit | STEMCELL Technologies | Cat#19844 |
| CellXVivo Human B cell expander | R&D system | Cat# CDK005 |
| EasySep human CD8+ T cell isolation kit | STEMCELL Technologies | Cat#17953 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat# 20020594 |
| Chromium Next GEM Single Cell 5’ Kit v2 | 10xgenomics | Cat#1000265 |
| Chromium Single Cell Human TCR Amplification Kit | 10xgenomics | Cat#1000252 |
| 5’ Feature Barcode Kit | 10xgenomics | Cat#1000256 |
| SPRISelect beads | Beckman Coulter | Cat# B23317 |
| DNA Clean & Concentrator-5 | Zymo | Cat#D4013 |
| Deposited data | ||
| Raw and processed sequencing data (single cell) | This paper | GEO database (GSE212998) |
| Raw and processed bulk RNA seq data | This paper | GEO database (GSE212998) |
| Experimental models: Cell lines | ||
| Raji | ATCC | CCL-86 |
| Ramos | Lingwood lab | N/A |
| HEK293T | ATCC | ATCC CRL-3216 |
| HLAKO HEK293T | This paper | N/A |
| Jurkat -76 | Davis Lab | N/A |
| Jurkat_CMV-TCR | Davis Lab | N/A |
| Jurkat_M1-TCR | Davis Lab | N/A |
| Ramos_HPV16-L2-BCR | This paper | N/A |
| Ramos_HER2-BCR | This paper | N/A |
| Ramos_SARS2-RBD-BCR | This paper | N/A |
| Jurkat_NYESO-1-TCR (1G4wt) | This paper | N/A |
| Jurkat_NYESO-1-TCR (1G4 a95:LY) | This paper | N/A |
| Jurkat-TCR5 | This paper | N/A |
| Jurkat_NYESO-1-TCR+mscarlet | This paper | N/A |
| Oligonucleotides | ||
| Primers for libraries preparation | This paper | Table S5 |
| shRNA oligo | This paper | Table S5 |
| CRISPR guide RNAs target HLA | This paper | Table S5 |
| Recombinant DNA | ||
| pMD2.G | addgene | Cat#12259 |
| psPAX2 | addgene | Cat#12260 |
| psPAX2-D64V-NC-MS2 | addgene | Cat#122944 |
| GFP-VPR | addgene | Cat#83374 |
| JWW-1 | addgene | Cat#66748 |
| pVITRO1-Trastuzumab-IgG1/κ | addgene | Cat#61883 |
| CMVTCRa_candidate5 | addgene | Cat#164999 |
| CMVTCRb_candidate5 | addgene | Cat#165000 |
| HER2 WT | addgene | Cat#16257 |
| pCCLc-MND-A0201-Mart1-SABR | addgene | Cat #119052 |
| HLA_A0101 | addgene | Cat#165009 |
| cEF.tk-GFP | addgene | Cat #33308 |
| pcDNA3.1-CD40L-mgSrtA | addgene | Cat#125795 |
| pcDNA3.1-CD40L-mutant | addgene | Cat #125796 |
| pMD2.G-VSV-G-mut | This paper | N/A |
| psPax2-MA-eGFP | This paper | N/A |
| psPax2-NC-eGFP | This paper | N/A |
| psPax2-NC-mNeon | This paper | N/A |
| plenti-sffv-tcr5-2a-hygro | This paper | N/A |
| plenti-EF1-NYESO_TCR-2a-hygro | This paper | N/A |
| PBCMV-HPV16_L2-BCR-2a-mCherry | This paper | N/A |
| plentiCMV-aCD19-Puro-2A-eGFP | This paper | N/A |
| plentiCMV-CD40L-Puro-2A-eGFP | This paper | N/A |
| plentiCMV-antigen-TM | This paper | N/A |
| plentiCMV-sp-HPV-TM | This paper | N/A |
| plentiCMV-sp-RBD-TM | This paper | N/A |
| plentiCMV-sp-HER2 | This paper | N/A |
| plentiCMV-sct-b2m-HLA-A0201 | This paper | N/A |
| plentiCMV-sct-p5-b2m-HLA-A0101 | This paper | N/A |
| plentiCMV10x-sct-b2m-HLA-A0201 | This paper | N/A |
| plentiCMV10x-sct-esp3i-b2m-HLA-A0201 | This paper | N/A |
| Software and algorithms | ||
| Cellranger | 10xgenomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| STAR | 60 | |
| Htseq-count | 61 | https://htseq.readthedocs.io/en/release_0.11.1/count.html |
| DESeq2 | 62 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Scanpy | 63 | https://scanpy.readthedocs.io/en/stable/ |
| Scirpy | 64 | https://github.com/scverse/scirpy |
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Flowjo | TreeStar | https://www.flowjo.com/ |
| Other | ||
HIGHLIGHTS.
ENTER displays ligands, deliver payloads, and records receptor specificity.
ENTER enables antigen-specific expansion or depletion of T or B cells.
ENTER-seq maps TCR specificity, clonality and cell state in single cells.
ENTER-seq of patient sample decodes antiviral T cell memory.
ACKNOWLEDGEMENT
Supported by NIH RM1-HG007735 (H.Y.C.), the Parker Institute for Cancer Immunotherapy (H.Y.C., A.T.S.), the Parker Bridge Fellow award from the Parker Institute for Cancer Immunotherapy and the V foundation (B.Y.), a Career Award for Medical Scientists from the Burroughs Wellcome Fund (A.T.S.), a Technology Impact Award from the Cancer Research Institute (A.T.S.), and an ASH Scholar Award from the American Society of Hematology (A.T.S.). J.A.B was supported by a Stanford Graduate Fellowship and the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1656518. H.Y.C. and M.M.D. are Investigators of the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS
A patent based on this work has been filed. H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio, Cartography Biosciences, Orbital Therapeutics, and is an advisor of 10x Genomics, Arsenal Biosciences, and Spring Discovery. A.T.S. is a co-founder of Cartography Biosciences and Immunai. K.R.P. is a co-founder of Cartography Biosciences. K.E.Y. is a consultant for Cartography Biosciences. J.A.B is a consultant for Foresite Capital. W.J.G. is a consultant and equity holder for 10x Genomics, Guardant Health, Quantapore and Ultima Genomics, and cofounder of Protillion Biosciences, and is named on patents describing ATAC-seq.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research. One or more of the authors identifies as a member of the LGBTQ+ community.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Davis MM, and Bjorkman PJ (1988). T-cell antigen receptor genes and T-cell recognition. Nature 334, 395–402. 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- 2.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, and Carlson CS (2009). Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 114, 4099–4107. 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joglekar AV, Leonard MT, Jeppson JD, Swift M, Li G, Wong S, Peng S, Zaretsky JM, Heath JR, Ribas A, et al. (2019). T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat Methods 16, 191–198. 10.1038/s41592-018-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kula T, Dezfulian MH, Wang CI, Abdelfattah NS, Hartman ZC, Wucherpfennig KW, Lyerly HK, and Elledge SJ (2019). T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell 178, 1016–1028.e13. 10.1016/j.cell.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee MN, and Meyerson M (2021). Antigen identification for HLA class I- and HLA class II-restricted T cell receptors using cytokine-capturing antigen-presenting cells. Sci Immunol 6, eabf4001. 10.1126/sciimmunol.abf4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li G, Bethune MT, Wong S, Joglekar AV, Leonard MT, Wang JK, Kim JT, Cheng D, Peng S, Zaretsky JM, et al. (2019). T cell antigen discovery via trogocytosis. Nat Methods 16, 183–190. 10.1038/s41592-018-0305-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birnbaum ME, Dong S, and Garcia KC (2012). Diversity-oriented approaches for interrogating T-cell receptor repertoire, ligand recognition, and function. Immunol Rev 250, 82–101. 10.1111/imr.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCutcheon M, Wehner N, Wensky A, Kushner M, Doan S, Hsiao L, Calabresi P, Ha T, Tran TV, Tate KM, et al. (1997). A sensitive ELISPOT assay to detect low-frequency human T lymphocytes. J Immunol Methods 210, 149–166. 10.1016/s0022-1759(97)00182-8. [DOI] [PubMed] [Google Scholar]
- 9.Zhang S-Q, Ma K-Y, Schonnesen AA, Zhang M, He C, Sun E, Williams CM, Jia W, and Jiang N (2018). High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat Biotechnol. 10.1038/nbt.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su F-Y, Zhao QH, Dahotre SN, Gamboa L, Bawage SS, Silva Trenkle AD, Zamat A, Phuengkham H, Ahmed R, Santangelo PJ, et al. (2022). In vivo mRNA delivery to virus-specific T cells by light-induced ligand exchange of MHC class I antigen-presenting nanoparticles. Sci Adv 8, eabm7950. 10.1126/sciadv.abm7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo X-ZJ, and Elledge SJ (2022). V-CARMA: A tool for the detection and modification of antigen-specific T cells. Proc Natl Acad Sci U S A 119, e2116277119. 10.1073/pnas.2116277119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burns JC, Friedmann T, Driever W, Burrascano M, and Yee JK (1993). Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci U S A 90, 8033–8037. 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finkelshtein D, Werman A, Novick D, Barak S, and Rubinstein M (2013). LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A 110, 7306–7311. 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nikolic J, Belot L, Raux H, Legrand P, Gaudin Y, and A Albertini A (2018). Structural basis for the recognition of LDL-receptor family members by VSV glycoprotein. Nat Commun 9, 1029. 10.1038/s41467-018-03432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jäger E, Chen YT, Drijfhout JW, Karbach J, Ringhoffer M, Jäger D, Arand M, Wada H, Noguchi Y, Stockert E, et al. (1998). Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med 187, 265–270. 10.1084/jem.187.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gotch F, Rothbard J, Howland K, Townsend A, and McMichael A (1987). Cytotoxic T lymphocytes recognize a fragment of influenza virus matrix protein in association with HLA-A2. Nature 326, 881–882. 10.1038/326881a0. [DOI] [PubMed] [Google Scholar]
- 17.Wills MR, Carmichael AJ, Mynard K, Jin X, Weekes MP, Plachter B, and Sissons JG (1996). The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J Virol 70, 7569–7579. 10.1128/JVI.70.11.7569-7579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Briney B, Inderbitzin A, Joyce C, and Burton DR (2019). Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566, 393–397. 10.1038/s41586-019-0879-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang JW, Jagu S, Wu W-H, Viscidi RP, Macgregor-Das A, Fogel JM, Kwak K, Daayana S, Kitchener H, Stern PL, et al. (2015). Seroepidemiology of Human Papillomavirus 16 (HPV16) L2 and Generation of L2-Specific Human Chimeric Monoclonal Antibodies. Clin Vaccine Immunol 22, 806–816. 10.1128/CVI.00799-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jalaguier P, Cantin R, Maaroufi H, and Tremblay MJ (2015). Selective acquisition of host-derived ICAM-1 by HIV-1 is a matrix-dependent process. J Virol 89, 323–336. 10.1128/JVI.02701-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gutierrez C, and Schiff R (2011). HER2: biology, detection, and clinical implications. Arch Pathol Lab Med 135, 55–62. 10.5858/2010-0454-RAR.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beltinger C, Fulda S, Kammertoens T, Meyer E, Uckert W, and Debatin K-M (1999). Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proceedings of the National Academy of Sciences 96, 8699–8704. 10.1073/pnas.96.15.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yonehara S, Ishii A, and Yonehara M (1989). A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J Exp Med 169, 1747–1756. 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bollard CM, Kuehnle I, Leen A, Rooney CM, and Heslop HE (2004). Adoptive immunotherapy for posttransplantation viral infections. Biol Blood Marrow Transplant 10, 143–155. 10.1016/j.bbmt.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 25.Einsele H, Kapp M, and Grigoleit GU (2008). CMV-specific T cell therapy. Blood Cells Mol Dis 40, 71–75. 10.1016/j.bcmd.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Gerdemann U, Katari UL, Papadopoulou A, Keirnan JM, Craddock JA, Liu H, Martinez CA, Kennedy-Nasser A, Leung KS, Gottschalk SM, et al. (2013). Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol Ther 21, 2113–2121. 10.1038/mt.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehmann AA, Zhang T, Reche PA, and Lehmann PV (2020). Discordance Between the Predicted Versus the Actually Recognized CD8+ T Cell Epitopes of HCMV pp65 Antigen and Aleatory Epitope Dominance. Front Immunol 11, 618428. 10.3389/fimmu.2020.618428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lübke M, Spalt S, Kowalewski DJ, Zimmermann C, Bauersfeld L, Nelde A, Bichmann L, Marcu A, Peper JK, Kohlbacher O, et al. (2020). Identification of HCMV-derived T cell epitopes in seropositive individuals through viral deletion models. J Exp Med 217, e20191164. 10.1084/jem.20191164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Solache A, Morgan CL, Dodi AI, Morte C, Scott I, Baboonian C, Zal B, Goldman J, Grundy JE, and Madrigal JA (1999). Identification of three HLA-A*0201-restricted cytotoxic T cell epitopes in the cytomegalovirus protein pp65 that are conserved between eight strains of the virus. J Immunol 163, 5512–5518. [PubMed] [Google Scholar]
- 30.Billerbeck E, Blum HE, and Thimme R (2007). Parallel expansion of human virus-specific FoxP3− effector memory and de novo-generated FoxP3+ regulatory CD8+ T cells upon antigen recognition in vitro. J Immunol 179, 1039–1048. 10.4049/jimmunol.179.2.1039. [DOI] [PubMed] [Google Scholar]
- 31.Churlaud G, Pitoiset F, Jebbawi F, Lorenzon R, Bellier B, Rosenzwajg M, and Klatzmann D (2015). Human and Mouse CD8(+)CD25(+)FOXP3(+) Regulatory T Cells at Steady State and during Interleukin-2 Therapy. Front Immunol 6, 171. 10.3389/fimmu.2015.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fontenot JD, Gavin MA, and Rudensky AY (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4, 330–336. 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 33.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, and Sakaguchi S (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275. 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 34.Vieyra-Lobato MR, Vela-Ojeda J, Montiel-Cervantes L, López-Santiago R, and Moreno-Lafont MC (2018). Description of CD8+ Regulatory T Lymphocytes and Their Specific Intervention in Graft-versus-Host and Infectious Diseases, Autoimmunity, and Cancer. J Immunol Res 2018, 3758713. 10.1155/2018/3758713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, Wheeler DK, Sette A, and Peters B (2019). The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 47, D339–D343. 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GMA, Papagno L, Ogg GS, King A, Lechner F, Spina CA, et al. (2002). Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med 8, 379–385. 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 37.Derhovanessian E, Maier AB, Hähnel K, Beck R, de Craen AJM, Slagboom EP, Westendorp RGJ, and Pawelec G (2011). Infection with cytomegalovirus but not herpes simplex virus induces the accumulation of late-differentiated CD4+ and CD8+ T-cells in humans. J Gen Virol 92, 2746–2756. 10.1099/vir.0.036004-0. [DOI] [PubMed] [Google Scholar]
- 38.Wildt S, and Gerngross TU (2005). The humanization of N-glycosylation pathways in yeast. Nat Rev Microbiol 3, 119–128. 10.1038/nrmicro1087. [DOI] [PubMed] [Google Scholar]
- 39.Wolfert MA, and Boons G-J (2013). Adaptive immune activation: glycosylation does matter. Nat Chem Biol 9, 776–784. 10.1038/nchembio.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garboczi DN, Hung DT, and Wiley DC (1992). HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci U S A 89, 3429–3433. 10.1073/pnas.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis MM, and Boyd SD (2019). Recent progress in the analysis of αβT cell and B cell receptor repertoires. Curr Opin Immunol 59, 109–114. 10.1016/j.coi.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchholz CJ, Friedel T, and Büning H (2015). Surface-Engineered Viral Vectors for Selective and Cell Type-Specific Gene Delivery. Trends Biotechnol 33, 777–790. 10.1016/j.tibtech.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 43.Srivastava A (2016). In vivo tissue-tropism of adeno-associated viral vectors. Curr Opin Virol 21, 75–80. 10.1016/j.coviro.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bentzen AK, and Hadrup SR (2017). Evolution of MHC-based technologies used for detection of antigen-responsive T cells. Cancer Immunol Immunother 66, 657–666. 10.1007/s00262-017-1971-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore MD, and Hu WS (2009). HIV-1 RNA dimerization: It takes two to tango. AIDS Rev 11, 91–102. [PMC free article] [PubMed] [Google Scholar]
- 46.Stano A, Leaman DP, Kim AS, Zhang L, Autin L, Ingale J, Gift SK, Truong J, Wyatt RT, Olson AJ, et al. (2017). Dense Array of Spikes on HIV-1 Virion Particles. J Virol 91, e00415–17. 10.1128/JVI.00415-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chandran SS, and Klebanoff CA (2019). T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol Rev 290, 127–147. 10.1111/imr.12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hinrichs CS, and Rosenberg SA (2014). Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev 257, 56–71. 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu Z, Ott PA, and Wu CJ (2018). Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol 18, 168–182. 10.1038/nri.2017.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ju B, Zhang Q, Ge J, Wang R, Sun J, Ge X, Yu J, Shan S, Zhou B, Song S, et al. (2020). Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 584, 115–119. 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- 51.Lee DSW, Rojas OL, and Gommerman JL (2021). B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discov 20, 179–199. 10.1038/s41573-020-00092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waldman AD, Fritz JM, and Lenardo MJ (2020). A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 20, 651–668. 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J, Medaer R, Stinissen P, Hafler D, and Raus J (1993). MHC-restricted depletion of human myelin basic protein-reactive T cells by T cell vaccination. Science 261, 1451–1454. 10.1126/science.7690157. [DOI] [PubMed] [Google Scholar]
- 54.Purcell AW, Ramarathinam SH, and Ternette N (2019). Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat Protoc 14, 1687–1707. 10.1038/s41596-019-0133-y. [DOI] [PubMed] [Google Scholar]
- 55.Reynisson B, Alvarez B, Paul S, Peters B, and Nielsen M (2020). NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res 48, W449–W454. 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobson CS, Reich AN, Gaglione S, Smith BE, Kim EJ, Dong J, Ronsard L, Okonkwo V, Lingwood D, Dougan M, et al. (2022). Antigen identification and high-throughput interaction mapping by reprogramming viral entry. Nat Methods 19, 449–460. 10.1038/s41592-022-01436-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma K-Y, Schonnesen AA, He C, Xia AY, Sun E, Chen E, Sebastian KR, Guo Y-W, Balderas R, Kulkarni-Date M, et al. (2021). High-throughput and high-dimensional single-cell analysis of antigen-specific CD8+ T cells. Nat Immunol 22, 1590–1598. 10.1038/s41590-021-01073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, Such L, Furness AJS, McGranahan N, Rosenthal R, et al. (2016). Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat Biotechnol 34, 1037–1045. 10.1038/nbt.3662. [DOI] [PubMed] [Google Scholar]
- 59.Rodenko B, Toebes M, Hadrup SR, van Esch WJE, Molenaar AM, Schumacher TNM, and Ovaa H (2006). Generation of peptide–MHC class I complexes through UV-mediated ligand exchange. Nat Protoc 1, 1120–1132. 10.1038/nprot.2006.121. [DOI] [PubMed] [Google Scholar]
- 60.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15. 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sturm G, Szabo T, Fotakis G, Haider M, Rieder D, Trajanoski Z, and Finotello F (2020). Scirpy: a Scanpy extension for analyzing single-cell T-cell receptor-sequencing data. Bioinformatics 36, 4817–4818. 10.1093/bioinformatics/btaa611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, Xu H, Morgan RA, Feldman SA, Johnson LA, et al. (2008). Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol 180, 6116–6131. 10.4049/jimmunol.180.9.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pasqual G, Chudnovskiy A, Tas JMJ, Agudelo M, Schweitzer LD, Cui A, Hacohen N, and Victora GD (2018). Monitoring T cell-dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature 553, 496–500. 10.1038/nature25442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang H, Sun M, Wang J, Zeng B, Cao X, Han Y, Tan S, and Gao GF (2021). Identification of NY-ESO-1157–165 Specific Murine T Cell Receptors With Distinct Recognition Pattern for Tumor Immunotherapy. Front Immunol 12, 644520. 10.3389/fimmu.2021.644520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Guzman RN, Wu ZR, Stalling CC, Pappalardo L, Borer PN, and Summers MF (1998). Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element. Science 279, 384–388. 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- 69.Kutluay SB, Zang T, Blanco-Melo D, Powell C, Jannain D, Errando M, and Bieniasz PD (2014). Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell 159, 1096–1109. 10.1016/j.cell.2014.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu X, Liu H, Xiao H, Kim J, Seshaiah P, Natsoulis G, Boeke JD, Hahn BH, and Kappes JC (1995). Targeting foreign proteins to human immunodeficiency virus particles via fusion with Vpr and Vpx. J Virol 69, 3389–3398. 10.1128/JVI.69.6.3389-3398.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Burnie J, and Guzzo C (2019). The Incorporation of Host Proteins into the External HIV-1 Envelope. Viruses 11, E85. 10.3390/v11010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burnie J, Tang VA, Welsh JA, Persaud AT, Thaya L, Jones JC, and Guzzo C (2020). Flow Virometry Quantification of Host Proteins on the Surface of HIV-1 Pseudovirus Particles. Viruses 12, E1296. 10.3390/v12111296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cantin R, Fortin JF, and Tremblay M (1996). The amount of host HLA-DR proteins acquired by HIV-1 is virus strain- and cell type-specific. Virology 218, 372–381. 10.1006/viro.1996.0206. [DOI] [PubMed] [Google Scholar]
- 74.Chertova E, Chertov O, Coren LV, Roser JD, Trubey CM, Bess JW, Sowder RC, Barsov E, Hood BL, Fisher RJ, et al. (2006). Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J Virol 80, 9039–9052. 10.1128/JVI.01013-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grover JR, Veatch SL, and Ono A (2015). Basic motifs target PSGL-1, CD43, and CD44 to plasma membrane sites where HIV-1 assembles. J Virol 89, 454–467. 10.1128/JVI.02178-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259. 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Crooks GE, Hon G, Chandonia J-M, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Flow cytometry analysis of the viral binding and fusion of scFv-CD19 displayed viruses on Raji B cells at different temperature before and after proteinase K treatment.
B. Bar plot showing the percentage of GFP+ cells from Figure S1A.
C. Schematic view of experimental set up. CD40 expressing Raji B cells were incubated with GFP viruses displaying either wild-type CD40 ligand (CD40L) or CD40L mutant (K142E, R202E) with decreased binding to its cognate receptor CD40.
D. Flow cytometry analysis of GFP signal in Raji B cells upon incubation with either wild-type CD40L or mutant CD40L displayed GFP viruses.
E. Bar plot showing the percentage of GFP+ cells from Figure S1C–D.
F. Schematic view of immunocapture assay. Magnetic beads were conjugated with anti-CD40L, anti-VSVG, and IgG antibodies and then incubated with CD40L displayed viruses. Viruses that were immunocaptured were subjected to viral RNA isolation and qRT-PCR of CD40L.
G. Bar plot showing the qRT-PCR result of enrichment of ENTER viruses by different antibody conjugated beads as in Figure S1F.
Data are represented as mean +/− SEM in B and G from two biological replicates, and in E from triplicates.
P-values were calculated by unpaired t-test. n.s. not significant p>0.05; * p<0.05; *** p<0.001
A. Flow cytometry analysis of HLA-A2 and B2M surface expression in wild-type HEK293T, HLA-KO HEK293T, and HLA-A2 reconstituted HLA-KO HEK293T cells.
B. Flow cytometry plots of CMV-pp65495–503-TCR T cells upon 2-hour incubation of HLA-A2-peptide displayed GFP viruses followed by staining of PE-tetramers. M1(58–66) (flu antigen) displayed viruses and M1(58–66) tetramers are negative controls.
C. Histogram plots (left) and Bar plots (right) showing tetramer intensity and CD3 surface expression of CMV-pp65495–503-TCR T cells.
D. Schematic view of experimental design (top) and flow cytometry analysis (bottom) of T cell mixing experiments. Flu-m158–66-TCR T cells were labeled by Cell Trace Violet dye and then mixed with NY-ESO-1157–165-TCR T cells at different ratio. Mixed T cell population was incubated with HLA-A2:m1 displayed GFP viruses for 2h and then subjected to flow cytometry. Representative flow cytometry plot showing mixing of two T cell population and GFP signal of T cells gated on Violet+ and Violet− population.
E. Bar plots showing sensitivity (left) and specificity (right) of ENTER from Figure S2D.
Data are represented as mean +/− SEM in C from two biological replicates and in E from triplicates.
A. Flow cytometry analysis of GFP+ cells among HER2-BCR+ B cells that were incubated with HER2 displayed viruses or RBD displayed viruses.
B. Bar plot showing the percentage of GFP+ cells as in Figure S3A.
C. Schematic view of experimental design (top) and flow cytometry analysis (bottom) of B cell mixing experiments. RBD-BCR+ B cells were labeled by Cell Trace violet and mixed with HPV-BCR+ B cells at different ratios. Mixed B cell population was incubated with GFP viruses displaying RBD antigen fused with ICAM1 TM domain and then subjected to flow cytometry. Representative flow cytometry plot showing mixing of two B cell population and GFP signal of B cells gated on Violet+ and Violet− population.
D. Bar plot showing the sensitivity of ENTER from Figure S3C.
E. Bar plot showing the specificity of ENTER from Figure S3C.
F. Bar plot showing the delivery efficiency of viruses with wild-type VSV-G in cell types with different antigen specificity. P-values were calculated by unpaired t-test. n.s. not significant P>=0.05.
G. Flow cytometry analysis of cargo delivery in T cell mixing experiment. mScarlet expressed NY-ESO-1 TCR+ T cells were mixed with CMV-pp65 TCR T cells and then infected with ENTER viruses displaying pp65495–503 antigen and carrying transgene containing HSV-TK and GFP.
H. Flow cytometry analysis of cargo delivery in T cell mixing experiment. mCherry expressed RBD BCR+ B cells were mixed with HER2 BCR+ B cells and then infected with ENTER viruses displaying HER2 antigen and carrying transgene containing HSV-TK and GFP.
I. Histogram plot showing the surface expression of FAS in T cells transduced with diverse FAS shRNAs or control shRNA.
J. Representative flow plot showing the Annexin V and 7-AAD gating of T cells upon anti-FAS induced apoptosis and cell death.
Data are represented as mean +/− SEM in B, D and E from triplicates, and in F from two replicates.
A. Flow cytometry analysis of GFP signal in T cells expressing TCRs (NY-ESO-1 TCR or CMV pp65-TCR) upon incubation with NY-ESO-1157–165 antigen peptide displayed GFP viruses whose viral RNA are either intact or inserted with sequencing capture tag.
B. Bar plot showing the percentage of GFP+ cells from Figure S4A.
C. Schematic view of experimental design. Donor isolated T cells were incubated with pp65495–503 displayed viruses carrying either GFP or mNeon fluorescence proteins and then stained with pp65495–503 tetramer and other antibodies followed by flow cytometry. We first gated on CD3+ CD8+ T cells to measure pp65-specific T cells using tetramer as a positive control, and then monitored GFP signals in pp65 tetramer+ T cells.
D. Flow cytometry analysis of tetramer and GFP signal in primary human T cells from donors with CMV infection as in Figure S4C.
E. Bar plot showing the percentage of GFP+ and GFP− cells among pp65 tetramer+ T cells from Figure S4D.
F. Representative flow plot showing the GFP+ cells among pp65 tetramer+ T cells after incubation with pp65495–503 viruses or negative control viruses (NY-ESO-1157–165)
G. Schematic view of experimental design. PBMCs were isolated from CMV seropositive HLA-A2+ donors and cultured with a pool of twelve different CMV antigen peptides (10 ug/mL) for 10 days. Primary T cells prior or post peptide-induced expansion were incubated with pp65 antigen displayed mNeon viruses for 2 hours and then stained with antibodies followed by flow cytometry.
H. Representative flow plot showing co-staining of pp65495–503 tetramer and pp65495–503 displayed mNeon viruses in pp65495–503 peptide enriched T cells post 15 days expansion. These T cells are gated on live CD8+ CD3+ T cells.
I. Representative flow cytometry plot showing the percentage of GFP+ T cells (stained by 12 pooled HLA-A2:CMV-antigen mNeon viruses) from 4 different CMV seropositive donors prior or post pooled CMV peptide-induced expansion as in Figure S4G.
J. MA plot showing the bulk RNA-seq analysis of CMV-pp65 TCR T cells incubated with pp65495–503 tetramer or pp65495–503 displayed ENTER viruses. Genes with log2 fold change and adjusted p value <0.01 are highlighted in red.
Data are represented as mean +/− SEM in B from two replicates and E from triplicates.
P-values were calculated by unpaired t-test. n.s. P>=0.05 * p<0.05
A. UMAP plots showing CMV-specific T cells (yellow) and bystander T cells (blue) via clustering before or after removal of ENTER-induced gene signature.
B. UMAP plot showing 10 clusters of human CD8 T cell subsets
C. UMAP plots showing amount of surface protein CD127 from CITE-seq, and expression of genes for naïve T cells (CCR7, SELL, and LEF1).
D. UMAP plots showing gene expression of cytolytic molecules.
E. UMAP plots showing gene expression of markers for MAIT cells
F. UMAP plots showing expression of marker genes for each subset/cluster.
G. UMAP plot showing donor origin.
H. Fraction of T cell subsets from 10 clusters in CMV antigen-specific T cells (ENTER+) and bystander T cells (ENTER−), separated by donor origin.
A. UMAP plots showing the clonal expansion size of CMV antigen-specific T cells (ENTER+) and bystander T cells (ENTER−).
B. TCR clonotypes of CMV antigen-specific T cells colored by donor. Each circle represents a clonotype with identical CDR3 nucleotide sequences. The size of circle represents the number of cells in each clonotype.
C. Scatter plots showing the correlation of clonal expansion size with cytotoxic gene score.
D. Scatter plots showing the correlation of clonal expansion size with exhaustion gene score or activation gene score.
E. A summary table of pp65495–503-specific TCR clones showing convergent TCR clonotypes with identical CDR3 amino acid sequences
F. Summary tables of US874–82 - and UL100200–208 -specific TCR clones.
G. Fraction of T cell subsets in pp65495–503-specific TCR clones, separated by CDR3 clones and colored by 10 clusters.
Coefficient r and p values in Figure S6C–D are calculated by Pearson correlation.
A. UMAP plots showing the expression of key genes associated with diverse functions (type-I IFN ISG, cytotoxicity, chemokine, and transcription factors)
B. UMAP plots showing the subset clustering of primary T cells directly isolated from patient blood sample before and after removal of ENTER-induced gene signature
C. UMAP embedding density plots showing the density of CMV antigen-specific T cells for different CMV antigen epitopes.
D. Bar plot showing the percentage of US874–82-specific T cells with shared TCR before and after peptide induced expansion.
E. The number of US874–82-specific T cells isolated from fresh PBMC before expansion, separated by donor and colored by TCR CDR3 sequence. TCR sequence shared before and after expansion are circled in black.
F. A summary table of top TCR clones of US874–82-specific T cells (identified from E) with cell number and frequency metrics before and after expansion.
G. The number of T cell subsets in pp65495–503-specific TCR clones, separated by CDR3 clones and colored by 13 clusters.
H. Heatmap showing the expression of genes associated with diverse functions among different TCR clone-specific T cells.
I. Density plots showing the expression of IL13 and IL4 in different TCR clone-specific T cells before and after peptide-induced expansion.
Data Availability Statement
All sequencing data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of publication. Accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.